Adrenal malignancies
1. What types of cancers occur in the adrenal glands?
Carcinomas may arise in the adrenal cortex (adrenocortical carcinoma [ACC]) or the adrenal medulla (malignant pheochromocytomas). Metastatic cancer from other sites may also be seen.
2. Do adrenocortical carcinomas produce hormones?
3. What are the clinical features of functioning adrenocortical carcinomas?
A functioning ACC secretes cortisol, androgens, estrogens, or aldosterone—alone or in combination. Cortisol overproduction is most common (∼45%) and results in Cushing syndrome. Androgen secretion (∼25%) causes hirsutism and virilization in women and precocious puberty in children but is often asymptomatic in men. Estrogen production causes menstrual disturbances in women and gynecomastia and hypogonadism in men. Excess aldosterone secretion causes hypertension and hypokalemia (Conn syndrome) (Fig. 29-1).
4. What are the clinical features of nonfunctioning adrenocortical carcinomas?
A nonfunctioning ACC manifests clinically as abdominal or flank pain or as an adrenal mass discovered on physical examination or incidentally during an imaging procedure.
5. What imaging procedure is best for evaluating an adrenal mass?
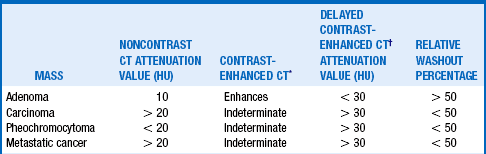
Computed tomography (CT) is the method of choice to determine the size and physical characteristics of the mass. Features that most strongly suggest ACC are size greater than 6 cm, heterogeneity, calcifications, irregular borders, local invasion, lymphadenopathy, and decreased lipid content. The last feature is assessed by signal attenuation, expressed in Hounsfield units (HU). An adrenal mass CT protocol is employed in many institutions; this consists of a non-contrast CT, a contrast-enhanced CT, a delayed contrast-enhanced CT, and calculation of the relative washout percentage (RWP) of enhancement. Table 29-1 shows the typical CT findings for some commonly seen adrenal masses. MRI can also be used to assess the size, features, and lipid content of adrenal masses but is more expensive. Fluorodeoxyglucose (18F) positron emission tomography (FDG-PET) or PET-CT fusion scanning may also be useful, especially for masses with high HU or low RWP on CT. High sensitivities and specificities have been reported with this technique using a standard uptake value (SUV) cutoff of 3.1. Other PET tracers, such as metomidate 11C, may offer even better sensitivity in the future.
6. What hormone tests should be used to evaluate an adrenal mass?
The goal is to determine whether the mass is producing hormones that may cause symptoms or that may be indicative of malignancy (androgens). Many experts recommend a focused evaluation consisting of an overnight 1-mg dexamethasone suppression test, measurement of plasma free metanephrines or fractionated urinary catecholamines and metanephrines, and, for hypertensive patients, measurement of plasma aldosterone and renin. Tests currently recommended by the European Network for the Study of Adrenal Tumors (ENSAT) are shown in Table 29-2.
TABLE 29-2.
HORMONAL EVALUATION OF THE INCIDENTAL ADRENAL MASS PROPOSED BY ENSAT
| Cortisol testing (3 of 4 tests) | Dexamethasone suppression test (1-mg) |
| Urine cortisol (24-hour) | |
| Serum cortisol, basal | |
| Plasma ACTH, basal | |
| Sex steroid testing (all) | Serum testosterone |
| Serum dehydroepiandrosterone sulfate (DHEA-S) | |
| Serum androstenedione | |
| Serum 17-OH progesterone | |
| Serum estradiol (men, postmenopausal women) | |
| Aldosterone testing (if hypertension present) | Plasma aldosterone |
| Plasma renin activity | |
| Serum potassium | |
| Pheochromocytoma testing (1 or 2 tests) | Plasma metanephrines |
| Urine metanephrines (24-hour) |
ENSAT (European Network for the Study of Adrenal Tumors).
