Acute Diabetic Emergencies, Glycemic Control, and Hypoglycemia
DIABETES MELLITUS: EPIDEMIOLOGY AND CLASSIFICATION
DIABETIC KETOACIDOSIS AND HYPERGLYCEMIC HYPEROSMOLAR STATE
Diabetes Mellitus: Epidemiology and Classification
Diabetes mellitus (DM) is the fourth to fifth most common cause of death in developed countries and is one of the most common noncommunicable diseases globally.1 DM affects nearly 27 million individuals in the United States and 366 million adults aged 20 to 79 years worldwide.2 Nearly half of those with diabetes are undiagnosed.2 The worldwide incidence of DM is expected to reach 552 million by 2030 with the prevalence of diabetes increasing in every country.2 Diabetes is the leading cause of kidney failure, nontraumatic lower limb amputations, and new cases of blindness among adults in the United States, and is a major cause of heart disease and stroke.3
Diabetes is generally classified as follows:
1. Type 1 diabetes—autoimmune β-cell destruction resulting in an absolute insulin deficiency
2. Type 2 diabetes—progressive defects resulting in increased insulin resistance and a relative insulin deficiency
3. Gestational diabetes—diabetes appearing during pregnancy
4. Diabetes due to other causes—such as genetic defects in β-cell function, genetic defects in insulin action, diseases of the exocrine pancreas (such as cystic fibrosis, pancreatitis, or pancreatectomy), and drug- or chemical-induced diabetes4
Type 2 diabetes accounts for 90% to 95% of DM in the United States.3 The epidemic of type 2 DM has been attributed to the increasing rates of obesity.
In patients with diabetes, absolute or relative insulin deficiency leads to hyperglycemia from decreased glucose utilization in peripheral tissues (especially skeletal muscle) and increased hepatic glucose output from glycogenolysis and gluconeogenesis.5 Proteolysis and lipolysis are also increased, providing the amino acids and free fatty acids that are the substrate for gluconeogenesis and alternative fuel sources such as ketones. The various actions of insulin are shown in Box 58.1.6
Diabetic Ketoacidosis and the Hyperglycemic Hyperosmolar State
Diabetic ketoacidosis (DKA) and hyperglycemic hyperosmolar state (HHS) are both part of an overlapping process of decompensated hyperglycemia.7 A significant imbalance between insulin and its counterregulatory hormones (especially glucagon) leads to DKA, a severe metabolic state of hyperglycemia and acidemia from abnormal metabolism of fats, lipids, and carbohydrates.8 DKA is common complication of type 1 DM and is also seen, albeit less frequently, in type 2 DM. The combined findings of serum glucose greater than 250 mg/dL, a pH less than 7.30 with an elevated anion gap, and elevated serum ketones are consistent with DKA.
HHS is a state of hyperglycemia associated with osmotic diuresis and dehydration. HHS is defined by glucose levels greater than 600 mg/dL, an elevated serum osmolality, a lack of significant acidemia or ketosis, and typically a change in mental status. HHS has replaced the term “hyperglycemic, hyperosmolar nonketotic coma” because patients may present with a mild elevation in urine or serum ketones and may have a variety of mental status changes other than coma. A careful evaluation of history, physical examination, and laboratory values helps differentiate DKA from HHS (Table 58.1) and assists in distinguishing DKA and HHS from other conditions with overlapping clinical presentation.9
Table 58.1
Diagnostic Criteria for Diabetic Ketoacidosis (DKA) and Hyperglycemic Hyperosmolar Syndrome (HHS)
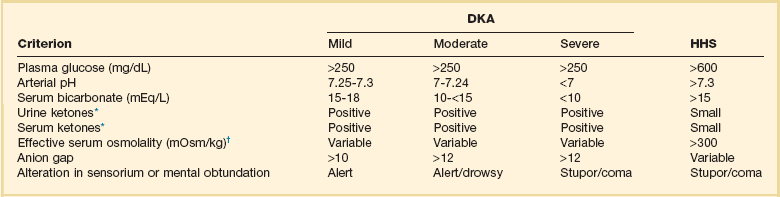
*Nitroprusside reaction method.
†Calculation: 2 [measured Na+] + [glucose]/18.
Copyright ©2004 American Diabetes Association. Modified from Kitabchi AE, Umpierrez GE, Murphy MB, et al: Hyperglycemic crises in diabetes. Diabetes Care 2004:27:S94-S102, used with permission from the American Diabetes Association.
Epidemiology
DKA has high morbidity rates, mortality rates, and financial costs despite numerous advances in the treatment of DM. The incidence of DKA in the United States is estimated at 5.9 to 12.9 cases per 100,000 people.10,11 The 120,000 patients admitted to the hospital for DKA each year12 account for approximately $1.4 billion in hospital reimbursement.13 DKA carries a mortality rate of 1% to 5%.9,14 Though the highest mortality rate is in the elderly and in patients with comorbid conditions, DKA remains the leading cause of death in diabetic patients below the age of 24.15
Although most patients will have a history of diabetes, 27% to 37% of patients with DKA carry no prior diagnosis.16,17 This is especially true in young children.18 Although the development of DKA is classically associated with type 1 DM, some patients lack the classic clinical presentation, autoimmune markers, or diminished β-cell reserve of type 1 diabetes.19–21 This subgroup, known as “ketosis-prone type 2 diabetes,” represents patients with type 2 DM who present with DKA and often are African American or Latino, male, middle-aged, and overweight or obese; have a family history of diabetes; and have newly diagnosed diabetes.22,23 Patients with ketosis-prone type 2 DM account for 1 in 5 cases of DKA.22 Patients with “classic” type 2 diabetes may also develop DKA in situations of extreme stress.
HHS occurs far less commonly than DKA, with an estimated prevalence of 1 in 1000,24 though the increasing incidence of type 2 DM is expected to increase the prevalence of HHS. HHS typically occurs in patients with type 2 DM and 30% to 40% cases of HHS may be the initial finding leading to diagnosis of type 2 DM.25,26 The mortality rate in HHS tends to be higher than in DKA, with the patient’s comorbid conditions and severity of presentation significantly affecting risk of death.27
Pathophysiology
The pathophysiology of DKA is driven by insulin deficiency and a rise in the counterregulatory hormones, particularly glucagon, but also epinephrine, cortisol, and growth hormone (Fig. 58.1).28,29 Insulin deficiency results in decreased glucose uptake by peripheral tissues, resulting in elevated serum glucose levels, and hyperglycemia worsens when an increase in glucagon (and an imbalanced insulin/glucagon ratio) results in unrestrained hepatic glycogenolysis and gluconeogenesis.9,24,28 Osmotic diuresis results in total body loss of water and electrolytes, causing intravascular volume depletion. Cellular dehydration occurs as water and electrolytes move from the intracellular to extracellular space. With volume depletion, impaired renal function limits the clearance of glucose and worsens the hyperglycemia.9,24 In an insulin-deficient state proteolysis and lipolysis drive accelerated gluconeogenesis as amino acids and free fatty acids are shunted to the liver.30 At the same time, oxidation of free fatty acids results in the production of the keto acids acetoacetate and β-hydroxybutyrate.9,24 These weak acids react with bicarbonate and diminish the body’s bicarbonate stores. The development of metabolic acidosis in DKA results when the increase in acid production exceeds the body’s buffering capacity.31
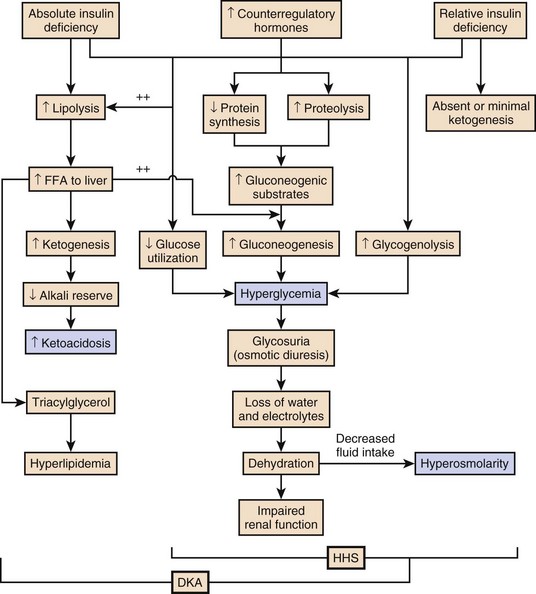
HHS is also caused by a relative insulin deficiency and a rise in counterregulatory hormones. Decreased peripheral glucose uptake and increased glycogenolysis and gluconeogenesis result in hyperglycemia followed by osmotic diuresis with volume depletion and dehydration. However, the amount of insulin is sufficient to limit the oxidation of free fatty acids,9 and significant keto acid production does not occur. In the absence of ketoacidosis, the process of dehydration is allowed to continue for longer than in DKA, and patients present with significant hyperosmolarity.
Precipitating Factors
The precipitating factors for the development of DKA and HHS are listed in the Table 58.2. Omission of antidiabetic medications is an important factor in the development of DKA and HHS. Omission of insulin may result in DKA in a patient with type 1 diabetes in a matter of hours. In contrast, the development of HHS (or DKA) in a patient with type 2 diabetes occurs over days to weeks in a patient omitting insulin or oral agents. In addition to the factors listed in the table, impaired thirst or lack of access to water, particularly in the elderly, will contribute to the development of HHS.32 Eating disorders may account for up to 20% of cases of recurrent DKA.9
Table 58.2
Causes of Diabetic Ketoacidosis and Hyperglycemic Hyperosmolar Syndrome
| Etiologic Factor/Condition | Diagnoses with Comments |
| Inadequate insulin | Nonadherence,25,193,194 inadequate insulin regimen,34 insulin pump failure or omission of insulin,195,196 new-onset diabetes197 |
| Infections | Any moderate to severe infectious process29,198 Most commonly, pneumonia, urinary tract infection, sepsis9 |
| Acute medical conditions | Myocardial infarction,9 pancreatitis,41 cerebrovascular accident,9 arterial thrombosis (mesenteric, iliac)199 |
| Interference with glucose metabolism or insulin secretion32 | Thiazide diuretics, corticosteroids,200 second-generation (atypical) antipsychotics,201 FK506,202 glucagon,203 sympathomimetic agents (albuterol, terbutaline, dobutamine)204 |
| Endocrine disturbance | Pheochromocytoma,205 hyperthyroidism,206 acromegaly,207 hemochromatosis208 |
| Substance abuse | Cocaine209 |
| Nutritional factors | Parenteral nutrition210 |
| Immune modulation | Interferon211 |
Superscript numbers refer to references provided online for this chapter.
Presentation
The presentation of DKA and HHS varies with the severity of the metabolic derangement and any intercurrent illnesses or comorbid conditions. DKA can occur in a patient without a history of diabetes who presents with only mild symptoms; a high index of suspicion is needed to make the diagnosis in this setting, particularly in children under the age of 6. A detailed evaluation should reveal precipitating factors, especially infection and medication nonadherence, which are common triggers of DKA and HHS (see Table 58.2).24 Patients may present with symptoms of hyperglycemia (polyuria, polyphagia, polydipsia, or blurred vision) or with nonspecific symptoms of fatigue, abdominal pain, nausea, vomiting, and headache. Mental status can vary from somnolence to lethargy and even coma as the acidosis or hyperosmolarity worsens. Volume depletion may be manifested by tachycardia, poor skin turgor, dry mucous membranes, and orthostatic hypotension. In patients with DKA, metabolic acidosis causes rapid deep breathing (known as Kussmaul respiration), and acetone (derived from acetoacetate) can be sensed as a fruity smell on the patient’s breath.
Diagnosis
The diagnosis of DKA is based on a serum glucose level greater than 250 mg/dL, presence of ketones, arterial pH less than 7.3, and a CO2 concentration less than 18 mmol/L.5,9 The anion gap will be elevated, reflecting the presence of the unmeasured keto acids. The laboratory findings and changes in mental status help the clinician gauge the severity of DKA and distinguish DKA from HHS. Although arterial blood had previously been the standard for determining pH, current evidence supports the use of venous pH as a less expensive, more accessible alternative.33,34 Exceptions to the typical presentation include pregnant patients or those with poor oral intake who may have a serum glucose value below 250 mg/dL, or even in the normal range.35,36 In patients with severe vomiting, the bicarbonate value may be normal or elevated.
The serum potassium level may also be normal, low, or elevated depending on factors such as the degree of acidosis or osmotic diuresis and the duration of the process. Insulin deficiency and acidosis decrease cellular uptake of potassium, although hyperosmolality enhances cellular efflux of potassium.37 Large amounts of potassium shift from the intracellular space to the extracellular space and are then lost in the urine. Regardless of the serum potassium level, all patients with DKA and HHS have substantial depletion of total body potassium.9,37,38
The serum osmolality (in mOsm/kg) can be estimated by the following calculation:
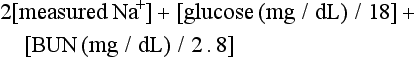
Serum osmolarity is generally normal or mildly elevated in DKA but is often profoundly elevated in patients with HHS.
