5 Acute Lung Injury
A wide variety of insults can produce acute lung damage, inclusive of those that injure the lungs directly. Early terms for diffuse acute lung injury occurring indirectly in the setting of overwhelming nonthoracic trauma accompanied by hypovolemia were “shock lung,” “postperfusion lung,” “traumatic wet lung,” and “congestive atelectasis.”1,2
In 1967, Ashbaugh and coworkers formally described a syndrome characterized by acute onset of severe respiratory distress after an identifiable injury. Clinical signs included dyspnea, reduced lung compliance, diffuse chest radiographic infiltrates, and hypoxemia refractory to supplementary oxygen.3 Today this sequence of clinical events is referred to as the acute respiratory distress syndrome (ARDS). The clinical course is rapid and the mortality rate is high, with more than one half of affected patients dying of respiratory failure within days to weeks.2,4,5 A recent meta-regression analysis performed by Zambon and Vincent6 of mortality rates from 72 published studies of ARDS identified a decrease of 1.1% per year for the period 1994 to 2006, with an overall pooled mortality rate for all studies of 43%.
The American-European Consensus Conference (AECC) formally defined ARDS in 1994 using the following criteria: acute onset; bilateral chest radiographic infiltrates; hypoxemia regardless of the positive end-expiratory pressure oxygen concentration, an arterial partial pressure of oxygen to inspired oxygen fraction ratio less than 200, and no evidence of left atrial hypertension.7 The AECC also agreed that ARDS represents the most severe form on a spectrum of disease conditions encompassed under the general term acute lung injury.
The histopathologic counterpart of ARDS is distinctive and referred to as diffuse alveolar damage (DAD). DAD is the most extreme manifestation of lung injury and can occur as a result of a large number of direct injuries to the lungs (e.g., infection). In this chapter, the emphasis is on DAD and less severe manifestations of acute lung injury. Some authors have considered organizing pneumonia to be a form of acute lung injury, but we and others believe that organization is a subacute phenomenon with a more protracted clinical course (extending over several days to weeks). Subacute organizing pneumonia of unknown etiology (previously known as idiopathic bronchiolitis obliterans organizing pneumonia)8 is discussed with the chronic diffuse diseases (see Chapter 7).
Diffuse Alveolar Damage: The Morphologic Prototype of Acute Lung Injury
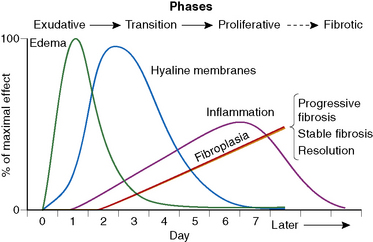
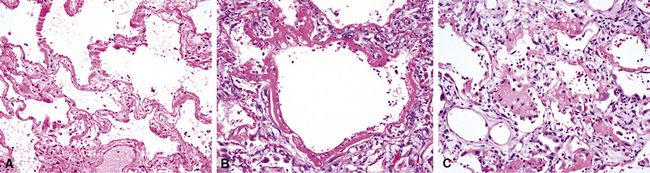
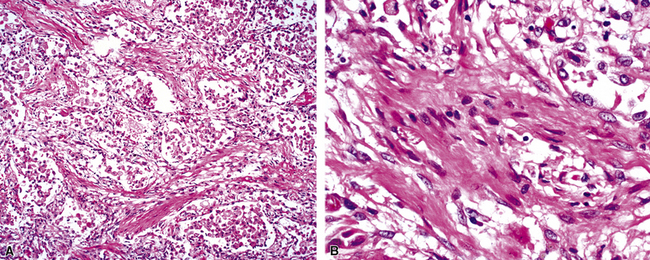
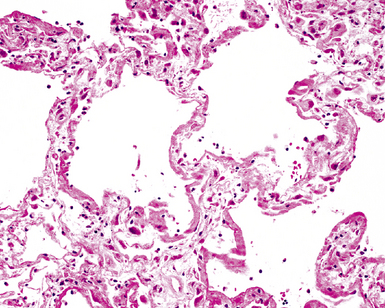
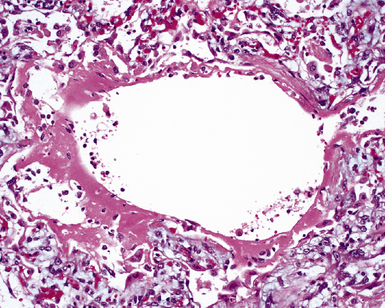
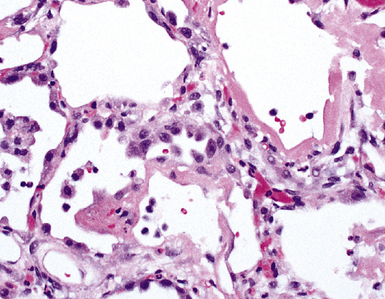
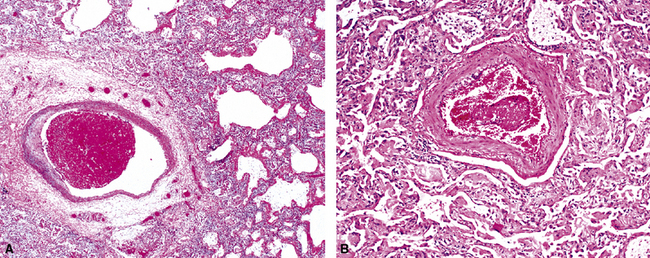
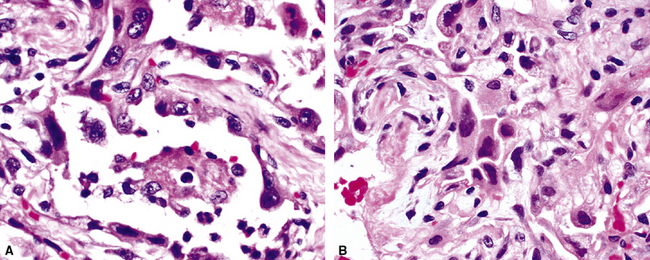
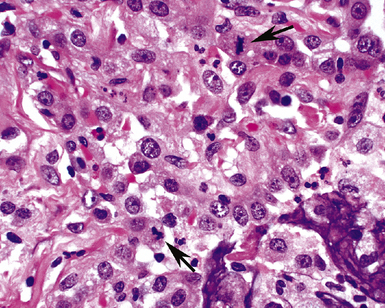
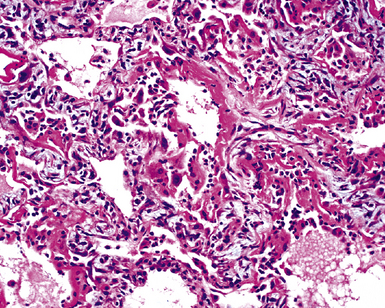
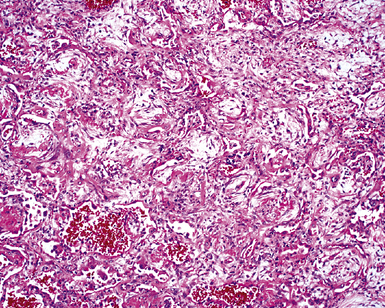
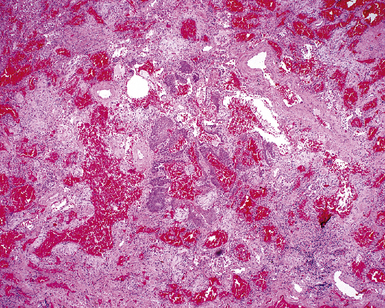
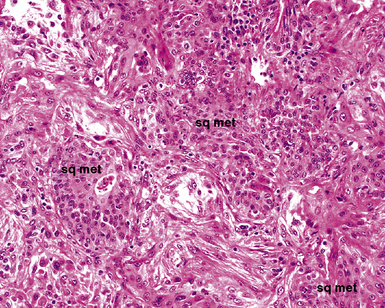
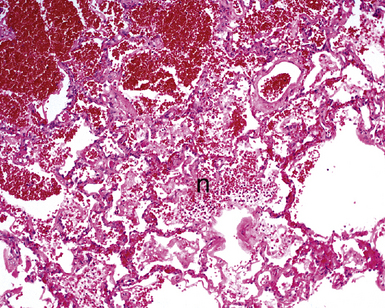
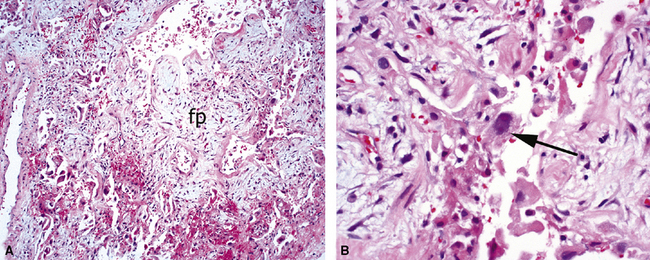
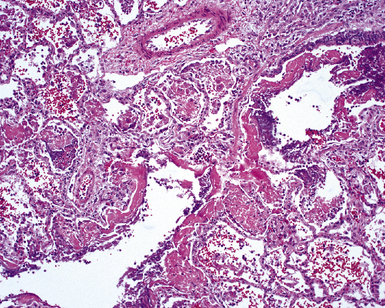
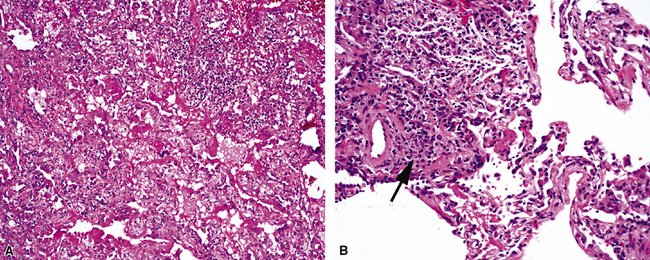
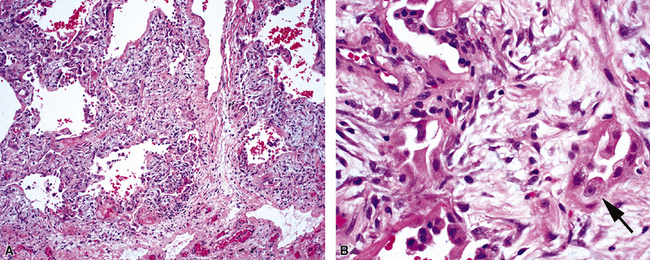
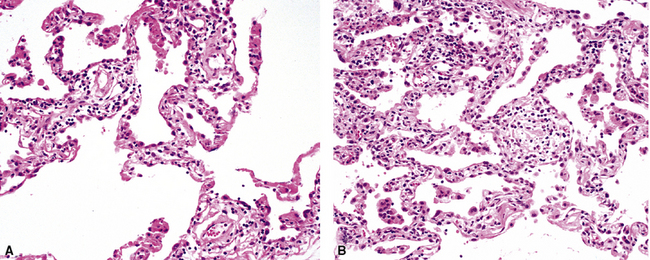
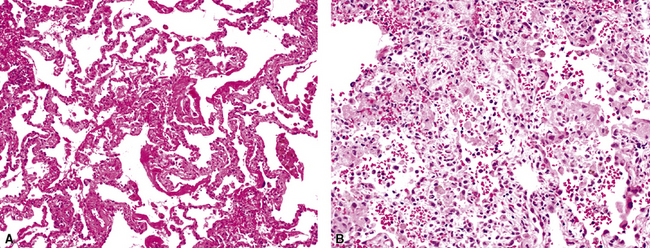
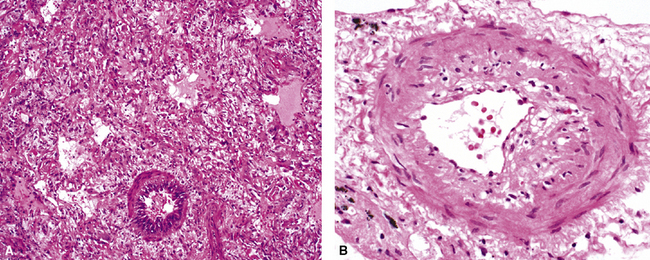
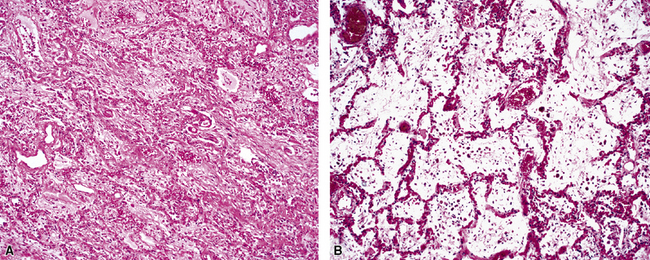
The causes of acute lung injury are numerous (Box 5-1). The lung reacts to various types of insults in similar ways, regardless of etiology. The resultant endothelial and alveolar epithelial cell injury is attended by fluid and cellular exudation. Subsequent reparative fibroblastic proliferation is accompanied by type II pneumocyte hyperplasia.4,9 The microscopic appearance depends on the time interval between insult and biopsy, and on the severity and extent of the injury. 2 DAD is the usual pathologic manifestation of ARDS and is the best-characterized prototype of acute lung injury. From studies of ARDS, the pathologic changes appear to proceed consistently through discrete but overlapping phases (Fig. 5-1)—an early exudative (acute) phase (Fig. 5-2A and B), a subacute proliferative (organizing) phase (see Fig. 5-2C), and a late fibrotic phase (Fig. 5-3).2,4,5,8,10 The exudative phase is most prominent in the first week of injury. The earliest changes include interstitial and intra-alveolar edema with variable amounts of hemorrhage and fibrin deposition (Fig. 5-4). Hyaline membranes (Fig. 5-5), the histologic hallmark of the exudative phase of ARDS, are most prominent at 3 to 7 days after injury. Minimal interstitial mononuclear inflammatory infiltrates (Fig. 5-6) and fibrin thrombi in