15 Adverse effects of drugs on the liver
Epidemiology
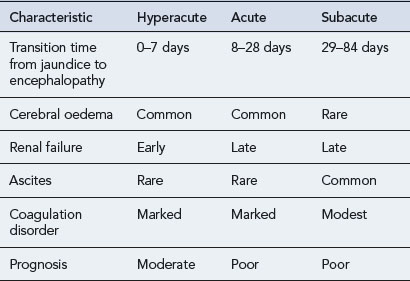
The incidence of drug-induced liver disease (DILD) has continued to rise steadily since the late 1960s, although the incidence of idiosyncratic reactions for most drugs remains low, occurring at therapeutic doses from 1 in every 1000 patients to 1 in every 100,000 patients. DILD is not usually life-threatening; however, for the small number of patients who develop drug-induced acute liver failure (ALF) the prognosis is poor, with a 60–80% mortality rate, unless they receive a liver transplant. The incidence and severity of DILD is shown in Fig. 15.1. It is estimated that 15–40% of ALF cases may be attributable to drugs. Classification of ALF suggests three classes: hyperacute, acute and subacute (Table 15.1).
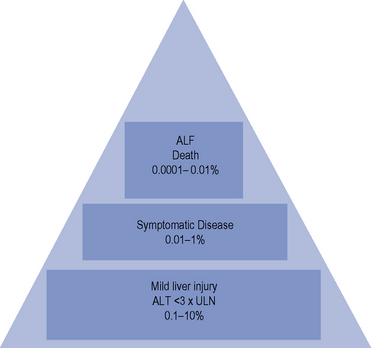
Table 15.1 Characteristics of the different types of acute liver failure (Richardson and O’Grady, 2002)
Many drugs cause elevated liver enzymes with apparently no clinically significant adverse effect, although in a few patients there may be significant hepatotoxicity. For example, isoniazidcauses elevated liver enzymes in 10–36% of patients taking the drug as a single agent. However, only 1% suffer significant hepatotoxicity, with the liver function tests (LFTs) of the majority returning to normal if therapy is discontinued. Other examples of drugs that elevate liver enzymes are shown in Table 15.2.
| Drug | Percentage of patients with increase in transaminases |
|---|---|
| Cefaclor | 11% |
| Cefixime | 0.7% |
| Ciprofloxacin | 5% |
| Chlorpromazine | 50% |
| Diclofenac | 15% |
| Donepezil | MHRA reportsa |
| Efavirenz | 4% |
| Heparin/LWMH | 5% |
| Isoniazid | 10–36% |
| Naproxen | 4% |
| Norfloxacin | 0.1% |
| Nevirapine | 12% |
| Niacin | 50% |
| Rifampicin | 15–30% |
| Sodium valproate | 11% |
| SSRIs | MHRA reportsa |
| Statins | 1–2% |
| Sulphonamides | 10% |
Risk factors
Concurrent diseases and pregnancy
Pre-existing renal disease, diabetes, pregnancy and poor nutrition may all affect the ability of the liver to metabolise drugs effectively and may put the patient at risk of developing liver damage. Table 15.3 summarises the host factors that may predispose a patient to drug hepatotoxicity.
