[level-membership-for-critical-care-medicine-category]44
Acute Pulmonary Embolism
PREVALENCE OF VENOUS THROMBOEMBOLISM IN INTENSIVE CARE UNIT PATIENTS
Clinical Presentation on Admission to the Intensive Care Unit
Recognition of Pulmonary Embolism During Intensive Care Unit Admission
DIAGNOSTIC TESTING FOR PULMONARY EMBOLISM
Arterial Blood Gas Measurement
Duplex Compression Ultrasonography
Ventilation-Perfusion Nuclear Scans
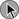
Pulmonary venous thromboembolism (VTE) and deep venous thrombosis (DVT) are different manifestations of the same disease. Despite adequate VTE prophylaxis in the intensive care unit (ICU) the presence of a VTE event is a constant threat to critically ill patients. Although the critical care clinician may encounter the embolization of air, fat, infected clots, amniotic fluid, tumor, and inorganic substances, by far VTE will be the most common pulmonary embolic condition encountered. Critical care clinicians will encounter life-threatening pulmonary embolism (PE) ranging from hemodynamically stable patients with varying degress of right ventricular dysfunction (RVD) to acute massive PE, defined as hemodynamic shock from acute PE, which represents the most serious manifestation along the spectrum of venous thromboembolic disease.1–3 According to population-based studies, the annual incidence of VTE in the United States has ranged from 200,000 to a recent estimate of 900,000 cases in which approximately 300,000 people die every year from acute PE.4,5 More recently, in 2006, 467,000 patients were hospitalized with DVT and 247,000 patients were admitted with acute PE.6 VTE is associated with a significant health care costs. Based on 2004 provider payments, the economic burden of VTE costs ranges between $5.8 billion and $7.8 billion.7
The mortality rate can exceed 58% in patients with acute PE presenting with shock, and most of these deaths occur within 1 hour of presentation.1,2 Acute PE is the third most common cause of death among hospitalized patients, and with an aging population the number of people with VTE is expected to increase. For these reasons, the U.S. Surgeon General issued a “Call to Action” in 2008, identifying VTE as a major public health problem.8
The development of DVT occurs largely in the lower extremities and can result in the sequelae of PE, post-thrombotic syndrome, and chronic thrombotic pulmonary hypertension.9 Venous thrombosis is associated with significant morbidity and mortality rates but can be prevented in most patients.2,3
Prevalence of Venous Thromboembolism in Intensive Care Unit Patients
ICU patients represent a heterogeneous population. In prospective screening studies for DVT, in the absence of thromboprophylaxis, the incidence of DVT in a medical-surgical ICU ranges from approximately 13% to 33%.10–12 In an autopsy review of six studies of 436 critically ill patients the incidence of PE ranged from 7% to 27% (mean 13%); PE contributed or directly caused death in 0% to 12% (mean 3%) of the patients. It is important to note that in a majority of the patients there was no antemortem suspicion of fatal PE.13
Contemporary prevalence of VTE that are admitted to or occur in the ICU setting can be obtained from large VTE clinical trials. Approximately 10% of patients entered into the PIOPED-I (Prospective Investigation of Pulmonary Embolism Diagnosis) study for possible PE were hospitalized in a medical or surgical critical care unit.14 In the recent international, multicenter EINSTEIN PE trial,15 which randomized 4823 symptomatic PE patients to rivaroxiban or enoxaparin and a vitamin K antagonist, 600 patients (12.4%) required ICU admission. Finally, in the PROTECT trial (Prophylaxis for Thromboembolism in Critical Care Trial)16 among 3764 critically ill patients enrolled in which a majority were on mechanical ventilator support and 40% were on vasopressor therapy, dalteparin and unfractionated heparin (UFH) were the agents used for VTE prophylaxis. The primary end point was proximal DVT presence. Using compression ultrasonography (CUS), 3.5% of the patients had proximal DVT on admission. The incidence of proximal DVT was 5.1% in the dalteparin group and 5.4% in the UFH group. The proportion of patients with pulmonary emboli was significantly lower with dalteparin (24 patients, 1.3%) than with UFH (43 patients, 2.3%).16
There is controversy over routine DVT screening in the ICU. As mentioned earlier, most ICU patients with DVT are asymptomatic and the clinical signs for DVT are nonspecific. CUS is time consuming and not cost effective. Efforts must be made to ensure adherence to current DVT prophylaxis guidelines. In ICU patients in whom these guidelines cannot not be fully implemented (intracranial, spinal, or leg injuries, or patients at increased bleeding risk) then routine CUS screening may be appropriate.17–21
Risk Factors
In the nineteenth century Virchow described stasis, vascular wall abnormalities, and hypercoagulability as general risk factors for the development of DVT.22
As illustrated in Table 44.1, patients admitted to the ICU possess multiple preexisting VTE risks factors on admission to the ICU, yet in the International Cooperative Pulmonary Embolism Registry (ICOPER) 20% of the patients had idiopathic or unprovoked PE.3
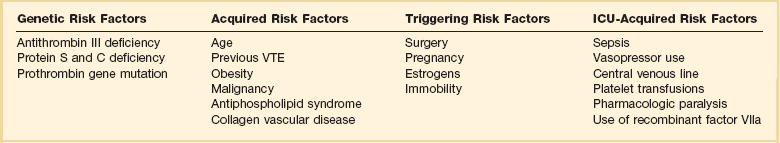
The interaction between patient-related and setting-related risk factors appears to be responsible for the development of the VTE.23–25 Patient-related risk factors are usually acquired and are longstanding. Examples of patient-related risk factors are age, history of prior VTE, malignancy, neurologic and medical illnesses that result in immobility, cardiopulmonary disorders, collagen vascular disease, and vasculitis. In addition, patient-related factors may include genetic and acquired thrombophilia and hormone replacement therapy and oral contraceptive therapy. Setting-related risk factors are usually temporary. Examples of setting-related risk factors include hospitalizations, nursing home placement, and admission to the ICU.
 Virchow has described several risk factors unique to the ICU setting that predispose patients to VTE events. In a prospective observational study of 93 medical-surgical ICU patients, the investigators identified several ICU-related factors that are associated with increased risk of VTE, including mechanical ventilation, immobility, femoral venous catheters, sedatives and paralytic drugs, and failure to prescribe VTE prophylaxis. Prolonged mobility in the ICU setting is the result of mechanical ventilation, sedative administration, and paralytic agents. These four factors (mechanical ventilation, immobility, sedation, and paralysis) may converge to greatly increase the risk of VTE.26 Sedation and paralysis also predispose to other ICU complications including increased duration of mechanical ventilation, and critical illness polyneuropathy.27,28
Virchow has described several risk factors unique to the ICU setting that predispose patients to VTE events. In a prospective observational study of 93 medical-surgical ICU patients, the investigators identified several ICU-related factors that are associated with increased risk of VTE, including mechanical ventilation, immobility, femoral venous catheters, sedatives and paralytic drugs, and failure to prescribe VTE prophylaxis. Prolonged mobility in the ICU setting is the result of mechanical ventilation, sedative administration, and paralytic agents. These four factors (mechanical ventilation, immobility, sedation, and paralysis) may converge to greatly increase the risk of VTE.26 Sedation and paralysis also predispose to other ICU complications including increased duration of mechanical ventilation, and critical illness polyneuropathy.27,28
Deep Venous Thrombosis
It is estimated that as high as 95% of clinically significant pulmonary emboli originate from the deep veins of the lower extremity. The proximal deep veins of the leg are the most common sites of origin of clots that embolize to the pulmonary circulation. A general consensus suggests that clinically significant proximal DVTs and risk for PEs originate by proximal extension of calf DVT.47,48 Venous thrombi are created in the setting of low flow and low shear stress. These thrombi consist of fibrin strands, red blood cells, and platelets. The thrombi form in the valve pockets of calf veins and extend to the proximal veins.48 After the thrombosis is formed there is raised venous capillary pressure, which increases the transcapillary filtration and the pressure rate, resulting in edema. In approximately 50% of patients, venous outflow obstruction decreases within 3 months by lysis and recanalization.49 Patients who have early edema are most likely to have residual thrombosis. Clots that continue to propagate have a greater risk to break apart and lead to embolization. This generally occurs more frequently in the first few days after clot formation. Clots that do not continue to propagate resolve by either fibrinolysis or organization. These processes generally occur within 7 days of clot formation.49,50
In general only 25% of patients with suspected DVT actually have it.47 The detection of DVT in critically ill patients is hampered by the patient’s inability to report symptoms and the unreliability of physical signs of DVT. CUS is noninvasive and highly sensitive and specific for the diagnosis of proximal lower extremity DVT.51 Full compressibility of either the femoral or popliteal veins excludes proximal DVT51 when compared to the gold standard demography; CUS has a sensitivity of 97% to 100% and a specificity of 98% to 99% for the detection of proximal thrombosis. Ultrasonography is less accurate in the diagnosis of distal calf vein thrombosis.51,52 Whether high-risk critically ill patients should be systematically screened for DVT using CUS, given the high incidence of DVT and limitation of clinical examination, is unresolved.13,17
Upper extremity deep venous thrombosis (UEDVT) accounts for approximately 4% of the venous thromboembolic events.53 Primary UEDVT makes up one third of all UEDVTs. Primary UEDVTs appear to be associated with inherited thrombophilic conditions, and the risk markedly increased to 14-fold when oral contraceptives were used.54 Catheter UEDVTs are more associated with the placement of a central venous catheter, and these patients are more likely to be inpatients than outpatients, as demonstrated by a large registry.55 In addition, patients with cancer have an increased occurrence of UEDVT when a central venous catheter is placed in these areas.55 Catheter-related UEDVT may have a higher pulmonary embolic potential than primary UEDVT.52
Pathophysiology
Nearly two thirds of patients who die from pulmonary thromboembolism die within 1 hour of presentation, but anatomically massive pulmonary emboli (>50% obstruction of the pulmonary circulation) are responsible for only half of the deaths. The term major pulmonary thromboembolism has been used to describe any pulmonary thromboembolus that results in a hemodynamically significant event.1 In patients with pulmonary thromboembolism, hemodynamic presentation is an important predictor of survival. The Urokinase Pulmonary Embolism Trial (UPET) demonstrated that the presence of hemodynamic decompensation was associated with a sevenfold increase in mortality rate.56 ICOPER confirmed these results by demonstrating a fourfold increase in mortality rate for those patients with hemodynamic instability.3 Because of the associations between outcome from pulmonary thromboembolism and shock or hypotension, aggressive intervention in patients thought to have pulmonary thromboembolism has given rise to the term golden hour, during which timely diagnosis and treatment are paramount.1
Hemodynamic Consequences
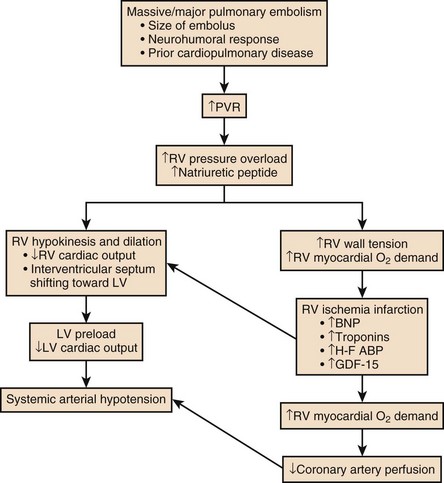
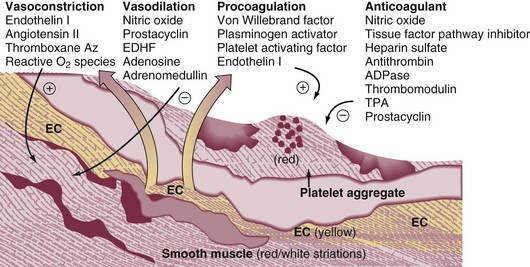
The principal pathophysiologic effects of pulmonary thromboembolism result from the acute impaction of material into pulmonary circulation and the resulting vascular obstruction and humoral mediator release as depicted in Figure 44.1.57 In nonthrombotic obstruction of the pulmonary vasculature, obliteration of 60% to 70% of the vascular tree is required to cause an elevation of the pulmonary artery pressure. In contrast, only 30% of the vascular tree must be obstructed in pulmonary thromboembolism for elevation of the pulmonary artery pressure to be achieved.58,59 Therefore, factors other than simple mechanical obstruction of the pulmonary vascular system play a role in the elevation of the pressures in the pulmonary vasculature during pulmonary thromboembolism.
The production of platelet-derived vasoconstrictors may play a role in augmenting the increase in pulmonary vascular resistance (PVR) (Fig. 44.2).57,58,60 Thromboxane A2 (TxA2) is a potent vasoconstrictor and is the end product of arachidonic acid metabolism. TxA2 is produced by endothelial cells, and even in greater quantities by platelets in response to platelet aggregation. It has been demonstrated that there is increased production of TxA2 in the early phase of PE, which contributes to the PVR elevation and may be attenuated by cyclooxygenase (COX) inhibition in animal models.57 Serotonin (5-hydroxytryptamine) is one of the most potent pulmonary vasoconstrictors. Infusion of serotonin in an animal model can actually simulate the signs and symptoms of PE. Like TxA2, platelets are the primary source of serotonin production in PE. Serotonin antagonism can reduce PVR.57,60
There appears to be nearly an exponential increase in VTE events with age.29 The elderly demonstrate an incidence of 300 to 500 cases/100,000 persons compared to 30 cases/100,000 of persons age 25 to 35 years.29 A longitudinal study of 855 men followed from the ages of 50 to 80 years demonstrated the cumulative incidence of a VTE event was 0.5% by the age of 50 years and increased to 10.7% by the age of 80 years.3 The risk of VTE recurrences increases with age. In the elderly the PE rates are rising faster than the DVT rates; thus, a VTE event in an elderly person is more likely to be a PE, with its potential lethality.3,30
Anderson and Spencer31 reviewed multiple risk factors associated with a diagnosis of VTE and found that the risk factors were not equal in their impact on VTE occurrence. The strongest risk factors were hip and leg fractures, major general surgery, hip or knee replacement, major trauma, and spinal cord injury. Moderate risk factors included arthroscopic knee surgery, central venous lines, chemotherapy, congestive heart failure, hormonal replacement or oral contraceptives, cancer, pregnancy, thrombophilia, and prior VTE.31 Heit and associates32 found that the hospital setting, nursing home, or other chronic facility confinement was an independent risk factor for the VTE. This is most likely reflective of the various degrees of immobility that these patients experience.
Several acquired and inherited hypercoagulable states have been shown to increase the risk of VTE and are collectively known as thrombophilia. Major acquired hypercoagulable conditions include malignancy, myeloproliferative disorders, and antiphospholipid syndrome (APS).33 DVT, the most common manifestation of the APS, occurs in 29% to 55% of patients with the syndrome, and about half of these patients have pulmonary emboli.34–36
The occurrence of a VTE event in a young adult alerts the clinician to the potential of an underlying inheritable thrombophilic condition. Nearly half the patients with a VTE event before the age of 45 have an associated inherited disorder.33 Factor V Leiden mutation causing resistance to activated protein C is the most common inherited risk factor. Factor V Leiden mutation is present in up to 5% of the normal population and is the most common cause of familial thromboembolism.33 Factor V Leiden mutation appears to be more associated with the development of DVT rather than PE.37 Primary or acquired deficiencies in protein C, protein S, and antithrombin III are other risk factors and their thrombotic expression is variable.33
Several primary disorders are associated with a high risk for fatal VTE. Patients frequently present with massive PE, and ICU admission and management is required. From the RIETE registry, risk factors of immobilization secondary to neurologic disease, cancer, and cardiopulmonary disorders had two- to threefold increased risk for fatal PE.38 In hospitalized cancer patients, VTE is the second leading cause of death.39 The risk of VTE in cancer patients undergoing surgery is three to five times greater than that in surgical patients without cancer. Moreover, cancer patients with symptomatic DVT exhibit a high risk of recurrent VTE.40 Heart failure is a frequent admission diagnosis in the coronary care unit. In patients with severe heart failure (ejection fractions less than 30%) PE is a major cause of morbidity and mortality rates.41,42 Among 198 patients with severe heart failure in the coronary care unit, 9.1% developed PE during their hospitalization, despite a majority receiving thromboprophylaxis.41 In an autopsy series of cardiac patients nearly a quarter had PE and the diagnosis was unsuspected in 82% of the patients.43 Acute exacerbation of chronic obstructive pulmonary disease (COPD) is a common reason for admission to the general hospital ward service and the ICU. Increase work of breathing leads to immobility, which is a risk factor of DVT. In a prospective cohort of 196 patients with COPD exacerbation, all patients had ultrasonography of the lower extremities performed. Among the 196 patients, 21 (10.7%) had DVT and the majority was asymptomatic.44 Moreover, hospitalized patients with unexplained exacerbations of COPD, when routinely evaluated, show demonstrated PE in 25% to 29%.45,46
As shown in Figure 44.1, the degree of mechanical obstruction, the interplay of neurohumoral mediators, and the patient’s underlying cardiopulmonary status will dictate the degree of RVD. A rapid rise in the pulmonary artery resistance produces a significant rise in right ventricular (RV) afterload, thereby causing RV dilatation. The dilation of the right ventricle results in shifting of the interventricular septum into the left ventricle, decreasing left ventricular (LV) preload.61–63 Moreover, RV dilation potentially promotes an increase in constrictive forces by the pericardium, leading to reduced LV compliance. Decreasing LV preload and compliance subsequently lead to lowering of the cardiac output and systemic hypotension with resultant decrease in aortic perfusion pressure and decreasing right coronary perfusion pressure.64,65 A compromised and burdened right ventricle is a direct result of oxygen demand outstripping oxygen supply, creating an ischemic environment for the right ventricle. The functional status of the cardiopulmonary system is very important in the initial hemodynamic presentation and is a major determinant of short- and long-term outcome.66 In patients without cardiopulmonary disease the anatomic and humoral obstruction must be 75% or greater to cause a mean pulmonary artery pressure (MPAP) of 40 mm Hg or higher, which is not sustainable; even the previously healthy heart may proceed to cardiovascular collapse and fail.66 Right atrial pressure (RAP) elevation occurs when the MPAP is 30 mm Hg or higher and obstruction exceeds 35% to 40%.67 Many patients who have a pulmonary embolic event have underlying cardiopulmonary disease. In these patients cardiopulmonary deterioration (CPD) may occur with a lesser degree of vascular obstruction.66 This is illustrated by the UKEP (Urokinase-Embolie Pulmonaire) Trial in which an initial presentation of shock with acute PE was seen in 56% of patients who had prior cardiopulmonary disease compared to 2% without cardiopulmonary disease. Obstruction of greater than 50% was uncommon.68
Respiratory Consequences
Hypoxemia is the most common gas exchange abnormality in patients. However, the PaO2 and alveolar-arterial (A-a) gradient have been reported to be normal in approximately 30% of patients with PE.69 Figure 44.2 summarizes the gas exchange abnormalities as a result of the pulmonary embolic event. Ventilation-perfusion mismatch and low mixed venous oxygen levels are the prevalent causes of hypoxemia.70,71 As pulmonary artery pressures increase, right-to-left flow across a patent foramen ovale (PFO) may occur. Endothelial damage may result from hypoxic exposure and further lead to pulmonary vasoconstriction. Hypoxemia may even promote a prothrombotic and antifibrinolytic effect.57
In acute PE an increase in alveolar dead space impairs CO2 elimination; however, the increase in minute ventilation results in hypocapnia. In patients with massive PE there can be a paradoxical elevation of the PaCO2 as a result of a marked increase in dead space ventilation.72 In PE patients because of an increase in alveolar dead space there is a reduction in alveolar CO2 content, which can be detected by capnography by a reduction in the capnographic waveform area. In one study, PE patients significantly demonstrated a reduction in the capnographic waveform area when compared to patients without PE.73 There is widening of the PETCO2-PaCO2 gradient. Bedside PETCO2 coupled with a Wells score has been shown to have a high negative predictive value.74
Clinical Presentation
The signs and symptoms of acute PE are at best nonspecific. The presence of syncope, current DVT, hemoptysis, leg swelling, active cancer, surgery, leg pain, and shock each marginally increases the probability of PE. Further, the absence of dyspnea or tachycardia marginally reduces the probability of PE.75
Approximately 50% of patients with acute PE present to the emergency department. In the Emperor Registry, among 1880 documented PE patients from 22 U.S. emergency departments, the most common presenting signs and symptoms were dyspnea at rest (50%), pleuritic chest pain (39%), dyspnea with exertion (24%), and syncope, which occurred in 5% as the initial presentation of acute PE. Only 58 patients (3%) had systolic blood pressures of 90 mm Hg or lower on presentation.76
Clinical prediction rules have been used in the emergency department setting to determine the pretest probability of acute PE.77 The most widely known prediction model has been the Wells score and it can allow for a dichotomous classification.78,79 Other prediction scores have been used, such as the Geneva and the modified Geneva.77,78 All prediction scores appear to have similar accuracy but are influenced by the local prevalence of PE. The clinical prediction scores are enhanced in identifying the patient with low probability of PE coupled with low D-dimer value.79
Clinical Presentation on Admission to the Intensive Care Unit
In patients admitted to the ICU with PE the clinical manifestations may be more demonstrative, as shown in Table 44.2. In the Urokinase Pulmonary Embolism Trial (UPET), the clinical features of massive PE were evaluated. Sudden unexplained onset of dyspnea was the most common symptom and was present in 80% of the patients. A majority of the patients had a well-defined risk factor. Tachypnea and tachycardia were present in 88% and 63% of the patients, respectively. Accentuated second heart sound was noted in 67%, and S3 or S4 gallop was present in 47%. Clinical shock was noted in 12%.80 Syncope can occur. Retrosternal nonpleuritic chest pain may mimic the pain experienced with a myocardial infarction and represent demand ischemia of the right ventricle. The differential diagnosis of pulseless electrical activity (PEA) is massive PE and should be expected when there is a known major risk factor.
Table 44.2
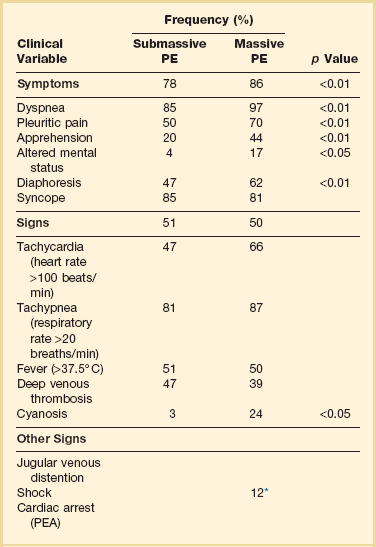
PE, pulmonary embolism; PEA, pulseless electrical activity.
*Adapted from Stein PD, Willis PW III, DeMets DL: History and physical examination in acute pulmonary embolism in patients without pre-existing cardiac or pulmonary disease. Am J Cardiol 1981;47;218-223.
