[level-membership-for-cardiothoracic-surgery-category]
Chapter 27 Acute Respiratory Failure
Acute respiratory failure is the inability to maintain adequate gas exchange. It is defined by abnormalities of arterial oxygen (Pao2) and carbon dioxide (Paco2) tensions. Hypoxemia may occur with a normal or low Paco2 (type 1 respiratory failure) or with an elevated Paco2 (type 2 respiratory failure). Respiratory failure is commonly associated with respiratory distress, which manifests as tachypnea, dyspnea, and the use of the accessory muscles of breathing.
Respiratory failure is relatively common in cardiac surgery patients and has a wide variety of causes (Table 27-1). The need for prolonged ventilatory support (>72 hours) occurs in approximately 6% of patients undergoing coronary artery bypass graft surgery,1 and reintubation is required in approximately 3% of patients.2
Table 27-1 Common Causes of Postoperative Respiratory Failure Hypoxemia in Patients After Cardiac Surgery
| On Arrival in the ICU and During Mechanical Ventilation |
| Pulmonary edema |
| Atelectasis/lobar collapse |
| Endobronchial intubation |
| Pneumothorax |
| Hemothorax |
| Ventilator dysynchrony |
| Bronchospasm |
| Low SVO2 (low cardiac output,* anemia) |
| Immediately Following Extubation |
| Residual sedative or opioid drugs |
| Residual neuromuscular blockade |
| Atelectasis |
| Upper airway obstruction/edema |
| Late Deterioration in Respiratory Function |
| Cardiac tamponade (following removal of pacing wires) |
| Pneumothorax (following removal of chest drains) |
| Left ventricular dysfunction |
| Arrhythmias (particularly atrial fibrillation) |
| Atelectasis and lobar collapse |
| Sepsis (pneumonia, mediastinitis) |
| Pulmonary embolus |
| Pulmonary edema |
ICU, intensive care unit.
* Causes of low cardiac output in the early period after cardiac surgery include all conditions listed in Table 20-4 with the exception of low systemic vascular resistance.
This chapter is divided into three sections: (1) mechanisms of respiratory failure; (2) diagnosis and treatment of respiratory failure; (3) specific causes of respiratory failure. Only an overview of respiratory failure is provided here. The physiological mechanisms of respiratory failure are explored in greater detail in Chapter 1, and the ventilatory treatment is described in Chapters 28 and Chapter 29.
MECHANISMS OF RESPIRATORY FAILURE
Central Nervous System and Neuromuscular Dysfunction
Dysfunction of the CNS or neuromuscular system can lead to hypoventilation, loss of protective airway reflexes, and impaired swallowing. Hypoventilation causes hypercarbia and, if severe, hypoxemia. Impaired swallowing and loss of protective airway reflexes predispose a patient to pulmonary aspiration.
Exacerbating Factors
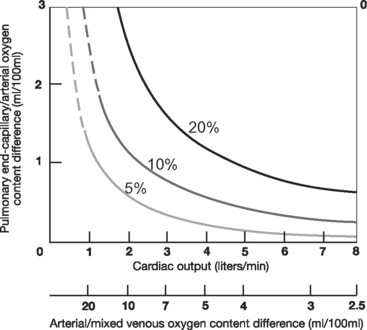
With normal lung function, the main determinant of Pao2 is alveolar oxygen tension (PAo2). However, with intrapulmonary shunting, systemic venous blood passes through the lungs without being oxygenated, and SVo2 becomes an important determinant of Pao2. Low SVo2 can arise from anemia, low cardiac output, and high metabolic rate. This is illustrated in Figure 27-1, which shows the relationship between cardiac output and arterial oxygenation at various levels of intrapulmonary shunting.
APPROACH TO PATIENTS WITH ACUTE RESPIRATORY DISTRESS
Diagnosis
Examination
If appropriate, a targeted neurologic assessment should be performed, focusing on level of consciousness (Glasgow coma scale), cough, gag reflex, and focal neurologic deficit (see Chapter 37).
Investigations
Blood Gas Analysis and Oximetry.
In addition to measurement of arterial oxygen saturation (Sao2), Pao2, and Paco2, useful information is obtained from (1) calculation of the alveolar-arterial (A-a) Po2 gradient, (2) measurement of Svo2, and (3) assessment of metabolic parameters (pH, bicarbonate, base excess, and lactate; see Chapters 1 and Chapter 31).
The A-a gradient is the difference between PAo2 and Pao2, where PAo2 is calculated from the alveolar air equation (see Equation 1-18). With healthy lungs breathing air, the A-a gradient is very low, less than 1 κPa (<7.5 mmHg). However, the A-a gradient is influenced by age and FIo2. At age 40, the upper limit of normal is about 3 κPa (23 mmHg), increasing to 5 κPa (38 mmHg) at age 80 (when breathing room air). To this must be added 0.75 to 1 κPa (6 to 7.6 mmHg) for every 10% increase in FIo2. Within these limitations, the A-a gradient can be used to differentiate hypoventilation (normal A-a gradient) from intrapulmonary shunting (elevated A-a gradient).
Chest Radiography.
The chest radiograph (see Chapter 6) is essential in the assessment of respiratory failure. If possible, the film should be obtained while the patient is erect because this position improves the diagnostic yield, particularly in terms of pleural collections and pneumothoraces. In a patient with respiratory failure, a normal chest radiograph is consistent with the diagnoses of hypoventilation, microatelectasis, low SVo2, and shunting across a PFO.