Semiquantitative nitroprusside assays for urine and serum ketones are commonly employed but test only for acetone and acetoacetate, not β-hydroxybutyrate. This can be problematic, although β-hydroxybutyrate is the predominant keto acid, accounting for 75% of the keto acid load in DKA.39 In addition, nitroprusside assays may remain positive long after resolution of the metabolic acidosis, so serial measurements may be misleading and are not recommended. Quantitative assays for β-hydroxybutyrate are available and may be useful for diagnosis, especially if the nitroprusside test is negative. A small pilot study found that a serum level above 3.5 mmol/L has 100% specificity for DKA.40 Unlike the nitroprusside assay, the β-hydroxybutyrate test can also be used to monitor the management of DKA because appropriate treatment will be accompanied by a progressive fall in β-hydroxybutyrate levels.
The diagnostic criteria that distinguish HHS from DKA include higher plasma glucose (>600 mg/dL), a lack of acidosis (pH > 7.3, CO2 > 18 mEq/L), more profound dehydration (serum osmolality > 320 mOsm/kg), and a change in mental status.9 Patients with HHS often have low levels of ketones (β-hydroxybutyrate between 0.3 and 3.0 mmol/L) compared with DKA patients, whose β-hydroxybutyrate levels will be greater than 3.0 mmol/L.
Initial laboratory studies should include electrolytes, phosphorus, blood urea nitrogen (BUN), creatinine, urinalysis, complete blood count with differential, and electrocardiogram (see Table 58.2). Further evaluation should be undertaken based on the patient’s presentation. A variety of laboratory abnormalities are frequently seen in DKA. Amylase, from both salivary and pancreatic sources, may be increased in up to 90% of patients with DKA. Lipase, normally a sensitive marker for pancreatitis, may also be elevated in DKA. However, the clinician should be aware that 10% to 15% of DKA patients do have concomitant pancreatitis.41,42 Leukocytosis of 10,000 to 15,000 white blood cells/mm3 is common, but levels greater than 25,000 cells/mm3 or the presence of greater than 10% band neutrophils should increase the clinical suspicion for an active infection. Elevated hemoglobin due to the volume depletion may be present. High liver function studies occur commonly, especially in patients with fatty liver,43 and mild increases in creatine kinase and troponin may occur in the absence of myocardial damage.44
Intercurrent illnesses contribute to the morbidity and mortality rates in a hyperglycemic emergency, and every patient with DKA or HHS should undergo a thorough assessment with a focus on infectious and cardiovascular causes.34 Appropriate management should be started immediately and continue concurrent to the treatment of the DKA and HHS.
Additional causes of acidosis or ketosis should be considered in the differential diagnosis including starvation ketosis; alcoholic ketoacidosis; lactic acidosis; intoxication from methanol, salicylate, or ethylene glycol; chronic renal failure; and rhabdomyolysis.34
Management
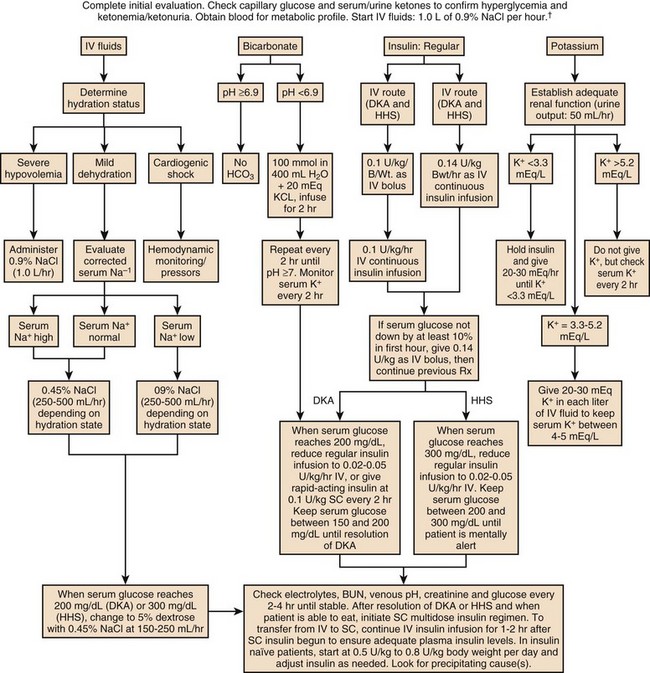
The treatment of HHS and DKA in adults and children follows similar principles of intravenous (IV) fluid, insulin, and electrolyte management. Children require more attention to weight-based regimens for fluid management in order to avoid cerebral edema.45 The details of the management of HHS and DKA in patients older than 20 years are detailed in Figure 58.2.
Fluid Replacement
Although hyperglycemia drives the cascade of osmotic diuresis, volume depletion, and renal insufficiency, the process is reversed with appropriate fluid replacement.46 In both DKA and HHS, normal saline solution should be started at 15 to 20 mL/kg body weight per hour initially (maximum of 1 L/hour), with hourly assessments of fluid status and urine output.46 The infusion rate can be lowered to 250 to 500 mL/hour once the blood pressure stabilizes.
Insulin
An initial bolus of regular insulin is typically given at 0.1 to 0.15 units/kg body weight followed by an IV insulin infusion at 0.1 unit/hour, though there is little evidence to support the need for an initial bolus.47,48 Higher dose insulin regimens are not beneficial.49,50 In patients receiving IV insulin, point of care testing should be performed every 1 to 2 hours and may be changed to every 2 hours once the patient is stable. Although subcutaneous (SC) and intramuscular (IM) insulin protocols can treat DKA or HHS, the initial response to glucose lowering is slower, and SC or IM regimens are generally reserved for emergent situations (i.e., a DKA patient with severe hyperkalemia and electrocardiographic [ECG] changes) in which IV access has been delayed.49,51
With the initial correction of volume depletion, particularly in HHS, the glucose level may fall precipitously. Thereafter, glucose levels should make a steady decline of 50 to 75 mg/dL each hour.34 There is no advantage to a more rapid improvement in the glucose level. The rate of insulin infusion may be adjusted to allow the glucose to trend downward at an appropriate rate. Once the glucose level decreases to less than 250 mg/dL, dextrose is added to the IV fluid to allow for the continued insulin infusion necessary to resolve the acidosis, because the fall in glucose will precede normalization of the acid-base status.9 By this point, the insulin infusion rate will usually have been adjusted to 0.02 to 0.05 unit/hour.
The insulin infusion should be continued until resolution of the hyperglycemic crisis. In DKA, resolution of ketoacidosis is indicated by a normalization of the anion gap or a drop in the β-hydroxybutyrate level to less than 3 mmol/L.52 In HHS, resolution is indicated by correction of the fluid and electrolyte abnormalities. Once the acute abnormalities have resolved and the glucose level has improved to less than 200 mg/dL, the patient can be transitioned to scheduled SC insulin. Patients should be placed on a physiologic insulin regimen, which includes a basal insulin (NPH, glargine, or detemir) and a mealtime, or “nutritional,” insulin (regular, lispro, aspart, or glulisine). NPH and regular insulin are associated with higher rates of hypoglycemia.53 The use of corrective (“sliding scale”) insulin as the sole SC insulin strategy is never appropriate in a patient recovering from DKA. Most protocols call for 80% to 100% of the calculated daily insulin requirements to be started based on a stable insulin infusion.54–57 The insulin doses are divided into 50% basal and 50% nutritional insulin.
Older age, higher IV insulin requirements, and greater blood glucose variation decrease the chance of a successful transition.56 Other potential pitfalls include discontinuing the IV insulin without initiating physiologic insulin, and over- or underestimating the 24-hour insulin requirements based on fluctuating insulin infusion rates. Despite pressures in a busy intensive care unit (ICU) to transition patients off IV insulin, the SC insulin needs to be calculated based on the relatively stable IV insulin requirements over 4 to 6 hours prior to the transition.57 IV insulin should not be discontinued until at least 2 hours after the start of basal insulin. A protocol for the successful transition from IV to SC insulin is listed in Box 58.2.58
Box 58.2
Important Steps in Transition from Insulin Infusion to Subcutaneous (SC) Insulin
Step 1: Is the patient stable enough for transition?
Step 2: Does the patient need to switch to scheduled SC insulin?

Step 3: If transition is needed, calculate a total daily dose (TDD) of insulin. The TDD is an estimate of the 24-hour insulin requirement when the patient is receiving full nutrition.
• Determine mean insulin infusion rate from last 6-8 hours.
• Calculate 24-hour insulin dose based on this rate, and reduce the 24-hour dose by 20%.
• Multiply hourly insulin infusion rate by 20 (80% of 24) to arrive at a safety-adjusted 24-hour insulin dose.
• Determine if this total is the TDD or basal dose based on current nutrition.
Step 4: Construct a basal/bolus regimen tailored to the patient’s nutritional situation, building safeguards for any changes in nutritional intake and uncertainties about reliability of intake. Several options are again available.
• Basal: glargine, detemir, or NPH
• Nutritional: The remainder of TDD is scheduled nutritional insulin in divided doses. In general, these doses need to be adjusted down for <100% nutritional intake, and the orders should allow for administering nutritional insulin just AFTER observed meal to allow an assessment of intake. Several options are available for estimating the initial doses:
Step 5: Be sure to give SC insulin BEFORE the infusion stops.
DM, diabetes mellitus; HbA1c, hemoglobin A1c (glycosylated hemoglobin).
Modified from O’Malley CW, Emanuele, M, Halasyamani L, Amin AN. Bridge over troubled waters: safe and effective transitions of the inpatient with hyperglycemia. J Hops Med 2008, 3(S5):S55-65.
Potassium
Patients with DKA or HHS may initially present with hyperkalemia or hypokalemia, each with significant potential cardiac consequences. Tall peaked T waves in the precordial leads are the first ECG sign of hyperkalemia as the potassium increases above 5.5 mmol/L. As potassium levels increase to greater than 6.5 mmol/L, additional ECG changes emerge: prolonged PR interval, decreased amplitude of P waves, and a widening of the QRS complex. As the potassium increases to greater than 8.0 mmol/L more ominous changes occur, including loss of P waves; intraventricular, fascicular, and bundle branch blocks; and widening of the QRS complex, ultimately resulting in asystole.59 Conversely, hypokalemia also increases the risk of cardiac arrhythmia. ECG changes in hypokalemia include broad flat T waves, ST-segment depression, and QT interval prolongation.60 Additionally, hypokalemia decreases myocardial contractility.60
Treatment with volume expansion and insulin will lower serum potassium levels as potassium shifts from the extracellular to intracellular space. Electrolytes should be monitored every 2 to 4 hours in the early stages of DKA.9,32 For a potassium concentration greater than 5 mmol/L, serum K+ testing should be repeated every 1 to 2 hours. Potassium replacement should be started once the potassium decreases into the normal range and the patient is producing urine. Once serum potassium is below 5 mmol/L, patients should receive KCl 5 mEq/hour via the IV fluid, and the rate can be increased to 10 mEq/hour if the serum K+ is below 4 mmol/L. Lower doses should be used if the patient has evidence of impaired renal function.
Bicarbonate
The metabolic acidosis of DKA impairs myocardial contractility, affects oxyhemoglobin dissociation and tissue oxygen delivery, inhibits intracellular enzymes, alters cellular metabolism, and may result in vital organ dysfunction.61 Correction of acidosis focuses on the correction of insulin deficiency and volume depletion, as well as any other underlying conditions that may be contributing to the acidosis. Although the administration of bicarbonate would appear to be a reasonable consideration, multiple trials of bicarbonate replacement in DKA patients have demonstrated no benefit.61,62 Furthermore, the use of IV bicarbonate may result in complications.34,40 Bicarbonate therapy has been associated with hypokalemia, intracellular acidosis, and tissue hypoxia.47 On the other hand, a potential benefit for bicarbonate therapy in those individuals with severe acidemia (pH < 6.9)62,63 has been suggested, though the literature is limited. In patients with pH less than 6.9, the use of sodium bicarbonate (100 mmol sodium bicarbonate in 400 mL sterile water with 20 mEq KCl, administered at a rate of 200 mL/hour for 2 hours, and then repeating every 2 hours) may be considered until the pH is greater than 6.9.9
Phosphate
Phosphate levels initially may be normal to elevated, but usually decline with treatment.64 There is no evidence to support the routine use of phosphate replacement in patients with DKA.65,66 Nevertheless, severe hypophosphatemia may impair oxygen delivery and cause muscle fatigue, and selective replacement is suggested in patients with phosphate levels less than 1.0 mg/dL, anemia, respiratory failure, or CHF.9,67 Correcting the phosphorus level improves tissue oxygenation and restores the body’s buffering capacity. Phosphate is generally replaced as a potassium salt.68 To avoid complications, particularly hypocalcemia, phosphorus replacement should be limited to 40 to 50 mmol of potassium phosphate at approximately 3 to 4 mmol/hour. The potassium being provided in other IV fluids may need to be adjusted to account for the potassium infused as part of the phosphorus replacement.68
Hyperchloremic Acidosis
The development of hyperchloremic acidosis occurs in DKA because the ability to completely regenerate bicarbonate is hampered by the urinary loss of keto acids, which normally act as substrate for the re-formation of bicarbonate. To maintain the appropriate balance of cations and anions, chloride ions from the infused saline will partially compensate for the lack of bicarbonate. As the ketoacidosis resolves, a hyperchloremic acidosis (with a normal anion gap) may ensue. This benign process resolves gradually in the hours to days after the saline infusion is reduced or stopped.68
Complications of the Management of Hyperglycemic Emergencies
Cerebral edema is the one of the most feared complications of hyperglycemic crisis. Classically, cerebral edema has been reported in children with DKA. It occurs in 0.5% to 1% of all DKA cases69,70 and carries a mortality rate of 20%. Survivors are at risk for residual neurologic problems.45 Although cerebral edema is occasionally seen in adults with HHS, it is rare in adults with DKA and children with HHS.71 Children who present with (1) low partial pressures of arterial carbon dioxide and (2) high serum urea nitrogen concentrations and (3) are treated with bicarbonate are at increased risk for cerebral edema. The exact cause of cerebral edema remains unknown. It does not appear to be associated with the initial level of hyperosmolarity or more rapid correction of the hyperosmolarity.72,73 Cerebral ischemia, and not changes in osmolality, may be the causal factor in patients who develop cerebral edema.73 Nevertheless, care should be taken to avoid rapid changes in glucose and osmolarity, especially in younger patients. The clinician should also be aware of the signs of cerebral edema, which include headache, persistent vomiting, hypertension, bradycardia, and neurologic changes.
