CHAPTER 93 Acute Liver Failure
DEFINITION
Acute liver failure is defined as the rapid development of hepatocellular dysfunction, specifically coagulopathy and mental status changes (encephalopathy) in a patient without known prior liver disease.1 Acute liver failure is a clinical syndrome that represents the final common pathway of severe liver injury resulting from numerous infectious, immunologic, metabolic, vascular, and infiltrative disorders. The mechanism of liver injury in acute liver failure is most often severe hepatocellular necrosis, as occurs with acetaminophen toxicity or viral hepatitis. Acute liver failure can also result from severe cellular or mitochondrial dysfunction, as occurs with some forms of drug toxicity (e.g., antiretroviral agents), Wilson disease, and acute fatty liver of pregnancy.2
Acute liver failure (or fulminant hepatic failure) originally was defined by an interval of eight weeks or less between the onset of illness and appearance of encephalopathy.3 In an attempt to improve the prediction of the prognosis and outcome, O’Grady and colleagues divided patients into three groups based on the time interval between the onset of jaundice and encephalopathy: those with hyperacute liver failure (seven days or less), those with acute liver failure (eight to 28 days), and those with subacute liver failure (four to 24 weeks).4 In general, patients with hyperacute liver failure are more likely to develop cerebral edema and to recover without liver transplantation. By contrast, patients with subacute or late-onset hepatic failure are more likely to present with evidence of portal hypertension such as ascites and to have a low rate of survival without transplantation.5–7 Although the duration of illness may help predict prognosis, the overlap among patients with acute liver failure with varying presentations is great, and the duration of symptoms is largely related to the cause of liver failure. The original definition of acute liver failure (encephalopathy and coagulopathy within eight weeks of the illness onset) is used in this chapter, because this definition is the most widely used in clinical studies and in criteria for liver transplantation in the United States.
CAUSES
The underlying cause of acute liver failure in an individual patient is established by the patient’s history, serologic and molecular diagnostic test results, and characteristic radiologic or histologic features. The predominant cause of acute liver failure differs markedly throughout the world. In the United States and other Western countries, medications, including acetaminophen, and idiosyncratic drug toxicity are the most commonly identified causes of acute liver failure (Table 93-1).8,9 In France, Japan, and India, severe acute HBV infection is a leading cause of acute liver failure.10–12 In addition to these causes, numerous other, often rare, conditions can lead to acute liver failure (Table 93-2).
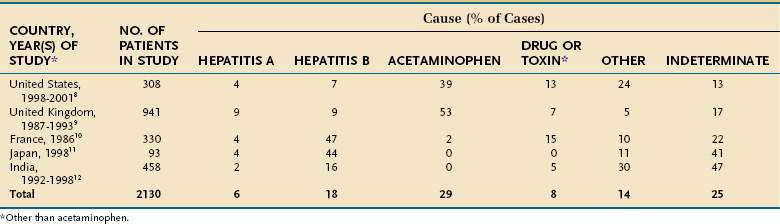
Table 93-2 Uncommon Causes of Acute Liver Failure
ACETAMINOPHEN TOXICITY
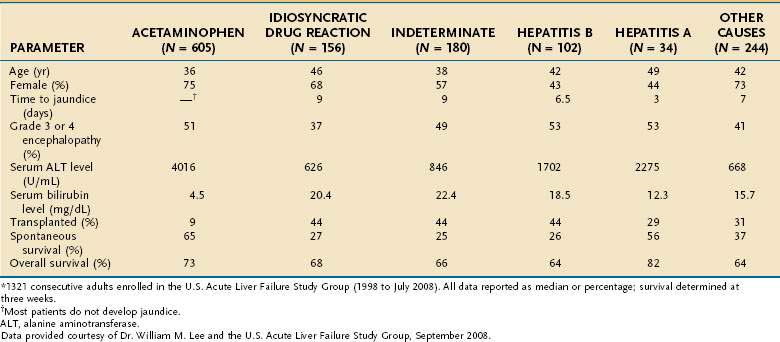
Acetaminophen is a dose-dependent hepatotoxin that, when ingested in excessive doses, can lead to life-threatening liver injury characterized by hypoprothrombinemia, towering aminotransferase elevations, and a normal or minimally elevated serum bilirubin level (Table 93-3). Measurement of serum acetaminophen levels is helpful in assessing the risk of hepatotoxicity following an acute overdose, but false-positive detection of acetaminophen in serum has been reported in some patients with deep jaundice if a colorimetric assay for acetaminophen is used.13 Because of the widespread availability of acetaminophen, intentional acetaminophen overdose (i.e., >10 g) is a common mode of attempted suicide, with over 60,000 cases reported each year in the United States.14 Although most patients who take an intentional overdose of acetaminophen recover, acute liver failure develops in a minority, and acetaminophen toxicity has become the leading cause of acute liver failure in both the United States and United Kingdom (Fig. 93-1).8,9 An increasing frequency of cases of unintentional acetaminophen overdose leading to acute liver failure has also been reported since the 1990s.15–17 In many such “therapeutic misadventures,” patients have ingested over-the-counter products containing acetaminophen as well as narcotic-acetaminophen congeners prescribed for an acute medical condition. Almost 10% of U.S. patients discharged from urban emergency rooms are given a prescription for an acetaminophen-narcotic congener, but most do not receive proper instruction regarding the need to reduce the dose or discontinue the use of other acetaminophen-based analgesics.18 Chronic heavy alcohol consumption may lower the threshold for acetaminophen toxicity in some patients by inducing cytochrome P450 enzyme activity (see Chapter 86).19 In addition, preexisting hepatic dysfunction and resulting glutathione depletion may predispose some patients to acetaminophen toxicity.20 Most patients with an unintentional overdose of acetaminophen have ingested large doses of acetaminophen-containing products over several days, but one study has suggested that ingestion of only 4 g of acetaminophen daily can lead to mild, transient serum aminotransferase elevations in 40% of healthy volunteers.21
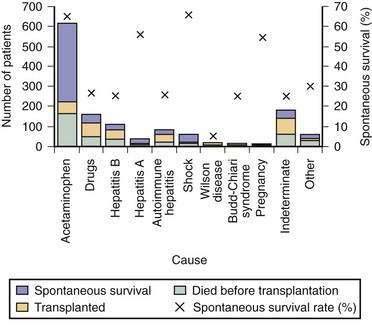
In the United Kingdom, restrictions on the quantity of acetaminophen dispensed, as well as blister packaging of products, were introduced in 1998 to reduce the incidence of acetaminophen toxicity. Since then, rates of hospitalization, acute liver failure, and the need for liver transplantation for acetaminophen toxicity have declined.22,23 In the United States, black box warnings on acetaminophen products and package labeling have been instituted, but the impact of these measures on the incidence and severity of acute liver failure caused by acetaminophen has not been evaluated. Additional measures that may prove useful include imposing limits on the number of tablets sold at one time, unbundling or reducing the dose of acetaminophen in prescription-narcotic congeners, and placing stronger warnings on package labeling.24,25
IDIOSYNCRATIC DRUG TOXICITY
Numerous prescription drugs, including various antibiotics, nonsteroidal anti-inflammatory drugs, and antiseizure medications, have been implicated in acute liver failure (see Chapter 86).26,27 In most cases, drug-induced acute liver failure is a rare and unpredictable event resulting from a metabolic idiosyncrasy that occurs in 1 in 10,000 to 1,000,000 patient–exposure years. Patients with drug-induced acute liver failure are frequently female (70%) and develop jaundice within six months of starting the suspected agent.8,26 Among 141 U.S. liver transplant recipients with drug-induced acute liver failure, isoniazid (16%), propylthiouracil (9%), phenytoin (7%), and valproic acid (7%) were the most commonly identified causative medications.28 Various over-the-counter herbal products and dietary supplements also have been associated with acute liver failure, including kava kava, weight loss supplements, and ephedra.29–31 In addition, severe hepatotoxicity has been associated with extracts of green tea, black cohosh, and adulterated traditional Chinese medicines (see Chapter 87).32–34 Unfortunately, herbal products and dietary supplements are not closely regulated during development, manufacturing, or marketing, and, in many mixtures, the hepatotoxic ingredient(s) cannot be identified.
Establishing a diagnosis of drug-induced acute liver failure is usually difficult because of the lack of specific laboratory markers, inability to rechallenge the patient, and limitations of available causality assessment instruments.35 In addition, less than 10% of patients have evidence of a hypersensitivity reaction associated with a rash or eosinophilia at presentation. The Drug-Induced Liver Injury Network (DILIN) was established to improve our understanding of the risk factors, mechanisms, and outcomes of drug-induced liver injury in the United States.36 The development of an evidence-based causality assessment instrument to assist with the early recognition and diagnosis of idiosyncratic drug-induced liver injury is a priority (see http://dilin.dcri.duke.edu for additional information). The primary treatment of drug-induced acute liver failure is to withdraw the culprit drug immediately once drug-induced liver injury is suspected. In selected patients with severe hepatotoxicity caused by α-methyldopa, nitrofurantoin, or minocycline and autoimmune features (i.e., hypergammaglobulinemia, autoantibodies, plasma cell infiltration on a liver biopsy specimen), treatment with glucocorticoids may be of benefit, but controlled studies are lacking.37,38 Overall, the outcome of drug-induced acute liver failure is poor, with a spontaneous survival rate of only 2% to 40% unless liver transplantation is performed emergently.
VIRAL INFECTIONS
Hepatotropic Viruses
Hepatitis A virus (HAV) and HBV infections are major causes of acute liver failure in many parts of the world, including India and other developing countries (see Table 93-1 and Chapters 77 and 78). Acute infection with HAV rarely leads to acute liver failure (<0.01% of cases), and, when it does, the prognosis is relatively good (see Fig. 93-1). Data from the U.S. Acute Liver Failure Study Group have indicated that the incidence of fulminant HAV infection is declining in parallel with the declining incidence of acute HAV in the general population because of increased immunization against HAV.39 Although HBV is the most common viral cause of acute liver failure, acute liver failure is an uncommon manifestation of acute HBV infection. Infection with hepatitis D virus (HDV), which requires coinfection with HBV, accounts for 4% of cases of acute liver failure in areas endemic for HBV.40 Older studies have suggested that virologic factors, including infection with precore or core promoter variants of HBV, may account for the development of acute liver failure in some patients.41 Subsequent studies have failed to demonstrate unique HBV mutants or variants associated with acute liver failure; the role of HBV genotypes in acute liver failure requires further investigation.42,43 Acute hepatitis E virus (HEV) infection is a leading cause of acute liver failure in India and other tropical countries but is rarely seen in Western countries (see Chapter 80).12,44 Pregnant women may be particularly prone to develop acute liver failure caused by HEV. European series have suggested that organ transplant recipients may be at increased risk of acquiring acute HEV infection and may develop chronic HEV infection.45,46 Hepatitis C virus (HCV) rarely causes acute liver failure.
