43 Acyanotic Congenital Heart Disease
Shunt Lesions
Atrial Septal Defect
Atrial septal defects (ASDs) constitute 5% to 10% of all congenital heart defects and occur in approximately one in 1500 live births. There are five types of ASDs (Figure 43-1). The most common type is the ostium secundum ASD, which results from a deficiency in septum primum, the thin membrane-like septum that normally closes the foramen ovale. The second most common type is the ostium primum ASD, which is a defect in the canal septum. This septum normally divides the common atrioventricular (AV) canal and in so doing completes the anterior portion of the atrial septum and the posterior portion of the ventricular septum while dividing the common AV valve into the tricuspid and mitral valve. Defects in this septum result in AV canal defects, which are discussed later in this chapter. The third type is the sinus venosus defect, which is not a defect in atrial septum per se but rather a communication between the two atria by way of a “straddling” venous structure, either a pulmonary vein or a vena cava. It is frequently associated with partial anomalous drainage of the right-sided pulmonary veins connected to the superior vena cava (SVC). Coronary sinus ASDs are the fourth type and again are not true defects in the atrial septum but rather the physiologic consequence of a partially or completely unroofed coronary sinus with left atrial to right atrial drainage through the coronary sinus ostium. The fifth type of ASD is that seen with juxtaposition of the atrial appendages. This is extremely rare and results from an absence or misplacement of septum secundum, which normally closes the foramen ovale.
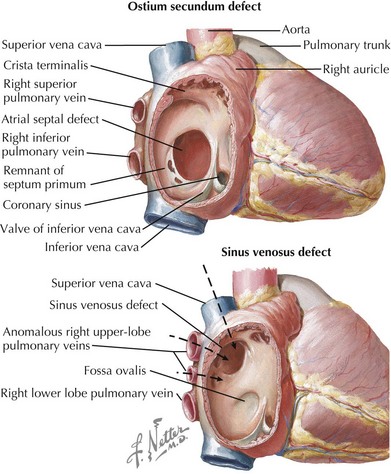
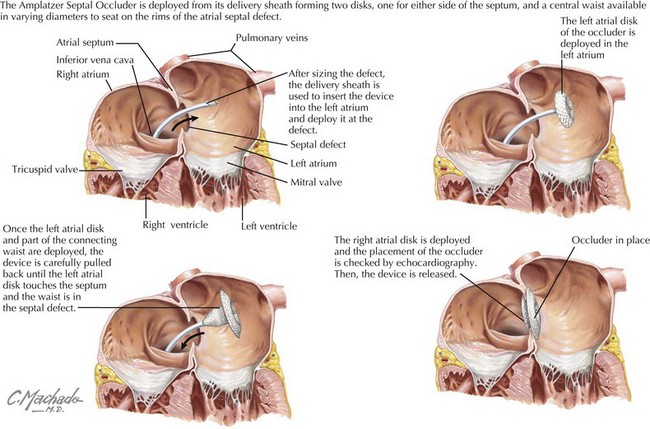
Ostium secundum defects may spontaneously close within the first 4 years of life, but the other types of ASDs usually do not. Options for repair include surgical closure or transcatheter device closure (Figure 43-2). Secundum defects with well-defined margins are the only type amenable to device closure. If left untreated into adulthood, ASDs can lead to pulmonary hypertension; exercise intolerance; atrial arrhythmias; increased risk of paradoxical embolus or stroke; and, late in life, to heart failure. Even when successfully closed in childhood, atrial arrhythmias may still occur decades later.
Ventricular Septal Defect
Ventricular septal defects (VSDs) account for about 20% of all congenital heart disease and occur in 2-10 of /1000 live births. The ventricular septum consists of the inlet (canal septum) posteriorly and inferiorly, running the full superoinferior length of the septal leaflet of the tricuspid valve; the infundibular, conal, or outlet septum superiorly, the muscular or trabecular septum; and the small, membranous septum at the junction of the other three (Figure 43-3). There are five types of VSDs that result from defects in or between these various components of the ventricular septum.
