[level-membership-for-basic-science-category]
Chapter 23 Adrenergic mechanisms and drugs
Adrenergic mechanisms
The discovery in 1895 of the hypertensive effect of adrenaline/epinephrine was initiated by Dr Oliver, a physician in practice, who conducted a series of experiments on his young son into whom he injected an extract of bovine suprarenal and detected a ‘definite narrowing of the radial artery’.1 The effect was confirmed in animals and led eventually to the isolation and synthesis of adrenaline/epinephrine in the early 1900s. Many related compounds were examined and, in 1910, Barger and Dale invented the word sympathomimetic2 and also pointed out that noradrenaline/norepinephrine mimicked the action of the sympathetic nervous system more closely than did adrenaline/epinephrine.
Classification of sympathomimetics
By mode of action
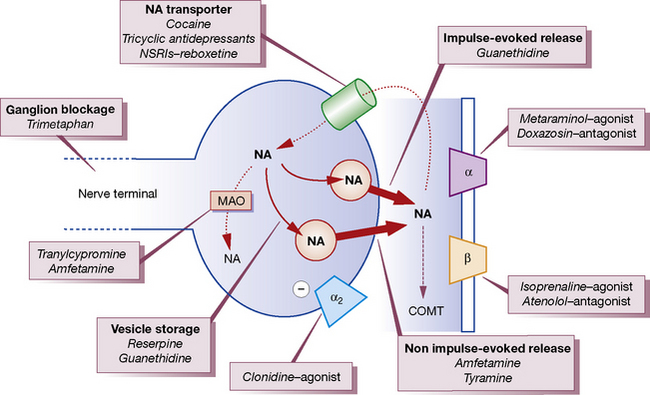
Noradrenaline/norepinephrine is synthesised and stored in vesicles within adrenergic nerve terminals (Fig. 23.1). The vesicles can be released from these stores by stimulating the nerve or by drugs (ephedrine, amfetamine). The noradrenaline/norepinephrine stores can also be replenished by intravenous infusion of noradrenaline/norepinephrine, and abolished by reserpine or by cutting the sympathetic nerve. Sympathomimetics may be classified on the basis of their sites of action (see Fig. 23.1) as acting:
1. Directly: adrenoceptor agonists, e.g. adrenaline/epinephrine, noradrenaline/norepinephrine, isoprenaline (isoproterenol), methoxamine, xylometazoline, metaraminol (entirely); and dopamine and phenylephrine (mainly).
2. Indirectly: by causing a release of preformed noradrenaline/norepinephrine from stores in nerve endings,3 e.g. amfetamines, tyramine; and ephedrine (largely).
3. By both mechanisms (1 and 2, though one usually predominates): other synthetic agents.
All of the above mechanisms operate in both the central and peripheral nervous systems, but discussion below will focus on agents that influence peripheral adrenergic mechanisms.
Tachyphylaxis
(rapidly diminishing response to repeated administration) is a particular feature of group 2 drugs. It reflects depletion of the ‘releasable’ pool of noradrenaline/norepinephrine from adrenergic nerve terminals that makes these agents less suitable as, for example, pressor agents than drugs in group 1. Longer-term tolerance (see p. 78) to the effects of direct sympathomimetics is much less of a clinical problem and reflects an alteration in adrenergic receptor density or coupling to second messenger systems.
Interactions of sympathomimetics
with other vasoactive drugs are complex. Some drugs block the reuptake mechanism for noradrenaline/norepinephrine in adrenergic nerve terminals and potentiate the pressor effects of noradrenaline/norepinephrine, e.g. cocaine, tricyclic antidepressants or highly noradrenaline/norepinephrine-selective reuptake inhibitors (NSRIs) such as reboxetine (see Fig. 23.1). Others deplete or destroy the intracellular stores within adrenergic nerve terminals (e.g. reserpine and guanethidine) and thus block the action of indirect sympathomimetics.
History
Subclassification of adrenoceptors is shown in Table 23.1.