The signs and symptoms of submassive PE will depend upon the degree of RVD. Dyspnea may be progressive over the last 4 to 6 days. Chest pain can be either pleuritic or nonpleuritic in nature. If pulmonary infarction occurs as result of distal embolization, then hemoptysis may occur. From the PIOPED II database, 92% of patients with main or lobar pulmonary emboli had dyspnea or tachycardia in contrast to segmental emboli dyspnea and tachycardia, where 65% had these findings.81,82
Recognition of Pulmonary Embolism During Intensive Care Unit Admission
When a patient is admitted to the ICU for a non-PE-related clinical diagnosis, the abrupt and unexplained occurrence of some combination of chest pain, dyspnea, tachypnea, hypotension, and hypoxemia should raise the suspicion of PE. Unexplained fever with and without mild leukocytosis could represent a possible VTE event.83 Hemoptysis can occur as a result of small distal pulmonary emboli causing alveolar hemorrhage. If PE occurs while the critically ill patient or postoperative patient is on mechanical ventilation, the clinical suspicion and conformation can be difficult. An increase in minute ventilation dead space or an increase in supplemental oxygen may be clues to an embolic event. If the patient has end-tidal CO2 (PETCO2) monitoring an increase in the PaCO2-PETCO2 gradient may occur.73
Diagnostic Testing for Pulmonary Embolism
Arterial Blood Gas Measurement
Hypoxemia and an increased alveolar-arterial gradient are usually seen in PE but can be normal in about 14% and are most likely the result of small embolic events.84,85 The initial acid-base disturbance is a respiratory alkalosis, which may evolve to a metabolic (lactic) acidosis as hypotension and shock ensue. A linear relationship between PE severity and PaO2 in patients without prior cardiopulmonary disease has been described. In the UPET and PIOPED trials 12% and 19% of patients, respectively, had PaO2 80 mm Hg or greater.86 Hypocarbia is usually present with acute PE because of hyperventilation, but hypercapnia can occur with large PEs producing increased dead space.87,72
Chest Radiography
The chest radiograph is abnormal in most cases of PE; however, radiographic findings possess inadequate sensitivity to rule out PE. Common radiographic abnormalities include atelectasis, pleural effusion, parenchymal opacities, and elevation of a hemidiaphragm. Radiographic findings of pulmonary infarction often show a wedge-shaped, pleural-based triangular opacity with an apex pointing toward the hilus (Hampton hump). Focal oligemia or decreased vascularity has been known as the Westermark sign. These findings are suggestive of PE but are infrequently prospectively observed.88,89 Radiographic findings of RVD may include a prominent central pulmonary artery (knuckle sign) and cardiomegaly. In one large prospective cohort of PE patients a normal chest radiograph was found in 24% of the patients.89 A normal-appearing chest radiograph in a patient with severe dyspnea and hypoxemia, is suggestive of a pulmonary embolic event.
Electrocardiography
In 1935 McGinn and White first described the association between acute PE and specific electrocardiogram (ECG) changes when they noted the S1Q3T3 pattern in seven patients with acute cor pulmonale.90 A normal ECG can occur with PE in a frequency ranging from 9% to 30%. The most common ECG abnormalities in the setting of PE are tachycardia and nonspecific ST-T wave abnormalities. The classic findings of right-sided heart strain and acute cor pulmonale are tall, peaked P waves in lead II (P pulmonale); right-axis deviation; right bundle branch block; and an S1Q3T3 pattern. Only 20% of patients with proven PE have any of these classic ECG abnormalities.91–93 T-wave inversion in the precordial leads representing subepicardial ischemia may be seen. The presence of this ECG finding is associated with increased degree of vascular obstruction and elevation of the pulmonary artery (PA) pressure.94 The new onset of atrial fibrillation has been thought to be associated with a pulmonary embolic event. In a recent study the occurrence of atrial fibrillation did not in general increase the probability of PE.95 However, if chest pain were the only symptom, the presence of atrial fibrillation increases the probability of PE (odds ratio [OR] 2.42, 95% confidence interval [CI] 0.97-6.07).
D-Dimer Assay
The serum D-dimer is a degradation product produced by plasmin-mediated proteases of cross-linked fibrin. D-dimer is measured by a variety of assays. These assays are categorized as quantitative, semiquantitative, qualitative rapid enzyme-linked immunosorbent assays (ELISAs); quantitative and semiquantitative latex; and whole-blood assays. The quanitative rapid ELISAs are the best assays to use in terms of sensitivity and likelihood ratio.96 When clinical prediction scores indicate that the patient has a low or moderate pretest probability of PE, D-dimer testing is useful to further define the likelihood of PE. Negative results on a high-sensitivity D-dimer test in a patient with a low pretest probability of PE (Wells rule) are associated with a low likelihood of VTE and reliably exclude PE.97 In a large prospective, randomized trial in patients with a low probability of PE who had negative D-dimer results, not performing additional diagnostic testing was not associated with an increased frequency of symptomatic VTE during the subsequent 6 months.98
D-dimer testing loses its ability to predict the likelihood of PE in older patients and in the presence of cancer, pregnancy, and various inflammatory or infectious disorders. If the pretest probability for PE is high, D-dimer testing should not be performed and objective testing should be done.96
Combining D-dimer results with measurement of the exhaled end-tidal ratio of carbon dioxide to oxygen (PETCO2/PO2) can be useful for diagnosis of PE. Kline and colleagues demonstrated in moderate-risk patients a positive D-dimer (>499 ng/mL) and a PETCO2/PO2 less than 0.28 significantly increased the probability of finding segmental or larger PE on computed tomography (CT) multidetector-row pulmonary angiography.99 In contrast, a PETCO2/PO2 greater than 0.45 predicted the absence of segmental or larger PE.
Duplex Compression Ultrasonography
Currently, DVT is largely diagnosed by duplex CUS. When using CUS, the ultrasonographic transducer pressed against veins shows they are easily compressed, but the muscular arteries are extremely resistant to compression. Where DVT is present, the veins do not collapse completely when pressure is applied using the ultrasonographic probe.100–102
Before an ultrasonographic scan can be considered negative, the entire deep venous system must be examined using centimeter-by-centimeter compression testing of every vessel. The risk of a DVT event following a single whole leg CUS in 3 months is 0.57% (95% CI 0.25-0.89).103
CUS is associated with a sensitivity of 97% for proximal DVT, 72% for distal DVT, and a specificity of 94%.104 In a meta-analysis of the role of CUS in DVT, CUS alone had a sensitivity of 93.8% (92.0-95.3%) for proximal DVT, 56.8% (49.0-66.4%) for distal DVT, and specificity of 97.8% (97.0-98.4%).105
Ventilation-Perfusion Nuclear Scans
The 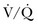 scan has a limited role in the diagnosis of acute PE in the ICU, although its diagnostic accuracy is no different from that in non–critically ill populations.106 Because of the advancing technology of CT scanning, the lung scan is used in the critical care units in special circumstances such as renal insufficiency or in the patient who is not intubated and capable of transport from the ICU to the nuclear medicine suite for a full view scan. If the institution has a portable lung scanner this can be used in the ICU but the number of images is reduced when compared to the standard
scan has a limited role in the diagnosis of acute PE in the ICU, although its diagnostic accuracy is no different from that in non–critically ill populations.106 Because of the advancing technology of CT scanning, the lung scan is used in the critical care units in special circumstances such as renal insufficiency or in the patient who is not intubated and capable of transport from the ICU to the nuclear medicine suite for a full view scan. If the institution has a portable lung scanner this can be used in the ICU but the number of images is reduced when compared to the standard 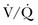 scan.
scan.
A high-probability scan, as shown in Figure 44.3, is sufficient diagnostic evidence of PE to begin anticoagulation therapy, and a normal 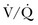 scan is considered sufficient evidence to exclude PE. However, the frequency of low- or intermediate-probability scans (indeterminate scans) can be as high as 50% to 70%, with a 10% to 50% probability of PE. Therefore, it is impossible to initiate anticoagulation therapy based on this indeterminate lung scan probability. In the first PIOPED study, only 40% of patients with PE had a high-probability
scan is considered sufficient evidence to exclude PE. However, the frequency of low- or intermediate-probability scans (indeterminate scans) can be as high as 50% to 70%, with a 10% to 50% probability of PE. Therefore, it is impossible to initiate anticoagulation therapy based on this indeterminate lung scan probability. In the first PIOPED study, only 40% of patients with PE had a high-probability 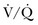 scan result, whereas another 40% of patients with PE had an indeterminate result and 14% had a low-probability result. A high-probability
scan result, whereas another 40% of patients with PE had an indeterminate result and 14% had a low-probability result. A high-probability 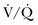 scan had a positive predictive value of 88% in patients without prior history of PE. The combination of a high clinical probability with the high-probability lung scan resulted in a 96% positive predictive value. The negative predictive value of a low-probability scan was 88% and improved to 96% when combined with a low clinical probability. Thus, a low-probability scan with a low clinical probability score rules out PE as definitively as a normal scan. Finally, 39% of the patients had intermediate scans and 32% of these patients had angiographically proven PE.14,107
scan had a positive predictive value of 88% in patients without prior history of PE. The combination of a high clinical probability with the high-probability lung scan resulted in a 96% positive predictive value. The negative predictive value of a low-probability scan was 88% and improved to 96% when combined with a low clinical probability. Thus, a low-probability scan with a low clinical probability score rules out PE as definitively as a normal scan. Finally, 39% of the patients had intermediate scans and 32% of these patients had angiographically proven PE.14,107
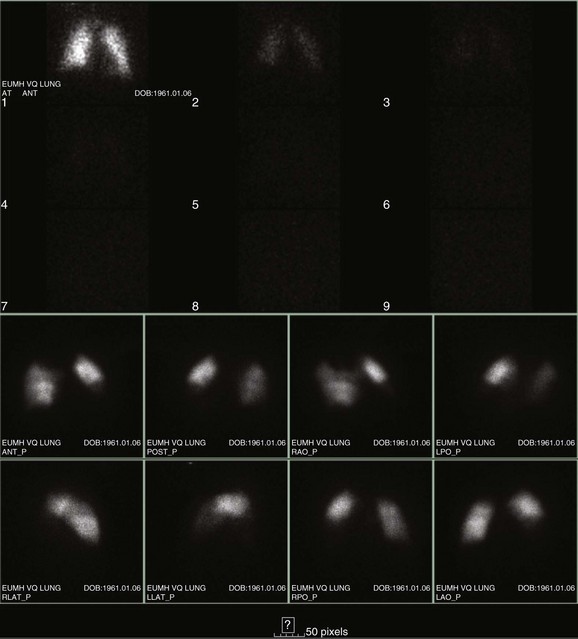
Preexisting cardiopulmonary disease diminishes the clinical utility of 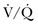 scans by increasing the prevalence of nondiagnostic scans. Nondiagnostic scans are more likely to occur in patients with preexisting cardiopulmonary disease. Lung scans are not useful to either establish or rule out the diagnosis of PE in more than 80% of cases. A similar disadvantage of
scans by increasing the prevalence of nondiagnostic scans. Nondiagnostic scans are more likely to occur in patients with preexisting cardiopulmonary disease. Lung scans are not useful to either establish or rule out the diagnosis of PE in more than 80% of cases. A similar disadvantage of 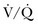 lung scans is noted in critically ill patients with prior cardiopulmonary disease. Nevertheless,
lung scans is noted in critically ill patients with prior cardiopulmonary disease. Nevertheless, 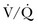 lung scans can be performed in the critically ill, even if patients are receiving mechanical ventilation, and chest radiographic abnormalities can be used as a surrogate of ventilation defects. Despite the limited ventilation component under positive-pressure respiration, the perfusion component is not significantly affected by mechanical ventilation.
lung scans can be performed in the critically ill, even if patients are receiving mechanical ventilation, and chest radiographic abnormalities can be used as a surrogate of ventilation defects. Despite the limited ventilation component under positive-pressure respiration, the perfusion component is not significantly affected by mechanical ventilation. 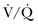 scans remain a useful tool in patients with contraindications to pulmonary angiography or CT angiography.108,109
scans remain a useful tool in patients with contraindications to pulmonary angiography or CT angiography.108,109
Multidetector Computed Tomography Scan of the Chest
The PIOPED II study firmly established the role of CT pulmonary angiography (CTPA) and largely replaced ventilation-perfusion (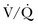 ) lung scintigraphy as the main imaging modality in suspected PE.110 The technology is continually evolving and its use is widespread. As illustrated in Figure 44.4, CTPA provides direct visualization of emboli throughout the pulmonary arterial vasculature. The advantage of CTPA over pulmonary angiography and lung scan is the discovery of an alternative diagnosis when PE is not found on CTPA. In one study, an alternative diagnosis was established in 57% of patients who did not have a PE.111 Currently, CTPA is the “gold standard” for PE diagnosis. A meta-analysis of 23 studies with 4657 patients with a negative CTPA who did not receive anticoagulation showed a 3-month rate of subsequent VTE of 1.4% (95% CI 1.1-1.8) and a 3-month rate of fatal PE of 0.51% (0.33-0.76), which compares favorably with the results noted after a normal “gold standard” invasive contrast pulmonary angiogram.112 The increased use of CTPA raises concerns about the increased incidence of cancer attributable to radiation.113 For this reason, combined use of CTPA and CT leg venography should not be performed because the latter adds little to diagnostic utility. In the PIOPED II trial, no patient with PE or DVT would have been undiagnosed if imaging of the pelvic and proximal leg veins had been omitted.107,110
) lung scintigraphy as the main imaging modality in suspected PE.110 The technology is continually evolving and its use is widespread. As illustrated in Figure 44.4, CTPA provides direct visualization of emboli throughout the pulmonary arterial vasculature. The advantage of CTPA over pulmonary angiography and lung scan is the discovery of an alternative diagnosis when PE is not found on CTPA. In one study, an alternative diagnosis was established in 57% of patients who did not have a PE.111 Currently, CTPA is the “gold standard” for PE diagnosis. A meta-analysis of 23 studies with 4657 patients with a negative CTPA who did not receive anticoagulation showed a 3-month rate of subsequent VTE of 1.4% (95% CI 1.1-1.8) and a 3-month rate of fatal PE of 0.51% (0.33-0.76), which compares favorably with the results noted after a normal “gold standard” invasive contrast pulmonary angiogram.112 The increased use of CTPA raises concerns about the increased incidence of cancer attributable to radiation.113 For this reason, combined use of CTPA and CT leg venography should not be performed because the latter adds little to diagnostic utility. In the PIOPED II trial, no patient with PE or DVT would have been undiagnosed if imaging of the pelvic and proximal leg veins had been omitted.107,110
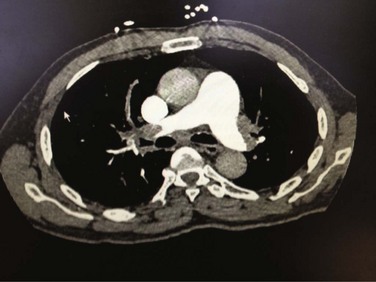
Figure 44.4 Extensive bilateral lobar pulmonary emboli.
Magnetic Resonance Imaging
In the PIOPED II study, CT angiography of the chest, as many as 25% of patients had a contraindication for the test. Preexisting renal failure and pregnancy were the primary conditions that prevented this mode from being used as the primary test for the evaluation of PE.110 Thus, gadolinium-enhanced magnetic resonance pulmonary angiography (MRA) could potentially be used to diagnose PE because it is devoid of radiation. The accuracy of this technique combined with magnetic resonance venography (MRV) has been studied in the prospective, multicenter PIOPED III accuracy study. The proportion of technically inadequate images ranged from 11% to 52% across the seven participating centers. Technically adequate MRA had a sensitivity of 78% and a specificity of 99%, whereas technically adequate MRA and MRV had a sensitivity of 92% and a specificity of 96%. However, 194 (52%) of 370 patients had technically inadequate results, which substantially restricts its clinical use.114
Risk Stratification
Death from an embolic event almost always results from hemodynamic deterioration due to an abrupt rise in pulmonary vascular resistance overwhelming the contractile power of the right ventricle. Progressive hemodynamic and respiratory deterioration characterizes the clinical presentation of major or massive PE and is seen in approximately 5% of PE patients.2 Patients can be categorized as having submassive PE. Risk stratification becomes important to identify this group of patients who present with relative hemodynamic stability but exhibit varying degrees of RVD. Risk stratification schemes in normotensive patients center on RV function assessment after an embolic event and identify patients who may be at risk for progressive hemodynamic deterioration. Approximately 50% of patients who are normotensive have varying degrees of RVD and 10% will die. The European Society of Cardiology recommends a classification of PE into high-risk and non–high-risk groups. The high-risk groups represent the massive or major PE presentations. The non–high-risk group is further subdivided into intermediate risk in which the mortality rate ranges between 3% and 15% and a low-risk group in which the mortality rate is less than 3%. Initial therapeutic interventions are greatly influenced by risk stratification that includes clinical scoring systems, laboratory data, and imaging.115–117
The presence of RVD is a predictor of the increased risk of embolic-related adverse events and mortality rates when compared with patients without RVD.2 Patients who present with circulatory shock usually have severe RVD and have mortality rates up to 65%.1,2,115 In PE patients with normal hemodynamics and evidence of RVD the mortality rates can range from 8% to 14%. Several registry studies have observed that the frequency of RVD in normotensive patients is approximately 40%.116,117 In PE patients with initial normal hemodynamics and no RVD the mortality rate is low (0-3%).
Clinical and Hemodynamic Parameters
The initial hemodynamic presentation of PE is the most powerful predictor of outcome, as illustrated in Figure 44.5. Arterial hypotension at the time of PE presentation is an early predictor of death. In the 2392 patients from the ICOPER study, the 90-day mortality rates were 52.4% in patients with systolic arterial blood pressures below 90 mm Hg, and 14.7% in those patients who had preserved arterial blood pressures. In addition, comorbid conditions increase the risk of adverse clinical events, even in the presence of anatomically small PE. In the ICOPER findings, age over 70, congestive heart failure, cancer, and chronic lung disease were identified as independent predictors of 3-month mortality risk, with approximately twofold increase in the risk of death.2 In one study, altered mental status at presentation was associated with a sevenfold increase in risk of adverse outcome. Altered mental status most likely reflected decreased cardiac perfusion or hypoxia. In the same study the initial presentation of shock was associated with a threefold increase in risk of 30-day adverse outcome (death, secondary shock, or recurrent PE).117
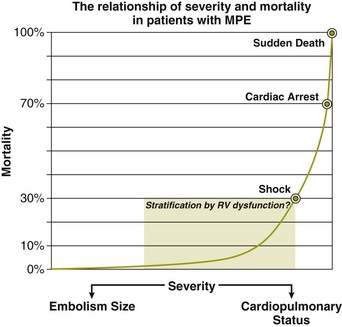
Patients can be clinically risk stratified by the use of a clinical scoring system known as the Pulmonary Embolism Severity Index (PESI), which also has a simplified version.118,119 This prognostic model was developed by Aujesky and colleagues (Table 44.3) using a large database of hospitalized PE patients from which they derived and validated a prediction rule that identified acute PE patients with low to high risk for fatal PE and adverse events. Using 11 variables of predictors, five distribution categories emerged looking at inpatient fatality, 30-day all-cause mortality rate, and the adverse outcomes of nonfatal cardiogenic shock or cardiopulmonary arrest. Class I to class III all-cause mortality rates ranged between 0% and 3.1%, although class IV to class V all-cause mortality rates were between 10% and 24%. This score has an excellent negative predictive value and readily identifies low-risk patients but suffers in the attempt to predict patients at high risk for adverse events.119 A more simplied PESI score has been developed and has a high negative prediction value.120
Table 44.3
Pulmonary Embolism Severity Index (PESI)
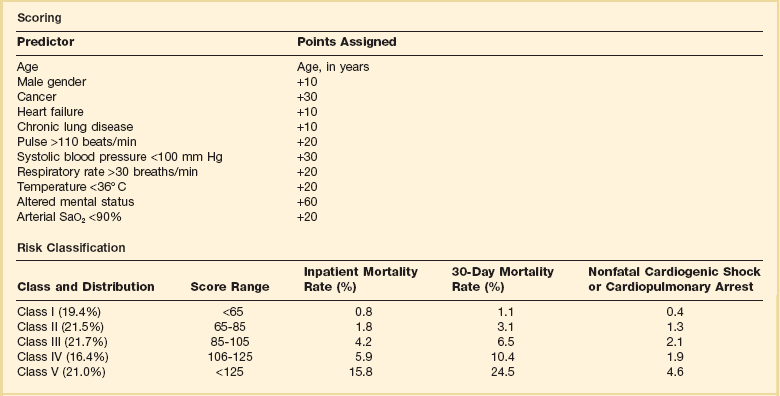
Adapted from Aujesky D, Obrosky DS, Stone RA, et al: Derivation and validation of a prognostic model for pulmonary embolism. Am J Respir Crit Care Med 2005;172:1041-1046.
In patients with symptomatic acute PE, the presence of DVT may have prognostic significance. In a prospective study by Jiménez and coworkers, among 707 patients diagnosed with acute PE, 51.2% had concomitant DVT and 10.9% had PE-specific fatality at a 3-month follow-up. Patients with PE with the presence of DVT at the time of diagnosis had an adjusted hazard ratio (HR) of 2.05% (95% CI 1.24 to 3.38) and PE-specific fatality-adjusted HR of 4.25 (95% CI 1.61 to 11.25). These findings were validated by a large international registry trial (RIETE) that again demonstrated a significant prediction of all-cause mortality rate and PE-specific mortality rate when the concomitant DVT was present during the acute embolic event. These results suggest that patients with symptomatic acute PE have an increase in all-cause mortality rate, PE-related death, and recurrent VTE over a 3-month period if at the time of PE a diagnosis of proximal DVT is also present.121
Electrocardiogram
Several studies have demonstrated that ECG signs of RV strain correlate with the presence of RVD.122,123 A study by Punukollu and associates showed that T-wave inversion in leads V1 to V3 had a specificity of 88% and diagnostic accuracy of 81% for RVD in acute PE.124 Daniel and colleagues developed an ECG scoring system based on typical features of acute PE such as presence of right bundle branch block (RBBB), T-wave inversions, and additional features of right-sided heart strain that predicted severe pulmonary hypertension (sPAP >50 mm Hg) with a specificity of 97.7% if the patient’s “ECG score” is 10 or greater.125 The ECG may also be a tool for risk stratification of normotensive acute PE patients. In a study by Vanni and coworkers among 306 patients with documented PE 130 (34%), patients demonstrated ECG findings of right-sided heart strain. In this study ECG findings of right-sided heart strain were defined as incomplete or complete RBBB, S1Q3T3, or inverted T waves in the precordial leads V1, V2, and V3. For this study only one of these ECG patterns had to be present. The investigators demonstrated that the presence of RV strain on the initial ECG was associated with clinical deterioration and death (HR 2.58) in normotensive PE patients.122
Cardiac Biomarkers
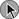 Biomarkers are relied on in the risk stratification of patients with PE. Tests for biomarkers are widely available and there is usually a quick turnaround from the clinical laboratory. Caution must be taken on the interpretation of these biomarkers. It is important for the critical care clinician to understand the clinical issues that impact the interpretation of the biomarkers and understand the negative and positive predictive values.115 Elevations of brain natriuretic peptide (BNP) and troponin occur in many patients with PE and correlate with magnitude of RVD.
Biomarkers are relied on in the risk stratification of patients with PE. Tests for biomarkers are widely available and there is usually a quick turnaround from the clinical laboratory. Caution must be taken on the interpretation of these biomarkers. It is important for the critical care clinician to understand the clinical issues that impact the interpretation of the biomarkers and understand the negative and positive predictive values.115 Elevations of brain natriuretic peptide (BNP) and troponin occur in many patients with PE and correlate with magnitude of RVD.
Echocardiography
The echocardiogram has become the main imaging modality to assess RV structure and function. In addition, pulmonary artery pressures can be estimated. Most hospitals in the United States have portable echocardiographic technology allowing for serial assessment of the impact of treatment on RVD. In hemodynamically stable patients with PE, 20% to 40% will have some echocardiographic finding of RVD.116,117 Box 44.1 illustrates the echocardiographic finding of acute PE. Toosi and colleagues performed a quantitative risk assessment of various echocardiographic measurements of RVD in acute PE. For in-patient mortality rate the positive predictive values range between 11% and 20% and the negative predictive values are 97% to 100%.140 The major drawback with the role of the echocardiographic definition of RVD of acute PE is that it lacks standardization. There is no clear agreement on which of the echocardiographic
Brain Natriurectic Peptide
The heart will release natriuretic peptides as result of ventricular volume overload. Ventricular stretch results in cellular production of proBNP. This peptide is subsequently cleaved to the biologically active BNP and the inactive N-terminal fragment (NT-proBNP).126 Both BNP and NT-proBNP can be measured clinically and have been validated as diagnostic and prognostic markers of ventricular stretch in multiple conditions, including congestive heart failure, pulmonary hypertension, and acute PE.127 Elevated concentrations of BNP distinguish patients with PE at higher risk of complicated in-hospital course and death from those with low BNP levels. Both BNP and NT-proBNP have been extensively investigated as prognostic markers in acute PE. In an early study, ten Wolde and colleagues127 demonstrated in a prospective cohort of normotensive acute PE patients that those who died within 90 days had a significantly increased BNP level at the time of diagnosis compared with those patients who did not die in follow-up (245 pg/mL vs. 30 pg/mL, respectively). In addition, using a cut-off point of 75 pg/mL, these investigators reported that BNP had a positive predictive value of PE-related fatality of 17%, whereas the negative predictive value for death was 99%.127 As will be seen for most of the prognostic markers for acute PE, the investigators noted that BNP was able to identify those at low risk of adverse outcomes but did not have good specificity (many false-positives) in identifying those who were likely to die from their PE.
