[level-membership-for-pulmolory-and-respiratory-category]
Chapter 36 Acute Respiratory Distress Syndrome
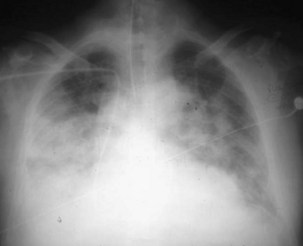
ARDS usually occurs in previously healthy people. Characteristically, there is a latent period between the insult and the development of the full-blown clinical syndrome, which usually is 18 to 24 hours in duration. After this interval, tachypnea, labored breathing, and cyanosis are observed, and arterial blood gas analysis confirms hypoxemia. The abnormalities in lung mechanics and oxygenation are assessed once the patient is intubated and receiving mechanical ventilation. The chest radiograph classically shows diffuse, bilateral, interstitial alveolar infiltrates (Figure 36-1). Resolution of the infiltrates, if it occurs at all, is much slower than with cardiogenic pulmonary edema.
Pathophysiology, Histopathology, and Etiology
The loss of epithelial integrity in ARDS has several consequences. First, under normal conditions, the epithelial barrier is much less permeable than the endothelial barrier; thus epithelial injury can contribute to alveolar flooding. Second, the loss of epithelial integrity and injury to type II cells serve to disrupt normal epithelial fluid transport, impairing the removal of edema fluid from the alveolar space. Third, injury to type II cells reduces the production and turnover of surfactant. Fourth, loss of epithelial barrier can lead to sepsis in patients with bacterial pneumonia. Finally, in severe alveolar epithelium injury, pulmonary fibrosis can develop. Independent of the clinical disorders associated with ARDS (Box 36-1), it is useful to think of the pathogenesis of ARDS as a result of two different pathways: a direct insult to lung cells and an indirect insult occurring as a result of an acute systemic inflammatory response. The host’s inflammatory response to the initial direct (pulmonary) or indirect (nonpulmonary) insult is a key factor in determining the development and progression of the acute injury to the lung. Despite ongoing elucidation of the role of cellular and humoral components of the inflammatory responses in the lung, the precise sequence of events leading to lung damage is still unknown. As with any form of inflammation, ALI during ARDS represents a complex process in which multiple cellular signaling pathways can propagate or inhibit damage to the lung.













Definition, Incidence, and Severity
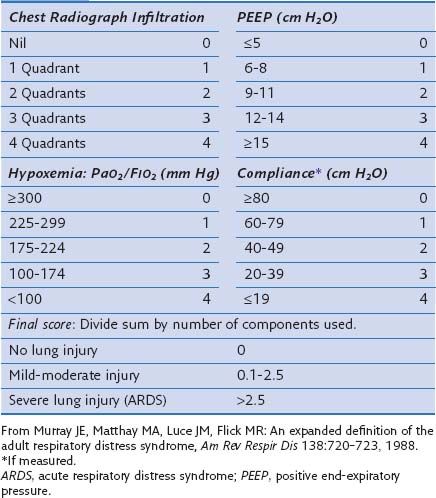
In an attempt to overcome some of these problems, Murray and colleagues proposed an expanded definition of ARDS that takes into account various pathophysiologic features of the syndrome. Their definition uses a “lung injury score” (LIS) to characterize the acute pulmonary damage by considering four components: assessment of the chest radiograph, degree of hypoxemia (determined as the ratio of arterial partial pressure of oxygen to the fraction of inspired oxygen, PaO2/FIO2), level of PEEP, and the value of lung compliance, when available (Table 36-1). The final injury score is obtained by dividing the total score by the number of components that were used. A score of 0 indicates no lung injury, a score of 1 to 2.5 indicates mild to moderate lung injury, and a score greater than 2.5 indicates severe lung injury or ARDS. The LIS is not specific for ARDS, however, and has not been validated, because it is not clear whether patients with identical LIS have similar degrees of lung injury. Furthermore, patients with a major component of cardiogenic edema may be mislabeled as having ARDS, and a postoperative patient with moderate atelectasis and mild fluid overload may fit the LIS criteria for ARDS.
Table 36-1 Lung Injury Scoring System
According to these guidelines, ALI exists when the PaO2/FIO2 ratio is 300 mm Hg or less regardless of the level of PEEP and FIO2, and ARDS is present when the PaO2/FIO2 ratio is 200 mm Hg or less regardless of the PEEP setting and FIO2 (Box 36-2). Although this definition formalized the criteria for the diagnosis of ARDS and is simple to apply in the clinical setting, it has been challenged over the years in several studies. Such definitions have limitations: The physiologic thresholds do not require standardized ventilatory support, and the use of PEEP can improve oxygenation indices sufficiently to convert the patient’s status from meeting the definition of ARDS to not meeting the ARDS definition and also can change the physiology in the lung such that the patient does not meet the criteria for ARDS. Therefore, the ARDS criteria may be met when the PaO2 is measured with zero PEEP but not when measured at a PEEP of 5 or 10 cm H2O, making patient comparisons difficult. Furthermore, most of the randomized controlled studies did not use the same definition for ARDS, nor did they evaluate the same ventilatory approaches. Diversity among ARDS definitions is apparent in a large number of studies (Table 36-2).