Other Viruses
Nonhepatotropic viruses, including Epstein-Barr virus (EBV), cytomegalovirus (CMV), varicella-zoster virus, herpes simplex virus (HSV), and parvovirus B-19 infection account for less than 1% of cases of acute liver failure in adults.47,48 Whether these rare causes of acute liver failure are the result of viral variants or an aberrant host immune response to the virus is unclear. Making a diagnosis of acute liver failure caused by a nonhepatotropic virus is often difficult and frequently requires histologic confirmation, as well as polymerase chain reaction testing for viral deoxyribonucleic acid (DNA) in the serum. Almost 50% of patients with HSV-related acute liver failure have no characteristic skin lesions at presentation; the mortality rate is high because the diagnosis is usually delayed.49 Severe acute EBV, CMV, and HSV infection should be considered as possible causes of acute liver failure, particularly in immunosuppressed patients, because they can be treated successfully with antiviral therapy (see Chapter 81).
MISCELLANEOUS CAUSES
Acute liver failure occasionally develops in pregnant women, particularly during the third trimester (see Table 93-2).50 Acute fatty liver of pregnancy occurs in 0.0008% of all pregnancies and is associated with preeclampsia in over 50% of cases (see Chapter 38).51 Some affected women have an inherited deficiency in a fatty acid oxidation enzyme that can be identified by genetic testing. Wilson disease, a rare autosomal recessive disorder characterized by impaired biliary excretion of copper, can present as acute liver failure in up to 25% of cases (see Chapter 75). Most of these patients present in the second or third decade of life and have prominent hemolysis, a low serum alkaline phosphatase level, an elevated serum aspartate aminotransferase (AST)–to–alanine aminotransferase (ALT) ratio, increased urinary copper excretion, and Kayser-Fleischer rings.52 Prompt recognition and listing for liver transplantation are essential, because the outcome is otherwise fatal. Infrequent causes of acute liver failure include mushroom (Amanita phalloides) poisoning (see Chapter 87), Budd-Chiari syndrome (see Chapter 83), autoimmune hepatitis (see Chapter 88), and malignant infiltration of the liver (see Chapter 35). All the miscellaneous causes of acute liver failure combined account for 5% to 30% of cases of acute liver failure (see Table 93-1).
INDETERMINATE ACUTE LIVER FAILURE
Acute liver failure of unknown cause, defined by negative serologic testing for hepatitis A, B, C, D, and E and the absence of other known causes, constitutes 15% to 44% of cases of acute liver failure worldwide (see Table 93-1). Because many of these patients present with a viral prodrome, the hope has been that new, more sensitive molecular laboratory methods would identify a viral cause of acute liver failure of unknown cause. Occult HBV infection has been identified in the sera or livers of some patients with acute liver failure of unknown cause by some41,53 but not other42,43 investigators. Although HCV has been implicated as a cause of acute liver failure in a few patients, it is an exceedingly rare cause of acute liver failure in Western countries.53–55 Togavirus-like particles have been identified by electron microscopy in 7 of 18 liver explants from patients who underwent transplantation for indeterminate acute liver failure but are unlikely to be responsible for a substantial portion of cryptogenic cases of acute liver failure.56 The transfusion-transmitted virus (TTV) was found in the sera of patients with acute liver failure in initial studies, but TTV infection is not thought to be pathogenic.57,58 Studies have failed to demonstrate a link between hepatitis G virus (GB agent, or GBV-C), parvovirus B19, or SEN virus and indeterminate acute liver failure (see Chapter 81).59–61
Using a highly sensitive and specific assay for serum acetaminophen-cysteine adducts, Davern and colleagues identified adducts in 7 of 36 (19%) patients presumed to have indeterminate acute liver failure; the adduct levels were similar to those in patients with known acetaminophen hepatotoxicity.62 These patients tended to have high serum aminotransferase levels and low serum bilirubin levels at presentation, features similar to those seen in patients with a known acetaminophen overdose (see Table 93-3). Whether acetaminophen was the primary cause of liver injury or a cofactor requires further study.63 Regardless, patients with indeterminate acute liver failure should be evaluated rapidly for liver transplantation because of the low likelihood of spontaneous recovery (see later).
CLINICAL FEATURES
The clinical features of acute liver failure may result from the loss of critical hepatocellular functions (e.g., protein synthesis, intermediary metabolism, detoxification) and from effects on organs other than the liver. The major complications of acute liver failure, as well as their pathogenesis and medical management, are outlined in Table 93-4. The initial presentation usually includes nonspecific complaints such as nausea, vomiting, and malaise, and jaundice usually develops soon after. Hepatocellular injury leads to impaired elimination of bilirubin, depressed synthesis of coagulation factors I, II, V, VII, IX, and X, and diminished synthesis of glucose. In addition, decreased uptake and increased generation of intracellular lactate occur as a result of anaerobic glycolysis. These derangements manifest clinically as jaundice, coagulopathy, hypoglycemia, and metabolic acidosis. In addition to portending liver failure, coagulopathy increases the risk of gastrointestinal and intracranial hemorrhage, hypoglycemia can contribute to brain injury, and acidosis can contribute to hypotension.
Table 93-4 Pathogenesis and Management of Major Complications of Acute Liver Failure
| COMPLICATION | PATHOGENESIS | MANAGEMENT |
|---|---|---|
| Hypoglycemia | Diminished hepatic glucose synthesis | Blood glucose monitoring |
| Intravenous glucose supplementation (10% or 20% dextrose) | ||
| Encephalopathy | Cerebral edema | CT scan (if advanced encephalopathy) |
| ICP monitoring (if stage 3 or 4 encephalopathy) | Elevate head of the bed > 30 degrees | |
| Consider osmotherapy (mannitol) or barbiturates | ||
| Treat other contributing factors (e.g., hypoglycemia, hypoxemia, fever) | ||
| Reduce fever (cooling blankets, antibiotics) | ||
| Avoid benzodiazepines and other sedative medications | ||
| ? Moderate hypothermia (see text) | ||
| Infections | Reduced immune function | Aseptic medical, nursing care |
| Invasive procedures | Daily surveillance cultures of blood, urine, and sputum | |
| High index of suspicion for bacterial and fungal infection | ||
| Preemptive antibiotics for gram-negative organisms, anaerobes, and skin flora | ||
| Consider antifungal therapy if patient worsens despite antibacterial coverage | ||
| Gastrointestinal hemorrhage | Stress ulceration | Nasogastric tube placement |
| Intravenous H2 receptor antagonist or proton pump inhibitor | ||
| Coagulopathy | Reduced clotting factor synthesis | Parenteral vitamin K |
| Thrombocytopenia | Platelet infusions for bleeding and before procedures | |
| Fibrinolysis | Plasma infusions for bleeding and before procedures | |
| Cryoprecipitate for bleeding with hypofibrinogenemia | ||
| Recombinant factor VIIa (?) (see text) | ||
| Hypotension | Hypovolemia | Hemodynamic monitoring of central venous pressures |
| Decreased vascular resistance | Volume repletion with blood or colloid | |
| α-Adrenergic agents | ||
| Respiratory failure | ARDS (DAD) | Hemodynamic monitoring of central venous pressures |
| Mechanical ventilation | ||
| Pancreatitis | ?Hypoxia | Supportive care, including supplemental oxygen if needed |
| Abdominal CT to exclude necrotizing pancreatitis | ||
| Renal failure | Hypovolemia | Hemodynamic monitoring of central venous pressures |
| Hepatorenal syndrome | Volume repletion with blood or colloid | |
| Acute tubular necrosis | Avoidance of nephrotoxic agents (e.g., aminoglycosides, NSAIDs, contrast dye) | |
| Oral N-acetylcysteine prior to intravenous contrast agent | ||
| Hemofiltration, dialysis |
ARDS, acute respiratory distress syndrome; CT, computed tomography; DAD, diffuse alveolar damage; ICP, intracranial pressure; NSAIDs, nonsteroidal anti-inflammatory drugs.
HEPATIC ENCEPHALOPATHY AND CEREBRAL EDEMA
Hepatic encephalopathy is a defining criterion for acute liver failure. Encephalopathy in acute liver failure is thought to arise primarily from the development of cerebral edema and resulting intracranial hypertension, rather than from portosystemic shunting of toxins. In addition to cerebral edema, many of the other complications of acute liver failure, including hypoglycemia, sepsis, fever, hypoxemia, and hypotension, may contribute to neurologic abnormalities. The staging of encephalopathy in acute liver failure is similar to that used for patients with cirrhosis (see Chapter 92). In stage 1, patients have subtle changes in affect, altered sleep patterns, or difficulties with concentration. Stage 2 is characterized by drowsiness, disorientation, and confusion. Stage 3 is marked by somnolence and incoherence. In stage 4, frank coma with minimal (4A) or no (4B) response to noxious stimuli is detected. On physical examination, many patients have asterixis or a tremor in stage 1 or 2, whereas hyperreflexia, clonus, and muscular rigidity are common in stages 3 and 4. Although worrisome, these upper motor neuron signs do not portend a poor prognosis and can reverse with recovery or replacement of the failing liver.