Normal levels of BNP have a high negative predictive value for an elevated level of BNP or NT-proBNP as a risk factor for short-term fatality and overall short-term complicated clinical outcome, and an indicator of RVD in patients with acute PE. This is illustrated by the study by Kucher and colleagues,128 who investigated the prognostic capabilities of NT-proBNP in a prospective cohort of acute PE patients. In those suffering an adverse event (defined as a combined end point of in-hospital death or need for escalation of care, including cardiopulmonary resuscitation, mechanical ventilation, vasopressor, or thrombolysis), NT-proBNP was significantly higher compared with those who did not experience an adverse event (median of 4250 pg/mL vs. 121 pg/mL, respectively). An NT-proBNP cut-off point of 500 pg/mL yielded a negative predictive value for adverse outcome of 97% with a corresponding positive predictive value of adverse outcome of 45%.128 In a meta-analysis of 13 studies by Klok and associates, they indeed demonstrate that increased BNP or proBNP was a risk factor for short-term fatality and adverse clinical events and a marker for RVD. However, the investigators caution against using this biomarker alone to justify more aggressive medical therapy, such as thrombolytic therapy.129
Cardiac Troponin
Serum troponins are widely available for the evaluation of diverse forms of myocardial injury. The assays are rapid and are well suited as a risk stratification tool. A number of studies and meta-analyses have evaluated the prognostic value of cardiac troponin I (TnI) or T (TnT) (determined by conventional assays) in acute PE.130–132 Overall, the elevation of cardiac troponin levels was associated with an adverse early outcome including fatality.
In hemodynamically (normotensive) stable patients with PE the incidence of elevated troponins can be as high as 40%.133 As with the management of acute myocardial infarction, in a patient with acute PE, troponin levels must be obtained serially. The first troponin level in patients with PE may be normal, and subsequently levels will increase. This was illustrated in the study by Ferrari and colleagues who investigated the time course of TnT elevation in patients with acute PE. In a prospective cohort, they found that whereas 50% of normotensive patients with acute PE were found to have an elevated TnT, 5% of these patients had an initial normal TnT that became abnormal at the 6-hour analysis; moreover, 25% of those with an elevated TnT (0.01 ng/mL) died during the hospitalization compared with no patient dying who had a TnT value less than 0.01 ng/mL.134
In a recent meta-analysis, Becattini and colleagues132 analyzed the prognostic usefulness of cardiac troponins in acute PE. In this meta-analysis of 20 studies, the investigators reported that 20% of patients with elevated troponins died, compared with 3.7% of patients with normal troponin levels. A high level of either TnT or TnI was associated with a fivefold increased risk of short-term death. The investigators noted that there were a multitude of different troponin assays and selected cut-off points used in the included reports, although results were generally consistent across studies. Most studies included in the meta-analysis defined an elevated troponin level as any value that was above the upper limit of normal for the individual troponin assay.132
Combining biomarkers with imaging may enhance the prognostication of normotensive patients with acute PE. In one study, the presence of an elevated troponin, and echocardiographic evidence of RVD and the presence of a proximal DVT was associated with an increased 30-day mortality rate.135
New generations of highly sensitive troponin (hsTnT) assays in patients with acute myocardial infarction have demonstrated excellent diagnostic performance and the potential to improve early risk stratification PE.136
Other cardiac biomarkers are released in the systemic circulation when the myocardium is injured. Heart type fatty acid binding protein (H-FABP) is a cytosolic protein that is within cardiac myocytes that is released faster than cardiac troponins during injury. Studies have demonstrated that H-FABP has a good negative predictive value for adverse outcomes and, in one study, a 28% positive predictive value in patients with acute PE.137,138 Growth differentiation factor (GDF)-15 is a distant member of the transforming growth factor-β family. Under normal conditions, myocardium does not produce GDF-15, but its cardiac expression sharply increases after pressure overload and ischemia. The prognostic value of GDF-15 was investigated in a study including 123 patients with acute PE.23 On multivariate analysis, cardiogenic shock on admission, cardiac TnI, and GDF-15 were independently associated with an adverse outcome at 30-day follow-up.139 Further studies are needed to confirm these preliminary results.
findings predicts adverse outcomes and fatality in normotensive submassive PE. However, an echocardiographic finding of right ventricle/left ventricle ratio of greater than 0.9 was found to be an independent risk factor of in-hospital death (OR 2.7% CI 1.7-6.0) in a large registry trial.141
In the ICOPER study that included 1035 PE patients with systolic arterial pressure 90 mm Hg or higher, an echocardiogram was performed within 24 hours of PE diagnosis. The incidence of RVD was 39% (405 patients). The 30-day survival rates were lower in patients with RV hypokinesis (84%) compared with those without RV hypokinesis (91%), HR 1.94 (CI, 1.41-3.16; p ≤ 0.001). The negative predictive value of 30-day mortality rate of echocardiographic confirmation of RVD was 91%. The positive predictive value was 16%.2
In the assessment of RVD, if cardiac biomarkers are elevated, how well does this correlate with RVD on transthoracic echocardiography? A study by Logeart and coworkers found that a BNP level of less than 100 pg/mL had a negative predictive value of 100% and TnI of less than 0.10 ng/mL had a negative predictive value of 67% for the presence of RVD on echocardiography. Combining a BNP of greater than 100 pg/mL and TnI of greater than 0.10 ng/mL had a positive predictive value of 80% for the presence of RVD by echocardiography.142
The echocardiogram can visualize free-floating right-sided heart thrombi. In the ICOPER study patients with this finding had a mortality rate of 21% compared with 11% in those without right-sided heart thrombi.143 The detection of a patent forman ovale or atrial septal defect by echocardiography may be a warning finding for the development of paradoxical embolization, which is associated with a risk of stroke and increased mortality rate.144
For the evaluation of circulatory shock transesophageal echocardiography (TEE) is a useful diagnostic modality. In patients with massive PE who cannot undergo CTPA, when TEE is performed, it has a 60% to 80% sensitivity and a 95% to 100% specificity to diagnose acute PE. TEE has a 9% to 95% sensitivity and a 100% specificity in main pulmonary artery PE.145 In patients with unexplained cardiac arrest or pulseless electrical activity (PEA), TEE has been shown to identify acute massive PE. In a small study of 25 patients with PEA, TEE demonstrated eight (36%) patients with massive PE.146
Computed Tomography Pulmonary Angiography
CT scans findings of right-sided heart dysfunction are illustrated in Figures 44.6A and B. The detection of RV enlargement by spiral chest CT has recently been evaluated in the risk stratification of patients with acute PE. Using measurements from a reconstructed CT four-chamber view, RV enlargement, defined as a ratio of RV to LV dimension of greater than 0.9, was a significant independent predictor of 30-day mortality rate.146 In a recent multicenter study of 460 consecutive patients with multidetector CT confirmed PE a majority of the study cohort were hemodynamically stable and 50% had RVD at the time of diagnosis. Again, an RV/LV ratio of 0.9 or greater provided good correlation with RVD demonstrated by echocardiography. In addition, RVD found on multidetector CT was an independent risk factor for death and clinical deterioration. The investigators propose that multidetector CT scan potentially can be the single procedure, not only for diagnosis of PE but also for risk stratification. However, this assumption will require more validation studies.112
In hemodynamically stable patients with acute PE, assessing RV/LV ratio may be important in not only risk stratification but also the location of the emboli. In a recent multicenter study of 516 hemodynamically stable PE patients central localization of emboli was found to be an independent mortality risk factor, although distal localization was inversely associated with adverse events. Thus, anatomic findings by CT scan may be important in assessing risk in hemodynamically stable patients with pulmonary embolus.147
Table 44.4 summarizes the various risk modalities used in risk stratification of acute PE.
The Management of Acute Pulmonary Embolism
Pharmacologic Management
The anticoagulation treatment for DVT and acute PE is essentially the same. Anticoagulation therapy does not dissolve the clot but prevents extension. Anticoagulation therapy indirectly decreases clot burden by allowing the natural fibrinolytic system to dissolve thrombus. When PE is suspected, if there is no contraindication, anticoagulation therapy should be started immediately while diagnostic studies are being performed.148
Unfractionated and Low-Molecular-Weight Heparin
Heparins act by binding to the natural anticoagulant antithrombin, thereby accelerating the inactivation of thrombin by antithrombin and several other activated coagulation factors This mechanism of action will prevent extension of the thrombus. UFH is usually administered as an initial bolus, followed by a continuous intravenous infusion. Because of a large individual difference in the binding of heparins to plasma proteins, the doses should be adjusted to the results of the activated partial thromboplastin time (aPTT) or the anti–factor Xa (anti-Xa) activity.149
The effectiveness of heparin therapy depends largely on achieving a critical therapeutic level of heparin within the first 24 hours of treatment. Nomogram dosing of heparin has been shown to achieve this goal.150 The critical therapeutic level of heparin is 1.5 times the baseline control value or the upper limit of normal range of the aPTT. This level of anticoagulation is expected to correspond to a heparin blood level of 0.2 to 0.4 U/mL by the protamine sulfate titration assay and 0.3 to 0.6 by the anti-Xa assay.151 If intravenous UFH is chosen, an initial bolus of 80 U/kg or 5000 U followed by an infusion of 18 U/kg/hour or 1300 U/hour should be given, with the goal of rapidly achieving and maintaining the aPTT at levels that correspond to therapeutic heparin levels. Fixed-dose and monitored regimens of subcutaneous UFH are available and are acceptable alternatives.148,150
Low-molecular-weight heparins (LMWHs) have many advantages over UFH. These agents have a greater bioavailability, can be administered by subcutaneous injections once or twice a day, and have a longer duration of anticoagulant effect. There is a lower risk of osteoporosis and immune-mediated thrombocytopenia.152 A fixed dose of LMWH can be used and laboratory monitoring is not necessary except in clinical circumstances such as morbid obesity, low weight (<40 kg), pregnancy, and renal insufficiency.153 Comparison trials between LMWH and UFH have demonstrated that LMWH is at least as effective and as safe as UFH, and there are no significant differences in recurrent thromboembolic events, major bleeding, or mortality risk between the two types of heparin.154 Caution must be used when administering LMWH in patients with renal insufficiency. LMWH is primarily cleared by the kidneys. Therefore, monitoring anti-Xa activity should be performed in patients with impaired renal function. If the creatinine clearance is less than 30 mL/minute, UFH should be used.155
LMWH heparin has a significantly lower incidence of heparin-induced thrombocytopenia (HIT), yet HIT can still occur with exposure to LMWH. Thus, LMWH is not an anticoagulant substitute for the patient with HIT requiring VTE prophylaxis or thrombosis treatment.156
LMWH can be administered safely in an outpatient setting. This has led to the development of programs in which clinically stable patients with PE are treated at home, at substantial cost savings.157 An international, open-label, randomized trial compared outpatient and inpatient treatment (both using the LMWH enoxaparin as initial therapy) of low-risk patients with acute PE and concluded that outpatient treatment was not inferior to inpatient treatment.158
Fondaparinux
Fondaparinux is a synthetic polysaccharide derived from the antithrombin binding region of heparin. Fondaparinux catalyzes factor Xa inactivation by antithrombin without inhibiting thrombin, and studies have shown that it is effective in the initial treatment and prophylaxis of the VTE. Fondaparinux has been shown to be just as effective and safe as UFH and enoxaparin.159 Fondaparinux dosing is based upon various weight ranges. For patients weighing less than 50 kg, 5 mg once daily is recommended. Patients weighing 50 to 100 kg are given 7.5 mg, and those weighing greater than 100 kg receive 10 mg daily. HIT is rare with the use of fondaparinux because of this lack of interaction with platelet factor 4 (PF4).148
Warfarin Therapy
The anticoagulant effect of warfarin (Coumadin) is mediated by the inhibition of vitamin K–dependent factors, which are II, VII, IX, and X. Vitamin K antagonists (VKAs) include substances with a short (acenocoumarol), intermediate (warfarin, fluindione), or long (phenprocoumone) half-life. For this reason, and because of genetically induced metabolic variability, the variable vitamin K content of food, a narrow therapeutic index, and interactions with other drugs, the effect on coagulation must be closely monitored. The peak effect does not occur until 36 to 72 hours after drug administration and the dosage is difficult to titrate. Administration of heparins or fondaparinux should overlap during at least 5 days with that of VKAs. After 5 days the parenteral drug can be stopped when the anticoagulant concentration induced by the VKA has achieved international normalized ratio (INR) between 2.0 and 3.0.148,160
The current American College of Chest Physicians (ACCP) guidelines for warfarin therapy suggest that the target INR is 2.5 even in high-risk patients (e.g., antiphospholipid syndrome). Because of its lack of validation, the ACCP does recommend pharmacogenetic testing. Warfarin should be started on day 1 or 2 that heparin is started. Usually warfarin 10 mg is administered and INR is adjusted to target INR. There is an increased risk of bleeding when antiplatelet drugs (acetylsalicylic acid, clopidogrel) are administered along with warfarin. In patients with acute coronary syndrome, mechanical valves, or recent coronary artery bypass or coronary stents, there is a likely benefit from the combination. Nonsteroidal anti-inflammatory drugs (NSAIDs) also increase the risk of bleeding when taken with warfarin and should be avoided.160
New Oral Anticoagulants
A new generation of anticoagulants is emerging with promising advantages over current therapy. These drugs have their effect on specific steps of the coagulation cascade. These agents are taken orally and do not require hematologic monitoring. The direct thrombin inhibitor dabigatran and factor Xa inhibitors rivaroxban, apixaban, and edoxaban are moving through the pipeline for Food and Drug Administration (FDA) drug approval. Rivaroxaban recently received U.S. FDA approval to expanded use of Xarelto (rivaroxaban) to include treating DVT or PE and to reduce the risk of recurrent DVT and PE following initial treatment. Two studies have demonstrated that rivaroxaban is as effective as warfarin.15,161 A major drawback, there is no reversal agent or antidote for hemorrhage induced by rivaroxaban.162
Duration of Anticoagulation Therapy
A patient with a first thromboembolic event occurring in the setting of reversible risk factors, such as immobilization, surgery, or trauma, should receive warfarin therapy for at least 3 months. No difference in the rate of recurrence was observed in either of two studies comparing 3 versus 6 months of anticoagulant therapy in patients with unprovoked first events.69,70 The current recommendation is anticoagulation for at least 3 months in these patients; the need for extending the duration of anticoagulation should be reevaluated at that time.163
The current ACCP guidelines recommend that all patients with unprovoked PE should undergo a risk-to-benefit evaluation to determine if long-term therapy is needed (grade 1C).163 Long-term treatment is recommended for these patients who do not have risk factors for bleeding and in whom accurate anticoagulant monitoring is possible (grade 1A). Patients who have PE and preexisting irreversible risk factors, such as deficiency of antithrombin III, protein S and C, factor V Leiden mutation, or the presence of antiphospholipid antibodies, should be placed on long-term anticoagulation.
Heparin-Induced Thrombocytopenia
HIT is a transient prothrombotic disorder initiated by heparin. The main features of HIT are (1) thrombocytopenia resulting from immunoglobulin G–mediated platelet activation and (2) in vivo thrombin generation and increased risk of venous and arterial thrombosis.164
HIT may manifest clinically as an extension of the thrombus or formation of new arterial thrombosis. HIT should be suspected whenever the patient’s platelet count falls to less than 100,000/µL or less than 50% of the baseline value, generally after 5 to 15 days of heparin therapy. The pretest probability for HIT can be determined by the application of the “4 Ts”: (1) Thrombocytopenia, (2) Timing, (3) Thrombosis, and (4) no oTher explanation of thrombocytopenia. At low scores the probability of the presence of platelet-activating HIT antibiodies is low; at high scores there is a moderate probability of the presence of the antibodies.164,165
The diagnosis of HIT is based upon assays that detect the presence of platelet-activating antibodies. The PF4-dependent enzyme immunoassay (EIA) test may lead to overdiagnosis of HIT. However, a negative EIA essentially rules out HIT. The serotonin release assay (SRA) is more specific for HIT than the PF4-dependent EIA. EIA is measured in optical density units (OD) and the strength of this measurement is a strong predictor of a positive SRA and confirmed HIT. In a patient with thrombocytopenia in whom the OD of EIA was less than 1.5 units the thrombocytopenia was not caused by HIT.164,165
The treatment of patients who develop HIT is to stop all heparin products, including catheter flushes and heparin-coated catheters, and to initiate an alternative, nonheparin anticoagulant, even when thrombosis is not clinically apparent. Preferred agents include direct thrombin inhibitors, such as lepirudin or argatroban. Start warfarin while the patient receives an alternative, nonheparin anticoagulant and only when the platelet count has recovered to at least 100,000/µL (preferably to 150,000/µL).148
Thrombolytic Therapy
The goal of thrombolytic therapy, by accelerating the conversion of plasminogen to plasmin, is to promote clot lysis and unload the right ventricle by decreasing pulmonary vascular obstruction. Thrombolysis has demonstrated a more rapid resolution of pulmonary vascular obstruction with improvement of RV hemodynamics within 2 hours after initiation of infusion. However, the controversy regarding thrombolytic therapy is the unloading of the right ventricle associated with improved mortality rate. In the larger original thrombolysis trials, streptokinase and urokinase were compared with heparin alone at infusion rates of 24 and 12 hours, respectively. These studies demonstrated rapid reperfusion of the pulmonary vasculature, reduction in pulmonary resistance, and no difference in mortality rate when compared to heparin.166,167
Currently three thrombolytics have FDA approval in the United States for acute PE: streptokinase, urokinase, and alteplase. Streptokinase and urokinase are nonselective agents activating both circulating and clot-bound plasminogen. Both agents have significant limitations; streptokinase is antigenic and may cause hypotension. Alteplase (tissue plasminogen activator [tPA]) is fibrin specific and is the thrombolytic of choice for the management of PE.168,169
The current ACCP key recommendations for the use of thrombolytic therapy in acute PE are noted here:163
1. In patients with acute PE associated with hypotension (e.g., systolic blood pressure <90 mm Hg) without a high bleeding risk, we suggest systemically administered thrombolytic therapy (grade 2C—weak recommendation [suggestion]/low quality or very low quality evidence).
2. In most patients with acute PE not associated with hypotension, we recommend against systemically administered thrombolytic therapy (grade 1C—strong recommendation/low quality or very low quality evidence).
3. In selected patients with acute PE without hypotension and with a low bleeding risk in whom there appears to be a high risk of developing hypotension, we suggest administration of thrombolytic therapy (grade 2C—weak recommendation [suggestion]/low quality or very low quality evidence).
4. When a thrombolytic agent is used, we suggest short infusion times (e.g., a 2-hour infusion) (grade 2C—weak recommendation [suggestion]/low quality or very low quality evidence).
5. In patients with acute PE when a thrombolytic agent is used, we suggest administration through a peripheral vein. Central administration is not deemed necessary (grade 2C—weak recommendation [suggestion]/low quality or very low quality evidence). Incidence of major hemorrhage from systemic thrombolytic administration can be as high as 20%, including a 3% to 5% risk of hemorrhagic intracranial complications.
Contraindications to systemic thrombolytic therapy in acute PE include an intracranial neoplasm, recent (i.e., <2 months) intracranial surgery or trauma, active or recent internal bleeding during the prior 6 months, history of a hemorrhagic stroke, bleeding diathesis, severe uncontrolled hypertension (i.e., systolic blood pressure >200 mm Hg or diastolic blood pressure >110 mm Hg), nonhemorrhagic stroke within the prior 2 months, surgery within the previous 10 days, and thrombocytopenia (i.e., <100,000 platelets/mL). Thrombolytic therapy may cause moderate bleeding in menstruating women, but it has not been associated with major hemorrhage. Therefore, menstruation is not a contraindication to thrombolytic therapy.170
Submassive Pulmonary Embolism with Hemodynamic Stability and Right Ventricular Dysfunction
Role of Thrombolytic Therapy in Normotensive Submassive Pulmonary Embolism with Right Ventricular Dysfunction
Patients with known or suspected PE must be treated initially with UFH infusion. It appears that RVD in normotensive PE patients is associated with an increased risk of short-term adverse events and death than in patients without RVD. A number of studies have suggested that thrombolytic therapy should be considered in these patients. In the Management Strategy and Prognosis of Pulmonary Embolism Registry, of 719 acute PE patients with normal systemic arterial blood pressure and echocardiographic evidence of RVD, 550 were treated with heparin alone and 169 patients received thrombolytic therapy. The subgroup analysis demonstrated that patients undergoing thrombolysis had a significantly lower in-house mortality rate at 4.1% in contrast to anticoagulation alone at 10.5%. PE recurrence was observed in 7.7% in the thrombolysis group and in 18.7% in patients receiving anticoagulation alone.2 Although this study demonstrated differences in terms of mortality rates and PE recurrence between the two groups, it must be emphasized that this was a registry trial and not a randomized clinical investigation Therefore, it was subject to unavoidable selection bias.
An important study that attempted to determine whether normotensive patients with PE-induced RVD should receive thrombolytic therapy produced some very interesting conclusions. In a prospective trial (MAPPET-3 [Management Strategies and Prognosis of Pulmonary Embolism-3 Trial]) of 256 patients with PE, the investigators randomized patients to receive heparin plus either recombinant tissue plasminogen activator (rtPA) or placebo. This study used a combined end point of in-hospital death or clinical deterioration (escalation of therapy needed). Clinical deterioration was defined as a requirement for either a catecholamine infusion to treat hypotension or shock, secondary or “rescue” thrombolysis, endotracheal intubation, cardiopulmonary resuscitation, or emergency catheter or surgical embolectomy. Initially the study results seem to support the use of thrombolytic therapy. Patients treated with anticoagulation alone were more likely to die or require treatment escalation than those who received rtPA (24.6% vs. 11.0%; p = 0.006), and importantly, there was no difference in the incidence of major bleeding or intracranial hemorrhage between the two treatment groups. The difference between the two groups was due to a more frequent need for secondary emergency thrombolysis in the heparin group, although the overall mortality rate was not affected by thrombolysis.171 One of the major problems with this study was the lack of a consistent definition of RVD. In this study various modalities were used to define RVD and included echocardiography, right-sided heart catheterization, and even ECG signs of RV strain. Nevertheless, this study has been the catalyst for a large multinational European trial currently under way to attempt to resolve the controversy surrounding the role of thrombolytic therapy in patients with PE who are normotensive but have RVD.
Role of Catheter-Directed Therapy in Normotensive Submassive Pulmonary Embolism with Right Ventricular Dysfunction
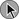 Catheter-directed therapy (CDT) has the potential to locally reduce clot burden in the pulmonary artery by mechanically disrupting the clot or locally infusing low-dose thrombolytics or both. In hemodynamically stable patients with RVD who have right and left pulmonary PE or main pulmonary artery clot CDT may be considered. However, CDT provides an additional option in the treatment of patients with life-threatening PE and a possible alternative to surgical embolectomy and systemic thrombolysis, especially in patients who are poor surgical candidates and have relative contraindications to systemic thrombolysis. Bleeding risk appears to be lower in CDT administered thrombolysis compared to systemic thrombolysis.172
Catheter-directed therapy (CDT) has the potential to locally reduce clot burden in the pulmonary artery by mechanically disrupting the clot or locally infusing low-dose thrombolytics or both. In hemodynamically stable patients with RVD who have right and left pulmonary PE or main pulmonary artery clot CDT may be considered. However, CDT provides an additional option in the treatment of patients with life-threatening PE and a possible alternative to surgical embolectomy and systemic thrombolysis, especially in patients who are poor surgical candidates and have relative contraindications to systemic thrombolysis. Bleeding risk appears to be lower in CDT administered thrombolysis compared to systemic thrombolysis.172
There are no large prospective randomized CDT trials for making evidence-based recommendations. In a meta-analysis of 594 patients with acute massive PE treated with CDT, clinical success was achieved in 86.5%, with success defined as the stabilization of hemodynamics, resolution of hypoxia, and survival to hospital discharge.173 In the same study, 96% of patients received CDT as the first adjunct to heparin with no previous systemic tPA infusion, and 33% of cases were initiated with mechanical treatment alone without local thrombolytic infusion. When injected locally, the required dose of tPA is likely to be lower compared with a full-dose systemic tPA infusion, and there is less risk for bleeding. Indeed, the rate of major complications from modern CDT has proved to be only 2.4%.