small pulmonary arteries (Fig. 5-7) also are seen. Type II pneumocyte hyperplasia (Fig. 5-8) begins by the end of this phase and persists through the proliferative phase. The reactive type II pneumocytes may demonstrate marked nuclear atypia, with numerous mitotic figures (Fig. 5-9). The proliferative phase begins at 1 week after the injury and is characterized by fibroblastic proliferation, seen mainly within the interstitium but also focally in the alveolar spaces (Fig. 5-10). The fibrosis consists of loose aggregates of fibroblasts admixed with scattered inflammatory cells (Fig. 5-11); collagen deposition is minimal. Reactive type II pneumocytes persist. Immature squamous metaplasia may occur (Fig. 5-12) in and around terminal bronchioles. The degree of cytologic atypia in this squamous epithelium can be so severe as to mimic malignancy (Fig. 5-13). The hyaline membranes are mostly resorbed by the late proliferative stage, but a few remnants may be observed along alveolar septa. Some cases of DAD resolve completely, with few residual morphologic effects, but in other cases, fibrosis may progress to extensive structural remodeling and honeycomb lung. As might be expected, a recent review of outcomes for 109 survivors of ARDS revealed persistent functional disability at 1 year after discharge from intensive care.11
Box 5-1 Etiology of Diffuse Alveolar Damage
Modified from Katzenstein A, Askin F, eds. Katzenstein and Askin’s Surgical Pathology of Non-Neoplastic Lung Disease, 3rd ed. Philadelphia: Saunders; 1997:16.
By definition, ARDS has a known inciting event. The foregoing description is based on a model of ARDS due to oxygen toxicity, wherein the evolution of histopathologic abnormalities can be studied over a defined time period.2,5 In practice, lung biopsy most often is performed in patients without a known cause or specific time of onset of injury. Moreover, with some causes of acute lung injury, the damage evolves over a protracted period of time, or the lung may be injured in repetitive fashion (e.g., with drug toxicity). In such circumstances, the pathologic changes do not necessarily progress sequentially through defined stages as in ARDS, so both acute and organizing phases may be encountered in the same biopsy specimen. The basic histopathologic elements of acute lung injury are presented in Box 5-2.
Acute fibrinous and organizing pneumonia (AFOP) is a recently recognized histologic pattern of acute lung injury with a clinical presentation similar to that of classic DAD, in terms of both potential etiologic disorders and outcome. It differs from DAD in that hyaline membranes are absent. The dominant feature is intra-alveolar fibrin “balls” or aggregates, typically in a patchy distribution. Organizing pneumonia in the form of luminal loose fibroblastic tissue is present surrounding the fibrin. The alveolar septa adjacent to areas of fibrin deposition show a variety of changes similar to those of DAD, such as septal edema, type II pneumocyte hyperplasia, and acute and chronic inflammatory infiltrates. The intervening lung shows minimal histologic changes. AFOP may represent a fibrinous variant of DAD. In some patients, both DAD and AFOP disease patterns may be present simultaneously.12,13
Specific Causes of Acute Lung Injury
Infection
Infection is one of the most common causes of acute lung injury. Among infectious organisms, viruses most consistently produce DAD.2,5 Occasionally, fungi (e.g., Pneumocystis) and bacteria (e.g., Legionella) also can cause infections manifesting as DAD. Some of the organisms that are well known to cause acute lung injury with characteristic histopathologic changes are discussed next.
Viral Infection
Influenza is a common cause of viral pneumonia. The histopathology ranges from mild organizing acute lung injury (resembling organizing pneumonia) in nonfatal cases to severe DAD with necrotizing tracheobronchitis (Fig. 5-14) in fatal cases.14,15 Specific viral cytopathic effects are not identifiable by light microscopy. On ultrastructural examination, intranuclear fibrillary inclusions may be seen in epithelial and endothelial cells.16
The coronavirus responsible for severe acute respiratory syndrome (SARS) produces the acute lung injury associated with this disorder.17–20 Both DAD and AFOP patterns have been identified in affected patients. On ultrastructural examination, involved lung tissue revealed numerous to moderate numbers of cytoplasmic viral particles in pneumocytes, many within membrane-bound vesicles.21–23 The virus particles were spherical and enveloped, with spike-like projections on the surface and coarse clumps of electron-dense material in the center. Most had sizes ranging from 60 to 95 nm in diameter, but some were as large as 180 nm.