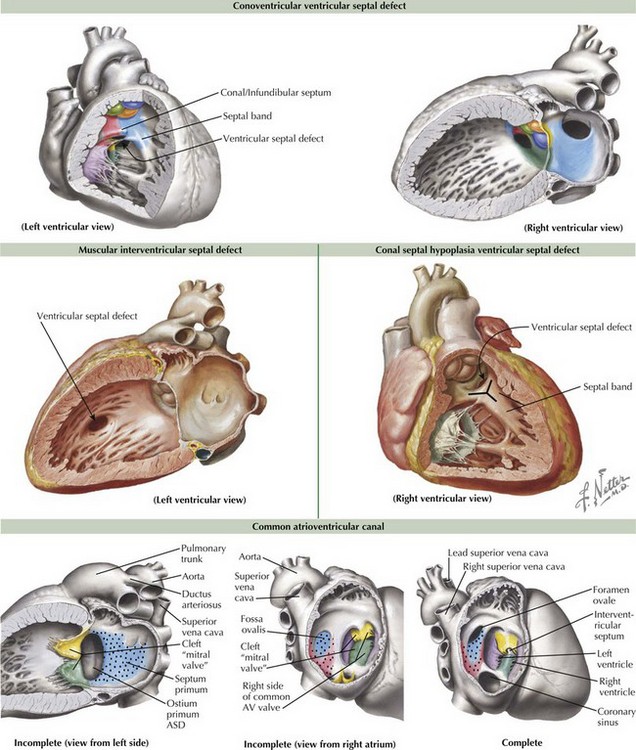
The most common type of VSDs are the conoventricular VSDs, which are defects between the conal or infundibular septum and the rest of the ventricular septum (see Figure 43-3). They may include the membranous septum, in which case they are a type of perimembranous VSD. These VSDs can be partially closed by tissue from the tricuspid valve, and many defects become smaller with time. Rarely, aortic regurgitation may occur because of prolapse of the right or noncoronary cusp into the VSD. Canal-type or inlet defects are usually seen in common AV canal defects (described more fully below) and occur from absence of the inlet septum; they extend along the full length of the AV valve. They may also be seen in straddling tricuspid valve and in some cases of transposition or double outlet right ventricle without AV valve abnormality. Malalignment and conal septal hypoplasia defects occur as a result of malalignment or absence of the conal or infundibular septum, respectively. Malalignment defects are seen in patients with tetralogy of Fallot (discussed in Chapter 44) and interrupted aortic arch along with other complex congenital lesions. Conal septal hypoplasia defects (see Figure 43-3) are sometimes referred to as subpulmonary or supracristal VSDs. They occur within the Y-shaped septal band beneath both semilunar valves and may be associated with prolapse of an aortic cusp resulting in aortic regurgitation. The second most common type of VSDs are called muscular VSDs, which are defects located anywhere other than those described above. These defects often spontaneously close if they are small to moderate in size (see Figure 43-3).
Common Atrioventricular Canal
A complete common AV canal consists of the above-mentioned septal deficiency, with the common AV valve suspended within the septal defect such that there is space proximal to the valve between the two atria (ostium primum ASD) and space distal to the valve between the two ventricles (canal-type VSD) (see Figure 43-3). An incomplete (or partial) AV canal has the same septal deficiency but has leaflet tissue dividing the valve orifice into two orifices and adhering to the crest of the ventricular septum such that there is no direct communication between ventricles. Thus, the entire septal defect, being proximal to the AV valve, is called an ostium primum ASD, and the morphology of the left side of the common AV valve is described as a cleft “mitral” because the two components of what should have formed the anterior leaflet of a mitral valve, remain separate or cleft. A transitional AV canal, similar to an incomplete canal, occurs when the AV valve attachments to the ventricular septum result in a restrictive VSD. The AV valve in this case also has two orifices. The primary defect in the canal septum remains the same, but the defects vary by degree of VSD closure by valve tissue.
