Chapter 35 Adrenal corticosteroids, antagonists, corticotropin
• Adrenocortical steroids and their synthetic analogues.













In 1855, Dr Thomas Addison, assisted in his observations by three colleagues, published his famous monograph ‘On the constitutional effects of disease on the suprarenal capsules’ (Addison’s disease). It was not until the late 1920s that the vital importance of the adrenal cortex was appreciated and the distinction made between the hormones secreted by the cortex and medulla.
Adrenal steroids and their synthetic analogues
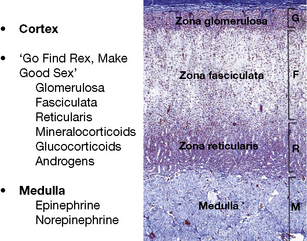
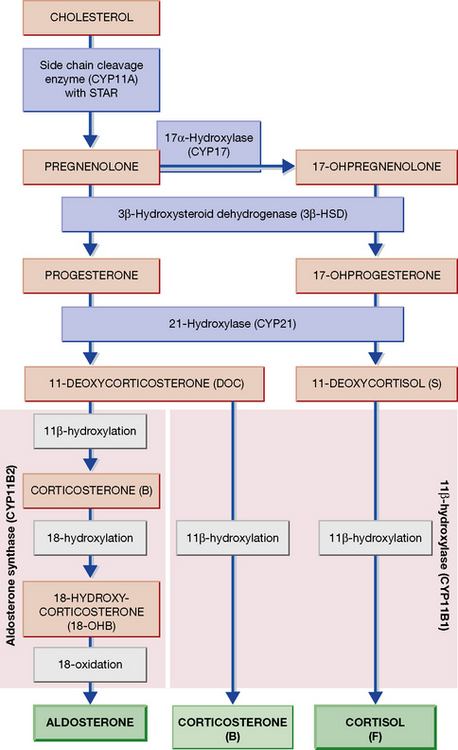
The adrenal is a composite endocrine gland, and each zone of the cortex synthesises a different predominant steroid; a mnemonic is offered in Figure 35.1, in which the first letter of each word is the first letter of each zone and its corresponding steroid product. The principal synthetic pathways are illustrated in Figure 35.2.
In the account that follows, the effects of hydrocortisone will be described and then other steroids in so far as they differ. In the context of this chapter, ‘adrenal steroid’ means a substance with hydrocortisone-like activity. Androgens are described in Chapter 38.
Mechanism of action
Glucocorticoids stimulate the cell through both a classical cytosolic receptor that, on binding with agonist, translocates to the nucleus, and an unidentified membrane-bound receptor (Fig. 35.3). The classical receptor is responsible for so-called genomic effects through either activation or repression of DNA transcription. Up-regulation of gene transcription occurs when the receptor dimerises on specific DNA glucocorticoid response elements (GREs) with consequent recruitment of coactivator proteins. Many of the undesired effects of glucocorticoid occur through this pathway.
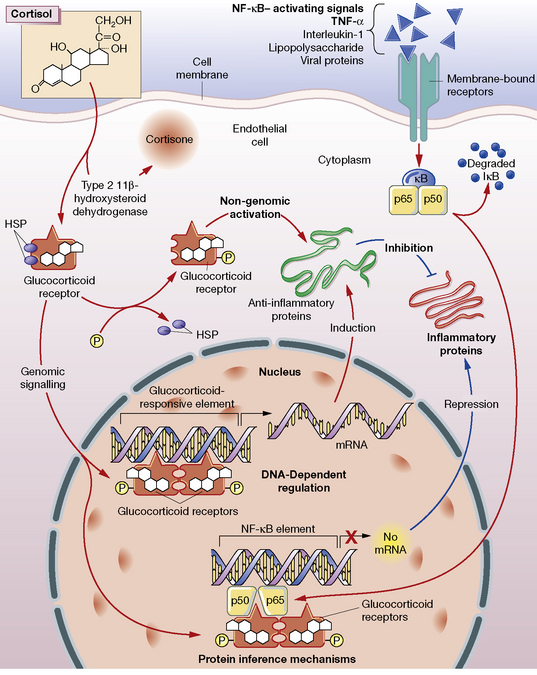
Repression of DNA transcription occurs at slightly lower cortisol concentrations than required for transactivation. Through protein–protein interaction, the glucocorticoid–receptor complex inactivates pro-inflammatory transcription factors such as nuclear factor (NF)-κB and activator protein 1 (AP-1), preventing their stimulation of inflammatory mediators: prostaglandins, leukotrienes, cytokines and platelet-activating factor. These mediators would normally contribute to increased vascular permeability and subsequent changes including oedema, leucocyte migration and fibrin deposition.1
On organic metabolism
• Carbohydrate metabolism. Glycogenolysis and gluconeogenesis are increased and peripheral glucose utilisation is decreased (due to insulin antagonism).