Hemodynamic Monitoring and Echocardiography.
Hemodynamic evaluation (see Chapter 8) is an integral part of the assessment of respiratory failure. A PAC allows for quantification of cardiac output, SVo2, and pulmonary artery wedge pressure (PAWP). PAWP helps to determine the cause of pulmonary edema (see subsequent material). Echocardiography (see Chapter 7) reliably identifies the cause of low cardiac output (see Chapter 20) and can be used to identify a PFO.
Treatment
Treatment of acute respiratory failure is both supportive and specific to the underlying condition. Specific treatments are discussed in specific causes of respiratory failure. Supportive treatments are listed subsequently. The treatment goals are outlined in Table 27-2.
Table 27-2 Therapeutic Aims for Managing Acute Respiratory Failure
| SaO2 ≥ 90% (approximately 7 kPa or 55 mmHg) |
| pH >7.25 |
| Respiratory rate <30 to 35/min* |
| Ability to speak in short sentences |
| Hemodynamic stability |
| Ability to protect airway† |
* Higher respiratory rates cannot usually be sustained indefinitely; exhaustion and respiratory arrest are likely to supervene.
† Respiratory failure severe enough to cause a fall in the level of consciousness such that the patient cannot obey commands mandates endotracheal intubation to protect against pulmonary aspiration.
Supplemental Oxygen
All patients who are hypoxemic (Pao2 <8 to 10 κPa or 60 to 75 mmHg) should receive supplemental oxygen by nasal cannula or face mask. This includes patients with COPD (see subsequent material). Oxygen is very effective in treating hypoxemia due to hypoventilation but is less effective when it is due to V/Q mismatch. Supplemental oxygen may mask severe hypoventilation, emphasizing the importance of monitoring respiratory rate, level of consciousness, and Paco2 (or end-tidal CO2) in addition to Spo2. Oxygen therapy is discussed further in Chapter 28.
Positive Pressure Ventilation
Positive pressure ventilation is used to improve gas exchange and reduce the work of breathing. It may be invasive (see Chapter 29) or noninvasive (see Chapter 28).
The need for ventilatory support depends on the severity of the respiratory failure, the rate of deterioration, the underlying disease process, and the need to protect the airway. When respiratory failure develops rapidly or occurs in the context of multiple organ dysfunction syndrome, early institution of invasive ventilation is indicated. However, if respiratory failure develops slowly and is not associated with severe metabolic derangement or other organ failure, urgent intubation may not be required, and other targeted therapy (e.g., antibiotics, physical therapy, diuretics) may be tried first. The signs of severe respiratory failure that, if not rapidly corrected, indicate the need for urgent intubation and ventilation are listed in Table 27-3.
Table 27-3 Features of Severe Respiratory Failure That Indicate the Need for Urgent Endotracheal Intubation
| Respiratory System |
| SaO2 ≤ 85% (approximately 6.5 kPa or 50 mmHg) with high-flow face-mask oxygen or an FIO2 ≥ 0.6 with noninvasive ventilation |
| Rising PaCo2* with respiratory acidemia (pH < 7.1) |
| Respiratory distress: respiratory rate >40/min, intercostal in-drawing, use of accessory muscles, the inability to speak more than one or two words between breaths |
| Cardiovascular System |
| Heart rate >120 beats/min |
| Falling blood pressure or cardiac output or escalating inotrope requirements |
| Increasing lactic acidosis |
| Central Nervous System |
| Confusion and agitation |
| Falling level of consciousness (GCS ≤ 8) |
GCS, Glasgow Coma Score (see Chapter 37).
* PaCo2 >10 kPa is associated with carbon dioxide narcosis.
Most causes of hypoxemia are improved by positive pressure ventilation but some, notably pneumothorax and right-to-left shunting across a PFO, may be made worse. Pneumothoraces should be sought clinically before positive pressure ventilation is instituted. If respiratory failure is part of the final stage of a terminal illness, intubation and ventilation are not appropriate, and symptomatic treatment of dyspnea should be instituted (see Chapter 39).
Treatment of Exacerbating Factors
Treatment of low Svo2 involves correcting anemia and low cardiac output and, if possible, reducing metabolic demands. Achieving an Svo2 above 70% early in patients who are septic is associated with improved outcome (see Chapter 35); however, no target value for patients who are not septic has been defined. Simple measures to reduce oxygen consumption (and carbon dioxide production) involve treating fever and avoiding overfeeding, particularly with carbohydrates (see Chapter 34). In patients who are critically unwell, neuromuscular paralysis and active cooling may be considered.
Cardiac Support
Acute treatment of cardiac failure involves diuretics, vasodilators, inotropic drugs, positive pressure ventilation, and intraaortic balloon counterpulsation (see Chapters 19 and Chapter 20). As this acute treatment is withdrawn, weaning from mechanical ventilation may be facilitated by the administration of angiotensin-converting enzyme inhibitors and vasodilators, which reduce left ventricular afterload.
SPECIFIC CAUSES OF RESPIRATORY FAILURE
Some of the causes of respiratory failure described subsequently, for example, cardiogenic pulmonary edema, are dealt with in greater detail elsewhere in the book. Other causes, including pulmonary embolism (see Chapter 23), ventilator-associated pneumonia (see Chapter 35), and mediastinitis (see Chapter 35), are dealt with exclusively in other chapters.