Measles virus produces a mild pneumonia in the normal host but can cause serious pneumonia in immunocompromised children. Histopathologic features of such infection include interstitial pneumonia, bronchitis and bronchiolitis, and DAD.24 The characteristic histologic feature is the presence of multinucleated giant cells (Fig. 5-15A) with characteristic eosinophilic intranuclear and intracytoplasmic inclusions.24–28 These cells are found in the alveolar spaces and within alveolar septa (see Fig. 5-15B). Viral inclusions are seen on ultrastructural examination as tightly packed tubules.28
Adenovirus is an important cause of lower respiratory tract disease in children,29,30 although adults (particularly those who are immunocompromised)31 and military recruits also are occasionally affected.32 The lung shows necrotizing bronchitis, or bronchiolitis, accompanied by DAD. The pathologic changes are more severe in bronchi, bronchioles, and peribronchiolar regions (Fig. 5-16A). Two types of inclusions can be observed in lung epithelial cells: An eosinophilic intranuclear inclusion with a halo usually is less conspicuous than the more readily identifiable “smudge cells” (see Fig. 5-16B). These latter cells are larger than normal and entirely basophilic, with no defined inclusion or halo evident by light microscopy.29 On ultrastructural examination, smudge cell inclusions are represented by arrays of hexagonal particles.33
Herpes simplex virus is mainly a cause of respiratory infection in the immunocompromised host. Two patterns of infection are recognized: airway spread resulting in necrotizing tracheobronchitis (Fig. 5-17) and blood-borne dissemination producing miliary necrotic parenchymal nodules. DAD and hemorrhage can occur in both forms.34,35 Characteristic inclusions may be seen in bronchial and alveolar epithelial cells (Fig. 5-18). The more obvious type is an intranuclear eosinophilic inclusion surrounded by clear halo (Cowdry A inclusion), and the other is represented by a basophilic to amphophilic ground-glass nucleus (Cowdry B inclusion). Rounded viral particles with double membranes are seen under the electron microscope.34,35
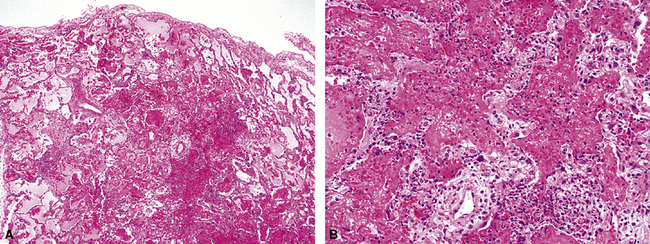
Figure 5-17 Diffuse alveolar damage in herpes simplex pneumonia. Herpesviridae viruses are capable of producing nodular necrotizing pneumonia (see Chapter 6). A, The nodular appearance of lung involved by herpes simplex pneumonia is evident, with zonal areas of hemorrhage and necrosis. B, A higher-magnification view of the hemorrhagic and necrotizing pneumonia.
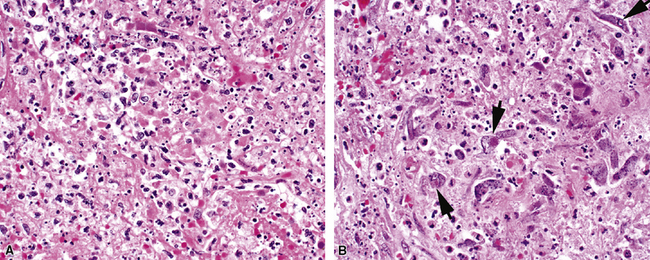
Varicella-zoster virus causes disease predominantly in children and is the agent of chickenpox.36 Pulmonary complications of chickenpox are rare in children with normal immunity (accounting for less than 1% of the cases). By contrast, however, pneumonia develops in 15% of adults with chickenpox; immunocompetent and immunocompromised persons are equally affected.32,36 The histopathologic picture in varicella pneumonia (Fig. 5-19) is similar to that in herpes simplex. Although identical intranuclear inclusions are reported to occur,32,36 these can be considerably more difficult to identify in chickenpox pneumonia.
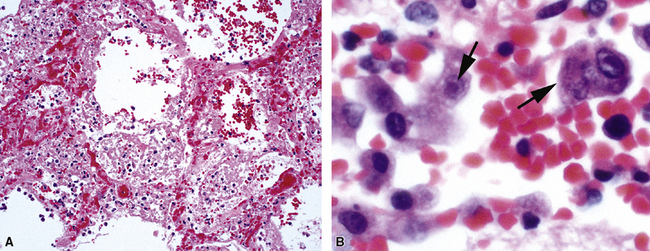
Cytomegalovirus is an important cause of symptomatic pneumonia in immunocompromised persons, especially those who have received bone marrow or solid organ transplants, and in patients with human immunodeficiency virus (HIV) infection.37–39 The histopathologic findings range from little or no inflammatory response to hemorrhagic nodules with necrosis (Fig. 5-20A) and DAD.37 The diagnostic histopathologic pattern, seen in endothelial cells, macrophages, and epithelial cells, consists of cellular enlargement, a prominent intranuclear inclusion, and an intracytoplasmic basophilic inclusion37 (see Fig. 5-20B).
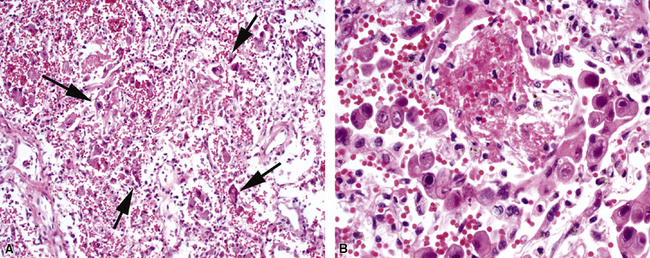
Hantavirus is a rare cause of acute lung injury.40–42 The infection produces alveolar edema, hyaline membranes, and atypical interstitial mononuclear inflammatory infiltrates40–42 (Fig. 5-21). Spherical membrane-bound viral particles have been found in the cytoplasm of endothelial cells by electron microscopy.
Fungal Infection
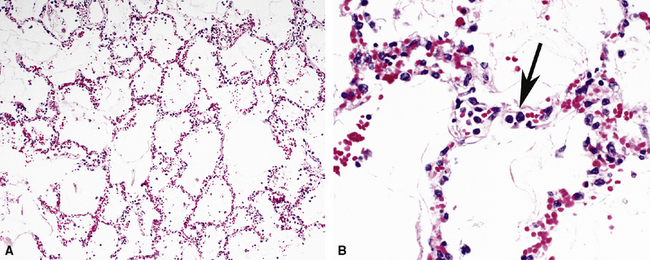
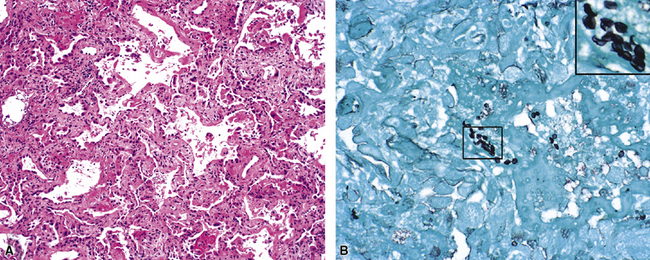
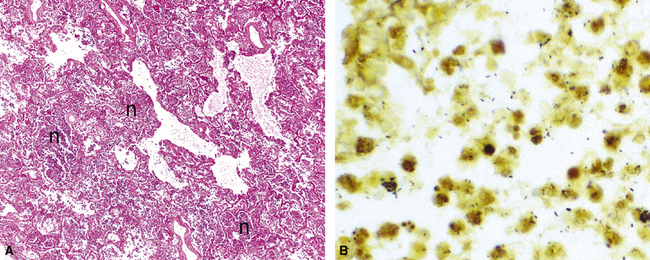
Pneumocystis jiroveci (previously known as Pneumocystis carinii) is the most common fungus to cause DAD.43–45 The histopathology of Pneumocystis infection in the setting of profound immunodeficiency is one of frothy intra-alveolar exudates (Fig. 5-22A) (so-called “alveolar casts”), with many organisms44,45 (see Fig. 5-22B). In the mildly immunocompromised patient, however, this feature is not observed, or the pathologic changes may be subtle. In such cases, several “atypical” manifestations have been described.43,45,46 DAD is the most dramatic of these atypical presentations (Fig. 5-23A), with the organisms present within hyaline membranes (see Fig. 5-23B) and in isolated intra-alveolar fibrin deposits.46 The Grocott methenamine silver method (GMS) is routinely used to stain the organisms, which typically are seen in small groups and clusters (see Figs. 5-22B and 5-23B).43,45,46
Bacterial Infection
Common bacterial pneumonias rarely cause DAD; however, this lung injury pattern has been described in legionnaires disease, Mycoplasma pneumonia, and rickettsial infection.47–51
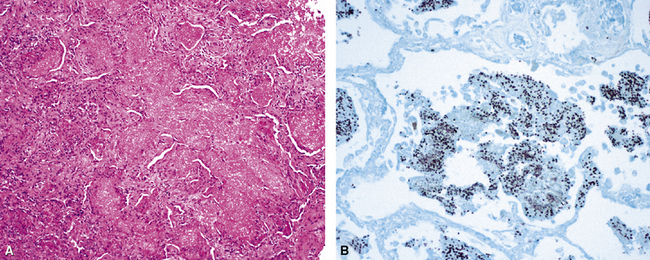
Legionella is a fastidious gram-negative bacillus that causes acute respiratory infection in elderly and immunodeficient individuals.47,48,51 The histopathologic pattern is that of a pyogenic necrotizing bronchopneumonia (Fig. 5-24A) affecting the respiratory bronchioles, alveolar ducts, and adjacent alveolar spaces. DAD is common.47,48,51 The rod-shaped organisms (see Fig. 5-24B) can be identified by Dieterle silver stain.51
Of note, in immunocompromised patients, any type of infection can cause DAD, with Pneumocystis pneumonia being the most common.28 For this reason, it is essential to use special stains (acid-fast bacilli [AFB] stains or GMS or Warthin-Starry silver stain, and so on) on every lung biopsy specimen exhibiting DAD.