Attention to the diagnosis and management of any infections or cardiovascular events is imperative. Other complications of the management of DKA and HHS include hypokalemia, hypophosphatemia, acute renal failure, and shock. Appropriate management of hyperglycemic emergencies with close attention to fluid and electrolyte balance minimizes these complications. Less common problems can include rhabdomyolysis,74 thrombosis and stroke,75 pneumomediastinum,76 and memory loss with decreased cognitive function in children.77
Prevention
Despite improvements in insulin treatment and glucose monitoring78 the incidence of DKA is increasing.3 Prevention is paramount. Health care providers should recognize signs of DM in all age groups. Patients with DM and their caregivers should be familiar with sick day recommendations (Box 58.3), and patients with type 1 DM should check urine ketones by dipstick if the glucose is greater than 240 mg/dL.79 Home measurement of serum ketones with a commercial glucometer may allow earlier detection of DKA and decreased hospital visits,80 though most meters lack this capability.
Nonadherence to medical regimens is often cited as the cause of recurrent DKA, and in fact, 5% of patients account for more than 25% of all cases of DKA.81 Health care providers need to recognize disparities in the health care system that limit access to appropriate DM care or otherwise contribute to nonadherence with insulin or other antidiabetic medications. Diabetes education enhances patient knowledge of appropriate monitoring and prevention of DKA,82 and diabetes educators are an excellent resource to help assess a patient’s barriers to optimal care including financial, social, and cultural issues.82
Glycemic Control in the Intensive Care Unit
Epidemiology and Significance of Hyperglycemia
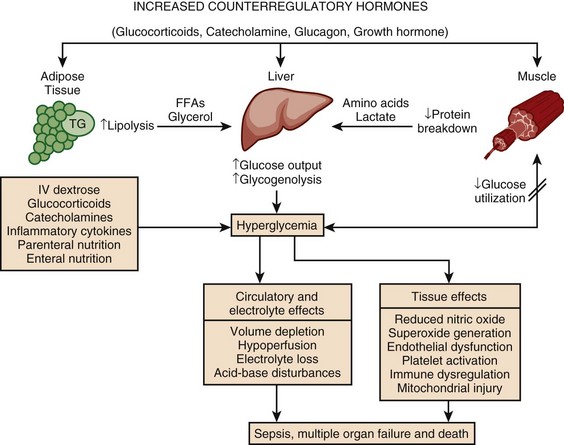
Hyperglycemia is a common occurrence in hospitalized patients and is associated with increased morbidity and mortality rates,83–85 including higher rates of surgical infections after cardiothoracic surgery,86,87 increased mortality rates after MI,88–90 and poor outcome in critically ill patients.91,92 Forty percent of patients in the ICU and 80% of those undergoing cardiac surgery have elevated glucose levels.93 Of hyperglycemic patients in the hospital, two thirds have a history of diabetes, although the remainder have no prior history.85 This latter group includes patients with previously unrecognized diabetes as well as those with “stress hyperglycemia” (patients who are normoglycemic on follow-up testing). Only 15% of patients with new-onset hyperglycemia have confirmed diabetes 1 year after the hospital admission.94 Hyperglycemic patients with no prior history of diabetes have a worse prognosis than patients with preexisting diabetes, including greater mortality rates after MI83 and a significantly increased 30-day and 1-year mortality rate.95 This is particularly true of patients with stress hyperglycemia, a condition of acutely elevated glucose related to illness-related increases in counterregulatory hormones seen (Fig. 58.3)93 in patients with otherwise normal glucose metabolism. Stress hyperglycemia is consistently associated with increased adjusted mortality rates for patients with unstable angina, acute MI, CHF, arrhythmia, ischemic and hemorrhagic stroke, gastrointestinal bleeding, acute renal failure, pneumonia, pulmonary embolism, and sepsis.96
Glucose variability, measured as the standard deviation of glucose excursions around the mean, adds to the metabolic burden.97 Glycemic variability may be a better predictor of morbidity and mortality rates than the level of hyperglycemia in patients with preexisting diabetes.98–100 In a retrospective analysis, the odds ratio for ICU death is significantly greater in patients with higher hourly change in glucose.101 Glycemic variability may be a better predictor of morbidity and mortality rates than the level of hyperglycemia in patients with preexisting diabetes.98–100
Synopsis of Trials of Glycemic Control
The benefits of improved glucose control were initially suggested by several retrospective or nonrandomized trials in patients with acute MI,102 with stroke,84 after coronary artery bypass surgery,87 and in the ICU.103 A landmark single-center prospective study in surgical ICU patients was published in 2001, in which intensive insulin therapy (IIT) was reported to decrease ICU and in-hospital mortality rates, as well as significant comorbid conditions such as bloodstream infections, acute renal failure requiring dialysis or hemofiltration, the median number of red blood cell transfusions, critical-illness polyneuropathy, and prolonged mechanical ventilation. The number of patients who received red-cell transfusions did not differ significantly between the two groups. However, the median number of transfusions in the intensive-treatment group was only half that in the conventional-treatment group. This difference was not due to more liberal use of transfusions in the conventional-treatment group, as indicated by the lower hemoglobin and hematocrit values in that group.104 Based on this study, many ICUs initiated IV insulin protocols designed to achieve glucose levels in the 70- to 110-mg/dL range. Soon, however, enthusiasm over the benefits of normoglycemia from IIT in the critically ill patient were tempered by (1) an inability to extend the initial results across multiple centers,105,106 (2) concern over the risk of hypoglycemia,105,106 (3) questions over the role of early nutrition,104 and (4) a less pronounced improvement for patients with preexisting diabetes.107 Additionally, nonrandomized and observational studies suggested a J-shaped curve for mortality rate related to glycemic control in the ICU with increasing mortality rate for glucose levels less than 90 mg/dL and greater than 120 mg/dL.96,103,108–110 In the NICE-SUGAR (Normoglycemia in Intensive Care Evaluation—Survival Using Glucose Algorithm Regulation) study,111 a randomized controlled, multicenter trial, in which ICU patients were randomized to intensive glycemic control or conventional glucose control, no mortality rate benefit was seen with IIT. In a meta-analysis of studies evaluating the impact of intensive glycemic control, including the results from NICE-SUGAR, the relative risk of death was not significantly different with IIT compared with conventional therapy, but the risk of severe hypoglycemia was significantly greater.112 Figure 58.4 summarizes key studies on the impact of intensive glycemic control.
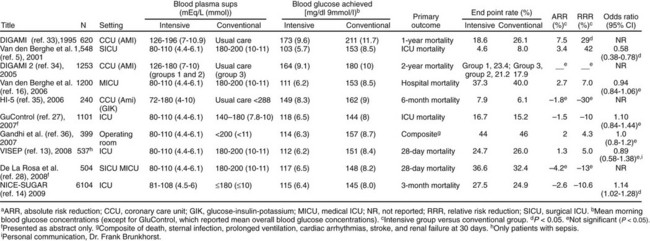
The foremost obstacle to the use of IIT is hypoglycemia. Patients managed with IIT for tight glycemic control have significantly greater rates and duration of hypoglycemia.112,113 Hypoglycemia from insulin therapy increases mortality risks in critically ill patients,105,114–118 which may mitigate any benefits of tight glucose control.104 As expected, use of glucose control algorithms modified for a higher glucose target range significantly reduces the risk of severe hypoglycemia.119,120
Intravenous Insulin Protocols
A standardized insulin protocol should be used to maintain goal glucose levels and reduce hypoglycemic episodes. Current practice requires the clinician to consider the multiple factors affecting glucose control, such as changes in nutrition, medication or medical acuity, and proactively respond to avoid large fluctuations in glucose levels. An effective IV insulin protocol allows for titration of IV insulin in the setting of changing glucose levels. Multiple published insulin protocols are available,54,57,121–126 though any protocol will need to be adapted and adjusted to fit the needs of the local environment. A multidisciplinary team consisting of intensivists, endocrinologists, nurses, and pharmacologists should be involved in the development, implementation, and continued evaluation of IV insulin protocols.
Technical Issues in Measuring Glucose
Accurate measurement of glucose in the ICU is essential to maintain glycemic control and limit the risk of severe hypoglycemia. The use of IIT magnified the importance of accurate point of care (POC) glucose monitoring. In the ICU measurements of glucose are affected by the clinical status of the patient, by certain medications, and by the type of blood sample.127 Anemia may falsely elevate POC glucose levels, which can mask hypoglycemia.128–130 This deserves attention given the high prevalence of anemia routinely encountered in ICUs.131 The accuracy of POC testing is also affected by several other common conditions (e.g., acidosis, hypothermia, and hypotension) and medications (dopamine, mannitol, acetaminophen, and pressor use).132–135 The rates of intrapatient variability in glucose measurements are lowest from arterial samples. Variability increases in venous sampling with the greatest variability in capillary blood. Arterial blood analysis correlates better with central laboratory glucose values than capillary blood measurements. During hypoglycemia, the discrepancy is exaggerated. POC capillary and whole blood samples from patients with poor peripheral perfusion overestimate the laboratory glucose level by 20% in a significant proportion of samples.136 The difference between capillary and arterial blood glucose measurements has led most insulin protocols for ICUs to recommend the use of arterial blood measurements when available.136
Subcutaneous electrochemical sensors can measure glucose levels in the interstitial fluid and report an average glucose level every 1 to 10 minutes, although a lag time of 8 to 18 minutes exists between blood glucose and interstitial glucose equilibration.137 Determining trends in glucose, the devices may provide earlier detection of hypoglycemia and hyperglycemia. Several small studies found excellent accuracy for continuous glucose monitoring (CGM) in the ICU as compared to POC testing.137–140 However, there is currently insufficient evidence to support the general use of CGM in the ICU.141
Current Guidelines for Glycemic Control
The current guidelines for glycemic control in the ICU have transitioned from declarations for tight glycemic control (based on observational and retrospective data and a single-site randomized controlled study) to more moderated goals taking into account the increased mortality rate associated with hypoglycemia and the limited benefit from tight glycemic control seen in other trials.105,106,142,143
The 2009 American Academy of Clinical Endocrinologists and American Diabetes Association Consensus Statement on Inpatient Glycemic Control122 recommends insulin therapy for persistent hyperglycemia at a threshold no greater than 180 mg/dL, with a target range for glucose levels in the ICU of 140 to 180 mg/dL. The guideline recommends treatment with IV insulin using an established protocol associated with low rates of hypoglycemia and frequent glucose monitoring.
The 2012 Surviving Sepsis Guidelines (2012)144 recommend:
• Initiating an IV insulin protocol in ICU patients with severe sepsis who have two consecutive blood glucose levels greater than 180 mg/dL
• Targeting a blood glucose level less than 180 mg/dL
• Providing a glucose calorie source to all patients on IV insulin
• Monitoring glucose levels every 1 to 2 hours until infusion rates are stable, then every 4 hours
• Interpreting POC capillary blood glucose testing with caution, keeping in mind that this may overestimate arterial blood glucose or plasma glucose
The Effect of Parenteral Nutrition, Enteral Nutrition, and Glucocorticoids on Glycemic Control
Enteral feeding preparations have high concentrations of carbohydrates, and are associated with an increase in glucose concentration.145,146 Similarly, the majority of patients with type 2 DM who are not on insulin prior to ICU admission require insulin to maintain glycemic control once parenteral nutrition is initiated.147 It is advisable to begin an IV or SC insulin regimen at the onset of total parenteral nutrition/peripheral parenteral nutrition (TPN/PPN) use,122 or alternatively, regular insulin can be added to parenteral nutrition. When nutrition is held or discontinued, the insulin dosing will usually need to be reduced, or dextrose-containing IV fluid started, to reduce the risk of hypoglycemia.