Atelectasis
In atelectasis, regions of collapsed, airless alveoli are present. These occur for two main reasons. First, microatelectasis occurs because of the closure of small airways due to the encroachment of closing capacity on the functional residual capacity (see Chapter 1). Once these small airways become obstructed, gas within distal alveoli is absorbed (absorption atelectasis), and alveolar collapse occurs. Microatelectasis is common in patients who are obese, elderly, anesthetized, in a supine position or who have reduced lung compliance. It tends to develop in dependent regions of the lung. Also, atelectasis occurs when mucus plugs medium-sized and large airways because of excessive secretions, impaired bronchial mucociliary function, and poor cough. Mucus plugging results in lobar, segmental, or sub segmental collapse. Collapse of an entire lung is usually caused by endobronchial intubation.
On examination, there may be dullness to percussion and reduced breath sounds over atelectatic lung, most commonly at the lung bases. Microatelectasis may not be apparent on a chest radiograph but is clearly seen on a computed tomography scan of the chest (Fig. 27-2). Lobar collapse is readily apparent on a chest radiograph (see Chapter 6).
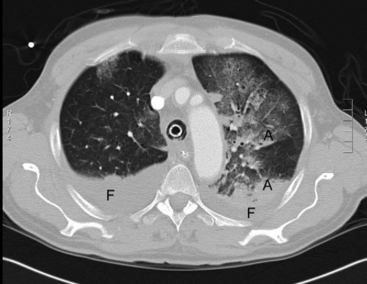
Treatment is directed toward reexpanding the collapsed alveoli. In spontaneously breathing patients treatment includes incentive spirometry, huffing and coughing exercises, saline nebulizers to aid sputum expectoration, and adequate analgesia. Ventilatory strategies include continuous positive airway pressure (CPAP; noninvasive ventilation) and positive end-expiratory pressure (PEEP; invasive ventilation). In invasively ventilated patients, mucus plugging in large airways may be ameliorated by tracheal suctioning and low-volume (10 ml) saline lavage. However, if the primary problem is microatelectasis or pulmonary edema, aggressive tracheal toilet may worsen gas exchange because of the repetitive loss of PEEP.
Pulmonary Edema
Pulmonary edema is an increase in extravascular lung water and may be cardiogenic or noncardiogenic, depending on whether pulmonary capillary hydrostatic pressure is elevated or normal. Cardiogenic pulmonary edema is defined as pulmonary edema in association with a PAWP greater than 18 mmHg. The causes of noncardiogenic pulmonary edema are listed in Table 27-4.
Table 27-4 Causes of Noncardiogenic Pulmonary Edema That May Be Encountered in the Cardiothoracic ICU
| Pulmonary Causes | Nonpulmonary Causes |
|---|---|
| Postpneumonectomy pulmonary edema | Massive blood transfusion |
| Pneumonia | TRALI |
| Pulmonary aspiration | Systemic inflammation secondary to CPB |
| Severe lung contusion secondary to chest trauma | Shock from any cause but particularly sepsis |
| Reperfusion injury following lung transplantation or thromboendarterectomy | Fat or amniotic fluid embolism |
| Drug toxicity | |
| Re-expansion pulmonary edema following large volume thoracocentesis | Mesenteric infarction |
| Acute pancreatitis | |
| Disseminated intravascular coagulation | |
| Negative pressure pulmonary edema following upper airway obstruction | |
| Neurogenic pulmonary edema |
ICU, intensive care unit; TRALI, transfusion related acute lung injury; CPB, cardiopulmonary bypass
In most circumstances, pulmonary edema can be differentiated from other causes of acute respiratory failure, and cardiogenic and noncardiogenic edema can be distinguished, on the basis of the history, the examination, and simple bedside tests (troponin, B-type natriuretic peptide, electrocardiogram, chest radiograph, and echocardiogram; see Chapter 19). Only very rarely is pulmonary artery catheterization required for diagnosis, but it may be useful to guide therapy.
Irrespective of the cause, pulmonary edema results in a clinical syndrome of hypoxemia and respiratory distress. Crackles and wheezes may be heard on auscultation of the chest. If expiratory wheeze is marked, it may easily be mistaken for bronchospasm. The chest radiograph typically demonstrates interstitial or alveolar shadowing (see Chapter 6). Certain radiographic features may help to distinguish cardiogenic from noncardiogenic pulmonary edema; these features are listed in Table 27-5 (see Figs. 6-8 and 6-17).
Table 27-5 Radiographic Features That May Distinguish Cardiogenic From Noncardiogenic Pulmonary Edema
| Radiographic Feature | Cardiogenic Edema | Noncardiogenic Edema |
|---|---|---|
| Heart size | Normal or greater than normal | Usually normal |
| Vascular distribution | Balanced or inverted | Normal or balanced |
| Distribution of edema | Even or central | Patchy or peripheral |
| Pleural effusions | Present | Not usually present |
| Peribronchial cuffing | Present | Not usually present |
| Septal lines | Present | Not usually present |
| Air bronchograms | Not usually present | Usually present |
Modified from Ware LB, Matthay MA: Clinical practice: acute pulmonary edema. N Engl J Med 353:2788-2796, 2005, with permission.
