17 Acute kidney injury
Definition and incidence
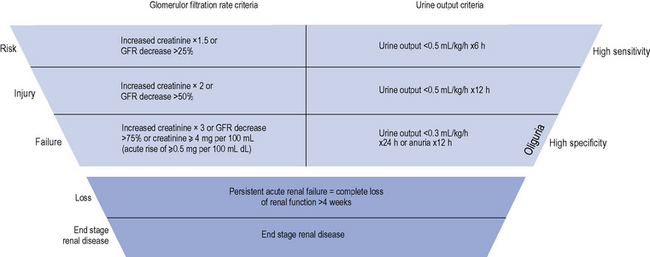
The diagnostic criteria for AKI is based on an increase in serum creatinine or the presence of oliguria (see Table 17.1). Criteria have recently been introduced for the definition and staging of the condition; the acronym RIFLE is used (Risk, Injury, Failure, Loss and End-stage renal disease (ESRD)), which is now becoming established in clinical practice (see Fig. 17.1).
| Acute kidney injury type | Typical % cases | Common aetiology |
|---|---|---|
| Pre-renal | 40–80 | Reversible ↓ renal perfusion through hypoperfusion |
| Intra-renal (including ATN) | 10–50 | Renal parenchymal injury |
| Post-renal | <10 | Urinary tract obstruction |
ATN, acute tubular necrosis.
Classification and causes
AKI is not a single disease state with a uniform aetiology, but a consequence of a range of different diseases and conditions. The most useful practical classification comprises three main groupings: (i) pre-renal, (ii) renal, or (iii) post-renal. More than one category may be present in an individual patient. Common causes of each type of AKI are outlined in Table 17.1.
Pre-renal acute kidney injury
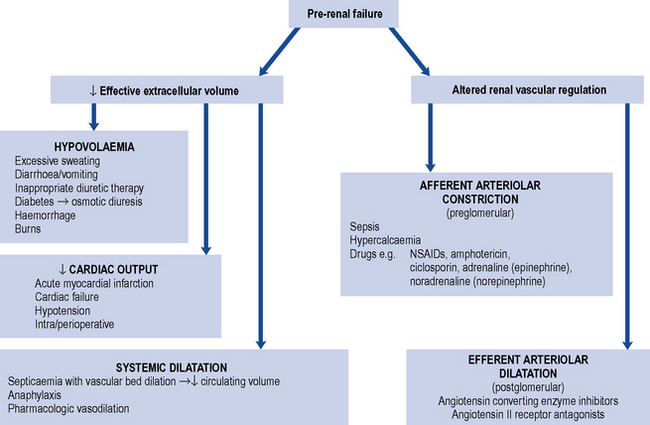
This is caused by impaired perfusion of the kidneys with blood, and is usually a consequence of decreased intravascular volumes (hypovolaemia) and/or decreased intravascular pressures. Some of the commonest causes of pre-renal AKI are summarised in Fig. 17.2. Perfusion of the kidneys at the level of the microvascular beds (glomerular and tubulo-interstitial) is usually maintained through wide variations in pressure and flow through highly efficient auto-regulatory pathways, such as the renin–angiotensin–aldosterone system (RAAS) and regulated prostaglandin synthesis. However, when the systolic blood pressure (BP) drops below 80 mmHg, AKI may develop. In individuals with chronic kidney disease (CKD) or in the elderly, this may occur at higher levels of systolic BP. Drugs that inhibit the RAAS, such as angiotension converting enzyme inhibitors (ACE inhibitors) and angiotensin receptor blockers (ARBs), or block the production of prostaglandins, such as non-steroidal anti-inflammatory drugs (NSAIDs), can pre-dispose to the development of pre-renal AKI. These are discussed in more detail below.
Intra-renal acute kidney injury
This is caused by a variety of causes (see Tables 17.1 and 17.2), most commonly (in >80% of cases) acute tubular necrosis (ATN). ATN occurs usually as a consequence of a combination of factors, including hypotension, often in the setting of sepsis and nephrotoxic agents including drugs or chemical poisons, or endogenous sources such as myoglobin or haemoglobin.
