CHAPTER 54 Adenocarcinoma and Other Tumors of the Stomach
EPIDEMIOLOGY
Gastric cancer remains the second leading cause of cancer mortality in the world,1 although the overall incidence is declining.2 The incidence of gastric cancer in Western countries has decreased dramatically over the past century.3 For example, gastric cancer mortality has decreased 86% since 1950 in the United States, and the incidence of gastric cancer has diminished four-fold since 1930 to approximately 8 cases per 100,000 people.4,5 As recently as 1930, gastric cancer was the leading cause of cancer mortality in the United States for men and the third leading cause for women.6 Gastric cancer is now the seventh leading cause of cancer mortality in the United States.5 It was estimated that in 2008, approximately 21,500 Americans would be diagnosed with gastric cancer and 10,880 would die of it.7
There is great geographic variation in gastric cancer incidence, with the highest incidence rates in the Far East (Fig. 54-1). Eastern Europe and Central and South America also have high incidence rates, and the lowest incidence rates are observed in North America, North Africa, South Asia, and Australia.6 Although gastric cancer was common in industrialized countries in the past, the latest epidemiologic data indicate that more than 60% of new cases of gastric cancer are in developing countries, reflecting a more rapid decline in developed countries.
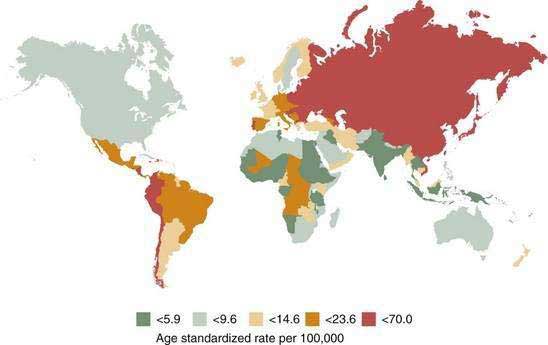
Figure 54-1. Worldwide incidence of gastric cancer in men.
(From Parkin DM. International variation. Oncogene 2004; 23:6239-40.)
In the United States, the median age of diagnosis is 71 years, with the highest proportion (28%) diagnosed between the ages of 75 and 84.8 In Japan, a country with a high incidence of gastric cancer, the mean age of diagnosis is roughly a decade earlier, perhaps reflecting lead-time bias due to widespread screening. The incidence of gastric cancer in men is approximately twice that in women (Table 54-1).3 The incidence of gastric cancer in blacks in the United States is nearly double that in whites.5 Native Americans and Hispanics also have a higher risk of development of gastric cancer than whites.9 Lower socioeconomic status is associated with a much higher incidence of gastric cancer.3 In the United States, the distribution of gastric cancer within the stomach is 39% in the proximal third, 17% in the middle third, 32% in the distal third, and 12% involving the entire stomach.10 In contrast to the pattern seen with noncardia tumors, the incidence rates of gastric cardia cancer are rising.2,11
Table 54-1 Gastric Cancer Incidence and Mortality Rates per 100,000 US Population by Race from 2001 to 2005
Dietary, environmental, and genetic risk factors for gastric adenocarcinoma are listed in Table 54-2, some of which are or may be protective.
Table 54-2 Risk Factors Including Protective Factors for Gastric Adenocarcinoma
| Definite | Helicobacter pylori infection |
| Chronic atrophic gastritis | |
| Intestinal metaplasia | |
| Dysplasia* | |
| Adenomatous gastric polyps* | |
| Cigarette smoking | |
| History of gastric surgery (esp. Billroth II)* | |
| Genetic factors | |
| Family history of gastric cancer (first-degree relative)* | |
| Familial adenomatous polyposis (fundic gland polyps)* | |
| Hereditary nonpolyposis colorectal cancer* | |
| Peutz-Jeghers syndrome* | |
| Juvenile polyposis* | |
| Probable | High intake of salt |
| Obesity (adenocarcinoma of cardia only) | |
| Snuff tobacco use | |
| History of gastric ulcer | |
| Pernicious anemia* | |
| Regular aspirin or NSAID use (protective) | |
| Possible | Low socioeconomic status |
| Ménétrier’s disease | |
| High intake of fresh fruits and vegetables (protective) | |
| High ascorbate intake (protective) | |
| Questionable | Hyperplastic and fundic gland polyps |
| High intake of nitrates | |
| High intake of green tea (protective) |
NSAID, nonsteroidal anti-inflammatory drug.
* Surveillance for cancer is suggested in patients with this risk factor.
ETIOLOGY AND PATHOGENESIS
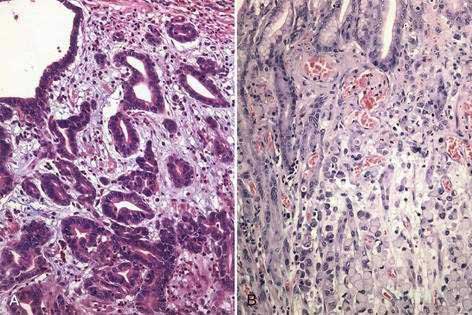
Gastric cancer can be subdivided using the Lauren classification into two distinct histologic subtypes with different epidemiologic and prognostic features (Fig. 54-2).12 The intestinal type of cancer is characterized by the formation of gland-like tubular structures with features reminiscent of intestinal glands. This type of gastric cancer is more closely linked to environmental and dietary risk factors, tends to be the predominant form in regions with a high incidence of gastric cancer, and is the form of cancer that is now declining worldwide. The diffuse type of cancer lacks glandular structure and consists of poorly cohesive cells that infiltrate the wall of the stomach. It is found at the same frequency throughout the world, occurs at a younger age, and is associated with a worse prognosis than the intestinal form. Extensive involvement of the stomach by the diffuse type can result in a rigid and thickened stomach, a condition referred to as linitis plastica. Adenocarcinoma of the stomach can also be classified into proximal tumors (gastric cardia and gastroesophageal junction) and distal tumors (fundus, body and antrum of the stomach). Distal tumors have been declining, whereas proximal tumors have been increasing (see Chapter 46).
It is now believed that the development of intestinal-type cancers occurs through a multistep process in which the normal mucosa is sequentially transformed into a hyperproliferative epithelium, followed by an early adenoma, late adenoma, and then carcinoma. In colon cancer, the evidence is strong that each step in the transition is associated with a specific gene mutation,13 but evidence that gastric cancer follows a comparable sequence of genetic events has been lacking. However, in gastric and colon cancer, it does appear that deoxyribonucleic acid (DNA) mutations are established over time in stem cells; in intestinal metaplasia these mutations spread through the stomach through a process involving crypt fission and monoclonal conversion of glands.14 The contention that the pathogenesis of intestinal-type gastric cancer is a multistep process is supported mainly by the observation that both atrophic gastritis and intestinal metaplasia are found in higher incidences in patients with intestinal-type cancer and in countries with a high incidence of gastric cancer (see Chapter 51).15
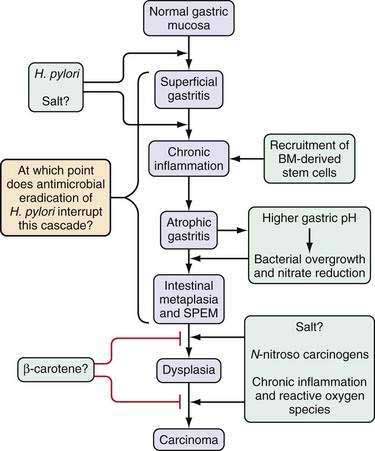
This multistep model of intestinal-type gastric cancer, developed in large part by Correa and colleagues,16,17 postulates that there is a temporal sequence of preneoplastic changes that eventually lead to the development of gastric cancer. A common feature of the initiation and progression to intestinal-type gastric cancer is chronic inflammation. Helicobacter pylori infection is the primary cause of gastric inflammation and the leading etiologic agent for gastric cancer (see Chapter 50). In a subset of patients infected with H. pylori, the inflammatory process leads to the development of atrophic gastritis (with loss of glandular tissue) followed by progression to intestinal metaplasia, dysplasia, early gastric cancer, and, eventually, advanced gastric cancer (Fig. 54-3). The current view is that all stages prior to the development of high-grade dysplasia are potentially reversible, although this concept is still somewhat controversial, it has been supported by a number of studies in animal models.18,19 Unlike the situation observed with colon cancer, the precise genes involved in each step of this progression are still not defined. Furthermore, during endoscopy the premalignant stages of gastric cancer are not as readily identifiable as those of colon cancer, and many gastric carcinomas are very heterogeneous, containing a large percentage of stromal cells. These stromal cells, which include cancer-associated fibroblasts known to promote tumor growth, have been reported to show distinct genetic and epigenetic changes that may confound tumor analysis.20 This feature makes characterization of the timing of specific gene mutations in gastric cancer difficult at best. Currently the role of chronic inflammation in the diffuse type of gastric cancer remains to be clarified, and the similarities if any to the proposed pathway in Figure 54-3 for the intestinal type of cancer.
HELICOBACTER PYLORI INFECTION
H. pylori is a gram-negative microaerophilic bacterium that infects nearly half of the world’s population and is recognized as the primary etiologic agent for gastric cancer (see Chapter 50). Indeed, H. pylori has been classified as a class I (or definite) carcinogen by the International Agency for Research on Cancer (IARC), a branch of the World Health Organization (WHO). Infection with H. pylori has been found in every population studied, although the prevalence is higher in developing countries.21,22
The natural history of chronic H. pylori infection includes three possible outcomes23: (1) superficial gastritis, in which most patients remaining asymptomatic; (2) duodenal ulcer phenotype, which occurs in 10% to 15% of infected subjects; and (3) gastric ulcer/gastric cancer phenotype, which is the least common in the United States. In general, the risk for gastric cancer is dependent on the types of gastritis, and an increased risk is associated with a low acid state. H. pylori–induced duodenal ulcer disease is associated with a high gastric acid output as well as a reduced risk for developing gastric cancer.24 Patients with H. pylori–associated gastric ulcer disease exhibit low gastric acid output, and their ulcers are typically associated with preneoplastic changes of atrophic gastritis and metaplasia. Overall, studies suggest that H. pylori–infected patients are at risk for development of chronic atrophic gastritis at a rate of 1% to 3% per year of infection.17,25,26 Thus, those patients who are genetically predisposed to forming atrophic gastritis in response to H. pylori infection are predisposed to gastric cancer. Although Helicobacter infection is associated with both intestinal-type and diffuse-type adenocarcinomas, the mechanisms responsible for the formation of intestinal-type adenocarcinoma have been better studied and are focused on here. The association of H. pylori with mucosa-associated lymphoid tissue (MALT) lymphoma is discussed briefly at the end of this chapter and in more detail in Chapter 29.
The increased risk of development of gastric adenocarcinoma due to H. pylori infection depends on multiple factors including the strain of bacteria, host genetic factors, the duration of infection, and the presence or absence of other environmental risk factors (e.g., poor diet, smoking, etc.). In a Japanese cohort of 1526 patients with peptic ulcer disease, nonulcer dyspepsia, and gastric hyperplasia, only those infected with H. pylori developed gastric adenocarcinoma during follow-up (2.9% vs. 0%, P < 0.001).27 Additional cohort studies from China and Taiwan have reported similar findings.28,29 At least in Western countries, the association between H. pylori and gastric cancer appears to be confined to noncardia gastric tumors.30
Potential mechanisms for H. pylori–induced gastric carcinogenesis include host factors, bacterial factors, environmental factors, and interactions among all three factors. Our latest understanding suggests that a combination of a virulent bacterial strain, a genetically permissive host, and a favorable gastric environment are necessary for disease to occur. The most important factor appears to be the induction of chronic inflammation by H. pylori infection. Several aspects of the inflammatory milieu have been implicated as carcinogens; they include increased oxidative stress and the formation of oxygen free radicals leading to DNA damage, increased CD4+ T cells and myeloid cells, and elevated proinflammatory cytokine production, all leading to accelerated cell turnover, reduced apoptosis, and the potential for faulty or incomplete DNA repair.31 Indeed, recent studies suggest that animals with deficient DNA repair mechanisms display more severe gastric dysplasia after chronic infection with H. pylori.32 Thus, evidence to date clearly indicates that the most important cofactor in the induction of Helicobacter-related disease is the host immune response. Indeed, chronic inflammation has been linked to a large number of cancers.
H. pylori infection leads to innate and adaptive immune responses (see Chapter 2). Initiation of the innate immune response to H. pylori is just beginning to be unraveled. Classically, the innate immune system consists of professional antigen presenting cells (APCs) such as macrophages, dendritic cells, and in some cases epithelial cells. Recent work supports a role for pattern recognition receptors (Toll-like receptors [TLRs]) in the initial response to Helicobacter colonization and the subsequent induction of the adaptive response. The most convincing evidence to date implicates TLR2 as the major TLR in Helicobacter species recognition.33 A role for TLRs 4, 5, and 9 remains more controversial.34–37 TLR4, along with CD14 and MD-2, serves as the receptor for Escherichia coli lipopolysaccharide (LPS) and probably H. pylori LPS and thus may be involved as well.
The C57BL/6 mouse is a susceptible inbred strain, in which initial colonization of the antrum by bacteria later spreads to the body or corpus, leading to severe chronic inflammation and increases in apoptosis (programmed cell death) and proliferation. The alterations in cellular turnover are associated with a loss of parietal and chief cells (atrophy), intestinal metaplasia, and dysplasia, followed by invasive gastric adenocarcinoma in mice 14 to 22 months after infection.38,39 Genetic manipulation of the C57BL/6-susceptible murine strain has facilitated detailed study and has thus led to a deeper understanding of genetic factors that promote murine gastric cancer, and in particular, the role of the adaptive immune response. For example, gastric Helicobacter infection in mice deficient in lymphocytes, including mice with recombinase-activating gene (RAG) deficiency, severe combined immunodeficiency, or T cell deficiency, does not result in tissue damage, cell lineage alterations, or the metaplasia-dysplasia-carcinoma sequence.40,41 Infection in B cell–deficient mice (which retain a normal T cell response) results in severe atrophy and metaplasia identical to that seen in infected wild-type mice.41 Taken together these studies underscore the crucial role of CD4+ T lymphocytes in orchestrating gastric neoplasia.
How do CD4+ T lymphocytes contribute to gastric cancer progression? Susceptible mouse strains, such as C57BL/6, mount a strong helper T cell type 1 (Th1) interferon-γ (IFN-γ), interleukin-12 (IL-12) type of immune response, whereas resistant strains, such as the BALB/c, have an opposite Th2 response (IL-4, IL-5).39,42,43 A Th2 response is associated with protection from mucosal damage despite the inability to eliminate bacterial colonization and in fact is often associated with higher bacterial colonization rates. Mouse strains such as the C3H, which has a mixed Th1/Th2 cytokine profile, show intermediate disease, suggesting that cytokines within an immune response interact to form a continuum of disease rather than discrete disease states. More recently, Th17 cells, which express IL-17, have been shown to be an important component of H. pylori–induced gastritis.
Although the composite immune milieu most likely dictates disease manifestations, studies are beginning to define the role of individual cytokines in the predisposition to disease. This is best illustrated in the IFN-γ knockout mice, in which a lack of IFN-γ protects infected mice from atrophy.39,43 On the other hand, mice lacking IL-10, a cytokine that acts to dampen an immune response, develop severe atrophic gastritis in response to infection.39–43 More recently, genetic murine models have illustrated the importance of the IL-6–IL-11 family of cytokines in the development of gastric cancer.44
Manipulation of the immune response within wild-type strains confirms the central role of the Th1/Th2 response in producing disease. For example, infection with the intestinal helminth Heligmosomoides polygyrus skews the immune response toward Th2 polarization and protects the C57BL/6 host from Helicobacter-induced atrophy and metaplasia.45 This mouse model mimics both the parasitic infection status and the paradoxical low gastric cancer–high H. pylori infection rates seen in areas of Africa, potentially explaining this apparent inconsistency. These observations in mice led to human studies in Africa and Latin America that confirmed that geographic regions with low gastric cancer rates had much higher Th2 relative to Th1 immune responses to H. pylori.46,47 In general, the increased Th2-type responses were found in areas where serum immunoglobulin E (IgE) levels were high and the prevalence of intestinal parasitism by helminths is greater than 50%. These findings further stress the importance of the host response to infection and suggest the possibility that manipulation of the genetically predetermined host cytokine profile in response to environmental challenges may lessen or exacerbate the disease process.
There is a great deal of genetic diversity between strains of H. pylori owing to point mutations, insertions, deletions, and base-pair substitutions within its genome. Several strains may infect a single individual, and existing strains can undergo mutations and change over time.48,49 Despite this genetic diversity, several genes are recognized as risk factors for gastric carcinoma, including the cag pathogenicity island, the vacA gene, and the babA2 gene.
The H. pylori genome is 1.65 million base pairs and codes for approximately 1500 genes, two thirds of which have been assigned biological roles.50 The function of the remaining one third of the genome remains obscure. Factors that contribute to carcinogenesis include those that enable the bacteria to effectively colonize the gastric mucosa, those that incite a more aggressive host immune response, and those that affect host cell-growth signaling pathways.
Motility toward epithelial cells of the stomach is a vital feature of H. pylori survival tactics. This function is ensured by several factors, including spiraling movement (FlaA and FlaB proteins), which are designed to navigate the thick gastric mucus and through efficient modifications of the extracellular matrix and mucus layer, thus decreasing viscosity and allowing bacterial penetration.51,52 In addition, H. pylori expresses a variety of genes that contribute to buffering of stomach acid in order to maintain a relatively neutral pH. This includes a urease gene cluster, consisting of seven genes, of which UreA/UreB complex (comprising the urease enzyme) codes for 10% of the protein of H. pylori and is vital for its survival.
A significant proportion (e.g., ≈20%) of H. pylori organisms can be found adherent to the surface of gastric mucous cells. Occasionally H. pylori can also be found intracellularly, particularly in preneoplastic and neoplastic lesions.53 Adhesion of the bacteria to the epithelial layer is facilitated by a large family of 32 related outer-membrane proteins (Hop proteins) that include the adhesins. One of the best-characterized adhesins is BabA, which is encoded by the strain-specific gene babA2, a member of a highly conserved family of outer membrane proteins. BabA binds to the fucosylated Lewis B blood group antigen on gastric epithelial cells and forms a scaffold apparatus that allows bacterial proteins to enter host epithelial cells. Bacterial strains that possess the babA2 gene adhere more tightly to epithelial cells, promote a more aggressive phenotype, and are associated with a higher incidence of gastric adenocarcinoma.54
The cag pathogenicity island is approximately 40 kb and contains 31 genes. The terminal gene of this island, cagA, is often used as a marker for the entire cag locus. Compared with cagA-negative (cag−) strains, cag-positive (cagA+) strains are associated with more severe inflammation, higher degrees of atrophic changes, and a greater chance of progressing to gastric adenocarcinoma.55–58 The estimated risk has ranged from an odds ratio of 2 to as high as 28.4.23 However, many of the genes adjacent to cagA code for a type 4 secretion system (TFSS), often viewed as a molecule needle that injects bacterial proteins (such as CagA) into host cells. The remarkable finding that CagA is injected into host cells, where it is phosphorylated by Src and c-Abl kinases, has raised the possibility that CagA could directly promote growth, migration, and transformation. Indeed, transgenic expression of H. pylori CagA induces gastrointestinal (GI) and hematopoietic neoplasms in mice.59 Other genes within the pathogenicity island are also believed to be important for disease (cagE or picB, cagG, cagH, cagI, cagL, cagM) because they appear to be required for in vitro epithelial cell cytokine release, although they do not seem to have as great an effect on immune cell cytokine activation as cagA.60–62 These findings may explain the attenuated inflammatory response and lower cancer risk with cagA− strains in vivo.63–66
All strains of H. pylori carry the vacA gene, which codes for a pore-forming vacuolating toxin, but expression of vacA differs according to allelic variation. Approximately 50% of H. pylori strains express the vacA protein, which has been shown to be a very powerful inhibitor of T cell activation in vitro.67 Although vacA and cagA map to different loci within the H. pylori genome, the vacA protein is commonly expressed in cagA+ strains. There are various forms of vacA, and the s1m1 strains are highly toxigenic. Other bacterial virulence factors, such as cagE, may play a role in the modulation of apoptosis and the host inflammatory response, thereby contributing to disease manifestations. Indeed, “virulent strains” (cagA+, cagE+, and VacA+ s1m1) appear to be more potent inducers of proinflammatory mediators than “nonvirulent strains” (cagA−, cagE−, and VacA−), possibly explaining the higher association of cagA+ strains with gastric cancer.68
DIETARY FACTORS
Numerous dietary factors have been implicated as risk factors for gastric cancer. The decline in gastric cancer rates has coincided with the widespread use of refrigeration and the concomitant higher intake of fresh fruits and vegetables and lower intake of pickled and salted foods. Use of refrigeration for more than 10 to 20 years has been associated with a decreased risk of gastric cancer.17,69 Lower temperatures reduce the rate of bacterial, fungal, and other contaminants of fresh food, as well as the bacterial formation of nitrites. Additionally, high intake of highly preserved foods may be associated with increased gastric cancer risk,70 likely because of higher contents of salt, nitrates, and polycyclic aromatic amines.71
Much attention has been given to the effects of high nitrate intake. When nitrates are reduced to nitrite by bacteria or macrophages, they can react with other nitrogenated substances to form N-nitroso compounds that are known mitogens and carcinogens.72,73 In rats, N-nitroso compounds have been shown to cause gastric cancer.74 However studies trying to link N-nitroso exposure to gastric cancer risk have been inconclusive, perhaps reflecting the fact that nitrate intake does not necessarily correlate with nitrosation levels.75,76 A Swedish cohort study found a nearly two-fold increased risk of gastric cancer associated with high dietary nitrate intake.70 However, separate large cohort studies from Europe did not demonstrate an association between nitrate intake and risk of gastric cancer.77,78
Another factor implicated in the development of gastric cancer is a diet high in salt (pickled foods, soy sauce, dried and salted fish and meat). High salt intake has been associated with higher rates of atrophic gastritis in humans and animals in the setting of Helicobacter infection79 and increases the mutagenicity of nitrosated food in animal models.17 High salt diets are associated with a roughly two-fold increased risk of gastric cancer.80,81 Cohort and case-control studies have also found an increased risk of gastric cancer associated with processed meat intake.70,82 Possible mechanisms include higher bacterial loads, up-regulation of H. pylori cagA expression, and increased cell proliferation and p21 expression.79,83,84
Epidemiologic studies have had inconsistent findings with regard to fruit and vegetable consumption and risk of gastric cancer. A cohort study from Japan found significantly decreased risks of gastric cancer associated with increased vegetable and fruit intake.85 A Swedish cohort study demonstrated a reduced risk of gastric cancer associated with high vegetable intake, but no association was seen with amount of fruit consumption.86 A large cohort study of nearly 500,000 adults in the United States and a separate nested case-control study from Europe failed to find an association between fruit and vegetable intake and gastric cancer risk.87,88
Other foods or dietary factors that have been implicated as potential risk factors for gastric cancer are high intake of fried food, foods high in fat, high intake of red meat, and aflatoxins.82,89–91 Diets with a high intake of fresh fish and antioxidants may be protective.90,92–94 However, there are insufficient data to make definitive conclusions regarding these factors.