Table 36-2 Definitions of Acute Respiratory Distress Syndrome in Several Published Reports
| Published Study | Criteria |
|---|---|
| Montgomery et al: Am Rev Respir Dis 132:485–489, 1985 | PaO2/FIO2 <150 mm Hg |
| PCP <18 mm Hg | |
| Villar et al: Am Rev Respir Dis 140:814–816, 1989 | PaO2 ≤75 mm Hg on FIO2 ≥0.5 |
| PCP <18 mm Hg | |
| Bone et al: Chest 96:849–851, 1989 | PaO2/FIO2 ≤150 mm Hg (with ZEEP) |
| OR PaO2/FIO2 ≤250 mm Hg with PEEP | |
| PCP ≤18 mm Hg | |
| Amato et al: N Engl J Med 338:347–354, 1998 | Lung injury score ≥2.5 and PCP <16 mm Hg |
| Stewart et al: N Engl J Med 338:355–361, 1998 | PaO2/FIO2 <250 mm Hg on PEEP of 5 cm H2O |
| Brochard et al: Am J Respir Crit Care Med 158:1831–1838, 1998 | Lung injury score >2.5 |
| Villar et al: Intensive Care Med 25:930–935, 1999 | PaO2/FIO2 ≤150 mm Hg on PEEP ≥5 cm H2O |
| ARDSNet: N Engl J Med 342:1301–1308, 2000 | AECC |
| Gattinoni et al: N Engl J Med 345:568–573, 2001 | PAO2/FIO2 ≤200 mm Hg on PEEP ≥5 cm H2O |
| PCP ≤18 mm Hg | |
| Villar et al: Crit Care Med 34:1311–1318, 2006 | PAO2/FIO2 ≤200 mm Hg on PEEP ≥5 cm H2O and FIO2 ≥0.5 |
| Meade et al: JAMA 299:637–645, 2008 | PAO2/FIO2 <250 mm Hg |
| Mercat et al: JAMA 299:646–655, 2008 | PAO2/FIO2 ≤200 mm Hg PCP ≤18 mm Hg |
AECC, American-European Consensus Conference; PCP, pulmonary capillary pressure; PEEP, positive end-expiratory pressure; ZEEP, zero PEEP.
Genetics
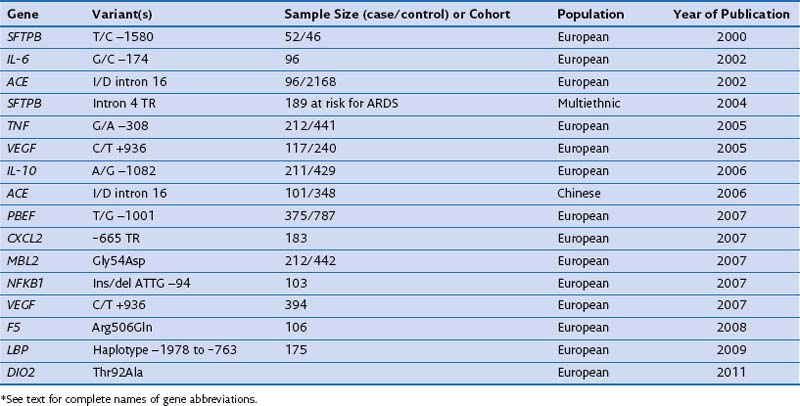
In case-control studies of patients with severe sepsis and ARDS, several investigators have explored variants of the gene encoding the lipopolysaccharide-binding protein (LBP) and serial measurements of the LBP in serum to relate them with risk gene variants. It has been reported that (1) a four-SNP risk haplotype of the LBP gene is associated with mean serum LBP concentrations within the first week of the disease process; (2) LBP levels at 48 hours are much higher in patients with ARDS than in those with ALI; and (3) a subsequent increase of LBP levels at 48 hours is associated with a four-fold increase in mortality rate. A positive association with ARDS susceptibility and/or outcome has been reported for several other genes, including surfactant pulmonary-associated protein B (SFTPB), angiotensin-converting enzyme (ACE), tumor necrosis factor (TNF), vascular endothelial growth factor (VEGF), IL-10, pre-B cell–enhancing factor (PBEF), chemokine CXC motif ligand 2 (CXCL2), mannose-binding lectin-2 (MBL2), myosin light chain kinase (MLCK), nuclear factor κ light polypeptide gene enhancer in B cells (NFKB1), coagulation factor V (F5), and type 2 deiodinase (DIO2) (Table 36-3). These genes are involved mainly in the response to external stimulus and cell signal transduction.
Ventilator-Induced Lung Injury
Ventilators are intended to deliver air or oxygen at tidal volumes and frequencies sufficient to provide adequate alveolar ventilation, to reduce the work of breathing, and to enhance oxygenation (see also Chapter 32). However, mechanical ventilation is a nonphysiologic process, and complications are associated with its application, including increased risk for pneumonia, impaired cardiac performance, and lung injury. During mechanical ventilation, pressures, gas volumes, ventilatory rates, and concentrations of inspired oxygen often are applied at levels that exceed those normally experienced by healthy lungs during spontaneous breathing. VILI is not a new concept—it is the designation historically applied to macroscopic injuries associated with alveolar rupture due to overdistention resulting from application of high inspiratory pressures. Clinical manifestations include interstitial emphysema, pneumothorax, pneumomediastinum, and pneumoperitoneum. The concept of VILI has shifted somewhat from pressure-induced (really volume-induced) injury to increased vascular permeability, accumulation of lung fluid, “atelectrauma,” and inflammation induced by mechanical ventilation.