Cardiogenic Pulmonary Edema
Patients with cardiogenic pulmonary edema (see Chapter 19) may have signs of cardiac failure such as distended neck veins, a third or fourth heart sound, an abnormal cardiac impulse, or a murmur. Cardiogenic pulmonary edema that occurs early following cardiac surgery may be due to a failed surgical repair, severe myocardial stunning, myocardial ischemia or infarction, or tachyarrhythmia. Persistent subacute pulmonary edema that limits weaning from mechanical ventilation is commonly due to left ventricular systolic or diastolic dysfunction, occult tamponade, or valvular regurgitation. Acute pulmonary edema that occurs following a myocardial infarction is usually caused by left ventricular dysfunction or mitral regurgitation. Flash pulmonary edema that occurs in a patient with hypertension may indicate left ventricular diastolic dysfunction. The diagnosis and treatment of cardiogenic pulmonary edema are discussed in Chapter 19.
Acute Lung Injury and Acute Respiratory Distress Syndrome
Acute lung injury (ALI) and acute respiratory distress syndrome (ARDS) are clinical syndromes that arise as a consequence of a wide range of pulmonary and extrapulmonary pathologies (see Table 27-4). Specific criteria for the diagnosis of ALI/ARDS have been developed.3 They are as follows:
In the acute phase, ALI/ARDS is characterized by increased permeability of the alveolar-capillary membrane and the accumulation of protein-rich fluid and inflammatory cells within the alveoli. To varying degrees, there is increased extravascular lung water, atelectasis, lobar collapse, and infection, which together result in reduced lung compliance and impaired gas exchange. Pulmonary vascular resistance is variably elevated. A proportion of patients go on to develop a chronic phase characterized by pulmonary fibrosis.
ALI/ARDS that occurs in the early period following cardiac surgery may be due to severe inflammatory response to cardiopulmonary bypass (see Chapter 2); transfusion-related acute lung injury (see Chapter 30), or massive transfusion (see Chapter 30). Later in the postoperative period, ALI/ARDS may occur as a consequence of distributive shock (sepsis, pancreatitis, mesenteric infarction) or pulmonary aspiration. ALI/ARDS can also occur following pneumonectomy (see Chapter 12) or following major pulmonary trauma. ALI/ARDS frequently occurs in the context of multiple organ dysfunction syndrome (see Chapter 2).
Treatment is primarily supportive and includes fluid restriction, mechanical ventilation using a lung-protective strategy (see Chapter 29), and support of other organ dysfunction. The role of adjuvant therapies, such as prone ventilation, inhaled nitric oxide, and corticosteroids, is discussed in Chapter 29.
Aspiration
Pulmonary aspiration usually occurs in patients who have one or more risk factors, such as an enteral feeding tube, gastroesophageal reflux, vomiting, reduced level of consciousness, or impaired swallowing. Impaired swallowing may be caused by a stroke, muscle weakness, or a tracheotomy tube. The presence of a cuffed endotracheal or tracheotomy tube does not reliably prevent aspiration, but may limit its severity. Pulmonary aspiration occurs in up to 20% of patients who undergo emergency endotracheal intubation.4
The clinical features of aspiration vary from a subclinical event detectable only by sensitive assays (e.g., the presence of pepsin in the tracheal secretions) to a symptomatic episode with choking, coughing, and recovery of fluid or particulate material from the trachea. Aspiration, particularly of gastric contents, can result in severe chemical pneumonitis, characterized by alveolar edema, bronchospasm, V/Q mismatch, and atelectasis. Severe hypoxemia and respiratory distress can develop very rapidly. Crackles and wheezes may be heard on chest auscultation. Fever and leukocytosis are common. The chest radiograph may show patchy infiltrates in dependent regions which, when the patient is in the supine position, are the posterior segments of the upper lobes and the superior segments of the lower lobes (Fig. 27-3). Secondary bacterial pneumonia or ARDS can supervene.
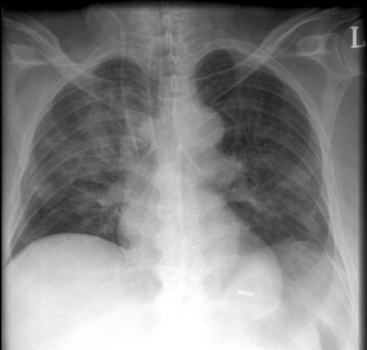
Guidelines for the treatment of major pulmonary aspiration in a critically unwell patient have been developed.5 In the first instance, the patient should be positioned on his or her side and the oropharynx should be suctioned. If particulate material is aspirated or the patient is in significant respiratory distress, endotracheal intubation followed by tracheal toilet is recommended. Removal of particulate matter may be aided by bronchoscopy. Bronchospasm should be treated by nebulized bronchodilators. Prophylactic antimicrobials are not recommended, but if pulmonary infiltrates do not clear or worsen, antimicrobial therapy like that used for ventilator-associated pneumonia (see Chapter 35) is indicated. Corticosteroids are not recommended.
Chronic Obstructive Pulmonary Disease
COPD incorporates three clinical entities: asthma, emphysema, and chronic bronchitis. Asthma is characterized by bronchiolar smooth muscle hypertrophy, bronchospasm, and increased airway secretions. Emphysema is characterized by loss of pulmonary elastic tissue, destruction of alveolar capillary units, and airway collapse. The main pathologic processes in chronic bronchitis are airway inflammation and excess secretions. In an individual patient, characteristics of all three conditions may coexist, but one usually predominates. Increased airflow resistance is common to all patients with COPD.