Cerebral edema is found in up to 80% of patients who die of acute liver failure and is almost universal in patients with coma.64 The pathogenesis of cerebral edema in acute liver failure is poorly understood. It has been proposed to result, in part, from the actions of intestine-derived neurotoxins that escape hepatic clearance and are released into the systemic circulation.65 The demonstration of swollen endothelial and astroglial cells in the brains of patients with acute liver failure suggests a potential role for cytotoxic edema, possibly resulting from increased brain glutamine levels. On the other hand, vacuolization in the basement membranes of capillaries, consistent with disruption of the blood-brain barrier, suggests a vasogenic mechanism of cerebral edema in acute liver failure. In any event, an increased production of glutamine in the central nervous system as a result of high circulating levels of ammonia and intracerebral lactate is believed to be critical to the pathogenesis of cerebral edema. In one study, arterial ammonia levels were associated with the risk of uncal herniation and death in patients with acute liver failure.66 Another study suggested that a lack of reduction in arterial ammonia levels over time was associated with an increased risk of progression to cerebral edema.67
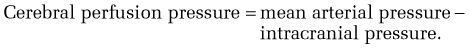
COAGULOPATHY AND BLEEDING
The liver is the major site of synthesis of coagulation factor and related inhibitory proteins. The reticuloendothelial cell system of the liver is involved in the clearance of activated clotting factors and their degradation products. Therefore, patients with acute liver failure frequently have a multifactorial coagulopathy and a resulting increased risk of bleeding and clotting. Laboratory features of fibrinolysis, hypofibrinogenemia, dysfibrinogenemia, and DIC are frequent in patients with acute liver failure.68 Thrombocytopenia also develops in most patients with acute liver failure and may be the result of increased destruction of platelets from a consumptive coagulopathy or reduced thrombopoietin production, bone marrow dysfunction, or the effects of medications.69 Clinically significant bleeding has been reported to occur in 10% to 20% of patients with acute liver failure; the most common sources are the upper gastrointestinal tract, nasopharynx, and skin puncture sites. Critically ill patients with acute liver failure have a particular propensity for gastrointestinal bleeding caused by acute portal hypertension, increased intracranial pressure, and coagulopathy.70
INFECTION
Bacterial infections may develop in as many as 80% of patients with acute liver failure, and bacteremia is present in up to 25%.65,71 Uncontrolled infection accounts for exclusion of approximately 25% of patients with acute liver failure from liver transplantation and approximately 40% of post-transplantation deaths. At least three factors place patients with acute liver failure at increased risk for infection. First, intestine-derived microorganisms may enter the systemic circulation from portal venous blood as a result of damage to hepatic macrophages (Kupffer cells). Second, impaired neutrophil function may result from reduced hepatocellular synthesis of acute-phase reactants, including components of the complement cascade. Third, patients with acute liver failure are often subjected to invasive procedures (e.g., intravascular and urethral catheterization, endotracheal intubation), and physical barriers to infection, including skin and airway, are thus breached. Indeed, the major sites of infection are the respiratory and urinary tracts. Not surprisingly, the most commonly isolated bacteria are staphylococcal and streptococcal species and gram-negative aerobes.72
Fungal infections develop in up to one third of patients with acute liver failure.73 Most of these infections are caused by Candida albicans and typically develop after the second or third week of hospitalization. Although Aspergillus infections were thought to be uncommon in the setting of acute liver failure, they may be more prevalent than previously appreciated, and aspergillosis may account for up to half of fatal infections in the post-transplantation period.74 Risk factors for fungal infections are renal failure, prolonged antibiotic therapy for bacterial infections, and use of invasive monitoring devices. Characteristically, fungal infection is associated with fever or leukocytosis refractory to broad-spectrum antibiotics. Patients in whom symptoms and signs of the systemic inflammatory response syndrome develop in association with bacterial or fungal infection are more likely to experience worsening encephalopathy and to die; this observation highlights the importance of infections in the outcome of acute liver failure.75,76
MULTIPLE ORGAN FAILURE SYNDROME
The multiple organ failure syndrome manifests clinically as peripheral vasodilatation with hypotension, pulmonary edema, renal failure, and DIC. Liver failure may trigger the microcirculatory derangements that underlie this syndrome by two mechanisms. First, polymerization of actin (released from dying hepatocytes) within the capillary lumen and platelet activation may cause endothelial injury.77 Second, impaired hepatic clearance may lead to the accumulation of vasoactive substances in the systemic circulation.78 Multiple organ failure syndrome is an important contributor to mortality and constitutes a major contraindication to liver transplantation.
Hypotension is observed frequently in patients with acute liver failure and can result from reduced vascular resistance or intravascular volume depletion. Subclinical cardiac injury, defined by elevated serum troponin levels, is common in patients with acute liver failure, and the level of troponin correlates with the risk of an adverse outcome.79 Acute pancreatitis may also develop in patients with acute liver failure, particularly those with an acetaminophen overdose, as a result of tissue hypoxia and hypoperfusion. In one series, 44% of patients who died of acute liver failure had acute pancreatitis.80 Acute pancreatitis is not a contraindication to liver transplantation unless evidence of extensive pancreatic necrosis is seen by computed tomography (CT).
Respiratory failure is commonly associated with acute liver failure. In one series, 37% of patients with acute liver failure had pulmonary edema,81 whereas acute respiratory distress syndrome (ARDS) was present in 33% of patients with acetaminophen-associated acute liver failure.82 Furthermore, ARDS was associated with intracranial hypertension, the requirement for vasopressor agents, and a higher rate of mortality.
The cause of renal failure, which develops in 30% to 50% of patients with acute liver failure, is usually multifactorial.78 Hepatorenal syndrome is often difficult to differentiate from intravascular volume depletion because both entities are associated with oliguria, azotemia, and a low fractional excretion of sodium. Acute tubular necrosis is associated with a 50% decrease in survival in patients with acetaminophen-induced acute liver failure,83 and the mortality rate is more than doubled in patients with multiple organ failure syndrome.84
PROGNOSIS
Patients with acute liver failure fall into two broad categories: (1) those in whom intensive medical care enables recovery of hepatic function through liver regeneration and (2) those who require liver transplantation to survive. Rapid identification of patients with an unfavorable prognosis is critical. The cause of acute liver failure and clinical presentation are important correlates of prognosis. For example, patients with acute liver failure caused by acetaminophen have a better prognosis than those with indeterminate acute liver failure (see Fig. 93-1).8 Similarly, patients who reach stage 3 or stage 4 encephalopathy tend to do worse than those who reach only stage 1 or stage 2 encephalopathy.4 In an individual patient, however, these indicators do not allow accurate prediction of the need for liver transplantation.
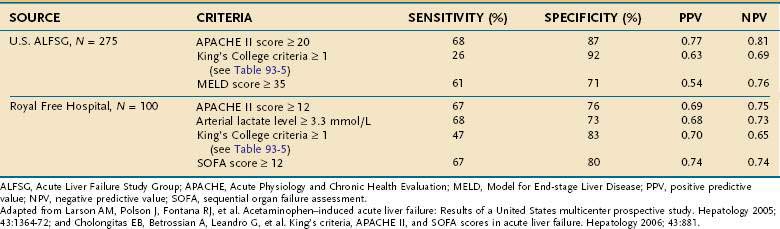
Investigators at King’s College in London have performed a multivariate analysis of clinical and biochemical variables and their relationship to mortality in 588 patients with acute liver failure.83 In this analysis, a distinction was made between patients with acetaminophen toxicity and those with other causes of acute liver failure (Table 93-5). For patients with acetaminophen-induced acute liver failure, the presence of any single adverse characteristic was associated with a mortality rate of at least 55%, and severe acidosis was associated with a mortality rate of 95%. Subsequently, the addition of arterial lactate levels to these predictors has been shown to improve the positive and negative predictive values in patients with acetaminophen-associated acute liver failure.85 Among patients with nonacetaminophen acute liver failure, the presence of any single adverse prognostic factor was associated with a mortality rate of 80%, and the presence of three adverse characteristics was associated with a mortality rate of more than 95%. These mortality rates vastly exceed those associated with liver transplantation. Therefore, the presence of any single indicator of a poor prognosis should prompt early referral to a liver transplantation center. These selection criteria are simple, and acquisition of the necessary data requires only brief history taking, routine laboratory studies, and serologic testing for HAV and HBV.86 Another study from the U.S. Acute Liver Failure Study Group has demonstrated a lower sensitivity rate and negative predictive value for these prognostic factors in 108 patients with acetaminophen-induced acute liver failure.15 Other groups have found that markers of organ failure, such as the Acute Physiology and Chronic Health Evaluation (APACHE) II score and Sequential Organ Failure Assessment (SOFA) index, are better predictors of outcome in patients with acetaminophen-related acute liver failure than the King’s College criteria and Model for End-stage Liver Disease (MELD) score (Table 93-6).16,87,88
Table 93-5 King’s College Criteria for Liver Transplantation in Acute Liver Failure83,85
| ACETAMINOPHEN CASES | NON-ACETAMINOPHEN CASES |
|---|---|
INR, international normalized ratio; PT, prothrombin time.
* Measured after fluid resuscitation.
Table 93-6 Criteria Used to Predict the Prognosis of Patients with Acetaminophen-Induced Acute Liver Failure
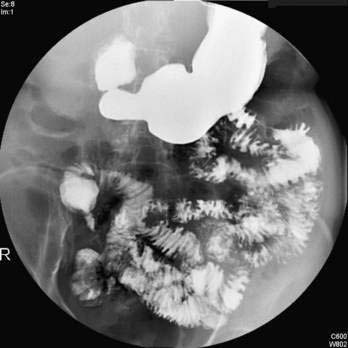
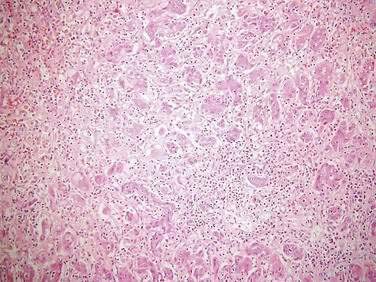
Liver histologic evaluation in acute liver failure is associated with substantial sampling error and potential complications and does not reliably predict outcome.89 Therefore, percutaneous or transjugular liver biopsy is not recommended for prognosis or staging purposes but can be helpful in confirming a diagnosis of malignant infiltration, autoimmune hepatitis, and nonhepatotropic viral infection (Fig. 93-2). The predictive value of serum Gc-globulin levels is comparable to the King’s College criteria, but the assay is technically difficult and not generally available.90 Other investigators have examined the prognostic usefulness of measuring plasma factor V levels (see Table 95-5)91 and hepatic volumetry,92 but these parameters do not appear to add significantly to the assessment of outcome. The U.S. Acute Liver Failure Study Group and others have reported on the potential use of elevated serum phosphate levels (>3.7 mg/dL) as a marker of impaired liver regeneration and poor prognosis in patients with acetaminophen-induced acute liver failure.93,94 Other groups, however, have failed to confirm the clinical usefulness of serum phosphate measurements in acute liver failure.95 Serial assessment of serum alpha fetoprotein levels, which correlate with hepatic regeneration, have been reported to be of value in predicting prognosis in patients with acute liver failure.96
TREATMENT
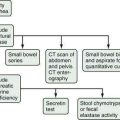
A variety of therapies have been proposed and studied in patients with acute liver failure, including glucocorticoid therapy, prostaglandin infusions, and exchange transfusions. Only liver transplantation, however, has permitted salvage of patients with irreversible liver failure. Unfortunately, many patients with irreversible acute liver failure do not undergo liver transplantation because of late referral, contraindications, or the lack of a donor liver. Therefore, patients with acute liver failure should be evaluated for liver transplantation as soon as possible and, if no contraindications are identified, placed on a liver transplant waiting list. If and when a donor organ becomes available, a patient listed for transplantation should be reassessed for the continued need for transplantation. An algorithm depicting the management of acute liver failure is shown in Figure 93-3.