Adapted from Lacroix A: Approach to the patient with adrenocortical carcinoma. J Clin Endocrinol Metab 95: 4812-4822, 2010
7. How should the incidentally discovered adrenal mass be managed?
Surgery is often recommended for tumors larger than 4 cm, for those showing significant growth on follow-up, and for those with evidence of excessive cortisol, androgen, estrogen, aldosterone, or catecholamine secretion. Nonfunctioning adrenal masses smaller than 4 cm should be reassessed in 6 months and then annually thereafter.
8. Describe a useful staging system for adrenocortical carcinoma.
TABLE 29-3.
STAGING SYSTEM FOR ADRENOCORTICAL CARCINOMA PROPOSED BY ENSAT IN 2008
| STAGE | TNM |
| I | T1, N0, M0 |
| II | T2, N0, M0 |
| III | T1-2, N1, M0 |
| T3-4, N0-1, M0 | |
| IV | T1-4, N0-1, M1 |
ENSAT (European Network for the Study of Adrenal Tumors).
Adapted from Lacroix A: Approach to the patient with adrenocortical carcinoma. J Clin Endocrinol Metab 95:4812–4822, 2010
9. Describe the initial treatment for an adrenocortical carcinoma.
Surgery is the initial treatment of choice for ACC. If the tumor resection is complete, adjuvant therapy with mitotane, an adrenocorticolytic agent, is recommended because of the high recurrence rate (∼60%). After an incomplete resection, adjuvant mitotane alone or combined with streptozotocin is recommended. Adjuvant tumor bed radiation therapy is also advised by some investigators. Follow-up imaging, with or without tumor marker measurement, should be performed every 3 months for 1 year, then every 6 months for 5 years, and then annually thereafter. In patients receiving mitotane, serum mitotane levels should be monitored and maintained in the 14 to 20 μg/mL range; concomitant glucocorticoid with or without mineralocorticoid replacement therapy is necessary in all patients except those with hypercortisolism. Patients with persistent hypercortisolism despite mitotane use may need additional therapy with an enzyme inhibitor (ketoconazole, metyrapone).
10. How should advanced, metastatic, and recurrent adrenal cortical carcinoma be managed?
Aggressive surgery, when possible, and radiation therapy may offer benefit, although usually only temporarily. Chemotherapy regimens using either mitotane plus etoposide, doxorubicin, and cisplatin (EDC) or mitotane plus streptozotocin have shown benefit, but superior response rates and better progression-free survival have been reported with mitotane-EDC. Targeted therapies utilizing tyrosine kinase inhibitors and growth factor receptor (insulin-like growth factor 1 receptor, epidermal growth factor receptor) inhibitors also show some promise. Targeted radionuclide therapy with iodometomidate 131I offers a novel approach with encouraging results reported thus far.
11. What is the prognosis for patients with adrenocortical carcinoma?
The mean survival is 15 months. The 5-year survival rate is less than 30%. Prognosis is improved by young age, small tumor size, localized disease, complete tumor resection, and nonfunctioning of the tumor. Table 29-4 shows 5-year disease-free survival rates by disease stage.
TABLE 29-4.
FIVE-YEAR DISEASE-FREE SURVIVAL FOR ADRENOCORTICAL CARCINOMA ACCORDING TO ENSAT 2008 STAGING SYSTEM
| STAGE | 5-YEAR DISEASE-FREE SURVIVAL |
| I | 82% |
| II | 61% |
| III | 50% |
| IV | 13% |
ENSAT (European Network for the Study of Adrenal Tumors).
Adapted from Lacroix A: Approach to the patient with adrenocortical carcinoma. J Clin Endocrinol Metab 95:4812-4822, 2010
12. How often are pheochromocytomas malignant?
13. What are the clinical features of a malignant pheochromocytoma?
Pheochromocytomas, whether benign or malignant, usually cause hypertension, headaches, sweating, and palpitations. They are diagnosed biochemically by the finding of increased levels of metanephrine or catecholamines in the plasma or urine. Malignant pheochromocytomas often do not differ clinically or histologically at presentation from those that are benign.