Mild to moderate COPD does not substantially increase the risk of death in patients undergoing cardiac surgery.6,7 However, the risk of adverse events, such as prolonged intubation, repeat intubation, and arrhythmias, is increased.8,9 Elderly patients (>75 years) with COPD who are being treated with corticosteroids have a very high perioperative risk.7
Pathophysiology
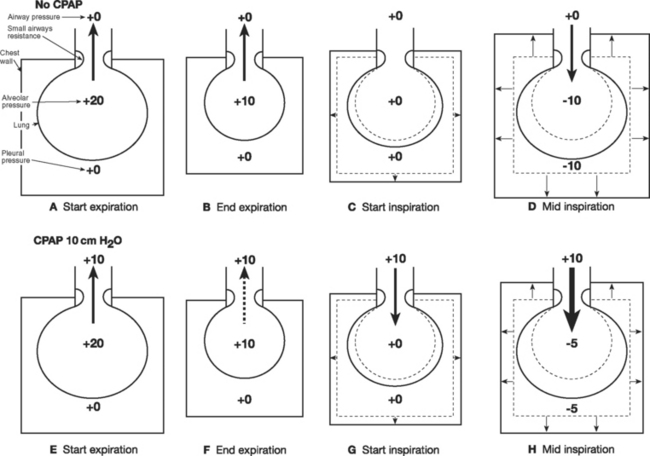
Increased airways resistance is spontaneously breathing patients results in increased work of breathing. Initially this occurs during inspiration but as airways resistance increases, expiration also becomes an active, energy-requiring process. (Expiration is normally passive; see Chapter 1 and Fig. 29-5F) When airway resistance is high, alveolar pressure may not fall to zero by the time expiration has finished, which leads to air trapping (auto-PEEP). There are two main effects of auto-PEEP. First, tidal ventilation occurs at higher lung volumes, which positions the lung on a less favorable part of the compliance curve (see Fig. 1-15). Second, the respiratory muscles must expend additional effort during inspiration to reduce alveolar pressure to zero before gas flow occurs. This effort is termed the inspiratory threshold load, and it is explained in Figure 27-4.
Besides increasing work of breathing, COPD also causes V/Q mismatch, which can lead to hypoxemia and hypercarbia. COPD patients with hypercarbic respiratory failure can experience worsening of their carbon dioxide retention when given supplemental oxygen. This has long been ascribed to inhibition of hypoxic ventilatory drive, and to a degree this is true. However, the majority of the increase in Paco2 is due to inhibition of hypoxic pulmonary vasoconstriction, which results in increased blood flow to poorly ventilated lung units and reduced flow to well-ventilated lung units. This redistribution of blood flow increases alveolar dead space, which leads to hypercarbia (see Chapter 1). Although real, the problem of oxygen-induced hypercarbia has been overstated. Severe respiratory compromise is rare and, as long as patients are carefully monitored, supplemental oxygen may be safely administered to patients with COPD.
Assessment and Treatment
Patients with COPD commonly have a barrel-shaped chest. The lungs are hyperresonant to percussion, and on auscultation breath sounds are reduced and expiratory wheezes and crackles may be heard. The chest radiograph typically shows lung hyperinflation with a reduced cardiothoracic ratio, flattened diaphragms, and sometimes bullae (Fig. 27-5). With severe COPD, there may be a compensated respiratory acidosis, indicative of chronically elevated Paco2, and an elevated hematocrit, indicative of chronic hypoxemia.
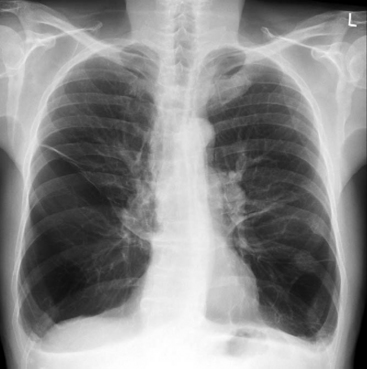
Treatment of postoperative decompensation involves: (1) general supportive measures, such as supplemental oxygen, physical therapy, effective analgesia, and ventilatory support; and (2) correction of precipitating causes, such as bronchospasm, pneumonia, and pneumothorax. Treatment of bronchospasm involves inhaled bronchodilators (see Chapter 28), intravenous corticosteroids and, occasionally, intravenous infusions of albuterol, aminophylline, or epinephrine. If there is evidence of pneumonia (see Chapter 35), intravenous antimicrobial therapy is appropriate. However, routine use of antibiotics for all exacerbations of COPD is not appropriate.10 Prior to commencing antibiotics, a sputum specimen should be sent for microscopy, culture, and sensitivity. Commonly encountered pathologic organisms include Streptococcus pneumoniae, Haemophilus influenzae, Moraxella catarrhalis and, occasionally, Pseudomonas aeruginosa and Klebsiella pneumoniae. Empiric antibiotic therapy should be guided by local prevalence and resistance patterns but is typically similar to that for ventilatorassociated pneumonia (see Chapter 35).
Ventilatory strategies for COPD are outlined in Chapters 28 and Chapter 29. Noninvasive ventilation can be very effective in treating acute hypercarbic respiratory failure secondary to COPD. In Figure 27-4 it can be seen that CPAP (and PEEP) reduce the inspiratory threshold load and therefore reduce the work of breathing in patients with auto-PEEP. If invasive ventilation is required, appropriate ventilator settings must be used to avoid causing barotrauma and dynamic lung hyperinflation, which can cause profound cardiovascular compromise.