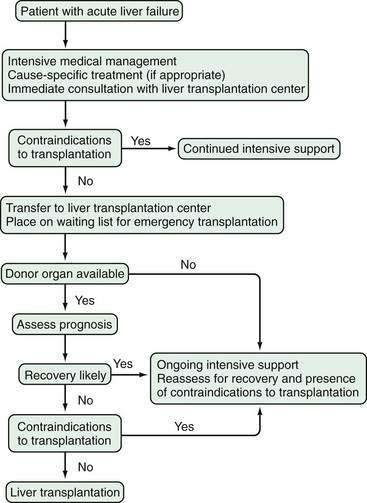
INITIAL EVALUATION AND MANAGEMENT
The initial management of a patient with acute liver failure should include rapid identification of the cause of acute liver failure, with an emphasis on treatable conditions. For example, acetaminophen toxicity is treated initially with gastric lavage, oral charcoal, and prompt administration of oral N-acetylcysteine (see Chapter 86).97 For patients with severe nausea and vomiting, an approved intravenous formulation of N-acetylcysteine can be administered safely in a monitored setting.98 Similarly, patients with Amanita mushroom poisoning should be treated with immediate gastric lavage and instillation of charcoal in an attempt to reduce the toxin load.99 In addition, hemodialysis can remove toxin from the serum, and intravenous penicillin, milk thistle (silymarin), and cytochrome c may further lower the enterohepatic toxin load (see Chapter 87); however, the clinical benefit of these measures is uncertain.100 HSV-induced acute liver failure has been reported to respond to intravenous acyclovir.101 Rapid delivery and supportive care constitute the treatment of choice for pregnant women with acute fatty liver of pregnancy, the hemolysis, elevated liver enzyme levels, and low platelet (HELLP) syndrome, and preeclampsia (see Chapter 38). The benefit of oral antiviral agents in patients with fulminant hepatitis B remains unproven; many experts advise prescribing a nucleoside analog, such as entecavir or lamivudine, in light of their favorable safety profiles, in the hope of salvaging liver function and reducing the level of viremia prior to liver transplantation (see Chapter 78).102–104
N-acetylcysteine has been proposed as a potential treatment for non–acetaminophen-related acute liver failure on the basis of studies demonstrating improvements in tissue oxygenation and systemic hemodynamics and the drug’s antioxidant properties.105 The U.S. Acute Liver Failure Study Group reported on a multicenter, randomized, controlled trial that compared a 72-hour infusion of intravenous N-acetylcysteine and placebo in 173 adult patients with acute liver failure.106 The overall patient survival at three weeks was similar in the two groups (70% N-acetylcysteine vs. 66% placebo, P = 0.28), but transplant-free survival was significantly better in patients who received N-acetylcysteine (40% vs. 27%, P = 0.04). The benefit of N-acetylcysteine appeared to be limited to the subgroup of patients with grade 1 or 2 encephalopathy at entry (52% vs. 31%, P = 0.021). In addition, a single-center retrospective study of 170 children with nonacetaminophen acute liver failure demonstrated that treatment with N-acetylcysteine was associated with a shorter length of hospital stay and a higher rate of spontaneous recovery.107 In this study, however, the untreated controls were not contemporaneous and had more severe illness at presentation. In both studies,106,107 N-acetylcysteine was generally well tolerated, with a low rate of side effects (e.g., rash, bronchospasm, arrhythmia). Additional studies of N-acetylcysteine for non–acetaminophen-related acute liver failure are in progress to identify which patients may benefit from this treatment.
All patients with acute liver failure should be cared for in an intensive care unit because they can deteriorate rapidly.104 Serial laboratory studies, including acid-base status, arterial ammonia levels, and INR, should be carried out to monitor the patient’s condition. Urgent transfer to a liver transplantation center is advisable early in the course, prior to the development of advanced encephalopathy or complications.108
ENCEPHALOPATHY AND CEREBRAL EDEMA
Encephalopathy associated with acute liver failure tends to be progressive, unless liver failure is reversed. Sedative-hypnotic drugs, which may exacerbate encephalopathy, should be avoided unless patients require mechanical ventilation. Lactulose is of uncertain benefit and may be associated with bowel ischemia. Reversible conditions that may contribute to altered mental status (e.g., hypoglycemia, hypoxemia) should be treated immediately. Hypoglycemia generally responds to parenteral administration of glucose. Similarly, underlying infection and sepsis should be treated aggressively with fluids and antibiotics, because systemic cytokines may alter brain function (see Table 93-4).
Patients with stage 3 or 4 encephalopathy should undergo elective endotracheal intubation and mechanical ventilation for protection of the airway, particularly before being transported to a liver transplantation center. Many mechanically ventilated patients are also deeply sedated or paralyzed, and evidence of generalized seizure activity that can worsen encephalopathy may be concealed. Therefore, continuous electroencephalographic monitoring of deeply sedated or paralyzed patients with acute liver failure has been proposed. Treatment of subclinical seizures with phenytoin or other antiepileptic medications is appropriate, but the efficacy of prophylactic therapy to prevent seizure activity has not been established.109
Intracranial hypertension can be suspected on the basis of noninvasive assessment or direct measurement, but noninvasive assessment by physical examination and radiologic imaging has important limitations. Impaired pupillary responses, posturing, or seizures, which may suggest the presence of intracranial hypertension, are not sensitive signs for intracranial hypertension, particularly when sedatives or neuromuscular blocking agents are used in mechanically ventilated patients. CT of the head is useful for identifying mass lesions, intracranial hemorrhage, and evidence of brainstem herniation because these conditions may affect clinical decision making. CT scans of the head should be obtained in all patients with advanced encephalopathy. Nevertheless, the correlation between CT evidence of cerebral edema and measured ICP is imperfect, with a sensitivity varying from 60% to 75%.110,111 Transcranial Doppler measurements of middle cerebral artery blood flow continue to improve, but further refinements are needed to make this noninvasive modality reliable enough for clinical decision-making.112
Monitoring of ICP represents the most accurate way to detect intracranial hypertension but has several potential limitations. First, placement of an ICP transducer requires correction of underlying coagulopathy. Second, the ICP transducer represents a potential portal of entry for infectious organisms. Third, placement of the transducer can precipitate intracranial hemorrhage, which can be fatal. The frequency of serious complications ranges from 4% to 20%; parenchymal catheters are associated with a higher rate of complications than subdural or epidural transducers.113 Nevertheless, ICP transducers can provide invaluable physiologic data that influence management and decisions regarding liver transplantation. In one study, 92 patients with advanced encephalopathy had ICP monitors placed and were compared with 239 unmonitored patients. The monitored patients tended to have more severe liver failure and multiorgan failure.114 In subjects listed for liver transplantation, ICP monitoring was associated with a greater number of medical treatments and procedural interventions. Although overall and post-transplantation survival rates were similar in the two groups, the monitored patients had a low rate of intracranial bleeding (i.e., 10%). The data suggest that future studies of therapies for acute liver failure should include ICP monitoring. For patients with refractory intracranial hypertension, technetium perfusion scans are useful to detect irreversible brain death, which may otherwise be unrecognized in a sedated patient.
Elevation of the head of the bed to at least 30 degrees from horizontal (and avoidance of the head-down position) is a simple measure to reduce ICP. If this maneuver fails, specific treatment is required. Osmotherapy and barbiturates are two options for treating intracranial hypertension. Osmotherapy with intravenous mannitol (0.5 to 1 g/kg) requires preserved renal function (or concomitant hemofiltration, if necessary) and effectively controls intracranial hypertension in approximately 60% of cases.115 Hypertonic saline may also be of value in patients with cerebral edema, but controlled trials are needed to confirm a benefit.116 Uncontrolled data have supported the use of intravenous thiopental, a barbiturate; its efficacy is similar to that of mannitol.117 Thiopental has two relative advantages—its onset of action is rapid, and its use does not require preserved renal function. Potential drawbacks of thiopental are a risk of hypotension and, of greater importance, the potential to mask clinical indicators of neurologic recovery or deterioration. In general, it is reasonable to use mannitol as first-line therapy and to reserve a barbiturate for patients with renal insufficiency or refractory intracranial hypertension. Glucocorticoids are of no benefit.113 Pilot studies have suggested that moderate hypothermia (e.g., 32° C to 33° C), achieved by the use of an external cooling blanket, may be of benefit in patients with acute liver failure and refractory cerebral edema.118,119 All patients treated with hypothermia require the placement of an ICP monitor because paralytic agents are required to prevent shivering. Moreover, the effect of moderate hypothermia on the risk of bleeding and infection require further study. A prophylactic role for hypothermia also requires further study.
COAGULOPATHY AND BLEEDING
Placement of a nasogastric tube to monitor gastrointestinal bleeding and gastric pH is recommended for intubated patients with acute liver failure. The risk of upper gastrointestinal hemorrhage can be reduced by intravenous administration of a histamine H2 receptor antagonist,120 and proton pump inhibitors probably have a similar benefit. Administration of subcutaneous vitamin K to attempt to reverse hypoprothrombinemia is also reasonable. Coagulation parameters, including the INR, plasma factor V level, platelet count, and plasma fibrinogen level, should be assessed serially in all patients with acute liver failure.
For patients with acute liver failure who undergo an invasive procedure and whose INR fails to improve with plasma, recombinant factor VIIa has been shown to be efficacious, but the optimal dose has not been established and use of this agent carries a risk of thrombosis.121 Cryoprecipitate, plasma, and platelets should be given to patients with hypofibrinogenemia, DIC, and active bleeding.
INFECTION
Clinical recognition of infection may be difficult because signs such as hypotension, leukocytosis, and acidosis may reflect the underlying liver failure. Therefore, daily surveillance cultures of blood, urine, and ascitic fluid are recommended in patients with acute liver failure. The advisability of prophylactic antibiotics in the setting of acute liver failure is debatable. On one hand, prophylactic antibiotics may delay the development of infections that limit the applicability of liver transplantation. On the other hand, antibiotics may increase the risk of superinfection with resistant bacteria or fungi. This issue has been addressed in a small randomized trial.122 Patients treated with prophylactic intravenous cefuroxime had a significant reduction in the rate of documented infections (from 61% to 32%) compared with those treated conservatively and a modest (but statistically insignificant) increase in the rate of survival (from 45% to 67%). Enteral decontamination with orally administered antibiotics (as well as systemic antibiotics) does not appear to alter the clinical outcome of patients with acute liver failure, compared with that observed with systemic antibiotics alone.123 The usefulness of systemic prophylactic antibiotics warrants further investigation. At the least, a high level of suspicion for infection and a low threshold for administering antibiotics are required in managing patients with acute liver failure. If infection is suspected, the choice of antibiotics should be based on the spectrum of likely bacterial pathogens (e.g., Staphylococcus, gram-negative aerobes) and local hospital patterns of microbial sensitivity. A reasonable empirical regimen is intravenous vancomycin and a third-generation cephalosporin or fluoroquinolone.