Table 17.2 Common clinical factors known to cause acute tubular necrosis
| Clinical factor | Mechanism |
|---|---|
| Hypoperfusion | Reduced oxygen/nutrient supply |
| Radiocontrast media | Medullary ischaemia may result from contrast media induced renal vasoconstriction. The high ionic load of contrast media may produce ischaemia particularly in diabetics and those with myeloma (who produce large quantities of light chain immunoglogulins) |
| Sepsis | Infection produces endotoxaemia and systemic inflammation in combination with a pre-renal state and nephrotoxins. The immunological response to sepsis involves release of vasoconstrictors and vasodilators (e.g. eicosanoids, nitric oxide) and damage to vascular endothelium with resultant thrombosis |
| Rhabdomyolysis | Damaged muscles release myoglobin, which can cause ATN through direct nephrotoxicity and by a reduction in blood flow in the outer medulla |
| Renal transplantation | The procedures and conditions encountered during renal transplantation can induce ischaemic ATN which can be difficult to distinguish from the nephrotoxic effects of immunosuppressive drug therapy used in these circumstances and rejection |
| Hepatorenal syndrome | Renal vasoconstriction is frequently seen in patients with end-stage liver disease. Progression to ATN is common |
| Nephrotoxins | |
| Aminoglycosides | Aminoglycosides are transported into tubular cells where they exert a direct nephrotoxic effect. Current dosage regimens recommend once daily doses, with frequent monitoring of drug levels, to minimise total uptake of aminoglycoside |
| Amphotericin | Amphotericin appears to cause direct nephrotoxicity by disturbing the permeability of tubular cells. The nephrotoxic effect is dose dependent and minimised by limiting total dose used, rate of infusion and by volume loading. These precautions also apply to newer liposomal formulations |
| Immunosuppressants | Ciclosporin and tacrolimus cause intra-renal vasoconstriction that may result in ischaemic ATN. The mechanism is unclear but enhanced by hypovolaemia and other nephrotoxic drugs |
| NSAIDs | Vasodilator prostaglandins, mainly E2, D2 and I2 (prostacyclin), produce an increase in blood flow to the glomerulus and medulla. In normal circumstances, they play no part in the maintenance of the renal circulation. However, increased amounts of vasoconstrictor substances arise in a variety of clinical conditions such as volume depletion, congestive cardiac failure or hepatic cirrhosis associated with ascites. Maintenance of renal blood flow then becomes more reliant on the release of vasodilatory prostaglandins. Inhibition of prostaglandin synthesis by NSAIDs may cause unopposed arteriolar vasoconstriction, leading to renal hypoperfusion |
| Cytotoxic chemotherapy | For example, cisplatin |
| Anaesthetic agents | Methoxyflurane, enflurane |
| Chemical poisons/naturally occurring poisons | Insecticides, herbicides, alkaloids from plants and fungi, reptile venoms |
Acute tubular necrosis
ATN is a diagnosis made by renal biopsy; the findings can include damage to the proximal tubule and the ascending limb of the loop of Henle, interstitial oedema and sparse infiltrating inflammatory cells. Whilst severe and sustained hypoperfusion can lead to ATN, it usually develops when there is a combination of factors including the presence of one or more of a range of nephrotoxins. These may arise exogenously from drugs or chemical poisons, or from endogenous sources such as haemoglobin, myoglobin, crystals (uric acid, phosphate) and toxic products from sepsis or tumours (see Table 17.2). Some endogenous toxins may be released as a direct consequence of drug exposure. For example, myoglobin may be released (rhabdomyolosis) following muscle injury or necrosis, hypoxia, infection or following drug treatment, for example, with fibrates and statins, particularly when both are used in combination. The mechanism of the subsequent damage to renal tissue is not understood fully but probably results from a combination of factors including hypoperfusion, haem-catalysed free radical tubular cytotoxicity and haem cast formation and precipitation leading to tubular injury.
Differentiating pre-renal from renal acute kidney injury
It is sometimes possible to distinguish between cases of pre-renal and renal AKI through examination of biochemical markers (see Table 17.3). In renal AKI, the kidneys are generally unable to retain Na+ owing to tubular damage. This can be demonstrated by calculating the fractional excretion of sodium (FENa); in practice this is not often done because it lacks sensitivity and specificity and may be difficult to interpret in the elderly who may have pre-existing concentrating defects.