Pneumothorax
Because of the elastic recoil of the lung, once the pleura (either parietal or visceral) is breached, air enters the chest cavity and the lung collapses, resulting in a “simple” pneumothorax (see Fig. 25-3). If the source of air entry is self-limited (e.g., during removal of chest drains), a small pneumothorax typically develops. Clinically, there may be signs of reduced chest movement and diminished air entry on the affected side. The diagnosis is easily missed, particularly in patients with COPD, who normally have quiet breath sounds.
If the site of air entry into the pleura functions as a one-way valve, a tension pneumothorax will develop (see Fig. 25-3). A tension pneumothorax may develop very rapidly in patients receiving positive pressure ventilation. Tension pneumothorax results in hyperinflation of the affected pleural space and compression of the mediastinal structures, resulting in obstruction of the venous return and, potentially, cardiorespiratory collapse. Immediate needle thoracocentesis may be lifesaving (see Chapter 40).
If there is clinical suspicion of a pneumothorax, a chest radiograph—ideally taken with the patient erect—must be obtained. In general, in a spontaneously breathing patient, pneumothoraces involving more than 25% of the lung field should be drained. In patients who are receiving positive pressure ventilation, all pneumothoraces should be drained.11 Following drainage, atelectasis or air leak may cause ongoing impairment of gas exchange.
Hemothorax and Pleural Effusion
Most hemothoraces that occur following cardiac surgery are a result of mediastinal or sternal bleeding that gravitates to an opened pleural cavity. Pleural effusions commonly develop a few days following surgery in patients with elevated systemic venous pressure. Large hemothoraces and pleural effusions are usually apparent on chest radiograph, particularly on erect films (see Fig. 6-18). Smaller collections are best diagnosed by ultrasound imaging of the pleural spaces. Ultrasound scanning allows for accurate localization of the collection and the diaphragm and may be used to guide drain placement. Fluid collections that are less than about 400 ml generally do not warrant draining except as a diagnostic procedure. As with pneumothoraces, significant atelectasis may coexist with pleural collections.
Pulmonary Hemorrhage
Bleeding within the parenchyma of the lung may occur due to surgical retraction or handling of the lung (e.g., during procedures on the descending thoracic aorta; see Chapter 11); thoracic trauma (e.g., pulmonary contusion; see Chapter 25); and a wide variety of medical conditions (see Chapter 26). Merely a few hundred milliliters of blood within the alveolar space can cause acute respiratory failure. Radiographically, pulmonary hemorrhage appears as alveolar shadowing, which may be localized to a specific lobe or lung.
Phrenic Nerve Injury
Phrenic nerve dysfunction is common following cardiac surgery; it can occur in up to 70% of cases (see Chapter 37). However, complete paresis of a hemidiaphragm occurs in less than 10% of patients.12,13 Phrenic nerve dysfunction is identified by the development of a raised hemidiaphragm on the chest radiograph following extubation (see Fig. 6-25). Phrenic nerve injury can cause, and also can be mistaken for, lower lobe atelectasis. In isolation, unilateral phrenic nerve palsy does not usually cause respiratory embarrassment, but if it is bilateral (very rare) or if it occurs in association with other pulmonary disease, it can contribute to severe respiratory compromise. The diagnosis is confirmed by identifying paradoxic motion of the diaphragm with ultrasound imaging. Treatment is supportive but occasionally, diaphragmatic plication may be performed.
Hypoventilation
Patients who are over sedated or whose airways are not protected should be intubated and ventilated. Naloxone (see Chapter 4) can be used to reverse the effects of opioids but can precipitate pain, agitation, hypertension, and myocardial ischemia. Flumazenil (see Chapter 4) may be used to reverse benzodiazepine-induced sedation. Although less problematic than naloxone, flumazenil can cause agitation, arrhythmias, and hypertension. If residual neuromuscular blockade is suspected, neuromuscular function should be assessed with a nerve stimulator (see Chapter 4), and if it is present, it may be reversed by administering neostigmine (see Chapter 4). Weakness that occurs in the setting of prolonged critical illness has a number of causes and is discussed in Chapter 37.
Intracardiac Shunting
Hypoxemia due to right-to-left intracardiac shunting occurs with certain congenital cardiac abnormalities (see Chapter 15). These conditions are usually well documented in a patient’s medical records. However, right-to-left shunting can also occur across a PFO in a patient who is not suspected of having a congenital cardiac defect. A PFO is present in about 25% of patients,14 and right-to-left shunting occurs when right atrial pressure exceeds left atrial pressure. Right ventricular dysfunction and patient-ventilator dysynchrony predispose to right-to-left shunting across a PFO. Hypoxemia may develop and resolve over a few seconds. Central venous pressure is usually high but there may be little else to suggest the diagnosis. A PFO may be identified by transesophageal echocardiography (see Chapter 7). The diagnosis and treatment of right ventricular dysfunction and patient ventilator dysynchrony are described in Chapters 20, 24 and 29.
1 Canver CC, Chanda J. Intraoperative and postoperative risk factors for respiratory failure after coronary bypass. Ann Thorac Surg. 2003;75:853-857. discussion 857-858
2 Engoren M, Buderer NF, Zacharias A, et al. Variables predicting reintubation after cardiac surgical procedures. Ann Thorac Surg. 1999;67:661-665.
3 Bernard GR, Artigas A, Brigham KL, et al. The American-European Consensus Conference on ARD S. Definitions, mechanisms, relevant outcomes, and clinical trial coordination. Am J Respir Crit Care Med. 1994;149:818-824.