MULTIPLE ORGAN FAILURE SYNDROME
The fundamental goal of management of multiple organ failure syndrome in patients with acute liver failure is similar to that in patients with other causes of multiple organ failure—to optimize arterial pressure and tissue oxygenation. Ideally, the mean arterial pressure (MAP) should be kept above 60 mm Hg to maintain cerebral perfusion.64 A central venous or right heart catheter may be useful for monitoring the patient’s intravascular volume status. Hypotension resulting from intravascular volume depletion should be corrected with blood or colloids. If hypotension is caused by reduced vascular resistance, administration of an α-adrenergic agonist may be useful. Although pressors can be used to maintain MAP within a physiologic range, they have the potential to impair tissue oxygenation further; terlipressin (a long-acting vasopressin analog not available in the United States) may worsen cerebral edema (see Chapters 90 and 92).124,125 Most experts recommend using norepinephrine or dopamine rather than vasopressin because of adverse effects of the latter on intracranial hypertension.125 In small short-term studies, N-acetylcysteine has been shown to improve tissue oxygenation without adverse effects on hemodynamics126; however, the impact of this agent on overall patient outcome has not yet been determined.
Endotracheal intubation and mechanical ventilation are frequently necessary for patients with acute liver failure. Hypoxemia can result from respiratory depression caused by coma or impaired gas exchange caused by ARDS or superimposed pneumonia. Vigorous suctioning and Valsalva maneuvers should be avoided to prevent surges in ICP. Patients with acute liver failure tolerate volume overload poorly in light of their propensity to develop ARDS and cerebral edema. Early measurement of the central venous or pulmonary capillary wedge pressure is preferable in oliguric patients to empirical administration of fluid boluses. If oliguria persists in the face of adequate central filling pressures, continuous renal replacement therapy should be initiated. Continuous venovenous hemofiltration has been shown to be superior to intermittent hemodialysis, with less hemodynamic instability and improved tissue oxygen delivery, in oliguric patients with acute liver failure.127 Nephrotoxic drugs such as aminoglycosides and nonsteroidal anti-inflammatory drugs should be avoided in all patients with acute liver failure, and appropriate precautions should be taken if intravenous contrast dye is required.
LIVER TRANSPLANTATION
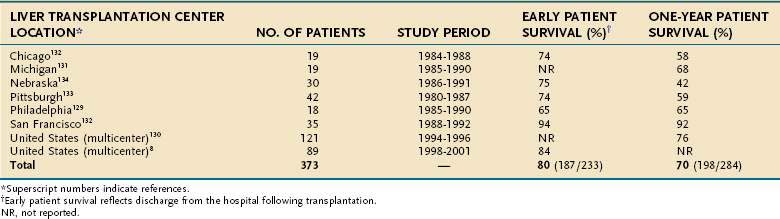
Liver transplantation has transformed the management of patients with acute liver failure and is discussed in greater detail in Chapter 95. Before the advent of liver transplantation, less than 30% of patients with acute liver failure survived. By contrast, survival rates for patients with acute liver failure who undergo liver transplantation have been substantially higher, with a short-term survival rate of 80% and a one-year survival rate of 70% when the results of several major transplantation centers are combined (Table 93-7).8,128–134 The decision to perform transplantation in a patient with acute liver failure must balance the likelihood of spontaneous recovery with the risks of surgery and long-term immunosuppression. Furthermore, contraindications to transplantation, particularly irreversible brain damage, active extrahepatic infection, or multiple organ failure syndrome, must be considered.
Although changes in the rules governing allocation of donor livers have shortened waiting times for patients with acute liver failure in the United States, the decision to place a patient on the waiting list for transplantation must still be made promptly.135,136 A patient’s clinical status needs to be assessed frequently to determine whether the patient is likely to recover or has developed a contraindication to transplantation. In one series, a contraindication that precluded transplantation developed in 22% of liver transplantation candidates, whereas 12% improved and were removed from the waiting list.8 The shorter median waiting times among liver transplant recipients compared with the times to exclusion for nontransplanted patients (three versus five days) highlights the critical, ongoing shortage of donor organs.
Because of the shortage of donor organs, patients with acute liver failure are more likely to receive an ABO-incompatible rather than an ABO-compatible or ABO-identical graft.137 In addition, marginal donor grafts that are older or steatotic are used more frequently in transplant recipients with acute liver failure than in recipients with other indications for transplantation. These factors may explain in part the higher rate of primary nonfunction and rejection among transplant recipients with acute liver failure compared with other recipients.138 Post-transplantation seronegative chronic hepatitis is also more common in transplant recipients with acute liver failure than in recipients with cirrhosis (41% versus 14% at one year).139 The long-term functional and cognitive outcomes of transplant recipients with acute liver failure have not been well studied but may be inferior to those of transplant recipients with cirrhosis.140
Live Donor Liver Transplantation
In countries in which cadaveric livers are not readily available, live donor liver transplantation has been performed successfully in highly selected patients with acute liver failure (see Chapter 95).141,142 In the United States, adult live donor liver transplantation has rarely been undertaken for acute liver failure, and a moratorium has been placed on using this procedure for patients with acute liver failure in New York. One study, however, has demonstrated that seven of ten live donor organ recipients with acute liver failure survived, compared with two of three cadaveric transplant recipients with acute liver failure.143 In addition, the frequency of complications in live donors to a patient with acute liver failure (50%) was similar to that reported in donors who were evaluated electively for donation to a transplant recipient with cirrhosis (33%). Nevertheless, because of concerns regarding the ability to evaluate donors safely in an accelerated time frame, the potential for coercion, and the potential for poorer outcomes, most experts recommend a whole-sized cadaveric transplant whenever possible in a patient with acute liver failure.
INVESTIGATIONAL APPROACHES
Treatment strategies, such as charcoal hemoperfusion and administration of prostaglandin E1, which showed promise in uncontrolled trials, have not been shown to be superior to standard care when studied in randomized studies.144,145 Plasmapheresis and hepatectomy have been suggested as a possible bridge to liver transplantation, but prospective trials have yet to be performed.146,147 Three additional forms of therapy may provide a bridge to liver transplantation or to regeneration of the native liver with spontaneous recovery—auxiliary liver transplantation, extracorporeal liver support devices, and hepatocyte transplantation.
AUXILIARY LIVER TRANSPLANTATION
Auxiliary liver transplantation, in which the donor graft is implanted orthotopically beside the surgically reduced native liver or heterotopically inferior to the native liver, has been investigated by a number of centers.148,149 The advantage of this procedure is that by providing a temporary auxiliary liver, the severely diseased native liver may be allowed to regenerate. Ideally, immunosuppression may then be gradually withdrawn, thereby allowing the transplanted liver to involute or be surgically removed. The usefulness of this operation is limited by technical complications as well as by the difficulty in predicting which patients with acute liver failure are likely to experience hepatic regeneration. This approach should only be undertaken in centers with specialized expertise.
EXTRACORPOREAL LIVER SUPPORT
Extracorporeal liver support devices fall into two broad categories, hemodiadsorption systems and bioartificial livers.150 Hemodiadsorption systems use hemodialysis in combination with perfusion of the patient’s plasma or blood through three hollow fiber filters impregnated with charcoal, resins, and albumin, respectively. Although these devices may remove circulating toxins, they do not replace other liver functions. Albumin dialysis (e.g., the molecular adsorbent recirculating system [MARS]) uses hemodialysis of whole blood in series with an albumin dialyzer and charcoal filter. This technology is simpler to use than plasmapheresis and has shown some promise in trials of cirrhotic patients with hepatic encephalopathy,151,152 but information regarding the safety and efficacy of these systems in patients with acute liver failure is limited.153,154
Bioartificial liver devices contain liver cells grown within specialized hollow fiber cartridges through which the patient’s plasma is perfused. The success of such devices depends largely on the mass of cells they contain, the extent to which these cells maintain liver-specific functions, and the duration for which these functions are maintained. Because the devices under clinical investigation contain only hepatocytes, derangements attributable to nonparenchymal cells, such as Kupffer cells and biliary epithelia, are not replaced. The results of the HepatAssist bioartificial liver device trial were reported in 2004.155 This device uses a dialysis cartridge loaded with approximately 100 g of cryopreserved porcine hepatocytes, or 7 billion cells, and also has a charcoal filter. The 85 patients with acute liver failure who were treated with the HepatAssist device did not experience an improvement in 30-day survival compared with the 86 patients with acute liver failure who received standard care (71% versus 62%, P = 0.26). Treatment with the device was well tolerated, and the rate of thrombocytopenia, hypotension, and other adverse events was not significantly greater in treated patients. Furthermore, there were no reports of inadvertent transmission of porcine retroviruses or development of xenogenic antibodies in treated patients.156 Although this pioneering trial failed to demonstrate a significant benefit in outcome with the HepatAssist device, the proof of concept that an extracorporeal device with porcine hepatocytes can be used and can lead to a trend toward improvement in metabolic, hemodynamic, and clinical parameters was realized. The development of devices with a larger hepatocyte mass, simplified circuitry, and differentiated hepatocyte function is eagerly awaited.
HEPATOCYTE TRANSPLANTATION
The potential role of hepatocyte transplantation in patients with acute liver failure is likely to be as a bridge to liver transplantation or regeneration.157 Human hepatocyte transplantation has demonstrated efficacy in preliminary studies of patients with metabolic disorders and decompensated cirrhosis.158,159 Stable expression of transplanted hepatocytes has been difficult to achieve. In one trial, three of six patients with acute liver failure survived 14, 20, and 52 days after transplantation of 109 to 1010 hepatocytes, representing 1% to 10% of normal liver cell mass.121 Although metabolic parameters improved within 72 hours of transplantation, transient respiratory insufficiency was observed in several patients. This report demonstrated that transplanted human hepatocytes can engraft into a regenerating liver, but further work on enhancing graft function, native liver regeneration, and cell delivery is needed.
Pluripotent hepatocyte stem cells derived from bone marrow may prove useful for hepatocyte transplantation. In one study, liver biopsy specimens from human recipients of gender-discordant bone marrow or liver transplants were analyzed for marrow-derived hepatocytes and cholangiocytes.160 Differences in the extent of engraftment were associated with the degree of allograft injury, thereby suggesting a possible role for bone marrow–derived stem cells in the treatment of severe acute hepatitis, acute liver failure, and metabolic defects. Further studies are needed to identify and isolate pluripotent liver stem cells from human bone marrow and clarify the factors that govern cellular differentiation and liver regeneration.161
Bernal W, Donaldson N, Wyncoll D, et al. Blood lactate as an early predictor of outcome in paracetamol-induced acute liver failure: A cohort study. Lancet. 2002;359:558-63. (Ref 85.)