• Protein metabolism. Anabolism (conversion of amino acids to protein) decreases but catabolism continues, so that there is a negative nitrogen balance with muscle wasting. The skin atrophies and this, with increased capillary fragility, causes bruising and striae. Healing of peptic ulcers or of wounds is delayed, as is fibrosis.
• Bone metabolism. Cortisol inhibits the number and function of osteoblasts, and the synthesis of collagen. Osteoporosis (reduction of bone protein matrix) is the main consequence of chronic glucocorticoid administration. Growth slows in children.
• Fat metabolism. Lipolysis is increased, and the secretion of leptin (the appetite suppressant) is inhibited. These actions lead to increased appetite and deposition of adipose tissue, particularly on shoulders, face and abdomen.
• Inflammatory response is depressed. Neutrophil and macrophage function are depressed, including the release of chemical mediators and the effects of these on capillaries.
• Allergic responses are suppressed. The antigen–antibody interaction is unaffected, but its injurious inflammatory consequences do not follow.
• Antibody production is lessened by heavy doses.
• Lymphoid tissue is reduced (including leukaemic lymphocytes).
• Renal excretion of urate is increased.
• Blood eosinophils reduce in number and neutrophils increase.
• Euphoria or psychotic states may occur, perhaps due to central nervous system (CNS) electrolyte changes.
• Anti-vitamin D action, see calciferol (p. 635).
• Reduction of hypercalcaemia, chiefly where this is due to excessive absorption of calcium from the gut (sarcoidosis, vitamin D intoxication).
• Urinary calcium excretion is increased and renal stones may form.
• Growth reduces where new cells are being added (growth in children), but not where they are replacing cells as in adult tissues.
• Suppression of hypothalamic–pituitary–adrenocortical feedback system (with delayed recovery) occurs with chronic use, so that abrupt withdrawal leaves the patient in a state of adrenocortical insufficiency.
Individual adrenal steroids
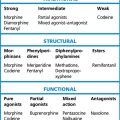
The relative potencies1 for glucocorticoid and mineralocorticoid (sodium-retaining) effects (Table 35.1) are central to the choice of agent in relation to clinical indication.
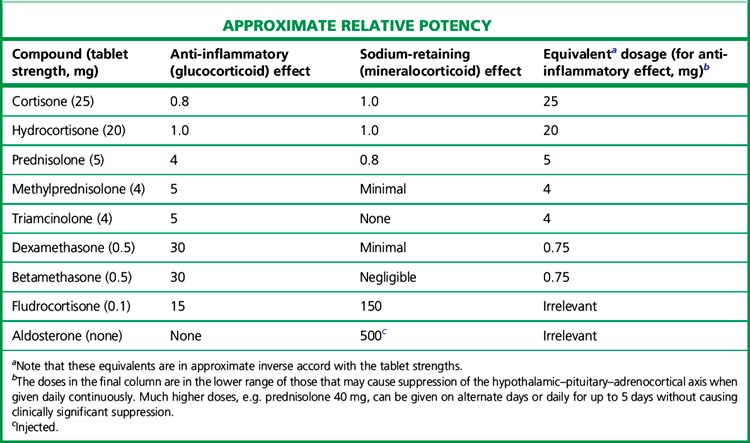
All drugs in Table 35.1 except aldosterone are active when swallowed, being protected from hepatic first-pass metabolism by high binding to plasma proteins. Some details of preparations and equivalent doses appear in the table. Injectable and topical forms are available (creams, suppositories, eye drops).
