[level-membership-for-allergy-and-immunology-category]
Chapter 17 AIDS, Secondary Immunodeficiency and Immunosuppression
• Nutrient deficiencies often lead to impaired immune responses. Malnutrition increases the risk of infant mortality from infection through reduction in cell-mediated immunity, including reduced numbers and function of CD4+ helper cells and a reduction in levels of secretory IgA. Trace elements, iron, selenium, copper, and zinc are also important in immunity. Lack of these elements can lead to diminished neutrophil killing of bacteria and fungi, susceptibility to viral infections, and diminished antibody responses. Vitamins A, B6, C, E, and folic acid are likewise important in overall resistance to infection. Proper diet and nutrition, therefore, reduce morbidity and mortality caused by infection.
• Some drugs selectively alter immune function. Immunomodulatory drugs can severely depress immune functions. These drugs are often necessary to treat solid organ transplant patients and those with an autoimmune disease. Although necessary in such settings, these drugs are often broad acting, thereby increasing patients’ susceptibility to a broad array of opportunistic infections caused by viruses, bacteria and fungi.
• HIV is a primary cause of immunodeficiency. Human immunodeficiency virus (HIV) is a retrovirus that predominantly targets CD4+ T cells. Acute infection depletes CD4 T cell subsets and transiently suppresses circulating CD4 T cell numbers before the immune system establishes partial control of the virus and the chronic phase of infection begins. Though patients can remain in the chronic phase for an average of 10 years, without anti-retroviral drug treatment, CD4 T cell levels gradually fall, resulting in loss of cell-mediated immunity and susceptibility to life-threatening opportunistic infections. This final stage, AIDS (acquired immunodeficiency syndrome), is marked by low CD4 T cell counts, high HIV plasma levels, reactivation of other latent infections, and often, virus-associated malignancies such as Kaposi’s sarcoma and non-Hodgkin’s lymphoma.
• Combination therapy for AIDS with inhibitors of HIV reverse transcriptase, protease, and entry are reasonably successful, but associated with long-term toxicities in almost 50% of persons. An effective vaccine remains an elusive goal, in part due to the rapid mutation rate of the virus during reverse transcription.
Overview
Nutrient deficiencies
Infection and malnutrition can exacerbate each other
• increasing metabolic demands;
• decreasing appetite, thereby lowering intake of nutrients; and
• with gastrointestinal infection, decreasing nutrient absorption.
Once this cycle begins, it is self-propagating as infection compromises immunity, which then leads to more infection and debility (Fig. 17.1). At a population level, this may lead to decreased productivity, further decreasing economic and food resources and, again, driving the malnutrition and immune deficiency loop.
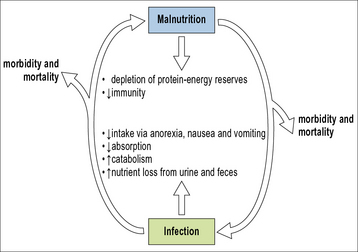
Protein–energy malnutrition and lymphocyte dysfunction
Nutrition also affects innate mechanisms of immunity
Poor nutrition also causes deficits in innate immune defenses, for example,
• a larger number of bacteria bind to epithelial cells of malnourished subjects;
• the production of certain inflammatory cytokines, such as IL-2 and TNFα, is decreased;
• opsonization is decreased, largely because of a reduction in levels of various complement components – C3, C5, and factor B.
Deficiencies in trace elements impact immunity

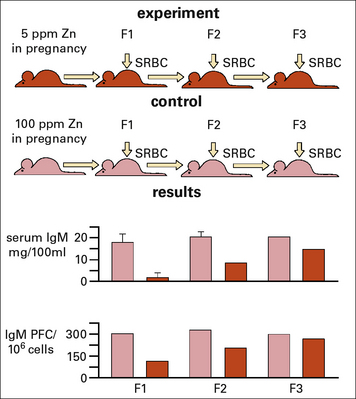
Zinc deficiency during pregnancy has also been shown to have an inter-generational effect on the levels of IgM and IgM producing B cells in the pups, and more surprisingly even in the 2nd generation (Fig. 17.w1).