Collagen Vascular Diseases
Systemic collagen vascular disorders are a well-known cause of diffuse lung disease.52–59 In some cases, lung involvement may be the first manifestation of the systemic disease, even without identifiable serologic evidence.57 Acute lung injury has been reported to occur in the following collagen vascular diseases.
Systemic Lupus Erythematosus
Pulmonary involvement in SLE may manifest as pleural disease, acute or chronic diffuse inflammatory lung disease, airway disease, or vascular disease (vasculitis and thromboembolic lesions). Acute lupus pneumonitis (ALP) is a form of fulminant interstitial disease (Fig. 5-25A) with a high mortality rate.52 Patients present with severe dyspnea, tachypnea, fever and arterial hypoxemia. ALP represents the first manifestation of SLE in approximately 50% of affected persons.52,58 The most common histopathologic feature of this acute disease is DAD. Alveolar hemorrhage, with capillaritis and small-vessel vasculitis (see Fig. 5-25B), and pulmonary edema also may be observed.52,57,60 Immunofluorescence studies demonstrate immune complexes in lung parenchyma, and both immune complexes and tubuloreticular inclusions may be seen on ultrastructural examination.57,58,60
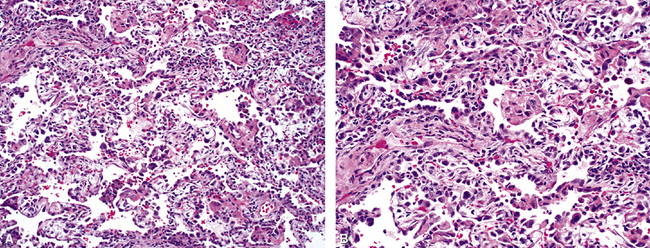
Rheumatoid Arthritis
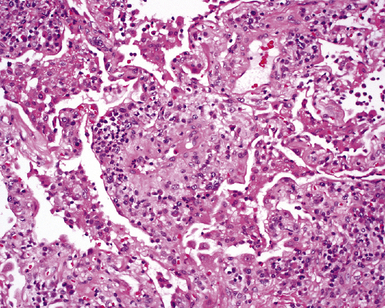
A significant percentage of patients with rheumatoid arthritis have lung disease.53,54,61–64 Many different morphologic patterns of lung disease in rheumatoid arthritis have been described,54,57,59 with the rheumatoid nodule being the most specific. Acute lung injury has been reported (Fig. 5-26), referred to as acute interstitial pneumonia in some publications65 and as DAD in others.54
Polymyositis/Dermatomyositis
Polymyositis/dermatomyositis, a systemic connective tissue disorder, is well known to be associated with interstitial lung disease.55,56 Three main clinical presentations are recognized: (1) acute fulminant respiratory distress resembling the so-called Hamman-Rich syndrome, (2) slowly progressive dyspnea, and (3) an asymptomatic form with abnormalities on radiologic and pulmonary function studies.59 Three major histopathologic patterns have been observed: DAD (Fig. 5-27A), organizing pneumonia (see Fig. 5-27B), and chronic fibrosis (see Fig. 5-27C)—the so-called usual interstitial pneumonia (UIP) pattern.66 The rapidly progressive clinical presentation is associated with a DAD histopathologic pattern on lung biopsy studies and carries the worst prognosis.56
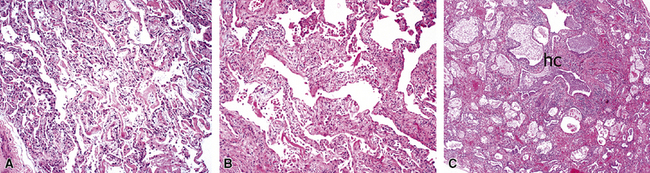
Figure 5-27 Diffuse alveolar damage (DAD) in polymyositis/dermatomyositis. All of the systemic connective tissue diseases can manifest with acute, subacute, and chronic lung disease. Three examples of diffuse lung disease accompanying polymyositis/dermatomyositis are presented: A, DAD; B, a subacute organizing pneumonia with an interstitial mononuclear infiltrate (nonspecific interstitial pneumonia [NSIP]-like; see Chapter 7); and C, a usual interstitial pneumonia (UIP)-like pattern of lung fibrosis with microscopic honeycomb remodeling (hc).
DAD associated with scleroderma and mixed connective disease also has been described.57,67
Drug Effect
Drugs can produce a wide range of pathologic effects with lung manifestations, and the causative agents are numerous.68–81 The spectrum of drug-induced lung disease runs the entire gamut from DAD to fibrosis. Between these two extremes, subacute clinical manifestations may include organizing pneumonia, chronic interstitial pneumonia, eosinophilic pneumonia, obliterative bronchiolitis, pulmonary hemorrhage, pulmonary edema, pulmonary hypertension, veno-occlusive disease, and granulomatous interstitial pneumonia.78,82,83
DAD is a common and dramatic manifestation of pulmonary drug toxicity.78 Many drugs are known to cause DAD.82 A few of the more common ones are discussed next. (Drug-related lung disease is also discussed in Chapter 7.)
Chemotherapeutic Agents
DAD frequently is caused by cytotoxic drugs, and the commonly implicated ones include bleomycin (Fig. 5-28), busulfan (Fig. 5-29), and carmustine.5,78,82 Patients usually present with dyspnea, cough, and diffuse pulmonary infiltrates.84–88 The histologic pattern most commonly is one of nonspecific acute lung injury with hyaline membranes, but some changes may be present to at least suggest a causative agent. For example, the presence of acute lung injury with associated atypical type II pneumocytes with markedly enlarged pleomorphic nuclei89 and prominent nucleoli (see Fig. 5-29) is characteristic for busulfan-induced pulmonary toxicity, and on ultrastructural examination, intranuclear tubular structures have been found in type II pneumocytes in association with administration of busulfan and bleomycin.89–92 In most cases, the possibility that a drug is the cause of DAD can only be inferred from the clinical history. Considerations in the differential diagnosis typically include other treatment-related injury or complication of therapy (e.g., concomitant irradiation or infection). For example, oxygen therapy is a well-recognized cause of DAD (Fig. 5-30) and also may exacerbate bleomycin-induced lung injury.93 Methotrexate (Fig. 5-31) is another commonly used cytotoxic drug that can cause acute and organizing DAD.94 Methotrexate also produces other distinctive patterns, such as granulomatous interstitial pneumonia (see Chapter 7) that is seldom seen in association with other commonly used chemotherapeutic agents. To complicate matters further, methotrexate also is used in the treatment of rheumatoid arthritis, a disease known to produce DAD independently as one of its pulmonary manifestations.57,62
Amiodarone
Amiodarone is a highly effective antiarrhythmic drug that is increasingly recognized as a cause of pulmonary toxicity.77,95–99 Because patients taking amiodarone have known cardiac disease, the clinical presentation often is complicated, with several superimposed processes potentially affecting the lungs in various ways. Clinical and radiologic considerations typically include congestive heart failure, pulmonary emboli, and acute lung injury from other causes.77,99
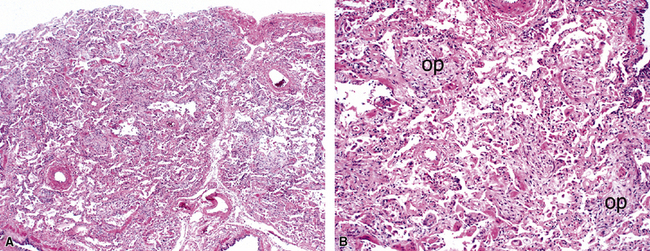
Distinctive features may be present on chest CT scans.77 The lung biopsy commonly shows acute and organizing lung injury (Fig. 5-32A). Other patterns include chronic interstitial pneumonitis with fibrosis and organizing pneumonia.97 Characteristically, type II pneumocytes and alveolar macrophages show finely vacuolated cytoplasm in response to amiodarone therapy (see Fig. 5-32B), but these changes alone are not evidence of toxicity because they also may be seen in patients taking amiodarone who do not have evidence of lung toxicity.95–98
Anti-inflammatory Drugs
Methotrexate and gold, common agents for treatment of rheumatoid arthritis, are frequently implicated in lung toxicity. Methotrexate is discussed earlier in this chapter. Organizing DAD (Fig. 5-33) and chronic interstitial pneumonia are commonly described pulmonary manifestations of so-called gold toxicity.74,76,100
Acute Eosinophilic Pneumonia
Acute eosinophilic pneumonia was first described in 1989101 and is characterized by acute respiratory failure, fever of days’ to weeks’ duration, diffuse pulmonary infiltrates on radiologic studies, and eosinophilia in bronchoalveolar lavage (BAL) fluid or lung biopsy specimens in the absence of infection, atopy, and asthma.102 Peripheral eosinophilia frequently is described but is not a consistent finding at initial presentation.103,104 Acute eosinophilic pneumonia is easily confused with acute interstitial pneumonia because both manifest as acute respiratory distress without an obvious underlying cause.102 Histologically, the disease is characterized by acute and organizing lung injury showing classic features (Fig. 5-34) of (1) alveolar septal edema, (2) eosinophilic air space macrophages, (3) tissue and air space eosinophils in variable numbers, and (4) marked reactive atypia of alveolar type II cells. Intra-alveolar fibroblastic proliferation (patchy organizing pneumonia) and inflammatory cells are present to a variable degree. Hyaline membranes and organizing intra-alveolar fibrin also may be present (Fig. 5-35). The most significant feature is the presence of interstitial and alveolar eosinophils. Infiltration of small blood vessels by eosinophils also may be seen. It is important to distinguish acute eosinophilic pneumonia from other causes of DAD, because patients typically benefit from systemic corticosteroid treatment, with prompt recovery. Before initiation of immunosuppressive therapy, however, infection should be rigorously excluded by culture and special stains, because parasitic and fungal infections also can manifest as tissue eosinophilia.