Glucocorticoid-induced hyperglycemia is common in the critically ill patient. Glucocorticoids decrease insulin sensitivity, impair glucose disposal, and increase hepatic insulin resistance.148 All patients should have regularly scheduled glucose monitoring for the first 48 hours after steroids are initiated. Subcutaneous insulin therapy may be started for glucocorticoid-induced hyperglycemia with an initial total daily dose (TDD) of 0.3 to 0.5 unit per kg body weight. IV insulin infusion is appropriate for moderate to severe hyperglycemia.149 In patients started on insulin for glucocorticoid-induced hyperglycemia, the insulin dose will need to be titrated for changes in steroid dose.
Subcutaneous Insulin Use in the Hospitalized Patient
Insulin is the therapy of choice for glucose control in the ICU. Oral agents and noninsulin injectable agents should be discontinued on admission. The multiple contraindications and the inability to make rapid adjustments for changes in clinical or nutritional status make noninsulin therapy poorly suited for the care of the critically ill patient. The onset, peak, and duration of action of the currently available insulin preparations are listed in (Table 58.3). Insulin can be categorized based on the physiologic profile: basal insulin is either long or intermediate duration; short- or rapid-acting insulin is used for prandial control and for the correction of hyperglycemia. The lack of a defined peak and prolonged action profile make glargine (Lantus) and detemir (Levemir) advantageous for use as a basal insulin. Glargine is typically dosed once daily and detemir one to two times daily. NPH has an intermediate duration of action with peak effect at 6 to 8 hours after dosing and is usually dosed twice daily.
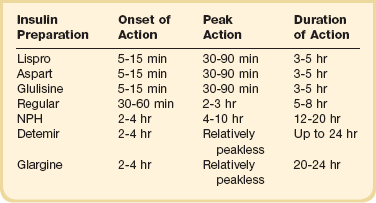
With a variety of insulin regimens available, the practitioner has several options available to manage hospitalized patients with diabetes or stress hyperglycemia. Patients who are not eating or who are receiving continuous nutrition (enteral or parenteral) should have a basal insulin in place. The basal insulin is given SC, or in the case of parenteral nutrition, regular insulin may be added to the PPN or TPN at an initial dose of 1 unit for each 15 g of dextrose.150 In a patient on continuous nutrition or NPO (nothing by mouth), glargine is dosed daily, detemir every 12 to 24 hours, or NPH every 8 to 12 hours. No research is available to support the use of one basal insulin over another in the hospitalized patient. The potential risk of NPH is the peak effect, although the longer duration of glargine and detemir may result in longer periods of hypoglycemia. Glucose monitoring should be performed every 4 to 8 hours, depending on the stability of the patient.
In patients tolerating scheduled meals, a prandial insulin should be added. Lispro, aspart, and glulisine each have a more physiologic profile with a quicker onset of action, time to peak, and shorter duration of action than regular insulin. The use of a rapid-acting insulin analog is associated with better glycemic control compared to regular insulin.151 In patients who are eating, glucose monitoring is usually performed before the meal and at bedtime.
1. A weight-based protocol152 can be utilized, typically starting at 0.3 to 0.6 unit/kg body weight depending on the patient’s weight and risk factors for insulin sensitivity (body mass index [BMI] < 25, hepatic insufficiency, renal insufficiency) or insulin resistance (morbid obesity, sepsis, glucocorticoids). A weight-based regimen is helpful in initiating an SC insulin regimen in a patient with new-onset hyperglycemia or a patient who is on a noninsulin antidiabetic regimen with or without basal insulin. Fifty percent of the dose is given as basal insulin, and the remaining 50% is divided into three mealtime boluses.
2. The patient’s home insulin basal-bolus dose can be restarted if the patient has good outpatient glucose control, based on a recent hemoglobin A1c (HbA1c) or a reliable history. The results of an HbA1c obtained in the past 3 months will give insight into the efficacy of the patient’s outpatient medical regimen, and may also help to determine if a patient without a history of diabetes has unrecognized DM versus stress hyperglycemia.
3. A premixed insulin regimen can be changed to basal-bolus by adding the TDD of insulin and then giving 50% as basal insulin and 50% as nutritional (mealtime) insulin.
4. An insulin regimen can be determined based on the IV insulin infusion requirements.
The use of correctional (sliding) scale insulin as sole therapy cannot be promoted for the patient with hyperglycemia. Reliance on sliding scale insulin is associated with worse glycemic control in hospitalized patients.153,154 In particular, patients with type 1 DM always require physiologic insulin dosing including a basal insulin to prevent the development of DKA. Correctional insulin doses may be used as an adjunct to a physiologic insulin regimen. Doses of correctional insulin should be aligned to the TDD of prescribed insulin; thus, a patient requiring 100 units of insulin a day will require more correctional dose insulin than a patient whose TDD is 30 units. The clinician can calculate the expected change in glucose for 1 unit of regular or rapid-acting insulin analog using the “Rule of 1500” formula: 1500/TDD = expected decrease in glucose (in mg/dL) for 1 unit of correctional dose insulin. For insulin-sensitive patients (e.g., type 1 DM, BMI < 25 kg/m2, end-stage renal disease [ESRD], or hepatic failure), 1800 rather than 1500 is used for the numerator.
Hypoglycemia
As noted in the previous section, hypoglycemia is the main deterrent to good glycemic control155 and is associated with higher rates of morbidity and mortality in the ICU.105,120,156,157 Under normal circumstances the fasting glucose is 70 to 90 mg/dL.158 Although only 1.5% of patients without DM or who are pre-DM will have clinically significant hypoglycemia,114 the critically ill patient is at enhanced risk of hypoglycemia due to underlying illness as well as the use of medications that can lower glucose.
Physiologic Response to Hypoglycemia
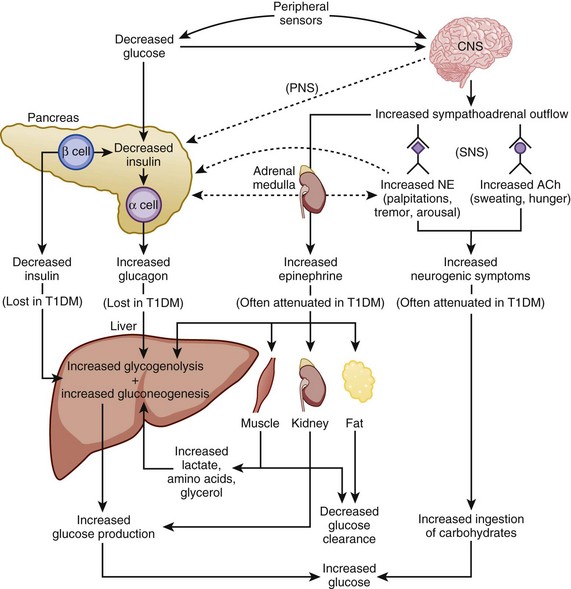
The physiologic response to hypoglycemia is mediated through neural and humoral pathways and that involves both a reduction in insulin and activation of the counterregulatory response159 (Fig. 58.5). The first humoral response to a decreasing glucose is inhibition of insulin secretion when the glucose level drops to less than 80 mg/dL.159,160 The drop in insulin level causes decreased peripheral glucose utilization, as well as increased glucose production through glycogenolysis and gluconeogenesis.161 Glucagon, epinephrine, and growth hormone secretion increases as glucose levels fall below 70 mg/dL.159,160 Cortisol release increases as the glucose drops below 60 mg/dL. Of the counterregulatory hormones, glucagon plays the key role in the minute-to-minute prevention of hypoglycemia.162 If glucagon secretion is diminished (e.g., in longstanding autoimmune diabetes), epinephrine release becomes critical for the reversal of hypoglycemia. Under normal circumstances, cortisol and growth hormone are not essential in the prevention of hypoglycemia but are necessary in the absence of glucagon.159 The stepwise response to hypoglycemia and the redundancy in the counterregulatory hormones suggest the physiologic importance placed on the prevention of hypoglycemia.159
Symptoms
The symptoms of hypoglycemia can be thought of as either sympathoadrenal or neuroglycopenic (Table 58.4). The sympathoadrenal symptoms are mediated through both β- and α-adrenergic and cholinergic pathways, and begin as the glucose decreases to less than 60 mg/dL.163 These symptoms include cholinergic-mediated complaints of diaphoresis and hunger and adrenergic-related symptoms of tremulousness, palpitations, and anxiety.163
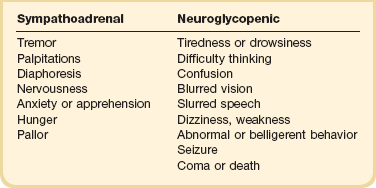
Neuroglycopenic symptoms represent the effects of low glucose on the brain. Although the brain is responsible for 50% of the body’s glucose use under physiologic conditions,164,165 it can neither generate glucose nor store glycogen. Thus, the brain requires a constant source of glucose.164,165 The onset of neuroglycopenic symptoms represents a drop in glucose below the critical level of glucose transport across the blood-brain barrier to meet the brain’s metabolic demands.166 Initial neuroglycopenic symptoms include the sensation of warmth, difficulty concentrating, weakness, fatigue, dizziness, difficulty speaking, and blurred vision.163 As glucose falls, these effects may progress to confusion, behavioral change, lethargy, seizure, and coma. The severity of symptoms may be altered by the degree of hypoglycemia, the rate of fall in glucose levels, and the blunting of the hypoglycemic response from either medication or habituation from recurrent hypoglycemia.165,167
Hypoglycemia in Patients with Diabetes or Who Receive Insulin
In patients with diabetes, hypoglycemia occurs with decreased or interrupted carbohydrate consumption, increased glucose utilization from elevated energy expenditure, or an increase in insulin to a degree that overwhelms the glucose counterregulatory system. The source of insulin may be exogenous, from an insulin injection or infusion, or endogenous, caused by an insulin secretagogue drug such as a sulfonylurea or a glinide. A detailed description of the onset, peak, and duration of the various types of insulin is given in the earlier section “Glycemic Control in the Intensive Care Unit.” Sulfonylurea drugs such as chlorpropamide, tolbutamide, and glyburide, which have extended half-lives and are renally metabolized, predispose to prolonged hypoglycemia, particularly in patients with renal insufficiency.168 Newer agents such as glipizide and glimepiride have dual hepatic and renal metabolism, which decreases the risk of prolonged hypoglycemia. The shorter duration of action of the glinides (repaglinide and nateglinide) reduce the risk of prolonged hypoglycemia. Diabetes medication affecting insulin resistance or carbohydrate consumption (i.e., metformin, thiazolidinediones, α-glucosidase inhibitors [AGIs], dipeptidyl peptidase IV inhibitors, amylin or incretin mimetics, bile acid sequestrants, or bromocriptine) do not typically cause clinically significant hypoglycemia unless taken in combination with insulin or an insulin secretagogue.
Hypoglycemic Unawareness (Hypoglycemia-Associated Autonomic Failure)
Recurrent episodes of hypoglycemia,169 particularly those that occur with sleep170,171 or exercise,172,173 blunt the normal response to hypoglycemia. This condition, known as hypoglycemia-associated autonomic failure (HAAF), reflects decreased adrenomedullary and sympathetic neural responses, and results in defective counterregulation and diminished symptoms of hypoglycemia, called hypoglycemic unawareness. The risk of severe hypoglycemia is markedly increased in patients with HAAF.174 Furthermore, the response to glucagon and epinephrine may already be blunted in the critically ill patient,158,175 further increasing the hypoglycemic risk.
Over time, patients with type 1 diabetes develop diminished α-cell production of glucagon, leaving these patients at even higher risk of hypoglycemia. The inability of patients with type 1 DM to appropriately regulate insulin and glucagon secretion is associated with a 25 times greater risk of iatrogenic hypoglycemia. The absence of both β-cell (insulin) and α-cell (glucagon)176 function also occurs following pancreatectomy and results in significant glycemic instability.176
Management of Hypoglycemia in Patients with Diabetes
A hypoglycemic patient with a history of diabetes should be evaluated for the frequency and severity of the hypoglycemic episodes, timing and dose of insulin or insulin secretagogues, missed or lower carbohydrate meals, increased physical activity, or new-onset hepatic or renal dysfunction. The risk of severe hypoglycemia increases with intensive glucose control.177 Although an HbA1c less than 7% can indicate excellent glucose control, it may also suggest an enhanced risk of hypoglycemia, particularly in the elderly.
Published protocols on the management of hypoglycemia are available.57,178,179 Patients who can eat should receive 15 to 20 g of carbohydrates with recheck of the glucose level in 15 to 20 minutes.180 If the glucose remains less than 70 mg/dL,178 the protocol should be repeated. A snack or small meal should follow once the hypoglycemic episode has resolved. Overtreatment with unmeasured carbohydrates such as fruit juice (with or without added sugar) increases the likelihood of rebound hyperglycemia and glycemic variability. Measured amounts of dextrose with glucose gel packets or glucose tablets offer more accurate dosing of carbohydrates. In the patient with IV access who is NPO or is unable to consume oral carbohydrates (including patients with impaired level of consciousness due to hypoglycemia), low glucose levels can be managed with 25 g of 50% dextrose (1 amp).159 The use of glucagon should be limited to those patients with neuroglycopenic symptoms who have no IV access. The glycemic response to IV dextrose or glucagon may lead to moderate hyperglycemia.