Table 17.3 Differentiating pre-renal from renal acute kidney injury
| Laboratory test | Pre-renal | Renal |
|---|---|---|
| Urine osmolality (mOsm/kg) | >500 | <400 |
| Urine sodium (mEq/L) | <20 | >40 |
| Urine/serum creatinine (μmol/L) | >40 | <20 |
| Urine/serum urea (μmol/L) | >8 | <3 |
| Fractional excretion of sodium (%) | <1 | >2 |
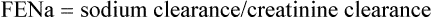
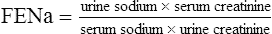
Clinical manifestations
Acute kidney injury with volume depletion
In those patients with volume depletion, a classic pathophysiological picture is likely to be present, with tachycardia, postural hypotension, reduced skin turgor and cold extremities (see Table 17.4). The most common sign in AKI is oliguria, where urine production falls to less than 0.5 mL/kg/h for several hours. This is below the volume of urine required to effectively excrete products of metabolism to maintain a physiological steady state. Therefore, the serum concentration of those substances normally excreted by the kidney will rise and differentially applies to all molecules up to a molecular weight of around 50 kDa. This includes serum creatinine, which at a molecular weight of 113 Da is normally freely filtered by the kidneys but with loss of kidney function the serum level climbs. Whilst the term uraemia is still in widespread use, it merely describes a surrogate for the overall metabolic disturbances that accompany AKI; these include excess potassium, hydrogen ions (acidosis) and phosphate in blood. Most cases of AKI are first identified by an abnormal blood test, though some patients may have symptoms that are specifically attributable to AKI; these include nausea, vomiting, diarrhoea, gastro-intestinal haemorrhage, muscle cramps and a declining level of consciousness.
| Volume depletion | Volume overload | |
|---|---|---|
| History | Thirst | Weight increase |
| Excessive fluid loss (vomiting or diarrhoea) | Orthopnoea/nocturnal dyspnoea | |
| Oliguria | ||
| Physical examination | Dry mucosae ↓ Skin elasticity |
Ankle swelling Oedema |
| Tachycardia | Jugular venous distension | |
| ↓ Blood pressure | Pulmonary crackles | |
| ↓ Jugular venous pressure | Pleural effusion |
Diagnosis and clinical evaluation
The assessment of renal function is described in detail in Chapter 18. However, unless a patient is at steady state, measurement of serum creatinine does not provide a reliable guide to renal function. For example, serum creatinine levels will usually rise by only 50–100 μmol/L per day following complete loss of renal function in a previously normal patient. These changes in serum creatinine are not sufficiently responsive to serve as a practical indicator of glomerular filtration rate, particularly in AKI in critical care scenarios.
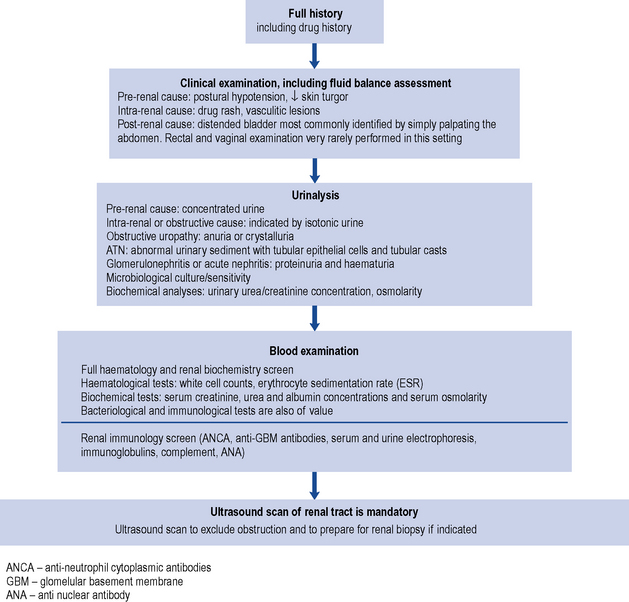
In the hospital situation, when AKI is detected incidentally, the cause(s) of the condition, such as fluid depletion (hypovolaemia), infection or the use of nephrotoxic drugs, are often apparent on close examination of the clinical history. The development of AKI in this setting is more likely to occur in people with pre-existing CKD. People with normal baseline kidney function usually need to sustain at least two separate triggers for the development of AKI; for example, hypovolaemia will rarely cause AKI in this setting, but when hypovolaemia occurs in the presence of nephrotoxic drugs then AKI may occur. In patients with pre-existing CKD, AKI (i.e. acute on chronic renal failure) can occur in patients with one trigger. By definition, the worse the baseline kidney function, the smaller the trigger required for the development of AKI. Irrespective of the presentation of AKI, it is wise to consider the complete differential diagnosis in all people; active exclusion of post-renal AKI and immune and inflammatory AKI should be considered in all cases. In AKI without an obvious precipitating pre-or post-renal cause, there is a greater need to consider these causes. Although the majority of patients have ATN, other causes such as rapidly progressive glomerulonephritis, interstitial nephritis, multiple myeloma or urinary tract obstruction must be screened for and systematically excluded. In addition to supportive care that is generic for all causes of AKI, disease-specific treatment may also be required. The investigation of AKI is outlined in Fig. 17.3.