Table 15.3 Examples of host factors that predispose to drug hepatotoxicity
| Host factor | Drug example |
|---|---|
| Pre-existing liver disease | Methotrexate, aspirin, sodium valproate |
| Age | |
| Older | Halothane, isoniazid, chlorpromazine, co-amoxiclav, nitrofuratoin |
| Younger | Aspirin, sodium valproate |
| Gender | |
| Female | Halothane, isoniazid, nitrofurantoin, flucloxacillin, chlorpromazine, erythromycin |
| Male | Sodium valproate (in prepubescent boys), co-amoxiclav |
| Genetics | Halothane, chlorpromazine, phenytoin, carbamazepine, phenobarbital, paracetamol, flucloxacillin |
| Enzyme induction | Paracetamol, halothane, isoniazid, sodium valproate |
| Polypharmacy | NSAIDs if used with other hepatotoxic drugs |
| Isoniazid with rifampicin or pyrazinamide | |
| Sodium valproate with phenytoin | |
| Paracetamol with zidovudine | |
| Concurrent diseases | |
| Diabetes mellitus | Methotrexate |
| Renal failure | Allopurinol, i.v. tetracycline |
| Malnutrition | Paracetamol |
| HIV positive with hepatitis C or B co-infection | Antiretroviral agents |
Aetiology
Drug-induced hepatotoxicity may present as an acute insult that may or may not progress to chronic disease, or it can present as an insidious development of chronic disease. The type of lesion may be cytotoxic (cellular destruction) or cholestatic (impaired bile flow). Cytotoxic damage may be further classified as necrotic (cell death) or steatic (fatty degeneration). The liver damage resulting from drug toxicity often presents as a mixed picture of cytotoxic and cholestatic injury. The mechanisms of drug-induced hepatic damage can be divided into intrinsic (type A) and idiosyncratic (type B) hepatotoxicity (Table 15.4). Intrinsic hepatotoxicity is predictable, dose-dependent and usually has a short latency period ranging from hours to weeks. The majority of individuals who take a toxic dose are affected and exhibit the same type of injury. Examples are paracetamol, salicylates, methotrexate and tetracycline. Other examples are presented in Table 15.5. Toxicity may be avoided by ensuring the doses listed are not exceeded.
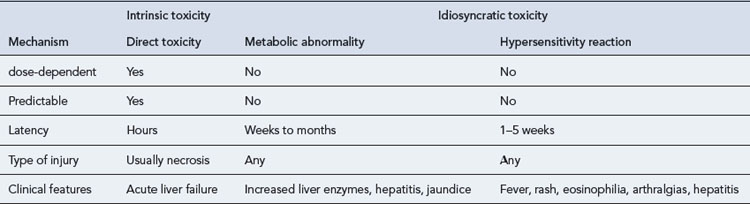
Table 15.5 Examples of dose-related drug-induced hepatotoxicity
| Drug | Toxic dose |
|---|---|
| Paracetamol | Single dose >10 g |
| Tetracycline | >2 g daily (oral), increased risk of toxicity in pregnancy and renal failure |
| Methotrexate | Weekly dose >15 mg |
| Cumulative dose >2 g in 3 years, increased risk of toxicity in pre-existing liver disease, alcohol abuse, diabetes | |
| 6-Mercaptopurine | >2.5 mg/kg |
| Vitamin A | Chronic use of 40,000 units daily |
| Cyclophosphamide | Daily dose >400 mg/m2 |
| Salicylates | Chronic use >2 g daily |
| Anabolic steroids | High dose >1 month |
| Oral contraceptive | Increased risk with higher oestrogen content, older preparations |
| Duration of treatment | |
| Iron | Single dose >1 g |