CIGARETTE SMOKING
Tobacco has long been established as a carcinogen, and numerous epidemiologic studies have demonstrated an association between cigarette smoking and risk of gastric cancer.95 Several large cohort studies from Europe and Asia have reported a significantly increased risk of gastric cancer among smokers.96–98 A meta-analysis found that compared with never smokers, current smokers had a 1.5-fold increased risk of gastric cancer for the cardia as well as noncardia region.99 The authors also reported an increased association with greater amounts of smoking.
Moist snuff is a smokeless tobacco product promoted as an alternative to cigarettes and has reportedly reduced levels of carcinogenic nitrosamines. However, results of a Swedish cohort study demonstrated a 1.4-fold increased risk of noncardia gastric cancer among regular snuff users.100 Snuff exposure also increases the rate of gastric carcinogenesis in H. pylori–infected mice.101
ALCOHOL
Multiple cohort and case-control studies from the United States and Europe have found no significant association between alcohol consumption and cardia or noncardia gastric cancer.98,102,103 A separate population-based case-control study in the United States also found no association between any alcohol use and risk of either cardia or noncardia gastric cancer, although a reduced risk was reported with wine consumption (cardia, odds ratio [OR] 0.6; 95% confidence interval [CI]: 0.5 to 0.9; noncardia, OR 0.7; 95% CI: 0.5 to 0.9).104
OBESITY
Obesity is a recognized risk factor for numerous gastrointestinal malignancies.105 Increased body mass index (BMI) appears to be associated with a mild to moderate increased risk of gastric cardia cancer but not for noncardia gastric cancer.106–110 Results of the National Institutes of Health–American Association of Retired Persons (NIH-AARP) Diet and Health Cohort Study demonstrated that marked obesity (BMI = 35 kg/m2) was associated with a significantly increased risk of gastric cardia cancer (hazard ratio, 2.46) but not with noncardia gastric cancer.106 A separate cohort study from the Netherlands also found an increased risk of cardia cancer with increasing BMI.107
INHERITED PREDISPOSITION
Overall, 10% of cases of gastric cancer appear to exhibit familial clustering,111 and family history is likely an independent risk factor for the disease even after controlling for H. pylori status.112,113 In a cohort study of relatives of patients with gastric cancer, siblings had a two-fold increased risk of gastric cancer, adjusted for H. pylori infection.114 In a case-control study from Japan, a positive family history was associated with a significantly increased odds of gastric cancer in women (OR, 5.1), but not in men.115 A study from Scandinavia showed that having a twin with gastric cancer conferred a markedly higher risk for the disease (hazard ratios, 9.9 for monozygotic twins and 6.6 for dizygotic twins), leading the researchers to calculate that heritable factors accounted for 28% of gastric cancers, compared with 10% for shared environmental factors and 62% for nonshared environmental factors.116
Some of the familial clustering seen with intestinal-type gastric cancer may be related to genetic factors that play a role in the host immune response to H. pylori infection. Data from South Korea indicate that individuals with a family history of gastric cancer more frequently have H. pylori infection as well as associated atrophic gastritis or intestinal metaplasia.117 In a case-control study from Scotland, relatives of patients with gastric cancer had a higher prevalence of atrophy and hypochlorhydria, but a similar prevalence of H. pylori infection, compared with controls.118 The greater prevalence of atrophy was confined to those patients with H. pylori infection, suggesting the possibility these individuals were perhaps exhibiting a more vigorous immune response to H. pylori. In a number of model systems, the development of gastric atrophy has been linked to a strong Th1 immune response.43,45,119 Thus, it was postulated that candidate disease-susceptibility genes for gastric atrophy and cancer might be genes that participate in the innate and adaptive immune responses to H. pylori infection. Inflammation is modulated by an array of pro- and anti-inflammatory cytokines, and several genetic polymorphisms have been described that influence cytokine response.
IL-1β is an important proinflammatory cytokine and a powerful inhibitor of acid secretion. Thus, the initial report in this area described in the setting of H. pylori infection an association between proinflammatory IL-1 gene cluster polymorphisms (IL-1B encoding IL-1β, and IL-1RN encoding its naturally occurring receptor antagonist, IL-1RA) and neoplastic progression. Individuals with the IL-1β-31*C or -511*T and IL-1RN*2/*2 genotypes were shown in the study to be at higher risk for development of H. pylori–dependent hypochlorhydria and gastric cancer.120 The increased risk of progression to cancer with these genotypes was in the two- to three-fold range compared with noninflammatory genotypes. The initial report was confirmed in other studies.121–125 Subsequently, Hwang and colleagues126 demonstrated that carriers of the IL-1β-511T/T genotype or the IL-1RN*2 allele had higher mucosal IL-1β levels than noncarriers, and they also confirmed the association between the -511T/T genotype and severe gastric inflammation and atrophy. The importance of IL-1β in carcinogenesis has now been demonstrated in a transgenic study, in which stomach-specific expression of human IL-1β in transgenic mice led to spontaneous gastric inflammation and cancer that correlated with early recruitment of myeloid-derived suppressor cells (MDSCs) to the stomach.127
Additional associations with gastric cancer risk have been reported for genetic polymorphisms in tumor necrosis factor-α (TNF-α) and IL-10. Proinflammatory genotypes of TNF-α and IL-10 each were associated with a two-fold higher risk of noncardia gastric cancer. When combined with proinflammatory genotypes of IL-1β and IL-1RN, patients with three or four high-risk genotypes showed a 27-fold greater risk of gastric cancer.128 Additional studies have shown that polymorphisms of the TLR4 gene also increase the risk of gastric cancer. Carriers of the TLR4+896G polymorphism had an 11-fold increased risk of hypochlorhydria, and significantly more severe gastric atrophy and inflammation.129 Accumulated evidence suggests that the genetic predisposition to gastric cancer may be largely determined by the TLR and cytokine responses to chronic Helicobacter infection.
The best described form of hereditary gastric cancer is the diffuse gastric cancer that is seen in the presence of a germline mutation in the gene CDH1, which encodes the cell adhesion molecule E-cadherin. A large New Zealand kindred was found to have a germline mutation in the E-cadherin gene, and similar mutations have been reported in several additional kindreds, all with diffuse-type gastric cancer.130–133 The age of onset of gastric cancer in individuals with CDH1 mutations is less than 40 years but can be highly variable, and the estimated lifetime risk of gastric cancer is close to 70%.134,135 Germline CDH1 mutations are also associated with familial lobular breast cancer.136,137
A small part of the familial clustering of gastric cancer can be attributed to other cancer syndromes (see Chapter 122). Patients with familial adenomatous polyposis (FAP) have a prevalence of gastric adenomas ranging from 35% to 100%, and their risk of gastric cancer is close to 10-fold higher than that of the general population.138 These cancers frequently arise from fundic gland polyps and develop at an early age.139,140 Patients with hereditary nonpolyposis colorectal cancer (HNPCC) syndrome have an approximately 11% risk of developing gastric cancer, predominantly of the intestinal type, with a mean age at diagnosis of 56 years.141 Patients with juvenile polyposis also have a 12% to 20% incidence of gastric cancer.142,143
GENETICS
Aneuploidy is common in gastric cancer (seen in 60% to 75% of cases), but cytogenetic studies have failed to identify any consistent chromosomal abnormality. Comparative genomic hybridization studies have shown that chromosome arms 4q, 5q, 9p, 17p, and 18q exhibit frequent decreases in DNA copy number, whereas chromosomes 8q, 17q and 20q often have increased DNA copy number.144
There is a consensus that TP53 is the most commonly mutated gene in gastric cancer (60% to 70% of gastric cancers) and that mutations in Ras, APC, and Myc are rare.145,146 Loss of heterozygosity (LOH) at the APC locus occurs more commonly. Another genetic abnormally found at high frequency includes deletion or suppression of the fragile histidine triad gene (FHIT) (60%), a tumor suppressor locus on chromosome 3p. Genes that normally inhibit entry into the cell cycle, such as p16 and p27, show diminished expression in nearly one half of gastric cancers.147–152 Absence of p27 expression is associated with a poorer prognosis.147,149 Absence of p16 expression is seen most commonly in poorly differentiated carcinomas but has no measurable impact on patient prognosis.153 Diminished expression of p16 and p27 occurs in the absence of detectable mutations and is believed to be secondary to hypermethylation of the respective genes.151 Many of these cancers show hypermethylation of a number of promoter regions, including the MLH1 promoter region, and show the high-level microsatellite instability (MSI) phenotype (see Chapter 3). Multiple tumor suppressor genes have been shown to be methylated in gastric cancers. Emerging evidence suggests that epigenetic changes, including global hypomethylation and promoter hypermethylation, occurs quite early in gastric carcinogenesis. In addition, it appears that DNA methylation changes also occur in the tumor-associated stromal fibroblasts, suggesting an important role for the tumor microenvironment.20
Overexpressions or amplifications of a number of growth factor pathways have been described, including cyclooxygenase-2 (COX-2) (70%), hepatocyte growth factor/scatter factor (HGF/SF) (60%), vascular endothelial growth factor (VEGF) (50%), c-met (50%), amplified in breast cancer-1 (AIB-1) (40%), and β-catenin (25%) (Table 54-3).154 Approximately 15% of gastric cancers have been reported to overexpress both epidermal growth factor (EGF) and EGF receptor (EGFR), consistent with an autocrine mechanism. Mutations in PIC3A, a gene that codes for a catalytic subunit of phosphotidylinositol 3-kinase (PI3K), has been found in up to 25% of gastric cancers analyzed.155 In addition, mutations in human protein tyrosine phosphatases (PTPs) were found by the same laboratory in 17% of gastric cancers, with the protein tyrosinase phosphatase–receptor type (PTPRT) the most frequently altered.156
| ABNORMALITIES | APPROXIMATE GENE FREQUENCY (%) |
|---|---|
| DNA aneuploidy | 60-75 |
| Microsatellite instability | 15-50 |
| Deletion/Suppression | |
| TP53 gene | 60-70 |
| Fragile histidine triad gene (FHIT) | 60 |
| Adenomatous polyposis coli (APC) gene LOH | 50 |
| Deleted in colorectal cancer (DCC) gene LOH | 50 |
| Decreased Expression Due to Hypermethylation | |
| p16 | ≈50 |
| TFF1 | ≈50 |
| p27 | <50 |
| MLH1 | 15-20 |
| E-cadherin | 50 |
| Amplification/Overexpression | |
| Cyclooxygenase-2 (COX-2) | 70 |
| Hepatocyte growth factor (HGF) | 60 |
| Vascular endothelial growth factor (VEGF) | 50 |
| c-Met | 50 |
| Amplified in breast cancer-1 (AIB-1) | 40 |
| Beta-catenin | 25 |
| EGF/EGFR | 15 |
| Mutations | |
| PI3K | 25 |
| PTPRT | 17 |
DNA, deoxyribonucleic acid; EGF, epidermal growth factor; EGFR, epidermal growth factor receptor; LOH, loss of heterozygosity; MLH1, human mutL homolog 1; PI3K, phosphatidylinositol 3-kinase; PTPRT, protein-tyrosine phosphatase receptor-type; TFF1, human trefoil factor 1.
Gastric-specific tumor suppressor genes TFF1 (Trefoil factor 1) and RUNX3 (Runt-related transcription factor 3), which have now been identified and may represent “gatekeepers” of the gastric cancer pathway, are logical targets for further study.157,158 Loss of TFF1 has been described in around 50% of gastric carcinomas, and TFF1 knockout mice develop spontaneous gastric antral tumors. Mutations of TFF1 also have been described, and these enhance gastric cancer cell invasion through signaling pathways that include PI3-K and phospholipase-C.159 TFF1 expression is repressed by STAT-3, and activation of STAT-3 is also emerging as a key pathway that leads to gastric cancer.44 RUNX3 most likely suppresses gastric epithelial growth by inducing p21 and Bim, attenuating Wnt signaling, and is altered in 82% of gastric cancers.160 Investigations into these genes and their contributions to the gastric cancer phenotype will prove valuable to our understanding of disease progression.
MSI in dinucleotide repeats secondary to defects in DNA mismatch repair genes, such as MLH1 and MLH2 (mutL homologs 1 and 2), have been implicated in the development of colorectal cancer, and in particular the HNPCC syndrome. Patients with HNPCC have an 11% incidence of gastric cancer, suggesting that MSI may also play a role in the development of gastric cancer.141 MSI is found in 15% to 50% of sporadic gastric cancers, with a higher prevalence in the intestinal type of cancer.161–166 Low-level microsatellite activity (e.g., MSI-low) can be found in 40% of areas of intestinal metaplasia in patients with gastric cancer166 and in 14% to 20% of adenomatous polyps.164,166,167 MSI-high (MSI-H) occurs in only 10% to 16% of gastric cancers. MSI is associated with the less frequent occurrence of TP53 mutations, well- to moderately well differentiated histology, and distal tumor location. Studies that have examined the effect of MSI on patient survival have shown inconsistent results.167,168 When the findings are taken together, it would appear that MSI does play a role in the pathogenesis of gastric cancer, likely before the development of intestinal metaplasia (see Fig. 54-3), and is most commonly due to methylation of the MLH1 promoter.
The data regarding the genetics of diffuse gastric cancer are less complete. Mutations in the E-cadherin (CDH1) gene have been linked to the development of the diffuse type of gastric cancer. Several kindreds, families with hereditary diffuse gastric cancer (HDGC) have been found to carry a germline mutation in the CDH1 gene, all with diffuse-type cancer.130–132,169,170 Further evidence supporting a role for E-cadherin in the pathogenesis of gastric cancer comes from studies showing that suppression of E-cadherin expression occurs in 51% of cancers, with a higher percentage found in diffuse-type cancers.171 Furthermore, E-cadherin underexpression is associated with higher rates of lymph node metastases and reduced survival.172,173 The overall rates of CDH1 mutations in gastric cancer are low, however, with the decreased expression of E-cadherin seen in gastric cancer likely secondary to hypermethylation of the CDH1 promoter, which occurs in 50% of gastric cancers and 83% of diffuse-type gastric cancers.174 E-cadherin is a transmembrane protein that connects to the actin cytoskeleton through α- and β-catenins to establish cell polarity and mediates homophilic cellular interactions.175,176 Decreased expression of E-cadherin is believed to promote dissociation of cancer cells from their cell matrix, enhancing the migration and invasion of gastric cancer cells. Expression of α-catenin is also decreased or absent in 68% of gastric cancers.177 Therefore, E-cadherin appears to act as a tumor suppressor gene that may be important in the pathogenesis of diffuse gastric cancer.
Perhaps as important as the genetic alterations acquired during the progression to gastric adenocarcinoma, is in what target cells do these changes occur? In order for a cell to accumulate the quantity of genetic changes necessary for autonomous growth, it must be long lived. For these reasons, the current thinking is that a resident tissue stem cell is the target of genetic mutations and becomes the “cancer stem cell” capable of autonomous growth and with metastatic potential. Work from our laboratory offers a new model for the cancer stem cell. Bone marrow–derived stem cells are capable of homing to injured and inflamed peripheral organs and differentiating into organ-appropriate cell lineages.178–181 In an environment of inflammation and altered growth signaling, these cells can differentiate aberrantly and become dysplastic and neoplastic, and we have shown they constitute the majority of cells within in situ as well as invasive gastric adenocarcinoma lesions.182 Although much work needs to be done to understand these findings completely, they offer an exciting possibility for new approaches to understanding and treating gastric and other inflammatory mediated cancers.
PREMALIGNANT CONDITIONS
CHRONIC ATROPHIC GASTRITIS
Chronic atrophic gastritis, which is defined as the loss of specialized glandular tissue in its appropriate region of the stomach, is an established early morphologic change that occurs along the sequence toward the development of gastric cancer.16,183 The presence of atrophic gastritis has an annual incidence of progression to gastric cancer of approximately 0.5% to 1%.184–187 The extent of atrophic gastritis within the stomach correlates with risk of progression to cancer.188–190
There are two forms of atrophic gastritis. The more common is multifocal atrophic gastritis (MAG), which is associated with H. pylori infection and more likely to be associated with metaplasia. The presence of H. pylori infection is associated with an approximately 10-fold increased risk of atrophic gastritis.191 There is considerable regional variation in the prevalence of atrophic gastritis in H. pylori–infected individuals, with a roughly 3-fold increase in Asian compared with Western countries.191,192 The second form of atrophic gastritis, corporal atrophic gastritis, is associated with antiparietal cell and intrinsic factor antibodies. This form of atrophy is confined to the body and fundus. Corporal atrophic gastritis is associated with pernicious anemia and an increased gastric cancer risk, albeit not as high as that seen with H. pylori–induced multifocal atrophic gastritis, owing most likely to a lesser degree of inflammation.185,193
Mechanisms underlying the increased risk of gastric cancer in the setting of gastric atrophy may be related to low acid output (achlorhydria), which predisposes to gastric bacterial overgrowth (with non-Helicobacter organisms), greater formation of N-nitroso compounds, and diminished ascorbate secretion into the gastric lumen.194 Additionally, serum gastrin levels are increased in response to the reduced acid output. Gastrin is a known growth factor for gastric mucosal cells, and sustained elevations of serum gastrin may contribute to abnormal growth and increased risk of neoplastic progression.195,196
INTESTINAL METAPLASIA
Intestinal metaplasia (IM) can be subdivided into three categories, as classified by Jass and Filipe and as discussed in Chapter 51.197 Type I is the complete form of IM, containing Paneth cells, goblet cells that secrete sialomucins, and absorptive epithelium with well-defined brush borders. Type I metaplasia does not raise the risk of gastric cancer. Type II or incomplete metaplasia contains few absorptive cells, few columnar intermediate cells, and goblet cells that express sialomucins. Type III is intermediate between type I and type II and contains properties of both.198 It is estimated that the presence of type II or III IM is associated with a 20-fold increased risk of gastric cancer. Early gastric cancer develops in 42% of patients with type III IM within five years of follow-up, suggesting that IM represents a precursor lesion for the intestinal form of gastric cancer.199 However, whether cancer arises from areas of IM or whether IM simply represents a marker for higher gastric cancer risk remains unclear. As is the case with atrophic gastritis, the prevalence of IM in H. pylori–infected individuals is higher in Asia (≈40%) as compared with the West.191,192
DYSPLASIA
Histologic assessment of gastric dysplasia and adenocarcinoma is based on the 2000 Vienna classification (Table 54-4).200 The prevalence of gastric dysplasia ranges from as low as 0.5% in low-risk areas201 to 20% in high-risk areas.202 Prospective studies have shown that low-grade dysplasia may regress in up to 60% of cases, whereas it progresses to high-grade dysplasia in 10% to 20% of cases (Fig. 54-4).203–205 High-grade dysplasia rarely regresses, and is associated with an annual incidence of progression to gastric cancer of 2% to 6%.205–207 In a prospective cohort study from the Netherlands, the presence of high-grade dysplasia was associated with a markedly increased risk of progression to cancer (adjusted hazard ratio, 40.1).206 High-grade dysplasia is often associated with synchronous cancer and can be uni- or multifocal.208
Table 54-4 Padova International Classification System for Gastric Dysplasia
| CATEGORY | DEFINITION | HISTOLOGIC DESCRIPTION |
|---|---|---|
| I | 1.0 Normal | Normal gastric architecture with absent or minimal inflammatory infiltrates. |
| 1.1 Reactive foveolar hyperplasia | The general architecture is well preserved, with evidence of hyperproliferative epithelium, enlarged nuclei, and mitotic figures. | |
| 1.2 Intestinal metaplasia | Type I. Closely resembles the morphology of the small intestine, with absorptive enterocytes, well-defined brush borders, and well-formed goblet cells. | |
| Type II. Incomplete metaplasia with irregular mucous vacuoles, absence of brush borders, and difficult-to-identify absorptive enterocytes. Cells secrete mainly sialomucins. | ||
| Type III. Same as type II except cells secrete mainly sulfomucins. | ||
| II | Indefinite for dysplasia | Inability to discern whether cells are neoplastic or non-neoplastic. Usually found in setting of inadequate biopsy specimens and presence of architectural distortion and nuclear atypia. |
| III | Noninvasive neoplasia | Phenotypically neoplastic epithelium confined to glandular structures inside the basement membrane. Includes adenomas. |
| Should be divided into “low grade” and “high grade.” | ||
| IV | Suspicious for invasive cancer | Presence of neoplastic epithelium, but where invasion cannot be clearly identified. |
| V | Invasive cancer | Invasive carcinoma. |
Adapted from Rugge M, Correa P, Dixon M, et al. Gastric dysplasia: The Padova International Classification. Am J Surg Pathol 2000; 24:167.