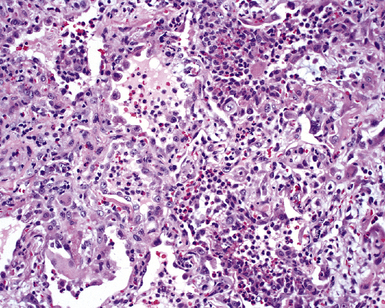
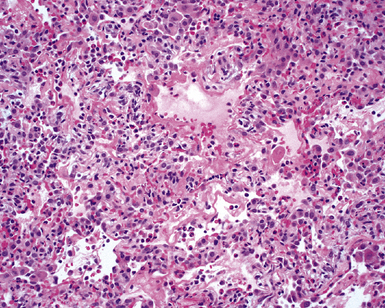
Acute Interstitial Pneumonia
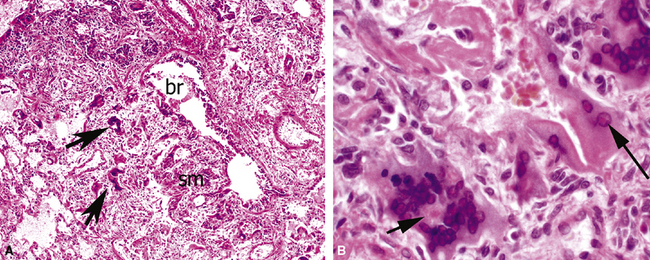
Acute interstitial pneumonia, also commonly referred to as Hamman-Rich syndrome, is a fulminant lung disease of unknown etiology occurring in previously healthy patients.105–107 Patients usually report a prodromal illness simulating viral infection of the upper respiratory tract, followed by rapidly progressive respiratory failure. The mortality rate is high, with death occurring weeks or months after the acute onset.105,107 The classic histopathologic pattern is that of acute and organizing DAD,105,107 with septal edema and hyaline membranes in the early phase and septal fibroblastic proliferation with reactive type II pneumocytes prominent in the organizing phase. In practice, a combination of acute and organizing changes (Fig. 5-36) often are seen in the lung at the time of biopsy.108 A variable degree of air space organization, mononuclear inflammatory infiltrates, thrombi in small pulmonary arteries, and reparative peribronchiolar squamous metaplasia also are seen in most cases.
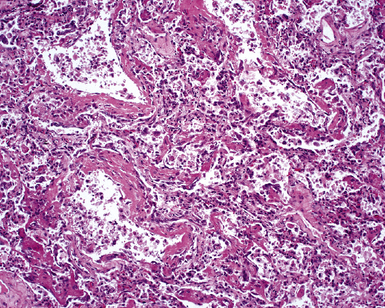
Because acute interstitial pneumonia is idiopathic, other specific causes of acute lung injury must be excluded before making this diagnosis. Considerations in the differential diagnosis include infection, collagen vascular disease, acute exacerbation of idiopathic pulmonary fibrosis (IPF), drug effect, and other causes of DAD.108 Most cases of DAD are not acute interstitial pneumonia, and detailed clinical information, radiologic findings (localized versus diffuse disease), serologic data, and microbiologic results will often point to or rule out a specific etiologic condition. Use of special stains applied to tissue sections or cytologic preparations (e.g., AFB, GMS or Warthin-Starry silver stain) also is essential to rule out infectious organisms in this setting.
Immunologically Mediated Pulmonary Hemorrhage and Vasculitis
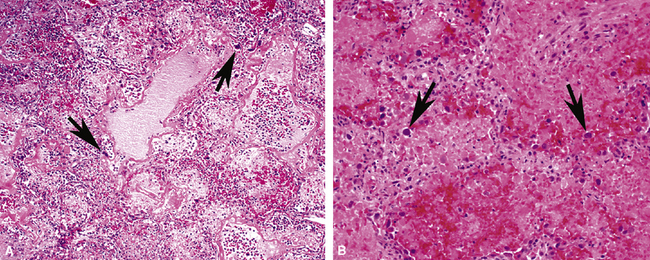
So-called “pulmonary hemorrhage syndromes” may feature the histopathologic changes of acute lung injury,109 in addition to the characteristic alveolar hemorrhage and hemosiderin-laden macrophages. In some patients, DAD may be the dominant histopathologic pattern.110 In the study by Lombard and colleagues in patients with Goodpasture syndrome, all showed acute lung injury ranging in distribution from focal to diffuse lung involvement.110 Histopathologic examination demonstrated typical acute and organizing DAD, with widened and edematous alveolar septa, fibroblastic proliferation, reactive type II pneumocytes, and, rarely, even hyaline membranes (Fig. 5-37). Alveolar hemorrhage, either focal or diffuse, was present in all cases. Capillaritis, an important finding indicating true alveolar hemorrhage,109 also was seen, as evidenced by marked septal neutrophilic infiltration. Capillaritis was absent in one case for which DAD was the dominant histopathologic pattern.
Microscopic polyangiitis can manifest as an acute interstitial pneumonia both clinically and histopathologically. Affected patients have vasculitis as the known cause of acute lung injury.111 Alveolar hemorrhage with arteritis, capillaritis and venulitis may be seen in some cases.111
Polyarteritis nodosa and vasculitis associated with systemic connective tissue disease (notably systemic lupus erythematosus and rheumatoid arthritis) can also show acute lung injury with alveolar hemorrhage as the dominant histopathologic finding.57,112
Radiation Pneumonitis
Radiation can produce both acute and chronic damage to the lung, manifesting as acute radiation pneumonitis and chronic progressive fibrosis, respectively.113 The effect is dependent on radiation dosage, total time of irradiation, and tissue volume irradiated. Concomitant chemotherapy and infections, which in themselves are causes of DAD, may potentiate the effect of radiation injury.5,79,114,115 Acute radiation pneumonitis manifests 1 to 2 months after radiation therapy.5,115 Clinical findings include dyspnea, cough, pleuritic pain, fever, and chest infiltrates. The lung biopsy specimen shows acute and organizing DAD.113,115 Markedly atypical type II pneumocytes with enlarged hyperchromatic nuclei and vacuolated cytoplasm constitute a hallmark of the disease (Fig. 5-38A), and increased numbers of alveolar macrophages are seen. Foamy cells are present in the intima and media of pulmonary blood vessels in some cases, and thrombosis (see Fig. 5-38B), with or without transmural fibrinoid necrosis, is common.79,116–118
Disease Presenting as Classic Acute Respiratory Distress Syndrome
By definition, ARDS must be associated with an identifiable inciting event. The histopathologic pattern is that of classic DAD. The histopathologic changes should be consistent with those expected for the time interval from the onset of clinical disease (see further on). In many cases, the ARDS may be caused by a combination of factors, each potentiating the other.4 For the purposes of illustration, a few thoroughly studied causes are discussed next.