Other caveats exist in the management of hypoglycemia. First, glucagon induces glycogenolysis and thus is not effective in patients who are glycogen-depleted such as those with alcohol abuse. Second, although mild or moderate cases of hypoglycemia related to insulin secretagogues respond well to treatment with oral carbohydrates or dextrose, the recommended treatment of sulfonylurea overdose is octreotide 100 µg,181 which decreases the insulin secretion. Third, AGIs block the digestion of disaccharides, and therefore, treatment of hypoglycemia with disaccharides such as sucrose (cane sugar) or lactose (in milk) will be ineffective in patients taking AGIs. Oral glucose (dextrose), a monosaccharide, should be used.
Prevention
Forethought is required to prevent hypoglycemia in the ICU, but the prevention of hypoglycemia can be forgotten in the midst of multiple competing interests. Intensivists need to remain attentive to the increased risk for hypoglycemia as the patient’s illness status, nutritional status, and medications change182 (Table 58.5). Particular attention should be given to the titration of insulin doses when steroids or epinephrine are being tapered.
Table 58.5
Risk Factors for Hypoglycemia in the Hospitalized Patient118,214
| Risk Factor | Prevention |
| Initiation of insulin | Individualized insulin dosing |
| Inappropriate timing of mealtime insulin | Mealtime insulin boluses up to 15 minutes before or 15 minutes after meal |
| Overuse or overdosing of correctional doses of insulin | Individualized insulin dosing based on total daily insulin requirements |
| Change in nutrition | Standardized orders to adjust insulin or add intravenous fluids with dextrose |
| Reduction in corticosteroid dose | Adjust insulin doses as steroid dose tapered |
A dependence on correctional dose insulin (“sliding scales”) rather than scheduled basal-bolus insulin regimens has become endemic in many hospitals. This practice is to be strongly discouraged. The reliance on correctional insulin as the sole glycemic management strategy is associated with poor glycemic control and increased hypoglycemia, although a physiologic, basal-bolus insulin regimen improves glycemic control.57
Hypoglycemia in Patients without Diabetes
Because of the tiered physiologic mechanisms to protect against low blood glucose, hypoglycemia is unusual in nondiabetic patients. Only 1.5% of hypoglycemic episodes occur in patients without diabetes or who are not receiving insulin,167 and most conditions or medications will not cause hypoglycemia unless multiple factors occur simultaneously.167 The acute management of hypoglycemia is similar in diabetic and nondiabetic individuals, but nondiabetic patients presenting with hypoglycemia require a careful diagnostic evaluation.
Diagnosis
All patients considered to have hypoglycemia should meet the criteria for “Whipple’s triad”: symptoms consistent with hypoglycemia, a low glucose level at the time of the symptoms, and prompt resolution of the symptoms with correction of hypoglycemia. The differential diagnosis of hypoglycemia can be broadly divided into those causes most commonly seen in otherwise healthy patients versus those seen in patients with underlying illness (Table 58.6). Essentially any critical illness can cause hypoglycemia, though this should remain a diagnosis of exclusion after consideration of any other potential causes.
Table 58.6
Causes of Hypoglycemia in Adults

From Cryer PE, Axelrod L, Grossman AB, et al: Evaluation and management of adult hypoglycemic disorders: An Endocrine Society Clinical Practice Guideline. J Clin Endocrinol Metab 2009;94(3):709-728.
A large number of medications have been reported to cause hypoglycemia (Table 58.7). Other than insulin and insulin secretagogues, most of these agents cause hypoglycemia only in rare instances.
Table 58.7
Examples of Medications Implicated as Causative Agents for Hypoglycemia
| Suspected Mechanism | Agent(s) |
| Accidental or malicious administration of antidiabetic agents | Insulins, sulfonylureas, glinides |
| Increased insulin or decreased clearance | Quinine, pentamidine, isoniazid, lithium, haloperidol, chloroquine, indomethacin214 |
| Decreased hepatic glucose output | Alcohol, unripe akee fruit |
| Autoimmune | Isoniazid, interferon-α, hydralazine, procainamide, sulfhydryl-containing drugs |
| Interactions with sulfonylureas | Phenylbutazone, colchicine, paracetamol |
| Beta cell toxicity | Pentaminide |
| Unknown mechanism | Sulfonamide, salicylates, warfarin, octreotide |
Ethanol inhibits gluconeogenesis167 by depleting hepatic nicotinamide adenine dinucleotide (NAD), impairs the counterregulatory actions of cortisol and growth hormone, and delays the response to glucagon and epinephrine. At particular risk of alcohol-induced hypoglycemia are alcoholic patients who are simultaneously malnourished and therefore glycogen-depleted as well.
Given the liver’s crucial role in glucose homeostasis, it is not surprising that patients with hepatic insufficiency, particularly acute liver disease, are susceptible to hypoglycemia. The kidneys are responsible for limited gluconeogenesis under normal conditions, though alterations in fasting glucose levels are not recognized until end-stage renal disease.183 Most renal patients with significant hypoglycemia are cachectic, and therefore lack the fat or muscle stores for gluconeogenesis.
In sepsis, an increased risk of hypoglycemia results from increased glucose utilization followed by decreased glucose production184 and decreased responsiveness to changes in insulin and glucagon.159,185 Cachexia is a rare cause of hypoglycemia,159,186 but has been reported in patients with severe muscle wasting or fat depletion who are therefore unable to generate glucose via gluconeogenesis. Malnourished patients are more likely to develop hypoglycemia in the setting of another disorder with increased risk of low blood sugar, such as renal insufficiency or alcohol abuse.
Insulinomas are rare (less than 1 in 250,000 patient-years) β-cell tumors that produce insulin. The tumors do not have an age or ethnic predominance, but do occur slightly more frequently in women.187 Patients present with recurrent hypoglycemia associated with neuroglycopenic symptoms. Increasingly, hypoglycemia has been reported in patients following gastric bypass with a roux-en-Y procedure. Typically, the episodes occur postprandially. The precise mechanism of this phenomenon is an area of active investigation.
Approach to the Nondiabetic Patient with Hypoglycemia
Patients in the ICU often have multiple reasons to become hypoglycemic. A standardized approach to the evaluation of hypoglycemia reduces the risk of unnecessary testing. A thorough history, physical examination, and review of laboratory results often give clues to the cause of the hypoglycemia.188 When a cause is evident, appropriate steps should be taken both to correct the offending problem and to increase nutritional support while the problem is being addressed. Once the acute hypoglycemic episode is treated, the appropriate steps should be taken to avoid recurrent hypoglycemia.
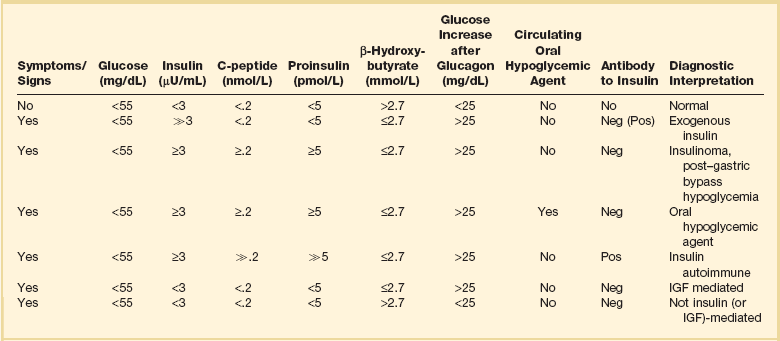
In a patient who satisfies Whipple’s triad but lacks an obvious cause for hypoglycemia, levels of glucose, cortisol, insulin, C-peptide, proinsulin, and β-hydroxybutyrate, as well as a urine screen for sulfonylurea, should be measured during an episode of spontaneous hypoglycemia.188–190 Some patients may need to have hypoglycemia provoked by a prolonged (up to 72 hours) fast.188,190,191 A protocol for a prolonged fast188,192 is listed in Figure 58.6,188 though this is not recommended in critically ill patients and should be deferred until the patient is stable. Response of glucose to IV injection of 1.0 mg glucagon may be useful in certain cases, and measurement of insulin antibodies is useful in cases in which antibody-induced hypoglycemia is suspected. An interpretation of laboratory values in hypoglycemia is presented in Table 58.8.188

References
1. International Diabetes Federation. IDF Diabetes Atlas: The Global Experience. 5th ed http://www.idf.org/diabetesatlas/5e/the-global-burden, 2011.
2. International Diabetes Federation. Diabetes Atlas. 5th ed http://www.idf.org/atlasmap/atlasmap, 2011.
3. Centers for Disease Control and Prevention. National diabetes fact sheet: National estimates and general information on diabetes and prediabetes in the United States, 2011. Atlanta, GA: U. S. Department of Health and Human Services, Centers for Disease Control and Prevention; 2011.
4. American Diabetes Association. Position statement—Standards of medical care in diabetes—2012. Diabetes Care. 2012; 35(Suppl 1):S11–S63.
5. Kahn, SE, Porte, D. The pathophysiology and genetics of type 2 diabetes. In: Inzucchi S, Porte D, Sherwin R, Baron A, eds. The Pathophysiology of Type 2 Diabetes Mellitus. The Diabetes Manual. New York: McGraw-Hill, 2005.
6. Barrett, KE, Barman, SM, Boitano, S, Brooks, H. Endocrine functions of the pancreas and regulation of carbohydrate metabolism. In Barrett KE, Barman SM, Boitano S, Brooks H, eds. : Ganong’s Review of Medical Physiology, 23rd ed, New York: McGraw-Hill, 2010.
7. Kitabchi, AE. Diabetes mellitus. In: Glew RH, Peters SP, eds. Clinical Studies in Medical Biochemistry. New York: Oxford University Press; 1987:102–117.
8. Delionback, E, Kreisberg, RA. Pathogenesis of diabetic ketoacidosis. In: Inzucchi S, Porte D, Sherwin R, Baron A, eds. The Pathophysiology of Type 2 Diabetes Mellitus. The Diabetes Manual. New York: McGraw-Hill, 2005.
9. Kitabchi, AE, Umpierrez, GE, Miles, JM, Fisher, JN. Hyperglycemic crises in adult patients with diabetes. Diabetes Care. 2009; 32(7):1335–1343.
10. Henriksen, OM, Røder, ME, Prahl, JB, Svendsen, OL. Diabetic ketoacidosis in Denmark: Incidence and mortality estimated from public health registries. Diabetes Res Clin Pract. 2007; 76(1):51–56.
11. Wang, ZH, Kihl-Selstam, E, Eriksson, JW. Ketoacidosis occurs in both type 1 and type 2 diabetes—A population-based study from Northern Sweden. Diabetic Med. 2008; 25(7):867–870.
12. Centers for Disease Control and Prevention (CDC), National Center for Health Statistics, Data from the National Hospital Discharge Survey and Division of Health Interview Statistics. U. S. Bureau of the Census, Washington, DC, Dec. 2008. http://www.cdc.gov/diabetes/statistics/dkafirst/fig1.htm
13. Kim, S. Burden of hospitalizations primarily due to uncontrolled diabetes: Implications of inadequate primary health care in the United States. Diabetes Care. 2007; 30:1281–1282.
14. Wang, J, Williams, DE, Narayan, KM, Geiss, LS. Declining death rates from hyperglycemic crisis among adults with diabetes, U. S., 1985-2002. Diabetes Care. 2006; 29(9):2018–2022.
15. Fishbein, HA, Faich, GA, Ellis, SE. Incidence and hospitalization patterns of insulin-dependent diabetes mellitus. Diabetes Care. 1982; 5(6):630–633.
16. Schober, E, Rami, B, Waldhoer, T, Austrian Diabetes Incidence Study Group. Diabetic ketoacidosis at diagnosis in Austrian children in 1989-2008: A population-based analysis. Diabetologia. 2010; 53(6):1057–1061.
17. Westphal, SA. The occurrence of diabetic ketoacidosis in non-insulin-dependent diabetes and newly diagnosed diabetic adults. Am J Med. 1996; 101(1):19–24.
18. Umpierrez, GE, Kitabchi, AE. Diabetic ketoacidosis: Risk factors and management strategies. Treat Endocrinol. 2003; 2(2):95–108.
19. Kim, MK, Lee, SH, Kim, JH, et al. Clinical characteristics of Korean patients with new-onset diabetes presenting with diabetic ketoacidosis. Diabetes Res Clin Pract. 2009; 85(1):e8–11.
20. Nyenwe, E, Loganathan, R, Blum, S, et al. Admissions for diabetic ketoacidosis in ethnic minority groups in a city hospital. Metabolism. 2007; 56(2):172–178.
21. Balasubramanyam, A, Nalini, R, Hampe, CS, Maldonado, M. Syndromes of ketosis-prone diabetes mellitus. Endocr Rev. 2008; 29(3):292–302.
22. Umpierrez, GE, Smiley, D, Kitabchi, AE. Narrative review: Ketosis-prone type 2 diabetes mellitus. Ann Intern Med. 2006; 144:350–357.
23. Newton, CA, Raskin, P. Diabetic ketoacidosis in type 1 and type 2 diabetes mellitus: Clinical and biochemical differences. Arch Intern Med. 2004; 164:1925–1931.