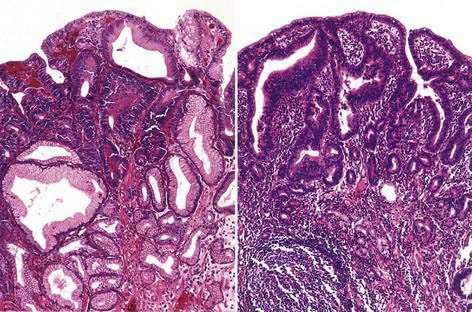
GASTRIC POLYPS
The prevalence of gastric polyps in the general population is approximately 0.8% to 2.4%.209,210 Gastric polyps consist predominantly of fundic gland polyps (≈50%), hyperplastic polyps (≈40%), and adenomatous polyps (≈10%).210,211 The clinical course of fundic gland polyps is generally benign, and they are detected with increasing frequency in the era of proton pump inhibitor (PPI) use. In a series of 599 consecutive patients who underwent upper endoscopy, use of PPIs for 5 years or longer was associated with a significantly increased risk of fundic gland polyps (hazard ratio, 3.8).212 The rate of malignant transformation of fundic gland polyps is generally quite low (≈1%) and confined to polyps larger than 1 cm.213 One notable exception to the benign nature of fundic gland polyps is in patients with FAP. In this group the prevalence of fundic gland polyps ranges from 51% to 88%, with dysplasia present in more than 40% of cases.139,140
Hyperplastic polyps are almost always benign lesions. The rare hyperplastic polyps that undergo malignant transformation often contain areas of intestinal metaplasia or dysplasia and typically form a well-differentiated intestinal-type cancer.213
In contrast to fundic gland and hyperplastic polyps, gastric adenomas undergo malignant transformation at a high rate. Gastric adenomas that were followed by serial endoscopy with biopsy were documented to progress to dysplasia and then to carcinoma in situ, which developed within 4 years of follow-up in approximately 11% of cases.214 Endoscopic biopsy of gastric polyps to find dysplasia or carcinoma is associated with significant sampling error.215 Therefore, it is suggested that all adenomas (and other polyps larger than 1 cm or those causing symptoms such as occult bleeding) be removed by endoscopic polypectomy. Decisions regarding surveillance intervals should be made on an individual basis.
PREVIOUS GASTRECTOMY
It has been reported by several groups that gastric surgery for benign conditions can predispose patients to a higher risk of gastric cancer, beginning 20 years after the surgery.216–219 The risk is greatest for those who underwent surgery before the age of 50 years, perhaps reflecting the long lag period necessary between the operation and the development of cancer.217 The cancers tend to occur at or near the surgical anastomosis on the gastric side; only rarely do they reside on the intestinal side of the anastomosis.220
Numerous theories have been proposed to explain the increased propensity for cancer to form at the surgical anastomosis site. They include hypochlorhydria resulting in gastric bacterial overgrowth, with increased production of nitrites, chronic enterogastric reflux of bile salts and pancreatic enzymes, which are potent gastric irritants, and atrophy of the remaining fundic mucosa secondary to low levels of antral hormones such as gastrin.17,221,222 The Billroth II operation predisposes to the development of cancer at a four-fold higher rate than a Billroth I procedure, suggesting that bile reflux may be a significant predisposing factor.217 H. pylori and associated intestinal metaplasia are found less frequently in postgastrectomy gastric cancers as compared to distal gastric cancers in the nonoperative stomach.223 It is unclear whether screening for gastric cancer in this population of patients in areas of low cancer incidence would be cost effective. With the advent of H. pylori eradication therapy as well as PPIs, the number of gastric resections for peptic ulcer disease has decreased dramatically, significantly reducing the effect of the postgastrectomy state as a risk factor for gastric cancer.
PEPTIC ULCER DISEASE
Large epidemiologic studies have demonstrated a consistently increased risk of gastric cancer in patients with a history of a gastric ulcer. In a cohort study of nearly 58,000 adults from Sweden who were followed for an average of nine years, a history of gastric ulcer was associated with a significant 1.8-fold increased risk of gastric cancer.224 Interestingly, a history of duodenal ulcer was associated with a significant 40% decreased risk of gastric cancer. A separate case control study of more than 90,000 U.S. veterans found a similarly increased risk of noncardia gastric cancer (but not cardia cancer) in patients with a history of gastric ulcer, and a corresponding decreased risk in those with a history of duodenal ulcer.225 The reasons for these disparate cancer risks are not entirely clear.
MÉNÉTRIER’S DISEASE (see Chapter 51)
In a review of case reports, 15% of patients with Ménétrier’s disease had associated gastric cancer,226 including several cases that documented a progression from dysplasia to cancer.227,228 Because of the rarity of Ménétrier’s disease, it has been difficult to study its relationship with gastric cancer in any controlled fashion, and no recommendations regarding endoscopic surveillance can be made.
SCREENING AND PREVENTION
SCREENING AND SURVEILLANCE
The majority of the literature regarding screening for gastric cancer comes from east Asia, where the prevalence of this disease is among the highest in the world.229 Since 1960, the Japanese have been performing mass screening, using upper GI barium studies followed by endoscopy if any suspicious lesions are found. Japanese researchers have reported a sensitivity of 66% to 90% and a specificity of 77% to 90% for this screening approach.230 Not surprisingly, studies from Japan have also shown that use of screening leads to a diagnosis of gastric cancer at earlier stages, with one study reporting that more than 50% of cases detected by screening were diagnosed as stage I.231 (Staging of gastric cancer is discussed later.) Long-term follow-up data from the Japanese Public Health Center cohort of more than 42,000 adults showed that subjects who underwent screening had a nearly 50% reduced risk of death from gastric cancer.232 A separate cohort study of 87,000 adults from Japan found a 25% to 35% risk reduction in death from gastric cancer among those who participated in gastric cancer screening.233 However, similar risk reductions were seen for death from all causes, casting a level of uncertainty on the true magnitude of benefit associated with screening with respect to preventing death from gastric cancer.
The serum pepsinogen (PG) test is increasingly used to screen for patients at highest risk for having preneoplastic gastric lesions.234 As discussed in Chapter 49, the stomach produces two types of pepsinogen: PGI and PGII. In the presence of atrophic gastritis, production of PGI from oxyntic glands is reduced, whereas PGII production remains relatively constant. Therefore, low serum pepsinogen I levels (<70 mg/L) and a serum pepsinogen I/II ratio less than 3 are useful for the identification of patients with atrophic gastritis.229 A prospective cohort study found that patients with low serum PGI concentrations had a significantly elevated risk of gastric cancer.235
Screening for gastric cancer with upper endoscopy is likely cost-effective in moderate- to high-risk populations, such as older Asian men.236 However, in populations with a lower incidence of gastric cancer, screening is less likely to have the same degree of benefit.
PREVENTION
Eradication of Helicobacter pylori
The effect of eradicating H. pylori on the subsequent risk of gastric cancer is not entirely clear. There is little question that chronic inflammation in a variety of organ systems can lead to malignancy237 and that H. pylori eradication can reduce or alleviate gastric inflammation. Studies in patients have demonstrated that H. pylori eradication can lead to decreased oxidative stress and cell proliferation.238 In addition, limited studies involving eradication of gastric Helicobacter organisms in Mongolian gerbils suggest that eradication can partially reverse atrophy and metaplasia and inhibit progression to gastric cancer.239–242 Studies in mice confirm the reversibility of metaplasia and prevention of gastric cancer with early eradication. With later eradication, cancer progression was slowed and cancer mortality dramatically decreased.18
Nevertheless, with regard to published trials in humans, conclusive evidence that treatment of H. pylori infection prevents gastric cancer is lacking, in part because of the rare endpoint—gastric cancer—needed for these studies. One approach has been to examine intermediate biomarkers such as gastric atrophy and intestinal metaplasia, which are generally considered premalignant lesions. Thus, a number of studies have looked at the effect of H. pylori eradication on these intermediate biomarkers, and a majority have shown a beneficial effect in preventing progression of gastric disease.243–247 In a randomized placebo-controlled trial of 587 Chinese patients with H. pylori infection, assignment to eradication therapy was associated with a significantly reduced risk of progression of intestinal metaplasia (odds ratio, 0.63).248 However, a randomized placebo-controlled trial of Mexican adults did not demonstrate any benefit of H. pylori eradication in the prevention of histologic progression.247
In an open-label randomized controlled trial of patients with resected early gastric cancer, patients were assigned to receive either H. pylori eradication therapy or placebo. After three years of follow-up, H. pylori eradication was associated with a reduced odds of development of metachronous gastric cancer (OR 0.35; 95% CI: 0.16 to 0.78).249 In a retrospective analysis of a cohort of Japanese patients who were treated for H. pylori, successful eradication (as compared to persistent infection) was associated with a significant 80% reduced risk of gastric cancer.250
A prospective, randomized placebo-controlled trial sought to determine whether H. pylori eradication in a high-risk population in China would reduce the incidence of gastric cancer.246 Although no overall benefit was seen in the group receiving H. pylori eradication, there was a clear reduction in gastric cancer incidence in a post hoc subgroup analysis of H. pylori carriers who did not have precancerous lesions (gastric atrophy, intestinal metaplasia, or dysplasia) at study initiation. It is possible that some of the patients in the eradication arm may have passed a “point of no return,” when cellular alterations had sufficiently accumulated to promote cancer.251 Further randomized trials are needed, but the evidence to date supports the notion that early eradication of H. pylori may prevent or delay progression to gastric cancer in high-risk patients.
In Western countries, gastric cancer prevention has not been extensively pursued due to the low prevalence of H. pylori and decreasing incidence of gastric cancer. However, a study by Parsonnet and colleagues252 suggested that screening and treatment of H. pylori infection would be cost-effective in the prevention of gastric cancer, particularly in high-risk populations, if it was assumed that treatment of H. pylori infection prevented 30% of attributable gastric cancers. Using a more conservative 10% reduction in gastric cancer risk, an analysis from the United Kingdom also concluded that H. pylori eradication was cost-effective.253
Antioxidants
Chronic inflammatory states such as H. pylori gastritis can result in the generation of free radicals derived from oxygen and nitrogen.254 These free radicals can promote carcinogenesis via numerous mechanisms, including direct DNA damage and inhibition of DNA repair mechanisms, inhibition of apoptosis, and activation of cellular proliferation pathways. Antioxidants, such as carotenoids and vitamins C and E, bind with reactive oxygen and nitrogen species to neutralize their damaging effects.
Epidemiologic data support a relationship between increased antioxidant intake and reduced risk of gastric cancer.255–259 In a nested case-control study from Japan, low plasma β-carotene levels were associated with an increased risk of gastric cancer.257 A case-control study from Korea found that elevated nitrate-to-antioxidant intake ratios were associated with an increased risk of gastric cancer.258 In a Swedish cohort study, high levels of vitamin A, retinol, and α- and β-carotene intake were associated with a 50% risk reduction in gastric cancer.259
However, in a randomized placebo-controlled trial of antioxidants (either vitamin A, C, or E) in patients with precancerous gastric lesions (nonatrophic or atrophic gastritis, intestinal metaplasia, or dysplasia), antioxidant supplementation did not result in either reduced histologic progression or increased histologic regression.260 A randomized controlled trial in China also found no effect of combined vitamin C, E, and selenium supplementation on the prevalence of a combined endpoint of atrophic gastritis, intestinal metaplasia, dysplasia, or cancer.261 Based on these results as well as those of the Beta Carotene and Retinol Efficacy Trial, in which subjects who received β-carotene and vitamin A had an increased risk of lung cancer,262 antioxidant supplementation for the prevention of gastric cancer cannot yet be recommended.
Aspirin and Nonsteroidal Anti-inflammatory Drugs
Among other effects, aspirin and other NSAIDs inhibit cyclooxygenases. COX-1 is constitutively expressed in the GI tract. COX-2 expression is generally not observed in normal GI mucosa, but has been described in multiple epithelial malignancies, including gastric cancer.263,264 COX-2 expression is associated with aggressive cell growth in human as well as mouse models of cancer265–268 and has been found to be overexpressed in 70% of gastric cancers.269 In this setting, COX-2 could promote the growth of tumors, inhibit apoptosis, and increase angiogenesis. COX-2 expression has been reported to be elevated in preneoplastic lesions, including intestinal metaplasia and dysplasia, and COX-2 levels appear to diminish after H. pylori eradication.270
Multiple epidemiologic studies have demonstrated a consistent association between NSAID use and a reduced risk of gastric cancer.271–274 In a case-control study from Los Angeles County, NSAID use for more than 5 years was associated with a significantly decreased risk of noncardia gastric cancer (OR, 0.61), and there was a significant dose-related effect.272 A nested case-control study using the General Practitioners Research Database found that long-term users of non-aspirin NSAIDs had a significantly reduced risk of gastric cancer (OR, 0.65), although there was no observed association between aspirin use and gastric cancer.274 A meta-analysis reported a significant association between any NSAID use and reduced risk of gastric cancer (OR, 0.78), with similar findings for both acetylsalicylic acid (ASA) and non-ASA NSAIDs.273
In a randomized controlled trial of H. pylori–negative patients with intestinal metaplasia, patients receiving the COX-2 selective inhibitor rofecoxib or placebo had no difference in the rate of regression of intestinal metaplasia after two years.275 This trial was limited by the relatively short follow-up period and use of premalignant endpoints. Further trials in high-risk patients are warranted to determine if NSAIDs are effective for gastric cancer prevention; at the present time they cannot be recommended due to their unproven efficacy and known side effects.
Green Tea
Green tea is widely consumed in Asian countries and is hypothesized to have protective effects against cancer of the upper digestive tract. Polyphenols and other metabolites present in green teas, such as epigallocatechin-3-gallate (EGCG) and other catechins, have a variety of antitumor effects, including induction of apoptosis, inhibition of tumor cell growth and proliferation, and reduction in COX-2 expression.276–278 EGCG also has antioxidant properties and may have anti-inflammatory properties as well.279,280
The majority of case-control studies have shown an inverse association between the risk of gastric cancer and the consumption of green tea.281–283 However, a population-based prospective cohort study in northern Japan found no association between green tea consumption and the risk of gastric cancer.284 A separate cohort study from Japan reported a reduced risk of gastric cancer in women with high green tea consumption, but no change in risk among men.285 Thus, prospective controlled trials are needed, and at present, green tea cannot be recommended as chemoprevention for gastric cancer.
CLINICAL FEATURES
Early gastric cancers are asymptomatic in up to 80% of cases. When symptoms occur they tend to mimic those of peptic ulcer disease. With advanced gastric cancer, the common symptoms are weight loss (≈60% of patients) and abdominal pain (≈50%).10 Other presenting symptoms include nausea, vomiting, anorexia, dysphagia, melena, and early satiety. Pyloric outlet obstruction can occur with tumors of the antrum and pylorus, and tumors of the cardia can cause dysphagia due to involvement of the lower esophageal sphincter and development of pseudoachalasia.286 Rarely, paraneoplastic syndromes occur, such as thrombophlebitis (Trousseau’s sign), neuropathies, nephrotic syndrome, and disseminated intravascular coagulation.287–289 Dermatologic paraneoplastic syndromes may occur uncommonly (see Chapter 22) and include hyperpigmented patches in the axilla (acanthosis nigricans) and the sudden onset of seborrheic dermatosis (senile warts) and pruritus (sign of Leser-Trélat).290
The physical examination is usually unremarkable. Cachexia and signs of bowel obstruction are the most common abnormal findings. Occasionally it is possible to detect an epigastric mass, hepatomegaly, ascites, and lower extremity edema.291 Laboratory studies are generally unrevealing until the cancer reaches advanced stages. Anemia and a positive test result for fecal occult blood may occur from chronic bleeding of an ulcerated mass. Hypoproteinemia can occur in patients with weight loss. Liver enzyme values, particularly alkaline phosphatase, can be elevated secondary to hepatic metastases.
Gastric cancer is metastatic at the time of diagnosis in 33% of cases.292 The most common sites of metastasis are the liver (40%) and peritoneum.293 Other sites of spread include the periumbilical area (Sister Mary Joseph’s nodule), left supraclavicular sentinel nodes (Virchow’s node), the pouch of Douglas (rectal shelf of Blumer), and the ovaries (Krukenberg’s tumor). Gastric cancer has also been reported to metastasize to the kidney, bladder, brain, bone, heart, thyroid, adrenal glands, and skin.291 There are reports of unusual presentations of metastatic disease, such as shoulder-hand syndrome from bone metastasis, diplopia and blindness from orbit and retinal metastases, and virilization due to Krukenberg’s tumors.294–297
DIAGNOSIS
ENDOSCOPY
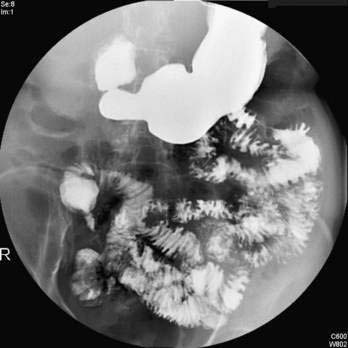
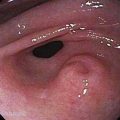
Esophagogastroduodenoscopy (EGD) is currently the procedure of choice for the diagnosis of gastric cancer (Fig. 54-5A). When a nonhealing gastric ulcer is found, at least six to eight biopsy specimens from the edge and base of the ulcer are recommended.298 As discussed in Chapter 13, the American Gastroenterological Association has recommended that an upper endoscopy be performed in patients who are older than 55 years with new-onset dyspepsia and in patients younger than 55 years who have “alarm” symptoms (weight loss, recurrent vomiting, dysphagia, evidence of bleeding, anemia).299 Dyspeptic patients in whom an empirical trial of proton pump inhibitors and eradication of H. pylori do not relieve symptoms should undergo prompt endoscopic evaluation as well. The basis for these recommendations is the low incidence of gastric cancer in individuals younger than 55 years. The yield of upper endoscopy for the detection of gastric cancer in patients with occult bleeding and a normal colonoscopy will vary based on the patient’s baseline risk of gastric cancer. Extensive involvement by the diffuse type of gastric cancer can manifest as a rigid and thickened stomach, also known as linitis plastica.