Table 23.1 Clinically relevant aspects of adrenoceptor functions and actions of agonists
| α1-Adrenoceptor effectsa | β-Adrenoceptor effects |
|---|---|
| Eye:b mydriasis | |
| Heart (β1, β2):c | |
| increased rate (SA node) | |
| increased automaticity (AV node and muscle) | |
| increased velocity in conducting tissue | |
| increased contractility of myocardium | |
| increased oxygen consumption; decreased refractory period of all tissues | |
| Arterioles: | Arterioles: |
| constriction (only slight in coronary and cerebral) | dilatation (β2) |
| Bronchi (β2): relaxation | |
| Anti-inflammatory effect: | |
| inhibition of release of autacoids (histamine, leukotrienes) from mast cells, e.g. asthma in type I allergy | |
| Uterus: contraction (pregnant) | Uterus (β2): relaxation (pregnant) |
| Skeletal muscle: tremor (β2) | |
| Skin: sweat, pilomotor | |
| Male ejaculation | |
| Blood platelet: aggregation | |
| Metabolic effect: | Metabolic effects: |
| hyperkalaemia | hypokalaemia (β2) |
| hepatic glycogenolysis (β2) | |
| lipolysis (β1, β2) | |
| Bladder sphincter: contraction | Bladder detrusor: relaxation |
| Intestinal smooth muscle relaxation is mediated by α and β adrenoceptors. α2-adrenoceptor effects:a α2 receptors on the nerve ending, i.e. presynaptic autoreceptors, mediate negative feedback which inhibits noradrenaline/norepinephrine release |
|
| Use of the term cardioselective to mean β1-receptor selective only, especially in the case of β-receptor blocking drugs, is no longer appropriate. | |
| Although in most species the β1 receptor is the only cardiac β receptor, this is not the case in humans. What is not generally appreciated is that the endogenous sympathetic neurotransmitter noradrenaline/norepinephrine has about a 20-fold selectivity for the β1 receptor – similar to that of the antagonist atenolol – with the consequence that under most circumstances, in most tissues, there is little or no β2-receptor stimulation to be affected by a non-selective β-blocker. Why asthmatics should be so sensitive to β-blockade is paradoxical: all the bronchial β receptors are β2, but the bronchi themselves are not innervated by noradrenergic fibres and the circulating adrenaline levels are, if anything, low in asthma. | |
a For the role of subtypes (α1 and α2), see prazosin.
b Effects on intraocular pressure involve both α and β adrenoceptors as well as cholinoceptors.
c Cardiac β1 receptors mediate effects of sympathetic nerve stimulation. Cardiac β2 receptors mediate effects of circulating adrenaline, when this is secreted at a sufficient rate, e.g. following myocardial infarction or in heart failure. Both receptors are coupled to the same intracellular signalling pathway (cyclic AMP production) and mediate the same biological effects.
Consequences of adrenoceptor activation
All adrenoceptors are members of the G-coupled family of receptor proteins, i.e. the receptor is coupled to its effector protein through special transduction proteins called G-proteins (themselves a large protein family). The effector protein differs among adrenoceptor subtypes. In the case of β-adrenoceptors, the effector is adenylyl cyclase and hence cyclic AMP is the second messenger molecule. For α-adrenoceptors, phospholipase C is the commonest effector protein and the second messenger here is inositol trisphosphate (IP3). It is the cascade of events initiated by the second messenger molecules that produces the variety of tissue effects shown in Table 23.1. Hence, specificity is provided by the receptor subtype, not the messengers.
Selectivity for adrenoceptors
The following classification of sympathomimetics and antagonists is based on selectivity for receptors and on use. But selectivity is relative, not absolute; some agonists act on both α and β receptors, some are partial agonists and, if sufficient drug is administered, many will extend their range. The same applies to selective antagonists (receptor blockers), e.g. a β1-selective-adrenoceptor blocker can cause severe exacerbation of asthma (a β2 effect), even at low dose. It is important to remember this because patients have died in the hands of doctors who have forgotten or been ignorant of it.4
Adrenoceptor agonists (see Table 23.1)
Effects of a sympathomimetic
To block all the effects of adrenaline/epinephrine and noradrenaline/norepinephrine, antagonists for both α and β receptors must be used. This can be a matter of practical importance, e.g. in phaeochromocytoma (see p. 419).