Q. In what ways can neutrophils and macrophages restrict iron availability for microorganisms?
A. Soluble proteins such as lactoferrin produced by neutrophils reduce the availability of free iron in the phagolysosome. Macrophages have ion pumps (e.g. nRAMP) in their phagosomal membrane that remove iron from the phagosome (see Chapter 15).
Vitamin deficiencies and immune function
Q. NFATc1, a member of the NFAT transcription factor family, is required for development of the B-1 B cell subset. Recent experiments using mice have shown that vitamin A deficiency severely reduces NFAT-c1 expression resulting in loss of the B-1 population. Why might this contribute to defects in mucosal immunity?
Until the advent of antibiotics, cod liver oil and sunlight, both sources of vitamin D, were used as primary treatments for TB. Vitamin D deficiency can lead to increased infection rates and recent studies have begun to elucidate some of the molecular mechanisms behind the anti-infective role of vitamin D. Many cell types express the vitamin D receptor (VDR), and while vitamin D metabolites may modulate adaptive immune responses, they can also enhance innate immunity. Importantly, particularly for TB, signaling via the VDR may enhance both cathelicidin and defensin expression, thus boosting macrophage anti-microbial activity (Chapter 7).
Immunodeficiency secondary to drug therapies
Several classes of drugs suppress immune function, either intentionally for therapeutic effect, or as an unwanted side effect.. For example, patients receiving organ transplants usually receive a variety of immunosuppressants to prevent rejection of the donor tissue and to treat graft-versus-host disease (GvHD, discussed in Chapter 21). Likewise, patients presenting with severe inflammatory, allergic, or autoimmune reactions often require therapeutic immunosuppression (Chapter 20 and Section 5). Pharmacological treatments that suppress immunity as a side effect include cancer treatments such as cytotoxic or anti-metabolite reagents that can also severely depress bone marrow hematopoiesis. Below, we will examine the different classes of immunosuppressive drugs commonly used and their impact on immune function.
Iatrogenic immune suppression post-organ transplantation
Due to genetic differences causing the immune system to perceive donor organs as foreign, recipients of organ transplants receive immunosuppressive regimens, often long-term. Essentially the goal of these treatments is to prevent an immune response against either the host or donor tissues while minimizing toxic side effects and susceptibility of the patient to infection. The primary effectors for both donor organ rejection and GvHD are T lymphocytes. Therefore, both prophylactic and therapeutic immunosuppressant drugs target the T cell branch of the immune system. We briefly summarize transplantation immunology and drugs that help prevent rejection below and Chapter 21 covers these subjects in more detail.
Approaches to suppress T cell-mediated damage include interfering with:
Drugs such as cyclosporin A and tacrolimus bind to cellular immunophilins and as a complex inhibit calcineurin (see Fig. 8.w1 ). This blockade dampens T cell signaling mediated by NFAT translocation and, in turn, decrease IL-2 and IFNγ production, impairing both T cell activation and proliferation. The drug sirolimus also binds an immunophilin; however, this interaction results in inhibition of the response to, rather than the production of IL-2, again blocking both proliferation and activation in some lymphocyte subsets.
). This blockade dampens T cell signaling mediated by NFAT translocation and, in turn, decrease IL-2 and IFNγ production, impairing both T cell activation and proliferation. The drug sirolimus also binds an immunophilin; however, this interaction results in inhibition of the response to, rather than the production of IL-2, again blocking both proliferation and activation in some lymphocyte subsets.
Glucocorticoids are powerful immune modulators
Patients can receive glucocorticoids:
• systemically, for example, during the early period immediately post-organ transplant (Chapter 21);
• locally, for example as an inhalant for treatment of asthma (Chapter 23);
• topically, for example as for treatment of poison ivy-induced contact hypersensitivity (Chapter 23).