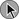 The three types of catheter-based interventions are (1) aspiration thrombectomy, (2) thrombus fragmentation, and (3) rheolytic thrombectomy. Each of these techniques has similar success rates in systematic reviews.172
The three types of catheter-based interventions are (1) aspiration thrombectomy, (2) thrombus fragmentation, and (3) rheolytic thrombectomy. Each of these techniques has similar success rates in systematic reviews.172
The infusion of tPA can be accomplished unilaterally or bilaterally at a total dose rate of 1.0 to 2.0 mg/hour depending on the degree of clot burden. In patients who may be at greater risk for bleeding or may require prolonged infusions greater than 24 hours, fibrinogen levels should be monitored. When the fibrinogen level is decreased below 150 to 200 mg/dL the infusion should be reduced or discontinued.172 The ACCP currently recommends that CDT be considered in selected highly compromised patients with PE who are unable to receive thrombolytic therapy because of bleeding risk. The decision to consider CDT therapy should be multidisciplinary in nature. Case-by-case assessment is based upon risk stratification findings and the patient’s risk of systemic thrombolysis and status as a candidate for surgical management.
Role of Inferior Vena Cava Filters in Patients with Submassive Pulmonary Embolism with Right Ventricular Dysfunction
The goal of the inferior vena cava (IVC) filter intervention is to present further potentially lethal embolic episodes from impacting on an already compromised right ventricle. The current IVC filters that are utilized are retrievable and usually placed by interventional radiology. There has been a marked increase in the use of IVC filters for treatment and prevention of the venous thromboembolic disease. Stein and colleagues showed that the use of IVC filters in the United States has increased from 2000 in 1979 to 92,000 in 2006.174
IVC filters are usually placed in patients with acute PE or DVT who develop active bleeding during anticoagulation and an existing contraindication to anticoagulation. IVC filters should also be considered in the following situations: (1) recurrent thromboembolism despite adequate anticoagulation, (2) the presence of a large free-floating caval thrombus, (3) chronic recurrent PE with pulmonary hypertension, and (4) patients who have had surgical embolectomy or pulmonary endarterectomy.175
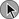 The Greenfield filter was introduced in 1973, and subsequently many types of permanent IVC filters have been developed; these filters are usually placed under fluoroscopic guidance.176 Retrievable IVC filters offer an attractive alternative to the standard permanent filters. These retrievable filters are inserted in patients who may have transient contraindications to anticoagulation or patients who have increased bleeding risks (e.g., trauma patients and neurosurgical patients) or in whom the RV hemodynamics normalize. These filters should be removed within 3 months after implantation; unfortunately, these filters are not removed in “real world” practice.175
The Greenfield filter was introduced in 1973, and subsequently many types of permanent IVC filters have been developed; these filters are usually placed under fluoroscopic guidance.176 Retrievable IVC filters offer an attractive alternative to the standard permanent filters. These retrievable filters are inserted in patients who may have transient contraindications to anticoagulation or patients who have increased bleeding risks (e.g., trauma patients and neurosurgical patients) or in whom the RV hemodynamics normalize. These filters should be removed within 3 months after implantation; unfortunately, these filters are not removed in “real world” practice.175
In patients with submassive PE and RVD who are normotensive the role of the retrievable IVC filter to prevent further embolization is controversial. In a small retrospective study adjunctive placement of an IVC filter, especially in patients with RV strain, resulted in no deaths. In patients who did not receive a filter the in-house mortality rate was 10.2%.179 Prospective studies are needed to further explore the role of retrievable IVC filters in this patient population.
Figure 44.7 illustrates the therapeutic options available for the management of submassive PE in normotensive patients with RVD. In summary, the goals of these interventions are to reduce clot burden and to prevent further embolic impact on a compromised right ventricle.
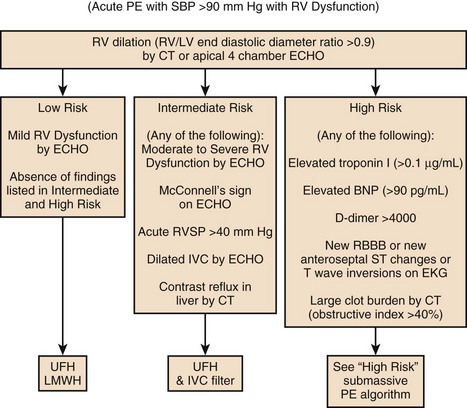
Massive Pulmonary Embolism
Respiratory Support
Although mechanical ventilation is often required in massive PE, the hemodynamic effects of mechanical ventilation may further aggravate the vicious circle of RVD and ischemia by augmenting RV afterload and further decreasing LV preload. Limiting the vasodilating hemodynamic effects of sedation and anesthesia needed for intubation is important. Intubating a patient with massive PE should not be prophylactic, and the risk versus benefit must be weighed carefully for each patient.180
Hemodynamic Support in Massive Pulmonary Embolism
Volume Resuscitation
Fluid should be administered in massive PE with caution. Fluid resuscitation may increase RV afterload precipitating RV failure as well as augment septal ballooning into the left ventricle, further decreasing cardiac output and worsening systemic hypotension. It is therefore recommended that small boluses of fluid (250-500 mL aliquots) be used initially to evaluate affect.181,182
Vasoactive Drugs
There are no randomized controlled studies among different vasoactive agents in massive PE. Norepinephrine (NE) which has both α-adrenergic and β-adrenergic effects, is superior to phenylephrine in a canine model of PE in terms of both improving cardiac output and improving myocardial perfusion.183 NE has been shown to increase cardiac contractility and RV perfusion pressure, thus increasing coronary blood flow. Therefore, NE has become the most frequently used vasoactive agent for hemodynamic support of massive PE.183,184 Dopamine, which possesses both β-adrenergic and, at sufficiently high doses, α-adrenergic agonist effects, appears to offer similar benefit. However, its use may be limited by the development of tachycardia.185
The 1998 PREPIC study and its 8-year follow-up suggested that IVC filters reduce the risk of PE, while increasing the risk of recurrent DVT, and do not reduce the all-cause mortality rate.177 Four hundred patients with proximal DVT were randomized to receive IVC filter or no IVC filter and all had received enoxaparin or UFH as a bridge to warfarin therapy, which was continued for at least 3 months. The primary end points were recurrent DVT, PE, major bleeding, and death. The patients were followed up at day 12, at 2 years, and then annually up to 8 years following randomization. At day 12, there were fewer PEs in the group that received filters (OR 0.22, 95% CI, 0.05-0.90). However, at year 2, there was no significant difference in PE development in the filter group compared with the no-filter group (OR 0.50, 95% CI, 0.19-1.33). Additionally, at year 2, the filter group was more likely to develop recurrent DVT (OR 1.87, 95% CI, 1.10-3.20). At year 8, there was a significant reduction in the number of PEs in the filter group versus the no-filter group (6.2% vs. 15.1%, p = 0.008). However, at 8-year follow-up, IVC filter use was associated with increased DVT (35.7% vs. 27.5%, p = 0.042). There was no difference in mortality rate between the two groups. In summary, the use of IVC filters was associated with decreased incidence of PE at 8 years, offset by higher rates of recurrent DVT and no overall mortality benefit.177,178 Importantly, the indications for IVC filter use in this study differ from the current ACCP guidelines; all patients were given concomitant anticoagulation for at least 3 months, which might not be possible in patients for whom the ACCP recommends IVC filters.
Inotropic Agents
Dobutamine, in combination with NE in the hypotensive patient, should be considered first-line therapy when treating cardiogenic shock secondary to a massive PE. In 10 patients with massive PE, dobutamine was able to increase cardiac index while reducing right atrial pressure, systemic vascular resistance, and PVR.186 Although phosphodiesterase inhibitors have been used to support the circulation in cardiogenic shock secondary to massive PE, these agents should be used with caution given the lack of literature support. Their significant vasodilating capabilities could potentially worsen hypoxemia due to their ameliorating effect on hypoxic vasoconstriction and potentially worsen systemic hypotension.187
Systemic Thrombolysis in Massive Pulmonary Embolism
If there are no contraindications, thrombolytic therapy is the treatment of choice for massive PE resulting in severe hemodynamic and respiratory compromise. This recommendation is sanctioned by the current ACCP guidelines.163 A number of studies have demonstrated the superiority of thrombolytic therapy in improving RV function (24-48 hours) when compared with heparin alone. The studies have not demonstrated an improvement in mortality rate with thrombolysis.166,167,188–190 One reason is that the studies were not large enough for mortality rate to be an outcome measurement. A meta-analysis by Wan and colleagues demonstrated that patients with massive PE had a reduction in mortality rate and PE recurrence and adverse events when compared with heparin alone (19.0% to 9.4%; OR, 0.45; 95% CI, 0.22-0.92).191 If thrombolytic therapy is given within 48 hours of the pulmonary embolic event there may be greater clinical improvement. However, thrombolysis may continue to be beneficial even in patients who have symptoms up to 2 weeks out from their initial PE symptoms.192
Massive PE can lead to cardiac arrest and the initial rhythm may include PEA and asystole. When cardiac arrest occurs, the mortality rate is in the range of 66% to 95%. Thrombolytic therapy given in boluses of 50 mg has been shown in case reports to be lifesaving.193
Inferior Vena Cava Filters in Massive Pulmonary Embolism
An observational study from an international registry that suggests that IVC filter placement in patients with massive PE (i.e., with hypotension) was associated with a reduction in combined end point of mortality rate and recurrent PE. This study cohort was small; however, it appears to be reasonable that IVC filters should be placed in patients with poor cardiopulmonary reserve.194
Unstable PE patients who are treated with standard anticoagulation therapy may benefit further with prophylactic placement of IVC filters. In a recent study by Stein and associates,195 the mortality rate in 38,000 unstable patients treated with standard anticoagulant therapy alone was 51% compared with 33% in 12,850 who had vena cava filters in addition to anticoagulants. This represents a 35% decrease in the case fatality rate when vena cava filters are combined with standard anticoagulant therapy.
Pulmonary Embolectomy in Massive Pulmonary Embolism
The indications for pulmonary embolectomy are patients with documented massive PE confirmed if possible by angiography (CTPA or pulmonary angiography) with persistent hemodynamic instability despite aggressive medical therapy including consideration of the local or systemic thrombolysis. Patients with refractory shock in which massive PE is strongly suspected should be transferred to a center where emergent surgical management for PE is performed. These patients must be urgently transferred to the operating room and on the operating table TEE should be performed to confirm the diagnosis. Despite these efforts, patients who present with cardiac arrest have a high surgical mortality rate.195,196
The mortality rate from pulmonary embolectomy ranges from 30% to 40%. There has been concern that medical therapy may delay definitive surgical therapy and may account for the high mortality rate. Among 1047 patients who underwent pulmonary embolectomy from 1961 to 1984 the average in-house mortality rate was 32%. Using the National Inpatient sample from 1999 to 2008, recently Stein and colleagues showed that the case fatality rates for unstable patients (shock or ventilator-dependent) with PE undergoing pulmonary embolectomy was 40% and in stable patients 24% with an overall case fatality rate of 28%. The case fatality rates were lower in patients with the primary diagnosis of PE who had few comorbid conditions. It was observed in both groups that patients with IVC filters had a lower case fatality rate than those who did not have filters.197,198 More recently the surgical mortality rates for pulmonary embolectomy have decreased at specialized centers (Fig. 44.8, pulmonary embolectomy). A multidisciplinary approach appears to be the key to diagnosis, treatment, and postoperative care, translating to reduced mortality and morbidity rates.199,200
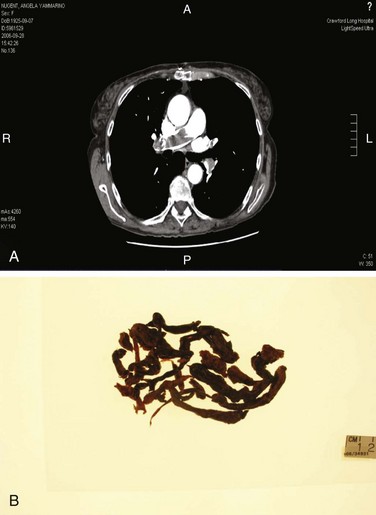
Endovascular Therapy in Massive Pulmonary Embolism
In patients with massive PE who are not candidates for either pulmonary embolectomy or systemic thrombolysis, if there is institutional expertise for endovascular management, this should be considered. The goal is to convert a massive PE into a submassive PE with resultant improvement in the patient’s hemodynamic and respiratory status. Initially CDT provides mechanical debulking of the clot, and if indicated because of persistent elevation of pulmonary artery pressures, infuse low-dose (3 mg/hour) tPA directly into the clot over a 12- to 24-hour period. CDT should be considered and the decision should be made quickly as part of a multidisciplinary discussion.172–203 Complications of CDT are rare, but when they occur they can be serious. They include perforation of the pulmonary artery or perforation of the atrium or ventricle with pericardial tamponade. These interventions should be performed only by clinicians who are experienced in catheter-based intervention and within institutions that have defined protocols and multidisciplinary teams to manage these critically ill patients.
The management of high-risk submassive PE is summarized in Figure 44.9. For the normotensive patient with PE it is important to quickly risk stratify and identify the patient with RVD and large clot burden. If patients in this subset manifest signs of RV injury with persistent elevations in troponin and echocardiographic evidence of RV overloading with impending RV failure, then systemic thrombolytic therapy, local thrombolytic therapy, and surgical management are management options to be considered. In the patient with massive PE, as with submassive PE with RVD, there must be multidisciplinary coordination between interventional cardiology and radiology and cardiothoracic surgery to quickly determine the best treatment approach. Both respiratory and hemodynamic support will allow some time to consider the various management options.
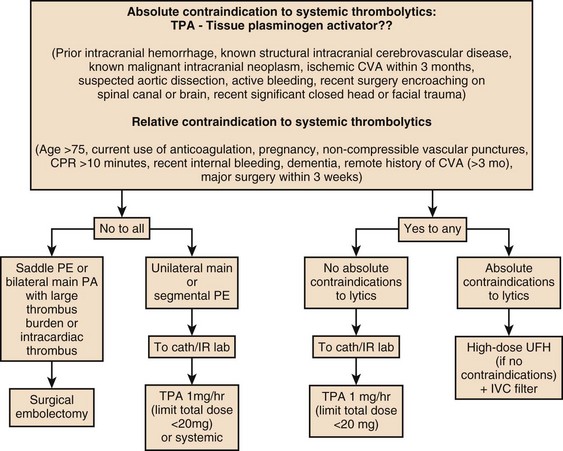
Special Populations
Pregnancy
The incidence of PE during pregnancy ranges between 0.3 and 1 per 1000 deliveries.204 PE is the leading cause of pregnancy-related maternal death in developed countries. In the United States, maternal death from PE ranks third after hemorrhage and hypertensive disorders.105,206 There is a fivefold increased risk of VTE in the pregnant woman because of venous stasis, a prothrombotic state as a result of increased levels of coagulation factors, reduced protein S, and fibrinolytic activity.207 The two major PE risk factors in pregnancy are a personal history of VTE and heritable thrombophilias.208 The risk of PE is higher in the postpartum period, particularly after a cesarean section. The clinical features of PE are no different in pregnancy compared with the nonpregnant state. There have been no prospective trials that have validated existing prediction models to exclude PE in pregnancy. It is been shown from a maternal death review that pregnant women who died from PE are over the age of 35, are obese (body mass index >30), and have chronic medical conditions.209
Diagnosis of Pulmonary Embolism in Pregnancy
Although D-dimer may be elevated in pregnancy, leading to a high rate of false positives, a normal D-dimer value has the same exclusion value for PE in pregnant women as in other patients with suspected PE. However, it has been suggested that D-dimer testing is not recommended for evaluation of suspected VTE in pregnancy or the early postpartum period. Therefore, it should be measured even though the probability of a negative result is lower than in other patients with suspected PE.207
An elevated D-dimer assay should be followed by CUS of the lower extremities. If CUS demonstrates DVT, then anticoagulation can occur and thoracic imaging is not necessary. If CUS is negative and the diagnosis is still suspected, then thoracic imaging is warranted. It is important to note that the upper limit of radiation with regard to the danger of injury to the fetus is considered to be 50 mSv (50,000 mGy), and all radiologic tests (CT and  ) fall well below this limit.208 In a pregnant or postpartum woman suspected of PE with a normal chest radiograph either a ventilation-perfusion scan or a perfusion scan alone will be sufficient. A normal lung scan essentially excludes the diagnosis of PE. Perfusion scanning compares favorably with CT as far as exposure of breast tissue to radiation is concerned.209 If the chest radiograph is abnormal and the lung scan is nondiagnostic, then CTPA is recommended.
) fall well below this limit.208 In a pregnant or postpartum woman suspected of PE with a normal chest radiograph either a ventilation-perfusion scan or a perfusion scan alone will be sufficient. A normal lung scan essentially excludes the diagnosis of PE. Perfusion scanning compares favorably with CT as far as exposure of breast tissue to radiation is concerned.209 If the chest radiograph is abnormal and the lung scan is nondiagnostic, then CTPA is recommended.
Treatment of Pulmonary Embolism in Pregnancy
The medical treatment of PE in pregnancy is heparin—either UFH or LWMH, neither of which crosses the placenta or is found in breast milk in any significant amount. As there are no specific data in the setting of pregnancy, treatment should consist of a weight-adjusted dose of LMWH. Adaptation according to anti-Xa monitoring may be considered in women at extremes of body weight or with renal disease. The heparin treatment should be given throughout the entire pregnancy. From limited data fondaparinux appears efficacious in pregnancy, but bleeding risk is not absent, and care is required when used as second-line therapy.210
The new oral anticoagulants are not to be used during pregnancy. Teratogenic effects, reduced fetal viability, hemorrhagic changes, and placental abnormalities have been found in both dabigatran and rivaroxaban. Rivaroxaban is secreted in breast milk.210 VKAs cross the placenta and are associated with a well-defined embryopathy during the first trimester. Administration of VKAs in the third trimester can result in fetal and neonatal hemorrhage as well as in placental abruption. Warfarin may be associated with central nervous system anomalies in any trimester in pregnancy.208
The management of labor and delivery requires particular attention. Epidural analgesia cannot be used unless LMWH is discontinued at least 12 hours before an epidural approach. Treatment can be resumed 12 to 24 hours after withdrawal of the epidural catheter. In any case, close collaboration among obstetrician, anesthetist, and critical care physician is recommended. After delivery, heparin treatment may be replaced by anticoagulation with VKA. Anticoagulant treatment should be administered for at least 3 months after delivery. VKAs can be given even to breastfeeding mothers.208 Thrombolytic therapy should be used when there is a life-threatening pulmonary embolic event. At the time of delivery, thrombolytic treatment should not be used except in extremely severe cases and if surgical embolectomy is not immediately available. Indications for IVC filters in pregnant women are similar to those in other patients with PE.
In summary, in pregnant women with a clinical suspicion of PE an accurate diagnosis is paramount, as a prolonged course of heparin is required. All diagnostic modalities, including CT scanning, may be used without significant risk to the fetus. LMWHs are recommended in confirmed PE; VKAs are not recommended during the first and third trimesters but could be considered with caution in the second trimester of pregnancy. Anticoagulant treatment should be administered for at least 3 months after delivery.208
Right-Sided Heart Thrombi
Echocardiographically documented right-sided heart thrombi occur in approximately 4% of patients with PE.25,143 These patients have more hemodynamic instability and have a higher mortality rate. In the ICOPER study in patient with acute PE and a free-floating right-sided heart thrombus, the mortality rate was 15% compared to 11% without such thrombi.143
In these patients, the treatment of choice is controversial. Thrombolytic therapy or surgical embolectomy have both been used and there have been multiple reports of successful results. In the ICOPER, thrombolytic treatment was the preferred option, but the 14-day mortality rate was above 20%.143 In contrast, excellent results with thrombolytic therapy were reported in a series of 16 patients, in which 50%, 75%, and 100% of clots disappeared from the right side of the heart within first 2, 12, and 24 hours after administration of thrombolytics. All patients survived 30 days.211 It appears from the literature that treatment with heparin alone is not a treatment option.
For right-sided heart thrombi crossing the interatrial septum (patent foramen ovale) surgical embolectomy is warranted (Fig. 44.10). Whichever therapy is selected, it should be implemented without delay: in the presence of unequivocal echocardiographic visualization of a mobile right-sided heart thrombus, no further diagnostic tests are needed.25,203,212,213
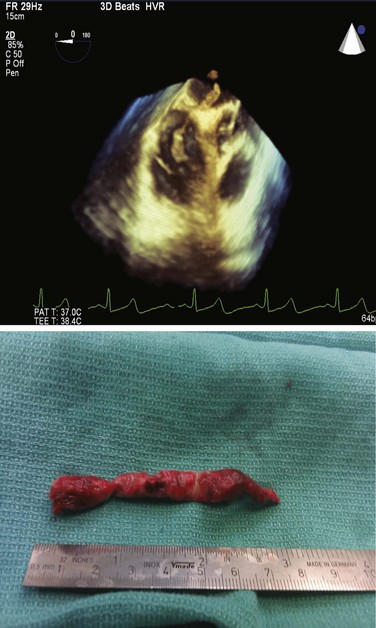
Chronic Thromboembolic Pulmonary Hypertension
 Chronic thromboembolic pulmonary hypertension (CTEPH) is an uncommon but serious sequela of PE.
Chronic thromboembolic pulmonary hypertension (CTEPH) is an uncommon but serious sequela of PE.