Physiological note
The termination of action of noradrenaline/norepinephrine released at nerve endings is by:
• reuptake into nerve endings by the noradrenaline/norepinephrine transporter where it is stored in vesicles or metabolised by monoamine oxidase (MAO) (see Fig. 23.1)
• diffusion away from the area of the nerve ending and the receptor (junctional cleft)
• metabolism (by extraneuronal MAO and catechol-O-methyltransferase, COMT).
Adverse effects
These may be deduced from their actions (Table 23.1, Fig. 23.2). Tissue necrosis due to intense vasoconstriction (α) around injection sites occurs as a result of leakage from intravenous infusions. The effects on the heart (β1) include tachycardia, palpitations, cardiac arrhythmias including ventricular tachycardia and fibrillation, and muscle tremor (β2). Sympathomimetic drugs should be used with great caution in patients with heart disease.
Sympathomimetics and plasma potassium
The hypokalaemic effect of administered (β2) sympathomimetics may be clinically important, particularly in patients with pre-existing hypokalaemia, e.g. due to intense adrenergic activity such as occurs in myocardial infarction,5 in fright (admission to hospital is accompanied by transient hypokalaemia), or with previous diuretic therapy, and patients taking digoxin. In such subjects the use of a sympathomimetic infusion or of an adrenaline/epinephrine-containing local anaesthetic may precipitate cardiac arrhythmia. Hypokalaemia may occur during treatment of severe asthma, particularly where the β2-receptor agonist is combined with theophylline.
Individual sympathomimetics
The actions are summarised in Table 23.1. The classic, mainly endogenous, substances will be described first despite their limited role in therapeutics, and then the more selective analogues that have largely replaced them.
Catecholamines
Traditionally catecholamines have had a dual nomenclature (as a consequence of a company patenting the name Adrenalin), broadly European and North American. The North American naming system has been chosen by the World Health Organization as recommended international non-proprietary names (rINNs) (see Ch. 7), and the European Union has directed member states to use rINNs. By exception, adrenaline and noradrenaline are the terms used in the titles of monographs in the European Pharmacopoeia and are thus the official names in the member states. Because uniformity has not yet been achieved, and because of the scientific literature, we use both names. For pharmacokinetics, see above.
Adrenaline/epinephrine
Adrenaline/epinephrine (α- and β-adrenoceptor effects) is used:
• as a vasoconstrictor with local anaesthetics (1 in 80 000 or weaker) to prolong their effects (about two-fold)
• as a topical mydriatic (sparing accommodation; it also lowers intraocular pressure)
• for severe allergic reactions, i.e. anaphylactic shock, intramuscularly or intravenously. The route must be chosen with care (for details, see p. 116). The subcutaneous route is not recommended as the intense vasoconstriction slows absorption.
Accidental overdose
with adrenaline/epinephrine occurs occasionally. It is rationally treated with propranolol to block the cardiac β effects (cardiac arrhythmia) and phentolamine or chlorpromazine to control the α effects on the peripheral circulation that will be prominent when the β effects are abolished. Labetalol (α + β blockade) is a good alternative. β-adrenoceptor block alone is hazardous as the then unopposed α-receptor vasoconstriction causes (severe) hypertension (see Phaeochromocytoma, p. 419). Use of other classes of antihypertensives is irrational and may even cause adrenaline/epinephrine release.
Dopamine
It may be mixed with dobutamine.
For CNS aspects of dopamine, agonists and antagonists, see Neuroleptics (p. 290) and Parkinsonism (p. 359).