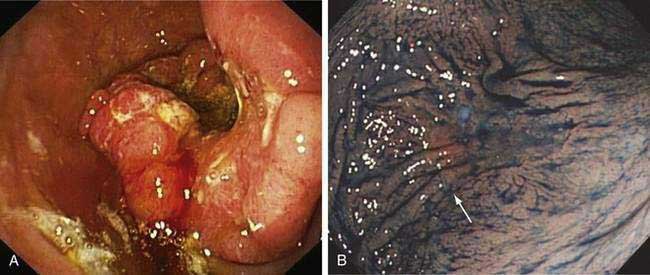
In Japan and other areas of high gastric cancer prevalence, chromoendoscopy, magnification endoscopy, and narrow band imaging are used alone or in combination as aids in the detection of early gastric cancer (see Fig. 54-5B). Distinct irregular mucosal surface and vascular patterns have been found to correlate with the presence of dysplasia and carcinoma.300 There are also ongoing investigations into the utility of newer techniques such as autofluorescence and confocal microendoscopy for the diagnosis of early gastric neoplasia.301,302
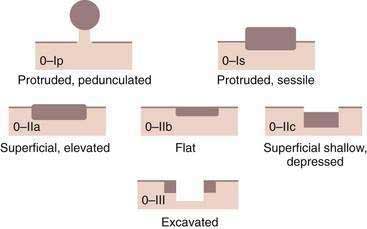
A classification system has been developed for early gastric cancer based on endoscopic appearance,303 the purpose of which is to assess early lesions for risk of submucosal invasion as well as risk of lymph node spread (Fig. 54-6). The three types include superficial polypoid (type 0-I), superficial flat/depressed (types 0-IIa to 0-IIc), and superficial excavated (type 0-III) lesions. The most commonly observed subtype is 0-IIc, the nonpolypoid depressed lesion.303 This classification system is used most often in Japan, where endoscopic mucosal resection and submucosal dissection are frequently performed for early gastric neoplasia.
UPPER GASTROINTESTINAL SERIES
Upper GI series has been largely replaced by upper endoscopy as the initial test of choice for the diagnosis of gastric cancer. Barium studies have been reported to have 60% to 70% sensitivity and 90% specificity for the detection of advanced gastric cancer.304
COMPUTED TOMOGRAPHY GASTROGRAPHY
Computed tomography (CT) colonography has gained significant attention for its potential role as a screening modality for colon polyps and colon cancer (see Chapter 123). CT gastrography has also been studied for the diagnosis of early gastric cancer. In a study of 39 patients from South Korea with early gastric cancer, CT gastrography had a sensitivity of 73% to 76% and good interobserver reliability (R = 0.84).305 Only small studies have been performed thus far using this imaging modality, and CT gastrography cannot yet be recommended for screening outside of the research setting.
SERUM MARKERS
To date no reliable serum marker has been identified with sufficient sensitivity and specificity for the diagnosis of gastric cancer. Low serum pepsinogen I levels, low ratios of serum pepsinogen I to pepsinogen II, and hypergastrinemia have been reported in patients with atrophic gastritis and intestinal metaplasia, but the utility for the detection of gastric cancer has been mixed.306,307 In a screening study of 17,000 Japanese men, a positive pepsinogen test (defined as a serum pepsinogen I <50 µg/L and a pepsinogen I/II <3), in combination with upper GI series, identified gastric cancer in 0.28% of subjects (≈1 in 350), and 88% of these were early gastric cancers.308 Additionally, 89% of the cancers identified by the pepsinogen test alone were early gastric cancers. The major limitation of this test is the low specificity for the diagnosis of gastric cancer.309
Serum carcinoembryonic antigen (CEA) and carbohydrate antigen (CA) 19-9 have been extensively studied for the diagnosis of gastric cancer. The sensitivities of these markers is quite low for early gastric cancer,310 and elevated levels are levels also seen in other epithelial malignancies. These tumor markers are frequently elevated in recurrent gastric cancer, especially in patients who had elevated levels prior to surgical resection.311 More recent studies have identified, among others, transforming growth factor-β1, CA 72-4, tumor M2-pyruvate kinase, hepatocyte growth factor, and others as potential markers for the diagnosis of gastric cancer.312–315 However, larger studies are required to determine their clinical utility.
CLASSIFICATION AND STAGING
Several classification systems further define gastric cancer and predict prognosis. As mentioned (see Fig. 54-2), gastric cancers can be subdivided into intestinal and diffuse types. Gastric cancer can also be divided into early and advanced lesions. Early gastric cancer is defined as a cancer that does not invade beyond the submucosa regardless of lymph node involvement. Early gastric cancer has a much higher prevalence in the Far East, especially Japan, and carries a very favorable prognosis, with five-year survival rates greater than 90% being reported in Asia and greater than 80% in Western countries.316–319
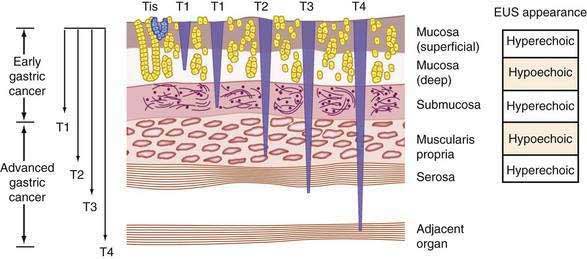
The most commonly used clinical staging classification system for gastric cancer is the TNM system, used by the International Union Against Cancer (UICC) and the American Joint Committee on Cancer (AJCC).320,321 In the TNM staging system, T (tumor) indicates the depth of penetration (Fig. 54-7): T1 denotes a tumor confined to the mucosa or submucosa, T2 denotes involvement of the muscularis propria, T3 denotes invasion through the serosa, and T4 denotes invasion through the serosa and into adjacent organs or structures. N (nodes) indicates the amount of lymph node invasion: N0 denotes no lymph node involvement, N1 denotes involvement of 1 to 6 regional lymph nodes, N2 denotes involvement of 7 to 15 regional lymph nodes, and N3 denotes involvement of more than 15 regional lymph nodes. M (metastasis) indicates the presence of metastases, with M0 denoting no metastases and M1 denoting distant metastases (Table 54-5).

METHODS OF STAGING
Endoscopic Ultrasonography
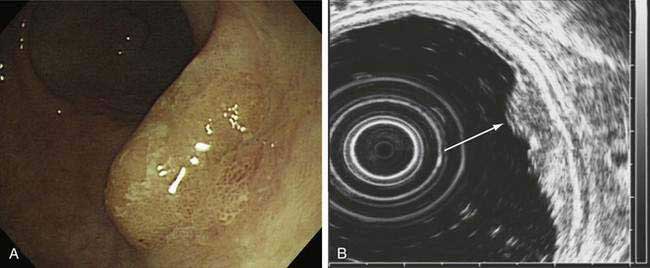
EUS allows the visualization of the five layers of the gastric wall. The superficial gastric mucosa is represented by an echogenic first layer, and the deeper mucosa by a hypoechogenic second layer; the submucosa is represented by an echogenic third layer, the muscularis propria as a hypoechogenic fourth layer, and the serosa as an echogenic fifth layer.322 Studies of the ability of EUS to determine T stage have reported an accuracy rate of 67% to 92%, with accuracy of determination of serosal involvement ranging from 78% to 100%.323 EUS is particularly useful for staging early gastric cancer, which can be potentially resected endoscopically (Fig. 54-8). In a study of 295 patients with early gastric cancer, high-frequency EUS was found to have 90% accuracy for differentiating between mucosal and submucosal tumor invasion.324
In terms of N staging, the rate of detection of perigastric lymph nodes with EUS is comparable to staging with CT, with a diagnostic accuracy ranging from 50% to 80%.325–329 A particular difficulty with N staging lies in the fact that many small lymph nodes can also harbor metastases, and thus understaging can occur. One study of 1253 lymph nodes in 31 patients with gastric cancer found that 55% of lymph nodes containing tumor were smaller than 5mm.330
EUS also has the ability to identify and biopsy submucosal lesions, such as gastric lymphomas and stromal tumors (discussed later and in Chapters 29 and 31). These lesions typically involve thickening of the submucosa and muscularis propria and may appear as gastric fold thickening on barium studies or endoscopy.
Computed Tomography
Advances in imaging technology have greatly improved the ability of CT to stage gastric tumors. Although not as extensively studied as EUS, multidetector row CT (MDCT), by which the wall of the stomach can be seen as three layers (an inner layer corresponding to the mucosa, an intermediate layer corresponding to the submucosa, and an outer layer of slightly higher attenuation corresponding to the muscularis propria and serosa), appears to have comparable accuracy to EUS in terms of T and N staging. The loss of fat planes between the gastric mass and an adjacent organ suggests tumor invasion. The accuracy of MDCT for overall T staging ranges from 77% to 89%, and discriminates serosal involvement with an accuracy of 83% to 100%.323 Accuracy with respect to N staging may be as high as 89% with MDCT.331,332 As with all other imaging modalities, CT has difficulty discerning lymph node metastases smaller than 5 mm. At present, the role of CT is mainly for the detection of distant metastases and as a complement to EUS for assessing regional lymph node involvement. It is not yet clear whether EUS or MDCT (or the combination) is superior for T and N staging in gastric cancer, and the underlying technology continues to evolve and improve.
Magnetic Resonance Imaging
MRI with gadolinium has also been used for gastric cancer staging. It is similar to CT in its advantages (ability to find distant metastases) and weaknesses (need for adequate gastric distention). The accuracy of MRI ranges from 90% to 93% for T staging and from 91% to 100% for N staging.323 However, given the small number of studies, MRI cannot yet be advocated as the test of choice for staging gastric cancer.
Positron Emission Tomography
Positron emission tomography (PET) is not very useful for staging in gastric cancer, largely due to the fact that most gastric adenocarcinomas have low fluorodeoxyglucose uptake.333
Restaging after Neoadjuvant Treatment
The accuracy of restaging gastric cancer after neoadjuvant chemotherapy decreases considerably. EUS has less than 50% accuracy for T and N restaging, and similarly disappointing results have been reported for post-treatment staging with CT.334 However, the use of preoperative staging to assess response to chemotherapy may correlate well with both overall and disease-free survival.335
PROGNOSIS AND TREATMENT
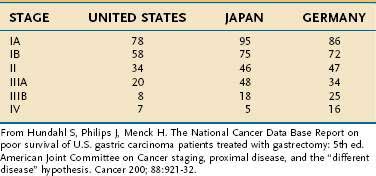
Overall, the five-year survival rate in the United States from gastric cancer is 24% compared with 64% for colon cancer.292 The TNM classification is used to stratify disease into four clinical stages (I through IV) to predict prognosis in patients treated with gastrectomy (Table 54-6).336 The survival data from Japanese studies are generally superior to those seen in Western countries, perhaps because of the preference in Japan for extended lymphadenectomy or because of less understaging than is found in Western countries.336
SURGICAL THERAPY
Surgery, and laparoscopy in particular, can be useful in the staging of cancer. Laparoscopy can help identify primary tumor resectability, peritoneal deposits, and appropriate candidates for neoadjuvant therapy. Laparoscopic peritoneal lavage has been used to detect occult intraperitoneal free cancer cells. A positive peritoneal lavage correlates significantly with eventual development of overt peritoneal metastases.337
In general, total gastrectomy is performed for proximal gastric tumors and diffuse gastric cancer, and partial gastrectomy is reserved for tumors in the distal stomach. Large, randomized multicenter trials performed in France and Italy compared subtotal with total gastrectomy for adenocarcinoma of the antrum and found no differences in five-year survival rates or operative mortality.338,339 Some centers have argued for performing a complete splenectomy with gastrectomy. However, several retrospective and prospective studies found that concurrent splenectomy increased morbidity and had either no effect on or worsened survival.340,341
The extent of lymphadenectomy accompanying the gastrectomy has been a subject of debate for many years. The Japanese advocate a more extensive lymph node dissection (D2 resection) than their Western counterparts (D1 resection) and have higher published survival rates. A D2 resection entails resection of the nodes of the celiac axis and the hepatoduodenal ligament in addition to the perigastric lymph nodes taken in a D1 procedure. The differences in reported survival rates may reflect the fact that the Japanese have a much higher incidence of early gastric cancer, and the more extensive lymph node dissection performed in Japan may find more positive lymph nodes, making survival rates of Japanese patients with N0 staging appear to be higher than those of their potentially understaged Western counterparts. A large prospective multicenter Dutch study of more than 1000 patients reported no significant difference in five-year survival, with higher rates of postoperative death and complications for D2 lymphadenectomy than for the more conservative D1 lymphadenectomy.342 A British prospective study of 400 patients likewise showed no benefit from more extensive surgery; five-year survival rates were 35% for D1 resection and 33% for D2 resection.340 At present, data are insufficient to support extended lymph node resection in centers outside Japan. To prevent understaging, the current recommendation is a minimum D1 lymphadenectomy with removal of at least 15 nodes.343
ENDOSCOPIC MUCOSAL RESECTION AND SUBMUCOSAL DISSECTION
Advances in endoscopic techniques have permitted endoscopic mucosal resection (EMR) and endoscopic submucosal dissection (ESD) to be used as curative therapies for select early gastric cancers (EGCs). This technique has been used widely for intestinal-type cancers in Japan, where studies have shown that only 3.5% of patients with EGCs smaller than 2 to 3 cm have lymph node involvement, making these lesions amenable to local therapy. Lesions larger than 4.5 cm have a greater than 50% chance of spread into the submucosa, are associated with “positive” nodes, and are therefore less likely to be endoscopically resectable.344
The following criteria have been suggested for choosing to perform EMR in gastric cancer: (1) the cancer is located in the mucosa and the lymph nodes are not involved, as indicated by EUS; (2) the maximum size of the tumor is less than 2 cm when the lesion is slightly elevated (type IIa) and less than 1 cm when the tumor is flat or slightly depressed (type IIb or IIc, respectively) without an ulcer scar (see Fig. 54-6); (3) there is no evidence of multiple gastric cancers or simultaneous abdominal cancers; and (4) the cancer is of the intestinal type.345
It is generally not possible to remove lesions larger than 1.5 to 2 cm en bloc by EMR. Endoscopic submucosal dissection is a technique developed in Japan and permits en bloc resection of larger early gastric cancers. With ESD, submucosal saline injection is performed, followed by the use of endoscopic electrosurgical knives to resect the entire tumor (Fig. 54-9).346 In addition to an R0 resection, ESD allows for more precise histopathologic assessment of depth of invasion and lymphovascular involvement, and permits appropriate assessment for risk of lymph node metastasis. If the preprocedure evaluation does not reveal regional lymph node involvement, much larger superficial lesions can be resected. Because experience with this technique has increased, en bloc resection rates are now reported to be greater than 90%, with local recurrence rates less than 3%.346 Due to the large size of some of the lesions being resected, the risk of perforation is relatively high (2% to 6%).347,348 However, perforations recognized early can generally be treated with closure using endoscopic clipping.346
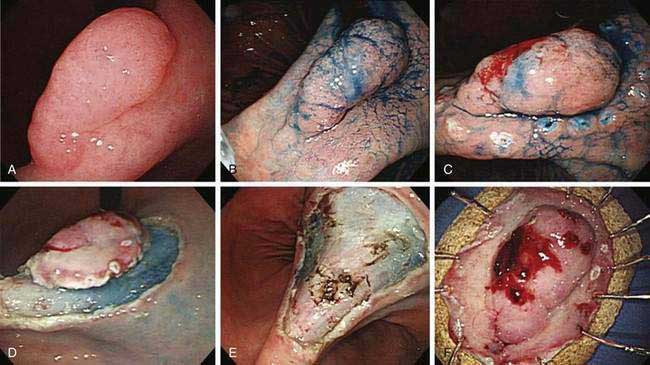
CHEMOTHERAPY
In Western countries, approximately 75% of patients with gastric cancer have disease that has spread to the perigastric lymph nodes or have distant metastases at the time of diagnosis.322 In such patients who have undergone potentially curative resection, recurrence rates are high. Unfortunately, gastric cancer appears to be fairly resistant to conventional chemotherapy. Numerous clinical trials have been performed evaluating the role of adjuvant chemotherapy following attempted curative resection for gastric cancer.349 The majority of the studies were inconclusive, but a series of meta-analyses of these trials suggested a 15% to 20% reduction in the risk of death in patients who received adjuvant chemotherapy.350,351 More recent randomized trials of cisplatin- or epirubicin-based adjuvant chemotherapy have largely failed to show a benefit.352,353
Neoadjuvant chemotherapy, on the other hand, does appear to benefit patients with resectable disease. In the United Kingdom MAGIC trial, 503 patients with gastric, gastroesophageal junction, or distal esophageal cancer were randomly assigned to undergo surgery alone or surgery with neoadjuvant epirubicin, cisplatin, and 5-fluorouracil (5-FU). Compared with surgery alone, the neoadjuvant group had significantly improved five-year survival (36% vs. 23%), progression-free survival (hazard ratio, 0.66), and overall survival (hazard ratio, 0.75).354 As a result, preoperative chemotherapy is now considered an acceptable treatment option for gastric cancer.
CHEMORADIATION
Combined chemoradiation after surgical resection appears to be effective at improving progression-free and overall survival in gastric cancer. The Intergroup Trial 0116 randomized 603 patients with gastric or gastroesophageal junction cancer to undergo surgery alone or surgery followed by 5-FU, leucovorin, and radiation therapy. Subjects in the surgery-alone group had shorter median survival time (27 months vs. 36 months) and worse overall- and relapse-free survivals (hazard ratios, 1.35 and 1.52, respectively).355 Following publication of the results of this study, adjuvant chemoradiation became the standard of care in the United States, although the optimal chemotherapy regimen is not yet clear. Early studies of the use of neoadjuvant chemoradiation have also shown promising results.356
INTRAPERITONEAL CHEMOTHERAPY
Because systemic chemotherapy is ineffective for peritoneal metastasis, intraperitoneal (IP) chemotherapy can be considered in patients whose tumors are resected for cure but have a high likelihood of microscopic residual disease. In a randomized trial of 248 patients with gastric cancer, postoperative hyperthermic IP chemotherapy was associated with improved overall survival compared with surgery alone.357 The treatment benefits were most pronounced in patients with stage III and IV disease, serosal invasion, and lymph node metastases. Although a second clinical trial reported similar results,358 other studies have failed to demonstrate a benefit of hyperthermic IP chemotherapy.359,360 A meta-analysis of studies of IP chemotherapy for patients with resectable gastric cancer reported a significantly reduced risk of death in patients who received hyperthermic IP chemotherapy (hazard ratio, 0.6).361 At present, the use of hyperthermic IP chemotherapy should be confined to patients enrolled in clinical trials, especially in Western countries.
UNRESECTABLE DISEASE
Unfortunately, up to one third of patients with gastric cancer will have unresectable disease at the time of diagnosis.292 Chemotherapy for locally advanced gastric cancer without distant metastases can result in shrinking of the tumor to the point at which successful curative resection is possible.362,363 Even when curative surgery is not possible, chemotherapy has been shown both to improve survival as well as quality of life compared to best supportive care in this group of patients.364 Several randomized clinical trials have demonstrated efficacy of multidrug cisplatin-based regimens.365,366 A newer clinical trial using the EOX regimen (epirubicin, oxaliplatin, and capecitabine) was found to be noninferior to cisplatin-based regimens, and had a median survival of 11.2 months.367 The benefit of the EOX regimen is the substitution of oxaliplatin and capecitabine for cisplatin and 5-FU, respectively, resulting in greater convenience, ease of administration, and potentially fewer side effects.
Patients with advanced gastric cancer of the distal antrum or pylorus are at risk for developing gastric outlet obstruction. Traditionally, surgical gastrojejunostomy was performed for relief of symptoms and to allow continued enteral nutrition. With the advent of endoscopic stents, duodenal stenting across the obstructing tumor has emerged as a nonsurgical alternative for palliation. The results of a literature review of studies evaluating gastrojejunostomy and stenting found no differences between rate of technical success (96% to 100%), early and late complications, and persistent symptoms.368 Recurrent obstructive symptoms were more common with stenting. Both gastrojejunostomy and endoscopic stenting are acceptable options for the relief of malignant gastric outlet obstruction. The decision should be based on the individual clinical scenario as well as the availability of appropriate surgical or endoscopic expertise.