CTEPH is an uncommon but serious sequela of PE. Certain patients with PE develop an aberrant and incomplete resolution of the thrombotic event, resulting in endothelialized thrombi and if left unchecked will result in the development of a distal, small vessel vasculopathy. This event results in progressive increases in RV afterload and pulmonary hypertension. When small distal vessel disease develops in patients with CTEPH the patient no longer is a candidate for surgical management.214 The reasons are unclear for this divergent pathway in thrombus resolution. Acquired and hereditary risk factors are suspected. For example, 20% of CTEPH patients have antiphospholipid syndrome and increased levels of factor VIII are found in CTEPH when compared with nonthrombotic pulmonary hypertension and control groups.203,212,213 Characteristics of the initial acute embolic event may be associated with subsequent development of CTEPH. Persistent RVD at discharge and persistent elevation of pulmonary artery pressures may play a role.214,215
In the past it was estimated that the incidence of CTEPH was only 0.1% to 0.5%.214 Recent prospective studies have shown that CTEPH is much more common, with an incidence ranging from 1% to 3.8% of patients who survived their acute pulmonary embolic event. It is estimated that in the United States 6000 to 22,000 new cases potentially may occur in each year.214,216–218
The  lung scan plays a central role in the evaluation of patients with pulmonary hypertension. The
lung scan plays a central role in the evaluation of patients with pulmonary hypertension. The  scan in a patient suspected with CTEPH will show the “mottled” appearance of subsegmental and segmental perfusion defects. Larger defects can also be seen.214 The lung scan is superior to CT angiography in screening patients for CTEPH. In a single center retrospective investigation comparing the lung scan with CT angiography and 227 patients with pulmonary hypertension demonstrated a sensitivity of 97.4% for
scan in a patient suspected with CTEPH will show the “mottled” appearance of subsegmental and segmental perfusion defects. Larger defects can also be seen.214 The lung scan is superior to CT angiography in screening patients for CTEPH. In a single center retrospective investigation comparing the lung scan with CT angiography and 227 patients with pulmonary hypertension demonstrated a sensitivity of 97.4% for  scanning but only 51% for CT angiography in detecting chronic thromboembolic disease.219
scanning but only 51% for CT angiography in detecting chronic thromboembolic disease.219
CT or MRA can be used to plan surgical intervention. Right-sided heart catheterization is required to determine the degree of hemodynamic impairment, and conventional pulmonary angiography can determine the proximal location of the thrombotic obstruction.214
Surgical removal of the obstructing material requires a true endarterectomy as opposed to a simple embolectomy. The current approach to pulmonary endarterectomy requires median sternotomy, cardiopulmonary bypass, and periods of hypothermic circulatory arrest in order to provide adequate visibility. The main pulmonary arteries are incised and the right endarterectomy level within the wall is defined. Thereafter, the plane is followed circumferentially down to the segmental and sometimes subsegmental branches of each lobar artery, a procedure that is performed with the help of special suction dissectors.217,218
The perioperative mortality rate is related to the severity of the disease, with a mortality rate of 4% in patients with a preoperative PVR less than 900 dynes/second/cm−5 and 20% in those with PVR above 1200 dynes/second/cm−5. The functional results of a successful pulmonary thromboendarterectomy are excellent and generally are sustained over time, with a 3-year survival rate of about 80%.219–221
The major causes of postoperative fatality from pulmonary endarterectomy are RV failure related to persistent pulmonary hypertension and acute lung injury. Lifelong anticoagulation and placement of the IVC filter are recommended. In patients who are deemed inoperable because of distal chronic thromboembolic disease or in whom the surgical mortality risk of endarterectomy is high, medical therapy that is developed for idiopathic pulmonary arterial hypertension has been studied in patients with CTEPH. It is important to identify patients as early as possible so they may have the opportunity for pulmonary endarterectomy because the hemodynamic and symptomatic benefits that medical therapy provide are modest.214,222,223
Prophylaxis
The VTE event is a common and recognized complication in critically ill patients. A VTE event can have dire consequences on the critically ill patient with preexisting cardiopulmonary compromise. All critically ill patients have significant risk factors for VTE and also are at higher risk for bleeding. Venous thromboembolic prophylaxis decreases the risk of VTE in ICU patients.17–21 DVT prophylaxis is effective at reducing the risk of death. In several meta-analyses of surgical patients, prophylactic measures reduced the development of thromboembolic events by more than 50% and also the VTE-attributable mortality rate.224
Lack of adherence to evidence based VTE prophylaxis guidelines has been problematic on a national and international scale.13,224 With the implementation of electronic medical admission order sets universal VTE prophylaxis can be achieved. Extended posthospitalization prophylaxis has been recommended in orthopedic surgical and medical patients. The advent of safe and effective prophylaxis strategies makes all critical care patients candidates for some form of preventive therapy on the basis of risk factors present in this population of patients. Several studies suggest that more than 85% of hospitalized patients are receiving some sort of DVT prophylaxis. VTE prophylactic measures are growing in ICUs with quality care initiatives targeting this intervention via bundled care pathways.225,226
Long-Term Prognosis
 Nearly 25% of patients with PE do not survive the first year after the diagnosis. A majority of these patients succumb to underlying conditions, such as cancer or chronic heart disease, rather than PE. After surviving the first year the patient continues facing bleeding complications from anticoagulation therapy, the risk of recurrent VTE, the development of cancer, and the development of CTEPH.227
Nearly 25% of patients with PE do not survive the first year after the diagnosis. A majority of these patients succumb to underlying conditions, such as cancer or chronic heart disease, rather than PE. After surviving the first year the patient continues facing bleeding complications from anticoagulation therapy, the risk of recurrent VTE, the development of cancer, and the development of CTEPH.227
Long-term prognosis may depend on whether the pulmonary embolic event was provoked or unprovoked (occurring in the absence of known risk factors). The prognosis of unprovoked PE may be more unfavorable than that for patients who experience provoked PE. Studies have shown that patients with provoked PE when compared with their unprovoked counterparts are at higher risk for recurrent PE, arterial cardiovascular events, the occurrence of cancer, and CTEPH.228–232
In a recent study Klok and colleagues investigated the long-term prognosis of acute PE in 308 unprovoked patients, 558 patients with provoked PE, and 334 patients without PE for a median follow-up period of 3.3 years. The mortality rate for the acute embolic event was 5.2%. The adjusted HR for recurrent VTE was increased for patients with unprovoked versus provoked PE (HR 2.1; 95% CI, 1.3-3.1) and unprovoked versus patients without PE (HR 10; 95% CI, 4.9-28). Recurrent PE was fatal in 19% of the patients and these fatalities usually occurred after 3 weeks from the initial PE episode. The risk for cancer was higher for the patients after unprovoked PE than for the patients with provoked (adjusted HR 4.4; 95% CI, 2.0-10) and without PE (adjusted HR 2.5; 95% CI, 1.1-2). Patients with unprovoked PE suffered severe cardiovascular disease two to three times more often than the patients from the other two study cohorts (adjusted HR 2.6; 95% CI, 1.5-3.8; and HR 2.4; 95% CI, 1.2-3.7). There was no difference in the pooled risk for adverse outcome of patients with unprovoked and provoked PE, although the risk on all separate end points except for malignancy-related death was markedly higher for the patients with unprovoked PE. From this study, in nearly 4 years after the embolic event, half of the patients diagnosed with acute PE died or were diagnosed with cancer, recurrent VTE, CTEPH, or arterial cardiovascular disease. This study sets forth a clarion call for studies to develop and evaluate accurate prediction strategies for duration of treatment and to provide further insight on clinical surveillance and personal prognostication for the patients who have survived their initial embolic event.233
References
1. Wood, KE. Major pulmonary embolism: Review of a pathophysiologic approach to the golden hour of hemodynamically significant pulmonary embolism. Chest. 2002; 121:877–905.
2. Tapson, VF. Acute pulmonary embolism. N Engl J Med. 2008; 358:1037–1052.
3. Goldhaber, SZ, Visani, L, De Rosa, M. Acute pulmonary embolism: Clinical outcomes in the International Cooperative Pulmonary Embolism Registry (ICOPER). Lancet. 1999; 353:1386–1389.
4. Silverstein, M, Heit, J, Mohr, D, et al. Trends in the incidence of deep-vein thrombosis and pulmonary embolism: A 25-year population-based study. Arch Intern Med. 1998; 158:585–593.
5. Heit, J. The epidemiology of venous thromboembolism in the community. Arterioscl Thromb Vasc Biol. 2008; 28:370.
6. Stein, PD, Matta, F. Acute pulmonary embolism. Curr Probl Cardiol. 2010; 35:314–376.
7. Spyropoulos, A, Lin, J. Direct medical costs of venous thromboembolism and subsequent hospital readmission rates: An administrative claims analysis from 30 managed care organizations. J Manage Care Pharm. 2007; 13:475–486.
8. The Surgeon General’s Call to Action to Prevent Deep Vein Thrombosis and Pulmonary Embolism Available at. Department of Health and Human Services, Washington, DC, 2008. http://www.surgeongeneral.gov/topics/deepvein/
9. Moser, K, Lemoine, J. Is embolic risk conditioned by location of deep-venous thrombosis? Ann Intern Med. 1981; 94:439.
10. Moser, KM, LeMoine, JR, Nachtwey, FJ, et al. Deep venous thrombosis and pulmonary embolism. Frequency in a respiratory intensive care unit. JAMA. 1981; 246:1422–1424.
11. Cade, JF. High risk of the critically ill for venous thromboembolism. Crit Care Med. 1982; 10:448–450.
12. Kapoor, M, Kupfer, YY, Tessler, S. Subcutaneous heparin prophylaxis significantly reduces the incidence of venous thromboembolic events in the critically ill. Crit Care Med. 1999; 27(Suppl):A69.
13. McLeod, AG, Geerts, W. Venous thromboembolism prophylaxis in critically ill patients. Crit Care Clin. 2011; 27:768–780.
14. The PIOPED Investigators. Value of the ventilation/perfusion scan in acute pulmonary embolism. Results of the prospective investigation of pulmonary embolism diagnosis (PIOPED). JAMA. 1990; 263:2753–2759.
15. EINSTEIN PE Investigators. Oral rivaroxaban for the treatment of symptomatic pulmonary embolism. N Engl J Med. 2012; 366(14):1287–1297.
16. The PROTECT Investigators. Dalteparin versus unfractionated heparin in critically ill patients. N Engl J Med. 2011; 364:1304–1314.
17. Crowther, MA, Cook, DJ. Thromboprophylaxis in medical-surgical critically ill patients. Curr Opin Crit Care. 2008; 14:520–523.
18. Chann, CM, Shorr, AF. Venous thromboembolic disease in the intensive care unit. Semin Respir Crit Care Med. 2010; 31:39–46.
19. Cook, D, Attia, J, Weaver, B, et al. Venous thromboembolic disease: An observational study in medical-surgical intensive care unit patients. J Crit Care. 2000; 15:127–132.
20. Crowther, MA, Cook, DJ, Griffith, LE, et al. Deep venous thrombosis: Clinically silent in the intensive care unit. J Crit Care. 2005; 20:334–340.
21. Geerts, WH, Bergqvist, D, Pineo, GF, et al. Prevention of venous thromboembolism: American College of Chest Physicians Evidence-Based Practice Guidelines, 8th ed. Chest. 2008; 133(Suppl):381S–453S.
22. Virchow, RLK. Gesammelte Abhandlungen zur Wissenschaftlichen. In: Thrombose and Emboli. Berlin: Buchhandlung GH-G; 1862.
23. Alikhan, R, Peters, F, Wilmott, R, Cohen, AT. Fatal pulmonary embolism in hospitalized patients: A necropsy review. J Clin Pathol. 2004; 57:1254–1257.
24. Heit, JA, O’Fallon, WM, Petterson, TM, et al. Relative impact of risk factors for deep vein thrombosis and pulmonary embolism: A population-based study. Arch Intern Med. 2002; 162:1245–1248.
25. Torbicki, A, Perrier, A, Konstantinides, S, et al. Guidelines on the diagnosis and management of acute pulmonary embolism: The Task Force for the Diagnosis and Management of Acute Pulmonary Embolism of the European Society of Cardiology (ESC). Eur Heart J. 2008; 29:2276–2315.
26. Kollef, MH, Levy, NT, Ahrens, TS, et al. The use of continuous intravenous sedation is associated with prolongation of mechanical ventilation. Chest. 1998; 114:541–548.
27. De Jonghe, BD, Cook, DJ, Sharshar, T, et al. Neuromuscular disorders in critically ill patients: A systematic review. Intensive Care Med. 1998; 24:1242–1250.
28. Stein, PD, Hull, RD, Kayali, F, et al. Venous thromboembolism according to age: The impact of an aging population. Arch Intern Med. 2004; 164:2260–2265.
29. White, RH. The epidemiology of venous thromboembolism. Circulation. 2003; 107:14–18.
30. Silverstein, RL, Bauer, KA, Cushman, M, et al. Venous thrombosis in the elderly: More questions than answers. Blood. 2007; 110:3097–3101.
31. Anderson, FA, Jr., Spencer, FA. Risk factors for venous thromboembolism. Circulation. 2003; 107:I9–I16.
32. Heit, JA, Silverstein, MD, Mohr, DN, et al. Risk factors for deep vein thrombosis and pulmonary embolism: A population-based case-control study. Arch Intern Med. 2000; 160(6):809–815.
33. Anderson, JAM, Weitz, JI. Hypercoagulable states. Crit Care Clin. 2011; 27:933–952.
34. Greaves, M. Antiphospholipid antibodies and thrombosis. Lancet. 1999; 353:1348–1353.
35. Levine, JS, Branch, DW, Rauch, J. The antiphospholipid syndrome. N Engl J Med. 2002; 346:752–763.
36. Asherson, RA, Khamashta, MA, Ordi-Ros, J, et al. The “primary” antiphospholipid syndrome: Major clinical and serological features. Medicine (Baltimore). 1989; 68:366–374.
37. Alarcon-Segovia, D, Perez-Vazquez, ME, Villa, AR, et al. Preliminary classification criteria for the antiphospholipid syndrome within systemic lupus erythematosus. Semin Arthritis Rheum. 1992; 21:275–286.
38. Vianna, JL, Khamashta, MA, Ordi-Ros, J, et al. Comparison of the primary and secondary antiphospholipid syndrome: A European Multicenter Study of 114 patients. Am J Med. 1994; 96:3–9.
39. Corral, J, Roldan, V, Vicente, V. Deep venous thrombosis or pulmonary embolism and factor V Leiden: Enigma or paradox. Haematologica. 2010; 95:863–866.
40. Laporte, S, Mismetti, P, Décousus, H, et al. Clinical predictors for fatal pulmonary embolism in 15,520 patients with venous thromboembolism. Findings from the Registro Informatizado de la Enfermedad TromboEmbolica venosa (RIETE) Registry. the RIETE Investigators: RIETE. Circulation. 2008; 117:1711–1716.
41. Khorana, AA, Francis, CW, Culakova, E, et al. Thromboembolism is a leading cause of death in cancer patients receiving outpatient chemotherapy. J Thromb Haemost. 2007; 5:632–634.
42. Lyman, GH, Khorana, AA, Falanga, A, et al. American Society of Clinical Oncology guideline: Recommendations for venous thromboembolism prophylaxis and treatment in patients with cancer. J Clin Oncol. 2007; 25:5490–5505.
43. Darze, ES, Latado, AL, Guimarães, AG, et al. Incidence and clinical predictors of pulmonary embolism in severe heart failure patients admitted to a coronary care unit. Chest. 2005; 128:2576–2580.
44. Stein, PD, Sostman, HD, Hull, RD, et al. Diagnosis of pulmonary embolism in the coronary care unit. Am J Cardiol. 2009; 103:881–886.
45. Pulido, T, Aranda, A, Zevallos, MA, et al. Pulmonary embolism as a cause of death in patients with heart failure. An autopsy study. Chest. 2006; 129:1282–1287.
46. Schonhfer, B, Kohler, B. Prevalence of deep-vein thrombosis in the leg in patients with acute exacerbation of chronic obstructive lung disease. Respiration. 1998; 65:173–177.
47. Kyrle, PA, Eichinger, S. Deep vein thrombosis. Lancet. 2005; 365:1163–1174.
48. Nicolaides, AN, Kakkar, VV, Field, ES, Renney, JT. The origin of deep vein thrombosis: A venographic study. Br J Radiol. 1971; 44:653–663.
49. Killewich, LA, Bedford, GR, Beach, KW, Strandness, DE, Jr. Spontaneous lysis of deep venous thrombi: Rate and outcome. J Vasc Surg. 1989; 9:89–97.
50. Dalen, JE, Bauds, JS, Brooke, HL, et al. Resolution rate of acute pulmonary embolism in man. N Engl J Med. 1969; 280:1194.
51. Lensing, AW, Prandoni, P, Brandjes, D, et al. Detection of deep-vein thrombosis by real-time B-mode ultrasonography. N Engl J Med. 1989; 320:342–345.
52. Quintavalla, R, Larini, P, Miselli, A, et al. Duplex ultrasound diagnosis of symptomatic proximal deep vein thrombosis of lower limbs. Eur J Radiol. 1992; 15:32–36.
53. Mustafa, S, Stein, PD, Patel, KC, et al. Upper extremity deep venous thrombosis. Chest. 2003; 123:1953–1956.
54. Martinelli, I, Battaglioli, T, Bucciarelli, P, et al. Risk factors and recurrence rate of primary deep vein thrombosis of the upper extremities. Circulation. 2004; 110:566–570.
55. Joffe, HV, Kucher, N, Tapson, VF, Goldhaber, SZ. Upper-extremity deep vein thrombosis: A prospective registry of 592 patients. for the Deep Vein Thrombosis (DVT) FREE Steering Committee. Circulation. 2004; 110:1605–1611.
56. Monreal, M, Lafoz, E, Ruiz, J, et al. Upper-extremity deep venous thrombosis and pulmonary embolism: A prospective study. Chest. 1991; 99:280–283.
57. Urokinase pulmonary embolism trial. Phase 1 results: A cooperative study. JAMA. 1970; 214(12):2163–2172.
58. Stratmann, G, Gregory, GA. Neurogenic and humoral vasoconstriction in acute pulmonary thromboembolism. Anesth Analg. 2003; 97:341–354.
59. Smulders, YM. Pathophysiology and treatment of haemodynamic instability in acute pulmonary embolism: The pivotal role of pulmonary vasoconstriction. Cardiovasc Res. 2000; 48:23–33.
60. Smith, G, Smith, AN. The role of serotonin in experimental pulmonary embolism. Surg Gynecol Obstet. 1955; 101:691–700.
61. Piazza, G, Goldhaber, SZ. The acutely decompensated right ventricle. Chest. 2005; 128:1836–1852.
62. Lualdi, JC, Goldhaber, SZ. Right ventricular dysfunction after acute pulmonary embolism: Pathophysiologic factors, detection, and therapeutic implications. Am Heart J. 1995; 130:1276–1282.
63. Goldhaber, SZ, Elliott, G. Acute pulmonary embolism: Part I, epidemiology, pathophysiology, and diagnosis. Circulation. 2003; 108:2726–2729.
64. Taylor, RR, Covell, JW, Sonnenblick, EH, Ross, J, Jr. Dependence of ventricular distensibility on filling of the opposite ventricle. Am J Physiol. 1967; 213:711–718.
65. Jardin, F, Dubourg, O, Gueret, P, et al. Quantitative two-dimensional echocardiography in massive pulmonary embolism: Emphasis on ventricular interdependence and leftward septal displacement. J Am Coll Cardiol. 1987; 10:1201–1206.
66. Benotti, JR, Dalen, LE. The natural history of pulmonary embolism. Clin Chest Med. 1984; 5:403–410.
67. McIntyre, KM, Sasahara, AA. The hemodynamic response to pulmonary embolism in patients without prior cardiopulmonary disease. Am J Cardiol. 1971; 28:288–294.
68. UKEP Study Research Group. The UKEP study: Multicentre clinical trial on two local regimens of urokinase in massive pulmonary embolism. Eur Heart J. 1987; 8:2–10.
69. Stein, PD, Beemath, A, Matta, F. Clinical characteristics of patients with acute pulmonary embolism: Data from PIOPED II. Am J Med. 2007; 120:871–879.
70. Santollcandro, A, Predlletto, R, Fomal, E. Mechanisms of hypoxemia and hypocapnia in pulmonary embolism. Am J Respir Crit Care Med. 1995; 152:336–347.
71. Elliot, CJ. Pulmonary physiology in pulmonary embolism. Chest. 1992; 101:163S–171S.
72. Bouchama, A, Curley, W, Al-Dossary, S, et al. Refractory hypercapnia complicating massive pulmonary embolism. Am Rev Respir Dis. 1988; 138:466–468.
73. Kline, JA, Arunachlam, M. Preliminary study of the capnogram waveform area to screen for pulmonary embolism. Ann Emerg Med. 1998; 32:289–296.
74. Hemnes, AR, Newman, AL, Rosenbaum, B, et al. Bedside end-tidal CO2 tension as a screening tool to exclude pulmonary embolism. Eur Respir J. 2010; 35:735–741.
75. West, J, Goodacre, S, Samson, F. The value of clinical features in the diagnosis of acute pulmonary embolism; systematic review and meta-analysis. Q J Med. 2007; 100:763–769.
76. Pollak, CV, Schreiber, D, Goldhaber, SZ, et al. Clinical characteristics, management and outcome of patients diagnosed with acute pulmonary embolism in the emergency department. J Am Coll Cardiol. 2011; 57:700–706.
77. Le Gal, G, Righini, M, Roy, PM, et al. Prediction of pulmonary embolism in the emergency department: The revised Geneva score. Ann Intern Med. 2006; 144:165–171.
78. Wells, PS, Anderson, DR, Rodger, M, et al. Derivation of a simple clinical model to categorize patients’ probability of pulmonary embolism: Increasing the model’s utility with the SimpliRED D-dimer. Thromb Haemost. 2000; 83:416–420.
79. Lucassen, W, Geersing, GJ, Petra, MG, et al. Clinical decision rules for excluding pulmonary embolism: A meta-analysis. Ann Intern Med. 2011; 155:448–460.
80. Bell, WR, Simon, TL, DeMets, DL. The clinical features of submassive and massive pulmonary emboli. Am J Med. 1977; 62:355–360.
81. Stein, PD, Henry, JW. Clinical characteristics of patients with acute pulmonary embolism stratified according to their presenting syndromes. Chest. 1997; 112:974–979.
82. Stein, PD, Willis, PW, III., DeMets, DL. History and physical examination in acute pulmonary embolism in patients without preexisting cardiac or pulmonary disease. Am J Cardiol. 1981; 47:218–223.
83. Stein, PD, Afzal, A, Henry, JW, Villareal, CG. Fever in acute pulmonary embolism. Chest. 2000; 117:39–42.
84. Stein, PD, Goldhaber, SZ, Henry, JW, et al. Arterial blood gas analysis in the assessment of suspected acute pulmonary embolism. Chest. 1996; 109:78–81.
85. Stein, PD, Goldhaber, SZ, Henry, JW. Alveolar-arterial oxygen gradient in the assessment of acute pulmonary embolism. Chest. 1995; 107:139–143.
86. Stein, PD. Arterial Blood Gases and the Alveolar-Arterial Oxygen Difference: Pulmonary Embolism. Baltimore, MD: Williams and Wilkins; 1996.
87. Goldberg, SK, Lipschutz, JB, Fein, AM, et al. Hypercapnia complicating massive pulmonary embolism. Crit Care Med. 1984; 12:686–688.
88. Worsley, DF, Alavi, A, Aronchick, JM, et al. Chest radiographic findings in patients with acute pulmonary embolism: Observations from the PIOPED study. Radiology. 1993; 189:133–136.
89. Elliot, C, Goldhaber, S, Visani, L, et al. Chest radiographs in acute pulmonary embolism. Chest. 2000; 118:33–38.
90. McGinn, S, White, PD. Acute cor pulmonale resulting from pulmonary embolism. JAMA. 1935; 104:1473.
91. Chan, CT, Vilke, GM, Pollack, M, Brady, WJ. Electrocardiographic manifestations: Pulmonary embolism. J Emerg Med. 2001; 21:263–270.
92. Ullman, E, Brady, WJ, Perron, AD, et al. Electrocardiographic manifestations of pulmonary embolism. Am J Emerg Med. 2001; 19:514–519.
93. Stein, PD, Terrin, ML, Hales, CA, et al. Clinical, laboratory, roentgenographic, and electrocardiographic findings in patients with acute pulmonary embolism and no pre-existing cardiac or pulmonary disease. Chest. 1991; 100:598–603.
94. Ferrari, E, Imbert, A, Chevalier, T. The ECG in pulmonary embolism: Predictive value of negative T waves in precordial leads—80 case reports. Chest. 1997; 111:537–543.
95. Gex, G, Gerstel, E, Righini, M, et al. Is atrial fibrillation associated with pulmonary embolism? J Thromb Haemost. 2012; 10:347–351.
96. Stein, PD, Hull, RD, Patel, KC, et al. D-dimer for the exclusion of acute venous thrombosis and pulmonary embolism: A systematic review. Ann Intern Med. 2004; 140:589–602.
97. Geersing, GJ, Erkens, PM, Lucassen, WA, et al. Safe exclusion of pulmonary embolism using the Wells rule and qualitative D-dimer testing in primary care prospective cohort study. BMJ. 2012; 345:e6564.
98. Kearon, C, Ginsberg, JS, Douketis, J, et al. An evaluation of D-dimer in the diagnosis of pulmonary embolism: A randomized trial. Ann Intern Med. 2006; 144:812–821.