24. Nugent, BW. Hyperosmolar hyperglycemic state. Emerg Med Clin North Am. 2005; 23(3):629–648.
25. Umpierrez, GE, Kelly, JP, Navarrete, JE, et al. Hyperglycemic crisis in urban blacks. Arch Intern Med. 1997; 157(6):669–675.
26. Trence, DL, Hirsch, IB. Hyperglycemic crises in diabetes mellitus type 2. Endocrinol Metab Clin North Am. 2001; 30(4):817–831.
27. Lorber, D. Nonketotic hypertonicity in diabetes mellitus. Med Clin North Am. 1995; 79:39–52.
28. Koul, P. Diabetic ketoacidosis: A current appraisal of pathophysiology and management. Clin Pediatr. 2009; 48(2):135–144.
29. Trachtenberg, D. Diabetic ketoacidosis. Am Fam Physician. 2005; 71:1705–1714.
30. Wyckoff, J, Abrahamson, MA. Diabetic ketoacidosis and hyperosmolar hyperglycemic state. In: Kahn CR, Weir GC, King GC, et al, eds. Joslin’s Diabetes Mellitus. 14th ed. Philadelphia: Lippincott Williams & Wilkins; 2004:887–899.
31. Dreesen, E, Meyer, AA. Acid-base disorders—Metabolic acidosis. In: Cameron JL, Cameron A, eds. Current Surgical Therapy. 10th ed. Philadelphia: WB Saunders; 2010:1136–1138.
32. Nyenwe, EA, Kitabchi, AE. Evidence-based management of hyperglycemic emergencies in diabetes mellitus. Diabetes Res Clin Pract. 2011; 94(3):340–351.
33. Ma, OJ, Rush, MD, Godfrey, MM, et al. Arterial blood gas results rarely influence emergency physician management of patients with suspected diabetic ketoacidosis. Acad Emerg Med. 2003; 10:836–841.
34. Wilson, JF. In clinic: Diabetic ketoacidosis. Ann Intern Med. 152(1), 2010. [ITC1-1–ITC1-15].
35. Chico, M, Levine, SN, Lewis, DF. Normoglycemic diabetic ketoacidosis in pregnancy. J Perinatol. 2008; 28(4):310–312.
36. Guo, RX, Yang, LZ, Li, LX, Zhao, XP. Diabetic ketoacidosis in pregnancy tends to occur at lower blood glucose levels: Case-control study and a case report of euglycemic diabetic ketoacidosis in pregnancy. J Obstet Gynecol Res. 2008; 34(3):324–330.
37. Mount, DB, Zandi-Nejad, K. Disorders of potassium balance. In: Taal MW, Glenn M, Chertow GM, et al, eds. Brenner and Rector’s The Kidney. 9th ed. Amsterdam: Elsevier; 2011:640–688.
38. Adrogue, HJ, Wilson, H, Boyd, AE, 3rd., et al. Plasma acid-base patterns in diabetic ketoacidosis. N Engl J Med. 1982; 307:1603–1610.
39. Samuelsson, U, Ludvigsson, J. When should determination of ketonemia be recommended? Diabetes Technol Ther. 2002; 4(5):645–650.
40. Charles, RA, Bee, YM, Eng, PH, Goh, SY. Point-of-care blood ketone testing: Screening for diabetic ketoacidosis at the emergency department. Singapore Med J. 2007; 48(11):986–989.
41. Nair, S, Yadav, D, Pitchumoni, CS. Association of diabetic ketoacidosis and acute pancreatitis: Observations in 100 consecutive episodes of DKA. Am J Gastroenterol. 2000; 95(10):2795–2800.
42. Yadav, D, Nair, S, Norkus, EP, Pitchumoni, CS. Nonspecific hyperamylasemia and hyperlipasemia in diabetic ketoacidosis: Incidence and correlation with biochemical abnormalities. Am J Gastroenterol. 2000; 95(11):3123–3128.
43. Takaike, H, Uchigata, Y, Iwamoto, Y, et al. Nationwide survey to compare the prevalence of transient elevation of liver transaminase during treatment of diabetic ketosis or ketoacidosis in new-onset acute and fulminant type 1 diabetes mellitus. Ann Med. 2008; 40(5):395–400.
44. Al-Mallah, M, Zuberi, O, Arida, M, Kim, HE. Positive troponin in diabetic ketoacidosis without evident acute coronary syndrome predicts adverse cardiac events. Clin Cardiol. 2008; 31(2):67–71.
45. Dunger, DB, Sperling, MA, Acerini, CL, et al. ESPE/LWPES consensus statement on diabetic ketoacidosis in children and adolescents. and ESPE and LWPES. Arch Dis Child. 2004; 89(2):188–194.
46. Hillman, K. Fluid resuscitation in diabetic emergencies—A reappraisal. Intensive Care Med. 1987; 13:4.
47. Kitabchi, AE, Umpierrez, GE, Fisher, JN, et al. Thirty years of personal experience in hyperglycemic crises: Diabetic ketoacidosis and hyperglycemic hyperosmolar state. J Clin Endocrinol Metab. 2008; 93:1541–1552.
48. Sacks, HS, Shahshahani, MN, Kitabchi, AE, et al. Similar responsiveness of diabetic ketoacidosis to low-dose insulin by intramuscular injection and albumin-free infusion. Ann Intern Med. 1979; 90:36–42.
49. Fisher, JN, Shahshahani, MN, Kitabchi, AE. Diabetic ketoacidosis: Low dose insulin therapy by various routes. N Engl J Med. 1977; 297:238–241.
50. Kitabchi, AE, Ayyagari, V, Guerra, SNO. Efficacy of low dose vs. conventional therapy of insulin for treatment of diabetic ketoacidosis. Ann Intern Med. 1976; 84:633–638.
51. Mazer, M, Chen, E. Is subcutaneous administration of rapid-acting insulin as effective as intravenous insulin for treating diabetic ketoacidosis? Ann Emerg Med. 2009; 53:259–263.
52. Sheikh-Ali, M, Karon, BS, Basu, A, et al. Can serum beta-hydroxybutyrate be used to diagnose diabetic ketoacidosis? Diabetes Care. 2008; 31:643–647.
53. Umpierrez, GE, Jones, S, Smiley, D, et al. Insulin analogs versus human insulin in the treatment of patients with diabetic ketoacidosis. Diabetes Care. 2009; 32:1164–1169.
54. DeSantis, AJ, Schmeltz, LR, Schmidt, K, et al. Inpatient management of hyperglycemia: The Northwestern experience. Endocr Pract. 2006; 12(5):491–505.
55. Avanzini, F, Marelli, G, Donzelli, W, et al. Desio Diabetes Diagram Study Group. Transition from intravenous to subcutaneous insulin: Effectiveness and safety of a standardized protocol and predictors of outcome in patients with acute coronary syndrome. Diabetes Care. 2011; 34(7):1445–1450.
56. Furnary, AP, Braithwaite, SS. Effects of outcome on in-hospital transition from intravenous insulin infusion to subcutaneous therapy. Am J Cardiol. 2006; 98:557–564.
57. Donaldson, S, Villanuueva, G, Rondinelli, L, Baldwin, D. Rush University guidelines and protocols for the management of hyperglycemia in hospitalized patients: Elimination of the sliding scale and improvement of glycemic control throughout the hospital. Diabetes Educ. 2006; 32(6):954–962.
58. O’Malley, CW, Emanuele, M, Halasyamani, L, Amin, AN. Bridge over troubled waters: Safe and effective transitions of the inpatient with hyperglycemia. Society of Hospital Medicine Glycemic Control Task Force. J Hosp Med. 2008; 3(Suppl 5):55–65.
59. Mattu, A, Brady, WJ, Robinson, DA. Electrocardiographic manifestations of hyperkalemia. Am J Emerg Med. 2000; 18:721–729.
60. Slovis, C, Jenkins, R. ABC of clinical electrocardiography: Conditions not primarily affecting the heart. BMJ. 2002; 324:1320–1323.
61. Chua, HR, Schneider, A, Bellomo, R. Bicarbonate in diabetic ketoacidosis—A systematic review. Ann Intensive Care. 2011; 1:23–34.
62. Morris, LR, Murphy, MB, Kitabchi, AE. Bicarbonate therapy in severe diabetic ketoacidosis. Ann Intern Med. 1986; 105:836–840.
63. Viallon, A, Zeni, F, Lafond, P, et al. Does bicarbonate therapy improve the management of severe diabetic ketoacidosis? Crit Care Med. 1999; 27:2690–2693.
64. Kebler, R, McDonald, FD, Cadnapaphornchai, P. Dynamic changes in serum phosphorus levels in diabetic ketoacidosis. Am J Med. 1985; 79:571.
65. Fisher, JN, Kitabchi, AE. A randomized study of phosphate therapy in the treatment of diabetic ketoacidosis. J Clin Endocrinol Metab. 1983; 57:177–180.
66. Wilson, HK, Keuer, SP, Lea, AS, et al. Phosphate therapy in diabetic ketoacidosis. Arch Intern Med. 1982; 142(3):517–520.
67. Kitabchi, AE, Umpierrez, GE, Murphy, MB, et al. Management of hyperglycemic crises. Diabetes Care. 2001; 24(1):131–153.
68. German, MS, Pancreatic hormones and diabetes mellitusGardner DG, Shoback D, eds. Greenspan’s Basic & Clinical Endocrinology. 9th ed. McGraw-Hill, New York, 2011. http://www.accessmedicine.com.libproxy2.umdnj.edu/content.aspx?aID=8407307
69. Glaser, N. Cerebral edema in children with diabetic ketoacidosis. Curr Diab Rep. 2001; 1(1):41–46.
70. Lawrence, SE, Cummings, EA, Gaboury, I, Daneman, D. Population-based study of incidence and risk factors for cerebral edema in pediatric diabetic ketoacidosis. J Pediatr. 2005; 146(5):688–692.
71. Haringhuizen, A, Tjan, DHT, Grool, A, et al. Fatal cerebral oedema in adult diabetic ketoacidosis. Neth J Med. 2010; 68:35–37.
72. Glaser, N, Barnett, P, McCaslin, I, et al. Risk factors for cerebral edema in children with diabetic ketoacidosis. The Pediatric Emergency Medicine Collaborative Research Committee of the American Academy of Pediatrics. N Engl J Med. 2001; 344(4):264–269.
73. Glaser, NS, Marcin, JP, Wootton-Gorges, SL, et al. Correlation of clinical and biochemical findings with diabetic ketoacidosis-related cerebral edema in children using magnetic resonance diffusion-weighted imaging. J Pediatr. 2008; 153(4):541–546.
74. Casteels, K, Beckers, D, Wouters, C, Van Geet, C. Rhabdomyolysis in diabetic ketoacidosis. Pediatr Diabetes. 2003; 4(1):29–31.
75. Carl, GF, Hoffman, WH, Passmore, GG, et al. Diabetic ketoacidosis promotes a prothrombotic state. Endocr Res. 2003; 29(1):73–82.
76. Weathers, LS, Brooks, WG, DeClue, TJ. Spontaneous pneumomediastinum in a patient with diabetic ketoacidosis: A potentially hidden complication. South Med J. 1995; 88(4):483–484.
77. Ghetti, S, Lee, JK, Sims, CE, et al. Diabetic ketoacidosis and memory dysfunction in children with type 1 diabetes. J Pediatr. 2010; 156(1):109–114.
78. Lin, SF, Lin, JD, Huang, YY. Diabetic ketoacidosis: Comparisons of patient characteristics, clinical presentations and outcomes today and 20 years ago. Chang Gung Med J. 2005; 28(1):24–30.
79. Weber, C, Kocher, S, Neeser, K, Joshi, SR. Prevention of diabetic ketoacidosis and self-monitoring of ketone bodies: An overview. Curr Med Res Opin. 2009; 25(5):1197–1207.
80. Laffel, LM, Wentzell, K, Loughlin, C, et al. Sick day management using blood 3-hydroxybutyrate (3-OHB) compared with urine ketone monitoring reduces hospital visits in young people with T1DM: A randomized clinical trial. Diabetic Med. 2006; 23:278–284.
81. Greene, S, Diabetic ketoacidosis in the young, how dangerous is it? Commentary. Arch Dis Child. 2001(85):21.
82. Funnell, MM, Brown, TL, Childs, BP, et al. National standards for diabetes self-management education. Diabetes Care. 2010; 33:S89–S96.
83. Capes, SE, Hunt, D, Malmberg, K, Gerstein, HC. Stress hyperglycaemia and increased risk of death after myocardial infarction in patients with and without diabetes: A systematic overview. Lancet. 2000; 355:773–778.
84. Capes, SE, Hunt, D, Malmberg, K, et al. Stress hyperglycemia and prognosis of stroke in nondiabetic and diabetic patients: A systematic overview. Stroke. 2001; 32:2426–2432.
85. Umpierrez, GE, Isaacs, SD, Bazargan, N, et al. Hyperglycemia: An independent marker of in-hospital mortality in patients with undiagnosed diabetes. J Clin Endocrinol Metab. 2002; 87(3):978–982.
86. Latham, R, Lancaster, AD, Covington, JF, et al. The association of diabetes and glucose control with surgical site infections among cardiothoracic surgery patients. Infect Control Hosp Epidemiol. 2001; 22:607–612.