Oxygen Toxicity and Inhalants
Oxygen is a well-known cause of ARDS and a useful model for all types of DAD.4,119,120 Oxygen toxicity also is important in that it is widely used in the care of patients, often in the setting of other injuries that can potentially cause ARDS, such as sepsis, shock, and trauma. Exposure to high concentrations of oxygen for prolonged periods can lead to characteristic pulmonary damage. In 1958, Pratt first noted pulmonary changes due to high concentrations of inspired oxygen.121 In 1967, Nash and colleagues described the sequential histopathologic changes of this injury,119 later reemphasized by Pratt.120 In neonates receiving oxygen for hyaline membrane disease, bronchopulmonary dysplasia was reported to occur. 122 As might be expected, the features of hyaline membrane disease in neonates and oxygen-induced DAD in adults are indistinguishable (see Fig. 5-30). Other inhalants such as chlorine gas, mercury vapor, carbon dioxide in high concentrations, and nitrogen mustard all have been reported to cause ARDS.2,4,5
Shock and Trauma
Massive extrapulmonary trauma and shock first became recognized as causes of unexplained respiratory failure during the wars of the second half of the 20th century. A variety of names were assigned to this wartime condition, including shock lung, congestive atelectasis, traumatic wet lung, Da Nang lung, respiratory insufficiency syndrome, post-traumatic pulmonary insufficiency, and progressive pulmonary consolidation.2 It became clear that shock of any cause (e.g., hypovolemia due to hemorrhage, cardiogenic shock, sepsis), could cause ARDS, and that in most cases, a number of factors come into play. In the typical presentation, dyspnea of rapid onset is accompanied by development of diffuse chest infiltrates several hours to days after an episode of shock. Once ARDS begins, the mortality rate is high.1,2,123
Ingested Toxins
Paraquat is a potent herbicide that causes the release of hydrogen peroxide and superoxide free radicals, resulting in damage to cell membranes.124–126 Oropharyngitis is the initial sign of poisoning, followed by impaired renal and liver function. Approximately 5 days later, ARDS develops. The histolopathologic pattern in most cases is one of organizing DAD (Fig. 5-39). The diagnosis is confirmed by tissue analysis for paraquat, which can be performed even on autopsy specimens. Other ingested toxins (e.g., kerosene, rapeseed oil) also have been reported to cause ARDS.5
Additional Features in the Differential Diagnosis of Acute Lung Injury
Acute lung injury is a pathologic pattern and by itself is a nonspecific finding. The following additional features often help narrow the list of possible causes (summarized in Table 5-1).
Table 5-1 Key Histopathologic Findings in Acute Lung Injury, with Possible Causes
| Finding | Possible Causes |
|---|---|
| Hyaline membranes | Infection, collagen vascular disease, drug toxicity, oxygen and inhalant toxicity, idiopathic (acute interstitial pneumonia); acute exacerbation of idiopathic pulmonary fibrosis (characteristic associated findings: background fibrosis and microscopic honeycombing) |
| Neutrophils and fibrinous exudates | Infection (viral, fungal, bacterial), alveolar hemorrhage |
| Diffuse alveolar hemorrhage (with or without capillaritis and small-vessel vasculitis) | Collagen vascular diseases (SLE, RA, MCTD, polymyositis/dermatomyositis, scleroderma), Goodpasture syndrome, microscopic polyangiitis, Wegener granulomatosis (organizing pneumonia—capillaritis variant) |
| Organizing pneumonia (alveolar organization) | Resolving infection, drug toxicity, collagen vascular diseases, idiopathic (cryptogenic organizing pneumonia); acute exacerbation of idiopathic pulmonary fibrosis |
| Fibrin and organization | Infection, drug toxicity, idiopathic (acute fibrinous and organizing pneumonitis), collagen vascular diseases; acute exacerbation of idiopathic pulmonary fibrosis |
| Alveolar eosinophils with fibrin | Infection, collagen vascular disease, drug toxicity; idiopathic acute eosinophilic pneumonia |
| Necrosis | Infection and infarction |
| Atypical cells | Infection (especially viral), radiation pneumonitis, chemotherapy-related changes (and effects of other drugs) |
| Foamy alveolar cells | Amiodarone and other drug toxicity, radiation pneumonitis |
MCTD, mixed connective tissue disease; RA, rheumatoid arthritis; SLE, systemic lupus erythematosus.
Clinicopathologic Correlation
Because the morphologic manifestations of acute diffuse lung disease may be relatively stereotypical, clinicopathologic correlation is often helpful in arriving at a specific diagnosis. A summary of the more important history and laboratory data pertinent to this correlation is presented in Box 5-3.
Box 5-3 Essential Information for Determining the Underlying Cause of Acute Lung Injury
ANA, antinuclear antibody; ANCA, anti-neutrophil cytoplasmic antibody; RF, rheumatoid factor.
1 Petty T. 41st Aspen Lung Conference: Overview. Chest. 1999;116:1S-2S.
2 Tomashefski J.Jr. Pulmonary pathology of acute respiratory distress syndrome. Clin Chest Med. 2000;21(3):435-466.
3 Ashbaugh D., Bigelow D.B., Petty T.L., Levine B.E. Acute respiratory distress in adults. Lancet. 1967;2:319-323.
4 Katzenstein A., Bloor C., Liebow A. Diffuse alveolar damage—the role of oxygen, shock and related factors. Am J Pathol. 1976;85:209-228.
5 Katzenstein A. Acute lung injury patterns: diffuse alveolar damage and bronchiolitis obliterans–organizing pneumonia. In: Katzenstein A., Askin F., editors. Katzenstein and Askin’s Surgical Pathology of Non-Neoplastic Lung Disease. Philadelphia: Saunders, 1997.
6 Zambon M., Vincent J.L. Mortality rates for patients with acute lung injury/ARDS have decreased over time. Chest. 2008;133(5):1120-1127.
7 Bernard G., Artigas A., Brigham K.L., et al. The American-European Consensus Conference on ARDS. Definitions, mechanisms, relevant outcomes, and clinical trial coordination. Am J Respir Crit Care Med. 1994;149:818-824.
8 Wright J. Adult respiratory distress syndrome. In: Thurlbeck W., Churg A., editors. Pathology of the Lung. New York: Thieme, 1995.
9 Bellingan G. The pulmonary physician in critical care 6: the pathogenesis of ALI/ARDS. Thorax. 2002;57:540-546.
10 Colby T., Lombard C., Yousem S.A., Kitaichi M. Atlas of Pulmonary Surgical Pathology. Philadelphia: Saunders, 1991.
11 Herridge M.S., Cheung A.M., Tansey C.M., et al. One-year outcomes in survivors of the acute respiratory distress syndrome. N Engl J Med. 2003;348(8):683-693.
12 Hwang D.M., Chamberlain D.W., Poutanen S.M., et al. Pulmonary pathology of severe acute respiratory syndrome in Toronto. Mod Pathol. 2005;18(1):1-10.
13 Cincotta D.R., Sebire N.J., Lim E., Peters M.J. Fatal acute fibrinous and organizing pneumonia in an infant: the histopathologic variability of acute respiratory distress syndrome. Pediatr Crit Care Med. 2007;8(4):378-382.
14 Oseasohn R., Adelson L., Kaji M. Clinicopathology study of 33 fatal cases of Asian influenza. N Engl J Med. 1959;260:509-518.
15 Yeldandi A., Colby T. Pathologic features of lung biopsy specimens from influenza pneumonia cases. Hum Pathol. 1994;25:47-53.
16 Tamura H., Aronson B. Intranuclear fibrillary inclusions in influenza pneumonia. Pathol Lab Med. 1978;102:252-257.
17 Cheung O.Y., Chan J.W., Ng C.K., Koo C.K. The spectrum of pathological changes in severe acute respiratory syndrome (SARS). Histopathology. 2004;45(2):119-124.
18 Franks T.J., Chong P.Y., Chui P., et al. Lung pathology of severe acute respiratory syndrome (SARS): a study of 8 autopsy cases from Singapore. Hum Pathol. 2003;34(8):743-748.
19 Hwang D., Chamberlain D.W., Poutanen S.M., et al. Pulmonary pathology of severe acute respiratory syndrome in Toronto. Mod Pathol. 2005;18:1-10.
20 Nicholls J.M., Poon L.L., Lee K.C., et al. Lung pathology of fatal severe acute respiratory syndrome. Lancet. 2003;361(9371):1773-1778.
21 Ksiazek T.G., Erdman D., Goldsmith C.S., et al. A novel coronavirus associated with severe acute respiratory syndrome. N Engl J Med. 2003;348(20):1953-1966.
22 Peiris J.S., Lai S.T., Poon L.L., et al. Coronavirus as a possible cause of severe acute respiratory syndrome. Lancet. 2003;361(9366):1319-1325.
23 Lee N., Hui D., Wu A., et al. A major outbreak of severe acute respiratory syndrome in Hong Kong. N Engl J Med. 2003;348(20):1986-1994.
24 Sobonya R.E., Hiller F.C., Pingleton W., Watanabe I. Fatal measles (rubeola) pneumonia in adults. Arch Pathol Lab Med. 1978;102:366-371.
25 Enders J.F., McCarthy K., Mitus A., Cheatham W.J. Isolation of measles virus at autopsy in cases of giant-cell pneumonia without rash. N Engl J Med. 1959;261:875-881.
26 Mitus A., Enders J.F., Craig J.M., Holloway A. Persistence of measles virus and depression of antibody formation in patients with giant-cell pneumonia after measles. N Engl J Med. 1959;261:882-889.
27 Haram K., Jacobsen J. Measles and its relationship to giant cell pneumonia (Hecht pneumonia). Acta Pathol Microbiol Immunol Scand [A]. 1973;81:761-769.
28 Katzenstein A. Infection. I. Unusual pneumonias. In: Katzenstein A., Askin F., editors. Katzenstein and Askin’s Surgical Pathology of Non-Neoplastic Lung Disease. Philadelphia: Saunders, 1997.
29 Becroft D. Histopathology of fatal adenovirus infection of the respiratory tract in young children. J Clin Pathol. 1967;20:561-569.