99. Kline, JA, Hogg, MM, Courtney, DM, et al. D-dimer and exhaled CO2/O2 to detect segmental pulmonary embolism in moderate-risk patients. Am J Respir Crit Care Med. 2010; 182:669–675.
100. Palareti, G, Cosmi, B, Legnani, C. Diagnosis of deep vein thrombosis. Semin Thromb Hemost. 2006; 32:659–672.
101. Tan, M, van Rooden, CJ, Westerbeek, RE, Huisman, MV. Diagnostic management of clinically suspected acute deep vein thrombosis. Br J Haematol. 2009; 146:347–360.
102. Fraser, JD, Anderson, DR. Deep venous thrombosis: Recent advances and optimal investigation with US. Radiology. 1999; 211:9–24.
103. Johnson, SA, Stevens, SM, Woller, SC, et al. Risk of deep vein thrombosis following a single negative whole-leg compression ultrasound: A systematic review and meta-analysis. JAMA. 2010; 303:438–445.
104. Kearon, C, Julian, JA, Newman, TE, Ginsberg, JS. Noninvasive diagnosis of deep venous thrombosis. McMaster Diagnostic Imaging Practice Guidelines Initiative. Ann Intern Med. 1998; 128:663–677.
105. Goodacre, S, Sampson, F, Thomas, S, et al. Systematic review and meta-analysis of the diagnostic accuracy of ultrasonography for deep vein thrombosis. BMC Medical Imaging. 2005; 5:6.
106. Henry, JW, Stein, PD, Gottschalk, A, et al. Scintigraphic lung scans and clinical assessment in critically ill patients with suspected acute pulmonary embolism. Chest. 1996; 109:462–466.
107. Clemens, S, Leeper, KV. Newer modalities for detection of pulmonary emboli. Am J Med. 2007; 120:S2–S12.
108. Stein, PD, Coleman, RE, Gottschalk, A, et al. Diagnostic utility of ventilation/perfusion lung scans in acute pulmonary embolism is not diminished by pre-existing cardiac or pulmonary disease. Chest. 1991; 100:604.
109. Lesser, BA, Leeper, KV, Jr., Stein, PD, et al. The diagnosis of acute pulmonary embolism in patients with chronic obstructive pulmonary disease. Chest. 1992; 102:17.
110. Stein, PD, Fowler, SE, Goodman, LR, et al. Multidetector computed tomography for acute pulmonary embolism. N Engl J Med. 2006; 354:2317–2327.
111. Remy-Jardin, M, Remy, J, Deschildre, F, et al. Diagnosis of pulmonary embolism with spiral CT: Comparison with pulmonary angiography and scintigraphy. Radiology. 1996; 200:699–706.
112. Becattini, C, Agnelli, G, Vedovati, MC, et al. Multidetector computed tomography for acute pulmonary embolism: Diagnosis and risk stratification in a single test. Eur Heart J. 2011; 32(13):1657–1663.
113. Hopper, KD, King, SH, Lobell, ME, et al. The breast: In-plane x-ray protection during diagnostic thoracic CT—Shielding with bismuth radioprotective garments. Radiology. 1997; 205:853–858.
114. Stein, PD, Chenevert, TL, Fowler, SE, et al. Gadolinium-enhanced magnetic resonance angiography for pulmonary embolism: A multicenter prospective study (PIOPED III). Ann Intern Med. 2010; 152:434–443.
115. Stamm, JA. Risk stratification for pulmonary embolism. Crit Care Clin. 2012; 28:301–321.
116. Sanchez, O, Planquette, B, Gosset-Wolmant, M, et al. Triaging in pulmonary embolism. Semin Respir Crit Care Med. 2012; 33:156–162.
117. Kasper, W, Konstantinides, S, Geibel, A, et al. Prognostic significance of right ventricular afterload stress detected by echocardiography in patients with clinically suspected pulmonary embolism. Heart. 1997; 77:346–349.
118. Sanchez, O, Trinquart, L, Caille, V, et al. Prognostic factors for pulmonary embolism: The PREP study, a prospective multicenter cohort study. Am J Respir Crit Care Med. 2010; 181:168–173.
119. Aujesky, D, Obrosky, DS, Stone, RA, et al. Derivation and validation of a prognostic model for pulmonary embolism. Am J Respir Crit Care Med. 2005; 172:1041–1046.
120. Jimenez, D, Aujesky, D, Moores, L, et al. Simplification of the pulmonary embolism severity index for prognostication in patients with acute symptomatic pulmonary embolism. Arch Intern Med. 2010; 170:1383–1389.
121. Jimenez, D, Aujesky, D, Diaz, G, et al. RIETE Investigators. Prognostic significance of deep vein thrombosis in patients presenting with acute symptomatic pulmonary embolism. Am J Respir Crit Care. 2010; 181:983–991.
122. Vanni, S, Polidori, G, Vergara, R. MD prognostic value of ECG among patients with acute pulmonary embolism and normal blood pressure. Am J Med. 2009; 122:257–264.
123. Toosi, MS, Merlino, JD, Leeper, KV. Electrocardiographic score and short term outcomes of acute pulmonary embolism. Am J Cardiol. 2007; 100:1172–1176.
124. Punukollu, G, Gowda, RM, Vasavada, BC, Khan, IA. Role of electrocardiography in identifying right ventricular dysfunction in acute pulmonary embolism. Am J Cardiol. 2005; 96:450–452.
125. Daniel, KR, Courtney, DM, Kline, JA. Assessment of cardiac stress from massive pulmonary embolism with 12-lead ECG. Chest. 2001; 120:474–481.
126. Daniels, LB, Maisel, AS. Natriuretic peptides. J Am Coll Cardiol. 2007; 50(25):2357–2368.
127. ten Wolde, M, Tulevski, II, Mulder, JW, et al. Brain natriuretic peptide as a predictor of adverse outcome in patients with pulmonary embolism. Circulation. 2003; 107(16):2082–2084.
128. Kucher, N, Printzen, G, Doernhoefer, T, et al. Low pro-brain natriuretic peptide levels predict benign clinical outcome in acute pulmonary embolism. Circulation. 2003; 107(12):1576–1578.
129. Klok, FA, Inge, CMM, Huisman, MV. Brain type natriuretic peptide levels in the prediction of adverse outcome in patients with pulmonary embolism. A systematic review and meta-analysis. Am J Respir Crit Care Med. 2008; 178:425–430.
130. Janata, K, Holzer, M, Laggner, AN, et al. Cardiac troponin T in the severity assessment of patients with pulmonary embolism: Cohort study. BMJ. 2003; 326:312–313.
131. Pruszczyk, P, Bochowicz, A, Torbicki, A, et al. Cardiac troponin T monitoring identifies high-risk group of normotensive patients with acute pulmonary embolism. Chest. 2003; 123:1947–1952.
132. Becattini, C, Vedovati, MC, Agnelli, G. Prognostic value of troponins in acute pulmonary embolism: A meta-analysis. Circulation. 2007; 116(4):427–433.
133. Horlander, KT, Leeper, KV. Troponin levels as a guide to treatment of pulmonary embolism. Curr Opin Pulm Med. 2003; 9:374–377.
134. Ferrari, E, Moceri, P, Crouzet, C, et al. Timing of troponin I measurement in pulmonary embolism. Heart. 2012; 98(9):732–735.
135. Jiménez, D, Aujesky, D, Moores, L, et al. Combinations of prognostic tools for identification of high-risk normotensive patients with acute symptomatic pulmonary embolism. Thorax. 2011; 66:75–81.
136. Lankeit, M, Friesen, D, Aschoff, J, Dellas, C. Highly sensitive troponin T assay in normotensive patients with acute pulmonary embolism. Eur Heart J. 2010; 31:1836–1844.
137. Boscheri, A, Wunderlich, C, Langer, M, et al. Correlation of heart-type fatty acid-binding protein with mortality and echocardiographic data in patients with pulmonary embolism at intermediate risk. Am Heart J. 2010; 160(2):294–300.
138. Dellas, C, Puls, M, Lankeit, M, et al. Elevated heart-type fatty acid-binding protein levels on admission predict an adverse outcome in normotensive patients with acute pulmonary embolism. J Am Coll Cardiol. 2010; 55(19):2150–2157.
139. Lankeit, M, Kempf, T, Dellas, C, et al. Growth differentiation factor-15 for prognostic assessment of patients with acute pulmonary embolism. Am J Respir Crit Care Med. 2008; 177(9):1018–1025.
140. Toosi, MS, Merlino, JD, Leeper, KV. Prognostic value of the shock index along with transthoracic echocardiography in risk stratification of patients with acute pulmonary embolism. Am J Cardiol. 2008; 101(5):700–705.
141. Frémont, B, Pacouret, G, Jacobi, D, et al. Prognostic value of echocardiographic right/left ventricular end-diastolic diameter ratio in patients with acute pulmonary embolism: Results from a monocenter registry of 1,416 patients. Chest. 2008; 133(2):358–362.
142. Logeart, D, Lecuyer, L, Thabut, G, et al. Biomarker-based strategy for screening right ventricular dysfunction in patients with non-massive pulmonary embolism. Intensive Care Med. 2007; 33(2):286–292.
143. Torbicki, A, Galie, N, Covezzoli, A, et al. Right heart thrombi in pulmonary embolism: Results from the International Cooperative Pulmonary Embolism Registry. J Am Coll Cardiol. 2003; 41:2245–2251.
144. Konstantinides, S, Geibel, A, Kasper, W, et al. Patent foramen ovale is an important predictor of adverse outcome in patients with major pulmonary embolism. Circulation. 1998; 97:1946–1951.
145. Leibowitz, D. Role of echocardiography in the diagnosis and treatment of acute pulmonary thromboembolism. J Am Soc Echocardiogr. 2001; 14:921–926.
146. Comess, KA, DeRook, FA, Russell, ML, et al. The incidence of pulmonary embolism in unexplained sudden cardiac arrest with pulseless electrical activity. Am J Med. 2000; 109:351–356.
147. Vedovati, MC, Becattini, C, Agnelli, G, et al. Multidetector computed tomography for acute pulmonary embolism: Embolic burden and clinical outcome. Chest. 2012; 142:1417–1424.
148. Tapson, V. Treatment of pulmonary embolism: Anticoagulation, thrombolytic therapy, and complications of therapy. Crit Care Clin. 2010; 27:825–839.
149. Garcia, DA, Baglin, TP, Weitz, JI. Parenteral anticoagulants: Antithrombotic therapy and revention of thrombosis 9th ed: American College of Chest Physicians evidence based—clinical practice guidelines. Chest. 2012; 141(Suppl 2):e24S–43S.
150. Raschke, RA, Reilly, BM, Guidry, JR, et al. The weight-based heparin dosing nomogram compared with a “standard care” nomogram. A randomized controlled trial. Ann Intern Med. 1993; 119:874–881.
151. Basu, D, Gallus, A, Hirsh, J, Cade, J. A prospective study of the value of monitoring heparin treatment with the activated partial thromboplastin time. N Engl J Med. 1972; 287(7):324–327.
152. Weitz, JI. Low-molecular-weight heparins. N Engl J Med. 1997; 337:688–698.
153. Wilson, SJ, Wilbur, K, Burton, E, et al. Effect of patient weight on the anticoagulant response to adjusted therapeutic dosage of low-molecular-weight heparin for the treatment of venous thromboembolism. Haemostasis. 2001; 31:42–48.
154. Monreal, M, Lafoz, E, Olive, A, et al. Comparison of subcutaneous unfractionated heparin with a low molecular weight heparin (Fragmin) in patients with venous thromboembolism and contraindications to coumarin. Thromb Haemost. 1994; 71(1):7–11.
155. Nagge, J, Crowther, M, Hirsh, J. Is impaired renal function a contraindication to the use of low-molecular-weight heparin? Arch Intern Med. 2002; 162:2605–2609.
156. Martel, N, Lee, J, Wells, PS. Risk for heparin-induced thrombocytopenia with unfractionated and low-molecular-weight heparin thromboprophylaxis: A meta-analysis. Blood. 2005; 106(8):2710–2715.
157. Squizzato, A, Galli, M, Dentali, F, Ageno, W. Outpatient treatment and early discharge of symptomatic pulmonary embolism: A systematic review. Eur Respir J. 2009; 33(5):1148–1155.
158. Aujesky, D, Roy, PM, Verschuren, F, et al. Outpatient versus inpatient treatment for patients with acute pulmonary embolism: An international, open-label, randomised, non-inferiority trial. Lancet. 2011; 378(9785):41–48.
159. Büller, HR, Davidson, BL, Decousus, H, et al. Fondaparinux or enoxaparin for the initial treatment of symptomatic deep venous thrombosis: A randomized trial. Matisse Investigators. Ann Intern Med. 2004; 140(11):867–873.
160. Guyatt, GH, Eikelboom, JW, Gould, MK, et al. Antithrombotic Therapy and Prevention of Thrombosis, 9th ed: American College of Chest Physicians Evidence-Based Clinical Practice Guidelines. Chest. 2012; 141(Suppl 2):e185S–e194S.
161. The EINSTEIN Investigators. Oral rivaroxaban for symptomatic venous thromboembolism. N Engl J Med. 2010; 363:2499–2510.
162. Kaatz, S, Kouides, PA, Garcia, DA, et al. Guidance on the emergent reversal of oral thrombin and factor Xa inhibitors. Am J Hematol. 2012; 87(Suppl 1):S141–S145.
163. Kearon, C, Akl, EA, Comerota, AJ, et al. Antithrombotic therapy for VTE disease: Antithrombotic Therapy and Prevention of Thrombosis, 9th ed: American College of Chest Physicians Evidence-Based Clinical Practice Guidelines. Chest. 2012; 141(Suppl 2):e419S–e494S.
164. Warkentin, TE. Heparin-induced thrombocytopenia in critically ill patients. Crit Care Clin. 2011; 27:805–823.
165. Lo, GK, Juhl, D, Warkentin, TE, et al. Evaluation of pretest clinical score (4 T’s) for the diagnosis of heparin-induced thrombocytopenia in two clinical settings. J Thromb Haemost. 2006; 4(4):759–765.
166. , UPET. Urokinase-Pulmonary Embolism Trial. Circulation. Suppl II, 1973:47–48.
167. USPET. Urokinase Streptokinase Embolism Trial: Phase II results. JAMA. 1974; 229:1606–1613.
168. Daley, MJ, Lat, I. Clinical controversies in thrombolytic therapy for the management of acute pulmonary embolism. Pharmacotherapy. 2012; 32:158–172.
169. British Thoracic Society Standards of Care Committee Pulmonary Embolism Guideline Development Group. British Thoracic Society guidelines for the management of suspected acute pulmonary embolism. Thorax. 2003; 58:470–483.
170. Tapson, V. Thrombolytic therapy in acute pulmonary embolism. Curr Opin Cardiol. 2012; 27:585–591.
171. Konstantinides, S, Geibel, A, Heusel, G, et al. Heparin plus alteplase compared with heparin alone in patients with submassive pulmonary embolism. N Engl J Med. 2002; 347:1143–1150.
172. Kuo, WT. Endovascular therapy for acute pulmonary embolism. J Vasc Interv Radiol. 2012; 23:167–179.
173. Kuo, WT, Gould, MK, Louie, JD, et al. Catheter-directed therapy for the treatment of massive pulmonary embolism: Systematic review and meta-analysis of modern techniques. J Vasc Interv Radiol. 2009; 20:1431–1440.
174. Stein, PD, Matta, F, Hull, RD. Increasing use of vena cava filters for prevention of pulmonary embolism. Am J Med. 2011; 124:655–661.
175. Tschoe, M, Kim, HS, Brotman, DJ, Streiff, MB. Retrievable vena cava filters: A clinical review. J Hosp Med. 2009; 4:441–448.
176. Greenfield, LJ, McCurdy, JR, Brown, PP, Elkins, RC. A new intracaval filter permitting continued flow and resolution of emboli. Surgery. 1973; 73:599–606.
177. Decousus, H, Leizorovicz, A, Parent, F, et al. A clinical trial of vena caval filters in the prevention of pulmonary embolism in patients with proximal deep-vein thrombosis. Prévention du Risque d’Embolie Pulmonaire par Interruption Cave Study Group. N Engl J Med. 1998; 338:409–415.
178. Decousus, H, Barral, F, Buchmuller-Cordier, A, et al. Participating centers eight-year follow-up of patients with permanent vena cava filters in the prevention of pulmonary embolism: The PREPIC randomization group. Circulation. 2005; 112:416–422.
179. Jha, VM, Lee-Llacer, J, Williams, J, et al. Adjunctive inferior vena cava filter placement for acute pulmonary embolism. Cardiovasc Intervent Radiol. 2010; 33:739–743.
180. Hoeper, MM, Granton, J. Intensive care unit management of patients with severe pulmonary hypertension and right heart failure. Am J Respir Crit Care Med. 2011; 184:1114–1124.
181. Mercat, A, Diehl, JL, Meyer, G, et al. Hemodynamic effects of fluid loading in acute massive pulmonary embolism. Crit Care Med. 1999; 27:540–544.
182. Molloy, WD, Lee, KY, Girling, L, et al. Treatment of shock in a canine model of pulmonary embolism. Am Rev Respir Dis. 1984; 130:870.
183. Hirsch, LJ, Rooney, MW, Wat, SS, et al. Norepinephrine and phenylephrine effects on right ventricular function in experimental canine pulmonary embolism. Chest. 1991; 100:796.
184. Ghigenone, M, Girling, L, Prewitt, RM. Volume expansion versus norepiephrine in the treatment of a low cardiac output state complicating an increase in right ventricular afterload in dogs. Anesthesiology. 1984; 185:381.
185. Layish, DT, Tapson, VF. Pharmacologic hemodynamic support in massive pulmonary embolism. Chest. 1997; 111:218–224.
186. Jardin, F, Genevray, B, Brun-Ney, D, Margairaz, A. Dobutamine: A hemodynamic evaluation in pulmonary embolism shock. Crit Care Med. 1985; 12:1009–1012.
187. Tanaka, H, Tajimi, K, Matsumoto, A, Kobayashi, K. Vasodilatory effects of milrinone on pulmonary vasculature in dogs with pulmonary hypertension due to pulmonary embolism: A comparison with those of dopamine and dobutamine. Clin Exp Pharmacol Physiol. 1990; 17(10):681–690.
188. Miller, GA, Sutton, GC, Kerr, IH, et al. Comparison of streptokinase and heparin in treatment of isolated acute massive pulmonary embolism. Br Heart J. 1971; 33:616.
189. Tibbutt, DA, Davies, JA, Anderson, JA, et al. Comparison by controlled clinical trial of streptokinase and heparin in treatment of life-threatening pulmonary embolism. Br Med J. 1974; 1(904):343–347.
190. Ly, B, Arnesen, H, Eie, H, et al. A controlled clinical trial of streptokinase and heparin in the treatment of major pulmonary embolism. Acta Med Scand. 1978; 203:465–470.
191. Wan, S, Quinlan, DJ, Agnelli, G, Eikelboom, JW. Thrombolysis compared with heparin for the initial treatment of pulmonary embolism: A meta-analysis of randomized controlled trials. Circulation. 2004; 110(6):744–749.
192. Daniels, LB, Parker, JA, Patel, SR, et al. Relation of duration of symptoms with response to thrombolytic therapy in pulmonary embolism. Am J Cardiol. 1997; 80:184–188.
193. Jantata, K, Holtzer, M, Kurkciyan, I. Major bleeding complications in cardiopulmonary resuscitation: The place of thrombolytic therapy in cardiac arrest due to massive pulmonary embolism. Resuscitation. 2003; 57(1):49–55.
194. Kasper, W, Konstantinides, S, Geibel, A, et al. Management strategies and determinants of outcome in acute major pulmonary embolism: Results of a multicenter registry. J Am Coll Cardiol. 1997; 30:1165–1171.
195. Stein, PD, Matta, F, Keyes, DC, Willyerd, GL. Impact of vena cava filters on in-hospital case fatality rates from pulmonary embolism. Am J Med. 2012; 125:478–484.
196. Dauphine, C, Omari, B. Pulmonary embolectomy for acute massive pulmonary embolism. Ann Thorac Surg. 2005; 79:1240–1244.
197. Stein, PD, Alnas, M, Beemath, A, Patel, NR. Outcome of pulmonary embolectomy. Am J Cardiol. 2007; 99:421–423.
198. Stein, PD, Matta, F. Case fatality rate with pulmonary embolectomy for acute pulmonary embolism. Am J Med. 2012; 125:471–477.
199. Leacche, M, Unic, D, Goldhaber, SZ, et al. Modern surgical treatment of massive pulmonary embolism: Results in 47 consecutive patients after rapid diagnosis and aggressive surgical approach. J Thorac Cardiovasc Surg. 2005; 129:1018–1023.
200. Kadner, A, Schmidli, J, Schonhoff, F, et al. Excellent outcome after surgical treatment of massive pulmonary embolism in critically ill patients. J Thorac Cardiovasc Surg. 2008; 136:448–451.
201. Kuo, WT, van den Bosch, MA, Hofmann, LV, et al. Catheter-directed embolectomy, fragmentation, and thrombolysis for the treatment of massive pulmonary embolism after failure of systemic thrombolysis. Chest. 2008; 134(2):250–254.
202. Lin, PH, Chen, H, Bechara, CF, Kougias, P. Endovascular interventions for acute pulmonary embolism. Perspect Vasc Surg Endovasc Ther. 2010; 22:171–182.
203. Jaff, MR, McMurty, S, Archer, SL, et al. Management of massive and submassive pulmonary embolism, iliofemoral deep venous thrombosis, and chronic thromboembolic pulmonary hypertension: A scientific statement from the American Heart Association. Circulation. 2011; 123(16):1788–1830.
204. Rutherford, SE, Phelan, JP. Deep venous thrombosis and pulmonary embolism in pregnancy. Obstet Gynecol Clin North Am. 1991; 18:345–370.
205. Berg, CJ, Chang, J, Callaghan, WM, Whitehead, SJ. Pregnancy-related mortality in the United States, 1991-1997. Obstet Gynecol. 2003; 101:289–296.
206. Pabinger, I, Grafenhofer, H. Thrombosis during pregnancy: Risk factors, diagnosis and treatment. Pathophysiol Haemost Thromb. 2002; 32:322–324.
207. Benson, MD. Pulmonary embolism in pregnancy. Consensus and controversies. Minerva Ginecol. 2012; 64:387–398.
208. Heyl, PS, Sappenfield, WM, Burch, D, et al. Pregnancy-related deaths due to pulmonary embolism: Findings from two state-based mortality reviews. Matern Child Health J. 2012.
209. LeChan, WS, Ray, JG, Murray, S, et al. Suspected pulmonary embolism in pregnancy: Clinical presentation, results of lung scanning, and subsequent maternal and pediatric outcomes. Arch Intern Med. 2002; 162:1170–1175.
210. Chan, WS, Lee, A, Spencer, FA, et al. D-dimer testing in pregnant patients: Towards determining the next “level” in the diagnosis of deep vein thrombosis. J Thromb Haemost. 2010; 8:1004–1011.
211. Chartier, L, Bera, J, Delomez, M, et al. Free floating thrombi in the right heart: Diagnosis, management, and prognostic indexes in 38 consecutive patients. Circulation. 1999; 99:2779–2783.
212. Chapoutot, L, Nazeyrollas, P, Metz, D, et al. Floating right heart thrombi and pulmonary embolism: Diagnosis, outcome and therapeutic management. Cardiology. 1996; 87:169–174.
213. Ferrari, E, Benhamou, M, Berthier, F, Baudouy, M. Mobile thrombi of the right heart in pulmonary embolism: Delayed disappearance after thrombolytic treatment. Chest. 2005; 127:1051–1053.
214. Fedullo, P, Kerr, KM, Kim, NH, Auger, WR. Chronic thromboembolic pulmonary hypertension. Am J Respir Crit Care Med. 2011; 183:1605–1613.
215. Wolf, M, Boyer-Neumann, C, Parent, F, et al. Thrombotic risk factors in pulmonary hypertension. Eur Respir J. 2000; 15:395–399.