Pathophysiology
The range of DILDs is illustrated in Table 15.6. Increased serum level of hepatobiliary enzymes without clinical liver disease occurs with variable frequency between drugs but for some agents it may occur in up to half the patients who receive a drug. This may reflect subclinical liver injury.
| Adverse reaction | Drugs associated with reaction |
|---|---|
| Hepatocellular necrosis | Paracetamol |
| Propylthiouracil | |
| Salicylates | |
| Iron salts | |
| Allopurinol | |
| Dantrolene | |
| Halothane | |
| Ketoconazole | |
| Isoniazid | |
| Mithramycin | |
| Cocaine | |
| ‘Ecstasy’ (methylenedioxymetamphetamine, MDMA) | |
| Fatty liver | Amiodarone |
| Tetracyclines | |
| Steroids | |
| Sodium valproate | |
| l-Asparaginase | |
| Cholestasis | Oral contraceptives |
| Carbimazole | |
| Anabolic steroids | |
| Ciclosporin | |
| Cholestasis with hepatitis | Chlorpromazine |
| Tricyclic antidepressants | |
| Erythromycin | |
| Flucloxacillin | |
| Co-amoxiclav | |
| ACE inhibitors | |
| Sulphonamides | |
| Sulphonylureas | |
| Phenytoin | |
| NSAIDs | |
| Cimetidine | |
| Ranitidine | |
| Trazodone | |
| Granulomatous hepatitis | Phenytoin |
| Allopurinol | |
| Carbamazepine | |
| Clofibrate | |
| Hydralazine | |
| Sulphonamides | |
| Sulphonylureas | |
| Acute hepatitis | Dantrolene |
| Isoniazid | |
| Phenytoin | |
| Chronic active hepatitis | Methyldopa |
| Nitrofurantoin | |
| Isoniazid | |
| Fibrosis and cirrhosis | Methotrexate |
| Methyldopa | |
| Vitamin A (dose-related) | |
| Vascular disorders | Azathioprine |
| Dactinomycin | |
| Dacarbazine |
Clinical manifestations
Treatment
Diagnosis
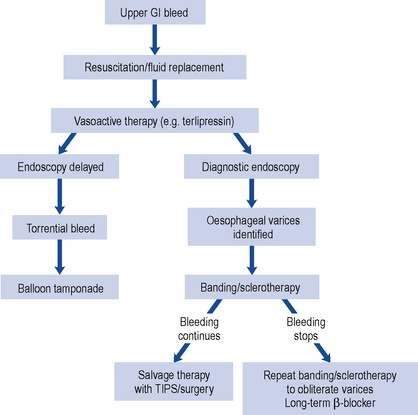
Drug-induced hepatic injury should be suspected in every patient with jaundice while ruling out other causes of liver disease by the clinical history and the results of investigations. The typical process in screening patients presenting with jaundice is outlined in Fig. 15.2 and the general approach to the differential diagnosis of acute hepatitis is set out in Fig. 15.3.
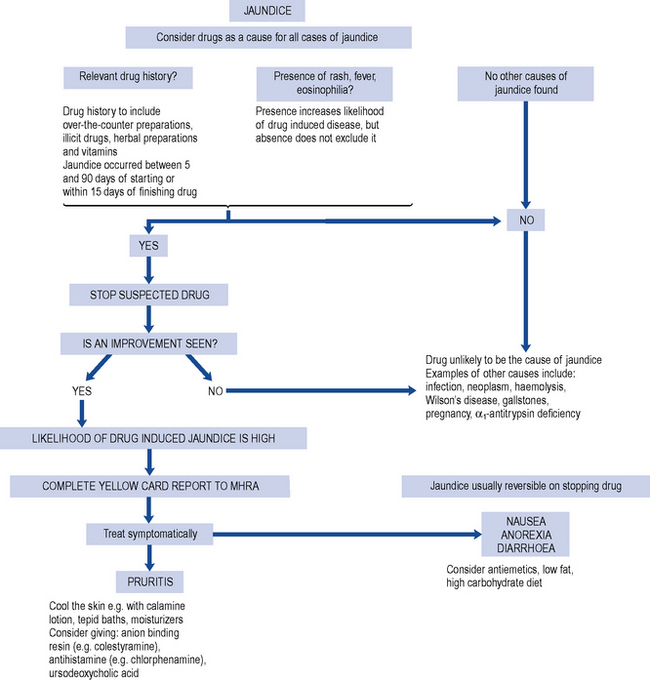
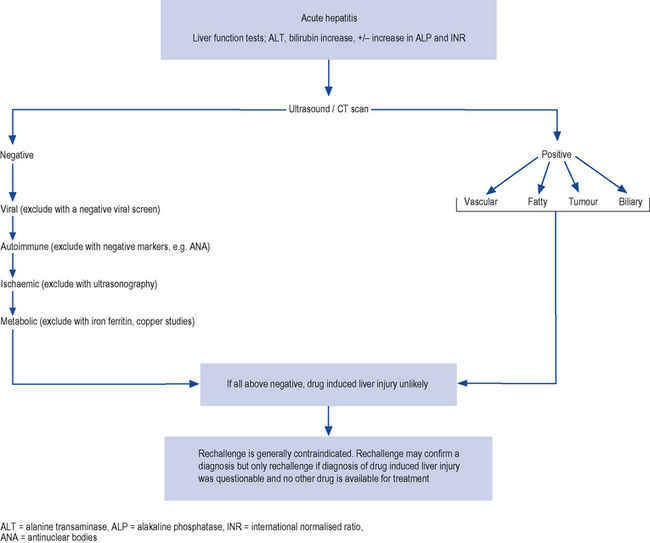
A detailed and thorough drug history, including use of oral contraceptives, over-the-counter medicines, vitamins, herbal preparations and illicit drug use, should be obtained. Examples of herbal and dietary preparations implicated in causing liver damage are listed in Box 15.1. Attention to the duration of treatment with a specific drug and the relationship to the onset of symptoms is important. The likelihood of a drug-related disease is greatest when the abnormality begins between 5 and 90 days after taking the first dose and within 15 days of taking the last dose. The latent period, that is, the time between starting therapy and the appearance ofsymptoms, may vary but for many drugs is sufficiently reproducible to be of some diagnostic value.