30 Becroft D. Bronchiolitis obliterans, bronchiectasis and other sequelae of adenovirus type 21 infection in young children. J Clin Pathol. 1971;24:72-79.
31 Zahradnik J., Spencer M., Porter D. Adenovirus infection in the immunocompromised patient. Am J Med. 1980;68:725-732.
32 Miller R. Viral infections of the respiratory tract. In: Thurlbeck W., Churg A., editors. Pathology of the Lung. 2nd ed. New York: Thieme; 1995:195-222.
33 Abbondanzo S., English C.K., Kagan E., McPherson R.A. Fatal adenovirus pneumonia in a newborn identified by electron microscopy and in-situ hybridization. Arch Pathol Lab Med. 1989;113:1349-1353.
34 Ramsey P., Fife K.H., Hackman R.C., et al. Herpes simplex virus pneumonia: clinical, virologic, and pathologic features in 20 patients. Ann Intern Med. 1982;97:813-820.
35 Graham B., Snell J.J. Herpes simplex virus infection of the adult lower respiratory tract. Medicine (Baltimore). 1983;62:384-393.
36 Pugh R.N., Omar R.I., Hossain M.M. Varicella infection and pneumonia among adults. Int J Infect Dis. 1998;2(4):205-210.
37 Craighead J. Cytomegalovirus pulmonary disease. Pathobiol Annu. 1975;5:197-220.
38 Beschorner W., Hutchins G.M., Burns W.H., et al. Cytomegalovirus pneumonia in bone marrow transplant recipients: miliary and diffuse patterns. Am Rev Respir Dis. 1980;122:107-114.
39 Winston D., Ho W., Champlin R. Cytomegalovirus after allogeneic bone marrow transplantation. Rev Infect Dis. 1992;12(suppl):S776-S792.
40 Colby T.V., Zaki S.R., Feddersen R.M., Nolte K.B. Hantavirus pulmonary syndrome is distinguishable from acute interstitial pneumonia. Arch Pathol Lab Med. 2000;124(10):1463-1466.
41 Duchin J., Koster F.T., Peters C.J., et al. Hantavirus pulmonary syndrome: a clinical description of 17 patients with a newly recognized disease. The Hantavirus Study Group. N Engl J Med. 1994;330:949-955.
42 Nolte K., Feddersen R.M., Foucar K., et al. Hantavirus pulmonary syndrome in the United States. A new pathological description of a disease caused by a new agent. Hum Pathol. 1995;26:110-120.
43 Weber W., Askin F., Dehner L. Lung biopsy in Pneumocystis carinii pneumonia. A histopathologic study of typical and atypical features. Am J Clin Pathol. 1977;67:11-19.
44 Ognibene F.P., Shelhamer J., Gill V., et al. The diagnosis of Pneumocystis carinii pneumonia in patients with the acquired immunodeficiency syndrome using subsegmental bronchoalveolar lavage. Am Rev Respir Dis. 1984;129:929-932.
45 Grimes M., LaPook J.D., Bar M.H., et al. Disseminated Pneumocystis carinii infection in a patient with acquired immunodeficiency syndrome. Hum Pathol. 1987;18:307-308.
46 Askin F., Katzenstein A. Pneumocystis infection masquerading as diffuse alveolar damage: a potential source of diagnostic error. Chest. 1979;4:420-422.
47 Blackmon J., Hicklin M., Chandler F. Legionnaires’ disease. Pathological and historical aspects of a new disease. Arch Pathol Lab Med. 1978;102:337-343.
48 Lattimen G., Rachman R., Scarlato M. Legionnaires’ disease pneumonia: histopathologic features and comparison with microbial and chemical pneumonias. Ann Clin Lab Sci. 1979;9:353-361.
49 Rollin S., Colby T., Clayton F. Open lung biopsy in Mycoplasma pneumoniae pneumonia. Arch Pathol Lab Med. 1986;110:34-41.
50 Torres A., de Celis M.R., Roisin R.R., et al. Adult respiratory distress syndrome in Q fever. Eur J Respir Dis. 1987;70:322-325.
51 Winn W.J., Myerowitz R. The pathology of the Legionella pneumonias. A review of 74 cases and the literature. Hum Pathol. 1981;12:401-422.
52 Matthay R., Schwarz M.I., Petty T.L., et al. Pulmonary manifestations of systemic lupus erythematosus: review of twelve cases of acute lupus pneumonitis. Medicine. 1974;54:397-409.
53 Hunninghake G., Fauci A. Pulmonary involvement in the collagen vascular diseases. Am Rev Respir Dis. 1979;119:471-503.
54 Yousem S., Colby T., Carrington C. Lung biopsy in rheumatoid arthritis. Am Rev Respir Dis. 1985;131:770-777.
55 Lakhanpal S., Lie J.T., Conn D.L., Martin W.J.2nd. Pulmonary disease in polymyositis/dermatomyositis: a clinicopathological analysis of 65 autopsy cases. Ann Rheum Dis. 1987;46:23-29.
56 Tazelaar H., Viggiano R.W., Pickersgill J., Colby T.V. Interstitial lung disease in polymyositis and dermatomyositis. Clinical features and prognosis as correlated with histologic findings. Am Rev Respir Dis. 1990;141:727-733.
57 Colby T. Pulmonary pathology in patients with systemic autoimmune disease. Clin Chest Med. 1998;19:587-612.
58 Quismorio F.Jr, Cheema G. Interstitial lung disease in systemic lupus erythematosus. Curr Opin Pulm Med. 2000;6:424-429.
59 Lamblin C., Bergoin C., Saelens T., Wallaert B. Interstitial lung disease in collagen vascular disease. Eur Respir J. 2001;18(suppl 32):69s-80s.
60 Myers J., Katzenstein A. Microangiitis in lupus-induced pulmonary hemorrhage. Am J Clin Pathol. 1986;85:552-556.
61 Walker W., Wright V. Pulmonary lesions and rheumatoid arthritis. Medicine (Baltimore). 1968;47:501-515.
62 Laitinen O., Nissilä M., Salorinne Y., Aalto P. Pulmonary involvement in patients with rheumatoid arthritis. Scand J Respir Dis. 1975;56:297-304.
63 Hakala M., Pääkkö P., Huhti E., et al. Open lung biopsy of patients with rheumatoid arthritis. Clin Rheumatol. 1990;9(4):452-460.
64 Gochuico B.R. Potential pathogenesis and clinical aspects of pulmonary fibrosis associated with rheumatoid arthritis. Am J Med Sci. 2001;321(1):83-88.
65 Pratt D., Schwartz M.I., May J.J., Dreisin R.B. Rapidly fatal pulmonary fibrosis: the accelerated variation of interstitial pneumonitis. Thorax. 1979;34:587-593.
66 Douglas W.W., Tazelaar H.D., Hartman T.E., et al. Polymyositis-dermatomyositis–associated interstitial lung disease. Am J Respir Crit Care Med. 2001;164(7):1182-1185.
67 Muir T., Tazelaar H.D., Colby T.V., Myers J.L. Organizing diffuse alveolar damage associated with progressive systemic sclerosis. Mayo Clin Proc. 1997;72:639-642.
68 Clarysse A., Cathey W.J., Cartwright G.E., Wintrobe M.M. Pulmonary disease complicating intermittent therapy with methotrexate. JAMA. 1969;209:1861-1864.
69 Bone R., Wolfe J., Sobonya R.E., et al. Desquamative interstitial pneumonia following chronic nitrofurantoin therapy. Chest. 1976;69(2):296-297.
70 Kruban Z. Pulmonary changes induced by amphophilic drugs. Environ Health Perspect. 1976;16:111-115.
71 Samuels M.L., Johnson D.E., Holoye P.Y., Lanzotti V.J. Large-dose bleomycin therapy and pulmonary toxicity. A possible role of prior radiotherapy. JAMA. 1976;235:1117-1120.
72 Kilburn K. Pulmonary disease induced by drugs. In: Fishman A.P., editor. Pulmonary Diseases and Disorders. New York: McGraw-Hill; 1980:707-724.
73 Williams T., Eidus L., Thomas P. Fibrosing alveolitis, bronchiolitis obliterans and sulfalazine therapy. Chest. 1982;81:766-768.
74 Schapira D., Nahir M., Scharf Y. Pulmonary injury induced by gold salts treatment. Med Interne. 1985;23(4):259-263.
75 Yousem S., Lifson J., Colby T. Chemotherapy-induced eosinophilic pneumonia. Relation to bleomycin. Chest. 1985;88(1):103-106.
76 Slingerland R., Hoogsteden H.C., Adriaansen H.J., et al. Gold-induced pneumonitis. Respiration. 1987;52(3):232-236.
77 Rosenow E.C.3rd, Myers J.L., Swensen S.J., Pisani R.J. Drug-induced pulmonary disease. An update. Chest. 1992;102:239-250.
78 Rossi S.E., Erasmus J.J., McAdams H.P., et al. Pulmonary drug toxicity: radiologic and pathologic manifestations. Radiographics. 2000;20(5):1245-1259.
79 Abid S., Malhotra V., Perry M. Radiation-induced and chemotherapy-induced pulmonary injury. Curr Opin Oncol. 2001;13(4):242-248.