Non-catecholamines
Salbutamol (see also Asthma)
Salmeterol
(Serevent) is a variant of salbutamol that has an additional binding site adjacent to the β2-adrenoceptor; this results in slow onset (15–30 min) and long duration of action (12–18 h) (see p. 474). This behaviour is distinct from the other widely used long-acting β2-agonist, formoterol, which has a rapid bronchodilator effect like salbutamol (within a few minutes).
Ephedrine
Ephedrine (t½ approx. 6 h) is a plant alkaloid6 with indirect sympathomimetic actions that resemble those of adrenaline/epinephrine peripherally and amfetamine centrally. Hence, (in adults) it produces increased alertness, anxiety, insomnia, tremor and nausea; children may be sleepy when taking it. In practice, central effects limit its use as a sympathomimetic in asthma.
Phenylpropanolamine (norephedrine) is similar but with fewer CNS effects. Prolonged administration of phenylpropanolamine to women as an anorectic has been associated with pulmonary valve abnormalities and stroke, leading to its withdrawal in some countries.6
Amfetamine (Benzedrine) and dexamfetamine (Dexedrine) act indirectly. They are seldom used for their peripheral effects, which are similar to those of ephedrine, but usually for their effects on the CNS (narcolepsy, attention deficit in children). (For a general account of amfetamine, see p. 343).
Shock
Definition
Shock is a state of inadequate organ perfusion (oxygen deficiency) sufficient adversely to affect cellular metabolism, causing the release of enzymes and vasoactive substances,7 i.e. it is a low flow or hypoperfusion state.
Treatment
• Treatment of the cause: bleeding, infection, adrenocortical deficiency.
• Replacement of any fluid lost from the circulation.
• Perfusion of vital organs (brain, heart, kidneys) and maintenance of the mean blood pressure.
Blood flow (oxygen delivery) rather than blood pressure is of the greatest immediate importance for the function of vital organs. A reasonable blood pressure is needed to ensure organ perfusion, but peripheral vasoconstriction may maintain a normal mean arterial pressure despite a very low cardiac output. Under these circumstances, blood flow to vital organs will be inadequate and multiple organ failure will ensue unless the patient is resuscitated adequately.
The decision on how to treat shock depends on assessment of the pathophysiology:
• whether cardiac output, and thus peripheral blood flow, is inadequate (low pulse volume, cold-constricted periphery)
• whether cardiac output is normal or high and peripheral blood flow is adequate (good pulse volume and warm dilated periphery), but there is maldistribution of blood
• whether the patient is hypovolaemic or not, or needs a cardiac inotropic agent, a vasoconstrictor or a vasodilator.
Types of shock
Septic shock
When septic shock is recognised, appropriate antimicrobials should be given in high dose immediately after taking blood for culture (see p. 174). Beyond that, the primary aim of treatment is to restore cardiac output and vital organ perfusion by increasing venous return to the heart, and to reverse the maldistribution of blood. Increasing intravascular volume will achieve this, guided by the central venous pressure to avoid overloading the heart. Oxygen is essential as there is often uneven pulmonary perfusion.
Choice of drug in shock
• Dobutamine is used when cardiac inotropic effect is the primary requirement.
• Adrenaline/epinephrine is used when a more potent inotrope than dobutamine is required, e.g. when the vasodilating action of dobutamine compromises mean arterial pressure.
• Noradrenaline/norepinephrine is used when vasoconstriction is the first priority, plus some cardiac inotropic effect, e.g. septic shock.
Additionally, recombinant activated protein C (APC) drotrecogin α can be given to reverse the procoagulant state of septic shock. Inflammatory conditions such as septic shock inhibit the generation of endogenous APC, which normally inactivates factors Va and VIIIa, with the result that production of these procoagulant factors is unchecked. Recombinant human APC improved survival of patients with septic shock and multi-organ failure in a large randomised controlled trial.8
Restoration of intravascular volume9
The choice of crystalloid or colloid for fluid resuscitation remains controversial. A Cochrane review of over 56 clinical trials with mortality data concluded that in critically ill patients there was no evidence that colloids offered superior survival over the use of crystalloids in patients following trauma, burns or surgery.10 Colloids are also much more expensive than crystalloids.