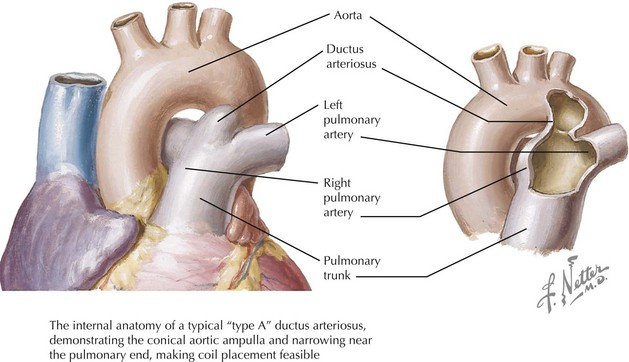
Patent Ductus Arteriosus
Failure of the ductus to close after birth results in a left-to-right shunt between the aorta and the pulmonary artery (Figure 43-4). The magnitude and direction of the shunt depend on the size of the open ductus and pulmonary versus systemic vascular resistance. For example, neonates with severe pulmonary hypertension will have a right-to-left shunt (pulmonary artery to aorta).
Nonshunt Lesions
Aortic Valve Disease
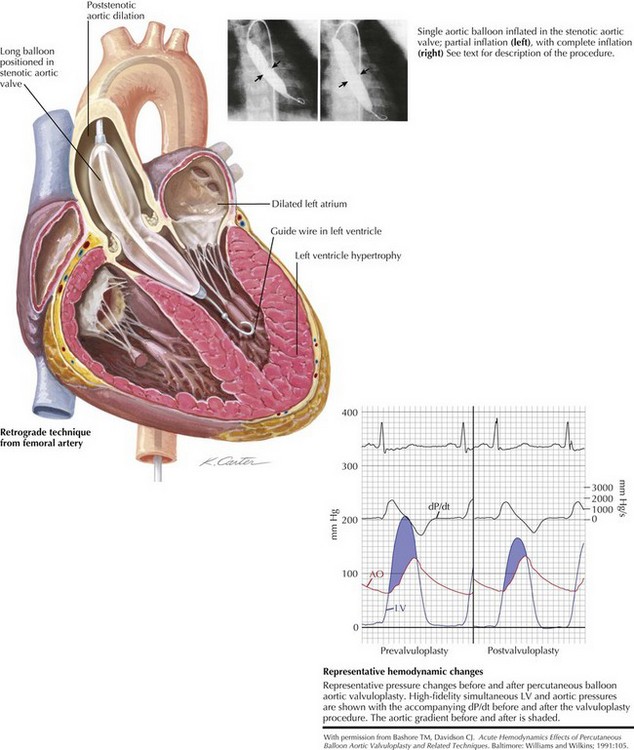
Intervention in the form of surgical valvotomy or balloon valvuloplasty is done for all infants with critical AS regardless of the gradient and in children with noncritical AS with a gradient greater than 50 to 60 mm Hg as measured by cardiac catheterization (Figure 43-5). The gradient upon which intervention is undertaken also depends on the presence of symptoms, changes on rest or exercise ECG, and the desire to play competitive sports. After intervention, the gradient is usually reduced, but resultant aortic regurgitation is not uncommon.
Aortic insufficiency (AI) very infrequently occurs as an isolated lesion. Instead, it can be seen in association with conoventricular VSDs with a prolapsed aortic leaflet, subaortic stenosis, bicuspid aortic valve, connective tissue disorders such as Marfan’s disease, or as a result of endocarditis. Rheumatic fever should always be considered in a patient with new-onset AI. The presentation and treatment of AI are discussed in Chapter 49.
Coarctation of the Aorta
Coarctation of the aorta is a discrete narrowing of the distal aortic arch opposite the entrance of the ductus arteriosus or the ligamentum (after ductal closure) (Figure 43-6). In some rare instances, it can be a narrowing of the abdominal aorta. It constitutes about 8% of all congenital heart defects. Boys are affected about four times more frequently than girls. Turner’s syndrome should be suspected in any girl with aortic coarctation. Coarctation is also frequently associated with left-sided lesions such as bicuspid aortic valve, AS, mitral valve abnormalities, and VSDs. It can also be associated with noncardiac abnormalities such as intracranial aneurysms.