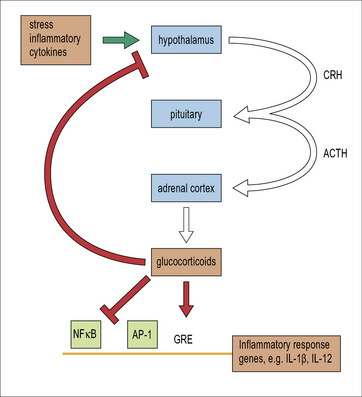
Glucocorticoids are naturally occurring steroids produced by the adrenal cortex. In response to chronic stress or to inflammatory cytokines, a cascade of hormone signals originating in the hypothalamus drives adrenal production of the immunomodulatory steroid, cortisol (Fig. 17.2 and see Fig. 11.17). Cortisol and its analogs are small steroid hormones that readily cross the cellular membrane and bind cytosolic glucocorticoid receptors. Activated glucocorticoid receptors enter the nucleus and can either directly bind DNA to affect gene transcription or regulate expression by disrupting other transcription factor complexes such as NFκB and AP-1 (Chapter 8).
Q. How would suppression of Fyn and Lck affect T cell responses?
A. During TCR signaling, both Lck and Fyn are important in activation of both phospholipase C and the MAP kinase cascades (see Fig. 8.w1 ). Thus, prevention of tyrosine kinase activity would suppress T cell activation in response to antigens.
). Thus, prevention of tyrosine kinase activity would suppress T cell activation in response to antigens.
Functional effects of steroid treatment
Within the adaptive branch of immunity, profound downregulation of the inflammatory cytokines and the response of T cells to these cytokines preferentially shift the adaptive immune profile from TH1 toward a TH2-type (Chapter 11). In particular, glucocorticoids suppress both DC production of IL-12 and T cell expression of the IL-12 receptor. In contrast, the effect of corticosteroids on B cell responses is less profound. Thus, overall, humoral immune responses dominate cell-mediated responses during glucocorticosteroid treatment.
Other causes of secondary immunodeficiencies
Human immunodeficieny virus causes AIDS
There are two main variants, HIV-1 and HIV-2:
• HIV-2 is endemic in West Africa and appears to be less pathogenic;
• HIV-1 has several subtypes (or clades), which are designated by the letters A through K, and the prevalence of the different clades varies by geographical region – over 90% of people infected with HIV-1 live in developing countries and spread is 80% by the sexual route.
HIV life cycle
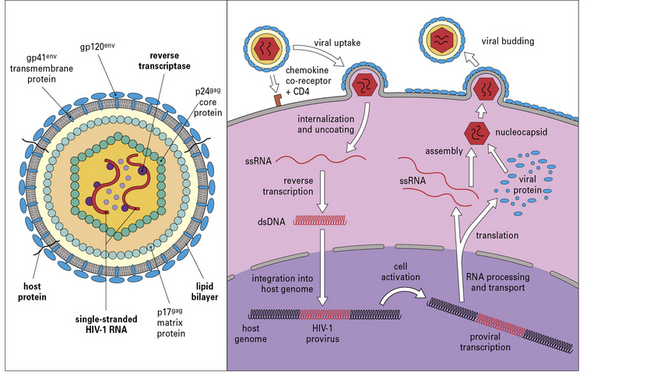
The HIV genome contains gag (core proteins), pol (reverse transcriptase, protease, and integrase enzymes), and env (envelope protein) genes (Fig. 17.3). In addition to these three main gene products, the virus encodes six regulatory and accessory proteins (Tat, Rev, Vpr, Vpu, Vif, and Nef). Alternatively spliced transcripts with overlapping open reading frames allow the coordinated expression of these proteins from the compact HIV genome.
Viral latency is associated with chronic infection
Without anti-retroviral treatment, HIV levels peak 3–4 weeks post-infection, then gradually drop and plateau (Fig. 17.4). This reflects a combination of the decrease in readily available activated targets and, perhaps more importantly, control by the innate and adaptive immune responses. There is usually a moderate rebound in circulating CD4 T cell numbers at this point, though recent studies indicate GALT CD4 T cell populations do not recover. Simultaneously, the virus establishes stable viral reservoirs. This first consists of cells supporting low-level viral replication in lymphoid and other tissues, likely with efficient cell-to-cell propagation of the virus. The second reservoir is within CD4 T cells in which the HIV genome is integrated as a provirus, yet remains latent as a silent infection without viral protein transcription. Subsequent T cell activation can then stimulate virus production.