Chronic orthostatic hypotension
As blood pressure can be considered a product of ‘volume’ and ‘vasoconstriction’, the logical initial treatment of orthostatic hypotension is to expand blood volume using a sodium-retaining adrenocortical steroid (fludrocortisone11) or desmopressin (see p. 485), plus elastic support stockings to reduce venous pooling of blood when erect.
Some of the variation reported in drug therapy may be due to differences in adrenergic function dependent on whether the degeneration is central, peripheral, preganglionic, postganglionic or due to age-related changes in the adrenoceptors on end-organs. In central autonomic degeneration – ‘multi-system atrophy’ – noradrenaline/norepinephrine is still present in peripheral sympathetic nerve endings. In these patients, an indirect-acting amine may be successful, and one patient titrated the amount of Bovril (a tyramine-rich meat extract drink) she required in order to stand up.12
Erythropoietin has also been used with success (it increases haematocrit and blood viscosity), but a cautionary note: increasing the haematocrit in this way is known to cause an excess of cardiovascular deaths in chronic renal failure patients and a significant thrombosis risk in cancer patients given erythropoietin.13
• The adrenergic arm of the autonomic system uses noradrenaline/norepinephrine as its neurotransmitter.
• Adrenaline/epinephrine, unlike noradrenaline/norepinephrine, is a circulating hormone.
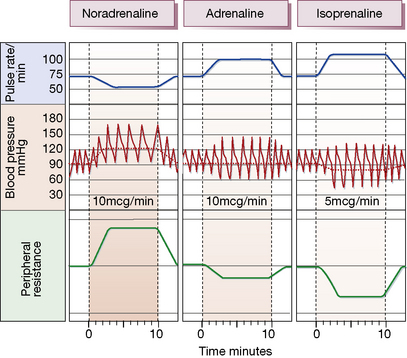
• These two catecholamines act on the same adrenoceptors: α1 and α2, which are blocked by phenoxybenzamine but not by propranolol, and β1 and β2, which are blocked by propranolol but not phenoxybenzamine. Noradrenaline/norepinephrine is a 20-fold weaker agonist at β2 receptors than is adrenaline/epinephrine.
• Distinction between receptor classes is made initially by defining the differing ability of two agonists (or antagonists) to mimic (or block) the effects of catecholamines.
• Often these differences correlate with a difference in receptor type on two different tissues: e.g. stimulation of cardiac contractility by β1 receptors, and bronchodilatation by β2 receptors.
• The distinction between α1 and α2 receptors corresponds to their principal location on blood vessels (causing vasoconstriction) and neurones, respectively.
• Catecholamines themselves can be used in therapy when rapid onset and offset are desired. Selective mimetics at each of the four main receptor subtypes are used for individual locations, e.g. α1 for nasal decongestion, α2 for systemic hypotension, α1 for septic shock, β2 for bronchoconstriction.
• Both α- and β-blockade are used in hypertension; selective β-blockade is used in angina and heart failure.
Ahlquist R.P. A study of adrenotropic receptors. Am. J. Physiol.. 1948;153:586–600.
Brown M.J. To β-block or better block? Br. Med. J.. 1995;311:701–702.
Brown S.G. Cardiovascular aspects of anaphylaxis: implications for treatment and diagnosis. Curr. Opin. Allergy Clin. Immunol.. 2005;5(4):359–364.
Evans T.W., Smithies M. ABC of intensive care. Organ dysfunction. Br. Med. J.. 1999;318:1606–1609.
Insel P.A. Adrenergic receptors – evolving concepts and clinical implications. N. Engl. J. Med.. 1996;334:580–585.
Moore F.A., McKinley B.A., Moore E.E., et al. The next generation in shock resuscitation. Lancet. 2004;363:1988–1996.
Rice T.W., Bernard G.R. Therapeutic intervention and targets for sepsis. Annu. Rev. Med.. 2005;56:225–248.