14. What clues suggest that a pheochromocytoma is malignant?
Malignancy is most strongly suggested by tumor size greater than 6 cm, evidence of extra-adrenal spread (usually to the lymph nodes, liver, lungs, or bones) and disproportionately increased plasma or urine levels of dopamine. Because malignant pheochromocytomas cannot be distinguished from benign ones histologically, the malignant character of some tumors may not become apparent until metastatic disease appears.
15. Which of the familial pheochromocytoma syndromes is most commonly associated with malignant pheochromocytomas?
Table 29-5 lists the four well-recognized familial pheochromocytoma and paraganglioma syndromes. Only the succinate dehydrogenase B mutation, an autosomal-dominantly inherited condition, is associated with malignant pheochromocytomas and paragangliomas.
TABLE 29-5.
GENETIC SYNDROMES ASSOCIATED WITH PHEOCHROMOCYTOMAS AND/OR PARAGANGLIOMAS
| SYNDROME | GENE MUTATION |
| Multiple endocrine neoplasia 2 | Ret |
| Von Hippel–Lindau syndrome | VHL |
| Neurofibromatosis 1 | NF-1 |
| Succinate dehydrogenase B mutation | SDH |
16. What are the best tests to localize metastatic pheochromocytomas?
CT or magnetic resonance imaging (MRI) localizes about 80% to 90% of metastatic pheochromocytomas. When these have negative results or are not feasible, functional imaging with metaiodobenzylguanidine (MIBG) 123I scintigraphy, octreotide scintigraphy (OctreoScan), and PET with [18F] fluorodeoxyglucose (FDG-PET) are the best current options. Other highly specific functional PET imaging agents, such as 6-[18F] fluorodopamine and 6-[18F] fluorodopa are in development but are not yet generally available.
17. What is the treatment for a malignant pheochromocytoma?
Surgery is the treatment of choice, when possible. Preoperatively, alpha-adrenergic blocking agents (phenoxybenzamine, prazosin) or calcium channel blockers are given to control blood pressure and to replete intravascular volume. Beta-blockers may then be added for reflex tachycardia or persistent hypertension. If surgery is not possible or successful, other available therapies are only palliative. Alpha-blockers, calcium channel blockers, and the catecholamine synthesis inhibitor α-methyltyrosine can provide blood pressure and symptom control. Therapies for which partial tumor responses have been reported include chemotherapy with cyclophosphamide, vincristine, and dacarbazine, and radionuclide therapy with 131I MIBG.
18. What is the prognosis for malignant pheochromocytoma?
19. What tumors metastasize to the adrenal glands?
The vascular adrenal glands are a common site of bilateral metastatic spread from cancers of the lung, breast, stomach, pancreas, colon, and kidney and from melanomas and lymphomas.
20. What is the clinical significance of metastatic disease to the adrenal glands?
Acute adrenal crises are rare. However, up to 33% of patients may have subtle adrenal insufficiency manifested by nonspecific symptoms and an inadequate response (peak cortisol level < 20 μg/dL) to a 250-μg cosyntropin stimulation test. These patients may experience improvement in well-being when given physiologic glucocorticoid replacement.
 KEY POINTS 1: ADRENAL MALIGNANCIES
KEY POINTS 1: ADRENAL MALIGNANCIES
1. Adrenal cortical carcinoma manifests with features of excess cortisol, androgens, estrogens, or aldosterone; with abdominal or flank pain; or as an incidentally discovered adrenal mass.
2. Malignant pheochromocytomas often manifest with features similar to those of benign pheochromocytomas (hypertension, headaches, palpitations, sweating).
3. Features suggesting that an adrenal tumor is malignant are size > 6 cm, heterogeneity, calcifications, irregular borders, local invasion, lymphadenopathy, decreased lipid content (Hounsfield units > 20), or elevations of serum androgens or urinary or plasma dopamine.