Bernal W, Hall C, Karvellas CJ, et al. Arterial ammonia and clinical risk factors for encephalopathy and intracranial hypertension in acute liver failure. Hepatology. 2007;46:1844-52. (Ref 67.)
Davern TJ, James LP, Hinson JA, et al. Measurement of serum acetaminophen-protein adducts in patients with acute liver failure. Gastroenterology. 2006;130:687-94. (Ref 62.)
Demetriou AA, Brown RS, Busuttil RW, et al. Prospective, randomized, multicenter controlled trial of a bioartificial liver in treating acute liver failure. Ann Surg. 2004;239:660-70. (Ref 155.)
Fontana RJ. Acute liver failure due to drugs. Sem Liv Dis. 2008;28:175-88. (Ref 27.)
Houlihan DD, Newsome PN. Critical review of clinical trials of bone marrow stem cells in liver Disease. Gastroenterology. 2008;135:438-50. (Ref 161.)
Kumar M, Satapathy S, Monga R, et al. A randomized controlled trial of lamivudine to treat acute hepatitis B. Hepatology. 2007;45:97-101. (Ref 102.)
Larson AM, Polson J, Fontana RJ, et al. Acetaminophen-induced acute liver failure: Results of a United States Multicenter prospective study. Hepatology. 2005;43:1364-72. (Ref 16.)
McDiarmid SV, Goodrich NP, Harper AM, et al. Liver transplantation for status 1: The consequences of good intentions. Liver Transpl. 2007;13:699-707. (Ref 136.)
O’Grady JG, Alexander GJ, Hayllar KM, et al. Early indicators of prognosis in fulminant hepatic failure. Gastroenterology. 1989;97:439-45. (Ref 83.)
Ostapowicz G, Fontana RJ, Schiodt FV, et al. Results of a prospective study of acute liver failure at 17 tertiary care centers in the United States. Ann Intern Med. 2002;137:947-54. (Ref 8.)
Stravitz RT, Kramer AH, Davern TJ, et al. Intensive care of patients with acute liver failure: Recommendations of the U.S. Acute Liver Failure Study Group. Crit Care Med. 2007;35:2498-508. (Ref 104.)
Vaquero J, Fontana RJ, Larson AM, et al. Complications and use of intracranial pressure monitoring in patients with acute liver failure and severe encephalopathy. Liver Transpl. 2005;11:1581-9. (Ref 114.)
Wade J, Rolando N, Philpott-Howard J, et al. Timing and a cause of bacterial infections in a liver intensive care unit. J Hosp Infection. 2003;53:144-6. (Ref 72.)
Watkins PB, Seeff LB. Drug-induced liver injury: Summary of a single-topic clinical research conference. Hepatology. 2006;43:618-31. (Ref 36.)
1. Hoofnagle JH, Carithers RLJr, Shapiro C, et al. Fulminant hepatic failure: Summary of a workshop. Hepatology. 1995;21:240-52.
2. Riordan SM, Williams R. Mechanisms of hepatocyte injury, multi-organ failure, and prognostic criteria in acute liver failure. Sem Liv Dis. 2003;23:203-15.
3. Trey C, Davidson C. The management of fulminant hepatic failure. Prog Liver Dis. 1970;3:292-98.
4. O’Grady JG, Schalm SW, Williams R. Acute liver failure: Redefining the syndromes. Lancet. 1993;342:273-5.
5. Dhiman RK, Makharia GK, Jain S, et al. Ascites and spontaneous bacterial peritonitis in fulminant hepatic failure. Am J Gastroenterol. 2000;95:233-8.
6. Gimson AE, O’Grady J, Ede RJ, et al. Late-onset hepatic failure: Clinical, serological and histological features. Hepatology. 1986;6:288-94.
7. O’Grady JG, Alexander GJ, Thick M, et al. Outcome of orthotopic liver transplantation in the aetiological and clinical variants of acute liver failure. Q J Med. 1988;68:817-24.
8. Ostapowicz G, Fontana RJ, Schiodt FV, et al. Results of a prospective study of acute liver failure at 17 tertiary care centers in the United States. Ann Intern Med. 2002;137:947-54.
9. Makin AJ, Wendon J, Williams R. A 7-year experience of severe acetaminophen-induced hepatotoxicity (1987-1993). Gastroenterology. 1995;109:1907-16.
10. Bernuau J, Rueff B, Benhamou JP. Fulminant and subfulminant liver failure: Definitions and causes. Semin Liv Dis. 1986;6:97-106.
11. Fujiwara K, Mochida S. Indications and criteria for liver transplantation for fulminant hepatic failure. J Gastroenterol. 2002;37:74-7.
12. Acharya SK, Panda SK, Saxena A, et al. Acute hepatic failure in India: A perspective from the east. J Gastroenterol Hepatology. 2000;15:473-9.
13. Polson J, Wians FH, Orsulak P, et al. False-positive acetaminophen concentrations in patients with liver injury. Clinica Chimica Acta. 2008;391:24-30.
14. Nourjah P, Ahmad SR, Karwoski C, et al. Estimates of acetaminophen (Paracetamol)-associated overdoses in the United States. Pharmacoepidemiol Drug Saf. 2006;15:398-405.
15. Zimmerman HJ, Maddrey WC. Acetaminophen (paracetamol) hepatotoxicity with regular intake of alcohol: Analysis of instances of therapeutic misadventure. Hepatology. 1995;22:767-73.
16. Larson AM, Polson J, Fontana RJ, et al. Acetaminophen-induced acute liver failure: Results of a United States Multicenter prospective study. Hepatology. 2005;43:1364-72.
17. Schiodt FV, Rochling FJ, Casey DL, et al. Acetaminophen toxicity in an urban county hospital. N Engl J Med. 1997;337:1112-17.
18. Osborne ZP, Bryant SM. Patients discharged with a prescription for acetaminophen-containing narcotic analgesics do not receive appropriate written instructions. Am J Emerg Med. 2003;21:48-50.
19. Schmidt LE, Dalhoff K, Poulsen HE. Acute versus chronic alcohol consumption in acetaminophen-induced hepatotoxicity. Hepatology. 2002;35:876-82.
20. Whitcomb DC, Block GD. Association of acetaminophen hepatotoxicity with fasting and ethanol use. JAMA. 1994;272:1845-50.
21. Watkins PB, Kaplowitz N, Slattery JT, et al. Aminotransferase elevations in healthy adults receiving 4 grams of acetaminophen daily. JAMA. 2006;296:87-93.
22. Bernal W. Changing patterns of causation and the use of liver transplantation in the United Kingdom. Sem Liv Disease. 2003;23:227-37.
23. Hawton K, Townsend E, Deeks J, et al. Effects of legislation restricting pack sizes of paracetamol and salicylate poisoning in the United Kingdom: Before and after study. BMJ. 2001;322:1203-7.
24. Fontana RJ, Adams PC. Unintentional acetaminophen overdose on the rise: Who is responsible? Can J Gastroenterol. 2006;20:319-24.
25. Lee WM. Acetaminophen toxicity: Changing perceptions on a social/medical issue. Hepatology. 2007;46:966-70.
26. Navarro VJ, Senior JR. Drug-related hepatotoxicity. N Engl J Med. 2006;354:731-9.
27. Fontana RJ. Acute liver failure due to drugs. Sem Liv Dis. 2008;28:175-88.
28. Russo MW, Shrestha R, Fried MW, et al. Liver transplantation for acute liver failure from drug-induced liver injury in the United States. Liver Transpl. 2004;10:1018-23.
29. Stickel F, Baumuller HM, Seitz K, et al. Hepatitis induced by kava. J Hepatology. 2003;39:62-7.
30. Favreau JT, Ryu ML, Braunstein G, et al. Severe hepatotoxicity associated with the dietary supplement Lipokinetix. Ann Intern Med. 2002;136:590-5.
31. Palmer ME, Haller C, McKinney PE, et al. Adverse events associated with dietary supplements: An observational study. Lancet. 2003;361:101-6.
32. Watt K, Molinari M, Kruszyna T, et al. Acute liver failure induced by green tea extracts: Case reports and review of the literature. Liver Transpl. 2006;12:1892-5.
33. Lyncy CR, Folkers ME, Hutson WR. Fulminant hepatic failure associated with the use of black cohosh: A case report. Liver Transpl. 2006;12:989-92.
34. Wai CT, Tan BH, Chan CL, et al. Drug-induced liver injury at an Asian center: A prospective study. Liver Int. 2007:465-74.
35. Lucena MI, Camargo R, Andrade RJ, et al. Comparison of two clinical scales for causality assessment in hepatotoxicity. Hepatology. 2001;33:123-30.
36. Watkins PB, Seeff LB. Drug-induced liver injury: Summary of a single-topic clinical research conference. Hepatology. 2006;43:618-31.
37. Sharp JR, Ishak KG, Zimmerman HJ. Chronic active hepatitis and severe hepatic necrosis associated with nitrofurantoin. Ann Intern Med. 1980;92:14-19.
38. Gough A, Chapman S, Wagstaff K, et al. Minocycline-induced autoimmune hepatitis and systemic lupus erythematous-like syndrome. BMJ. 1996;312:169-72.
39. Taylor R, Davern T, Munoz S, et al. Fulminant hepatitis A virus infection in the United States: Incidence, prognosis, and outcomes. Hepatology. 2006;44:1589-97.
40. Jaiswal SP, Chitnis DS, Artwani KK, et al. Prevalence of anti-delta antibodies in central India. Trop Gastroenterol. 1999;20:29-32.
41. Sato S, Suzuki K, Akahane Y, et al. Hepatitis B virus strains with mutations in the core promoter in patients with fulminant hepatitis. Ann Int Med. 1995;122:241-8.
42. Teo EK, Ostapowicz G, Hussain M, et al. Hepatitis B Infection in patients with acute liver failure in the United States. Hepatology. 2001;33:972-6.
43. Wai CT, Fontana RJ. Clinical significance of hepatitis B virus genotypes, variants, and mutants. Clinics Liv Dis. 2004;8:321-52.
44. Lee WM, Brown KE, Young NS, et al. Brief report: No evidence for parvovirus B19 or hepatitis E virus as a cause of acute liver failure. Dig Dis Sci. 2006;51:1712-15.
45. Kamar N, Selves J, Mansuy JM, et al. Hepatitis E virus and chronic hepatitis in organ-transplant recipients. N Engl J Med. 2008;358:811-17.
46. Haagsma EB, van den Berg AP, Porte RJ, et al. Chronic hepatitis E virus infection in liver transplant recipients. Liver Transpl. 2008;14:547-53.