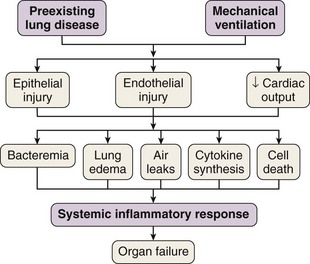
In 1998 Tremblay and Slutsky coined the term biotrauma to describe the pulmonary and systemic inflammatory response triggered by lung cell distention, alveolar disruption, and/or necrosis after the application of mechanical ventilation. Although a Consensus Conference in 1994 recommended that plateau pressure generally should be limited to 35 cm H2O, little change in ventilator practice occurred until publication of an ARDS Network study demonstrating that a lung-protective strategy using a tidal volume of 6 mL/kg of predicted body weight and moderate levels of PEEP decreased mortality in patients with ALI. This study confirmed that VILI was not just an interesting experimental entity but also was an important clinical entity. This recognition led to the widespread, albeit not universal, use of lung-protective strategies in patients with ALI. Unequivocal evidence from both experimental and clinical data has proved that mechanical ventilation can cause or aggravate ALI. Many of the pathophysiologic consequences of VILI mimic those of ARDS. A number of specific forms of injury caused by the trauma of mechanical ventilation have been identified: barotrauma, volutrauma, atelectrauma, and biotrauma. Current experimental and clinical evidence supports a link between VILI and the development of extrapulmonary organ dysfunction, by mechanisms similar to those whereby most severe cases of sepsis are clinically manifested (Figure 36-2).
Pathophysiology of Ventilator-Induced Lung Injury
Lung volumes for all mammals scale with a common function based on body mass. In spontaneously breathing mammals, tidal volumes are approximately 6 to 7 mL/kg of body weight (Figure 36-3), yet historically, tidal volumes of 12 to 15 mL/kg were used in mechanically ventilated patients with acute respiratory failure, and peak alveolar pressures were allowed to increase above 40 cm H2O. This “one tidal volume fits all” approach was formulated in the 1960s, because anesthesiologists and critical care pioneers had demonstrated that ventilation with small tidal volume resulted in a gradual loss of lung volume (i.e., atelectasis) and hypoxemia. Large-tidal-volume ventilation was useful to prevent this negative physiologic consequence. Since the mid-1970s, experimental studies have described the onset of pulmonary edema in rats and larger mammals ventilated with high peak alveolar pressures.
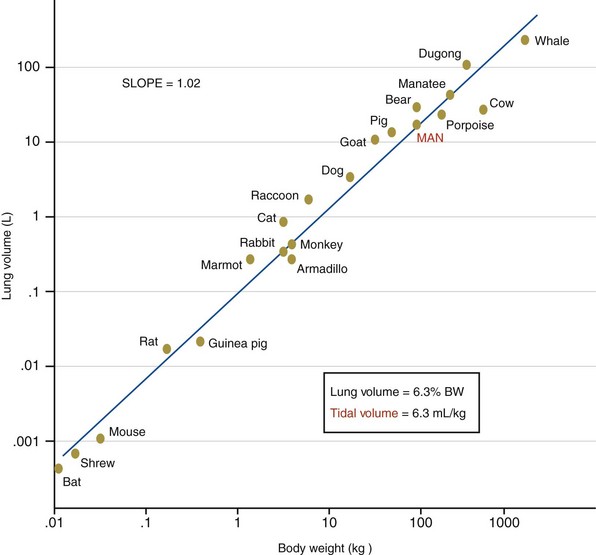
Figure 36-3 Scaling of the lung in mammals. BW, body weight.
(From Villar J, Kacmarek RM, Hedenstierna G: From ventilator-induced lung injury to physician-induced lung injury: why the reluctance to use small tidal volumes? Acta Anaesth Scand 48:267–271, 2004.)
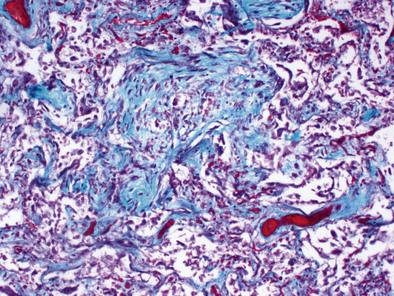
Mechanical ventilation with large tidal volumes results in extracellular matrix remodeling in patients. This effect suggests a potential contributory role of mechanical ventilation in causing lung fibrosis (Figure 36-4). In elective surgical patients, mechanical ventilation with a tidal volume of 12 mL/kg and no PEEP promote procoagulant changes, leading to fibrin deposition within airways; such changes can be prevented by use of low VT and high-level PEEP.
Multiple System Organ Dysfunction: A Common End for Ventilator-Induced Lung Injury and Sepsis
Multiple system organ dysfunction is a cumulative sequence of organ dysfunctions in patients suffering from the same diseases often found in patients with ARDS. Still to be resolved is why in most studies on sepsis, at least a third of the patients do not have a truly identified source of the septic process. Among several possible reasons, one of the most frequently offered is that antibiotics, administered early, have suppressed the growth of bacteria, as borne out by results of blood culture. It is plausible that mechanical ventilation could be partially responsible for the development of sepsis or a sepsis-like syndrome in critically ill patients, even if blood cultures are negative. Several investigators have suggested that the mechanical ventilation–induced inflammatory response may be a contributing factor in multiple system organ dysfunction by initiating or propagating a malignant, systemic inflammatory response (see Figure 36-2). ARDS and acute kidney injury are frequent complications in critically ill patients with multiple system organ dysfunction. Although the functional changes seen in septic patients are known to affect primarily the lungs, the cardiovascular system, and the kidneys, the gut has long been established as an important component of the immune barrier. An emerging consensus implicates the gut flora in the pathogenesis of sepsis. The gut is a reservoir of pathogens and pathogen-associated molecular patterns from microbes, and translocation of these molecular patterns and toxins other than endotoxin also induces a systemic inflammatory response (probably through Toll-like receptors and NFκB signaling pathways), contributing to the development of a systemic infection and multiple organ dysfunction in critically ill patients.