4 Ufberg JW, Bushra JS, Karras DJ, et al. Aspiration of gastric contents: association with prehospital intubation. Am J Emerg Med. 2005;23:379-382.
5 McClave SA, DeMeo MT, DeLegge MH, et al. North American Summit on Aspiration in the Critically Ill Patient: consensus statement. J Parenter Enter Nutr. 2002;26:S80-S85.
6 Medalion B, Katz MG, Cohen AJ, et al. Long-term beneficial effect of coronary artery bypass grafting in patients with COPD. Chest. 2004;125:56-62.
7 Samuels LE, Kaufman MS, Morris RJ, et al. Coronary artery bypass grafting in patients with COPD. Chest. 1998;113:878-882.
8 Cohen A, Katz M, Katz R, et al. Chronic obstructive pulmonary disease in patients undergoing coronary artery bypass grafting. J Thorac Cardiovasc Surg. 1995;109:574-581.
9 Mathew JP, Fontes ML, Tudor IC, et al. A multicenter risk index for atrial fibrillation after cardiac surgery. JAMA. 2004;291:1720-1729.
10 Wilson R, Grossman R. Introduction: the role of bacterial infection in chronic bronchitis. Semin Respir Infect. 2000;15:1-6.
11 Laws D, Neville E, Duffy J. BTS guidelines for the insertion of a chest drain. Thorax. 2003;58(suppl 2):ii53-ii59.
12 Wilcox P, Baile EM, Hards J, et al. Phrenic nerve function and its relationship to atelectasis after coronary artery bypass surgery. Chest. 1988;93:693-698.
13 Markand ON, Moorthy SS, Mahomed Y, et al. Postoperative phrenic nerve palsy in patients with open-heart surgery. Ann Thorac Surg. 1985;39:68-73.
14 Hagen PT, Scholz DG, Edwards WD. Incidence and size of patent foramen ovale during the first 10 decades of life: an autopsy study of 965 normal hearts. Mayo Clin Proc. 1984;59:17-20.
15 Nunn JF. Applied Respiratory Physiology, ed 3, Butterworth: Philadelphia, 1987.
16 Ware LB, Matthay MA. Clinical practice: acute pulmonary edema. N Engl J Med. 2005;353:2788-2796.
[/level-membership-for-cardiothoracic-surgery-category][not-level-membership-for-cardiothoracic-surgery-category]
Chapter 27 Acute Respiratory Failure
Acute respiratory failure is the inability to maintain adequate gas exchange. It is defined by abnormalities of arterial oxygen (Pao2) and carbon dioxide (Paco2) tensions. Hypoxemia may occur with a normal or low Paco2 (type 1 respiratory failure) or with an elevated Paco2 (type 2 respiratory failure). Respiratory failure is commonly associated with respiratory distress, which manifests as tachypnea, dyspnea, and the use of the accessory muscles of breathing.
Respiratory failure is relatively common in cardiac surgery patients and has a wide variety of causes (Table 27-1). The need for prolonged ventilatory support (>72 hours) occurs in approximately 6% of patients undergoing coronary artery bypass graft surgery,1 and reintubation is required in approximately 3% of patients.2
Table 27-1 Common Causes of Postoperative Respiratory Failure Hypoxemia in Patients After Cardiac Surgery
| On Arrival in the ICU and During Mechanical Ventilation |
| Pulmonary edema |
| Atelectasis/lobar collapse |
| Endobronchial intubation |
| Pneumothorax |
| Hemothorax |
| Ventilator dysynchrony |
| Bronchospasm |
| Low SVO2 (low cardiac output,* anemia) |
| Immediately Following Extubation |
| Residual sedative or opioid drugs |
| Residual neuromuscular blockade |
| Atelectasis |
| Upper airway obstruction/edema |
| Late Deterioration in Respiratory Function |
| Cardiac tamponade (following removal of pacing wires) |
| Pneumothorax (following removal of chest drains) |
| Left ventricular dysfunction |
| Arrhythmias (particularly atrial fibrillation) |
| Atelectasis and lobar collapse |
| Sepsis (pneumonia, mediastinitis) |
| Pulmonary embolus |
| Pulmonary edema |
ICU, intensive care unit.
* Causes of low cardiac output in the early period after cardiac surgery include all conditions listed in Table 20-4 with the exception of low systemic vascular resistance.
This chapter is divided into three sections: (1) mechanisms of respiratory failure; (2) diagnosis and treatment of respiratory failure; (3) specific causes of respiratory failure. Only an overview of respiratory failure is provided here. The physiological mechanisms of respiratory failure are explored in greater detail in Chapter 1, and the ventilatory treatment is described in Chapters 28 and Chapter 29.
MECHANISMS OF RESPIRATORY FAILURE
Central Nervous System and Neuromuscular Dysfunction
Dysfunction of the CNS or neuromuscular system can lead to hypoventilation, loss of protective airway reflexes, and impaired swallowing. Hypoventilation causes hypercarbia and, if severe, hypoxemia. Impaired swallowing and loss of protective airway reflexes predispose a patient to pulmonary aspiration.
Exacerbating Factors
With normal lung function, the main determinant of Pao2 is alveolar oxygen tension (PAo2). However, with intrapulmonary shunting, systemic venous blood passes through the lungs without being oxygenated, and SVo2 becomes an important determinant of Pao2. Low SVo2 can arise from anemia, low cardiac output, and high metabolic rate. This is illustrated in Figure 27-1, which shows the relationship between cardiac output and arterial oxygenation at various levels of intrapulmonary shunting.