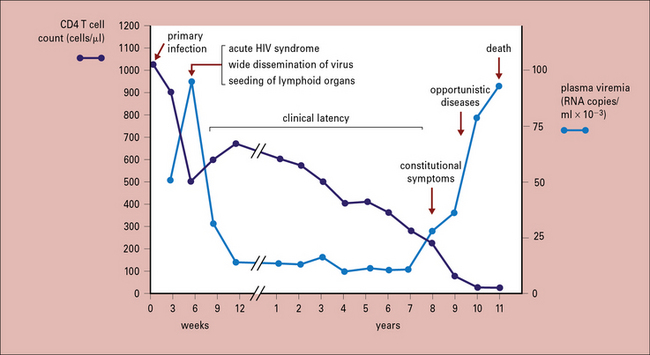
Immune dysfunction results from the direct effects of HIV and impairment of CD4 T cells
The chronic phase of HIV infection is marked by persistent generalized immune activation. Infected persons present with B cell polyclonal activation and hypergammaglobulinemia. Attachment of gp120 to mannose binding lectin and to subsets of Ig+ B cells contributes to this activation. Inflammatory cytokines such as IFNα, IFNγ, IL-18, IL-15, and TNFα are elevated during acute infection. Finally, increased translocation of bacteria through the gut barrier, due to extensive HIV infection within the GALT and lamina propria, increases circulating LPS levels, activating many immune effectors via Toll-like receptors. In patients with high viral load, HIV-specific CTL typically contain low levels of intracellular perforin (see Figs 10.10 and 10.12) and have a poor proliferative capacity. During this chronic phase, CD8 T cells often express PD-1, a receptor associated with programmed cell death (Chapter 8). The points above are all consistent with persistent activation and eventual exhaustion of the supply of anti-HIV CD8 T cells that worsens over time.
AIDS is the final stage of HIV infection and disease
Progression to this clinical stage includes a CD4 T cell count of <200/μL. As blood CD4 T cell counts gradually decline during the chronic phase, patients become susceptible to opportunistic infection and malignancies (see Fig. 17.4). Below 500 CD4 cells/μL, less severe conditions such as oral candidiasis, recurrent herpes virus outbreaks (e.g. shingles from varicella zoster virus and anogenital herpes from herpes simplex virus), and pneumococcal infections occur. CD4 T cell levels below 200/μL are associated with increased risk of life-threatening infections and malignancies including Pneumocystis jirovecii pneumonia and Kaposi’s sarcoma, respectively. With CD4 levels below 50/μL, patients become vulnerable to additional systemic infection with organisms such as Mycobacterium avium complex. The three main organ systems affected are the respiratory system, gastrointestinal tract, and central nervous system.
Pneumonia Pneumocystis jirovecii (previously P. carinii) is the most common opportunistic respiratory infection (Fig. 17.5), but pulmonary bacterial infections, including Mycobacterium tuberculosis, also occur. Protozoa (cryptosporidia and microsporidia) are the most common pathogens isolated in patients with diarrhoea and weight loss (see Fig. 17.5). Enteric bacteria such as Salmonella and Campylobacter spp. may also afflict AIDS patients.