Ventilatory Management for Acute Respiratory Distress Syndrome
The effect of different lung-protective ventilatory strategies in patients with ALI or ARDS has been investigated in randomized controlled trials. In the late 1990s, four such trials were conducted to evaluate the benefit of low tidal ventilation in ARDS patients compared with traditional tidal volume ventilation (Table 36-4). Only one of these trials, performed in Brazil by Amato and colleagues, showed a significant reduction in mortality in the experimental treatment group. Patients randomized to receive tidal volumes of 6 mL/kg of actual body weight or lower and driving pressures of less than 20 cm H2O were significantly less likely to die during the 28-day study period than were patients randomized to receive the traditional 12 mL/kg of actual body weight tidal volumes and unlimited driving pressures. Because all four studies had limited statistical power owing to small sample sizes, a large trial was needed to definitively determine the effect of low-tidal-volume ventilation in patients with ARDS.
Table 36-4 Randomized Controlled Trials Evaluating Pressure- and Volume-Limited (PVL) Ventilation for Patients With Acute Respiratory Distress Syndrome (ARDS)
1. High tidal volumes and high plateau pressures are to be avoided.
2. Tidal volume size should be based on predicted body weight, calculated from gender and height—50 + 0.91 × (height in cm − 152.4) for men and 45.5 + 0.91 × (height in cm − 152.4) for women—rather than on actual body weight.
3. Tidal volumes should be systematically adjusted (from 4 to 8 mL/kg of predicted body weight) to maintain a plateau pressure of 30 cm H2O or less.
4. The respiratory rate should be titrated as needed (over a range of 10 to 35 breaths/minute) to maintain a pH of 7.3 to 7.45.
5. An appropriate combination of FIO2 and PEEP should be used to achieve adequate oxygenation (PaO2 of 55 to 85 mm Hg, or SpO2 of 90% or greater).
The Use of Positive End-Expiratory Pressure in Acute Respiratory Distress Syndrome
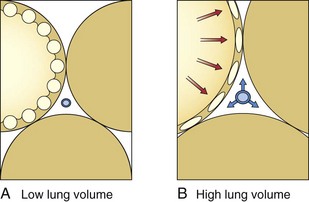
PEEP has become an essential component of the care of many critically ill patients who require ventilatory support. The rationale for the use of PEEP in ALI coincides with the theoretical basis for loss of lung volume and compliance in patients with ARDS. With the application of PEEP, the baseline end-expiratory pressure in mechanically ventilated patients is elevated above atmospheric pressure. The application of PEEP is expected to improve lung mechanics and gas exchange, because it maintains recruited lung volume open. PEEP should be utilized in patients with ARDS to decrease the proportion of nonaerated lung, resulting in improved oxygenation. PEEP prevents complete alveolar collapse and improves oxygenation by increasing functional residual capacity, probably by preventing airway closure and keeping open the previously recruited unventilated alveoli. Conversely, the increase in functional residual capacity also may increase lung compliance. In general, PEEP is applied to improve oxygenation, which usually is not observed except with a concomitant increase in functional residual capacity. The application of PEEP is expected to increase PaO2 and to decrease intrapulmonary shunt, alveolar-arterial O2 pressure difference, and arteriovenous O2 content difference because greater lung volumes are recruited. Four mechanisms have been proposed to explain the improved pulmonary function and gas exchange with PEEP: (1) increased functional residual capacity; (2) alveolar recruitment; (3) redistribution of extravascular lung water; and (4) improved ventilation-perfusion matching. The increase in lung volume is the result of three separate effects. First, PEEP increases lung volume as a result of distention of already patent airways and alveoli by an amount dependent on system compliance. Therefore, the stiffer the system, the smaller the volume change. Second, application of PEEP prevents alveolar collapse during expiration. Dependent, small airways tend to collapse at low lung volumes. Third, PEEP levels above 10 cm H2O recruit collapsed alveoli in the acutely injured lung. Alveolar recruitment describes reinflation of previously collapsed alveoli. The application of PEEP allows recruitment of flooded alveoli and improves oxygenation without diminishing lung water content, supporting the conclusion that PEEP redistributes lung water from alveoli to the perivascular space. In general, by increasing intraalveolar pressure, PEEP moves fluid from the interstitial space of alveolar vessels to the interstitial space around extraalveolar vessels. In addition, PEEP increases the transmural pressure across corner (in the peribronchial area) and extraalveolar vessels, thereby increasing flux into the interstitial space (Figure 36-5).
Although PEEP is well recognized to improve oxygenation in selected patients, its beneficial effects on morbidity and mortality have not been conclusively demonstrated. Early observations that PEEP greatly improves oxygenation in patients with ARDS led to its widespread use in such patients, but the level of PEEP needed to achieve maximum benefit with minimal complications has never been established. Traditionally, PEEP values of 5 to 12 cm H2O have been used in the ventilation of patients with ARDS. In the ARMA trial, PEEP levels in both the experimental and control groups were adjusted according to the required FIO2 and ranged between 5 cm H2O (when FIO2 was 0.3) and 18 to 24 cm H2O (when FIO2 was 1.0) (Table 36-5). On average, patients in the low-tidal-volume ventilation group were treated with levels of PEEP that were no different than those utilized in the high-tidal-volume group. However, it remains unclear whether these values are ideal because randomized controlled trials have not clearly shown that higher levels of PEEP lead to a reduction in mortality rate.