Fluorinated corticosteroids (triamcinolone, fludrocortisone)
Spironolactone
(see p. 550) is a competitive mineralocorticoid (aldosterone) antagonist. It is used in the treatment of primary hyperaldosteronism, as a diuretic in resistant hypertension, and when severe oedema is due to secondary hyperaldosteronism, e.g. cirrhosis, congestive cardiac failure. Long-term treatment increases survival in cardiac failure, possibly through blocking the fibrotic effect of aldosterone upon the heart. The dose of spironolactone is limited by its anti-androgen activity. Eplerenone has greater selectivity than spironolactone for the mineralocorticoid than androgen receptor, but has lower efficacy in blocking aldosterone and may need to be used together with amiloride.
Beclometasone, budesonide, fluticasone, mometasone and ciclesonide
are potent soluble steroids suitable for use by inhalation for asthma (see p. 474) and intranasally for hay fever. Patients swallow about 90% of an inhalation dose, which is then largely inactivated by hepatic first-pass. The drugs are listed in order of development; some newer agents possess properties (first-pass metabolism, high protein binding and lipophilicity) that may increase pulmonary residence time and reduce systemic effects. The main protection against these effects is simply that absorption through mouth, lungs and gut is low relative to the amounts used in systemic administration. The risk of suppression of the hypothalamic–pituitary–adrenal (HPA) axis is infrequent and dose dependent. The greatest risk of suppression appears to occur with high-dose fluticasone usage in children, who may present with hypoglycaemia and acute adrenal insufficiency.
Dosage schedules
Choice of adrenal steroid: summary
• For oral replacement therapy in adrenocortical insufficiency, use hydrocortisone as the glucocorticoid. In primary adrenal failure (Addison’s disease and congenital adrenal hyperplasia), use fludrocortisone as the mineralocorticoid to replace aldosterone.
• For anti-inflammatory and antiallergic (immunosuppressive) effect, use prednisolone or dexamethasone. For inhalation, more potent adrenal steroids are required, e.g. beclometasone or budesonide.
• In diagnostic testing use dexamethasone. In adults with congenital adrenal hyperplasia use hydrocortisone, prednisolone or dexamethasone and hydrocortisone in children.
Adverse effects of systemic adrenal steroid pharmacotherapy
The principal adverse effects of chronic corticosteroid administration are:
Precautions during chronic adrenal steroid therapy
• carry a steroid card giving details of therapy
• be impressed with the importance of compliance
• know what to do if they develop an intercurrent illness or other severe stress, i.e. double their next dose and consult their doctor. If a patient omits a dose, a replacement dose should be taken as soon as possible to maintain the same total daily intake, because every patient should be taking the minimum dose necessary to control the disease
• have access to parenteral hydrocortisone (their own supply for i.m. injection, if urgent medical referral is not always possible).
Membership of one of the self-help patient groups can be helpful.
Dosage and routes of administration
No single schedule suits every case, but examples appear below.
Systemic commencing doses:
• For a serious disease such as systemic lupus, dermatomyositis: prednisolone up to 0.5–1.5 mg/kg daily, orally in divided doses. There is no evidence of increased benefit above 60–80 mg/day.
• If the condition is life-threatening, give prednisolone up to 60 mg, or its equivalent of another steroid. Cyclophosphamide or azathioprine (see p. 250) are valuable adjuncts which may enhance the initial control of the disease and have a sparing effect on the maintenance dose of prednisolone.
• Alternatively, high dose pulses (methylprednisolone 1.0 g i.v. daily for 3 days) may be used, followed by oral maintenance with prednisolone and/or a steroid-sparing agent (above).