87. Furnary, AP, Gao, G, Grunkemeier, GL, et al. Continuous insulin infusion reduces mortality in patients with diabetes undergoing coronary artery bypass grafting. J Thorac Cardiovasc Surg. 2003; 125:1007–1021.
88. Ishihara, M, Kojima, S, Sakamoto, T, et al. Acute hyperglycemia is associated with adverse outcome after acute myocardial infarction in the coronary intervention era. Am Heart J. 2005; 150:814–820.
89. Norhammar, A, Tenerz, A, Nilsson, G, et al. Glucose metabolism in patients with acute myocardial infarction and no previous diagnosis of diabetes mellitus: A prospective study. Lancet. 2002; 359:2140–2144.
90. Sala, J, Masiá, R, González de Molina, FJ, et al. Short-term mortality of myocardial infarction patients with diabetes or hyperglycaemia during admission. J Epidemiol Community Health. 2002; 56:707–712.
91. Mizock, BA. Alterations in fuel metabolism in critical illness: Hyperglycaemia. Best Pract Res Clin Endocrinol Metab. 2001; 15:533–551.
92. Pittas, AG, Siegel, RD, Lau, J. Insulin therapy for critically ill hospitalized patients. A meta-analysis of randomized controlled trials. Arch Intern Med. 2004; 164:2005–2011.
93. Farrokhi, F, Smiley, D, Umpierrez, GE. Glycemic control in non-diabetic critically ill patients. Best Pract Res Clin Endocrinol Metab. 2011; 25:813–824.
94. Wexler, DJ, Nathan, DM, Grant, RW, et al. Prevalence of elevated hemoglobin A1c among patients admitted to the hospital without a diagnosis of diabetes. J Clin Endocrinol Metab. 2008; 93:4238–4244.
95. Kosiborod, M, Rathore, SS, Inzucchi, SE, et al. Admission glucose and mortality in elderly patients hospitalized with acute myocardial infarction: Implications for patients with and without recognized diabetes. Circulation. 2005; 111:3078–3086.
96. Falciglia, M, Freyberg, RW, Almenoff, PL, et al. Hyperglycemia-related mortality in critically ill patients varies with admission diagnosis. Crit Care Med. 2009; 37(12):3001–3009.
97. Monnier, L, Mas, E, Ginet, C, et al. Activation of oxidative stress by acute glucose fluctuations compared with sustained chronic hyperglycemia in patients with type 2 diabetes. JAMA. 2006; 295:1681–1687.
98. Egi, M, Bellomo, R, Stachowski, E, et al. Variability of blood glucose concentration and short-term mortality in critically ill patients. Anesthesiology. 2006; 105:244–252.
99. Krinsley, JS, Meyfroidt, G, van den Berghe, G, et al. The impact of premorbid diabetic status on the relationship between the three domains of glycemic control and mortality in critically ill patients. Curr Opin Clin Nutr Metab Care. 2012; 15(2):151–160.
100. Meyfroidt, G, Keenan, DM, Wang, X, et al. Dynamic characteristics of blood glucose time series during the course of critical illness: Effects of intensive insulin therapy and relative association with mortality. Crit Care Med. 2010; 38:1021–1029.
101. Hermanides, J, Vriesendorp, TM, Bosman, RJ, et al. Glucose variability is associated with intensive care unit mortality. Crit Care Med. 2010; 38:838–842.
102. Malmberg, K, Ryden, L, Efendic, S, et al. Randomized trial of insulin-glucose infusion followed by subcutaneous insulin treatment in diabetic patients with acute myocardial infarction (DIGAMI study): Effects on mortality at 1 year. J Am Coll Cardiol. 1995; 26:57–65.
103. Krinsley, JS. Effect of an intensive glucose management protocol on the mortality of critically ill adult patients. Mayo Clin Proc. 2004; 79:992–1000.
104. Van den Berghe, G, Wouters, P, Weekers, F, et al. Intensive insulin therapy in the critically ill patients. N Engl J Med. 2001; 345:1359–1367.
105. Brunkhorst, FM, Engel, C, Bloos, F, et al. Intensive insulin therapy and pentastarch resuscitation in severe sepsis. N Engl J Med. 2008; 358:125–139.
106. Devos, P, Preiser, JC, Mélot, C, et al. Impact of tight glucose control by intensive insulin therapy on ICU mortality and the rate of hypoglycaemia: Final results of the Glucontrol study. Intensive Care Med. 2007; 33:S189.
107. Vanhorebeek, I, Langouche, L, Van den Berghe, G. Tight glucose control: What is the evidence? Crit Care Med. 2007; 35:S496–S502.
108. Krinsley, JS. Association between hyperglycemia and increased hospital mortality in a heterogeneous population of critically ill patients. Mayo Clin Proc. 2003; 78:1471–1478.
109. Kosiborod, M, Inzucchi, SE, Krumholz, HM, et al. Glucometrics in patients hospitalized with acute myocardial infarction: Defining the optimal outcomes based measure of risk. Circulation. 2008; 117:1018–1027.
110. Scalea, TM, Bochicchio, GV, Bochicchio, KM, et al. Tight glycemic control in critically injured trauma patients. Ann Surg. 2007; 246:605–610.
111. NICE-SUGAR Study InvestigatorsFinfer, S, Chittock, DR, Su, SY, et al. Intensive versus conventional glucose control in critically ill patients. N Engl J Med. 2009; 360(13):1283–1297.
112. Griesdale, DE, de Souza, RJ, van Dam, RM, et al. Intensive insulin therapy and mortality among critically ill patients: A meta-analysis including NICE-SUGAR study data. Can Med Assoc J. 2009; 180(8):821–827.
113. Preiser, JC, Devos, P, Ruiz-Santana, S, et al. A prospective randomised multi-centre controlled trial on tight glucose control by intensive insulin therapy in adult intensive care units: The Glucontrol study. Intensive Care Med. 2009; 35(10):1738–1748.
114. Bagshaw, SM, Bellomo, R, Jacka, MJ, et al. The impact of early hypoglycemia and blood glucose variability on outcome in critical illness. Crit Care. 2009; 13:R91.
115. Bagshaw, SM, Egi, M, George, C, et al. Early blood glucose control and mortality in critically ill patients in Australia. Crit Care Med. 2009; 37:463–470.
116. Egi, M, Bellomo, R, Stachowski, E, et al. Hypoglycemia and outcome in critically ill patients. Mayo Clin Proc. 2010; 85:217–224.
117. Hermanides, J, Bosman, RJ, Vriesendorp, TM, et al. Hypoglycemia is associated with intensive care unit mortality. Crit Care Med. 2010; 38:1430–1434.
118. Krinsley, JS, Grover, A. Severe hypoglycemia in critically ill patients: Risk factors and outcomes. Crit Care Med. 2007; 35(10):2262–2267.
119. Pattan, V, Parsaik, A, Brown, JK, et al. Glucose control in Mayo Clinic intensive care units. J Diabetes Sci Technol. 2011; 5(6):1420–1426.
120. Kutcher, ME, Pepper, MB, Morabito, D, et al. Finding the sweet spot: Identification of optimal glucose levels in critically injured patients. J Trauma. 2011; 71(5):1108–1114.
121. Moghissi, ES. Insulin strategies for managing inpatient and outpatient hyperglycemia and diabetes. Mt Sinai J Med. 2008; 75:558–566.
122. Moghissi, ES, Korytkowski, MT, DiNardo, M, et al. American Association of Clinical Endocrinologists and American Diabetes Association consensus statement on inpatient glycemic control. Diabetes Care. 2009; 32(6):1119–1131.
123. Goldberg, PA, Siegel, MD, Sherwin, RS, et al. Implementation of a safe and effective insulin infusion protocol in a medical intensive care unit. Diabetes Care. 2004; 27:461–467.
124. Furnary, AP, Wu, Y, Bookin, SO. Effect of hyperglycemia and continuous intravenous insulin infusions on outcomes of cardiac surgical procedures: The Portland Diabetic Project. Endocr Pract. 2004; 10(Suppl 2):21–33.
125. Nazer, LH, Chow, SL, Moghissi, ES. Insulin infusion protocols for critically ill patients: A highlight of differences and similarities. Endocr Pract. 2007; 13:137–146.
126. Rea, RS, Donihi, AC, Bobeck, M, et al. Implementing an intravenous insulin infusion protocol in the intensive care unit. Am J Health Syst Pharm. 2007; 64:385–395.
127. Deschy, A, Vuagnat, AC, Ghazali, AD. Accuracy of bedside glucometry in critically ill patients: Influence of clinical characteristics and perfusion index. Mayo Clin Proc. 2008; 83:400–405.
128. Karon, BS, Griesmann, L, Scott, R, et al. Evaluation of the impact of hematocrit and other interference on the accuracy of hospital based glucose meters. Diabetes Technol Ther. 2008; 10:111–120.
129. Pidcoke, HF, Wade, CE, Mann, EA, et al. Anemia causes hypoglycemia in intensive care unit patients due to error in single-channel glucometers: Methods of reducing patient risk. Crit Care Med. 2010; 38:471–476.
130. Tang, Z, Lee, JH, Louie, RF, Kost, J. Effects of different hematocrit levels on glucose measurements with handheld meters for point-of-care testing. Arch Pathol Lab Med. 2000; 124(8):1135–1140.
131. Vincent, JL, Baron, JF, Reinhart, K, et al. Anemia and blood transfusion in critically ill patients. JAMA. 2002; 288:1499–1507.
132. Ray, JG, Hamielec, C, Mastracci, T. Pilot study of the accuracy of bedside glucometry in the intensive care unit. Crit Care Med. 2001; 29:2205–2207.
133. Tang, Z, Du, X, Louie, RF, Kost, GJ. Effects of pH on glucose measurements with handheld glucose meters and a portable glucose analyzer for point-of-care testing. Arch Pathol Lab Med. 2000; 124(4):577–582.
134. Tang, Z, Louie, RF, Payes, M, et al. Oxygen effects on glucose measurements with a reference analyzer and three handheld meters. Diabetes Technol Ther. 2000; 2(3):349–362.
135. Haupt, A, Berg, B, Paschen, P, et al. The effects of skin temperature and testing site on blood glucose measurements taken by a modern blood glucose monitoring device. Diabetes Technol Ther. 2005; 7:597–601.
136. Kanji, S, Buffie, J, Hutton, B, et al. Reliability of point-of-care testing for glucose measurement in critically ill adults. Crit Care Med. 2005; 33:2778–2785.
137. Rabiee, A, Andreasik, RN, Abu-Hamdah, R, et al. Numerical and clinical accuracy of a continuous glucose monitoring system during intravenous insulin therapy in the surgical and burn intensive care units. J Diabetes Sci Technol. 2009; 3:951–959.
138. Goldberg, PA, Siegel, MD, Russell, RR, et al. Experience with the continuous glucose monitoring system in a medical intensive care unit. Diabetes Technol Ther. 2004; 6:339–347.
139. Price, GC, Stevenson, K, Walsh, TS. Evaluation of a continuous glucose monitor in an unselected general intensive care population. Crit Care Resuscitation. 2008; 10:209–216.
140. Yamashita, K, Okabayashi, T, Yokoyama, T, et al. Accuracy and reliability of continuous blood glucose monitor in post-surgical patients. Acta Anaesthesiol Scand. 2009; 53:66–71.
141. Klonoff, DC, Buckingham, B, Christiansen, JS, et al. Continuous glucose monitoring: An Endocrine Society clinical practice guideline. J Clin Endocrinol Metab. 2011; 96(10):2968–2979.
142. Cheung, NW, Wong, VW, McLean, M. The Hyperglycemia: Intensive Insulin Infusion in Infarction (HI-5) study; a randomized controlled trial of insulin infusion therapy for myocardial infarction. Diabetes Care. 2006; 29:765–770.
143. Gandhi, GY, Nuttall, GA, Abel, MD, et al. Intensive intraoperative insulin therapy versus conventional glucose management during cardiac surgery: A randomized trial. Ann Intern Med. 2007; 146:233–243.
144. Dellinger, RP, Levy, MM, Rhodes, A, et al. Surviving sepsis campaign: International guidelines for management of severe sepsis and septic shock: 2012. Crit Care Med. 2013; 41(2):580–637.
145. Arinzon, Z, Shabat, S, Shuval, I, et al. Prevalence of diabetes mellitus in elderly patients received enteral nutrition longterm care service. Arch Gerontol Geriatr. 2008; 47:383–393.
146. Pancorbo-Hidalgo, PL, García-Fernandez, FP, Ramírez-Pérez, C. Complications associated with enteral nutrition by nasogastric tube in an internal medicine unit. J Clin Nurs. 2001; 10:482–490.
147. Rosmarin, DK, Wardlaw, GM, Mirtallo, J. Hyperglycemia associated with high, continuous infusion rates of total parenteral nutrition dextrose. Nutr Clin Pract. 1996; 11:151–156.
148. Strohmayer, EA, Krakoff, LR. Glucocorticoids and cardiovascular risk factors. Endocrinol Metab Clin North Am. 2011; 40(2):409–417.