Russell J.A. Management of sepsis. N. Engl. J. Med.. 2006;355:1699–1713.
Small K.M., McGraw D.W., Liggett S.B. Pharmacology and physiology of human adrenergic receptor polymorphisms. Annu. Rev. Pharmacol. Toxicol.. 2003;43:381–411.
1 Dale H 1938 Edinburgh Medical Journal 45:461.
2 ‘Compounds which … simulate the effects of sympathetic nerves not only with varying intensity but with varying precision … a term … seems needed to indicate the types of action common to these bases. We propose to call it “sympathomimetic”. A term which indicates the relation of the action to innervation by the sympathetic system, without involving any theoretical preconception as to the meaning of that relation or the precise mechanism of the action’ (Barger G, Dale H H 1910 Journal of Physiology XLI:19–50).
3 Fatal hypertension can occur when this class of agent is taken by a patient treated with a monoamine oxidase inhibitor. In addition, remember that large amounts of tyramine are contained in certain food items (cheese, red wine and marmite), forming the basis of the pressor ‘cheese reaction’ in these patients (see p. 403).
4 Although it is simplest to regard the selectivity of a drug as relative, being lost at higher doses, strictly speaking it is the benefits of the receptor selectivity of an agonist or antagonist that are dose-dependent. A 10-fold selectivity of an agonist at the β1 receptor, for instance, is a property of the agonist that is independent of dose, and means simply that 10 times less of the agonist is required to activate this receptor compared with the β2 subtype.
5 Normal subjects, infused with intravenous adrenaline/epinephrine in amounts that approximate to those found in the plasma after severe myocardial infarction, show a fall in plasma potassium concentration of about 0.8 mmol/L (Brown M J, Brown D C, Murphy M B 1983 Hypokalemia from beta2-receptor stimulation by circulating epinephrine. New England Journal of Medicine 309:1414–1419).
6 Ephedra alkaloids are found in Chinese herbal remedies (ma huang) and in guarana-derived caffeine products which are widely consumed as appetite suppressants or for energy enhancement. These have been associated with stroke and seizures only rarely. The relationship of phenylpropanolamine consumption and haemorrhagic stroke seems clearer and has led to the suspension of its sale in the USA (Fleming G A 2000 The FDA, regulation, and the risk of stroke. New England Journal of Medicine 243:1886–1887).
7 In fact, a cocktail of substances (autacoids) – kinins, prostaglandins, leukotrienes, histamine, endorphins, serotonin, vasopressin – has been implicated. In endotoxic shock, the toxin also induces synthesis of nitric oxide, the endogenous vasodilator, in several types of cell other than the endothelial cells that are normally its main source.
8 Bernard G D, Vincent J L, Laterre P F et al 2001 Efficacy and safety of recombinant human activated protein C for severe sepsis. New England Journal of Medicine 344:699–709.
9 Nolan J 2001 Fluid resuscitation for the trauma patient. Resuscitation 48:57–69.
10 Perel P, Roberts I, Pearson M 2007 Colloids versus crystalloids for fluid resuscitation in critically ill patients. Cochrane Database of Systematic Reviews 2007, Issue 4. Art. No.: CD000567. DOI: 10.1002/14651858.CD000567.pub3. Available online at: http://mrw.interscience.wiley.com/cochrane/clsysrev/articles/CD000567/frame.html (accessed 2 August 2010)
11 Effective doses may not restore blood volume and may work by sensitising vascular adrenoceptors.
12 Karet F E, Dickerson J E C, Brown J et al 1994 Bovril and moclobemide: a novel therapeutic strategy for central autonomic failure. Lancet 344:1263–1265.