Idiosyncratic reactions may also need to be considered and the literature consulted for previous reports. A key component of secondary prevention is the reporting of all suspected hepatic drug reactions to the appropriate monitoring agency, particularly for newer agents with fewer published cases. Many drugs are approved and licensed before the idiosyncratic reactions are identified as there is little chance of detecting the reaction in the phase III studies which typically involve 300 patients. To detect a single case of clinically significant hepatic injury due to a drug with 95% confidence, the number of patients included in the trial must be about three times the incidence of the reaction. Idiosyncratic reactions occur in about 1 in 10,000 patients so to detect the reaction, the clinical trial would have to study 30,000 patients. Practice points for diagnosing DILD are shown in Box 15.2.
Withdrawal
Once drug-induced hepatotoxicity has been recognised as a possibility, therapy should be stopped. If the patient is receiving more than one potentially hepatotoxic drug, all drugsshould be stopped. Withdrawal of the agent usually results in recovery that begins within a few days. However, LFTs may take many months or even years to return to normal. Co-amoxiclav and phenytoin are examples of drugs that have been associated with a worsening of the patient’s condition for several weeks after withdrawal, and a protracted recovery period of several months. Examples of drugs associated with chronic liver injury are shown in Box 15.3.
Management
If clinical or laboratory signs of hepatic failure appear, hospitalisation is mandatory.
Pruritus
The main symptom of drug-induced cholestasis is pruritus due to high systemic concentrations of bile acids deposited in tissues. General measures include light clothing (avoid wool) and cooling the skin with tepid baths or calamine lotion, and a general moisturising agent such as aqueous cream. The management of liver-induced pruritus is discussed in more detail in Chapter 16.
Long-term treatment
When the DILD is under control, consideration will have to be given to the treatment of the original condition for which the implicated drug was prescribed. In many cases, drug therapy will still be required and caution must, therefore, be exercised, as drugs with similar chemical structures may cause similar hepatotoxicity (Table 15.7).
| Problem | Action | |
|---|---|---|
| Phenothiazines | Cross-sensitivity | Avoid |
| Tricyclics | Cross-sensitivity | Avoid |
| NSAIDs | Cross-sensitivity | Avoid |
| Isoniazid, pyrazinamide | Chemically-related | Avoid |
| Halothane | Avoid enflurane | Isoflurane appears safe |
Paracetamol-induced hepatotoxicity
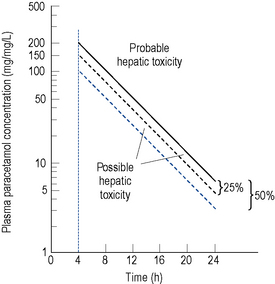
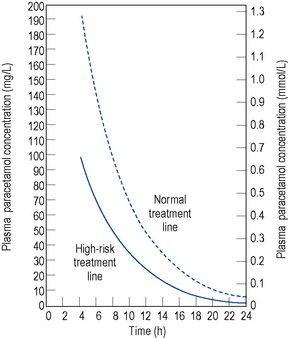
Ingestion of doses as low as 10–15 g of paracetamol have been reported to cause severe hepatocellular necrosis. Removal of unabsorbed paracetamol by gastric lavage may be worth-while if more than 150 mg/kg body weight has been taken and the patient presents within 4 h of ingestion. Activated charcoal may also be administered to reduce further absorption of paracetamol and facilitate removal of unmetabolised paracetamol from extracellular fluids. This may lessen the effect of any methionine given. A plasma paracetamol concentration should be taken as soon as possible but not within 4 h of ingestion due to the fact that a misleading and low level may be obtained because of continuing absorption and distribution of the drug. The plasma concentration measured should be compared with a standard nomogram reference line of a plot of plasma paracetamol concentration against time in hours after ingestion. This may be a semilogarithmic (Fig. 15.4) or linear (Fig. 15.5) plot. Generally, administration of intravenous acetylcysteine is the treatment of choice for paracetamol overdose when the blood paracetamol level is in the range predictive of possible or probable liver injury (see Fig. 15.4). Patients allergic to acetylcysteine may receive oral methionine.