47. Pishvaian AC, Bahrain M, Lewis JH. Fatal varicella-zoster hepatitis presenting with severe abdominal pain: A case report and review of the literature. Dig Dis Sci. 2006;51:1221-5.
48. Roque-Alfonso AM, Bralet MP, Ichai P, et al. Chickenpox-associated fulminant hepatitis that led to liver transplantation in a 63 year-old woman. Liver Transpl. 2008;14:1309-12.
49. Norvell JP, Blei AT, Jovanovic BD, et al. Herpes simplex virus hepatitis: An analysis of the published literature and institutional cases. Liver Transpl. 2007;13:1428-34.
50. Knox TA, Olans LB. Liver disease in pregnancy. N Engl J Med. 1996;335:569-76.
51. Ibdah JA, Bennett MJ, Rinaldo P, et al. A fetal fatty-acid oxidation disorder as a cause of liver disease in pregnant women. N Engl J Med. 1999;340:1723-31.
52. Korman JD, Volenberg I, Balko J, et al. Screening for Wilson disease in acute liver failure: A comparison of currently available diagnostic tests. Hepatology. 2008;48:1167-74.
53. Feray C, Gigou M, Samuel D, et al. Hepatitis C virus RNA and hepatitis B virus DNA in serum and liver of patients with fulminant hepatitis. Gastroenterology. 1993;104:549-55.
54. Kuwada SK, Patel VM, Hollinger FB, et al. Non-A, non-B fulminant hepatitis is also non-E and non-C. Am J Gastroenterol. 1994;89:57-61.
55. Farci P, Alter HJ, Shimoda A, et al. Hepatitis C virus–associated fulminant hepatic failure. N Engl J Med. 1996;335:631-4.
56. Fagan EA, Ellis DS, Tovey GM, et al. Toga virus-like particles in acute liver failure attributed to sporadic non-A, non-B hepatitis and recurrence after liver transplantation. J Med Virol. 1992;38:71-7.
57. Charlton M, Adjei P, Poterucha J, et al. TT-virus infection in North American blood donors, patients with fulminant hepatic failure, and cryptogenic cirrhosis. Hepatology. 1998;28:839-42.
58. Das K, Kar P, Gupta RK, et al. Role of transfusion-transmitted virus in acute viral hepatitis and fulminant hepatic failure of unknown cause. J Gastroenterol Hepatol. 2004;19:406-12.
59. Hadziyannis SJ. Fulminant hepatitis and the new G/GBV-C flavivirus. J Viral Hepat. 1998;5:15-19.
60. Karetnyi YV, Beck PR, Markin RS, et al. Human parvovirus B19 infection in acute fulminant liver failure. Arch Virol. 1999;144:1713-24.
61. Umemura T, Tanaka E, Ostapowicz G, et al. Investigation of SEN virus infection in patients with cryptogenic acute liver failure, hepatitis-associated aplastic anemia, or acute and chronic nonA-E hepatitis. J Inf Dis. 2003;188:1545-52.
62. Davern TJ, James LP, Hinson JA, et al. Measurement of serum acetaminophen-protein adducts in patients with acute liver failure. Gastroenterology. 2006;130:687-94.
63. Polson J, Ocama P, Larson AM, et al. Role of acetaminophen in acute liver failure due to viral hepatitis [abstract]. Hepatology. 2003;38(Suppl 1):544A.
64. Blei AT, Larsen FS. Pathophysiology of cerebral edema in fulminant hepatic failure. J Hepatol. 1999;31:771-6.
65. Rolando N, Harvey F, Brahm J, et al. Prospective study of bacterial infection in acute liver failure: An analysis of fifty patients. Hepatology. 1990;11:49-53.
66. Clemmesen JO, Larsen FS, Kondrup J, et al. Cerebral herniation in patients with acute liver failure is correlated with arterial ammonia concentration. Hepatology. 1999;29:648-53.
67. Bernal W, Hall C, Karvellas CJ, et al. Arterial ammonia and clinical risk factors for encephalopathy and intracranial hypertension in acute liver failure. Hepatology. 2007;46:1844-52.
68. Pereira SP, Langley PG, Williams R. The management of abnormalities of hemostasis in acute liver failure. Sem Liv Dis. 1996;16:403-14.
69. Schiodt FV, Balko J, Schilsky M, et al. Thrombopoietin in acute liver failure. Hepatology. 2003;37:558-61.
70. Cook DJ, Fuller HD, Guyatt GH, et al. Risk factors for gastrointestinal bleeding in critically ill patients. Canadian Critical Care Trials Group. N Engl J Med. 1994;330:377-81.
71. Wyke RJ, Canalese JC, Gimson AE, Williams R. Bacteraemia in patients with fulminant hepatic failure. Liver. 1982;2:45-52.
72. Wade J, Rolando N, Philpott-Howard J, et al. Timing and a cause of bacterial infections in a liver intensive care unit. J Hosp Infection. 2003;53:144-6.
73. Rolando N, Harvey F, Brahm J, et al. Fungal infection: A common, unrecognised complication of acute liver failure. J Hepatol. 1991;12:1-9.
74. Castells A, Salmeron JM, Navasa M, et al. Liver transplantation for acute liver failure: Analysis of applicability. Gastroenterology. 1993;105:532-8.
75. Rolando N, Wade J, Davalos M, et al. The systemic inflammatory response syndrome in acute liver failure. Hepatology. 2000;32:734-9.
76. Vaquero J, Polson J, Chung C, et al. Infection and the progression of hepatic encephalopathy in acute liver failure. Gastroenterology. 2003;125:755-64.
77. Lee WM, Galbraith RM. The extracellular actin-scavenger system and actin toxicity. N Engl J Med. 1992;326:1335-41.
78. Bihari DJ, Gimson AE, Williams R. Cardiovascular, pulmonary and renal complications of fulminant hepatic failure. Semin Liver Dis. 1986;6:119-28.
79. Parekh NK, Hynan LS, DeLemos J, et al. Elevated troponin I levels in acute liver failure: Is myocardial injury an integral part of acute liver failure? Hepatology. 2007;45:1489-95.
80. Parbhoo SP, Welch J, Sherlock S. Acute pancreatitis in patients with fulminant hepatic failure. Gut. 1973;14:428.
81. Trewby PN, Warren R, Contini S, et al. Incidence and pathophysiology of pulmonary edema in fulminant hepatic failure. Gastroenterology. 1978;74:859-65.
82. Baudouin SV, Howdle P, O’Grady JG, Webster NR. Acute lung injury in fulminant hepatic failure following paracetamol poisoning. Thorax. 1995;50:399-402.
83. O’Grady JG, Alexander GJ, Hayllar KM, et al. Early indicators of prognosis in fulminant hepatic failure. Gastroenterology. 1989;97:439-45.
84. Pitre J, Soubrane O, Dousset B, et al. How valid is emergency liver transplantation for acute liver necrosis in patients with multiple-organ failure? Liver Transpl Surg. 1996;2:1-7.
85. Bernal W, Donaldson N, Wyncoll D, et al. Blood lactate as an early predictor of outcome in paracetamol-induced acute liver failure: A cohort study. Lancet. 2002;359:558-63.
86. Anand AC, Nightingale P, Neuberger JM. Early indicators of prognosis in fulminant hepatic failure: An assessment of the King’s criteria. J Hepatol. 1997;26:62-8.
87. Fontana RJ. Acute liver failure including acetaminophen overdose. Med Clin N Am. 2008;92:761-94.
88. Cholongitas EB, Betrossian A, Leandro G, et al. King’s criteria, APACHE II, and SOFA scores in acute liver failure. Hepatology. 2006;43:881.
89. Hanau C, Munoz SJ, Rubin R. Histopathological heterogeneity in fulminant hepatic failure. Hepatology. 1995;21:345-51.
90. Schiodt FV, Rossaro L, Stravitz RT, et al. Gc-globulin and prognosis in acute liver failure. Liver Transpl. 2005;11:123-7.
91. Pauwels A, Mostefa-Kara N, Florent C, Lévy VG. Emergency liver transplantation for acute liver failure. Evaluation of London and Clichy criteria. J Hepatol. 1993;17:124-7.
92. Sekiyama K, Yoshiba M, Inoue K, Sugata F. Prognostic value of hepatic volumetry in fulminant hepatic failure. Dig Dis Sci. 1994;39:240-4.
93. Davern TJ, Brown RS, Shakil AO, et al. Serum phosphate as a predictor of clinical outcome in acetaminophen-induced acute liver failure [abstract]. Hepatology. 2003;38:212A.
94. Schmidt LE, Dalhoff K. Serum phosphate is an early predictor of outcome in severe acetaminophen-induced hepatotoxicity. Hepatology. 2002;36:659-63.
95. Macquillan GC, Seyam MS, Nightingale P, et al. Blood lactate but not serum phosphate levels can predict patient outcome in fulminant hepatic failure. Liver Transpl. 2005;11:1073-9.
96. Schmidt LE, Dalhoff K. Alpha-fetoprotein is a predictor of outcome in acetaminophen-induced liver injury. Hepatology. 2005;41:26-31.
97. Harrison PM, Keays R, Bray GP, et al. Improved outcome of paracetamol-induced fulminant hepatic failure by late administration of acetylcysteine. Lancet. 1990;335:1572-3.
98. Kao LW, Kirk MA, Furbee RB, et al. What is the rate of adverse events after oral N-acetylcysteine administered by the intravenous route to patients with suspected acetaminophen poisoning? Ann Emerg Med. 2003;42:741-50.
99. Busi C, Fiume L, Costantino D, et al. Amanita toxins in gastroduodenal fluid of patients poisoned by the mushroom, Amanita phalloides. N Engl J Med. 1979;300:800.
100. Bartoloni SO, Giannini A, Botti P, et al. Amanita poisoning: A clinical histopathological study of 64 cases of intoxication. Hepatogastroenterology. 1985;32:229-31.
101. Klein NA, Mabie WC, Shaver DC, et al. Herpes simplex virus hepatitis in pregnancy. Two patients successfully treated with acyclovir. Gastroenterology. 1991;100:239-44.
102. Kumar M, Satapathy S, Monga R, et al. A randomized controlled trial of lamivudine to treat acute hepatitis B. Hepatology. 2007;45:97-101.
103. Seremba E, Sanders C, Jain M, et al. Use of nucleoside analogues in HBV related acute liver failure [abstract]. Hepatology. 2007;46(Suppl 1):70.
104. Stravitz RT, Kramer AH, Davern TJ, et al. Intensive care of patients with acute liver failure: Recommendations of the U.S. Acute Liver Failure Study Group. Crit Care Med. 2007;35:2498-508.
105. Walsh TS, Hopton P, Phillips BJ, et al. The effect of N-acetylcysteine on oxygen transport and uptake in patients with fulminant hepatic failure. Hepatology. 1998;27:1332-40.