13 Drüeke TB, Locatelli F, Clyne N et al 2006 Normalization of hemoglobin level in patients with chronic kidney disease and anemia. New England Journal of Medicine 355(20): 2071–2084. And FDA advisory: http://www.fda.gov/Drugs/DrugSafety/PostmarketDrugSafetyInformationforPatientsandProviders/ucm126485.htm (accessed 2 August 2010)
[/level-membership-for-basic-science-category][not-level-membership-for-basic-science-category]
Chapter 23 Adrenergic mechanisms and drugs
Adrenergic mechanisms
The discovery in 1895 of the hypertensive effect of adrenaline/epinephrine was initiated by Dr Oliver, a physician in practice, who conducted a series of experiments on his young son into whom he injected an extract of bovine suprarenal and detected a ‘definite narrowing of the radial artery’.1 The effect was confirmed in animals and led eventually to the isolation and synthesis of adrenaline/epinephrine in the early 1900s. Many related compounds were examined and, in 1910, Barger and Dale invented the word sympathomimetic2 and also pointed out that noradrenaline/norepinephrine mimicked the action of the sympathetic nervous system more closely than did adrenaline/epinephrine.
Classification of sympathomimetics
By mode of action
Noradrenaline/norepinephrine is synthesised and stored in vesicles within adrenergic nerve terminals (Fig. 23.1). The vesicles can be released from these stores by stimulating the nerve or by drugs (ephedrine, amfetamine). The noradrenaline/norepinephrine stores can also be replenished by intravenous infusion of noradrenaline/norepinephrine, and abolished by reserpine or by cutting the sympathetic nerve. Sympathomimetics may be classified on the basis of their sites of action (see Fig. 23.1) as acting:
1. Directly: adrenoceptor agonists, e.g. adrenaline/epinephrine, noradrenaline/norepinephrine, isoprenaline (isoproterenol), methoxamine, xylometazoline, metaraminol (entirely); and dopamine and phenylephrine (mainly).
2. Indirectly: by causing a release of preformed noradrenaline/norepinephrine from stores in nerve endings,3 e.g. amfetamines, tyramine; and ephedrine (largely).
3. By both mechanisms (1 and 2, though one usually predominates): other synthetic agents.
All of the above mechanisms operate in both the central and peripheral nervous systems, but discussion below will focus on agents that influence peripheral adrenergic mechanisms.
Tachyphylaxis
(rapidly diminishing response to repeated administration) is a particular feature of group 2 drugs. It reflects depletion of the ‘releasable’ pool of noradrenaline/norepinephrine from adrenergic nerve terminals that makes these agents less suitable as, for example, pressor agents than drugs in group 1. Longer-term tolerance (see p. 78) to the effects of direct sympathomimetics is much less of a clinical problem and reflects an alteration in adrenergic receptor density or coupling to second messenger systems.
Interactions of sympathomimetics
with other vasoactive drugs are complex. Some drugs block the reuptake mechanism for noradrenaline/norepinephrine in adrenergic nerve terminals and potentiate the pressor effects of noradrenaline/norepinephrine, e.g. cocaine, tricyclic antidepressants or highly noradrenaline/norepinephrine-selective reuptake inhibitors (NSRIs) such as reboxetine (see Fig. 23.1). Others deplete or destroy the intracellular stores within adrenergic nerve terminals (e.g. reserpine and guanethidine) and thus block the action of indirect sympathomimetics.
History
Subclassification of adrenoceptors is shown in Table 23.1.