GASTRIC LYMPHOMA
Gastric lymphomas account for approximately 3% of all gastric malignancies, and can be subdivided into those in which the stomach is the primary site of involvement and those with disseminated nodal disease and secondary involvement of the stomach. More than 95% of gastric lymphomas are non-Hodgkin’s lymphomas. Gastric lymphoma is the most common form of extranodal non-Hodgkin’s lymphoma, accounting for more than 30% of all cases.369,370
The clinical manifestations of gastric lymphoma are similar to those of gastric adenocarcinoma. Patients tend to be asymptomatic in the early stages. With more advanced disease, patients may present with nonspecific complaints including abdominal pain, nausea, vomiting, anorexia, weight loss, bleeding, fever, night sweats, swelling, and diarrhea.371 On endoscopy, gastric lymphomas manifest in a variety of ways and can appear as fungating lesions, polypoid masses, thickened gastric folds, or ulcerating lesions. Because of frequent involvement of the submucosa, deep biopsy specimens, obtained by snare technique, mucosal resection, or use of jumbo forceps, are often needed for a pathologic diagnosis. Endoscopic ultrasonography is essential to document extent of disease and may be more useful than CT in the evaluation of perigastric lymph node involvement. Other imaging modalities, such as CT and MRI, may be of value in determining involvement of the liver and spleen as well as of distant lymph nodes. Gastric lymphoma is discussed in detail in Chapter 29.
GASTRIC CARCINOID TUMORS
Gastric carcinoid tumors (Fig. 54-10) account for 7% of all GI carcinoids and 0.2% of all gastric neoplasms.372 In the stomach, the well-differentiated tumors are mainly of enterochromaffin-like (ECL) cell origin, with a small minority being of other endocrine cell types.373 Although gastric carcinoids often contain neuroendocrine peptides, carcinoid syndrome does not occur unless there is hepatic involvement. The large majority of gastric carcinoid tumors express somatostatin receptors, and somatostatin receptor scintigraphy is a useful test to evaluate extent of and spread of disease. Classification, staging, prognosis, and therapy of gastric carcinoids are discussed in detail in Chapter 31.
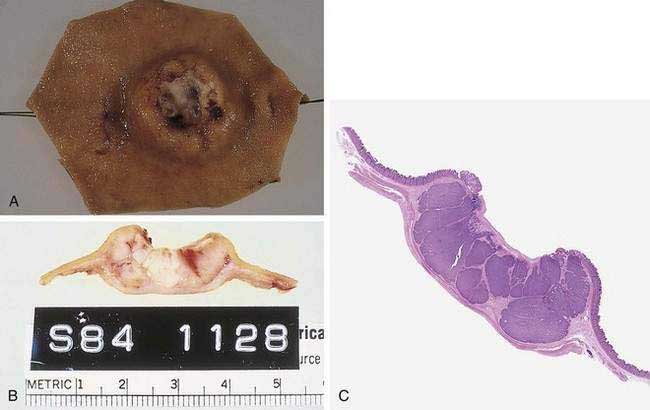
GASTROINTESTINAL STROMAL TUMORS
Gastrointestinal stromal tumors (GISTs) are intra-abdominal nonepithelial (mesenchymal) tumors that can develop throughout the GI tract. Of all GISTs in the GI tract, 50% to 60% occur in the stomach (Fig. 54-11).374 They are discussed in detail in Chapter 30.
Figure 54-11. Pathology of gastrointestinal stromal tumor (GIST). A, GISTs frequently have a heterogeneous cut surface with areas of hemorrhage. B, Histologically these tumors are composed of plump spindle cells with prominent cytoplasmic vacuoles enmeshed within a myxoid stroma (see also Chapter 30). (Hematoxylin and eosin.)
MISCELLANEOUS TUMORS
Metastatic disease to the stomach can occur with melanoma and with primary tumors of the breast, lung, ovary, liver, colon, and testis, with breast cancer being the most common.375 Other rare malignant tumors that can involve the stomach are Kaposi’s sarcoma (see Chapter 33), myenteric schwannoma, glomus tumor, small cell carcinoma, and parietal cell carcinoma.376–379 Miscellaneous benign tumors can involve the stomach and include pancreatic rests, xanthelasma, and fundic gland cysts.
Cunningham D, Allum WH, Stenning SP, et al. Perioperative chemotherapy versus surgery alone for resectable gastroesophageal cancer. N Engl J Med. 2006;355:11-20. (Ref 354.)
de Vries AC, van Grieken NC, Looman CW, et al. Gastric cancer risk in patients with premalignant gastric lesions: A nationwide cohort study in the Netherlands. Gastroenterology. 2008;134:945-52. (Ref 206.)
El-Omar EM, Carrington M, Chow WH, et al. Interleukin-1 polymorphisms associated with increased risk of gastric cancer. Nature. 2000;404:398-402. (Ref 120.)
Fukase K, Kato M, Kikuchi S, et al. Effect of eradication of Helicobacter pylori on incidence of metachronous gastric carcinoma after endoscopic resection of early gastric cancer: An open-label, randomised controlled trial. Lancet. 2008;372:392-7. (Ref 249.)
Hartgrink HH, van de Velde CJ, Putter H, et al. Extended lymph node dissection for gastric cancer: Who may benefit? Final results of the randomized Dutch gastric cancer group trial. J Clin Oncol. 2004;22:2069-77. (Ref 342.)
Kaurah P, MacMillan A, Boyd N, et al. Founder and recurrent CDH1 mutations in families with hereditary diffuse gastric cancer. JAMA. 2007;297:2360-72. (Ref 133.)
Lichtenstein P, Holm NV, Verkasalo PK, et al. Environmental and heritable factors in the causation of cancer—Analyses of cohorts of twins from Sweden, Denmark, and Finland. N Engl J Med. 2000;343:78-85. (Ref 116.)
Macdonald JS, Smalley SR, Benedetti J, et al. Chemoradiotherapy after surgery compared with surgery alone for adenocarcinoma of the stomach or gastroesophageal junction. N Engl J Med. 2001;345:725-30. (Ref 355.)
McDonald SA, Greaves LC, Gutierrez-Gonzalez L, et al. Mechanisms of field cancerization in the human stomach: The expansion and spread of mutated gastric stem cells. Gastroenterology. 2008;134:500-10. (Ref 14.)
Ohnishi N, Yuasa H, Tanaka S, et al. Transgenic expression of Helicobacter pylori CagA induces gastrointestinal and hematopoietic neoplasms in mouse. Proc Natl Acad Sci U S A. 2008;105:1003-8. (Ref 59.)
Plummer M, Vivas J, Lopez G, et al. Chemoprevention of precancerous gastric lesions with antioxidant vitamin supplementation: A randomized trial in a high-risk population. J Natl Cancer Inst. 2007;99:137-46. (Ref 260.)
Soetikno R, Kaltenbach T, Yeh R, Gotoda T. Endoscopic mucosal resection for early cancers of the upper gastrointestinal tract. J Clin Oncol. 2005;23:4490-8. (Ref 345.)
Tu S, Bhagat G, Cui G, et al. Overexpression of interleukin-1beta induces gastric inflammation and cancer and mobilizes myeloid-derived suppressor cells in mice. Cancer Cell. 2008;14:408-19. (Ref 127.)
Uemura N, Okamoto S, Yamamoto S, et al. Helicobacter pylori infection and the development of gastric cancer. N Engl J Med. 2001;345:784-9. (Ref 27.)
Wong BC, Lam SK, Wong WM, et al. Helicobacter pylori eradication to prevent gastric cancer in a high-risk region of China: A randomized controlled trial. JAMA. 2004;291:187-94. (Ref 246.)
1. . Global burden of disease (GBD), 2002 estimates. World Health Organization, 2008. Accessed July 11, 2008, at www.who.int/healthinfo/bodgbd2002/en/index.html
2. . IARC World Cancer Report. International Agency for Research on Cancer, 2006. Accessed July 11, 2008, at www.iarc.fr/IARCPress/pdfs/wcr/index.php
3. Crew KD, Neugut AI. Epidemiology of gastric cancer. World J Gastroenterol. 2006;12:354-62.
4. . Summary of changes in cancer mortality, 1950-2005. National Cancer Institute, 2008. Accessed July 11, 2008, at http://seer.cancer.gov/cgi-bin/csr/1975_2005
5. . Age-Adjusted SEER Incidence and US Death Rates and 5-Year Relative Survival Rates. National Cancer Institute, 2008. Accessed at http://seer.cancer.gov/cgi-bin/csr/1975_2005
6. Parkin DM. International variation. Oncogene. 2004;23:6329-40.
7. . Estimated new cancer cases and deaths for 2008. National Cancer Institute, 2008. Accessed July 11, 2008, at http://seer.cancer.gov/cgi-bin/csr/1975_2005
8. . SEER cancer stat fact sheets: Cancer of the stomach. National Cancer Institute, 2008. Accessed at http://seer.cancer.gov/statfacts/html/stomach.html
9. Wiggins CL, Becker TM, Key CR, Samet JM. Stomach cancer among New Mexico’s American Indians, Hispanic whites, and non-Hispanic whites. Cancer Res. 1989;49:1595-9.
10. Wanebo HJ, Kennedy BJ, Chmiel J, et al. Cancer of the stomach. A patient care study by the American College of Surgeons. Ann Surg. 1993;218:583-92.
11. Locke GR3rd, Talley NJ, Carpenter HA, et al. Changes in the site- and histology-specific incidence of gastric cancer during a 50-year period. Gastroenterology. 1995;109:1750-6.
12. Munoz N, Correa P, Cuello C, Duque E. Histologic types of gastric carcinoma in high- and low-risk areas. Int J Cancer. 1968;3:809-18.
13. Fearon ER. Molecular genetics of colorectal cancer. Ann N Y Acad Sci. 1995;768:101-10.
14. McDonald SA, Greaves LC, Gutierrez-Gonzalez L, et al. Mechanisms of field cancerization in the human stomach: The expansion and spread of mutated gastric stem cells. Gastroenterology. 2008;134:500-10.
15. Munoz N, Matko I. Histological types of gastric cancer and its relationship with intestinal metaplasia. Recent Results Cancer Res. 1972;39:99-105.
16. Correa P. Human gastric carcinogenesis: A multistep and multifactorial process—First American Cancer Society Award Lecture on Cancer Epidemiology and Prevention. Cancer Res. 1992;52:6735-40.
17. Correa P, Haenszel W, Cuello C, et al. Gastric precancerous process in a high risk population: Cohort follow-up. Cancer Res. 1990;50:4737-40.
18. Cai X, Carlson J, Stoicov C, et al. Helicobacter felis eradication restores normal architecture and inhibits gastric cancer progression in C57BL/6 mice. Gastroenterology. 2005;128:1937-52.
19. Lee CW, Rickman B, Rogers AB, et al. Helicobacter pylori eradication prevents progression of gastric cancer in hypergastrinemic INS-GAS mice. Cancer Res. 2008;68:3540-8.
20. Jiang L, Gonda TA, Gamble MV, et al. Global hypomethylation of genomic DNA in cancer-associated myofibroblasts. Cancer Res. 2009. (in press)
21. Everhart JE. Recent developments in the epidemiology of Helicobacter pylori. Gastroenterol Clin North Am. 2000;29:559-78.
22. Ernst PB, Gold BD. The disease spectrum of Helicobacter pylori: The immunopathogenesis of gastroduodenal ulcer and gastric cancer. Annu Rev Microbiol. 2000;54:615-40.
23. Amieva MR, El-Omar EM. Host-bacterial interactions in Helicobacter pylori infection. Gastroenterology. 2008;134:306-23.
24. Ahmed A, Smoot D, Littleton G, et al. Helicobacter pylori inhibits gastric cell cycle progression. Microbe Infect. 2000;2:1159-69.
25. Suganuma M, Kurusu M, Okabe S, et al. Helicobacter pylori membrane protein 1: A new carcinogenic factor of Helicobacter pylori. Cancer Res. 2001;61:6356-9.
26. Baik SC, Youn HS, Chung MH, et al. Increased oxidative DNA damage in Helicobacter pylori–infected human gastric mucosa. Cancer Res. 1996;56:1279-82.
27. Uemura N, Okamoto S, Yamamoto S, et al. Helicobacter pylori infection and the development of gastric cancer. N Engl J Med. 2001;345:784-9.
28. Hsu PI, Lai KH, Hsu PN, et al. Helicobacter pylori infection and the risk of gastric malignancy. Am J Gastroenterol. 2007;102:725-30.
29. Kamangar F, Qiao YL, Blaser MJ, et al. Helicobacter pylori and oesophageal and gastric cancers in a prospective study in China. Br J Cancer. 2007;96:172-6.
30. Kamangar F, Dawsey SM, Blaser MJ, et al. Opposing risks of gastric cardia and noncardia gastric adenocarcinomas associated with Helicobacter pylori seropositivity. J Natl Cancer Inst. 2006;98:1445-52.
31. Williams CS, Smalley W, DuBois RN. Aspirin use and potential mechanisms for colorectal cancer prevention. J Clin Invest. 1997;100:1325-9.
32. Meira LB, Bugni JM, Green SL, et al. DNA damage induced by chronic inflammation contributes to colon carcinogenesis in mice. J Clin Invest. 2008;118:2516-25.
33. Mandell L, Moran AP, Cocchiarella A, et al. Intact gram-negative Helicobacter pylori, Helicobacter felis, and Helicobacter hepaticus bacteria activate innate immunity via toll-like receptor 2 but not toll-like receptor 4. Infect Immun. 2004;72:6446-54.
34. Raghavan S, Nystrom J, Fredriksson M, et al. Orally administered CpG oligodeoxynucleotide induces production of CXC and CC chemokines in the gastric mucosa and suppresses bacterial colonization in a mouse model of Helicobacter pylori infection. Infect Immun. 2003;71:7014-22.
35. Kawahara T, Kuwano Y, Teshima-Kondo S, et al. Helicobacter pylori lipopolysaccharide from type I, but not type II strains, stimulates apoptosis of cultured gastric mucosal cells. J Med Invest. 2001;48:167-74.
36. Kawahara T, Teshima S, Oka A, et al. Type I Helicobacter pylori lipopolysaccharide stimulates toll-like receptor 4 and activates mitogen oxidase 1 in gastric pit cells. Infect Immun. 2001;69:4382-9.
37. Smith MFJr, Mitchell A, Li G, et al. Toll-like receptor (TLR) 2 and TLR5, but not TLR4, are required for Helicobacter pylori–induced NF-kappa B activation and chemokine expression by epithelial cells. J Biol Chem. 2003;278:32552-60.
38. Wang TC, Goldenring JR, Dangler C, et al. Mice lacking secretory phospholipase A2 show altered apoptosis and differentiation with Helicobacter felis infection. Gastroenterology. 1998;114:675-89.
39. Sutton P, Kolesnikow T, Danon S, Wilson J, Lee A. Dominant nonresponsiveness to Helicobacter pylori infection is associated with production of interleukin 10 but not gamma interferon. Infect Immun. 2000;68:4802-4.
40. Eaton KA, Mefford M, Thevenot T. The role of T cell subsets and cytokines in the pathogenesis of Helicobacter pylori gastritis in mice. J Immunol. 2001;166:7456-61.
41. Roth KA, Kapadia SB, Martin SM, Lorenz RG. Cellular immune responses are essential for the development of Helicobacter felis–associated gastric pathology. J Immunol. 1999;163:1490-7.
42. Fox JG, Sheppard BJ, Dangler CA, et al. Germ-line p53-targeted disruption inhibits Helicobacter-induced premalignant lesions and invasive gastric carcinoma through down-regulation of Th1 proinflammatory responses. Cancer Res. 2002;62:696-702.
43. Smythies LE, Waites KB, Lindsey JR, et al. Helicobacter pylori-induced mucosal inflammation is Th1 mediated and exacerbated in IL-4, but not IFN-gamma, gene-deficient mice. J Immunol. 2000;165:1022-9.
44. Ernst M, Najdovska M, Grail D, et al. STAT3 and STAT1 mediate IL-11-dependent and inflammation-associated gastric tumorigenesis in gp130 receptor mutant mice. J Clin Invest. 2008;118:1727-38.
45. Fox JG, Beck P, Dangler CA, et al. Concurrent enteric helminth infection modulates inflammation and gastric immune responses and reduces Helicobacter-induced gastric atrophy. Nat Med. 2000;6:536-42.
46. Segal I, Ally R, Mitchell H. Helicobacter pylori—An African perspective. QJM. 2001;94:561-5.
47. Whary MT, Sundina N, Bravo LE, et al. Intestinal helminthiasis in Colombian children promotes a Th2 response to Helicobacter pylori: Possible implications for gastric carcinogenesis. Cancer Epidemiol Biomarkers Prev. 2005;14:1464-9.
48. Blaser MJ, Berg DE. Helicobacter pylori genetic diversity and risk of human disease. J Clin Invest. 2001;107:767-73.
49. Israel DA, Salama N, Krishna U, et al. Helicobacter pylori genetic diversity within the gastric niche of a single human host. Proc Natl Acad Sci U S A. 2001;98:14625-30.
50. Tomb JF, White O, Kerlavage AR, et al. The complete genome sequence of the gastric pathogen Helicobacter pylori. Nature. 1997;388:539-47.
51. Kostrzynska M, Betts JD, Austin JW, Trust TJ. Identification, characterization, and spatial localization of two flagellin species in Helicobacter pylori flagella. J Bacteriol. 1991;173:937-46.
52. Kavermann H, Burns BP, Angermuller K, et al. Identification and characterization of Helicobacter pylori genes essential for gastric colonization. J Exp Med. 2003;197:813-22.
53. Necchi V, Candusso ME, Tava F, et al. Intracellular, intercellular, and stromal invasion of gastric mucosa, preneoplastic lesions, and cancer by Helicobacter pylori. Gastroenterology. 2007;132:1009-23.
54. Oliveira AG, Santos A, Guerra JB, et al. babA2- and cagA-positive Helicobacter pylori strains are associated with duodenal ulcer and gastric carcinoma in Brazil. J Clin Microbiol. 2003;41:3964-6.
55. Crabtree JE, Taylor JD, Wyatt JI, et al. Mucosal IgA recognition of Helicobacter pylori 120 kDa protein, peptic ulceration, and gastric pathology. Lancet. 1991;338(8763):332-5.
56. Kuipers EJ, Perez-Perez GI, Meuwissen SG, Blaser MJ. Helicobacter pylori and atrophic gastritis: importance of the cagA status. J Natl Cancer Inst. 1995;87(23):1777-80.
57. Blaser MJ, Perez-Perez GI, Kleanthous H, et al. Infection with Helicobacter pylori strains possessing cagA is associated with an increased risk of developing adenocarcinoma of the stomach. Cancer Res. 1995;55:2111-15.
58. Parsonnet J, Friedman GD, Orentreich N, Vogelman H. Risk for gastric cancer in people with CagA positive or CagA negative Helicobacter pylori infection. Gut. 1997;40:297-301.
59. Ohnishi N, Yuasa H, Tanaka S, et al. Transgenic expression of Helicobacter pylori CagA induces gastrointestinal and hematopoietic neoplasms in mouse. Proc Natl Acad Sci U S A. 2008;105:1003-8.
60. Sharma SA, Tummuru MK, Miller GG, Blaser MJ. Interleukin-8 response of gastric epithelial cell lines to Helicobacter pylori stimulation in vitro. Infect Immun. 1995;63:1681-87.
61. Tummuru MK, Sharma SA, Blaser MJ. Helicobacter pylori picB, a homologue of the Bordetella pertussis toxin secretion protein, is required for induction of IL-8 in gastric epithelial cells. Mol Microbiol. 1995;18:867-76.
62. Glocker E, Lange C, Covacci A, Bereswill S, Kist M, Pahl HL. Proteins encoded by the cag pathogenicity island of Helicobacter pylori are required for NF-kappaB activation. Infect Immun. 1998;66:2346-8.
63. Peek RMJr, Miller GG, Tham KT, et al. Heightened inflammatory response and cytokine expression in vivo to cagA+ Helicobacter pylori strains. Lab Invest. 1995;73:760-70.
64. Yamaoka Y, Kita M, Kodama T, et al. Helicobacter pylori cagA gene and expression of cytokine messenger RNA in gastric mucosa. Gastroenterology. 1996;110:1744-52.
65. Ogura K, Maeda S, Nakao M, et al. Virulence factors of Helicobacter pylori responsible for gastric diseases in Mongolian gerbil. J Exp Med. 2000;192:1601-10.
66. Israel DA, Salama N, Arnold CN, et al. Helicobacter pylori strain-specific differences in genetic content, identified by microarray, influence host inflammatory responses. J Clin Invest. 2001;107:611-20.
67. Gebert B, Fischer W, Weiss E, et al. Helicobacter pylori vacuolating cytotoxin inhibits T lymphocyte activation. Science. 2003;301:1099-102.
68. Peek RMJr, Blaser MJ. Helicobacter pylori and gastrointestinal tract adenocarcinomas. Nat Rev Cancer. 2002;2:28-37.
69. Hansson LE, Engstrand L, Nyren O, et al. Helicobacter pylori infection: Independent risk indicator of gastric adenocarcinoma. Gastroenterology. 1993;105:1098-103.