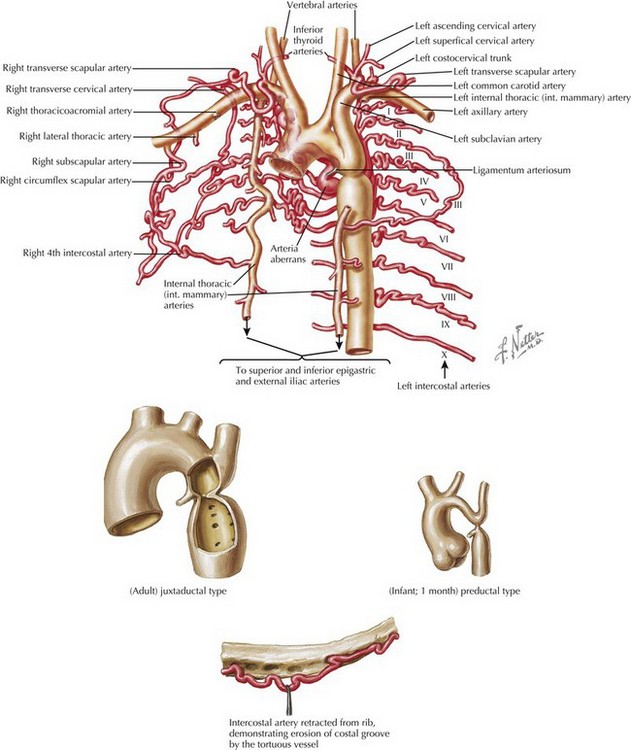
In neonates with critical or severe coarctation, the presentation is usually shock. Although they too sometimes have discrete juxtaductal coarctation, infants usually have arch hypoplasia proximal to the entrance of the ductus arteriosus (see Figure 43-6) along with intracardiac abnormalities. With slightly less severe coarctation, infants can present with symptoms of CHF, notably feeding intolerance. Children who have adapted to the gradual development of discrete coarctation are often asymptomatic despite upper extremity hypertension but may experience claudication; cold extremities; and rarely, chest pain with exercise.
Pulmonary Valve Disease
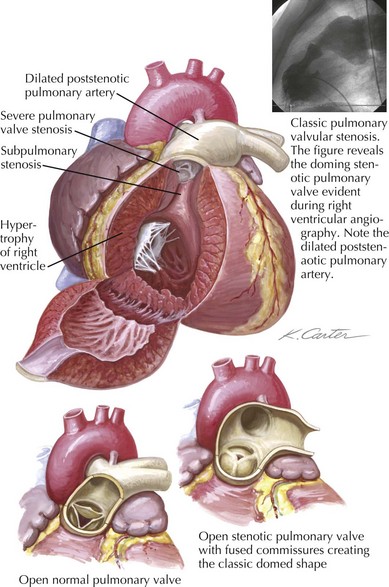
Pulmonary stenosis (PS) occurs in about 8% of children with congenital heart disease or seven in 100,000 live births. PS can be valvular, subvalvular, or supravalvular. Valvular PS is the most common (90%) and is usually seen with varying degrees of leaflet fusion of all three commissures (Figure 43-7). Dysplastic pulmonary valve abnormalities can be seen in association with Noonan’s syndrome. Supravalvular stenosis is very rarely an isolated finding and is associated with Williams syndrome, Alagille syndrome, or LEOPARD (lentigines, ECG conduction abnormalities, ocular hypertelorism, pulmonary stenosis, abnormal genitalia, retarded growth, and sensorineural deafness) syndrome. Subvalvar PS is rare in isolation and is typically part of tetralogy of Fallot or is caused by an anomalous muscle bundle of the right ventricle (so-called double-chambered RV) associated with a conoventricular VSD.
Mitral Valve Disease
Mitral regurgitation (MR) is also commonly found as a complication of rheumatic heart disease but can occur with structural abnormalities as well. Isolated cleft mitral valve is a rare form of congenital MR. Mitral valve prolapse (MVP) may also result in MR caused by posterior movement of one leaflet into the left atrium during systole (Figure 43-8). Although unusual in infants, MVP can be seen in patients with connective tissue disorders such as Marfan and Ehlers-Danlos syndromes. Bacterial endocarditis of the mitral valve may also result in MR.