Patient care
Patient counselling
The challenge for all members of the health care team is to alert patients to the potentially toxic effects of drugs without creating so much concern that they fail to comply with vital medication. For the limited number of drugs presented in Table 15.8, careful monitoring of LFTs during the first 6 months of treatment is advisable, although not always practical. Thereafter, regular monitoring of LFTs is appropriate in patients who are at greater risk of hepatotoxicity. Such patients would include those with known liver disease, those taking other hepatotoxic drugs, those aged over 40 years, and heavy alcohol consumers. Surveillance should be particularly frequent in the first 2 months of treatment. In patients with no risk factors and normal pretreatment liver function, LFTs need only be repeated if fever, malaise, vomiting, jaundice or unexplained deterioration during treatment occurs.
Table 15.8 Examples of drugs where regular monitoring of liver function is recommended
| Drug | Baseline measurementa | Frequency of monitoring |
|---|---|---|
| Anti-TB therapy (isoniazid, rifampicin, pyrazinamide) | Yes | Patients with pre-existing chronic liver disease: check LFTs regularly, every week for the first 2 weeks, then twice a week for the first 2 months. |
| Patients with normal liver function tests and no evidence of pre-existing liver disease: regular monitoring is not necessary but LFTs repeated if signs of liver dysfunction develop, for example, fever, malaise, vomiting or jaundice. | ||
| Patients with raised pretreatment hepatic transaminases: Two or more times normal: check LFTs weekly for 2 weeks, then twice a week until normal. Under two times normal: check LFTs at 2 weeks. If these transaminases have fallen, further tests are only needed if symptoms occur | ||
| Amiodarone | Yes | LFTs checked every 6 months |
| Cyproterone | Yes | Recheck if any symptoms |
| Dantrolene | Yes | Repeat LFTs after first 6 weeks of therapy |
| Itraconazole | Yes | Monitor LFTs if therapy continues for more than 1 month. Recheck if any symptoms |
| Ketoconazole | Yes | LFTs checked on weeks 2 and 4 of therapy and then every month |
| Methotrexate | Yes | LFTs checked every 2 weeks for the first 2 months, then monthly for 4 months, then every 3 months |
| Methyldopa | Yes | Check LFTs at intervals during the first 6–12 weeks of treatment |
| Micafungin | Yes | Periodic monitoring of LFTs recommended. Recheck if any symptoms |
| Nevirapine | Yes | Check LFTs every 2 weeks for the first 2 months, then at month 3 and then regularly |
| Pioglitazone | Yes | Periodic monitoring of LFTs recommended. Recheck if any symptoms |
| Rosiglitazone | Yes | Periodic monitoring of LFTs recommended. Recheck if any symptoms. |
| Sodium valproate | Yes | Check LFTs regularly during the first 6 months of therapy |
| Statins | Yes | LFTs checked 12 weeks after initiation or after a dose increase and periodically thereafter |
| Sulfasalazine | Yes | LFTs checked every 2 weeks for the first 2 months, then monthly for 4 months then every 3 months |
| Tipranavir | Yes | Check LFTs on weeks 2, 4 and 8 of treatment and then every 2–3 months |
| Vildagliptin | Yes | LFTs checked every 3 months for the first year and then periodically |
a Baseline and subsequent liver function tests (LFTs) difficult to interpret in critically ill patients as LFTs will be affected by multiple factors
Minimising the risk of DILD
Practice points for patient care and minimising the risk of DILD are outlined in Box 15.4.