70. Larsson SC, Bergkvist L, Wolk A. Processed meat consumption, dietary nitrosamines and stomach cancer risk in a cohort of Swedish women. Int J Cancer. 2006;119:915-19.
71. Bartsch H. N-nitroso compounds and human cancer: Where do we stand? IARC Sci Publ. 1991:1-10.
72. Mowat C, Carswell A, Wirz A, McColl KE. Omeprazole and dietary nitrate independently affect levels of vitamin C and nitrite in gastric juice. Gastroenterology. 1999;116:813-22.
73. Oldreive C, Rice-Evans C. The mechanisms for nitration and nitrotyrosine formation in vitro and in vivo: Impact of diet. Free Radic Res. 2001;35:215-31.
74. Sugimura T, Fujimura S, Baba T. Tumor production in the glandular stomach and alimentary tract of the rat by N-methyl-N′-nitro-N-nitrosoguanidine. Cancer Res. 1970;30:455-65.
75. Bartsch H, Ohshima H, Pignatelli B, Calmels S. Human exposure to endogenous N-nitroso compounds: Quantitative estimates in subjects at high risk for cancer of the oral cavity, oesophagus, stomach and urinary bladder. Cancer Surv. 1989;8:335-62.
76. Knekt P, Jarvinen R, Dich J, Hakulinen T. Risk of colorectal and other gastro-intestinal cancers after exposure to nitrate, nitrite and N-nitroso compounds: A follow-up study. Int J Cancer. 1999;80:852-6.
77. Jakszyn P, Bingham S, Pera G, et al. Endogenous versus exogenous exposure to N-nitroso compounds and gastric cancer risk in the European Prospective Investigation into Cancer and Nutrition (EPIC-EURGAST) study. Carcinogenesis. 2006;27:1497-501.
78. van Loon AJ, Botterweck AA, Goldbohm RA, et al. Intake of nitrate and nitrite and the risk of gastric cancer: A prospective cohort study. Br J Cancer. 1998;78:129-35.
79. Fox JG, Dangler CA, Taylor NS, et al. High-salt diet induces gastric epithelial hyperplasia and parietal cell loss, and enhances Helicobacter pylori colonization in C57BL/6 mice. Cancer Res. 1999;59:4823-8.
80. Ramon JM, Serra L, Cerdo C, Oromi J. Dietary factors and gastric cancer risk. A case-control study in Spain. Cancer. 1993;71:1731-5.
81. Nazario CM, Szklo M, Diamond E, et al. Salt and gastric cancer: A case-control study in Puerto Rico. Int J Epidemiol. 1993;22:790-7.
82. Gonzalez CA, Jakszyn P, Pera G, et al. Meat intake and risk of stomach and esophageal adenocarcinoma within the European Prospective Investigation Into Cancer and Nutrition (EPIC). J Natl Cancer Inst. 2006;98:345-54.
83. Loh JT, Torres VJ, Cover TL. Regulation of Helicobacter pylori cagA expression in response to salt. Cancer Res. 2007;67:4709-15.
84. Gamboa-Dominguez A, Ubbelohde T, Saqui-Salces M, et al. Salt and stress synergize H. pylori–induced gastric lesions, cell proliferation, and p21 expression in Mongolian gerbils. Dig Dis Sci. 2007;52:1517-26.
85. Kobayashi M, Tsubono Y, Sasazuki S, et al. Vegetables, fruit and risk of gastric cancer in Japan: A 10-year follow-up of the JPHC Study Cohort I. Int J Cancer. 2002;102:39-44.
86. Larsson SC, Bergkvist L, Wolk A. Fruit and vegetable consumption and incidence of gastric cancer: A prospective study. Cancer Epidemiol Biomarkers Prev. 2006;15:1998-2001.
87. Gonzalez CA, Pera G, Agudo A, et al. Fruit and vegetable intake and the risk of stomach and oesophagus adenocarcinoma in the European Prospective Investigation into Cancer and Nutrition (EPIC-EURGAST). Int J Cancer. 2006;118:2559-66.
88. Freedman ND, Subar AF, Hollenbeck AR, et al. Fruit and vegetable intake and gastric cancer risk in a large United States prospective cohort study. Cancer Causes Control. 2008;19:459-67.
89. Tavani A, La Vecchia C, Gallus S, et al. Red meat intake and cancer risk: A study in Italy. Int J Cancer. 2000;86:425-8.
90. Ji BT, Chow WH, Yang G, et al. Dietary habits and stomach cancer in Shanghai, China. Int J Cancer. 1998;76:659-64.
91. Ward MH, Lopez-Carrillo L. Dietary factors and the risk of gastric cancer in Mexico City. Am J Epidemiol. 1999;149:925-32.
92. Ito LS, Inoue M, Tajima K, et al. Dietary factors and the risk of gastric cancer among Japanese women: A comparison between the differentiated and non-differentiated subtypes. Ann Epidemiol. 2003:1324-31.
93. Botterweck AA, van den Brandt PA, Goldbohm RA. Vitamins, carotenoids, dietary fiber, and the risk of gastric carcinoma: Results from a prospective study after 6.3 years of follow-up. Cancer. 2000;88:737-48.
94. Ekstrom AM, Serafini M, Nyren O, et al. Dietary antioxidant intake and the risk of cardia cancer and noncardia cancer of the intestinal and diffuse types: A population-based case-control study in Sweden. Int J Cancer. 2000;87:133-40.
95. Kuper H, Boffetta P, Adami HO. Tobacco use and cancer causation: Association by tumour type. J Intern Med. 2002;252:206-24.
96. Gonzalez CA, Pera G, Agudo A, et al. Smoking and the risk of gastric cancer in the European Prospective Investigation Into Cancer and Nutrition (EPIC). Int J Cancer. 2003;107:629-34.
97. Sung NY, Choi KS, Park EC, et al. Smoking, alcohol and gastric cancer risk in Korean men: The National Health Insurance Corporation Study. Br J Cancer. 2007;97:700-4.
98. Sjodahl K, Lu Y, Nilsen TI, et al. Smoking and alcohol drinking in relation to risk of gastric cancer: A population-based, prospective cohort study. Int J Cancer. 2007;120:128-32.
99. Ladeiras-Lopes R, Pereira AK, Nogueira A, et al. Smoking and gastric cancer: Systematic review and meta-analysis of cohort studies. Cancer Causes Control. 2008;19:689-701.
100. Zendehdel K, Nyren O, Luo J, et al. Risk of gastroesophageal cancer among smokers and users of Scandinavian moist snuff. Int J Cancer. 2008;122:1095-9.
101. Stenstrom B, Zhao CM, Rogers AB, et al. Swedish moist snuff accelerates gastric cancer development in Helicobacter pylori–infected wild-type and gastrin transgenic mice. Carcinogenesis. 2007;28:2041-6.
102. Freedman ND, Abnet CC, Leitzmann MF, et al. A prospective study of tobacco, alcohol, and the risk of esophageal and gastric cancer subtypes. Am J Epidemiol. 2007;165:1424-33.
103. Larsson SC, Giovannucci E, Wolk A. Alcoholic beverage consumption and gastric cancer risk: a prospective population-based study in women. Int J Cancer. 2007;120:373-7.
104. Gammon MD, Schoenberg JB, Ahsan H, et al. Tobacco, alcohol, and socioeconomic status and adenocarcinomas of the esophagus and gastric cardia. J Natl Cancer Inst. 1997;89:1277-84.
105. Renehan AG, Tyson M, Egger M, et al. Body-mass index and incidence of cancer: a systematic review and meta-analysis of prospective observational studies. Lancet. 2008;371:569-78.
106. Abnet CC, Freedman ND, Hollenbeck AR, et al. A prospective study of BMI and risk of oesophageal and gastric adenocarcinoma. Eur J Cancer. 2008;44:465-71.
107. Merry AH, Schouten LJ, Goldbohm RA, van den Brandt PA. Body mass index, height and risk of adenocarcinoma of the oesophagus and gastric cardia: A prospective cohort study. Gut. 2007;56:1503-11.
108. MacInnis RJ, English DR, Hopper JL, Giles GG. Body size and composition and the risk of gastric and oesophageal adenocarcinoma. Int J Cancer. 2006;118:2628-31.
109. Chow WH, Blot WJ, Vaughan TL, et al. Body mass index and risk of adenocarcinomas of the esophagus and gastric cardia. J Natl Cancer Inst. 1998;90:150-5.
110. Lindblad M, Rodriguez LA, Lagergren J. Body mass, tobacco and alcohol and risk of esophageal, gastric cardia, and gastric non-cardia adenocarcinoma among men and women in a nested case-control study. Cancer Causes Control. 2005;16:285-94.
111. Zanghieri G, Di Gregorio C, Sacchetti C, et al. Familial occurrence of gastric cancer in the 2-year experience of a population-based registry. Cancer. 1990;66:2047-51.
112. Brenner H, Arndt V, Sturmer T, et al. Individual and joint contribution of family history and Helicobacter pylori infection to the risk of gastric carcinoma. Cancer. 2000;88:274-9.
113. Parsonnet J. When heredity is infectious. Gastroenterology. 2000;118:222-4.
114. Leung WK, Ng EK, Chan WY, et al. Risk factors associated with the development of intestinal metaplasia in first-degree relatives of gastric cancer patients. Cancer Epidemiol Biomarkers Prev. 2005;14:2982-6.
115. Yatsuya H, Toyoshima H, Tamakoshi A, et al. Individual and joint impact of family history and Helicobacter pylori infection on the risk of stomach cancer: a nested case-control study. Br J Cancer. 2004;91:929-34.
116. Lichtenstein P, Holm NV, Verkasalo PK, et al. Environmental and heritable factors in the causation of cancer—Analyses of cohorts of twins from Sweden, Denmark, and Finland. N Engl J Med. 2000;343:78-85.
117. Chang YW, Han YS, Lee DK, et al. Role of Helicobacter pylori infection among offspring or siblings of gastric cancer patients. Int J Cancer. 2002;101:469-74.
118. El-Omar EM, Oien K, Murray LS, et al. Increased prevalence of precancerous changes in relatives of gastric cancer patients: critical role of H. pylori. Gastroenterology. 2000;118:22-30.
119. Stoicov C, Whary M, Rogers AB, et al. Coinfection modulates inflammatory responses and clinical outcome of Helicobacter felis and Toxoplasma gondii infections. J Immunol. 2004;173:3329-36.
120. El-Omar EM, Carrington M, Chow WH, et al. Interleukin-1 polymorphisms associated with increased risk of gastric cancer. Nature. 2000;404:398-402.
121. Figueiredo C, Machado JC, Pharoah P, et al. Helicobacter pylori and interleukin 1 genotyping: An opportunity to identify high-risk individuals for gastric carcinoma. J Natl Cancer Inst. 2002;94:1680-7.
122. Furuta T, El-Omar EM, Xiao F, et al. Interleukin 1beta polymorphisms increase risk of hypochlorhydria and atrophic gastritis and reduce risk of duodenal ulcer recurrence in Japan. Gastroenterology. 2002;123:92-105.
123. Machado JC, Figueiredo C, Canedo P, et al. A proinflammatory genetic profile increases the risk for chronic atrophic gastritis and gastric carcinoma. Gastroenterology. 2003;125:364-71.
124. Zeng ZR, Hu PJ, Hu S, et al. Association of interleukin 1B gene polymorphism and gastric cancers in high and low prevalence regions in China. Gut. 2003;52:1684-9.
125. Rad R, Prinz C, Neu B, et al. Synergistic effect of Helicobacter pylori virulence factors and interleukin-1 polymorphisms for the development of severe histological changes in the gastric mucosa. J Infect Dis. 2003;188:272-81.
126. Hwang IR, Hsu PI, Peterson LE, et al. Interleukin-6 genetic polymorphisms are not related to Helicobacter pylori-associated gastroduodenal diseases. Helicobacter. 2003;8:142-8.
127. Tu S, Bhagat G, Cui G, et al. Overexpression of interleukin-1beta induces gastric inflammation and cancer and mobilizes myeloid-derived suppressor cells in mice. Cancer Cell. 2008;14:408-19.
128. El-Omar EM, Rabkin CS, Gammon MD, et al. Increased risk of noncardia gastric cancer associated with proinflammatory cytokine gene polymorphisms. Gastroenterology. 2003;124:1193-201.
129. Hold GL, Rabkin CS, Chow WH, et al. A functional polymorphism of toll-like receptor 4 gene increases risk of gastric carcinoma and its precursors. Gastroenterology. 2007;132:905-12.
130. Guilford P, Hopkins J, Harraway J, et al. E-cadherin germline mutations in familial gastric cancer. Nature. 1998;392:402-5.
131. Richards FM, McKee SA, Rajpar MH, et al. Germline E-cadherin gene (CDH1) mutations predispose to familial gastric cancer and colorectal cancer. Hum Mol Genet. 1999;8:607-10.
132. Shinmura K, Kohno T, Takahashi M, et al. Familial gastric cancer: clinicopathological characteristics, RER phenotype and germline p53 and E-cadherin mutations. Carcinogenesis. 1999;20:1127-31.
133. Kaurah P, MacMillan A, Boyd N, et al. Founder and recurrent CDH1 mutations in families with hereditary diffuse gastric cancer. JAMA. 2007;297:2360-72.
134. Lynch HT, Kaurah P, Wirtzfeld D, et al. Hereditary diffuse gastric cancer: Diagnosis, genetic counseling, and prophylactic total gastrectomy. Cancer. 2008;112:2655-63.
135. Pharoah PD, Guilford P, Caldas C. Incidence of gastric cancer and breast cancer in CDH1 (E-cadherin) mutation carriers from hereditary diffuse gastric cancer families. Gastroenterology. 2001;121:1348-53.
136. Masciari S, Larsson N, Senz J, et al. Germline E-cadherin mutations in familial lobular breast cancer. J Med Genet. 2007;44:726-31.
137. Schrader KA, Masciari S, Boyd N, et al. Hereditary diffuse gastric cancer: Association with lobular breast cancer. Fam Cancer. 2008;7:73-82.
138. Zwick A, Munir M, Ryan CK, et al. Gastric adenocarcinoma and dysplasia in fundic gland polyps of a patient with attenuated adenomatous polyposis coli. Gastroenterology. 1997;113:659-63.
139. Bianchi LK, Burke CA, Bennett AE, et al. Fundic gland polyp dysplasia is common in familial adenomatous polyposis. Clin Gastroenterol Hepatol. 2008;6:180-5.
140. Attard TM, Cuffari C, Tajouri T, et al. Multicenter experience with upper gastrointestinal polyps in pediatric patients with familial adenomatous polyposis. Am J Gastroenterol. 2004;99:681-6.
141. Aarnio M, Salovaara R, Aaltonen LA, et al. Features of gastric cancer in hereditary non-polyposis colorectal cancer syndrome. Int J Cancer. 1997;74:551-5.
142. Howe JR, Mitros FA, Summers RW. The risk of gastrointestinal carcinoma in familial juvenile polyposis. Ann Surg Oncol. 1998;5:751-6.
143. Coburn MC, Pricolo VE, DeLuca FG, Bland KI. Malignant potential in intestinal juvenile polyposis syndromes. Ann Surg Oncol. 1995;2:386-91.
144. Kimura Y, Noguchi T, Kawahara K, et al. Genetic alterations in 102 primary gastric cancers by comparative genomic hybridization: Gain of 20q and loss of 18q are associated with tumor progression. Mod Pathol. 2004;17:1328-37.
145. Kim JH, Takahashi T, Chiba I, et al. Occurrence of p53 gene abnormalities in gastric carcinoma tumors and cell lines. J Natl Cancer Inst. 1991;83:938-43.
146. Victor T, Du Toit R, Jordaan AM, et al. No evidence for point mutations in codons 12, 13, and 61 of the ras gene in a high-incidence area for esophageal and gastric cancers. Cancer Res. 1990;50:4911-14.
147. Han S, Kim HY, Park K, et al. Expression of p27Kip1 and cyclin D1 proteins is inversely correlated and is associated with poor clinical outcome in human gastric cancer. J Surg Oncol. 1999;71:147-54.
148. Kim DH, Lee HI, Nam ES, et al. Reduced expression of the cell-cycle inhibitor p27Kip1 is associated with progression and lymph node metastasis of gastric carcinoma. Histopathology. 2000;36:245-51.
149. Takano Y, Kato Y, van Diest PJ, et al. Cyclin D2 overexpression and lack of p27 correlate positively and cyclin E inversely with a poor prognosis in gastric cancer cases. Am J Pathol. 2000;156:585-94.
150. Myung N, Kim MR, Chung IP, et al. Loss of p16 and p27 is associated with progression of human gastric cancer. Cancer Lett. 2000;153:129-36.
151. Shim YH, Kang GH, Ro JY. Correlation of p16 hypermethylation with p16 protein loss in sporadic gastric carcinomas. Lab Invest. 2000;80:689-95.
152. Schneider BG, Gulley ML, Eagan P, et al. Loss of p16/CDKN2A tumor suppressor protein in gastric adenocarcinoma is associated with Epstein-Barr virus and anatomic location in the body of the stomach. Hum Pathol. 2000;31:45-50.
153. Tsujie M, Yamamoto H, Tomita N, et al. Expression of tumor suppressor gene p16(INK4) products in primary gastric cancer. Oncology. 2000;58:126-36.
154. Stemmermann G, Heffelfinger SC, Noffsinger A, et al. The molecular biology of esophageal and gastric cancer and their precursors: Oncogenes, tumor suppressor genes, and growth factors. Hum Pathol. 1994;25:968-81.
155. Samuels Y, Wang Z, Bardelli A, et al. High frequency of mutations of the PIK3CA gene in human cancers. Science. 2004;304:554.
156. Wang Z, Shen D, Parsons DW, et al. Mutational analysis of the tyrosine phosphatome in colorectal cancers. Science. 2004;304:1164-6.
157. Park WS, Oh RR, Park JY, et al. Somatic mutations of the trefoil factor family 1 gene in gastric cancer. Gastroenterology. 2000;119:691-8.
158. Li QL, Ito K, Sakakura C, et al. Causal relationship between the loss of RUNX3 expression and gastric cancer. Cell. 2002;109:113-24.
159. Yio X, Diamond M, Zhang JY, et al. Trefoil factor family-1 mutations enhance gastric cancer cell invasion through distinct signaling pathways. Gastroenterology. 2006;130:1696-706.
160. Ito K, Lim AC, Salto-Tellez M, et al. RUNX3 attenuates beta-catenin/T cell factors in intestinal tumorigenesis. Cancer Cell. 2008;14:226-37.
161. Mironov NM, Aguelon MA, Potapova GI, et al. Alterations of (CA)n DNA repeats and tumor suppressor genes in human gastric cancer. Cancer Res. 1994;54:41-4.
162. Rhyu MG, Park WS, Meltzer SJ. Microsatellite instability occurs frequently in human gastric carcinoma. Oncogene. 1994;9:29-32.
163. Fang DC, Jass JR, Wang DX, et al. Infrequent loss of heterozygosity of APC/MCC and DCC genes in gastric cancer showing DNA microsatellite instability. J Clin Pathol. 1999;52:504-8.
164. Isogaki J, Shinmura K, Yin W, et al. Microsatellite instability and K-ras mutations in gastric adenomas, with reference to associated gastric cancers. Cancer Detect Prev. 1999;23:204-14.
165. Habano W, Sugai T, Nakamura SI, et al. Microsatellite instability and mutation of mitochondrial and nuclear DNA in gastric carcinoma. Gastroenterology. 2000;118:835-41.
166. Leung WK, Kim JJ, Kim JG, et al. Microsatellite instability in gastric intestinal metaplasia in patients with and without gastric cancer. Am J Pathol. 2000;156:537-43.
167. Yamamoto H, Perez-Piteira J, Yoshida T, et al. Gastric cancers of the microsatellite mutator phenotype display characteristic genetic and clinical features. Gastroenterology. 1999;116:1348-57.
168. Paulson TG, Wright FA, Parker BA, et al. Microsatellite instability correlates with reduced survival and poor disease prognosis in breast cancer. Cancer Res. 1996;56:4021-6.
169. Keller G, Vogelsang H, Becker I, et al. Diffuse type gastric and lobular breast carcinoma in a familial gastric cancer patient with an E-cadherin germline mutation. Am J Pathol. 1999;155:337-42.
170. Yoon KA, Ku JL, Yang HK, et al. Germline mutations of E-cadherin gene in Korean familial gastric cancer patients. J Hum Genet. 1999;44:177-80.
171. Xiangming C, Hokita S, Natsugoe S, et al. Co-occurrence of reduced expression of alpha-catenin and overexpression of p53 is a predictor of lymph node metastasis in early gastric cancer. Oncology. 1999;57:131-7.