106. Lee WM, Hynan LS, Rossaro L, et al. Intravenous N-acetylcysteine improves transplant-free survival in early stage non-acetaminophen acute liver failure. Gastroenterology. 2009;137:856-64.
107. Kortsalioudaki C, Taylor RM, Cheeseman P, et al. Safety and efficacy of N-acetylcysteine in children with non–acetaminophen-induced acute liver failure. Liver Transpl. 2008;14:25-30.
108. Ananthakrishnan AN, McGinley EL, Saeian K. Effect of hospital volume and teaching status on outcomes of acute liver failure. Liver Transpl. 2008;14:1347-56.
109. Ellis AJ, Wendon JA, Williams R. Subclinical seizure activity and prophylactic phenytoin infusion in acute liver failure: A controlled clinical trial. Hepatology. 2000;32:536-41.
110. Lidofsky SD, Bass NM, Prager MC, et al. Intracranial pressure monitoring and liver transplantation for fulminant hepatic failure. Hepatology. 1992;16:1-7.
111. Muñoz SJ, Robinson M, Northrup B, et al. Elevated intracranial pressure and computed tomography of the brain in fulminant hepatocellular failure. Hepatology. 1991;13:209-12.
112. Aggarwal S, Brooks DM, Kang Y, et al. Noninvasive monitoring of cerebral perfusion pressure in patients with acute liver failure using transcranial doppler ultrasonography. Liver Transpl. 2008;14:1048-57.
113. Blei AT, Olafsson S, Webster S, Levy R. Complications of intracranial pressure monitoring in fulminant hepatic failure. Lancet. 1993;341:157-8.
114. Vaquero J, Fontana RJ, Larson AM, et al. Complications and use of intracranial pressure monitoring in patients with acute liver failure and severe encephalopathy. Liver Transpl. 2005;11:1581-9.
115. Canalese J, Gimson AE, Davis C, et al. Controlled trial of dexamethasone and mannitol for the cerebral oedema of fulminant hepatic failure. Gut. 1982;23:625-9.
116. Murphy N, Auzinger G, Bernel W, et al. The effect of hypertonic sodium chloride on intracranial pressure in patients with acute liver failure. Hepatology. 2004;39:464-70.
117. Forbes A, Alexander GJ, O’Grady JG, et al. Thiopental infusion in the treatment of intracranial hypertension complicating fulminant hepatic failure. Hepatology. 1989;10:306-10.
118. Jalan R, Olde Damink SWM, Deutz NEP, et al. Restoration of cerebral blood flow autoregulation and reactivity to carbon dioxide in acute liver failure by moderate hypothermia. Hepatology. 2001;34:50-4.
119. Jalan R, Olde Damink SWM, Deutz NEP, et al. Moderate hypothermia prevents cerebral hyperemia and increase in intracranial pressure in patients undergoing liver transplantation for acute liver failure. Transplantation. 2003;75:2034-9.
120. Martin LF, Booth FV, Karlstadt RG, et al. Continuous intravenous cimetidine decreases stress-related upper gastrointestinal hemorrhage without promoting pneumonia. Crit Care Med. 1993;21:19-30.
121. Shami VM, Caldwell SH, Hespenheide EE, et al. Recombinant activated factor VII for coagulopathy in fulminant hepatic failure compared with conventional therapy. Liver Transpl. 2003;9:138-43.
122. Rolando N, Gimson A, Wade J, et al. Prospective controlled trial of selective parenteral and enteral antimicrobial regimen in fulminant liver failure. Hepatology. 1993;17:196-201.
123. Rolando N, Wade JJ, Stangou A, et al. Prospective study comparing the efficacy of prophylactic parenteral antimicrobials, with or without enteral decontamination, in patients with acute liver failure. Liver Transpl Surg. 1996;2:8-13.
124. Wendon JA, Harrison PM, Keays R, et al. Effects of vasopressor agents and epoprostenol on systemic hemodynamics and oxygen transport in fulminant hepatic failure. Hepatology. 1992;15:1067-71.
125. Shawcross DL, Davies NA, Mookerjee RP, et al. Worsening of cerebral hyperemia by the administration of terlipressin in acute liver failure with severe encephalopathy. Hepatology. 2004;39:471-5.
126. Harrison PM, Wendon JA, Gimson AE, et al. Improvement by acetylcysteine of hemodynamics and oxygen transport in fulminant hepatic failure. N Engl J Med. 1991;324:1852-7.
127. Davenport A, Will EJ, Davidson AM. Improved cardiovascular stability during continuous modes of renal replacement therapy in critically ill patients with acute hepatic and renal failure. Crit Care Med. 1993;21:328-38.
128. Muñoz SJ, Moritz MJ, Martin P, et al. Liver transplantation for fulminant hepatic failure. Transplant Proc. 1993;25:1773-5.
129. Ascher NL, Lake JR, Emond J, et al. Liver transplantation for fulminant hepatic failure. Arch Surg. 1993;128:677-82.
130. Schiodt FV, Atillasoy E, Shakil AO, et al. Cause and outcome for 295 patients with acute liver failure in the United States. Liver Transpl Surg. 1999;5:29-34.
131. Campbell DA, Ham JM, McCurry KR. Liver transplant for fulminant hepatic failure. American Surg. 1991;57:546-9.
132. Emond JC, Aran PP, Whitington PF, et al. Liver transplantation in the management of fulminant hepatic failure. Gastroenterology. 1989;96:583-9.
133. Iwatsuki S, Steiber AC, Marsh HW, et al. Liver transplantation for fulminant hepatic failure. Transplant Proc. 1989;21:2431-4.
134. Schafer DF, Shaw BWJr. Fulminant hepatic failure and orthotopic liver transplantation. Semin Liver Dis. 1989;9:189-94.
135. Higgins PDR, Fontana RJ. Liver transplantation in acute liver failure. Panminerva Medica. 2002;52:93-7.
136. McDiarmid SV, Goodrich NP, Harper AM, et al. Liver transplantation for status 1: The consequences of good intentions. Liver Transpl. 2007;13:699-707.
137. Detre K, Belle S, Beringer K, et al. Liver transplantation for fulminant hepatic failure in the United States: October 1987 through December 1991. Clin Transpl. 1994;8:274-80.
138. Devlin J, Williams R. Transplantation for fulminant hepatic failure. Transplantation. 1996;62:151-5.
139. Mohamed R, Hubscher SG, Mirza DF, et al. Postransplantation chronic hepatitis in fulminant hepatic failure. Hepatology. 1997;25:1003-7.
140. Jackson EW, Zacks S, Zinn S, et al. Delayed neuropsychologic dysfunction after liver transplantation for acute liver failure: A matched, case-controlled study. Liver Transpl. 2002;8:932-6.
141. Uemoto S, Inomata Y, Sakurai T, et al. Living donor liver transplantation for fulminant hepatic failure. Transplantation. 2000;70:152-7.
142. Miwa S, Hashikura Y, Mita A, et al. Living-related liver transplantation for patients with fulminant and subfulminant hepatic failure. Hepatology. 1999;30:1521-6.
143. Campsen J, Blei AT, Emond JC, et al. Outcomes of living donor liver transplantation for acute liver failure: The adult-to-adult living donor Transplantation Cohort Study. Liver Transpl. 2008;14:1273-80.
144. O’Grady JG, Gimson AE, O’Brien CJ, et al. Controlled trials of charcoal hemoperfusion and prognostic factors in fulminant hepatic failure. Gastroenterology. 1988;94:1186-92.
145. Sterling RK, Luketic VA, Sanyal AJ, et al. Treatment of fulminant hepatic failure with intravenous prostaglandin E1. Liver Transpl Surg. 1998;4:424-31.
146. Larsen FS, Hansen BA, Jorgensen LG, et al. High-volume plasmapheresis and acute liver transplantation in fulminant hepatic failure. Transplant Proc. 1994;26:1788.
147. Ejlersen E, Larsen FS, Pott F, et al. Hepatectomy corrects cerebral hyperperfusion in fulminant hepatic failure. Transplant Proc. 1994;26:1794-5.
148. Sudan DL, Shaw BWJr, Fox IJ, Langnas AN. Long-term follow-up of auxiliary orthotopic liver transplantation for the treatment of fulminant hepatic failure. Surgery. 1997;122:771-7.
149. van Hoek B, de Boer J, Boudjema K, et al. Auxiliary versus orthotopic liver transplantation for acute liver failure. EURALT Study Group. European Auxiliary Liver Transplant Registry. J Hepatol. 1999;30:699-705.
150. Allen JW, Hassanein T, Bhatia SN. Advances in bioartificial liver devices. Hepatology. 2001;34:447-55.
151. Heemann U, Treichel U, Loock J, et al. Albumin dialysis in cirrhosis with superimposed acute liver injury: A prospective, controlled study. Hepatology. 2003;36:949-58.
152. Hassanein TI, Tofteng F, Brown RS, et al. Randomized controlled study of extracorporeal albumin dialysis for hepatic encephalopathy in advanced cirrhosis. Hepatology. 2007;46:1853-62.
153. Schmidt LE, Wang LP, Hansen BA, et al. Systemic hemodynamic effects of treatment with the molecular adsorbents recirculating system in patients with hyperacute liver failure: A prospective, controlled trial. Liver Transpl. 2003;9:290-7.
154. Rifai K, Ernst T, Kretschmer U, et al. The Prometheus device for extracorporeal support of combined liver and renal failure. Blood Purif. 2005;23:298-302.
155. Demetriou AA, Brown RS, Busuttil RW, et al. Prospective, randomized, multicenter controlled trial of a bioartificial liver in treating acute liver failure. Ann Surg. 2004;239:660-70.
156. Pitkin Z, Switzer W, Chapman L. An interim analysis of PERV infection in 74 patients treated with a bioartificial liver in a prospective, randomized, multicenter controlled trial [abstract]. Hepatology. 2001;34:249A.
157. Strom SC, Fisher RA, Thompson MT, et al. Hepatocyte transplantation as a bridge to orthotopic liver transplantation in terminal liver failure. Transplantation. 1997;63:559-69.
158. Fox I, Chowdhury J, Kaufman S, et al. Treatment of the Crigler-Najjar syndrome type I with hepatocyte transplantation. NEJM. 1998;338:1422-6.
159. Bilir B, Guinette D, Karrer F, et al. Hepatocyte transplantation in acute liver failure. Liver Transpl. 2000;6:32-40.
160. Petersen BE, Bowen WC, Patrene KD, et al. Bone marrow as a potential source of hepatic oval cells. Science. 1999;284:1168-70.
161. Houlihan DD, Newsome PN. Critical review of clinical trials of bone marrow stem cells in liver disease. Gastroenterology. 2008;135:438-50.