[level-membership-for-surgery-category]Chapter 49
Acute Deep Venous Thrombosis
Pathophysiology and Natural History
Jordan P. Knepper, Thomas W. Wakefield
The complications of acute venous thromboembolism (VTE), including deep venous thrombosis (DVT), pulmonary embolism (PE), and the postthrombotic syndrome, are the most common preventable cause of hospital death1 and a source of substantial long-term morbidity.2–4 The impact on health is so great, the Surgeon General of the United States issued a Call to Action to aid in combating VTE.5 An understanding of the epidemiology, pathophysiology, and natural history of VTE is essential in guiding prophylaxis, diagnosis, and treatment. In addition, recognizing underlying risk factors and the multifactorial nature of VTE may aid in the identification of situations likely to provoke thrombosis in both high-risk individuals and those with unexplained thromboembolism. Furthermore, understanding of the natural history of VTE is important to define the relative risks and benefits of anticoagulation as well as the duration of treatment in individual patients.
Intimate to the pathophysiology is the triad initially described by Rudolf Virchow (stasis, endothelial damage or injury, and hypercoagulability). However, an improved understanding of the coagulation and fibrinolytic systems as well as of the role of the vascular endothelium and the inflammatory response in thrombosis and hemostasis has led to the identification of new prethrombotic conditions and also a more thorough appreciation of previously recognized risk factors. It now appears that many venous thrombi arise from the convergence of several risk factors on a background of an imbalance between coagulation and fibrinolysis. Similar factors define the balance between recanalization of the venous lumen and recurrent thrombotic events that may be important determinants of long-term outcome after an episode of acute venous thrombosis.
Epidemiology
Incidence
The incidence of recurrent fatal and nonfatal VTE is estimated to exceed 900,000 cases annually in the United States alone.6 A 35-year population-based study using the Rochester Epidemiology Project database of Olmsted County, Minnesota, demonstrated an overall average age- and sex-adjusted annual VTE incidence of 122 per 100,000 person-years (DVT, 56 per 100,000; PE, 66 per 100,000).7 This study also demonstrated higher age-adjusted rates among men than among women (134 vs 115 per 100,000 respectively). First-time VTE is approximated to occur in about 250,000 U.S. white individuals annually.8 Compared with other racial populations, whites have a lower incidence of VTE than African Americans (104 vs 141 per 100,000) and a higher incidence of VTE than Hispanics and Asian/Pacific Islanders combined (104 vs 21 per 100,000).9 However, the problem of VTE is not just isolated to the United States; it is a global issue. The estimates of VTE across the European Union were 684,019 cases of DVT and 434,723 cases of PE, with 543,454 VTE-related fatalities.10
Populations Affected
Although estimated incidences of VTE in the community and hospital have been well published, those individuals who are at risk for DVT are less well studied. One large epidemiologic study addressed this issue using the Seventh American College of Chest Physicians Conference on Antithrombotic and Thrombolytic Therapy guidelines for VTE prevention.11 Of the estimated 38 million patient discharges in 2003, 31% were considered to be at risk for VTE because of a major surgery or a medical illness.12
Similarly, the incidence of VTE varies with the population studied, the use of thromboprophylaxis, the intensity of screening, and the accuracy of the diagnostic test employed. For example, individuals with acute spinal cord injury who were screened systematically with venography demonstrated DVT at a rate of 81%.13 However, medical-surgical intensive care unit (ICU) patients who received thromboprophylaxis had a DVT rate reported at 10% to 18% compared with a rate of 25% to 32% for those who were not given DVT prophylaxis.14 Interestingly, the risk of VTE in the critically ill is not limited to the time actually spent in the ICU. A single-center study noted that of the VTEs diagnosed in the critically ill, 64% were diagnosed with a VTE after discharge from the ICU; prolonged immobility after discharge from the ICU may have contributed to the high rate of DVT.15 Similarly, prolonged immobility contributes to increased rates of VTE in another population of patients, nursing home residents.16 In summary, it appears that medical-surgical ICU patients are at lower risk for DVT compared with acute spinal cord injury, trauma, or neurosurgery patients; at a comparable risk to that of patients who have had major orthopedic surgery; and at higher risk than that of medical-surgical ward patients.14,17 Furthermore, a more recent study noted a high (15.2%) rate of DVT in critically ill trauma patients within the first week that did not vary regardless of whether prophylaxis was used.18
Recurrence
VTE is a devastating acute disease process, but it may be manifested chronically. Nearly 30% of patients have a recurrence in a 10-year time span.19 Recurrences are more likely with the same event type as that of the incident event, that is to say, those who initially developed a PE are more likely to develop another PE instead of a DVT.20
Mortality
The severity of PE is shown in 30-year autopsy studies, which demonstrated a 26% incidence of PE in hospitalized patients, of which 9% were fatal. This translates to a 1% incidence of PE and a 0.36% incidence of death from PE in all hospitalized patients per year.21
Risk Factors
DVT occurring in the setting of a recognized risk factor is often defined as a secondary event; in the absence of risk factors, it is termed primary or idiopathic.22 Known risk factors for DVT are listed in Table 49-1.