172. Kawanishi K, Doki Y, Shiozaki H, et al. Correlation between loss of E-cadherin expression and overexpression of autocrine motility factor receptor in association with progression of human gastric cancers. Am J Clin Pathol. 2000;113:266-74.
173. Shiozaki H, Oka H, Inoue M, et al. E-cadherin mediated adhesion system in cancer cells. Cancer. 1996;77:1605-13.
174. Tamura G, Yin J, Wang S, et al. E-cadherin gene promoter hypermethylation in primary human gastric carcinomas. J Natl Cancer Inst. 2000;92:569-73.
175. Batlle E, Henderson JT, Beghtel H, et al. Beta-catenin and TCF mediate cell positioning in the intestinal epithelium by controlling the expression of EphB/ephrinB. Cell. 2002;111:251-63.
176. Ouko L, Ziegler TR, Gu LH, et al. Wnt11 signaling promotes proliferation, transformation, and migration of IEC6 intestinal epithelial cells. J Biol Chem. 2004;279:26707-15.
177. Yu J, Ebert MP, Miehlke S, et al. alpha-catenin expression is decreased in human gastric cancers and in the gastric mucosa of first degree relatives. Gut. 2000;46:639-44.
178. Jiang Y, Jahagirdar BN, Reinhardt RL, et al. Pluripotency of mesenchymal stem cells derived from adult marrow. Nature. 2002;418:41-9.
179. Petersen BE, Bowen WC, Patrene KD, et al. Bone marrow as a potential source of hepatic oval cells. Science. 1999;284:1168-70.
180. Krause DS, Theise ND, Collector MI, et al. Multi-organ, multi-lineage engraftment by a single bone marrow-derived stem cell. Cell. 2001;105:369-77.
181. Schwartz RE, Reyes M, Koodie L, et al. Multipotent adult progenitor cells from bone marrow differentiate into functional hepatocyte-like cells. J Clin Invest. 2002;109:1291-302.
182. Houghton J, Stoicov C, Nomura S, et al. Gastric cancer originating from bone marrow-derived cells. Science. 2004;306:1568-71.
183. Dixon MF, Genta RM, Yardley JH, Correa P. Classification and grading of gastritis. The updated Sydney system. International Workshop on the Histopathology of Gastritis, Houston 1994. Am J Surg Pathol. 1996;20:1161-81.
184. Lahner E, Bordi C, Cattaruzza MS, et al. Long-term follow-up in atrophic body gastritis patients: Atrophy and intestinal metaplasia are persistent lesions irrespective of Helicobacter pylori infection. Aliment Pharmacol Ther. 2005;22:471-81.
185. Kokkola A, Sjoblom SM, Haapiainen R, et al. The risk of gastric carcinoma and carcinoid tumours in patients with pernicious anaemia. A prospective follow-up study. Scand J Gastroenterol. 1998;33:88-92.
186. Siurala M, Lehtola J, Ihamaki T. Atrophic gastritis and its sequelae. Results of 19-23 years’ follow-up examinations. Scand J Gastroenterol. 1974;9:441-6.
187. de Vries AC, Haringsma J, Kuipers EJ. The detection, surveillance and treatment of premalignant gastric lesions related to Helicobacter pylori infection. Helicobacter. 2007;12:1-15.
188. Kato I, Tominaga S, Ito Y, et al. A prospective study of atrophic gastritis and stomach cancer risk. Jpn J Cancer Res. 1992;83:1137-42.
189. Tatsuta M, Iishi H, Nakaizumi A, et al. Fundal atrophic gastritis as a risk factor for gastric cancer. Int J Cancer. 1993;53:70-4.
190. Cassaro M, Rugge M, Gutierrez O, et al. Topographic patterns of intestinal metaplasia and gastric cancer. Am J Gastroenterol. 2000;95:1431-8.
191. Kuipers EJ, Uyterlinde AM, Pena AS, et al. Long-term sequelae of Helicobacter pylori gastritis. Lancet. 1995;345:1525-8.
192. Asaka M, Sugiyama T, Nobuta A, et al. Atrophic gastritis and intestinal metaplasia in Japan: Results of a large multicenter study. Helicobacter. 2001;6:294-9.
193. Schafer LW, Larson DE, Melton LJ3rd, et al. Risk of development of gastric carcinoma in patients with pernicious anemia: a population-based study in Rochester, Minnesota. Mayo Clin Proc. 1985;60:444-8.
194. Sobala GM, Schorah CJ, Sanderson M, et al. Ascorbic acid in the human stomach. Gastroenterology. 1989;97:357-63.
195. Wang TC, Dangler CA, Chen D, et al. Synergistic interaction between hypergastrinemia and Helicobacter infection in a mouse model of gastric cancer. Gastroenterology. 2000;118:36-47.
196. Watson SA, Grabowska AM, El-Zaatari M, Takhar A. Gastrin—Active participant or bystander in gastric carcinogenesis? Nat Rev Cancer. 2006;6:936-46.
197. Jass JR, Filipe MI. The mucin profiles of normal gastric mucosa, intestinal metaplasia and its variants and gastric carcinoma. Histochem J. 1981;13:931-9.
198. Leung WK, Sung JJ. Review article: Intestinal metaplasia and gastric carcinogenesis. Aliment Pharmacol Ther. 2002;16:1209-16.
199. Rokkas T, Filipe MI, Sladen GE. Detection of an increased incidence of early gastric cancer in patients with intestinal metaplasia type III who are closely followed up. Gut. 1991;32:1110-13.
200. Rugge M, Correa P, Dixon MF, et al. Gastric dysplasia: The Padova international classification. Am J Surg Pathol. 2000;24:167-76.
201. de Vries AC, Kuipers EJ. Epidemiology of premalignant gastric lesions: implications for the development of screening and surveillance strategies. Helicobacter. 2007;12(Suppl 2):22-31.
202. You WC, Blot WJ, Li JY, et al. Precancerous gastric lesions in a population at high risk of stomach cancer. Cancer Res. 1993;53:1317-21.
203. Rugge M, Farinati F, Di Mario F, et al. Gastric epithelial dysplasia: A prospective multicenter follow-up study from the Interdisciplinary Group on Gastric Epithelial Dysplasia. Hum Pathol. 1991;22:1002-8.
204. Di Gregorio C, Morandi P, Fante R, De Gaetani C. Gastric dysplasia. A follow-up study. Am J Gastroenterol. 1993;88:1714-9.
205. Rugge M, Cassaro M, Di Mario F, et al. The long term outcome of gastric non-invasive neoplasia. Gut. 2003;52:1111-16.
206. de Vries AC, van Grieken NC, Looman CW, et al. Gastric cancer risk in patients with premalignant gastric lesions: A nationwide cohort study in the Netherlands. Gastroenterology. 2008;134:945-52.
207. Lansdown M, Quirke P, Dixon MF, et al. High grade dysplasia of the gastric mucosa: A marker for gastric carcinoma. Gut. 1990;31:977-83.
208. Kokkola A, Haapiainen R, Laxen F, et al. Risk of gastric carcinoma in patients with mucosal dysplasia associated with atrophic gastritis: a follow up study. J Clin Pathol. 1996;49:979-84.
209. Voutilainen M, Mantynen T, Kunnamo I, et al. Impact of clinical symptoms and referral volume on endoscopy for detecting peptic ulcer and gastric neoplasms. Scand J Gastroenterol. 2003;38:109-13.
210. Burt RW. Gastric fundic gland polyps. Gastroenterology. 2003;125:1462-9.
211. Borch K, Skarsgard J, Franzen L, et al. Benign gastric polyps: morphological and functional origin. Dig Dis Sci. 2003;48:1292-7.
212. Jalving M, Koornstra JJ, Wesseling J, et al. Increased risk of fundic gland polyps during long-term proton pump inhibitor therapy. Aliment Pharmacol Ther. 2006;24:1341-8.
213. Zea-Iriarte WL, Sekine I, Itsuno M, et al. Carcinoma in gastric hyperplastic polyps. A phenotypic study. Dig Dis Sci. 1996;41:377-86.
214. Kamiya T, Morishita T, Asakura H, et al. Long-term follow-up study on gastric adenoma and its relation to gastric protruded carcinoma. Cancer. 1982;50:2496-503.
215. Muehldorfer SM, Stolte M, Martus P, et al. Diagnostic accuracy of forceps biopsy versus polypectomy for gastric polyps: A prospective multicentre study. Gut. 2002;50:465-70.
216. Caygill CP, Hill MJ, Kirkham JS, Northfield TC. Mortality from gastric cancer following gastric surgery for peptic ulcer. Lancet. 1986;1:929-31.
217. Viste A, Bjornestad E, Opheim P, et al. Risk of carcinoma following gastric operations for benign disease. A historical cohort study of 3470 patients. Lancet. 1986;2:502-5.
218. Lundegardh G, Adami HO, Helmick C, et al. Stomach cancer after partial gastrectomy for benign ulcer disease. N Engl J Med. 1988;319:195-200.
219. Dubrow R. Gastric cancer following peptic ulcer surgery. J Natl Cancer Inst. 1993;85:1268-70.
220. Domellof L, Eriksson S, Janunger KG. Carcinoma and possible precancerous changes of the gastric stump after billroth II resection. Gastroenterology. 1977;73:462-8.
221. Greenlee HB, Vivit R, Paez J, Dietz A. Bacterial flora of the jejunum following peptic ulcer surgery. Arch Surg. 1971;102:260-5.
222. Dewar P, Dixon MF, Johnston D. Bile reflux and degree of gastritis in patients with gastric ulcer: Before and after operation. J Surg Res. 1984;37:277-84.
223. MacDonald WC, Owen DA. Gastric carcinoma after surgical treatment of peptic ulcer: An analysis of morphologic features and a comparison with cancer in the nonoperated stomach. Cancer. 2001;91:1732-8.
224. Hansson LE, Nyren O, Hsing AW, et al. The risk of stomach cancer in patients with gastric or duodenal ulcer disease. N Engl J Med. 1996;335:242-9.
225. Molloy RM, Sonnenberg A. Relation between gastric cancer and previous peptic ulcer disease. Gut. 1997;40:247-52.
226. Case records of the Massachusetts General Hospital. Case 2-1988. A 55-year-old man with innumerable gastric polyps and recent melena. N Engl J Med. 1988;318:100-9.
227. Scharschmidt BF. The natural history of hypertrophic gastrophy (Menetrier’s disease). Report of a case with 16 year follow-up and review of 120 cases from the literature. Am J Med. 1977;63:644-52.
228. Wood MG, Bates C, Brown RC, Losowsky MS. Intramucosal carcinoma of the gastric antrum complicating Menetrier’s disease. J Clin Pathol. 1983;36:1071-5.
229. Leung WK, Wu MS, Kakugawa Y, et al. Screening for gastric cancer in Asia: Current evidence and practice. Lancet Oncol. 2008;9:279-87.
230. Watanabe H, Mai M, Shimoda T, et al. Meeting report of the 72nd Japanese Gastric Cancer Congress. Gastric Cancer. 2000;3:1-8.
231. Kunisaki C, Ishino J, Nakajima S, et al. Outcomes of mass screening for gastric carcinoma. Ann Surg Oncol. 2006;13:221-8.
232. Lee KJ, Inoue M, Otani T, et al. Gastric cancer screening and subsequent risk of gastric cancer: A large-scale population-based cohort study, with a 13-year follow-up in Japan. Int J Cancer. 2006;118:2315-21.
233. Mizoue T, Yoshimura T, Tokui N, et al. Prospective study of screening for stomach cancer in Japan. Int J Cancer. 2003;106:103-7.
234. Miki K. Gastric cancer screening using the serum pepsinogen test method. Gastric Cancer. 2006;9:245-53.
235. Watabe H, Mitsushima T, Yamaji Y, et al. Predicting the development of gastric cancer from combining Helicobacter pylori antibodies and serum pepsinogen status: A prospective endoscopic cohort study. Gut. 2005;54:764-8.
236. Dan YY, So JB, Yeoh KG. Endoscopic screening for gastric cancer. Clin Gastroenterol Hepatol. 2006;4:709-16.
237. Rogers AB, Fox JG. Inflammation and cancer. I. Rodent models of infectious gastrointestinal and liver cancer. Am J Physiol Gastrointest Liver Physiol. 2004;286:G361-6.
238. Moss SF. The carcinogenic effect of H. pylori on the gastric epithelial cell. J Physiol Pharmacol. 1999;50:847-56.
239. Shimizu N, Ikehara Y, Inada K, et al. Eradication diminishes enhancing effects of Helicobacter pylori infection on glandular stomach carcinogenesis in Mongolian gerbils. Cancer Res. 2000;60:1512-14.
240. Nozaki K, Shimizu N, Ikehara Y, et al. Effect of early eradication on Helicobacter pylori–related gastric carcinogenesis in Mongolian gerbils. Cancer Sci. 2003;94:235-9.
241. Nozaki K, Shimizu N, Tsukamoto T, et al. Reversibility of heterotopic proliferative glands in glandular stomach of Helicobacter pylori–infected Mongolian gerbils on eradication. Jpn J Cancer Res. 2002;93:374-81.
242. Keto Y, Ebata M, Okabe S. Gastric mucosal changes induced by long term infection with Helicobacter pylori in Mongolian gerbils: Effects of bacteria eradication. J Physiol Paris. 2001;95:429-36.
243. Zivny J, Wang TC, Yantiss R, et al. Role of therapy or monitoring in preventing progression to gastric cancer. J Clin Gastroenterol. 2003;36(5 Suppl):S50-60.
244. Correa P, Fontham ET, Bravo JC, et al. Chemoprevention of gastric dysplasia: randomized trial of antioxidant supplements and anti-Helicobacter pylori therapy. J Natl Cancer Inst. 2000;92:1881-8.
245. Sung JJ, Lin SR, Ching JY, et al. Atrophy and intestinal metaplasia one year after cure of H. pylori infection: A prospective, randomized study. Gastroenterology. 2000;119:7-14.
246. Wong BC, Lam SK, Wong WM, et al. Helicobacter pylori eradication to prevent gastric cancer in a high-risk region of China: A randomized controlled trial. Jama. 2004;291:187-94.
247. Ley C, Mohar A, Guarner J, et al. Helicobacter pylori eradication and gastric preneoplastic conditions: A randomized, double-blind, placebo-controlled trial. Cancer Epidemiol Biomarkers Prev. 2004;13:4-10.
248. Leung WK, Lin SR, Ching JY, et al. Factors predicting progression of gastric intestinal metaplasia: Results of a randomised trial on Helicobacter pylori eradication. Gut. 2004;53:1244-9.
249. Fukase K, Kato M, Kikuchi S, et al. Effect of eradication of Helicobacter pylori on incidence of metachronous gastric carcinoma after endoscopic resection of early gastric cancer: An open-label, randomised controlled trial. Lancet. 2008;372:392-7.
250. Takenaka R, Okada H, Kato J, et al. Helicobacter pylori eradication reduced the incidence of gastric cancer, especially of the intestinal type. Aliment Pharmacol Ther. 2007;25:805-12.
251. Dixon MF. Prospects for intervention in gastric carcinogenesis: reversibility of gastric atrophy and intestinal metaplasia. Gut. 2001;49:2-4.
252. Parsonnet J, Harris RA, Hack HM, Owens DK. Modelling cost-effectiveness of Helicobacter pylori screening to prevent gastric cancer: A mandate for clinical trials. Lancet. 1996;348:150-4.
253. Mason J, Axon AT, Forman D, et al. The cost-effectiveness of population Helicobacter pylori screening and treatment: A Markov model using economic data from a randomized controlled trial. Aliment Pharmacol Ther. 2002;16:559-68.
254. Federico A, Morgillo F, Tuccillo C, et al. Chronic inflammation and oxidative stress in human carcinogenesis. Int J Cancer. 2007;121:2381-6.
255. Jenab M, Riboli E, Ferrari P, et al. Plasma and dietary carotenoid, retinol and tocopherol levels and the risk of gastric adenocarcinomas in the European prospective investigation into cancer and nutrition. Br J Cancer. 2006;95:406-15.
256. Jenab M, Riboli E, Ferrari P, et al. Plasma and dietary vitamin C levels and risk of gastric cancer in the European Prospective Investigation into Cancer and Nutrition (EPIC-EURGAST). Carcinogenesis. 2006;27:2250-7.
257. Persson C, Sasazuki S, Inoue M, et al. Plasma levels of carotenoids, retinol and tocopherol and the risk of gastric cancer in Japan: A nested case-control study. Carcinogenesis. 2008;29:1042-8.
258. Ja Kim H, Lee SS, Choi BY, Kim MK. Nitrate intake relative to antioxidant vitamin intake affects gastric cancer risk: A case-control study in Korea. Nutr Cancer. 2007;59:185-91.
259. Larsson SC, Bergkvist L, Naslund I, et al. Vitamin A, retinol, and carotenoids and the risk of gastric cancer: A prospective cohort study. Am J Clin Nutr. 2007;85:497-503.
260. Plummer M, Vivas J, Lopez G, et al. Chemoprevention of precancerous gastric lesions with antioxidant vitamin supplementation: A randomized trial in a high-risk population. J Natl Cancer Inst. 2007;99:137-46.
261. You WC, Brown LM, Zhang L, et al. Randomized double-blind factorial trial of three treatments to reduce the prevalence of precancerous gastric lesions. J Natl Cancer Inst. 2006;98:974-83.
262. Omenn GS, Goodman GE, Thornquist MD, et al. Effects of a combination of beta carotene and vitamin A on lung cancer and cardiovascular disease. N Engl J Med. 1996;334:1150-5.
263. Howe LR, Dannenberg AJ. A role for cyclooxygenase-2 inhibitors in the prevention and treatment of cancer. Semin Oncol. 2002;29(3 Suppl 11):111-19.
264. Saukkonen K, Rintahaka J, Sivula A, et al. Cyclooxygenase-2 and gastric carcinogenesis. Apmis. 2003;111:915-25.
265. Ristimaki A, Honkanen N, Jankala H, et al. Expression of cyclooxygenase-2 in human gastric carcinoma. Cancer Res. 1997;57:1276-80.
266. Murata H, Kawano S, Tsuji S, et al. Cyclooxygenase-2 overexpression enhances lymphatic invasion and metastasis in human gastric carcinoma. Am J Gastroenterol. 1999;94:451-5.
267. Sawaoka H, Kawano S, Tsuji S, et al. Cyclooxygenase-2 inhibitors suppress the growth of gastric cancer xenografts via induction of apoptosis in nude mice. Am J Physiol. 1998;274:G1061-7.
268. Yamamoto H, Itoh F, Fukushima H, et al. Overexpression of cyclooxygenase-2 protein is less frequent in gastric cancers with microsatellite instability. Int J Cancer. 1999;84:400-3.
269. van Rees BP, Saukkonen K, Ristimaki A, et al. Cyclooxygenase-2 expression during carcinogenesis in the human stomach. J Pathol. 2002;196:171-9.
270. Sung JJ, Leung WK, Go MY, et al. Cyclooxygenase-2 expression in Helicobacter pylori–associated premalignant and malignant gastric lesions. Am J Pathol. 2000;157:729-35.
271. Akre K, Ekstrom AM, Signorello LB, et al. Aspirin and risk for gastric cancer: A population-based case-control study in Sweden. Br J Cancer. 2001;84:965-8.
272. Duan L, Wu AH, Sullivan-Halley J, Bernstein L. Nonsteroidal anti-inflammatory drugs and risk of esophageal and gastric adenocarcinomas in Los Angeles County. Cancer Epidemiol Biomarkers Prev. 2008;17:126-34.
273. Wang WH, Huang JQ, Zheng GF, et al. Non-steroidal anti-inflammatory drug use and the risk of gastric cancer: A systematic review and meta-analysis. J Natl Cancer Inst. 2003;95:1784-91.
274. Lindblad M, Lagergren J, Garcia Rodriguez LA. Nonsteroidal anti-inflammatory drugs and risk of esophageal and gastric cancer. Cancer Epidemiol Biomarkers Prev. 2005;14:444-50.
275. Leung WK, Ng EK, Chan FK, et al. Effects of long-term rofecoxib on gastric intestinal metaplasia: results of a randomized controlled trial. Clin Cancer Res. 2006;12:4766-72.
276. Masuda M, Suzui M, Weinstein IB. Effects of epigallocatechin-3-gallate on growth, epidermal growth factor receptor signaling pathways, gene expression, and chemosensitivity in human head and neck squamous cell carcinoma cell lines. Clin Cancer Res. 2001;7:4220-9.
277. Ahmad N, Feyes DK, Nieminen AL, Agarwal R, Mukhtar H. Green tea constituent epigallocatechin-3-gallate and induction of apoptosis and cell cycle arrest in human carcinoma cells. J Natl Cancer Inst. 1997;89:1881-6.