APPROACH TO PATIENTS WITH ACUTE RESPIRATORY DISTRESS
Diagnosis
Examination
If appropriate, a targeted neurologic assessment should be performed, focusing on level of consciousness (Glasgow coma scale), cough, gag reflex, and focal neurologic deficit (see Chapter 37).
Investigations
Blood Gas Analysis and Oximetry.
In addition to measurement of arterial oxygen saturation (Sao2), Pao2, and Paco2, useful information is obtained from (1) calculation of the alveolar-arterial (A-a) Po2 gradient, (2) measurement of Svo2, and (3) assessment of metabolic parameters (pH, bicarbonate, base excess, and lactate; see Chapters 1 and Chapter 31).
The A-a gradient is the difference between PAo2 and Pao2, where PAo2 is calculated from the alveolar air equation (see Equation 1-18). With healthy lungs breathing air, the A-a gradient is very low, less than 1 κPa (<7.5 mmHg). However, the A-a gradient is influenced by age and FIo2. At age 40, the upper limit of normal is about 3 κPa (23 mmHg), increasing to 5 κPa (38 mmHg) at age 80 (when breathing room air). To this must be added 0.75 to 1 κPa (6 to 7.6 mmHg) for every 10% increase in FIo2. Within these limitations, the A-a gradient can be used to differentiate hypoventilation (normal A-a gradient) from intrapulmonary shunting (elevated A-a gradient).
Chest Radiography.
The chest radiograph (see Chapter 6) is essential in the assessment of respiratory failure. If possible, the film should be obtained while the patient is erect because this position improves the diagnostic yield, particularly in terms of pleural collections and pneumothoraces. In a patient with respiratory failure, a normal chest radiograph is consistent with the diagnoses of hypoventilation, microatelectasis, low SVo2, and shunting across a PFO.
Hemodynamic Monitoring and Echocardiography.
Hemodynamic evaluation (see Chapter 8) is an integral part of the assessment of respiratory failure. A PAC allows for quantification of cardiac output, SVo2, and pulmonary artery wedge pressure (PAWP). PAWP helps to determine the cause of pulmonary edema (see subsequent material). Echocardiography (see Chapter 7) reliably identifies the cause of low cardiac output (see Chapter 20) and can be used to identify a PFO.
Treatment
Treatment of acute respiratory failure is both supportive and specific to the underlying condition. Specific treatments are discussed in specific causes of respiratory failure. Supportive treatments are listed subsequently. The treatment goals are outlined in Table 27-2.
Table 27-2 Therapeutic Aims for Managing Acute Respiratory Failure
| SaO2 ≥ 90% (approximately 7 kPa or 55 mmHg) |
| pH >7.25 |
| Respiratory rate <30 to 35/min* |
| Ability to speak in short sentences |
| Hemodynamic stability |
| Ability to protect airway† |
* Higher respiratory rates cannot usually be sustained indefinitely; exhaustion and respiratory arrest are likely to supervene.
† Respiratory failure severe enough to cause a fall in the level of consciousness such that the patient cannot obey commands mandates endotracheal intubation to protect against pulmonary aspiration.
Supplemental Oxygen
All patients who are hypoxemic (Pao2 <8 to 10 κPa or 60 to 75 mmHg) should receive supplemental oxygen by nasal cannula or face mask. This includes patients with COPD (see subsequent material). Oxygen is very effective in treating hypoxemia due to hypoventilation but is less effective when it is due to V/Q mismatch. Supplemental oxygen may mask severe hypoventilation, emphasizing the importance of monitoring respiratory rate, level of consciousness, and Paco2 (or end-tidal CO2) in addition to Spo2. Oxygen therapy is discussed further in Chapter 28.
Positive Pressure Ventilation
Positive pressure ventilation is used to improve gas exchange and reduce the work of breathing. It may be invasive (see Chapter 29) or noninvasive (see Chapter 28).
The need for ventilatory support depends on the severity of the respiratory failure, the rate of deterioration, the underlying disease process, and the need to protect the airway. When respiratory failure develops rapidly or occurs in the context of multiple organ dysfunction syndrome, early institution of invasive ventilation is indicated. However, if respiratory failure develops slowly and is not associated with severe metabolic derangement or other organ failure, urgent intubation may not be required, and other targeted therapy (e.g., antibiotics, physical therapy, diuretics) may be tried first. The signs of severe respiratory failure that, if not rapidly corrected, indicate the need for urgent intubation and ventilation are listed in Table 27-3.
Table 27-3 Features of Severe Respiratory Failure That Indicate the Need for Urgent Endotracheal Intubation
| Respiratory System |
| SaO2 ≤ 85% (approximately 6.5 kPa or 50 mmHg) with high-flow face-mask oxygen or an FIO2 ≥ 0.6 with noninvasive ventilation |
| Rising PaCo2* with respiratory acidemia (pH < 7.1) |
| Respiratory distress: respiratory rate >40/min, intercostal in-drawing, use of accessory muscles, the inability to speak more than one or two words between breaths |
| Cardiovascular System |
| Heart rate >120 beats/min |
| Falling blood pressure or cardiac output or escalating inotrope requirements |
| Increasing lactic acidosis |
| Central Nervous System |
| Confusion and agitation |