Monitoring fluid balance in acute kidney disease
Course and prognosis
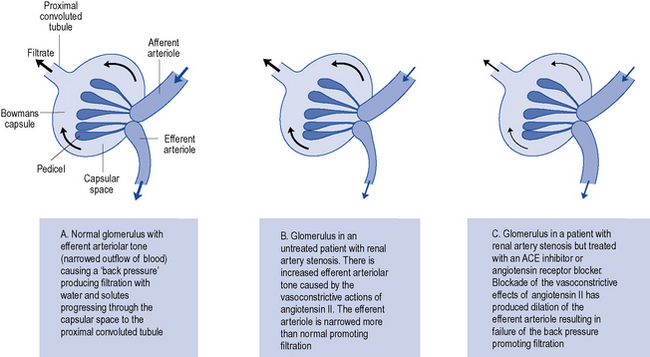
ACE inhibitors and angiotensin receptor blockers in acute kidney injury
ACE inhibitor use is, however, absolutely contraindicated when a patient has bilateral renal artery stenosis, or renal artery stenosis in a patient with a single functioning kidney. If an ACE inhibitor or ARB is initiated under these circumstances then pre-renal AKI may ensue. This may occur since the renin–angiotensin system is stimulated by low renal perfusion resulting from stenotic lesions in the arteries supplying the kidneys, most often at the origin of the renal artery from the abdominal aorta. Angiotensin II is produced which causes renal vasoconstriction, in part, through increased efferent arteriolar tone. This creates a ‘back pressure’ which paradoxically maintains glomerular filtration pressure in an otherwise poorly perfused kidney. If angiotensin II production is inhibited by an ACE inhibitor, or the effect is blocked by an ARB, then efferent arteriole dilatation will result. Since increased efferent vascular tone maintains filtration in such patients, then the overall result of ACE inhibitor or ARB therapy will be to reduce or shut down filtration at the glomerulus and put the patient at risk of pre-renal AKI (see Fig. 17.4).
Management
Early preventive and supportive strategies
Establishing and maintaining an adequate diuresis
Dopamine
Historically, dopamine has been recommended at low dose to improve renal blood flow and urine output. Dopamine at low dose acts as a renal vasodilator in normal kidneys, but in renal failure it is a renal vasoconstrictor even at a low dose. This translates into no demonstrable clinical benefit and it should no longer be used. Dopamine has alpha and beta adrenergic effects. Recently, fenoldapam, a pure dopaminergic D1 agonist has been investigated in small scale clinical trials; the results have shown a trend towards benefit in recovery of renal function from AKI. However, larger trials are needed to identify if fenoldapam has a role in routine clinical practice (Kellum et al., 2008).
Non-dialysis treatment of established acute kidney injury
Hyperkalaemia
Dietary potassium should be restricted to less than 40 mmol/day and potassium supplements and potassium-sparing diuretics removed from the treatment schedule. Emergency treatment is necessary if the serum potassium level reaches 7.0 mmol/L (normal range 3.5–5.5 mmol/L) or if there are the progressive changes in the electrocardiogram (ECG) associated with hyperkalaemia. These include tall, peaked T waves, reduced P waves with increased QRS complexes or the ‘sine wave’ appearance that often presages cardiac arrest (see Chapter 18, Fig. 18.10).
Emergency treatment of hyperkalaemia consists of the following:
Hyperphosphataemia
As phosphate is normally excreted by the kidney, hyperphosphataemia can occur in AKI but rarely requires treatment. Should it become necessary to treat, phosphate-binding agents may be used to retain phosphate ions in the gut. The most commonly used agents are calcium containing such as calcium carbonate or calcium acetate and are given with food. For further information see Chapter18.