278. Masuda M, Suzui M, Lim JT, Weinstein IB. Epigallocatechin-3-gallate inhibits activation of HER-2/neu and downstream signaling pathways in human head and neck and breast carcinoma cells. Clin Cancer Res. 2003;9:3486-91.
279. Saffari Y, Sadrzadeh SM. Green tea metabolite EGCG protects membranes against oxidative damage in vitro. Life Sci. 2004;74:1513-18.
280. D’Alessandro T, Prasain J, Benton MR, et al. Polyphenols, inflammatory response, and cancer prevention: chlorination of isoflavones by human neutrophils. J Nutr. 2003;133(11 Suppl 1):3773S-7S.
281. Yu GP, Hsieh CC, Wang LY, et al. Green-tea consumption and risk of stomach cancer: A population-based case-control study in Shanghai, China. Cancer Causes Control. 1995;6:532-8.
282. Inoue M, Tajima K, Hirose K, et al. Tea and coffee consumption and the risk of digestive tract cancers: Data from a comparative case-referent study in Japan. Cancer Causes Control. 1998;9:209-16.
283. Ji BT, Chow WH, Yang G, et al. The influence of cigarette smoking, alcohol, and green tea consumption on the risk of carcinoma of the cardia and distal stomach in Shanghai, China. Cancer. 1996;77:2449-57.
284. Tsubono Y, Nishino Y, Komatsu S, et al. Green tea and the risk of gastric cancer in Japan. N Engl J Med. 2001;344:632-6.
285. Sasazuki S, Inoue M, Hanaoka T, et al. Green tea consumption and subsequent risk of gastric cancer by subsite: the JPHC Study. Cancer Causes Control. 2004;15:483-91.
286. Tucker HJ, Snape WJJr, Cohen S. Achalasia secondary to carcinoma: Manometric and clinical features. Ann Intern Med. 1978;89:315-18.
287. Wakashin M, Wakashin Y, Iesato K, et al. Association of gastric cancer and nephrotic syndrome. An immunologic study in three patients. Gastroenterology. 1980;78:749-56.
288. Gragasin-Saviano L, Isaacson E, Stuck RM. Pedal gangrene secondary to disseminated intravascular coagulation with gastric carcinoma. J Am Podiatr Med Assoc. 2002;92:149-52.
289. Croft PB, Wilkinson M. The incidence of carcinomatous neuromyopathy in patients with various types of carcinoma. Brain. 1965;88:427-34.
290. Yeh JS, Munn SE, Plunkett TA, et al. Coexistence of acanthosis nigricans and the sign of Leser-Trelat in a patient with gastric adenocarcinoma: A case report and literature review. J Am Acad Dermatol. 2000;42:357-62.
291. Dupont JBJr, Lee JR, Burton GR, Cohn IJr. Adenocarcinoma of the stomach: Review of 1,497 cases. Cancer. 1978;41:941-7.
292. Jemal A, Siegel R, Ward E, et al. Cancer statistics, 2007. CA Cancer J Clin. 2007;57:43-66.
293. Hess KR, Varadhachary GR, Taylor SH, et al. Metastatic patterns in adenocarcinoma. Cancer. 2006;106:1624-33.
294. Hornung M, Vogel P, Schubert T, Schlitt HJ, Bolder U. A case of virilization induced by a Krukenberg tumor from gastric cancer. World J Surg Oncol. 2008;6:19.
295. Massarotti M, Ciocia G, Ceriani R, et al. Metastatic gastric cancer presenting with shoulder-hand syndrome: A case report. J Med Case Reports. 2008;2:240.
296. Yunker JJ, Vicinanzo MG, Braswell RA, et al. Unusual presentation of gastric adenocarcinoma metastatic to the orbit. Ophthal Plast Reconstr Surg. 2006;22:490-1.
297. Saornil MA, Blanco G, Sarasa JL, et al. Isolated metastasis of gastric adenocarcinoma to the retina: First presentation of systemic disease. Acta Ophthalmol Scand. 2004;82:86-8.
298. Farley DR, Donohue JH. Early gastric cancer. Surg Clin North Am. 1992;72:401-21.
299. Talley NJ. American Gastroenterological Association medical position statement: Evaluation of dyspepsia. Gastroenterology. 2005;129:1753-5.
300. Tanaka K, Toyoda H, Kadowaki S, et al. Surface pattern classification by enhanced-magnification endoscopy for identifying early gastric cancers. Gastrointest Endosc. 2008;67:430-7.
301. Kato M, Kaise M, Yonezawa J, et al. Autofluorescence endoscopy versus conventional white light endoscopy for the detection of superficial gastric neoplasia: A prospective comparative study. Endoscopy. 2007;39:937-41.
302. Kitabatake S, Niwa Y, Miyahara R, et al. Confocal endomicroscopy for the diagnosis of gastric cancer in vivo. Endoscopy. 2006;38:1110-14.
303. The Paris endoscopic classification of superficial neoplastic lesions: Esophagus, stomach, and colon. Gastrointest Endosc. 2003;58:S3-28.
304. Archer AG, Grant DC. Recent developments in diagnostic radiology of primary and recurrent gastric cancer. Cancer Treat Res. 1991;55:107-31.
305. Shin KS, Kim SH, Han JK, et al. Three-dimensional MDCT gastrography compared with axial CT for the detection of early gastric cancer. J Comput Assist Tomogr. 2007;31:741-9.
306. Kitahara F, Kobayashi K, Sato T, et al. Accuracy of screening for gastric cancer using serum pepsinogen concentrations. Gut. 1999;44:693-7.
307. Westerveld BD, Pals G, Lamers CB, et al. Clinical significance of pepsinogen A isozymogens, serum pepsinogen A and C levels, and serum gastrin levels. Cancer. 1987;59:952-8.
308. Ohata H, Oka M, Yanaoka K, et al. Gastric cancer screening of a high-risk population in Japan using serum pepsinogen and barium digital radiography. Cancer Sci. 2005;96:713-20.
309. Mukoubayashi C, Yanaoka K, Ohata H, et al. Serum pepsinogen and gastric cancer screening. Intern Med. 2007;46:261-6.
310. Fernandez-Fernandez L, Tejero E, Tieso A, et al. Receiver operating characteristic (ROC) curve analysis of the tumor markers CEA, CA 19-9 and CA 72-4 in gastric cancer. Int Surg. 1996;81:400-2.
311. Takahashi Y, Takeuchi T, Sakamoto J, et al. The usefulness of CEA and/or CA19-9 in monitoring for recurrence in gastric cancer patients: A prospective clinical study. Gastric Cancer. 2003;6:142-5.
312. Li X, Yue ZC, Zhang YY, et al. Elevated serum level and gene polymorphisms of TGF-beta1 in gastric cancer. J Clin Lab Anal. 2008;22:164-71.
313. Goral V, Yesilbagdan H, Kaplan A, Sit D. Evaluation of CA 72-4 as a new tumor marker in patients with gastric cancer. Hepatogastroenterology. 2007;54:1272-5.
314. Kumar Y, Tapuria N, Kirmani N, Davidson BR. Tumour M2-pyruvate kinase: a gastrointestinal cancer marker. Eur J Gastroenterol Hepatol. 2007;19:265-76.
315. Tanaka K, Miki C, Wakuda R, et al. Circulating level of hepatocyte growth factor as a useful tumor marker in patients with early-stage gastric carcinoma. Scand J Gastroenterol. 2004;39:754-60.
316. Fujiwara M, Kodera Y, Misawa K, et al. Longterm outcomes of early-stage gastric carcinoma patients treated with laparoscopy-assisted surgery. J Am Coll Surg. 2008;206:138-43.
317. Kitano S, Shiraishi N, Uyama I, et al. A multicenter study on oncologic outcome of laparoscopic gastrectomy for early cancer in Japan. Ann Surg. 2007;245:68-72.
318. Borie F, Plaisant N, Millat B, et al. Appropriate gastric resection with lymph node dissection for early gastric cancer. Ann Surg Oncol. 2004;11:512-17.
319. Yamashita K, Sakuramoto S, Kikuchi S, et al. Validation of staging systems for gastric cancer. Gastric Cancer. 2008;11:111-18.
320. Greene FL, Page DL, Fleming ID, et al. AJCC cancer staging manual, 6th ed. New York: Springer; 2002.
321. Sobin LH, Wittekind C. TNM classification of malignant tumors (UICC). New York: John Wiley; 1997.
322. Nicholson DA, Shorvon PJ. Review article: Endoscopic ultrasound of the stomach. Br J Radiol. 1993;66:487-92.
323. Kwee RM, Kwee TC. Imaging in local staging of gastric cancer: A systematic review. J Clin Oncol. 2007;25:2107-16.
324. Yoshida S, Tanaka S, Kunihiro K, et al. Diagnostic ability of high-frequency ultrasound probe sonography in staging early gastric cancer, especially for submucosal invasion. Abdom Imaging. 2005;30:518-23.
325. Kuntz C, Herfarth C. Imaging diagnosis for staging of gastric cancer. Semin Surg Oncol. 1999;17:96-102.
326. Habermann CR, Weiss F, Riecken R, et al. Preoperative staging of gastric adenocarcinoma: comparison of helical CT and endoscopic US. Radiology. 2004;230:465-71.
327. Polkowski M, Palucki J, Wronska E, et al. Endosonography versus helical computed tomography for locoregional staging of gastric cancer. Endoscopy. 2004;36:617-23.
328. Wang JY, Hsieh JS, Huang YS, et al. Endoscopic ultrasonography for preoperative locoregional staging and assessment of resectability in gastric cancer. Clin Imaging. 1998;22:355-9.
329. Xi WD, Zhao C, Ren GS. Endoscopic ultrasonography in preoperative staging of gastric cancer: Determination of tumor invasion depth, nodal involvement and surgical resectability. World J Gastroenterol. 2003;9:254-7.
330. Monig SP, Zirbes TK, Schroder W, et al. Staging of gastric cancer: Correlation of lymph node size and metastatic infiltration. AJR Am J Roentgenol. 1999;173:365-7.
331. Yang DM, Kim HC, Jin W, et al. 64 multidetector-row computed tomography for preoperative evaluation of gastric cancer: Histological correlation. J Comput Assist Tomogr. 2007;31:98-103.
332. Bhandari S, Shim CS, Kim JH, et al. Usefulness of three-dimensional, multidetector row CT (virtual gastroscopy and multiplanar reconstruction) in the evaluation of gastric cancer: A comparison with conventional endoscopy, EUS, and histopathology. Gastrointest Endosc. 2004;59:619-26.
333. Lim JS, Yun MJ, Kim MJ, et al. CT and PET in stomach cancer: Preoperative staging and monitoring of response to therapy. Radiographics. 2006;26:143-56.
334. Park SR, Lee JS, Kim CG, et al. Endoscopic ultrasound and computed tomography in restaging and predicting prognosis after neoadjuvant chemotherapy in patients with locally advanced gastric cancer. Cancer. 2008;112:2368-76.
335. Patel PR, Mansfield PF, Crane CH, et al. Clinical stage after preoperative chemoradiation is a better predictor of patient outcome than the baseline stage for localized gastric cancer. Cancer. 2007;110:989-95.
336. Hundahl SA, Phillips JL, Menck HR. The National Cancer Data Base Report on poor survival of U.S. gastric carcinoma patients treated with gastrectomy: Fifth edition American Joint Committee on Cancer staging, proximal disease, and the “different disease” hypothesis. Cancer. 2000;88:921-32.
337. Nakagawa S, Nashimoto A, Yabusaki H. Role of staging laparoscopy with peritoneal lavage cytology in the treatment of locally advanced gastric cancer. Gastric Cancer. 2007;10:29-34.
338. Gouzi JL, Huguier M, Fagniez PL, et al. Total versus subtotal gastrectomy for adenocarcinoma of the gastric antrum. A French prospective controlled study. Ann Surg. 1989;209:162-6.
339. Bozzetti F, Marubini E, Bonfanti G, et al. Subtotal versus total gastrectomy for gastric cancer: Five-year survival rates in a multicenter randomized Italian trial. Italian Gastrointestinal Tumor Study Group. Ann Surg. 1999;230:170-8.
340. Cuschieri A, Weeden S, Fielding J, et al. Patient survival after D1 and D2 resections for gastric cancer: Long-term results of the MRC randomized surgical trial. Surgical Co-operative Group. Br J Cancer. 1999;79:1522-30.
341. Yu W, Choi GS, Chung HY. Randomized clinical trial of splenectomy versus splenic preservation in patients with proximal gastric cancer. Br J Surg. 2006;93:559-63.
342. Hartgrink HH, van de Velde CJ, Putter H, et al. Extended lymph node dissection for gastric cancer: who may benefit? Final results of the randomized Dutch gastric cancer group trial. J Clin Oncol. 2004;22:2069-77.
343. Van Cutsem E, Van de Velde C, Roth A, et al. Expert opinion on management of gastric and gastro-oesophageal junction adenocarcinoma on behalf of the European Organisation for Research and Treatment of Cancer (EORTC)-gastrointestinal cancer group. Eur J Cancer. 2008;44:182-94.
344. Yamao T, Shirao K, Ono H, et al. Risk factors for lymph node metastasis from intramucosal gastric carcinoma. Cancer. 1996;77:602-6.
345. Soetikno R, Kaltenbach T, Yeh R, Gotoda T. Endoscopic mucosal resection for early cancers of the upper gastrointestinal tract. J Clin Oncol. 2005;23:4490-8.
346. Kakushima N, Fujishiro M. Endoscopic submucosal dissection for gastrointestinal neoplasms. World J Gastroenterol. 2008;14:2962-7.
347. Imagawa A, Okada H, Kawahara Y, et al. Endoscopic submucosal dissection for early gastric cancer: Results and degrees of technical difficulty as well as success. Endoscopy. 2006;38:987-90.
348. Onozato Y, Ishihara H, Iizuka H, et al. Endoscopic submucosal dissection for early gastric cancers and large flat adenomas. Endoscopy. 2006;38:980-6.
349. De Vita F, Giuliani F, Galizia G, et al. Neo-adjuvant and adjuvant chemotherapy of gastric cancer. Ann Oncol. 2007;18(Suppl 6):vi120-3.
350. Earle CC, Maroun JA. Adjuvant chemotherapy after curative resection for gastric cancer in non-Asian patients: Revisiting a meta-analysis of randomised trials. Eur J Cancer. 1999;35:1059-64.
351. Janunger KG, Hafstrom L, Glimelius B. Chemotherapy in gastric cancer: A review and updated meta-analysis. Eur J Surg. 2002;168:597-608.
352. Di Costanzo F, Gasperoni S, Manzione L, et al. Adjuvant chemotherapy in completely resected gastric cancer: A randomized phase III trial conducted by GOIRC. J Natl Cancer Inst. 2008;100:388-98.
353. Bouche O, Ychou M, Burtin P, et al. Adjuvant chemotherapy with 5-fluorouracil and cisplatin compared with surgery alone for gastric cancer: 7-year results of the FFCD randomized phase III trial (8801). Ann Oncol. 2005;16:1488-97.
354. Cunningham D, Allum WH, Stenning SP, et al. Perioperative chemotherapy versus surgery alone for resectable gastroesophageal cancer. N Engl J Med. 2006;355:11-20.
355. Macdonald JS, Smalley SR, Benedetti J, et al. Chemoradiotherapy after surgery compared with surgery alone for adenocarcinoma of the stomach or gastroesophageal junction. N Engl J Med. 2001;345:725-30.
356. Ajani JA, Winter K, Okawara GS, et al. Phase II trial of preoperative chemoradiation in patients with localized gastric adenocarcinoma (RTOG 9904): Quality of combined modality therapy and pathologic response. J Clin Oncol. 2006;24:3953-8.
357. Yu W, Whang I, Chung HY, et al. Indications for early postoperative intraperitoneal chemotherapy of advanced gastric cancer: Results of a prospective randomized trial. World J Surg. 2001;25:985-90.
358. Yonemura Y, de Aretxabala X, Fujimura T, et al. Intraoperative chemohyperthermic peritoneal perfusion as an adjuvant to gastric cancer: Final results of a randomized controlled study. Hepatogastroenterology. 2001;48:1776-82.
359. Sautner T, Hofbauer F, Depisch D, et al. Adjuvant intraperitoneal cisplatin chemotherapy does not improve long-term survival after surgery for advanced gastric cancer. J Clin Oncol. 1994;12:970-4.
360. Rosen HR, Jatzko G, Repse S, et al. Adjuvant intraperitoneal chemotherapy with carbon-adsorbed mitomycin in patients with gastric cancer: Results of a randomized multicenter trial of the Austrian Working Group for Surgical Oncology. J Clin Oncol. 1998;16:2733-8.
361. Yan TD, Black D, Sugarbaker PH, et al. A systematic review and meta-analysis of the randomized controlled trials on adjuvant intraperitoneal chemotherapy for resectable gastric cancer. Ann Surg Oncol. 2007;14:2702-13.
362. Cascinu S, Scartozzi M, Labianca R, et al. High curative resection rate with weekly cisplatin, 5-fluorouracil, epidoxorubicin, 6S-leucovorin, glutathione, and filgrastim in patients with locally advanced, unresectable gastric cancer: a report from the Italian Group for the Study of Digestive Tract Cancer (GISCAD). Br J Cancer. 2004;90:1521-5.
363. Schuhmacher CP, Fink U, Becker K, et al. Neoadjuvant therapy for patients with locally advanced gastric carcinoma with etoposide, doxorubicin, and cisplatinum. Closing results after 5 years of follow-up. Cancer. 2001;91:918-27.
364. Cervantes A, Rosello S, Roda D, Rodriguez-Braun E. The treatment of advanced gastric cancer: current strategies and future perspectives. Ann Oncol. 2008;19(Suppl 5):v103-7.
365. Ross P, Nicolson M, Cunningham D, et al. Prospective randomized trial comparing mitomycin, cisplatin, and protracted venous-infusion fluorouracil (PVI 5-FU) With epirubicin, cisplatin, and PVI 5-FU in advanced esophagogastric cancer. J Clin Oncol. 2002;20:1996-2004.
366. Ajani JA, Moiseyenko VM, Tjulandin S, et al. Clinical benefit with docetaxel plus fluorouracil and cisplatin compared with cisplatin and fluorouracil in a phase III trial of advanced gastric or gastroesophageal cancer adenocarcinoma: the V-325 Study Group. J Clin Oncol. 2007;25:3205-9.
367. Cunningham D, Starling N, Rao S, et al. Capecitabine and oxaliplatin for advanced esophagogastric cancer. N Engl J Med. 2008;358:36-46.
368. Jeurnink SM, van Eijck CH, Steyerberg EW, et al. Stent versus gastrojejunostomy for the palliation of gastric outlet obstruction: a systematic review. BMC Gastroenterol. 2007;7:18.
369. Azab MB, Henry-Amar M, Rougier P, et al. Prognostic factors in primary gastrointestinal non-Hodgkin’s lymphoma. A multivariate analysis, report of 106 cases, and review of the literature. Cancer. 1989;64:1208-17.
370. Amer MH, el-Akkad S. Gastrointestinal lymphoma in adults: clinical features and management of 300 cases. Gastroenterology. 1994;106:846-58.
371. Kolve M, Fischbach W, Greiner A, Wilms K. Differences in endoscopic and clinicopathological features of primary and secondary gastric non-Hodgkin’s lymphoma. German Gastrointestinal Lymphoma Study Group. Gastrointest Endosc. 1999;49:307-15.
372. Maggard MA, O’Connell JB, Ko CY. Updated population-based review of carcinoid tumors. Ann Surg. 2004;240:117-22.
373. Burkitt MD, Pritchard DM. Review article: Pathogenesis and management of gastric carcinoid tumours. Aliment Pharmacol Ther. 2006;24:1305-20.
374. Katz SC, DeMatteo RP. Gastrointestinal stromal tumors and leiomyosarcomas. J Surg Oncol. 2008;97:350-9.
375. Campoli PM, Ejima FH, Cardoso DM, et al. Metastatic cancer to the stomach. Gastric Cancer. 2006;9:19-25.
376. Stein L, Urban MI, O’Connell D, et al. The spectrum of human immunodeficiency virus-associated cancers in a South African black population: Results from a case-control study, 1995-2004. Int J Cancer. 2008;122:2260-5.
377. Hong HS, Ha HK, Won HJ, et al. Gastric schwannomas: Radiological features with endoscopic and pathological correlation. Clin Radiol. 2008;63:536-42.
378. Otsuji E, Yamaguchi T, Taniguchi H, et al. Malignant endocrine carcinoma of the stomach. Hepatogastroenterology. 2000;47:601-4.
379. Yang GY, Liao J, Cassai ND, et al. Parietal cell carcinoma of gastric cardia: Immunophenotype and ultrastructure. Ultrastruct Pathol. 2003;27:87-94.