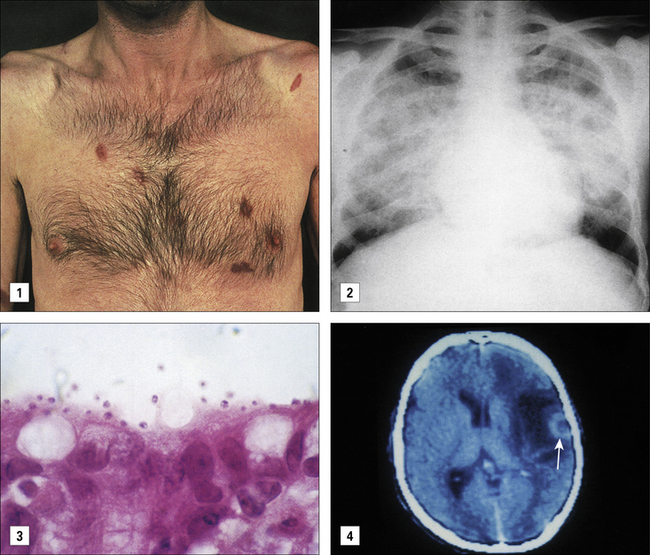
Neurological complications in AIDS are due to direct effects of HIV infection, opportunistic infections, or lymphoma. AIDS-related dementia once affected between 10–40% of patients with other manifestations of AIDS, but with more effective antiviral treatment has become less common. Neurological involvement can be due to a number of pathogens. Cryptococcus neoformans is a fungus and is the most common cause of AIDS-related meningitis. Toxoplasmosis, a protozoal infection, causes cysts in the brain and neurological deficit (see Fig. 17.5). Cytomegalovirus reactivation may cause inflammation of the retina, brain, and spinal cord and its nerve roots, and a polyomavirus (JC virus), which infects oligodendrocytes in the brain, produces a rapidly fatal demyelinating disease – progressive multifocal leukoencephalopathy.
Kaposi’s sarcoma (KS), caused by infection with KS-associated herpes virus (KSHV), is the most common AIDS-associated malignancy (Fig. 17.5). KSHV infections, similar to CMV herpesvirus infections, are often asymptomatic in individuals with competent T cell immunity. With HIV co-infection, however, KSHV titers increase and KS emerges with multifocal lesions (see Fig. 17.5) of mixed cellularity often resulting in widespread involvement of skin, mucous membranes, viscera (gut and lungs) and lymph nodes. KSHV infection can also lead to development of B cell lymphomas, affecting the brain, gut, bone marrow, lymph nodes, spleen and body cavities such as the pericardial and pleural spaces. KSHV also causes two B cell lymphoproliferative diseases, multicentric Castleman’s disease and primary effusion lymphoma.
An effective vaccine remains an elusive goal
Critical thinking: Secondary immunodeficiency (see p. 438 for explanations)
On examination he was underweight and had enlarged lymph nodes in the neck, axillae, and groin. Plaques of Candida albicans were visible in his throat. There were abnormal breath sounds in his lungs. The results of his blood tests are shown in Table 1.
| Investigation | Result (normal range) |
|---|---|
| hemoglobin (g/dL) | 12.8 (13.5–18.0) |
| platelet count (× 109/L) | 128 (150–400) |
| white cell count (× 109/L) | 6.2 (4.0–11.0) |
| neutrophils (× 109/L) | 5.4 (2.0–7.5) |
| eosinophils (× 109/L) | 0.24 (0.4–0.44) |
| total lymphocytes (× 109/L) | 0.75 (1.6–3.5) |
| T lymphocytes CD4+ (× 109/L) CD8+ (× 109/L) |
0.12 (0.7–1.1) 0.42 (0.5–0.9) |
| B lymphocytes (× 109/L) | 0.11 (0.2–0.5) |
| arterial blood gases PaO2 (kPa) PaCO2 (kPa) pH HCO3 base excess |
7.8 (>10.6) 5.52 (4.7–6.0) 7.39 (7.35–7.45) 25.6 –0.9 |
| ECG | normal |
| chest radiography | bilateral diffuse interstitial shadowing |
| bronchoscopy with bronchoalveolar lavage | positive for Pneumocystis jirovecii |
Because of his sexual history, the patient was counseled about having a human immunodeficiency virus (HIV) test, and consented. An enzyme-linked immunosorbent assay (ELISA, see Method box 3.2, Fig. 2 ) was positive for anti-HIV antibodies and a polymerase chain reaction (PCR) demonstrated HIV-1 RNA in the plasma.
) was positive for anti-HIV antibodies and a polymerase chain reaction (PCR) demonstrated HIV-1 RNA in the plasma.