216. Fedullo, PF, Auger, WR, Kerr, KM, et al. Chronic thromboembolic pulmonary hypertension. N Engl J Med. 2001; 345:1465–1472.
217. Pengo, V, Lensing, AWA, Prins, MH, et al. Incidence of chronic thromboembolic pulmonary hypertension after pulmonary embolism. Thromboembolic Pulmonary Hypertension Study Group. N Engl J Med. 2004; 350:2257–2264.
218. Becattini, C, Agnelli, G, Pesavento, R, et al. Incidence of chronic thromboembolic pulmonary hypertension after a first episode of pulmonary embolism. Chest. 2006; 130:172–175.
219. Tunariu, N, Gibbs, SJ, Win, Z, et al. Ventilation-perfusion scintigraphy is more sensitive than multidetector CTPA in detecting chronic thromboembolic pulmonary disease as a treatable cause of pulmonary hypertension. J Nucl Med. 2007; 48:680–684.
220. Madani, MM, Jamieson, SW. Technical advances of pulmonary endarterectomy for chronic thromboembolic pulmonary hypertension. Semin Cardiovasc Thorac Surg. 2006; 18:243–249.
221. Jamieson, SW, Kapelanski, DP, Sakakibara, et al. Pulmonary endarterectomy: Experience and lessons learned in 1,500 cases. Ann Thorac Surg. 2003; 76:1457–1462.
222. Mayer, E, Dahm, M, Hake, U, et al. Mid-term results of pulmonary thromboendarterectomy for chronic thromboembolic pulmonary hypertension. Ann Thorac Surg. 1996; 61:1788–1792.
223. Archibald, CJ, Auger, WR, Fedullo, PF, et al. Long-term outcome after pulmonary thromboendarterectomy. Am J Respir Crit Care Med. 1999; 160:523–528.
224. Collins, R, Scrimgeour, A, Yusuf, S, et al. Reduction on fatal pulmonary embolism and venous thrombosis by perioperative administration of subcutaneous heparin. Overview of results of randomized trials in general, orthopedic, and urologic surgery. N Engl J Med. 1988; 337:657–662.
225. McMullin, J, Cook, D, Griffith, L, et al. Minimizing errors of omission: Behavioural Reinforcement of Heparin to Avert Venous Emboli: The BEHAVE Study. Crit Care Med. 2006; 34:694–699.
226. Boddi, M, Barbani, F, Abbate, R, et al. Reduction in deep vein thrombosis incidence in intensive care after a clinician education program. J Thromb Haemost. 2010; 8:121–128.
227. Eichinger, S, Weltermann, A, Minar, E, et al. Symptomatic pulmonary embolism and the risk of recurrent venous thromboembolism. Arch Intern Med. 2004; 164:92–96.
228. Carson, JL, Kelley, MA, Duff, A, et al. The clinical course of pulmonary embolism. N Engl J Med. 1992; 326:1240–1245.
229. Sørensen, HT, Mellemkjaer, L, Steffensen, FH, et al. The risk of a diagnosis of cancer after primary deep venous thrombosis or pulmonary embolism. N Engl J Med. 1998; 338:1169–1173.
230. Christiansen, SC, Cannegieter, SC, Koster, T, et al. Thrombophilia, clinical factors, and recurrent venous thrombotic events. JAMA. 2005; 293:2352–2361.
231. Prandoni, P, Lensing, AW, Büller, HR, et al. Deep-vein thrombosis and the incidence of subsequent symptomatic cancer. N Engl J Med. 1992; 327:1128–1133.
232. Klok, FA, Mos, ICM, Broek, L, et al. Risk of arterial cardiovascular events in patients after pulmonary embolism. Blood. 2009; 114:1484–1488.
233. Klok, FA, Zondag, W, van Kralingen, KW, et al. Patient outcomes after acute pulmonary embolism. A pooled survival analysis of different adverse events. Am J Respir Crit Care Med. 2010; 181:501–506.
Attia, J, Ray, JG, Cook, DJ, et al. Deep vein thrombosis and its prevention in critically ill adults. Arch Intern Med. 2001; 161:1268–1279.
Belenkie, I, Dani, R, Smith, ER, Tyberg, JV. Ventricular interaction during experimental acute pulmonary embolism. Circulation. 1988; 78:761–768.
Bonderman, D, Turecek, PL, Jakowitsch, J, et al. High prevalence of elevated clotting factor VIII in chronic thromboembolic pulmonary hypertension. Thromb Haemost. 2003; 90:372–376.
Bonderman, D, Wilkens, H, Wakounig, S, et al. Risk factors for chronic thromboembolic pulmonary hypertension. Eur Respir J. 2009; 33:325–331.
Cook, DJ, Crowther, M, Meade, M, et al. Deep venous thrombosis in medical surgical critically ill patients: Prevalence, incidence and risk factors. Crit Care Med. 2005; 33:1565–1571.
Cutts, BA, Dasgupta, D, Hunt, BJ. New directions in the diagnosis and treatment of pulmonary embolism in pregnancy. Am J Obstet Gynecol. 2013; 208(2):102–108.
Geerts, WH, Code, KI, Jay, RM, et al. A prospective study of venous thromboembolism after major trauma. N Engl J Med. 1994; 331:1601–1606.
Hirsch, DR, Ingenito, EP, Goldhaber, SZ. Prevalence of deep venous thrombosis among patients in medical intensive care. JAMA. 1995; 271:335–337.
Ibrahim, EH, Iregui, M, Prentice, D, et al. Deep vein thrombosis during prolonged mechanical ventilation despite prophylaxis. Crit Care Med. 2002; 30:771–774.
Kudsk, KA, Fabian, TC, Baum, S. Silent deep venous thrombosis in immobilized multiple trauma patients. Am J Surg. 1989; 158:515–519.
Marik, PE, Andrews, L, Maini, B. The incidence of deep venous thrombosis in ICU patients. Chest. 1997; 111:661–664.
McIntyre, KM, Sasahara, AA. Determinants of right ventricular function and hemodynamics after pulmonary embolism. Chest. 1974; 65(5):534–543.
Mclintock, C, Brighton, T, Chunilal, S, et al. Recommendations for the diagnosis and treatment of deep venous thrombosis and pulmonary embolism in pregnancy and the postpartum period. Aust N Z J Obstet Gynaecol. 2012; 52(1):14–22.
Scales, DC, Dainty, K, Hales, B, et al. A multifaceted intervention for quality improvement in a network of intensive care units. JAMA. 2011; 305:363–372.
Scarsbrook, AF, Bradley, KM, Gleeson, FV. Perfusion scintigraphy: Diagnostic utility in pregnant women with suspected pulmonary embolic disease. Eur Radiol. 2007; 17:2554–2560.
Schoepf, UJ, Kucher, N, Kipfmueller, F, et al. Right ventricular enlargement on chest computed tomography: A predictor of early death in acute pulmonary embolism. Circulation. 2004; 110:3276–3280.
Shultz, DJ, Brasel, KJ, Washinton, L. Incidence of asymptomatic pulmonary embolism in moderately to severe injured trauma patients. J Trauma. 2004; 56:727–733.
Stein, PD, Beemath, A, Meyers, FA, et al. Pulmonary embolism and deep venous thrombosis in patients hospitalized with chronic obstructive pulmonary disease. J Cardiovasc Med. 2007; 8:253–257.
Tillie-Leblond, I, Marquette, CH, Perhez, T, et al. Pulmonary embolism in patients with unexplained exacerbation of chronic obstructive pulmonary disease: Prevalence and risk factors. Ann Intern Med. 2006; 144:390–396.
[/level-membership-for-critical-care-medicine-category][not-level-membership-for-critical-care-medicine-category]44
Acute Pulmonary Embolism
PREVALENCE OF VENOUS THROMBOEMBOLISM IN INTENSIVE CARE UNIT PATIENTS
Clinical Presentation on Admission to the Intensive Care Unit
Recognition of Pulmonary Embolism During Intensive Care Unit Admission
DIAGNOSTIC TESTING FOR PULMONARY EMBOLISM
Arterial Blood Gas Measurement
Duplex Compression Ultrasonography
Ventilation-Perfusion Nuclear Scans
Pulmonary venous thromboembolism (VTE) and deep venous thrombosis (DVT) are different manifestations of the same disease. Despite adequate VTE prophylaxis in the intensive care unit (ICU) the presence of a VTE event is a constant threat to critically ill patients. Although the critical care clinician may encounter the embolization of air, fat, infected clots, amniotic fluid, tumor, and inorganic substances, by far VTE will be the most common pulmonary embolic condition encountered. Critical care clinicians will encounter life-threatening pulmonary embolism (PE) ranging from hemodynamically stable patients with varying degress of right ventricular dysfunction (RVD) to acute massive PE, defined as hemodynamic shock from acute PE, which represents the most serious manifestation along the spectrum of venous thromboembolic disease.1–3 According to population-based studies, the annual incidence of VTE in the United States has ranged from 200,000 to a recent estimate of 900,000 cases in which approximately 300,000 people die every year from acute PE.4,5 More recently, in 2006, 467,000 patients were hospitalized with DVT and 247,000 patients were admitted with acute PE.6 VTE is associated with a significant health care costs. Based on 2004 provider payments, the economic burden of VTE costs ranges between $5.8 billion and $7.8 billion.7
The mortality rate can exceed 58% in patients with acute PE presenting with shock, and most of these deaths occur within 1 hour of presentation.1,2 Acute PE is the third most common cause of death among hospitalized patients, and with an aging population the number of people with VTE is expected to increase. For these reasons, the U.S. Surgeon General issued a “Call to Action” in 2008, identifying VTE as a major public health problem.8
The development of DVT occurs largely in the lower extremities and can result in the sequelae of PE, post-thrombotic syndrome, and chronic thrombotic pulmonary hypertension.9 Venous thrombosis is associated with significant morbidity and mortality rates but can be prevented in most patients.2,3
Prevalence of Venous Thromboembolism in Intensive Care Unit Patients
ICU patients represent a heterogeneous population. In prospective screening studies for DVT, in the absence of thromboprophylaxis, the incidence of DVT in a medical-surgical ICU ranges from approximately 13% to 33%.10–12 In an autopsy review of six studies of 436 critically ill patients the incidence of PE ranged from 7% to 27% (mean 13%); PE contributed or directly caused death in 0% to 12% (mean 3%) of the patients. It is important to note that in a majority of the patients there was no antemortem suspicion of fatal PE.13
Contemporary prevalence of VTE that are admitted to or occur in the ICU setting can be obtained from large VTE clinical trials. Approximately 10% of patients entered into the PIOPED-I (Prospective Investigation of Pulmonary Embolism Diagnosis) study for possible PE were hospitalized in a medical or surgical critical care unit.14 In the recent international, multicenter EINSTEIN PE trial,15 which randomized 4823 symptomatic PE patients to rivaroxiban or enoxaparin and a vitamin K antagonist, 600 patients (12.4%) required ICU admission. Finally, in the PROTECT trial (Prophylaxis for Thromboembolism in Critical Care Trial)16 among 3764 critically ill patients enrolled in which a majority were on mechanical ventilator support and 40% were on vasopressor therapy, dalteparin and unfractionated heparin (UFH) were the agents used for VTE prophylaxis. The primary end point was proximal DVT presence. Using compression ultrasonography (CUS), 3.5% of the patients had proximal DVT on admission. The incidence of proximal DVT was 5.1% in the dalteparin group and 5.4% in the UFH group. The proportion of patients with pulmonary emboli was significantly lower with dalteparin (24 patients, 1.3%) than with UFH (43 patients, 2.3%).16
There is controversy over routine DVT screening in the ICU. As mentioned earlier, most ICU patients with DVT are asymptomatic and the clinical signs for DVT are nonspecific. CUS is time consuming and not cost effective. Efforts must be made to ensure adherence to current DVT prophylaxis guidelines. In ICU patients in whom these guidelines cannot not be fully implemented (intracranial, spinal, or leg injuries, or patients at increased bleeding risk) then routine CUS screening may be appropriate.17–21
Risk Factors
In the nineteenth century Virchow described stasis, vascular wall abnormalities, and hypercoagulability as general risk factors for the development of DVT.22
As illustrated in Table 44.1, patients admitted to the ICU possess multiple preexisting VTE risks factors on admission to the ICU, yet in the International Cooperative Pulmonary Embolism Registry (ICOPER) 20% of the patients had idiopathic or unprovoked PE.3
The interaction between patient-related and setting-related risk factors appears to be responsible for the development of the VTE.23–25 Patient-related risk factors are usually acquired and are longstanding. Examples of patient-related risk factors are age, history of prior VTE, malignancy, neurologic and medical illnesses that result in immobility, cardiopulmonary disorders, collagen vascular disease, and vasculitis. In addition, patient-related factors may include genetic and acquired thrombophilia and hormone replacement therapy and oral contraceptive therapy. Setting-related risk factors are usually temporary. Examples of setting-related risk factors include hospitalizations, nursing home placement, and admission to the ICU.
Virchow has described several risk factors unique to the ICU setting that predispose patients to VTE events. In a prospective observational study of 93 medical-surgical ICU patients, the investigators identified several ICU-related factors that are associated with increased risk of VTE, including mechanical ventilation, immobility, femoral venous catheters, sedatives and paralytic drugs, and failure to prescribe VTE prophylaxis. Prolonged mobility in the ICU setting is the result of mechanical ventilation, sedative administration, and paralytic agents. These four factors (mechanical ventilation, immobility, sedation, and paralysis) may converge to greatly increase the risk of VTE.26 Sedation and paralysis also predispose to other ICU complications including increased duration of mechanical ventilation, and critical illness polyneuropathy.27,28
Deep Venous Thrombosis
It is estimated that as high as 95% of clinically significant pulmonary emboli originate from the deep veins of the lower extremity. The proximal deep veins of the leg are the most common sites of origin of clots that embolize to the pulmonary circulation. A general consensus suggests that clinically significant proximal DVTs and risk for PEs originate by proximal extension of calf DVT.47,48 Venous thrombi are created in the setting of low flow and low shear stress. These thrombi consist of fibrin strands, red blood cells, and platelets. The thrombi form in the valve pockets of calf veins and extend to the proximal veins.48 After the thrombosis is formed there is raised venous capillary pressure, which increases the transcapillary filtration and the pressure rate, resulting in edema. In approximately 50% of patients, venous outflow obstruction decreases within 3 months by lysis and recanalization.49 Patients who have early edema are most likely to have residual thrombosis. Clots that continue to propagate have a greater risk to break apart and lead to embolization. This generally occurs more frequently in the first few days after clot formation. Clots that do not continue to propagate resolve by either fibrinolysis or organization. These processes generally occur within 7 days of clot formation.49,50
In general only 25% of patients with suspected DVT actually have it.47 The detection of DVT in critically ill patients is hampered by the patient’s inability to report symptoms and the unreliability of physical signs of DVT. CUS is noninvasive and highly sensitive and specific for the diagnosis of proximal lower extremity DVT.51 Full compressibility of either the femoral or popliteal veins excludes proximal DVT51 when compared to the gold standard demography; CUS has a sensitivity of 97% to 100% and a specificity of 98% to 99% for the detection of proximal thrombosis. Ultrasonography is less accurate in the diagnosis of distal calf vein thrombosis.51,52 Whether high-risk critically ill patients should be systematically screened for DVT using CUS, given the high incidence of DVT and limitation of clinical examination, is unresolved.13,17
Upper extremity deep venous thrombosis (UEDVT) accounts for approximately 4% of the venous thromboembolic events.53 Primary UEDVT makes up one third of all UEDVTs. Primary UEDVTs appear to be associated with inherited thrombophilic conditions, and the risk markedly increased to 14-fold when oral contraceptives were used.54 Catheter UEDVTs are more associated with the placement of a central venous catheter, and these patients are more likely to be inpatients than outpatients, as demonstrated by a large registry.55 In addition, patients with cancer have an increased occurrence of UEDVT when a central venous catheter is placed in these areas.55 Catheter-related UEDVT may have a higher pulmonary embolic potential than primary UEDVT.52
Pathophysiology
Nearly two thirds of patients who die from pulmonary thromboembolism die within 1 hour of presentation, but anatomically massive pulmonary emboli (>50% obstruction of the pulmonary circulation) are responsible for only half of the deaths. The term major pulmonary thromboembolism has been used to describe any pulmonary thromboembolus that results in a hemodynamically significant event.1 In patients with pulmonary thromboembolism, hemodynamic presentation is an important predictor of survival. The Urokinase Pulmonary Embolism Trial (UPET) demonstrated that the presence of hemodynamic decompensation was associated with a sevenfold increase in mortality rate.56 ICOPER confirmed these results by demonstrating a fourfold increase in mortality rate for those patients with hemodynamic instability.3 Because of the associations between outcome from pulmonary thromboembolism and shock or hypotension, aggressive intervention in patients thought to have pulmonary thromboembolism has given rise to the term golden hour, during which timely diagnosis and treatment are paramount.1
Hemodynamic Consequences
The principal pathophysiologic effects of pulmonary thromboembolism result from the acute impaction of material into pulmonary circulation and the resulting vascular obstruction and humoral mediator release as depicted in Figure 44.1.57 In nonthrombotic obstruction of the pulmonary vasculature, obliteration of 60% to 70% of the vascular tree is required to cause an elevation of the pulmonary artery pressure. In contrast, only 30% of the vascular tree must be obstructed in pulmonary thromboembolism for elevation of the pulmonary artery pressure to be achieved.58,59 Therefore, factors other than simple mechanical obstruction of the pulmonary vascular system play a role in the elevation of the pressures in the pulmonary vasculature during pulmonary thromboembolism.
The production of platelet-derived vasoconstrictors may play a role in augmenting the increase in pulmonary vascular resistance (PVR) (Fig. 44.2).57,58,60 Thromboxane A2 (TxA2) is a potent vasoconstrictor and is the end product of arachidonic acid metabolism. TxA2 is produced by endothelial cells, and even in greater quantities by platelets in response to platelet aggregation. It has been demonstrated that there is increased production of TxA2 in the early phase of PE, which contributes to the PVR elevation and may be attenuated by cyclooxygenase (COX) inhibition in animal models.57 Serotonin (5-hydroxytryptamine) is one of the most potent pulmonary vasoconstrictors. Infusion of serotonin in an animal model can actually simulate the signs and symptoms of PE. Like TxA2, platelets are the primary source of serotonin production in PE. Serotonin antagonism can reduce PVR.57,60
There appears to be nearly an exponential increase in VTE events with age.29 The elderly demonstrate an incidence of 300 to 500 cases/100,000 persons compared to 30 cases/100,000 of persons age 25 to 35 years.29 A longitudinal study of 855 men followed from the ages of 50 to 80 years demonstrated the cumulative incidence of a VTE event was 0.5% by the age of 50 years and increased to 10.7% by the age of 80 years.3 The risk of VTE recurrences increases with age. In the elderly the PE rates are rising faster than the DVT rates; thus, a VTE event in an elderly person is more likely to be a PE, with its potential lethality.3,30
Anderson and Spencer31 reviewed multiple risk factors associated with a diagnosis of VTE and found that the risk factors were not equal in their impact on VTE occurrence. The strongest risk factors were hip and leg fractures, major general surgery, hip or knee replacement, major trauma, and spinal cord injury. Moderate risk factors included arthroscopic knee surgery, central venous lines, chemotherapy, congestive heart failure, hormonal replacement or oral contraceptives, cancer, pregnancy, thrombophilia, and prior VTE.31 Heit and associates32 found that the hospital setting, nursing home, or other chronic facility confinement was an independent risk factor for the VTE. This is most likely reflective of the various degrees of immobility that these patients experience.
Several acquired and inherited hypercoagulable states have been shown to increase the risk of VTE and are collectively known as thrombophilia. Major acquired hypercoagulable conditions include malignancy, myeloproliferative disorders, and antiphospholipid syndrome (APS).33 DVT, the most common manifestation of the APS, occurs in 29% to 55% of patients with the syndrome, and about half of these patients have pulmonary emboli.34–36
The occurrence of a VTE event in a young adult alerts the clinician to the potential of an underlying inheritable thrombophilic condition. Nearly half the patients with a VTE event before the age of 45 have an associated inherited disorder.33 Factor V Leiden mutation causing resistance to activated protein C is the most common inherited risk factor. Factor V Leiden mutation is present in up to 5% of the normal population and is the most common cause of familial thromboembolism.33 Factor V Leiden mutation appears to be more associated with the development of DVT rather than PE.37 Primary or acquired deficiencies in protein C, protein S, and antithrombin III are other risk factors and their thrombotic expression is variable.33
Several primary disorders are associated with a high risk for fatal VTE. Patients frequently present with massive PE, and ICU admission and management is required. From the RIETE registry, risk factors of immobilization secondary to neurologic disease, cancer, and cardiopulmonary disorders had two- to threefold increased risk for fatal PE.38 In hospitalized cancer patients, VTE is the second leading cause of death.39 The risk of VTE in cancer patients undergoing surgery is three to five times greater than that in surgical patients without cancer. Moreover, cancer patients with symptomatic DVT exhibit a high risk of recurrent VTE.40 Heart failure is a frequent admission diagnosis in the coronary care unit. In patients with severe heart failure (ejection fractions less than 30%) PE is a major cause of morbidity and mortality rates.41,42 Among 198 patients with severe heart failure in the coronary care unit, 9.1% developed PE during their hospitalization, despite a majority receiving thromboprophylaxis.41 In an autopsy series of cardiac patients nearly a quarter had PE and the diagnosis was unsuspected in 82% of the patients.43 Acute exacerbation of chronic obstructive pulmonary disease (COPD) is a common reason for admission to the general hospital ward service and the ICU. Increase work of breathing leads to immobility, which is a risk factor of DVT. In a prospective cohort of 196 patients with COPD exacerbation, all patients had ultrasonography of the lower extremities performed. Among the 196 patients, 21 (10.7%) had DVT and the majority was asymptomatic.44 Moreover, hospitalized patients with unexplained exacerbations of COPD, when routinely evaluated, show demonstrated PE in 25% to 29%.45,46
As shown in Figure 44.1, the degree of mechanical obstruction, the interplay of neurohumoral mediators, and the patient’s underlying cardiopulmonary status will dictate the degree of RVD. A rapid rise in the pulmonary artery resistance produces a significant rise in right ventricular (RV) afterload, thereby causing RV dilatation. The dilation of the right ventricle results in shifting of the interventricular septum into the left ventricle, decreasing left ventricular (LV) preload.61–63 Moreover, RV dilation potentially promotes an increase in constrictive forces by the pericardium, leading to reduced LV compliance. Decreasing LV preload and compliance subsequently lead to lowering of the cardiac output and systemic hypotension with resultant decrease in aortic perfusion pressure and decreasing right coronary perfusion pressure.64,65 A compromised and burdened right ventricle is a direct result of oxygen demand outstripping oxygen supply, creating an ischemic environment for the right ventricle. The functional status of the cardiopulmonary system is very important in the initial hemodynamic presentation and is a major determinant of short- and long-term outcome.66 In patients without cardiopulmonary disease the anatomic and humoral obstruction must be 75% or greater to cause a mean pulmonary artery pressure (MPAP) of 40 mm Hg or higher, which is not sustainable; even the previously healthy heart may proceed to cardiovascular collapse and fail.66 Right atrial pressure (RAP) elevation occurs when the MPAP is 30 mm Hg or higher and obstruction exceeds 35% to 40%.67 Many patients who have a pulmonary embolic event have underlying cardiopulmonary disease. In these patients cardiopulmonary deterioration (CPD) may occur with a lesser degree of vascular obstruction.66 This is illustrated by the UKEP (Urokinase-Embolie Pulmonaire) Trial in which an initial presentation of shock with acute PE was seen in 56% of patients who had prior cardiopulmonary disease compared to 2% without cardiopulmonary disease. Obstruction of greater than 50% was uncommon.68
Respiratory Consequences
Hypoxemia is the most common gas exchange abnormality in patients. However, the PaO2 and alveolar-arterial (A-a) gradient have been reported to be normal in approximately 30% of patients with PE.69 Figure 44.2 summarizes the gas exchange abnormalities as a result of the pulmonary embolic event. Ventilation-perfusion mismatch and low mixed venous oxygen levels are the prevalent causes of hypoxemia.70,71 As pulmonary artery pressures increase, right-to-left flow across a patent foramen ovale (PFO) may occur. Endothelial damage may result from hypoxic exposure and further lead to pulmonary vasoconstriction. Hypoxemia may even promote a prothrombotic and antifibrinolytic effect.57
In acute PE an increase in alveolar dead space impairs CO2 elimination; however, the increase in minute ventilation results in hypocapnia. In patients with massive PE there can be a paradoxical elevation of the PaCO2 as a result of a marked increase in dead space ventilation.72 In PE patients because of an increase in alveolar dead space there is a reduction in alveolar CO2 content, which can be detected by capnography by a reduction in the capnographic waveform area. In one study, PE patients significantly demonstrated a reduction in the capnographic waveform area when compared to patients without PE.73 There is widening of the PETCO2-PaCO2 gradient. Bedside PETCO2 coupled with a Wells score has been shown to have a high negative predictive value.74
Clinical Presentation
The signs and symptoms of acute PE are at best nonspecific. The presence of syncope, current DVT, hemoptysis, leg swelling, active cancer, surgery, leg pain, and shock each marginally increases the probability of PE. Further, the absence of dyspnea or tachycardia marginally reduces the probability of PE.75
Approximately 50% of patients with acute PE present to the emergency department. In the Emperor Registry, among 1880 documented PE patients from 22 U.S. emergency departments, the most common presenting signs and symptoms were dyspnea at rest (50%), pleuritic chest pain (39%), dyspnea with exertion (24%), and syncope, which occurred in 5% as the initial presentation of acute PE. Only 58 patients (3%) had systolic blood pressures of 90 mm Hg or lower on presentation.76
Clinical prediction rules have been used in the emergency department setting to determine the pretest probability of acute PE.77 The most widely known prediction model has been the Wells score and it can allow for a dichotomous classification.78,79 Other prediction scores have been used, such as the Geneva and the modified Geneva.77,78 All prediction scores appear to have similar accuracy but are influenced by the local prevalence of PE. The clinical prediction scores are enhanced in identifying the patient with low probability of PE coupled with low D-dimer value.79
Clinical Presentation on Admission to the Intensive Care Unit
In patients admitted to the ICU with PE the clinical manifestations may be more demonstrative, as shown in Table 44.2. In the Urokinase Pulmonary Embolism Trial (UPET), the clinical features of massive PE were evaluated. Sudden unexplained onset of dyspnea was the most common symptom and was present in 80% of the patients. A majority of the patients had a well-defined risk factor. Tachypnea and tachycardia were present in 88% and 63% of the patients, respectively. Accentuated second heart sound was noted in 67%, and S3 or S4 gallop was present in 47%. Clinical shock was noted in 12%.80 Syncope can occur. Retrosternal nonpleuritic chest pain may mimic the pain experienced with a myocardial infarction and represent demand ischemia of the right ventricle. The differential diagnosis of pulseless electrical activity (PEA) is massive PE and should be expected when there is a known major risk factor.