• For less dangerous disease, e.g. rheumatoid arthritis, give prednisolone 7.5–10.0 mg daily, adjusted later according to the response.
• In particular cases, including replacement of adrenal insufficiency, the dosage appears in the account of the disease.
• For continuous therapy, give the minimum amount to produce the desired effect. Imperfect control may have to be accepted by the patient if full control, e.g. of rheumatoid arthritis, though obtainable, involves doses that will lead to toxicity, e.g. osteoporosis, if continued for years.
Uses of adrenocortical steroids
Replacement therapy
Acute adrenocortical insufficiency (Addisonian crisis)
• An intravenous infusion of sodium chloride solution (0.9%) is set up immediately and a second 100 mg hydrocortisone is added to the first litre, which should be given as quickly as the cardiovascular status permits (2–3 L of sodium chloride may be infused in the first 1–2 h and 5–6 L may be needed in the first 24 h).
• The patient should then receive hydrocortisone 50 mg i.v. or i.m. 6-hourly for 24–72 h; thereafter a total of 30–40 mg/day orally in two or three divided doses usually suffices.
Chronic primary adrenocortical insufficiency (Addison’s disease)
Hydrocortisone is given orally (15–25 mg total daily) in two to three divided doses, according to the algorithm in Figure 35.4. Some patients working under increased physical activity or mental stress may require higher doses up to 40 mg daily. The aim is to mimic the natural diurnal rhythm of secretion. All patients also require mineralocorticoid replacement, and fludrocortisone; 50–200 micrograms orally once a day is the usual dose.
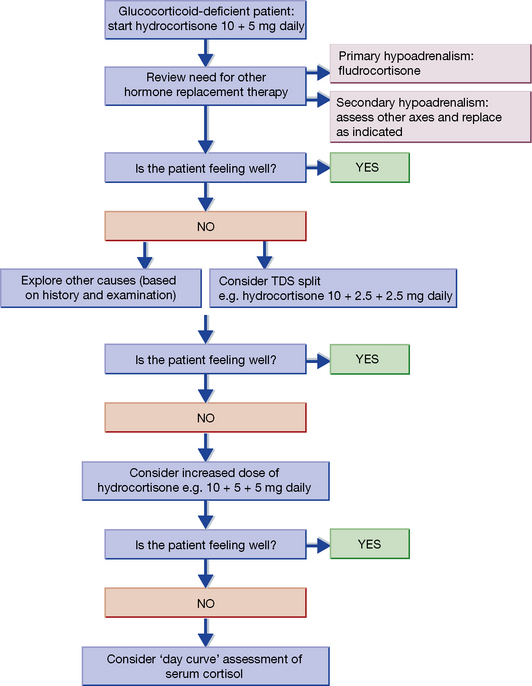
If there is vomiting, the parenteral replacement hormone must be given without delay.
Chronic secondary adrenocortical insufficiency
This occurs in hypopituitarism. The need for hydrocortisone may be less than in primary insufficiency, especially when ACTH deficiency is partial. Some patients with borderline adrenocortical insufficiency may require steroid supplementation only during periods of stress, such as infection or surgery. Mineralocorticoid replacement is seldom required, for the pituitary has little control over aldosterone production. Other pituitary replacement is given as appropriate (see p. 596).
Pharmacotherapy
Further specific uses
Adrenal steroids are used in nearly all cases of the following:
• Exfoliative dermatitis and pemphigus, if severe.
• Connective tissue diseases, if severe, e.g. lupus erythematosus (systemic), polyarteritis nodosa, polymyalgia rheumatica and cranial giant cell arteritis (urgent therapy to save sight), dermatomyositis.
• Acute lymphatic leukaemia (see Ch. 31).
• Acquired haemolytic anaemia.
• Severe allergic reactions of all kinds, e.g. serum sickness, angioedema, trichiniasis. Alone, they will not control acute manifestations of anaphylactic shock as they do not act quickly enough.
• Acute spinal cord injury: early, brief, and high dose (to reduce the oedema/inflammation).