Within 3 months he was seen again in accident and emergency with blurred vision and ‘flashing lights’ in his eyes. He was shown to have an infection of his retina with cytomegalovirus and was treated with injections of ganciclovir. The CD4 count at this time was 0.04 × 109/L. While receiving this treatment the patient became increasingly unwell and semiconscious. Investigations at this time are shown Table 2.
Table 2 Results of investigations 3 months after presentation
| Investigation | Result (normal range) |
|---|---|
| hemoglobin (g/dL) | 10.4 (13.5–18.0) |
| platelet count (× 109/L) | 104 (150–400) |
| white cell count (× 109/L) | 4.1 (4.0–11.0) |
| neutrophils (× 109/L) | 4.2 (2.0–7.5) |
| eosinophils (× 109/L) | 0.24 (0.4–0.44) |
| total lymphocytes (× 109/L) | 0.62 (1.6–3.5) |
| T lymphocytes CD4+ (× 109/L) CD8+ (× 109/L) |
0.03 (0.7–1.1) 0.40 (0.5–0.9) |
| B lymphocytes (× 109/L) | 0.09 (0.2–0.5) |
| chest radiography | minimal areas of diffuse shadowing |
| blood culture | negative |
| blood glucose (mmol/L) | 7.6 (<10.0) |
| CSF from lumbar puncture Appearance |
turbid |
| white cells (polymorphs/mm3) | 2500 |
| protein (g/L) | 4.2 (0.15–0.45) |
| glucose (mmol/L) | 4.5 (> 60% blood glucose) |
| Indian ink stain | positive for cryptococcus |
Altfeld M., Allen T.M., Yu X.G., et al. HIV-1 superinfection despite broad CD8+ T cell responses containing replication of the primary virus. Nature. 2002;420:434–439.
Brenchley J.M., Schacker T.W., Ruff L.E., et al. CD4+ T cell depletion during all stages of HIV disease occurs predominantly in the gastrointestinal tract. J Exp Med. 2004;200:749–759.
Cunningham-Rundles S., McNeeley D.F., Moon A. Mechanisms of nutrient modulation of the immune response. J Allergy Clin Immunol. 2005;115:1119–1128.
Day C.L., Walker B.D. Progress in defining CD4 helper cell responses in chronic viral infections. J Exp Med. 2003;198:1773–1777.
International HIV Controllers Study. The major genetic determinants of HIV-a control affect HLA class I peptide presentation. Science. 2010;330:1551–1557.
McElrath M.J., Haynes B.F. Induction of immunity to human immunodeficiency virus type-1 by vaccination. Immunity. 2010;33:542–554.
Migueles S.A., Laborico A.C., Shupert W.L., et al. HIV-specific CD8+ T cell proliferation is coupled to perforin expression and is maintained in nonprogressors. Nat Immunol. 2002;3:1061–1068.
Paczesny S., Hanauer D., Sun Y., Reddy P. New perspectives on the biology of acute GVHD. Bone Marrow Transplant. 2010;45:1–11.
Stahn C., Löwenberg M., Hommes D.W., Buttgereit F. Molecular mechanisms of glucocorticoid action and selective glucocorticoid receptor agonists. Mol Cell Endocrinol. 2007;275:71–78.
[/level-membership-for-allergy-and-immunology-category][not-level-membership-for-allergy-and-immunology-category]
Chapter 17 AIDS, Secondary Immunodeficiency and Immunosuppression
• Nutrient deficiencies often lead to impaired immune responses. Malnutrition increases the risk of infant mortality from infection through reduction in cell-mediated immunity, including reduced numbers and function of CD4+ helper cells and a reduction in levels of secretory IgA. Trace elements, iron, selenium, copper, and zinc are also important in immunity. Lack of these elements can lead to diminished neutrophil killing of bacteria and fungi, susceptibility to viral infections, and diminished antibody responses. Vitamins A, B6, C, E, and folic acid are likewise important in overall resistance to infection. Proper diet and nutrition, therefore, reduce morbidity and mortality caused by infection.
• Some drugs selectively alter immune function. Immunomodulatory drugs can severely depress immune functions. These drugs are often necessary to treat solid organ transplant patients and those with an autoimmune disease. Although necessary in such settings, these drugs are often broad acting, thereby increasing patients’ susceptibility to a broad array of opportunistic infections caused by viruses, bacteria and fungi.