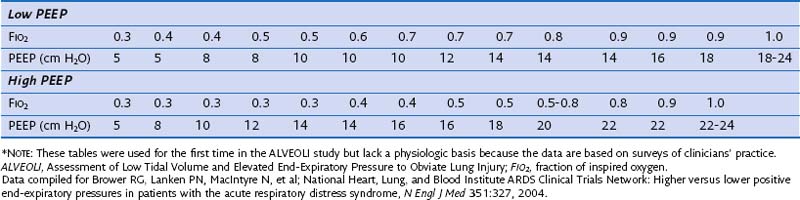
Several randomized controlled trials have evaluated the efficacy of high levels of PEEP in the treatment of ARDS. Thus far, a total of seven published randomized controlled trials have examined the effects of higher levels of PEEP in patients with ALI or ARDS, or both. Those trials have tested higher versus lower PEEP strategies during low-tidal-volume ventilation and lower tidal volume and PEEP titrated to above the lower inflection point of the individual pressure volume curve versus higher-tidal-volume ventilation and lower PEEP (see Table 36-4). At first look, the pooled results of a metaanalysis of these trials suggest that the application of either low or high PEEP levels in patients with ALI or ARDS does not influence outcome. However, this is not what the data show. First, because the ALI-ARDS population was not homogeneous in all the trials, the benefit of higher levels of PEEP could not be appropriately evaluated. Second, in some trials, a PEEP level of 5 cm H2O was a permissible setting in the higher PEEP group. Therefore, a patient with a PaO2 of 60 mm Hg on a FIO2 of 0.3 and 5 cm H2O of PEEP satisfied the AECC inclusion criteria. It is difficult to accept that in these patients there is a need to test the effects of high levels of PEEP. Finally, some trials (e.g., the Assessment of Low Tidal Volume and Elevated End-Expiratory Pressure to Obviate Lung Injury [ALVEOLI] and Expiratory Pressure [ExPress] trials) (see Table 36-4) included patients with ALI (without ARDS), who did not benefit and more often experienced adverse effects from higher levels of PEEP, suggesting that both trials failed to focus on the patients at highest risk. If the subjects in a trial have a very low risk for the condition that the intervention is hypothesized to prevent, the trial—regardless of sample size—will not verify the value of the experimental intervention under study. In the study of a protective ventilation strategy by Amato and Villar and their respective colleagues, PEEP was significantly higher in the intervention group than in the control group (13 to 14 cm H2O versus 9 cm H2O, respectively). Although both trials reported a significantly lower mortality in the intervention group, these findings cannot be solely attributed to higher levels of PEEP, because the intervention strategies in these trials also included both low tidal volumes and high levels of PEEP.
The speculation of many investigators regarding the lack of expected benefit from higher PEEP in the ALVEOLI, Lung Open Ventilation (LOV), and ExPress trials (see Table 36-4) is that in a substantial proportion of patients in those trials, the severity of lung injury was modest. In two other trials, Amato and colleagues and Villar and associates enrolled only patients with severe, established ARDS, and both groups found that the application of higher levels of PEEP was associated with a better outcome. By contrast, in the rest of the trials, the investigators studied an unselected, mixed population of ALI and ARDS and, as a result, missed the opportunity to test whether the use of higher levels of PEEP is beneficial in patients with persistent ARDS. Patients in the ALVEOLI, LOV, and ExPress trials had similar PaO2/FIO2 ratios at study entry (about 140 ± 50 mm Hg). It is conceivable, however, that a disproportionate number of patients meeting ALI criteria on standard ventilatory settings ended up in the control group, negating the beneficial effect of the treatment because of a lower mortality rate (20% or less in most series). A critical review of the two most recently published trials on the effects of PEEP in ARDS leads to an alternative interpretation. In both trials (the LOV study and the ExPress trial), patients who received higher PEEP were less likely to require rescue therapy. In addition, a lower PEEP was associated with significantly fewer ventilator-free and organ failure-free days, which may be the reason for a 4% mortality difference in favor of the high PEEP groups (P = .10).