Renal replacement therapy
Forms of renal replacement therapy
The common types of renal replacement therapy used in clinical practice are:
Haemodialysis
In haemodialysis, the form of access used in AKI is a dialysis line. This is placed in a vein (the jugular, femoral or sub-clavian), which has an arterial lumen through which the blood is removed from the patient and a venous lumen by which it is returned to the patient after passing through a dialyser. The terms arterial and venous lumen can be misleading as both lumens are situated in the same vein. They are part of the same line which bifurcates and has two lumens, the longer lumen is the ‘arterial’ lumen and the shorter the ‘venous’ lumen. Heparin is added to the blood as it leaves the body to prevent the dialyser clotting. Blood is then actively pumped through the artificial kidney before being returned to the patient (Fig. 17.5). In those patients at high risk of haemorrhage, the amount of heparin used can be reduced or even avoided altogether. The dialyser consists of a cartridge comprising either a bundle of hollow tubes (hollow fibre dialyser) or a series of parallel flat plates (flat-plate dialyser) made of a synthetic semi-permeable membrane. Flat-plate dialysers are now rarely used. Dialysis fluid flows around the membrane countercurrent (opposite) to the flow of blood in order to maximise diffusion gradients. The dialysis solution is essentially a mixture of electrolytes in water with a composition approximating to extracellular fluid into which solutes diffuse. The ionic concentration of the dialysis fluid can be manipulated to control the rate and extent of electrolyte transfer. Calcium and bicarbonate concentrations can also be increased in dialysis fluid to promote diffusion into blood as replacement therapy. By manipulating the hydrostatic pressure of the dialysate and blood circuits, the extent and rate of water removal by ultrafiltration can be controlled.
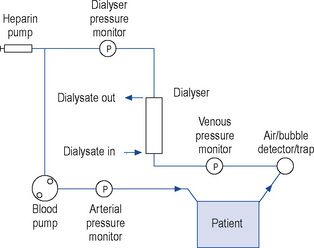
Haemodialysis can be performed in either intermittent or continuous schedules. The latter regimen is preferable in the critical care situation, providing 24-h control, and minimising swings in blood volume and electrolyte composition that are found using intermittent regimens. The haemodialysis described in this section is indistinguishable from that used as maintenance therapy for many patients with end stage renal failure, the method of access in this group is often via an arterio-venous fistula (see Chapter 18).
Haemodiafiltration
Haemodiafiltration is a technique that combines the ability to clear small molecules, as in haemodialysis, with the large molecule clearance of haemofiltration. It is, however, more expensive than traditional haemodialysis, but does offer potential benefits. Whilst some studies suggest that haemodialfiltration may provide a clinical benefit compared to haemofiltration or haemodialysis, this is controversial (Rabindranath et al., 2006). However, enhanced combined control of fluid and solute removal provided by this technique is likely to be increasingly used over the next decade.
Acute peritoneal dialysis
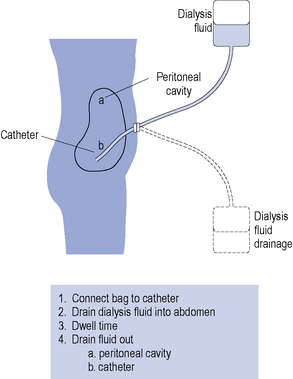
Acute peritoneal dialysis is rarely used now for AKI except in circumstances where haemodialysis is unavailable. A semi-rigid catheter is inserted into the abdominal cavity. Warmed sterile peritoneal dialysis fluid (typically 1–2 L) is instilled into the abdomen, left for a period of about 30 min (dwell time) and then drained into a collecting bag (Fig. 17.6). This procedure may be performed manually or by semiautomatic equipment. The process may be repeated up to 20 times a day, depending on the condition of the patient.
Drug dosage in renal replacement therapy
Whether a drug is significantly removed by dialysis or haemofiltration is an important clinical issue. Drugs that are not removed may well require dose reduction to avoid accumulation and minimise toxic effects. Alternatively, drug removal may be significant and require a dosage supplement to ensure an adequate therapeutic effect is maintained. In general, since haemodialysis, peritoneal dialysis and haemofiltration depend on filtration, the process of drug removal can be considered analoguous to glomerular filtration. Table 17.5 gives an indication of approximate clearances of common renal replacement therapies, which for continuous regimens provide an estimate for the creatinine clearance of the system.
Table 17.5 Approximate clearances of common renal replacement therapies
| Renal replacement therapy | Clearance rate (mL/min) |
|---|---|
| Intermittent haemodialysis | 150–200 |
| Intermittent haemofiltration | 100–150 |
| Acute intermittent peritoneal dialysis | 10–20 |
| Continuous haemofiltration | 5–15 |
For peritoneal dialysis other factors come into play and include:
Factors affecting drug use
Excretion

The volume of distribution may be altered but generally remains unchanged.