• HIV is a primary cause of immunodeficiency. Human immunodeficiency virus (HIV) is a retrovirus that predominantly targets CD4+ T cells. Acute infection depletes CD4 T cell subsets and transiently suppresses circulating CD4 T cell numbers before the immune system establishes partial control of the virus and the chronic phase of infection begins. Though patients can remain in the chronic phase for an average of 10 years, without anti-retroviral drug treatment, CD4 T cell levels gradually fall, resulting in loss of cell-mediated immunity and susceptibility to life-threatening opportunistic infections. This final stage, AIDS (acquired immunodeficiency syndrome), is marked by low CD4 T cell counts, high HIV plasma levels, reactivation of other latent infections, and often, virus-associated malignancies such as Kaposi’s sarcoma and non-Hodgkin’s lymphoma.
• Combination therapy for AIDS with inhibitors of HIV reverse transcriptase, protease, and entry are reasonably successful, but associated with long-term toxicities in almost 50% of persons. An effective vaccine remains an elusive goal, in part due to the rapid mutation rate of the virus during reverse transcription.
Overview
Nutrient deficiencies
Infection and malnutrition can exacerbate each other
• increasing metabolic demands;
• decreasing appetite, thereby lowering intake of nutrients; and
• with gastrointestinal infection, decreasing nutrient absorption.
Once this cycle begins, it is self-propagating as infection compromises immunity, which then leads to more infection and debility (Fig. 17.1). At a population level, this may lead to decreased productivity, further decreasing economic and food resources and, again, driving the malnutrition and immune deficiency loop.
Protein–energy malnutrition and lymphocyte dysfunction
Nutrition also affects innate mechanisms of immunity
Poor nutrition also causes deficits in innate immune defenses, for example,
• a larger number of bacteria bind to epithelial cells of malnourished subjects;
• the production of certain inflammatory cytokines, such as IL-2 and TNFα, is decreased;
• opsonization is decreased, largely because of a reduction in levels of various complement components – C3, C5, and factor B.
Deficiencies in trace elements impact immunity
Zinc deficiency during pregnancy has also been shown to have an inter-generational effect on the levels of IgM and IgM producing B cells in the pups, and more surprisingly even in the 2nd generation (Fig. 17.w1).
Q. In what ways can neutrophils and macrophages restrict iron availability for microorganisms?
A. Soluble proteins such as lactoferrin produced by neutrophils reduce the availability of free iron in the phagolysosome. Macrophages have ion pumps (e.g. nRAMP) in their phagosomal membrane that remove iron from the phagosome (see Chapter 15).
Vitamin deficiencies and immune function
Q. NFATc1, a member of the NFAT transcription factor family, is required for development of the B-1 B cell subset. Recent experiments using mice have shown that vitamin A deficiency severely reduces NFAT-c1 expression resulting in loss of the B-1 population. Why might this contribute to defects in mucosal immunity?
Until the advent of antibiotics, cod liver oil and sunlight, both sources of vitamin D, were used as primary treatments for TB. Vitamin D deficiency can lead to increased infection rates and recent studies have begun to elucidate some of the molecular mechanisms behind the anti-infective role of vitamin D. Many cell types express the vitamin D receptor (VDR), and while vitamin D metabolites may modulate adaptive immune responses, they can also enhance innate immunity. Importantly, particularly for TB, signaling via the VDR may enhance both cathelicidin and defensin expression, thus boosting macrophage anti-microbial activity (Chapter 7).
Immunodeficiency secondary to drug therapies
Several classes of drugs suppress immune function, either intentionally for therapeutic effect, or as an unwanted side effect.. For example, patients receiving organ transplants usually receive a variety of immunosuppressants to prevent rejection of the donor tissue and to treat graft-versus-host disease (GvHD, discussed in Chapter 21). Likewise, patients presenting with severe inflammatory, allergic, or autoimmune reactions often require therapeutic immunosuppression (Chapter 20
[/not-level-membership-for-allergy-and-immunology-category]