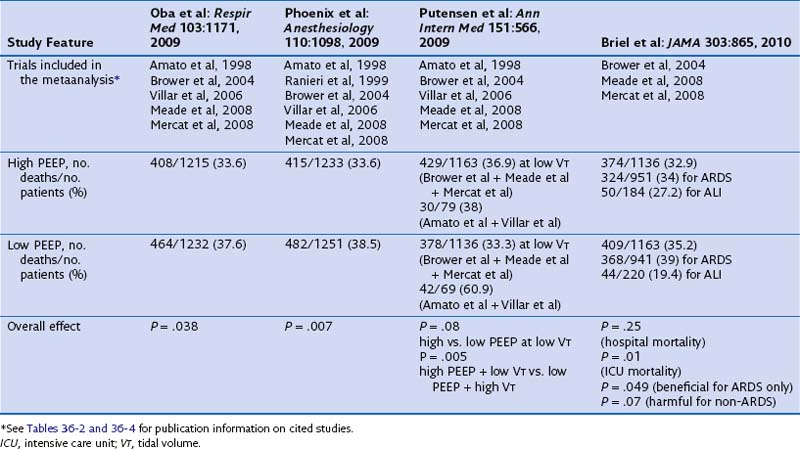
Of note, current published metaanalyses including some or all of the available trials reported conflicting results and are biased because they do not deal with all comparisons between tidal volume and PEEP (Table 36-6). The first metaanalysis from Oba and colleagues, which considered five trials (reported by Amato, Brower, Meade, Mercat, and Villar and their co-workers), showed a decrease in in-hospital mortality (P = .03) and a trend toward significance for the 28-day mortality (P = .06) with higher PEEP and without an increase in barotrauma. A second meta-analysis, by Phoenix and colleagues, included an additional study (by Ranieri and co-workers) and showed a reduction in mortality (pooling together the in-hospital and the 28-day mortality rates, P = .007) with higher PEEP. In a subsequent metaanalysis, Putensen and colleagues analyzed the five trials included in the Oba metaanalysis. Data from three of these trials—those reported by Brower, Meade, and Mercat and their co-workers—were pooled together for comparison of lower versus higher PEEP at low tidal volume ventilation. The remaining two studies (by Amato and Villar and their co-workers) were used for the comparison of higher tidal volume and lower PEEP versus lower tidal volume and higher PEEP. Despite opposite results from the two previous metaanalyses, the comparison between lower and higher levels of PEEP at low tidal volume ventilation showed similar hospital mortality rates (P = .08). However, the risk of in-hospital mortality was reduced only with the combination of a lower tidal volume and higher PEEP, compared with higher tidal volume and lower PEEP (P = .005). Thus, the beneficial effect of PEEP may have been due to a simultaneous reduction in the tidal volume and an increase in the level of PEEP. Finally, a fourth metaanalysis from Briel and colleagues (see Table 36-6) was performed on the individual data for 1136 patients (with higher PEEP) and 1163 patients (with lower PEEP) from trials that compared lower and higher PEEP values at a low-tidal-volume ventilation. The individual data analysis showed that a nonsignificant difference was found in hospital mortality rates between the higher and lower PEEP groups (32.9% versus 35.2%; P = .25). In conclusion, these metaanalyses of individual patient data suggest that higher levels of PEEP may be associated with lower mortality in patients who meet ARDS criteria, whereas such a benefit is unlikely in patients who have less severe lung injury.
Rescue Strategies for Refractory Hypoxemia
Supportive Treatment
Controversies and Pitfalls
The use of simple thresholds for the diagnosis of disease processes of increasing prevalence in the general population is common. This is the case with the use of blood sugar for diabetes and hemoglobin for anemia. It appears improbable that in the case of ARDS, a biomarker alone will resolve this issue. Instead, a clinical prediction model or a combination of such a predictor model with a biomarker would provide a better definition of ALI and ARDS (Figure 36-6). Ideally, such a biomarker should be (1) 100% sensitive, (2) 100% specific, (3) easy to measure in blood, exhaled air, or any other biologic sample, (4) affected by treatment, and (5) cost-effective. There have been recent efforts to identify biologic markers in pulmonary edema fluid and in blood collected from patients with and without ARDS. It has been postulated that owing to increased permeability of the alveolar-capillary barrier, proteins leak into the circulation. Patients at risk for ARDS who have higher levels of interleukin-8 in bronchoalveolar lavage fluid subsequently progress to ARDS. Serial LBP measurements may offer a clinically useful biomarker for identification of patients likely to experience the worst outcomes and with the highest probability for development of sepsis-induced ARDS. A combination of biologic markers that reflect endothelial and epithelial pulmonary injury, inflammation, and coagulation will be superior to clinical predictors or biomarkers alone for predicting mortality or stratifying patients with ALI or ARDS. Finally, serial elevation of plasma levels of functionally relevant cell proteins (above a critical threshold) could be used for the differential diagnosis for ALI and ARDS. The measure of those biomarkers may inform clinicians of the development of ALI or ARDS.
Ashbaugh DG, Bigelow DB, Petty TL, Levine BE. Acute respiratory distress in adults. Lancet. 1967;2:319–323.
Bernard GR, Artigas A, Brigham KL, et al. Consensus Committee: The American-European Consensus Conference on ARDS. Am J Respir Crit Care Med. 1994;149:818–824.
Briel M, Meade M, Mercat A, et al. Higher vs lower positive end-expiratory pressure in patients with acute lung injury and acute respiratory distress syndrome: systematic review and meta-analysis. JAMA. 2010;303:865–873.
Murray JE, Matthay MA, Luce JM, Flick MR. An expanded definition of the adult respiratory distress syndrome. Am Rev Respir Dis. 1988;138:720–723.
Sud S, Friedrich JO, Taccone P, et al. Prone ventilation reduces mortality in patients with acute respiratory failure and severe hypoxemia: systematic review and meta-analysis. Intensive Care Med. 2010;36:585–599.
Tomashefski JFJr. Pulmonary pathology of acute respiratory distress syndrome. Clin Chest Med. 2000;21:435–466.
2000 Ventilation with lower tidal volumes as compared with traditional tidal volumes for acute lung injury and the acute respiratory distress syndrome. The Acute Respiratory Distress Syndrome Network. N Engl J Med. 2000;342:1301–1308.
Villar J, Kacmarek RM, Pérez-Méndez L, Aguirre-Jaime A. A high positive end-expiratory pressure, low tidal volume ventilatory strategy improves outcome in persistent acute respiratory distress syndrome: a randomized, controlled trial. Crit Care Med. 2006;34:1311–1318.
Villar J, Pérez-Méndez L, Basaldúa S, et al. HELP network: Age, plateau pressure and PaO2/FIO2 at ARDS onset predict outcome. Respir Care. 56, 2011.