149. Umpierrez, GE, Hellman, R, Korytkowski, MT, et al. Management of hyperglycemia in hospitalized patients in non-critical care setting: An Endocrine Society clinical practice guideline. J Clin Endocrinol Metab. 2012; 97(1):16–38.
150. Jakoby, MG, 4th., Nannapaneni, N. An insulin protocol for management of hyperglycemia in patients receiving parenteral nutrition is superior to ad hoc management. JPEN J Parenter Enteral Nutr. 2012; 36(2):183–188.
151. Siebenhofer, A, Plank, J, Berghold, A, et al. Short acting insulin analogues versus regular human insulin in patients with diabetes mellitus. Cochrane Database Syst Rev. (2):2006.
152. Umpierrez, GE, Smiley, D, Zisman, A, et al. Randomized study of basal-bolus insulin therapy in the inpatient management of patients with type 2 diabetes (RABBIT 2 trial). Diabetes Care. 2007; 30(9):2181–2186.
153. Gearhart, JG, Duncan, JL, 3rd., Replogle, WH, et al. Efficacy of sliding-scale insulin therapy: A comparison with prospective regimens. Fam Pract Res J. 1994; 14(4):313–322.
154. Umpierrez, GE, Palacio, A, Smiley, D. Sliding scale insulin use: Myth or insanity? Am J Med. 2007; 120(7):563–567.
155. Cryer, PE. Hypoglycaemia: The limiting factor in the glycaemic management of type I and type II diabetes. Diabetologica. 2002; 45(7):937–948.
156. D’Ancona, G, Bertuzzi, F, Sacchi, L, et al. Iatrogenic hypoglycemia secondary to tight glucose control is an independent determinant for mortality and cardiac morbidity. Eur J Cardiothorac Surg. 2011; 40(2):360–366.
157. Egi, M, Bellomo, R, Stachowski, E, et al. Blood glucose concentration and outcome of critical illness: The impact of diabetes. Crit Care Med. 2008; 36:2249–2255.
158. Cryer, PE. The prevention and correction of hypoglycemia. In: Jefferson LS, Cherrington AD, eds. Handbook of Physiology. Sec. 7, The Endocrine System. Vol. II, The Endocrine Pancreas and Regulation of Metabolism. New York: Oxford University Press; 2001:1057–1092.
159. Cryer, PE. Glucose homeostasis and hypoglycemia. In: Kronenberg HM, Melmed S, Polonsky KS, Larsen PR, eds. Williams Textbook of Endocrinology. 12th ed. Philadelphia: WB Saunders; 2008:1552–1577.
160. Cryer, PE. Mechanisms of sympathoadrenal failure and hypoglycemia in diabetes. J Clin Invest. 2006; 116:1470–1473.
161. Clarke, WL, Santiago, JV, Thomas, L. Adrenergic mechanisms in the recovery from hypoglycemia in man: Adrenergic blockade. Am J Physiol. 1979; 236:E147–E152.
162. Taborsky, GJ, Jr., Mundinger, TO. Minireview: The role of the autonomic nervous system in mediating the glucagon response to hypoglycemia. Endocrinology. 2012; 153(3):1055–1062.
163. Towler, DA, Havlin, CE, Craft, S, et al. Mechanism of awareness of hypoglycemia: Perception of neurogenic [predominantly cholinergic] rather than neuroglycopenic symptoms. Diabetes. 1993; 42:1791–1798.
164. Clark, DD, Sokoloff, L. Circulation and energy metabolism of the brain. In: Siegel G, Agranoff B, Albers RW, Molinoff P, eds. Basic Neurochemistry: Molecular, Cellular and Medical Aspects. 5th ed. New York: Raven Press; 1994:645–680.
165. Cryer, PE. The pathophysiology of hypoglycaemia in diabetes. Diabetes Nutr Metab Clin Exper. 2002; 15(5):330–333.
166. Auer, R, Hugh, J, Cosgrove, E, Curry, B. Neuropathologic findings in three cases of profound hypoglycemia. Clin Neuropathol. 1989; 8(2):63–68.
167. Cryer, PE. Hypoglycemia in Diabetes: Pathophysiology, Prevalence and Prevention. Alexandria, VA: American Diabetes Association; 2009.
168. Gonzalez, RR, Zweig, S, Rao, J, et al. Octreotide therapy for recurrent refractory hypoglycemia due to sulfonylurea in diabetes-related kidney failure. Endocr Pract. 2007; 13(4):417–423.
169. Dagogo-Jack, SE, Craft, S, Cryer, PE. Hypoglycemia-associated autonomic failure in insulin-dependent diabetes mellitus: Recent antecedent hypoglycemia reduces autonomic responses to, symptoms of, and defense against subsequent hypoglycemia. J Clin Invest. 1993; 91:819–828.
170. Jones, TW, Porter, P, Sherwin, RS, et al. Decreased epinephrine responses to hypoglycemia during sleep. N Engl J Med. 1998; 338:1657–1662.
171. Banarer, S, Cryer, PE. Sleep-related hypoglycemia-associated autonomic failure in type 1 diabetes: Reduced awakening from sleep during hypoglycemia. Diabetes. 2003; 52:1195–1203.
172. Ertl, AC, Davis, SN. Evidence for a vicious cycle of exercise and hypoglycemia in type 1 diabetes mellitus. Diabetes Metab Res Rev. 2004; 20:124–130.
173. Sandoval, DA, Guy, DL, Richardson, MA, et al. Effects of low and moderate antecedent exercise on counter regulatory responses to subsequent hypoglycemia in type 1 diabetes. Diabetes. 2004; 53:1798–1806.
174. Geddes, J, Schopman, JE, Zammitt, NN, Frier, BM. Prevalence of impaired awareness of hypoglycaemia in adults with type 1 diabetes. Diabetic Med. 2008; 25:501–504.
175. Cryer, PE. Hypoglycaemia: The limiting factor in the glycaemic management of the critically ill? Diabetologia. 2006; 49:1722–1725.
176. Dresler, CM, Fortner, JG, McDermott, K, Bajorunas, R. Metabolic consequences of (regional) total pancreatectomy. Ann Surg. 1991; 214:131–140.
177. The Diabetes Control and Complications Trial Research Group. The effect of intensive treatment of diabetes on the development and progression of long-term complications in insulin-dependent diabetes mellitus. N Engl J Med. 1993; 329(14):977–986.
178. McEuen, JA, Gardner, KP, Barnachea, DF, et al. Cultivating quality: An evidence-based protocol for managing hypoglycemia. Am J Nurs. 2010; 110(7):40–45.
179. Tomky, D. Detection, prevention, and treatment of hypoglycemia in the hospital. Diabetes Spectrum. 2005; 18(1):39–44.
180. van Noord, C, Cos, S, Wulkan, RW, et al. Protocolised inpatient care of diabetes mellitus. Neth J Med. 2012; 70(9):406–410.
181. Dougherty, PP, Klein-Schwartz, W. Octreotide’s role in the management of sulfonylurea-induced hypoglycemia. J Med Toxicol. 2010; 6(2):199–206.
182. Braithwaite, SS, Buie, MM, Thompson, CL, et al. Hospital hypoglycemia: Not only treatment but also prevention. Endocr Pract. 2004; 10(Suppl 2):89–99.
183. Mitrakou, A. Kidney: Its impact on glucose homeostasis and hormonal regulation. Diabetes Res Clin Pract. 2011; 93(Suppl 1):S66–S72.
184. Maitra, SR, Wojnar, MM, Lang, CH. Alterations in tissue glucose uptake during the hyperglycemic and hypoglycemic phases of sepsis. Shock. 2000; 13:379–385.
185. Hargrove, DM, Lang, CH, Bagby, GJ, et al. Epinephrine-induced increase in glucose turnover is diminished during sepsis. Metabolism. 1989; 38:1070–1076.
186. Wharton, B. Hypoglycaemia in children with kwashiorkor. Lancet. 1970; 1:171–173.
187. Service, FJ, McMahon, MM, O’Brien, PC, Ballard, DJ. Functioning insulinoma—Incidence, recurrence, and long-term survival of patients: A 60-year study. Mayo Clin Proc. 1991; 66:711–719.
188. Cryer, PE, Axelrod, L, Grossman, AB, et al. Evaluation and management of adult hypoglycemic disorders: An Endocrine Society Clinical Practice Guideline. J Clin Endocrinol Metab. 2009; 94(3):709–728.
189. Guettier, JM, Gorden, P. Hypoglycemia. Endocrinol Metab Clin North Am. 2006; 35(4):753–766.
190. Service, F. Diagnostic approach to adults with hypoglycemic disorders. Endocrinol Metab Clin. 1999; 28(3):519–532.
191. Service, F. Hypoglycemic disorders. N Engl J Med. 1995; 332:1144–1152.
192. Service, F. Classification of hypoglycemic disorders. Endocrinol Metab Clin North Am. 1999; 28(3):501–517.
193. Musey, VC, Lee, JK, Crawford, R, et al. Diabetes in urban African-Americans. I. Cessation of insulin therapy is the major precipitating cause of diabetic ketoacidosis. Diabetes Care. 1995; 18:483–489.
194. Polonsky, WH, Anderson, BJ, Lohrer, PA, et al. Insulin omission in women with IDDM. Diabetes Care. 1994; 17:1178–1185.
195. Weinert, LS, Scheffel, RH, Severo, MD, et al. Precipitating factors of diabetic ketoacidosis at a public hospital in a middle-income country. Diabetes Res Clin Pract. 2012; 96(1):29–34.
196. Randall, L, Begovic, J, Hudson, M, et al. Recurrent diabetic ketoacidosis in inner-city minority patients: Behavioral, socioeconomic, and psychosocial factors. Diabetes Care. 2011; 34(9):1891–1896.
197. Klingensmith, GJ, Tamborlane, WV, Wood, J, et al. Diabetic ketoacidosis at diabetes onset: Still an all too common threat in youth. J Pediatr. 2013; 162(2):330–334.
198. Eledrisi, MS, Alshanti, MS, Shah, MF, et al. Overview of the diagnosis and management of diabetic ketoacidosis. Am J Med Sci. 2006; 331(5):243–251.
199. Hamblin, PS, Topliss, DJ, Chosich, N, et al. Deaths associated with diabetic ketoacidosis and hyperosmolar coma. 1973-1988. Med J Aust. 1989; 151:439.
200. Alavi, IA, Sharma, BK, Pillay, VK. Steroid-induced diabetic ketoacidosis. Am J Med Sci. 1971; 262:15–23.
201. Wilson, DR, D’Souza, L, Sarkar, N, et al. New-onset diabetes and ketoacidosis with atypical antipsychotics. Schizophr Res. 2003; 59:1–6.
202. Toyonaga, T, Kondo, T, Miyamura, N, et al. Sudden onset of diabetes with ketoacidosis in a patient treated with FK506/tacrolimus. Diabetes Res Clin Pract. 2002; 56:13–18.
203. Tyler, J, Walsh, CH, Baddeley, RM, Down, RH. Diabetic ketoacidosis following glucagon therapy in acute pancreatitis. A case report. Ir Med J. 1977; 70:488–489.
204. Tibaldi, JM, Lorber, DL, Nerenberg, A. Diabetic ketoacidosis and insulin resistance with subcutaneous terbutaline infusion: A case report. Am J Obstet Gynecol. 1990; 163:509–510.
205. Isotani, H, Fujimura, Y, Furukawa, K, Morita, K. Diabetic ketoacidosis associated with the pheochromocytoma of youth. Diabetes Res Clin Pract. 1996; 34(1):57–60.
206. Cooppan, R, Kozak, GP. Hyperthyroidism and diabetes mellitus. An analysis of 70 patients. Arch Intern Med. 1980; 140:370–373.
207. Kopff, B, Mucha, S, Wolffenbuttel, BH, Drzewoski, J. Diabetic ketoacidosis in a patient with acromegaly. Med Sci Monit. 2001; 7:142–147.
208. Pasternak, DP. Hemochromatosis presenting as diabetic ketoacidosis with extreme hyperglycemia. West J Med. 1974; 120:244–246.
209. Nyenwe, EA, Loganathan, RS, Blum, S, et al. Active use of cocaine: An independent risk factor for recurrent diabetic ketoacidosis in a city hospital. Endocr Pract. 2007; 13(1):22–29.
210. Sypniewski, E, Jr., Mirtallo, JM, Schneider, PJ. Hyperosmolar, hyperglycemic, nonketotic coma in a patient receiving home total parenteral nutrient therapy. Clin Pharm. 1987; 6(1):69–73.
211. Mofredj, A, Howaizi, M, Grasset, D, et al. Diabetes mellitus during interferon therapy for chronic viral hepatitis. Dig Dis Sci. 2002; 47:1649–1654.
212. American Diabetes Association. Living with diabetes. Accessed at www.diabetes.org/living-with-diabetes/treatment-and-care/my-healthcare-team/when-youre-sick.html.
213. Nilsen, M, Sick day management of people with diabetes. Diabetic Lifestyle 2012; . http://www.diabeticlifestyle.com/everyday-life/sick-day-management-people-diabetes
214. Clement, S, Braithwaite, SS, Magee, MF, et al. Management of diabetes and hyperglycemia in hospitals. Diabetes Care. 2004; 27(2):553–591.