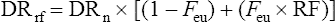
where DRrf is the dosing rate in renal failure, DRn is the normal dosing rate, RF is the extent of renal impairment = patient’s creatinine clearance (mL/min)/ideal creatinine clearance (120 mL/min) and Feu is the fraction of drug normally excreted unchanged in the urine. For example, when RF = 0.2 and Feu = 0.5, 60% of the normal dosing rate should be given.
Nephrotoxicity
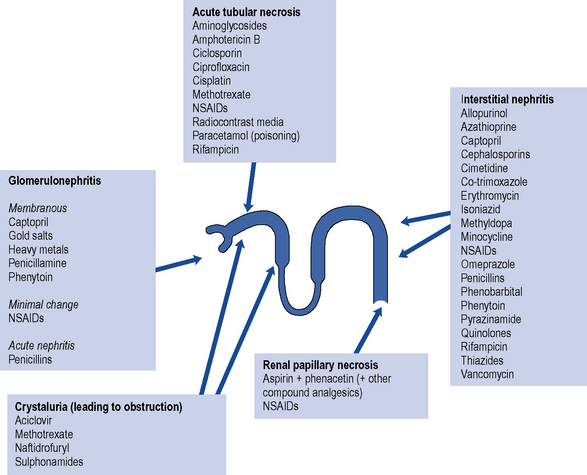
The list of potentially nephrotoxic drugs is long. Although the commonest serious forms of renal damage are interstitial nephritis and glomerulonephritis, the majority of drugs only cause damage by hypersensitivity reactions and are safe in many patients. Some drugs, however, are directly nephrotoxic, and their effects on the kidney are more predictable. Such drugs include aminoglycosides, amphotericin, colistin, the polymixins and ciclosporin. The use of any drug with recognised nephrotoxic potential should be avoided where possible. This is particularly true in patients with pre-existing renal impairement or renal failure. Figure 17.7 summarises the most important and common adverse effects of drugs on renal function, indicating the likely regions of the nephron in which damage occurs. Additional information on adverse effects can be found in Hems and Currie (2005).
Inevitably, occasions will arise when the use of potentially nephrotoxic drugs becomes necessary, and on these occasions constant monitoring of renal function is essential. In conclusion, when selecting a drug for a patient with renal failure, an agent should be chosen that approaches the ideal characteristics listed in Box 17.1.
Answers
Case 17.2
On examination he was found to be dehydrated and serum biochemistry revealed the following:
| Reference range | ||
| Sodium | 147 mmol/L | (135–145) |
| Potassium | 6.1 mmol/L | (3.5–5.0) |
| Calcium | 1.72 mmol/L | (2.20–2.55) |
| Phosphate | 2.0 mmol/L | (0.9–1.5) |
| Creatinine | 485 μmol/L | (50–120) |
| Creatinine kinase | 120,000 IU/L | (<200) |
Case 17.3
| Reference range | ||
| Sodium | 138 mmol/L | (135–145) |
| Potassium | 7.2 mmol/L | (3.5–5.0) |
| Bicarbonate | 19 mmol/L | (22–31) |
| Urea | 32.1 mmol/L | (3.0–6.5) |
| Creatinine | 572 μmol/L | (50–120) |
| pH | 7.28 | (7.36–7.44) |
Answer
The emergency treatment should include:
Hems S., Currie A. Renal disorders. In Lee A., editor: Adverse Drug Reactions, second ed., London: Pharmaceutical Press, 2005.
Kellum J., Leblanc M., Venkataraman R. Acute Renal Failure Clinical. Evidence. 2008;09:2001. BMJ Publishing Group, London. Available at http://clinicalevidence.bmj.com/ceweb/conditions/knd/2001/2001_contribdetails.jsp Accessed Sep. 2010.
Rabindranath K.S., Strippoli G.F.M., Daly C., et al. Haemodiafiltration, haemofiltration and haemodialysis for end-stage kidney disease. Cochrane Database of Systematic Reviews. 2006. Issue 4. Art No. CD006258. doi:10.1002/14651858.CD006258. Available at http://www2.cochrane.org/reviews/en/ab006258.html Accessed Sep. 2010
Dishart M.K., Kellum J.A. An evaluation of pharmacological strategies for the prevention and treatment of acute renal failure. Drugs. 2000;59:79-91.
Short A., Cumming A. ABC of intensive care: renal support. Br. Med. J.. 1999;319:41-44.
Steddon S., Ashman N., Chesser A., Cunningham J. Oxford Handbook of Nephrology and Hypertension. Oxford: Oxford University Press; 2007.