4. Surgery is the treatment of choice for all malignant adrenal tumors; mitotane ± tumor bed radiation therapy is the recommended adjuvant therapy for adrenocortical carcinomas.
5. Incidentally discovered adrenal masses should be evaluated for evidence of malignancy (size > 6 cm or progressive growth) and excess hormone secretion (cortisol, androgens, estrogens, aldosterone, catecholamines).
Abiven, G, Coste, J, Groussin, L, et al, Clinical and biological features in the prognosis of adrenocortical cancer. poor outcome of cortisol-secreting tumors in a series of 202 consecutive patients. J Clin Endocrinol Metab 2006;91:2650–2655.
Allolio, B, Fassnacht, M, Clinical review. Adrenocortical carcinomaclinical update. J Clin Endocrinol Metab 2006;91:2027–2037.
Assie, G, Antoni, G, Tissier, F, et al. Prognostic parameters of metastatic adrenocortical carcinoma. J Clin Endocrinol Metab. 2007;92:148–154.
Fassnacht, M, Hahner, S, Polat, B, et al. Efficacy of adjuvant radiotherapy of the tumor bed on local recurrence of adrenocortical carcinoma. J Clin Endocrinol Metab. 2006;91:4501–4504.
Fassnacht, M, Johanssen, S, Fenske, W, et al. Improved survival in patients with stage II adrenocortical carcinoma followed up prospectively by specialized centers. J Clin Endocrinol Metab. 2010;95:4925–4932.
Fassnacht, M, Terzolo, M, Allolio, B, et al. Combination chemotherapy in advanced adrenocortical carcinoma. N Engl J Med. 2012;366:2189–2197.
Groussin, L, Bonardel, G, Silvera, S, et al, 18F-Fluorodeoxyglucose positron emission tomography for the diagnosis of adrenocortical tumors. a prospective study in 77 operated patients. J Clin Endocrinol Metab 2009;94:1713–1722.
Hahner, S, Kreissl, MC, Fassnacht, M, et al. 131-I Iodometomidate for targeted radionuclide therapy of advanced adrenocortical carcinoma. J Clin Endocrinol Metab. 2012;97:914–922.
Hermsen, IG, Fassnacht, M, Terzolo, M, et al, Plasma concentrations of o,p′DDD, o,p′DDA, and o,p′DDE as predictors of tumor response to mitotane in adrenocortical carcinoma. results of a retrospective ENSAT Multicenter Study. J Clin Endocrinol Metab 2011;96:1844–1851.
Kirschner, LS, Review. Emerging treatment strategies for adrenocortical carcinomaa new hope. J Clin Endocrinol Metab 2006;91:14–21.
Lacroix, A. Approach to the patient with adrenocortical carcinoma. J Clin Endocrinol Metab. 2010;95:4812–4822.
Leboulleux, S, Dromain, G, Bonniaud, G, et al, Diagnostic and prognostic value of 18-fluorodeoxyglucose positron emission tomography in adrenocortical carcinoma. a prospective comparison with computed tomography. J Clin Endocrinol Metab 2006;91:920–925.
Mackie, GC, Shulkin, BL, Ribiero, RC, et al. Use of [F18] fluorodeoxyglucose positron emission tomography in evaluating locally recurrent and metastatic adrenocortical carcinoma. J Clin Endocrinol Metab. 2006;91:2665–2671.
Pacak, K, Eisenhofer, G, Ahlman, H, et al, Pheochromocytoma. recommendations for clinical practice from the First International Symposium. Nat Clin Pract Endocrinol Metab 2007;3:92–102.
Terzolo, M, Angeli, A, Fassnacht, M, et al. Adjuvant mitotane treatment for adrenocortical carcinoma. N Engl J Med. 2007;356:2372–2380.
Young, WF. The incidentally discovered adrenal mass. N Engl J Med. 2007;356:601–610.