80 Fassas A., Gojo I., Rapoport A., et al. Pulmonary toxicity syndrome following CDEP (cyclophosphamide, dexamethasone, etoposide, cisplatin) chemotherapy. Bone Marrow Transplant. 2001;28(4):399-403.
81 Erasmus J., McAdams H., Rossi S. Drug-induced lung injury. Semin Roentgenol. 2002;37(1):72-81.
82 Myers J. Pathology of drug-induced lung disease. In: Katzenstein A., Askin F., editors. Katzenstein and Askin’s Surgical Pathology of Non-Neoplastic Lung Disease. Philadelphia: Saunders, 1997.
83 Cleverley J.R., Screaton N.J., Hiorns M.P., et al. Drug-induced lung disease: High-resolution CT and histological findings. Clin Radiol. 2002;57:292-299.
84 Cooper J.Jr, White D., Mathay R. Drug-induced pulmonary disease (Parts 1 and 2). Am Rev Respir Dis. 1986;133:321-338. 488–502
85 Limper A.H., Rosenow E.C.3rd. Drug-induced interstitial lung disease. Curr Opin Pulm Med. 1996;2(5):396-404.
86 Copper J.A.Jr. Drug-induced lung disease. Adv Intern Med. 1997;42:231-268.
87 Camus P.H., Foucher P., Bonniaud P.H., Ask K. Drug-induced infiltrative lung disease. Eur Respir J. 2001;32(suppl):93s-100s.
88 Ozkan M., Dweik R.A., Ahmad M. Drug-induced lung disease. Cleve Clin J Med. 2001;68(9):782-785. 789–795
89 Littler W.A., Kay J.M., Hasleton P.S., Heath D. Busulphan lung. Thorax. 1969;24(6):639-655.
90 Koss L., Melamed M., Mayer K. The effect of bulsufan on human epithelia. Am J Clin Pathol. 1965;44:385-397.
91 Feingold M., Koss L. Effect of long-term administration of bulsufan. Arch Intern Med. 1969;124:66-71.
92 Gyorkey F., Gyorkey P., Sinkovies J. Origin and significance of intranuclear tubular inclusions in type II pulmonary alveolar epithelial cells of patients with bleomycin and bulsufan toxicity. Ultrastruct Pathol. 1980;1:211-221.
93 Ingrassia T.S.3rd, Ryu J.H., Trastek V.F., Rosenow E.C.3rd. Oxygen-exacerbated bleomycin pulmonary toxicity. Mayo Clin Proc. 1991;66:173-178.
94 Imokawa S., Colby T.V., Leslie K.O., Helmers R.A. Methotrexate pneumonitis: review of the literature and histopathological findings in nine patients. Eur Respir J. 2000;15:373-381.
95 Dean P.J., Groshart K.D., Porterfield J.G., et al. Amiodarone-associated pulmonary toxicity: a clinical and pathologic study of eleven cases. Am J Clin Pathol. 1987;87:7-13.
96 Kennedy J.I., Myers J.L., Plumb V.J., Fulmer J.D. Amiodarone pulmonary toxicity. Clinical, radiologic, and pathologic correlations. Arch Intern Med. 1987;147(1):50-55.
97 Myers J.L., Kennedy J.I., Plumb V.J. Amiodarone lung: pathologic findings in clinically toxic patients. Hum Pathol. 1987;18(4):349-354.
98 Martin W.2nd, Rosenow E.3rd. Amiodarone pulmonary toxicity. Recognition and pathogenesis (Part I). Chest. 1988;93:1067-1075.
99 Donaldson L., Grant I.S., Naysmith M.R., Thomas J.S. Acute amiodarone-induced lung toxicity. Intensive Care Med. 1998;24(6):626-630.
100 Blancas R., Moreno J.L., Martín F., et al. Alveolar-interstitial pneumopathy after gold-salts compounds administration, requiring mechanical ventilation. Intensive Care Med. 1998;24(10):1110-1112.
101 Allen J.N., Pacht E.R., Gadek J.E., Davis W.B. Acute eosinophilic pneumonia as a reversible cause of noninfectious respiratory failure. N Engl J Med. 1989;321:569-574.
102 Tazelaar H.D., Linz L.J., Colby T.V., et al. Acute eosinophilic pneumonia: histopathologic findings in nine patients. Am J Respir Crit Care Med. 1997;155:296-302.
103 Hayakawa H., Sato A., Toyoshima M. A clinical study of idiopathic eosinophilic pneumonia. Chest. 1994;105:1462-1466.
104 Pope-Harman A.L., Davis W.B., Allen E.D., et al. Acute eosinophilic pneumonia: a review of 12 cases. Chest. 1994;106:156s.
105 Hamman L., Rich A. Acute diffuse interstitial fibrosis of the lungs. Bull Johns Hopkins Hosp. 1944;74:177-212.
106 Katzenstein A., Myers J., Mazur M. Acute interstitial pneumonia. A clinicopathologic, ultrastructural, and cell kinetic study. Am J Surg Pathol. 1986;10:256-267.
107 Olson J., Colby T., Elliott C. Hamman-Rich syndrome revisited. Mayo Clin Proc. 1990;65:1538-1548.
108 Bouros D., Nicholson A.C., Polychronopoulos V., du Bois R.M. Acute interstitial pneumonia. Eur Respir J. 2000;15:412-418.
109 Colby T.V., Fukuoka J., Ewaskow S.P., et al. Pathologic approach to pulmonary hemorrhage. Ann Diagn Pathol. 2001;5:309-319.
110 Lombard C., Colby T., Elliott C. Surgical pathology of the lung in anti-basement membrane antibody–associated Goodpasture syndrome. Hum Pathol. 1989;20:445-451.
111 Akikusa B., Kondo Y., Irabu N., et al. Six cases of microscopic polyarteritis exhibiting acute interstitial pneumonia. Pathol Int. 1995;45:580-588.
112 Matsumoto T., Homma S., Okada M., et al. The lung in polyarteritis nodosa: a pathologic study of 10 cases. Hum Pathol. 1993;24:717-724.
113 Fajardo L., Berthrong M. Radiation injury in surgical pathology. Part I. Am J Surg Pathol. 1978;2:159-199.
114 Einhorn L., Krause M., Hornback N., Furnas B. Enhanced pulmonary toxicity with bleomycin and radiotherapy in oat cell lung cancer. Cancer. 1976;37:2414-2416.
115 Flint A., Colby T. Diffuse alveolar damage. In: Surgical Pathology of Diffuse Infiltrative Lung Disease. Orlando: Grune and Stratton; 1987.
116 Gross N. Pulmonary effects of radiation therapy. Ann Intern Med. 1977;86:81-92.
117 Fajardo L. Pathology of Radiation Injury, Vol 1. Masson Publishing, New York, 1982.
118 Coggle J., Lambert B., Moores S. Radiation effects in the lung. Environ Health Perspect. 1986;70:261-291.
119 Nash G., Blennerhassett J., Pontoppidan H. Pulmonary lesions associated with oxygen therapy and artifical ventilation. N Engl J Med. 1967;276:368-374.
120 Pratt P. Pathology of pulmonary oxygen toxicity. Am Rev Respir Dis. 1974;110(suppl):51-57.
121 Pratt P. Pulmonary capillary proliferation induced by oxygen. Am J Pathol. 1958;34:1033-1050.
122 Northway W.Jr, Rosan R., Porter D. Pulmonary disease following respirator therapy of hyaline-membrane disease: bronchopulmonary dysplasia. N Engl J Med. 1967;276:357-368.
123 Milberg J.A., Davis D.R., Steinberg K.P., Hudson L.D. Improved survival of patients with acute respiratory distress syndrome (ARDS): 1983–1993. JAMA. 1995;273(4):306-309.
124 Anderson C. Paraquat and the lung. Australas Radiol. 1970;14:409-412.
125 Dearden L.C., Fairshter R.D., McRae D.M., et al. Pulmonary ultrastructure of the late aspects of human paraquat poisoning. Am J Pathol. 1978;93:667-680.
126 Fairshter R. Paraquat poisoning. An update. West J Med. 1978;128:56-58.
127 Chian C.F., Chang F.Y. Acute respiratory distress syndrome in Mycoplasma pneumonia: a case report and review. J Microbiol Immunol Infect. 1999;32(1):52-56.
128 Weber W., Akin F., Dehner L. Lung biopsy in Pneumocystis carinii pneumonia: a histolopathologic study of typical and atypical features. Am J Clin Pathol. 1977;67:11-19.
129 Yousem S., Colby T., Gaensler E. Respiratory bronchiolitis–associated interstitial lung disease and its relationship to desquamative interstitial pneumonia. Mayo Clin Proc. 1989;64:1373-1380.
130 Everard M., Milner A. The respiratory syncytial virus and its role in acute bronchiolitis. Eur J Pediatr. 1992;151(9):638-651.
131 Ebsen M., Anhenn O., Roder C., Morgenroth K. Morphology of adenovirus type-3 infection of human respiratory epithelial cells in vitro. Virchows Arch. 2002;440(5):512-518.
132 Knodoh Y., Taniguchi H., Kawabata Y., et al. Acute exacerbation in idiopathic pulmonary fibrosis. Analysis of clinical and pathologic findings in three cases. Chest. 1993;103:1808-1812.