Table 49-1
Risk Factors for Acute Deep Venous Thrombosis and Pulmonary Embolism
| Risk Factor | Odds Ratio | 95% Confidence Interval |
| Hospitalization | ||
| With recent surgery | 21.72 | 9.44-49.93 |
| Without recent surgery | 7.98 | 4.49-14.18 |
| Trauma | 12.69 | 4.06-39.66 |
| Malignant neoplasm | ||
| With chemotherapy | 6.53 | 2.11-20.23 |
| Without chemotherapy | 4.05 | 1.93-8.52 |
| Prior central venous catheter or pacemaker | 5.55 | 1.57-19.58 |
| Prior superficial vein thrombosis | 4.32 | 1.76-10.61 |
| Neurologic disease with extremity paresis | 3.04 | 1.25-7.38 |
| Varicose veins | ||
| Age 45 y | 4.19 | 1.56-11.30 |
| Age 60 y | 1.93 | 1.03-3.61 |
| Age 75 y | 0.88 | 0.55-1.43 |
| Congestive heart failure | ||
| Thromboembolism not categorized as a cause of death at postmortem examination | 9.64 | 2.44-38.10 |
| Thromboembolism categorized as a cause of death at postmortem examination | 1.36 | 0.69-2.68 |
Modified from Heit JA, et al: Risk factors for deep vein thrombosis and pulmonary embolism: a population-based case-control study. Arch Intern Med 160:809, 2000.
The high incidence of acute DVT in hospitalized patients, the availability of objective diagnostic tests, and the existence of clinical trials evaluating prophylactic measures have helped to identify high-risk groups in this population. In contrast, the risk factors for acute DVT are less well defined in population-based studies. Nonetheless, substantive differences have been noted in the distribution of risk factors between inpatients and outpatients.23 Malignant disease, surgery, and trauma within the previous 3 months remain significant risk factors for outpatient thrombosis, whereas the frequency of surgery and malignant disease is higher among inpatients with DVT.24,25 Approximately 47% of outpatients with a documented DVT have one or more recognized risk factors,24 and the incidence of VTE increases with the number of risk factors.26 Although some investigators have found the risk of acute DVT to be significantly higher only for those with three or more risk factors,25 others have reported the relative risk to increase from 2.4 in those with one risk factor to more than 20 in patients who have three or more risk factors.27 Heit et al28,29 identified several risk factors for VTE, including increasing age, male gender, surgery, trauma, hospital or nursing home confinement, malignant disease, neurologic disease with extremity paresis, central venous catheter or transvenous pacemaker, prior superficial vein thrombosis, and varicose veins. Among women, additional risk factors include pregnancy, oral contraceptives, and hormone replacement therapy. Independent predictors for recurrent DVT, which occurs in 30% of people within 10 years, include increasing age, obesity, malignant neoplasm, and extremity paresis.
Age
VTE occurs in all ages, although a higher incidence has consistently been associated with advanced age. Some investigators have identified cutoff points at which the risk of DVT is raised, whereas others have reported a relative increase with advancing age. In a community-based study of phlebographically documented DVT, the yearly incidence of DVT was noted to increase progressively from almost 0 in childhood to 7.65 cases per 1000 in men and 8.22 cases per 1000 in women older than 80 years.30 The incidence of DVT increased 30-fold from those aged 30 years to those older than 80 years. Rosendaal27 similarly noted an incidence of 0.006 per 1000 children younger than 14 years, which rose to 0.7 per 1000 among adults 40 to 54 years old. Furthermore, Hansson et al31 found the prevalence of objectively documented thromboembolic events among men to increase from 0.5% at the age of 50 years to 3.8% at the age of 80 years.