Table 23.1 Clinically relevant aspects of adrenoceptor functions and actions of agonists
| α1-Adrenoceptor effectsa | β-Adrenoceptor effects |
|---|---|
| Eye:b mydriasis | |
| Heart (β1, β2):c | |
| increased rate (SA node) | |
| increased automaticity (AV node and muscle) | |
| increased velocity in conducting tissue | |
| increased contractility of myocardium | |
| increased oxygen consumption; decreased refractory period of all tissues | |
| Arterioles: | Arterioles: |
| constriction (only slight in coronary and cerebral) | dilatation (β2) |
| Bronchi (β2): relaxation | |
| Anti-inflammatory effect: | |
| inhibition of release of autacoids (histamine, leukotrienes) from mast cells, e.g. asthma in type I allergy | |
| Uterus: contraction (pregnant) | Uterus (β2): relaxation (pregnant) |
| Skeletal muscle: tremor (β2) | |
| Skin: sweat, pilomotor | |
| Male ejaculation | |
| Blood platelet: aggregation | |
| Metabolic effect: | Metabolic effects: |
| hyperkalaemia | hypokalaemia (β2) |
| hepatic glycogenolysis (β2) | |
| lipolysis (β1, β2) | |
| Bladder sphincter: contraction | Bladder detrusor: relaxation |
| Intestinal smooth muscle relaxation is mediated by α and β adrenoceptors. α2-adrenoceptor effects:a α2 receptors on the nerve ending, i.e. presynaptic autoreceptors, mediate negative feedback which inhibits noradrenaline/norepinephrine release |
|
| Use of the term cardioselective to mean β1-receptor selective only, especially in the case of β-receptor blocking drugs, is no longer appropriate. | |
| Although in most species the β1 receptor is the only cardiac β receptor, this is not the case in humans. What is not generally appreciated is that the endogenous sympathetic neurotransmitter noradrenaline/norepinephrine has about a 20-fold selectivity for the β1 receptor – similar to that of the antagonist atenolol – with the consequence that under most circumstances, in most tissues, there is little or no β2-receptor stimulation to be affected by a non-selective β-blocker. Why asthmatics should be so sensitive to β-blockade is paradoxical: all the bronchial β receptors are β2, but the bronchi themselves are not innervated by noradrenergic fibres and the circulating adrenaline levels are, if anything, low in asthma. | |
a For the role of subtypes (α1 and α2), see prazosin.
b Effects on intraocular pressure involve both α and β adrenoceptors as well as cholinoceptors.
c Cardiac β1 receptors mediate effects of sympathetic nerve stimulation. Cardiac β2 receptors mediate effects of circulating adrenaline, when this is secreted at a sufficient rate, e.g. following myocardial infarction or in heart failure. Both receptors are coupled to the same intracellular signalling pathway (cyclic AMP production) and mediate the same biological effects.
Consequences of adrenoceptor activation
All adrenoceptors are members of the G-coupled family of receptor proteins, i.e. the receptor is coupled to its effector protein through special transduction proteins called G-proteins (themselves a large protein family). The effector protein differs among adrenoceptor subtypes. In the case of β-adrenoceptors, the effector is adenylyl cyclase and hence cyclic AMP is the second messenger molecule. For α-adrenoceptors, phospholipase C is the commonest effector protein and the second messenger here is inositol trisphosphate (IP3). It is the cascade of events initiated by the second messenger molecules that produces the variety of tissue effects shown in Table 23.1. Hence, specificity is provided by the receptor subtype, not the messengers.
Selectivity for adrenoceptors
The following classification of sympathomimetics and antagonists is based on selectivity for receptors and on use. But selectivity is relative, not absolute; some agonists act on both α and β receptors, some are partial agonists and, if sufficient drug is administered, many will extend their range. The same applies to selective antagonists (receptor blockers), e.g. a β1-selective-adrenoceptor blocker can cause severe exacerbation of asthma (a β2 effect), even at low dose. It is important to remember this because patients have died in the hands of doctors who have forgotten or been ignorant of it.4
Adrenoceptor agonists (see Table 23.1)
Effects of a sympathomimetic
To block all the effects of adrenaline/epinephrine and noradrenaline/norepinephrine, antagonists for both α and β receptors must be used. This can be a matter of practical importance, e.g. in phaeochromocytoma (see p. 419).
Physiological note
The termination of action of noradrenaline/norepinephrine released at nerve endings is by:
• reuptake into nerve endings by the noradrenaline/norepinephrine transporter where it is stored in vesicles or metabolised by monoamine oxidase (MAO) (see Fig. 23.1)
• diffusion away from the area of the nerve ending and the receptor (junctional cleft)
• metabolism (by extraneuronal MAO and catechol-O-methyltransferase, COMT).
[/not-level-membership-for-basic-science-category]