Box 15.4 Practice points for minimising the risk of drug-induced liver disease (DILD)
Case 15.1
| Actual value (normal range) | |
|---|---|
| Paracetamol | 18 mg/mL |
| Albumin | 26 g/dL (30–50 g/L) |
| Alanine transaminase | 5435 units/L (0–50 units/L) |
| Bilirubin | 50 µmol/L (<17 µmol/L) |
| Alakline phosphatase | 66 units/L (30–135 units/L) |
| Prothrombin time | 57 s (9.8–12.6 s) |
| Creatinine | 133 (35–125 µmol/L) |
| Urea | 5.6 (0–7.5 mmol/L) |
| Actual value (normal range) | |
|---|---|
| Albumin | 22 g/dL (30–50 g/L) |
| Alanine transaminase | 12,477 units/L (0–50 units/L) |
| Bilirubin | 71 µmol/L (<17 µmol/L) |
| Alakline phosphatase | 73 units/L (30–135 units/L) |
| Prothrombin time | 90.8 s (9.8–12.6 s) |
| Arterial pH | 7.226 (7.350–7.450) |
| Lactate | 6.9 (0.4–2.2 mmol/L) |
| Creatinine | 336 (35–125 µmol/L) |
| Urea | 7.4 (0–7.5 mmol/L) |
Answers
| Prothrombin time | >36 s |
| Creatinine | >200 µmol/L |
| pH | <7.3 |
| Encephalopathy | Present |
| Cerebral oedema | Present |
| Time from onset of jaundice to encephalopathy | 0–7 days |
Answers
Case 15.3
| Actual value (normal range) | |
|---|---|
| Albumin | 30 g/dL (30–50 g/L) |
| Alanine transaminase | 350 units/L (0–50 units/L) |
| Bilirubin | 90 µmol/L (<17 µmol/L) |
| Alkaline phosphatase | 180 units/L (30–135 units/L) |
Answers
Brok J., Buckley N., Gluud C. Interventions for paracetamol (acetaminophen) overdose. Cochrane Database Syst. Rev.. (2):2006. Art No. CD003328. doi:10.1002/14651858.CD003328.pub2
Gomez-Dominguez E., Gisbert J., Moreno-Monteagudo J., et al. A pilot study of atorvastatin treatment in dyslipemid, non alcoholic fatty liver patients. Aliment. Pharmacol. Ther.. 2006;23:1643-1647.
Martínez E., Blanco J.L., Arnaiz J.A., et al. Hepatotoxicity in HIV-1-infected patients receiving nevirapine-containing antiretroviral therapy. AIDS. 2001;15:1261-1268.
McKenney J., Davidson M.H., Jacobson T.A., et al. Final conclusions and recommendations of the National Lipid Association Statin Safety Assessment Task Force. Am. J. Cardiol.. 2006;97:89C-94C.
Richardson P., O’Grady J. Acute liver disease. Hosp. Pharm.. 2002;9:131-136.
Soriano V., Puoti M., Garcia-Gasco P., et al. Antiretroviral drugs and liver injury. AIDS. 2008;22:1-13.
Bell L.N., Chalasani N. Epidemiology of idiosyncratic drug-induced liver injury. Semin. Liver Dis.. 2009;29:337-347.
Bjornsson E. Long term follow up of patients with mild to moderate drug induced liver injury. Aliment. Pharmacol. Ther.. 2007;26:79-85. Available at: http://www.medscape.com/viewarticle/558756
Bjornsson E. The natural history of drug-induced liver injury. Semin. Liver Dis.. 2009;24:357-363. Available at: http://www.medscape.com/viewarticle/711396
Heard K.J. Acetylcysteine for acetaminophen poisoning. N. Engl. J. Med.. 2008;359:285-292.
Lee W.L. Drug induced hepatotoxicity. N. Engl. J. Med.. 2003;349:474-485.
North-Lewis P., editor. Drugs and the Liver. London: Pharmaceutical Press, 2008.
Suzuki A., Andrade R.J., Bjornsson E., et al. Drugs associated with hepatotoxicity and their reporting frequency of liver adverse events in VigiBase: unified list based on international collaborative work. Drug Saf.. 2010;33:503-522.
Watkins P. Biomarkers for the diagnosis and management of drug induced liver injury. Semin. Liver Dis.. 2009;24:393-399. Available at: http://www.medscape.com/viewarticle/711398