The influence of age on the incidence of VTE is likely to be multifactorial. The number of thrombotic risk factors increases with age, three or more risk factors being present in only 3% of hospitalized patients younger than 40 years but in 30% of those 40 years and older.26 Interestingly, it also appears that the number of risk factors required to precipitate thrombosis decreases with age.27 This may be related to an acquired prethrombotic state associated with aging as higher levels of thrombin activation markers are found among older people.32 Advanced age also has been associated with anatomic changes in the soleal veins and more pronounced stasis in the venous valve pockets.33,34 We have also noted biologic changes with aging in our research laboratory, such as elevations in P-selectin, tissue factor, and procoagulant microparticles, supporting the effect of age on venous thrombogenesis.
Venous diseases, including VTE, are usually regarded as rare in young children,3 with an incidence of 0.006 per 1000 children younger than 14 years,27 whereas the incidence in hospitalized children younger than 18 years has been estimated to be 0.05%.35 Early mobilization and discharge may partially explain the lower incidence in children.35 However, the diagnosis is often not considered in pediatric patients, and few studies have systematically evaluated children for DVT. Over time, the number of recognized cases in children hospitalized has increased from 0.3 to 28.8 per 10,000 from 1992 to 2005.36
VTE in children is almost always associated with recognized thrombotic risk factors,27,37–39 and multiple risk factors are often required to precipitate thrombosis.27 Venous thrombosis is more common in those children hospitalized in the ICU, those with spinal cord injuries, and those undergoing prolonged orthopedic immobilization, although the incidence is substantially lower than in corresponding adult populations. DVT may occur in as many as 3.7% of pediatric patients immobilized in halo-femoral traction for preoperative treatment of scoliosis,40 4% of children hospitalized in the ICU,41 and 10% of children with spinal cord injuries.42,43 Symptomatic postoperative DVT is regarded as unusual in children, although there are few data from studies employing routine surveillance,44 and autopsy-identified PE is approximately four times more frequent in pediatric patients who have undergone surgery than in the general pediatric medical population.37 Other thrombotic risk factors in hospitalized children are local infection and trauma, immobilization,39 inherited hypercoagulable states,27 oral contraceptive use,45 lower limb paresis,40 and use of femoral venous catheters.46 In addition, the risk factors for inpatient DVT in children include catheters and concomitant severe respiratory, oncologic, and infectious diseases; outpatient DVT is often associated with a prior DVT and thrombophilia.36
Immobilization
Many of the clinical presentations of thromboembolism support immobilization as a risk factor for VTE. In addition, stasis in the soleal veins and behind the valve cusps is exacerbated by advancing age and inactivity of the calf muscle pump,34 both of which are associated with an increased risk of DVT. The prevalence of lower extremity DVT in autopsy studies also parallels the duration of bed rest, with an increase during the first 3 days of confinement and a rapid rise to high levels after 2 weeks. DVT was found in 15% of patients dying after 0 to 7 days of bed rest, in comparison with 79% to 94% of those dying after 2 to 12 weeks.33 Preoperative immobilization is also associated with a twofold increase in risk of postoperative DVT,47 and DVT among stroke patients is significantly more common in paralyzed or paretic extremities (53% of limbs) than in nonparalyzed limbs (7%).48 Patients with neurologic disease and extremity paresis or paralysis have a threefold higher risk for DVT and PE that appears to be independent of hospital confinement.28
Travel
Immobilization as a thrombotic risk factor extends to include prolonged travel, particularly the “economy class syndrome,” which occurs in people who have sat in a cramped position during extended aircraft flights.49 Several case series have reported the occurrence of PE in relation to extended travel,49–54 although none has rigorously examined the prevalence relative to that of the general population, and few have thoroughly reported the presence of other risk factors. A high prevalence of preexisting venous disease and other thrombotic risk factors in this group of patients has sometimes been noted.32,54 The question of prolonged travel as a risk factor is moderated by observations that extreme duration of venous stasis alone may fail to produce thrombosis55 and that no consistent rheologic or prothrombotic changes have been demonstrated during prolonged travel.32,56 However, PE is the second leading cause of travel-related death, accounting for 18% of 61 deaths, suggesting that a relationship cannot be excluded.53 After a consensus meeting, the World Health Organization published the following conclusions57:
• An association probably exists between air travel and venous thrombosis.