• Autoimmune active chronic hepatitis: a corticosteroid improves well-being, liver function and histological findings; prednisolone will benefit some 80% and should be continued in the long term, as most patients relapse if the drug is withdrawn.
Adrenal steroids are used in some cases of the following:
• Ulcerative colitis and proctitis.
• Regional enteritis (Crohn’s disease).
• Hay fever (allergic rhinitis); also some bronchitics with marked airways obstruction.
• Sarcoidosis. If there is hypercalcaemia or threat to a major organ, e.g. eye, adrenal steroid administration is urgent. Pulmonary fibrosis may be delayed and CNS manifestations may improve.
• Acute mountain/altitude sickness, to reduce cerebral oedema.
• Prevention of adverse reaction to radiocontrast media in patients who have had a previous severe reaction.
• Blood diseases due to circulating antibodies, e.g. thrombocytopenic purpura (there may also be a decrease in capillary fragility with lessening of purpura even though thrombocytes remain few); agranulocytosis.
• Eye diseases. Allergic diseases and non-granulomatous inflammation of the uveal tract. Bacterial and viral infections may be made worse and use of corticosteroids to suppress inflammation of infection is generally undesirable, is best left to ophthalmologists and must be accompanied by effective chemotherapy; this is of the greatest importance in herpes virus infection. Corneal integrity should be checked before use (by instilling a drop of fluorescein). Prolonged use of corticosteroid eye drops causes glaucoma in 1 in 20 of the population (a genetic trait). Application is generally as hydrocortisone, prednisolone or fluorometholone drops, or subconjunctival injection.
• Nephrotic syndrome. Patients with minimal change disease respond well to daily prednisolone, 2 mg/kg, for 6 weeks followed by 1.5 mg/kg on alternate days. Relapses are treated with the higher dose until proteinuria is trace level only, and then the lower dose for a month. Multiple relapses then require a tapering dose over several months rather than complete withdrawal. In such patients, steroid sparing agents, ciclosporin, tacrolimus, and mycophenolate, may be used to reduce long-term dosing.
• A variety of skin diseases, such as eczema. Severe cases may involve occlusive dressings if a systemic effect is undesirable, though absorption can be substantial (see Ch. 17).
• Aphthous ulcers. Hydrocortisone 2.5 mg oromucosal tablets are allowed to dissolve next to the ulcer; beclometasone dipropionate inhaler 50-100 micrograms may be sprayed on the oral mucosal, or betamethasone soluble talet 500 miligrams dissolved in water may be used, without swallowing, as a mouth wash. Triamcinolone acetonide in Orabase, or fluocinonide gel covered by Orabase may be used where available. Early initiation of treatment may accelerate healing.
• Acute gout resistant to other drugs (see p. 255).
• Hypercalcaemia of sarcoidosis and of vitamin D intoxication responds to prednisolone 30 mg daily (or its equivalent of other adrenal steroid) for 10 days. Hypercalcaemia of myeloma and some other malignancies responds more variably. Hyperparathyroid hypercalcaemia does not respond (see p. 638).
• Raised intracranial pressure due to cerebral oedema, e.g. in cerebral tumour or encephalitis. This is probably an anti-inflammatory effect, which reduces vascular permeability and acts in 12–24 h. Give dexamethasone 10 mg i.m. or i.v. (or equivalent) initially and then 4 mg 6-hourly by the appropriate route, reducing the dose after 2–4 days and withdrawing over 5–7 days. Much higher doses may be used in palliation of inoperable cerebral tumour.
• Preterm labour: (to mother) to enhance fetal lung maturation.
• Aspiration of gastric acid (Mendelsohn’s syndrome).
Withdrawal of pharmacotherapy
The longer the duration of therapy, the slower must be the withdrawal.
After use for 2 weeks, for rapid withdrawal, a 50% reduction in dose each day is reasonable.