Villar J, Pérez-Méndez L, Kacmarek RM. Current definitions of acute lung injury and the acute respiratory distress syndrome do not reflect their true severity and outcome. Intensive Care Med. 1999;25:930–935.
Villar J, Pérez-Méndez L, López J, et al. HELP Network: An early PEEP/FIO2 trial identifies different degrees of lung injury in patients with acute respiratory distress syndrome. Am J Respir Crit Care Med. 2007;176:795–804.
Ware LB, Matthay MA. The acute respiratory distress syndrome. N Engl J Med. 2000;342:1334–1349.
[/level-membership-for-pulmolory-and-respiratory-category][not-level-membership-for-pulmolory-and-respiratory-category]
Chapter 36 Acute Respiratory Distress Syndrome
ARDS usually occurs in previously healthy people. Characteristically, there is a latent period between the insult and the development of the full-blown clinical syndrome, which usually is 18 to 24 hours in duration. After this interval, tachypnea, labored breathing, and cyanosis are observed, and arterial blood gas analysis confirms hypoxemia. The abnormalities in lung mechanics and oxygenation are assessed once the patient is intubated and receiving mechanical ventilation. The chest radiograph classically shows diffuse, bilateral, interstitial alveolar infiltrates (Figure 36-1). Resolution of the infiltrates, if it occurs at all, is much slower than with cardiogenic pulmonary edema.
Pathophysiology, Histopathology, and Etiology
The loss of epithelial integrity in ARDS has several consequences. First, under normal conditions, the epithelial barrier is much less permeable than the endothelial barrier; thus epithelial injury can contribute to alveolar flooding. Second, the loss of epithelial integrity and injury to type II cells serve to disrupt normal epithelial fluid transport, impairing the removal of edema fluid from the alveolar space. Third, injury to type II cells reduces the production and turnover of surfactant. Fourth, loss of epithelial barrier can lead to sepsis in patients with bacterial pneumonia. Finally, in severe alveolar epithelium injury, pulmonary fibrosis can develop. Independent of the clinical disorders associated with ARDS (Box 36-1), it is useful to think of the pathogenesis of ARDS as a result of two different pathways: a direct insult to lung cells and an indirect insult occurring as a result of an acute systemic inflammatory response. The host’s inflammatory response to the initial direct (pulmonary) or indirect (nonpulmonary) insult is a key factor in determining the development and progression of the acute injury to the lung. Despite ongoing elucidation of the role of cellular and humoral components of the inflammatory responses in the lung, the precise sequence of events leading to lung damage is still unknown. As with any form of inflammation, ALI during ARDS represents a complex process in which multiple cellular signaling pathways can propagate or inhibit damage to the lung.
Definition, Incidence, and Severity
In an attempt to overcome some of these problems, Murray and colleagues proposed an expanded definition of ARDS that takes into account various pathophysiologic features of the syndrome. Their definition uses a “lung injury score” (LIS) to characterize the acute pulmonary damage by considering four components: assessment of the chest radiograph, degree of hypoxemia (determined as the ratio of arterial partial pressure of oxygen to the fraction of inspired oxygen, PaO2/FIO2), level of PEEP, and the value of lung compliance, when available (Table 36-1). The final injury score is obtained by dividing the total score by the number of components that were used. A score of 0 indicates no lung injury, a score of 1 to 2.5 indicates mild to moderate lung injury, and a score greater than 2.5 indicates severe lung injury or ARDS. The LIS is not specific for ARDS, however, and has not been validated, because it is not clear whether patients with identical LIS have similar degrees of lung injury. Furthermore, patients with a major component of cardiogenic edema may be mislabeled as having ARDS, and a postoperative patient with moderate atelectasis and mild fluid overload may fit the LIS criteria for ARDS.
Table 36-1 Lung Injury Scoring System
According to these guidelines, ALI exists when the PaO2/FIO2 ratio is 300 mm Hg or less regardless of the level of PEEP and FIO2, and ARDS is present when the PaO2/FIO2 ratio is 200 mm Hg or less regardless of the PEEP setting and FIO2 (Box 36-2). Although this definition formalized the criteria for the diagnosis of ARDS and is simple to apply in the clinical setting, it has been challenged over the years in several studies. Such definitions have limitations: The physiologic thresholds do not require standardized ventilatory support, and the use of PEEP can improve oxygenation indices sufficiently to convert the patient’s status from meeting the definition of ARDS to not meeting the ARDS definition and also can change the physiology in the lung such that the patient does not meet the criteria for ARDS. Therefore, the ARDS criteria may be met when the PaO2 is measured with zero PEEP but not when measured at a PEEP of 5 or 10 cm H2O, making patient comparisons difficult. Furthermore, most of the randomized controlled studies did not use the same definition for ARDS, nor did they evaluate the same ventilatory approaches. Diversity among ARDS definitions is apparent in a large number of studies (Table 36-2).