Table 44.2
PE, pulmonary embolism; PEA, pulseless electrical activity.
*Adapted from Stein PD, Willis PW III, DeMets DL: History and physical examination in acute pulmonary embolism in patients without pre-existing cardiac or pulmonary disease. Am J Cardiol 1981;47;218-223.
The signs and symptoms of submassive PE will depend upon the degree of RVD. Dyspnea may be progressive over the last 4 to 6 days. Chest pain can be either pleuritic or nonpleuritic in nature. If pulmonary infarction occurs as result of distal embolization, then hemoptysis may occur. From the PIOPED II database, 92% of patients with main or lobar pulmonary emboli had dyspnea or tachycardia in contrast to segmental emboli dyspnea and tachycardia, where 65% had these findings.81,82
Recognition of Pulmonary Embolism During Intensive Care Unit Admission
When a patient is admitted to the ICU for a non-PE-related clinical diagnosis, the abrupt and unexplained occurrence of some combination of chest pain, dyspnea, tachypnea, hypotension, and hypoxemia should raise the suspicion of PE. Unexplained fever with and without mild leukocytosis could represent a possible VTE event.83 Hemoptysis can occur as a result of small distal pulmonary emboli causing alveolar hemorrhage. If PE occurs while the critically ill patient or postoperative patient is on mechanical ventilation, the clinical suspicion and conformation can be difficult. An increase in minute ventilation dead space or an increase in supplemental oxygen may be clues to an embolic event. If the patient has end-tidal CO2 (PETCO2) monitoring an increase in the PaCO2-PETCO2 gradient may occur.73
Diagnostic Testing for Pulmonary Embolism
Arterial Blood Gas Measurement
Hypoxemia and an increased alveolar-arterial gradient are usually seen in PE but can be normal in about 14% and are most likely the result of small embolic events.84,85 The initial acid-base disturbance is a respiratory alkalosis, which may evolve to a metabolic (lactic) acidosis as hypotension and shock ensue. A linear relationship between PE severity and PaO2 in patients without prior cardiopulmonary disease has been described. In the UPET and PIOPED trials 12% and 19% of patients, respectively, had PaO2 80 mm Hg or greater.86 Hypocarbia is usually present with acute PE because of hyperventilation, but hypercapnia can occur with large PEs producing increased dead space.87,72
Chest Radiography
The chest radiograph is abnormal in most cases of PE; however, radiographic findings possess inadequate sensitivity to rule out PE. Common radiographic abnormalities include atelectasis, pleural effusion, parenchymal opacities, and elevation of a hemidiaphragm. Radiographic findings of pulmonary infarction often show a wedge-shaped, pleural-based triangular opacity with an apex pointing toward the hilus (Hampton hump). Focal oligemia or decreased vascularity has been known as the Westermark sign. These findings are suggestive of PE but are infrequently prospectively observed.88,89 Radiographic findings of RVD may include a prominent central pulmonary artery (knuckle sign) and cardiomegaly. In one large prospective cohort of PE patients a normal chest radiograph was found in 24% of the patients.89 A normal-appearing chest radiograph in a patient with severe dyspnea and hypoxemia, is suggestive of a pulmonary embolic event.
Electrocardiography
In 1935 McGinn and White first described the association between acute PE and specific electrocardiogram (ECG) changes when they noted the S1Q3T3 pattern in seven patients with acute cor pulmonale.90 A normal ECG can occur with PE in a frequency ranging from 9% to 30%. The most common ECG abnormalities in the setting of PE are tachycardia and nonspecific ST-T wave abnormalities. The classic findings of right-sided heart strain and acute cor pulmonale are tall, peaked P waves in lead II (P pulmonale); right-axis deviation; right bundle branch block; and an S1Q3T3 pattern. Only 20% of patients with proven PE have any of these classic ECG abnormalities.91–93 T-wave inversion in the precordial leads representing subepicardial ischemia may be seen. The presence of this ECG finding is associated with increased degree of vascular obstruction and elevation of the pulmonary artery (PA) pressure.94 The new onset of atrial fibrillation has been thought to be associated with a pulmonary embolic event. In a recent study the occurrence of atrial fibrillation did not in general increase the probability of PE.95 However, if chest pain were the only symptom, the presence of atrial fibrillation increases the probability of PE (odds ratio [OR] 2.42, 95% confidence interval [CI] 0.97-6.07).
D-Dimer Assay
The serum D-dimer is a degradation product produced by plasmin-mediated proteases of cross-linked fibrin. D-dimer is measured by a variety of assays. These assays are categorized as quantitative, semiquantitative, qualitative rapid enzyme-linked immunosorbent assays (ELISAs); quantitative and semiquantitative latex; and whole-blood assays. The quanitative rapid ELISAs are the best assays to use in terms of sensitivity and likelihood ratio.96 When clinical prediction scores indicate that the patient has a low or moderate pretest probability of PE, D-dimer testing is useful to further define the likelihood of PE. Negative results on a high-sensitivity D-dimer test in a patient with a low pretest probability of PE (Wells rule) are associated with a low likelihood of VTE and reliably exclude PE.97 In a large prospective, randomized trial in patients with a low probability of PE who had negative D-dimer results, not performing additional diagnostic testing was not associated with an increased frequency of symptomatic VTE during the subsequent 6 months.98
D-dimer testing loses its ability to predict the likelihood of PE in older patients and in the presence of cancer, pregnancy, and various inflammatory or infectious disorders. If the pretest probability for PE is high, D-dimer testing should not be performed and objective testing should be done.96
Combining D-dimer results with measurement of the exhaled end-tidal ratio of carbon dioxide to oxygen (PETCO2/PO2) can be useful for diagnosis of PE. Kline and colleagues demonstrated in moderate-risk patients a positive D-dimer (>499 ng/mL) and a PETCO2/PO2 less than 0.28 significantly increased the probability of finding segmental or larger PE on computed tomography (CT) multidetector-row pulmonary angiography.99 In contrast, a PETCO2/PO2 greater than 0.45 predicted the absence of segmental or larger PE.
Duplex Compression Ultrasonography
Currently, DVT is largely diagnosed by duplex CUS. When using CUS, the ultrasonographic transducer pressed against veins shows they are easily compressed, but the muscular arteries are extremely resistant to compression. Where DVT is present, the veins do not collapse completely when pressure is applied using the ultrasonographic probe.100–102
Before an ultrasonographic scan can be considered negative, the entire deep venous system must be examined using centimeter-by-centimeter compression testing of every vessel. The risk of a DVT event following a single whole leg CUS in 3 months is 0.57% (95% CI 0.25-0.89).103
CUS is associated with a sensitivity of 97% for proximal DVT, 72% for distal DVT, and a specificity of 94%.104 In a meta-analysis of the role of CUS in DVT, CUS alone had a sensitivity of 93.8% (92.0-95.3%) for proximal DVT, 56.8% (49.0-66.4%) for distal DVT, and specificity of 97.8% (97.0-98.4%).105
Ventilation-Perfusion Nuclear Scans
The scan has a limited role in the diagnosis of acute PE in the ICU, although its diagnostic accuracy is no different from that in non–critically ill populations.106 Because of the advancing technology of CT scanning, the lung scan is used in the critical care units in special circumstances such as renal insufficiency or in the patient who is not intubated and capable of transport from the ICU to the nuclear medicine suite for a full view scan. If the institution has a portable lung scanner this can be used in the ICU but the number of images is reduced when compared to the standard scan.
A high-probability scan, as shown in Figure 44.3, is sufficient diagnostic evidence of PE to begin anticoagulation therapy, and a normal scan is considered sufficient evidence to exclude PE. However, the frequency of low- or intermediate-probability scans (indeterminate scans) can be as high as 50% to 70%, with a 10% to 50% probability of PE. Therefore, it is impossible to initiate anticoagulation therapy based on this indeterminate lung scan probability. In the first PIOPED study, only 40% of patients with PE had a high-probability scan result, whereas another 40% of patients with PE had an indeterminate result and 14% had a low-probability result. A high-probability scan had a positive predictive value of 88% in patients without prior history of PE. The combination of a high clinical probability with the high-probability lung scan resulted in a 96% positive predictive value. The negative predictive value of a low-probability scan was 88% and improved to 96% when combined with a low clinical probability. Thus, a low-probability scan with a low clinical probability score rules out PE as definitively as a normal scan. Finally, 39% of the patients had intermediate scans and 32% of these patients had angiographically proven PE.14,107
Preexisting cardiopulmonary disease diminishes the clinical utility of scans by increasing the prevalence of nondiagnostic scans. Nondiagnostic scans are more likely to occur in patients with preexisting cardiopulmonary disease. Lung scans are not useful to either establish or rule out the diagnosis of PE in more than 80% of cases. A similar disadvantage of lung scans is noted in critically ill patients with prior cardiopulmonary disease. Nevertheless, lung scans can be performed in the critically ill, even if patients are receiving mechanical ventilation, and chest radiographic abnormalities can be used as a surrogate of ventilation defects. Despite the limited ventilation component under positive-pressure respiration, the perfusion component is not significantly affected by mechanical ventilation. scans remain a useful tool in patients with contraindications to pulmonary angiography or CT angiography.108,109
Multidetector Computed Tomography Scan of the Chest
The PIOPED II study firmly established the role of CT pulmonary angiography (CTPA) and largely replaced ventilation-perfusion () lung scintigraphy as the main imaging modality in suspected PE.110 The technology is continually evolving and its use is widespread. As illustrated in Figure 44.4, CTPA provides direct visualization of emboli throughout the pulmonary arterial vasculature. The advantage of CTPA over pulmonary angiography and lung scan is the discovery of an alternative diagnosis when PE is not found on CTPA. In one study, an alternative diagnosis was established in 57% of patients who did not have a PE.111 Currently, CTPA is the “gold standard” for PE diagnosis. A meta-analysis of 23 studies with 4657 patients with a negative CTPA who did not receive anticoagulation showed a 3-month rate of subsequent VTE of 1.4% (95% CI 1.1-1.8) and a 3-month rate of fatal PE of 0.51% (0.33-0.76), which compares favorably with the results noted after a normal “gold standard” invasive contrast pulmonary angiogram.112 The increased use of CTPA raises concerns about the increased incidence of cancer attributable to radiation.113 For this reason, combined use of CTPA and CT leg venography should not be performed because the latter adds little to diagnostic utility. In the PIOPED II trial, no patient with PE or DVT would have been undiagnosed if imaging of the pelvic and proximal leg veins had been omitted.107,110
Figure 44.4 Extensive bilateral lobar pulmonary emboli.
Magnetic Resonance Imaging
In the PIOPED II study, CT angiography of the chest, as many as 25% of patients had a contraindication for the test. Preexisting renal failure and pregnancy were the primary conditions that prevented this mode from being used as the primary test for the evaluation of PE.110 Thus, gadolinium-enhanced magnetic resonance pulmonary angiography (MRA) could potentially be used to diagnose PE because it is devoid of radiation. The accuracy of this technique combined with magnetic resonance venography (MRV) has been studied in the prospective, multicenter PIOPED III accuracy study. The proportion of technically inadequate images ranged from 11% to 52% across the seven participating centers. Technically adequate MRA had a sensitivity of 78% and a specificity of 99%, whereas technically adequate MRA and MRV had a sensitivity of 92% and a specificity of 96%. However, 194 (52%) of 370 patients had technically inadequate results, which substantially restricts its clinical use.114
Risk Stratification
Death from an embolic event almost always results from hemodynamic deterioration due to an abrupt rise in pulmonary vascular resistance overwhelming the contractile power of the right ventricle. Progressive hemodynamic and respiratory deterioration characterizes the clinical presentation of major or massive PE and is seen in approximately 5% of PE patients.2 Patients can be categorized as having submassive PE. Risk stratification becomes important to identify this group of patients who present with relative hemodynamic stability but exhibit varying degrees of RVD. Risk stratification schemes in normotensive patients center on RV function assessment after an embolic event and identify patients who may be at risk for progressive hemodynamic deterioration. Approximately 50% of patients who are normotensive have varying degrees of RVD and 10% will die. The European Society of Cardiology recommends a classification of PE into high-risk and non–high-risk groups. The high-risk groups represent the massive or major PE presentations. The non–high-risk group is further subdivided into intermediate risk in which the mortality rate ranges between 3% and 15% and a low-risk group in which the mortality rate is less than 3%. Initial therapeutic interventions are greatly influenced by risk stratification that includes clinical scoring systems, laboratory data, and imaging.115–117
The presence of RVD is a predictor of the increased risk of embolic-related adverse events and mortality rates when compared with patients without RVD.2 Patients who present with circulatory shock usually have severe RVD and have mortality rates up to 65%.1,2,115 In PE patients with normal hemodynamics and evidence of RVD the mortality rates can range from 8% to 14%. Several registry studies have observed that the frequency of RVD in normotensive patients is approximately 40%.116,117 In PE patients with initial normal hemodynamics and no RVD the mortality rate is low (0-3%).
Clinical and Hemodynamic Parameters
The initial hemodynamic presentation of PE is the most powerful predictor of outcome, as illustrated in Figure 44.5. Arterial hypotension at the time of PE presentation is an early predictor of death. In the 2392 patients from the ICOPER study, the 90-day mortality rates were 52.4% in patients with systolic arterial blood pressures below 90 mm Hg, and 14.7% in those patients who had preserved arterial blood pressures. In addition, comorbid conditions increase the risk of adverse clinical events, even in the presence of anatomically small PE. In the ICOPER findings, age over 70, congestive heart failure, cancer, and chronic lung disease were identified as independent predictors of 3-month mortality risk, with approximately twofold increase in the risk of death.2 In one study, altered mental status at presentation was associated with a sevenfold increase in risk of adverse outcome. Altered mental status most likely reflected decreased cardiac perfusion or hypoxia. In the same study the initial presentation of shock was associated with a threefold increase in risk of 30-day adverse outcome (death, secondary shock, or recurrent PE).117
Patients can be clinically risk stratified by the use of a clinical scoring system known as the Pulmonary Embolism Severity Index (PESI), which also has a simplified version.118,119 This prognostic model was developed by Aujesky and colleagues (Table 44.3) using a large database of hospitalized PE patients from which they derived and validated a prediction rule that identified acute PE patients with low to high risk for fatal PE and adverse events. Using 11 variables of predictors, five distribution categories emerged looking at inpatient fatality, 30-day all-cause mortality rate, and the adverse outcomes of nonfatal cardiogenic shock or cardiopulmonary arrest. Class I to class III all-cause mortality rates ranged between 0% and 3.1%, although class IV to class V all-cause mortality rates were between 10% and 24%. This score has an excellent negative predictive value and readily identifies low-risk patients but suffers in the attempt to predict patients at high risk for adverse events.119 A more simplied PESI score has been developed and has a high negative prediction value.120
Table 44.3
Pulmonary Embolism Severity Index (PESI)
Adapted from Aujesky D, Obrosky DS, Stone RA, et al: Derivation and validation of a prognostic model for pulmonary embolism. Am J Respir Crit Care Med 2005;172:1041-1046.
In patients with symptomatic acute PE, the presence of DVT may have prognostic significance. In a prospective study by Jiménez and coworkers, among 707 patients diagnosed with acute PE, 51.2% had concomitant DVT and 10.9% had PE-specific fatality at a 3-month follow-up. Patients with PE with the presence of DVT at the time of diagnosis had an adjusted hazard ratio (HR) of 2.05% (95% CI 1.24 to 3.38) and PE-specific fatality-adjusted HR of 4.25 (95% CI 1.61 to 11.25). These findings were validated by a large international registry trial (RIETE) that again demonstrated a significant prediction of all-cause mortality rate and PE-specific mortality rate when the concomitant DVT was present during the acute embolic event. These results suggest that patients with symptomatic acute PE have an increase in all-cause mortality rate, PE-related death, and recurrent VTE over a 3-month period if at the time of PE a diagnosis of proximal DVT is also present.121
Electrocardiogram
Several studies have demonstrated that ECG signs of RV strain correlate with the presence of RVD.122,123 A study by Punukollu and associates showed that T-wave inversion in leads V1 to V3 had a specificity of 88% and diagnostic accuracy of 81% for RVD in acute PE.124 Daniel and colleagues developed an ECG scoring system based on typical features of acute PE such as presence of right bundle branch block (RBBB), T-wave inversions, and additional features of right-sided heart strain that predicted severe pulmonary hypertension (sPAP >50 mm Hg) with a specificity of 97.7% if the patient’s “ECG score” is 10 or greater.125 The ECG may also be a tool for risk stratification of normotensive acute PE patients. In a study by Vanni and coworkers among 306 patients with documented PE 130 (34%), patients demonstrated ECG findings of right-sided heart strain. In this study ECG findings of right-sided heart strain were defined as incomplete or complete RBBB, S1Q3T3, or inverted T waves in the precordial leads V1, V2, and V3. For this study only one of these ECG patterns had to be present. The investigators demonstrated that the presence of RV strain on the initial ECG was associated with clinical deterioration and death (HR 2.58) in normotensive PE patients.122
Cardiac Biomarkers
Biomarkers are relied on in the risk stratification of patients with PE. Tests for biomarkers are widely available and there is usually a quick turnaround from the clinical laboratory. Caution must be taken on the interpretation of these biomarkers. It is important for the critical care clinician to understand the clinical issues that impact the interpretation of the biomarkers and understand the negative and positive predictive values.115 Elevations of brain natriuretic peptide (BNP) and troponin occur in many patients with PE and correlate with magnitude of RVD.
Echocardiography
The echocardiogram has become the main imaging modality to assess RV structure and function. In addition, pulmonary artery pressures can be estimated. Most hospitals in the United States have portable echocardiographic technology allowing for serial assessment of the impact of treatment on RVD. In hemodynamically stable patients with PE, 20% to 40% will have some echocardiographic finding of RVD.116,117 Box 44.1 illustrates the echocardiographic finding of acute PE. Toosi and colleagues performed a quantitative risk assessment of various echocardiographic measurements of RVD in acute PE. For in-patient mortality rate the positive predictive values range between 11% and 20% and the negative predictive values are 97% to 100%.140 The major drawback with the role of the echocardiographic definition of RVD of acute PE is that it lacks standardization. There is no clear agreement on which of the echocardiographic
Brain Natriurectic Peptide
The heart will release natriuretic peptides as result of ventricular volume overload. Ventricular stretch results in cellular production of proBNP. This peptide is subsequently cleaved to the biologically active BNP and the inactive N-terminal fragment (NT-proBNP).126 Both BNP and NT-proBNP can be measured clinically and have been validated as diagnostic and prognostic markers of ventricular stretch in multiple conditions, including congestive heart failure, pulmonary hypertension, and acute PE.127 Elevated concentrations of BNP distinguish patients with PE at higher risk of complicated in-hospital course and death from those with low BNP levels. Both BNP and NT-proBNP have been extensively investigated as prognostic markers in acute PE. In an early study, ten Wolde and colleagues127 demonstrated in a prospective cohort of normotensive acute PE patients that those who died within 90 days had a significantly increased BNP level at the time of diagnosis compared with those patients who did not die in follow-up (245 pg/mL vs. 30 pg/mL, respectively). In addition, using a cut-off point of 75 pg/mL, these investigators reported that BNP had a positive predictive value of PE-related fatality of 17%, whereas the negative predictive value for death was 99%.127 As will be seen for most of the prognostic markers for acute PE, the investigators noted that BNP was able to identify those at low risk of adverse outcomes but did not have good specificity (many false-positives) in identifying those who were likely to die from their PE.
Normal levels of BNP have a high negative predictive value for an elevated level of BNP or NT-proBNP as a risk factor for short-term fatality and overall short-term complicated clinical outcome, and an indicator of RVD in patients with acute PE. This is illustrated by the study by Kucher and colleagues,128 who investigated the prognostic capabilities of NT-proBNP in a prospective cohort of acute PE patients. In those suffering an adverse event (defined as a combined end point of in-hospital death or need for escalation of care, including cardiopulmonary resuscitation, mechanical ventilation, vasopressor, or thrombolysis), NT-proBNP was significantly higher compared with those who did not experience an adverse event (median of 4250 pg/mL vs. 121 pg/mL, respectively). An NT-proBNP cut-off point of 500 pg/mL yielded a negative predictive value for adverse outcome of 97% with a corresponding positive predictive value of adverse outcome of 45%.128 In a meta-analysis of 13 studies by Klok and associates, they indeed demonstrate that increased BNP or proBNP was a risk factor for short-term fatality and adverse clinical events and a marker for RVD. However, the investigators caution against using this biomarker alone to justify more aggressive medical therapy, such as thrombolytic therapy.129
Cardiac Troponin
Serum troponins are widely available for the evaluation of diverse forms of myocardial injury. The assays are rapid and are well suited as a risk stratification tool. A number of studies and meta-analyses have evaluated the prognostic value of cardiac troponin I (TnI) or T (TnT) (determined by conventional assays) in acute PE.130
[/not-level-membership-for-critical-care-medicine-category]