There are many reports of collapse, even coma, occurring within a few hours of omission of adrenal steroid therapy, e.g. due to patients’ ignorance of the risk to which their physicians are exposing them, or failure to carry their tablets with them. Patients must be instructed on the hazards of omitting therapy and, during intercurrent disease, intramuscular preparations should be freely used. For anaesthesia and surgery in adrenocortical insufficiency, see page 566.
Competitive antagonism of adrenal steroids
Spironolactone antagonises the sodium-retaining effect of aldosterone and other mineralocorticoids. It is used to treat primary and secondary hyperaldosteronism (see p. 563).
Adrenocorticotrophic hormone (ACTH) (corticotropin)
Preparations
• Tetracosactide Injection is supplied as an ampoule for injection i.v., i.m. or s.c.
• Tetracosactide Zinc Injection (Synacthen Depot) i.m. in which the hormone is adsorbed on to zinc phosphate from which it is slowly released. This is the form used in the long tetracosactide test.
• Physiological concentrations of cortisol are essential for supporting the circulation and glucose production. Physiological concentrations of aldosterone are essential to prevent excessive sodium loss.
• For systemic pharmacological uses, prednisolone or other synthetic adrenocorticosteroids are used because they are more selective glucocorticoids, i.e. have less sodium-retaining activity.
• For local administration (skin, lung), more potent, fluorinated steroids may be required.
• Glucocorticoids inhibit the transcriptional activation of many of the inflammatory cytokines, giving them a versatile role in the treatment of many types of inflammation.
• Fludrocortisone is a valuable treatment for many sodium-losing states, and for most causes of autonomic neuropathy.
• Corticotropin is used to test the capacity of the adrenal gland to produce cortisol.
Arlt W. Junior doctors’ working hours and the circadian rhythm of hormones. Clin. Med. (Northfield Il). 2006;6:127–129.
Arlt W. The approach to the adult with newly diagnosed adrenal insufficiency. J. Clin. Endocrinol. Metab.. 2009;94:1059–1067.
Barnes P.J., Adcock I.M. Glucocorticoid resistance in inflammatory diseases. Lancet. 2009;373:1905–1917.
Buttgereit F., Burmester G.R., Lipworth B.J. Optimised glucocorticoid therapy: the sharpening of an old spear. Lancet. 2005;365:801–803.
Cooper M.S., Stewart P.M. Corticosteroid insufficiency in acutely ill patients. N. Engl. J. Med.. 2003;348:727–734.
Hench P.S., et al. The effect of a hormone of the adrenal cortex (17-hydroxy-11-dehydrocorticosterone: Compound E) and of pituitary adrenocorticotropic hormone on rheumatoid arthritis. Proceedings of the Staff Meetings of the Mayo Clinic. 1949;24:181–277. (acute rheumatism)
The classic studies of the first clinical use of an adrenocortical steroid in inflammatory disease. See also page 301 for an account by E C Kendall of the biochemical and pharmaceutical background to the clinical studies. Kendall writes of his collaboration with Hench, ‘he can now say “17-hydroxy-11-dehydrocorticosterone” and in turn I can say “the arthritis of lupus erythematosus”. In sophisticated circles, however, I prefer to say, “the arthritis of L.E”.’
Hochhaus G. New developments in corticosteroids. Proc. Am. Thorac. Soc.. 2004;1:269–274.
Lipworth B.J. Therapeutic implications of non-genomic glucocorticoid activity. Lancet. 2000;356:87–88.
Løvås K., Husebye E. Addison’s disease. Lancet. 2005;365:2058–2061.
Newell-Price J., Bertagna X., Grossman A.B., Nieman L.K. Cushing’s syndrome. Lancet. 2006;367:1605–1617.
1 Potency (the weight of drug in relation to its effect) rather than efficacy (strength of response); see page 83. If a large enough dose of a glucocorticoid, e.g. prednisolone, were administered, the Na+ retention would be almost as great as that caused by a mineralocorticoid. This is why, in practice, different (more selective, and potent) glucocorticoids, not higher doses of prednisolone, need to be used when maximal stimulation of glucocorticoid receptors is desired, e.g. in the treatment of acute transplant rejections.