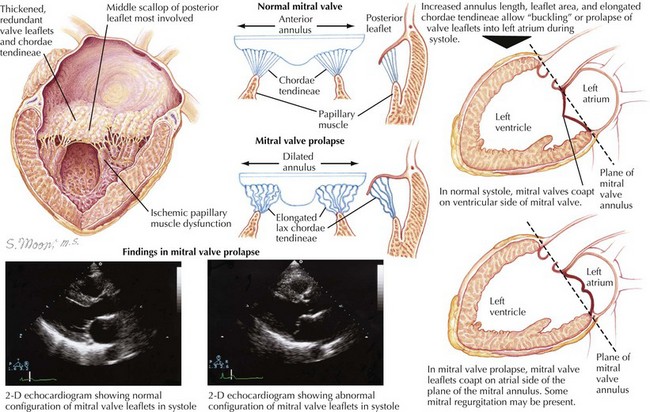
Mitral valve disease caused by rheumatic heart disease is discussed in further detail in Chapter 49.
Vascular Ring
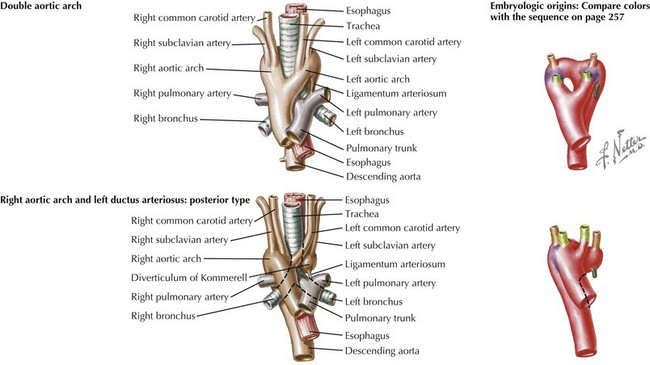
Vascular rings are anomalies of the aortic arch that can cause compression of the trachea, esophagus, or both. Infants may present with “noisy breathing” or with stridor. Respiratory distress associated concomitant upper respiratory infections is common. Older children and toddlers might present with swallowing difficulties, although asthma without family history and unresponsive to medical management warrants consideration of a vascular ring. Double aortic arch and right aortic arch with a retroesophageal diverticulum of Kommerell are the two most common types of vascular rings (Figure 43-9). If a vascular ring is suspected, chest radiography can determine arch sidedness and sometimes indentation of the trachea (double aortic arch). In addition, a barium esophagram can reveal a large posterior indentation on the esophagus (virtually all rings). Definitive diagnosis can be made by MRI. Surgical repair is indicated in any symptomatic patient. Symptoms do not always improve immediately and may persist for up to 1 year after repair.
Allen HD, Driscoll DJ, Shaddy RE, Feltes TF, editors. Moss and Adams’ Heart Disease in Infants, Children, and Adolescents Including the Fetus and Young Adult, ed 7, Philadelphia: Lippincott Williams & Wilkins, 2008.
Borer JS, Bonow RO. Contemporary approach to aortic and mitral regurgitation. Circulation. 2003;108(20):2432-2438.
Campbell M. Natural history of atrial septal defects. Br Heart J. 1970;32:820-826.
Chiappa E. The impact of prenatal diagnosis of congenital heart disease on pediatric cardiology and cardiac surgery. J Cardiovasc Med. 2007;8(1):12-16.
Graham TPJr, Bricker JT, James FW, Strong WB. 26th Bethesda conference: recommendations for determining eligibility for competition in athletes with cardiovascular abnormalities. Task Force 1: congenital heart disease. Med Sci Sports Exerc. 1994;26(10 suppl):S246-S253.
McMahon CJ, Feltes TF, Fraley JK, et al. Natural history of growth of secundum atrial septal defects and implications for transcatheter closure. Heart. 2002;87(3):256-259.
Pinto NM, Marino BS, Wernovsky G, et al. Obesity is a common comorbidity in children with congenital and acquired heart disease. Pediatrics. 2007;120(5):e1157-e1164.
Rudolph AM. The effects of postnatal circulatory adjustments in congenital heart disease. Pediatrics. 1965;36:763-772.