Table 36-2 Definitions of Acute Respiratory Distress Syndrome in Several Published Reports
| Published Study | Criteria |
|---|---|
| Montgomery et al: Am Rev Respir Dis 132:485–489, 1985 | PaO2/FIO2 <150 mm Hg |
| PCP <18 mm Hg | |
| Villar et al: Am Rev Respir Dis 140:814–816, 1989 | PaO2 ≤75 mm Hg on FIO2 ≥0.5 |
| PCP <18 mm Hg | |
| Bone et al: Chest 96:849–851, 1989 | PaO2/FIO2 ≤150 mm Hg (with ZEEP) |
| OR PaO2/FIO2 ≤250 mm Hg with PEEP | |
| PCP ≤18 mm Hg | |
| Amato et al: N Engl J Med 338:347–354, 1998 | Lung injury score ≥2.5 and PCP <16 mm Hg |
| Stewart et al: N Engl J Med 338:355–361, 1998 | PaO2/FIO2 <250 mm Hg on PEEP of 5 cm H2O |
| Brochard et al: Am J Respir Crit Care Med 158:1831–1838, 1998 | Lung injury score >2.5 |
| Villar et al: Intensive Care Med 25:930–935, 1999 | PaO2/FIO2 ≤150 mm Hg on PEEP ≥5 cm H2O |
| ARDSNet: N Engl J Med 342:1301–1308, 2000 | AECC |
| Gattinoni et al: N Engl J Med 345:568–573, 2001 | PAO2/FIO2 ≤200 mm Hg on PEEP ≥5 cm H2O |
| PCP ≤18 mm Hg | |
| Villar et al: Crit Care Med 34:1311–1318, 2006 | PAO2/FIO2 ≤200 mm Hg on PEEP ≥5 cm H2O and FIO2 ≥0.5 |
| Meade et al: JAMA 299:637–645, 2008 | PAO2/FIO2 <250 mm Hg |
| Mercat et al: JAMA 299:646–655, 2008 | PAO2/FIO2 ≤200 mm Hg PCP ≤18 mm Hg |
AECC, American-European Consensus Conference; PCP, pulmonary capillary pressure; PEEP, positive end-expiratory pressure; ZEEP, zero PEEP.
Genetics
In case-control studies of patients with severe sepsis and ARDS, several investigators have explored variants of the gene encoding the lipopolysaccharide-binding protein (LBP) and serial measurements of the LBP in serum to relate them with risk gene variants. It has been reported that (1) a four-SNP risk haplotype of the LBP gene is associated with mean serum LBP concentrations within the first week of the disease process; (2) LBP levels at 48 hours are much higher in patients with ARDS than in those with ALI; and (3) a subsequent increase of LBP levels at 48 hours is associated with a four-fold increase in mortality rate. A positive association with ARDS susceptibility and/or outcome has been reported for several other genes, including surfactant pulmonary-associated protein B (SFTPB), angiotensin-converting enzyme (ACE), tumor necrosis factor (TNF), vascular endothelial growth factor (VEGF), IL-10, pre-B cell–enhancing factor (PBEF), chemokine CXC motif ligand 2 (CXCL2), mannose-binding lectin-2 (MBL2), myosin light chain kinase (MLCK), nuclear factor κ light polypeptide gene enhancer in B cells (NFKB1), coagulation factor V (F5), and type 2 deiodinase (DIO2) (Table 36-3). These genes are involved mainly in the response to external stimulus and cell signal transduction.
Ventilator-Induced Lung Injury
Ventilators are intended to deliver air or oxygen at tidal volumes and frequencies sufficient to provide adequate alveolar ventilation, to reduce the work of breathing, and to enhance oxygenation (see also Chapter 32). However, mechanical ventilation is a nonphysiologic process, and complications are associated with its application, including increased risk for pneumonia, impaired cardiac performance, and lung injury. During mechanical ventilation, pressures, gas volumes, ventilatory rates, and concentrations of inspired oxygen often are applied at levels that exceed those normally experienced by healthy lungs during spontaneous breathing. VILI is not a new concept—it is the designation historically applied to macroscopic injuries associated with alveolar rupture due to overdistention resulting from application of high inspiratory pressures. Clinical manifestations include interstitial emphysema, pneumothorax, pneumomediastinum, and pneumoperitoneum. The concept of VILI has shifted somewhat from pressure-induced (really volume-induced) injury to increased vascular permeability, accumulation of lung fluid, “atelectrauma,” and inflammation induced by mechanical ventilation.
In 1998 Tremblay and Slutsky coined the term biotrauma to describe the pulmonary and systemic inflammatory response triggered by lung cell distention, alveolar disruption, and/or necrosis after the application of mechanical ventilation. Although a Consensus Conference in 1994 recommended that plateau pressure generally should be limited to 35 cm H2O, little change in ventilator practice occurred until publication of an ARDS Network study demonstrating that a lung-protective strategy using a tidal volume of 6 mL/kg of predicted body weight and moderate levels of PEEP decreased mortality in patients with ALI. This study confirmed that VILI was not just an interesting experimental entity but also was an important clinical entity. This recognition led to the widespread, albeit not universal, use of lung-protective strategies in patients with ALI. Unequivocal evidence from both experimental and clinical data has proved that mechanical ventilation can cause or aggravate ALI. Many of the pathophysiologic consequences of VILI mimic those of ARDS. A number of specific forms of injury caused by the trauma of mechanical ventilation have been identified: barotrauma, volutrauma, atelectrauma, and biotrauma. Current experimental and clinical evidence supports a link between VILI and the development of extrapulmonary organ dysfunction, by mechanisms similar to those whereby most severe cases of sepsis are clinically manifested (Figure 36-2).
Pathophysiology of Ventilator-Induced Lung Injury
Lung volumes for all mammals scale with a common function based on body mass. In spontaneously breathing mammals, tidal volumes are approximately 6 to 7 mL/kg of body weight (Figure 36-3), yet historically, tidal volumes of 12 to 15 mL/kg were used in mechanically ventilated patients with acute respiratory failure, and peak alveolar pressures were allowed to increase above 40 cm H2
[/not-level-membership-for-pulmolory-and-respiratory-category]