• Similar links may exist for other forms of travel.
• The available evidence does not permit an estimation of actual risk.
More evidence of a connection between travel and DVT and PE has accumulated. In a case-control study, Ferrari et al58 found that long-distance travel increased the risk of DVT with an odds ratio (OR) of 4.0, and Samama59 made similar observations (OR, 2.3). Scurr et al60 found a 10% risk of calf DVT in patients who traveled without compression stockings. Lapostolle et al61 observed that during an 86-month period, 56 of 135.3 million airline passengers had severe PE. The frequency among those who traveled more than 5000 km was 150 times as high as the frequency among those who traveled less than 5000 km. In another case-control study, Paganin et al62 observed a high incidence of VTE in patients with risk factors for DVT who traveled long distances. In particular, history of previous VTE (OR, 63.3), recent trauma (OR, 13.6), presence of varicose veins (OR, 10), obesity (OR, 9.6), immobility during flight (OR, 9.3), and cardiac disease (OR, 8.9) increased the risk of DVT. These investigators concluded that low mobility during flight was a striking modifiable risk factor for development of PE and that travelers with risk factors should increase their mobility.63
Hughes et al64 noted that the frequency of VTE is increased even in travelers otherwise at low to moderate risk. In a prospective study that involved 878 long-distance air travelers, the frequency of VTE was 1.0% (9 of 878; 95% confidence interval, 0.5-1.9), which included 4 cases of PE and 5 of DVT. Two of the patients traveled in business class, five used aspirin, and four wore compression stockings.
History of Venous Thromboembolism
Approximately 23% to 26% of patients presenting with acute DVT have a previous history of thrombosis,23,30 and histologic studies confirm that acute thrombi are often associated with fibrous remnants of previous thrombi in the same or nearby veins.65 Depending on sex and age, population-based studies have demonstrated that recurrent thromboembolism occurs in 2% to 9% of people with a previous episode of thromboembolism.3
The risk of recurrent thromboembolism is higher among patients with idiopathic DVT.66 In addition, primary hypercoagulability appears to have a significant role in many recurrences. Simioni et al66 reported the cumulative incidence of recurrent thrombosis among patients who are heterozygous for the factor V Leiden mutation to be 40% at 8 years of follow-up, 2.4-fold higher than in patients without the mutation, although the importance of heterozygous factor V Leiden to recurrent thrombosis has been questioned.67 den Heijer et al68 estimated that 17% of recurrent thromboembolic events may be due to hyperhomocysteinemia. A similar relationship between impaired fibrinolysis and recurrent DVT has been suggested by several investigators, although the methodologic validity of these findings has been questioned.69
Malignant Disease
Approximately 20% of all first-time VTE events are associated with malignant disease.70 An estimated 1 in 200 individuals with malignant disease will develop either DVT or PE, a fourfold higher risk than in those without malignant disease.28 Considering all-cause mortality of in-hospital death for cancer patients, one in seven will die of PE.71 Unfortunately, the discovery of an occult malignant neoplasm associated with an otherwise first-time idiopathic VTE is as high as 12% to 17% in some series.72,73 In another 5% to 11% of patients, malignant disease appears within 1 to 2 years of presentation for DVT.72–74 Related to this, several series have documented a significantly higher risk of malignant disease in patients with idiopathic DVT.72,75 Among such patients, 7.6% have been noted to have a malignant neoplasm during follow-up, with an odds ratio of 2.3 in comparison with those with secondary thrombosis.73 The incidence of occult malignant disease diagnosed within 6 to 12 months of an idiopathic DVT is 2.2 to 5.3 times higher than that expected from general population estimates.76,77 Furthermore, in individuals with recurrent idiopathic DVT, a malignant neoplasm can be identified 17% of the time.73 A correlation with location of tumor reveals that the highest rates of VTE are associated with pancreatic malignant neoplasms, followed by kidney, ovary, lung, and stomach malignant neoplasms.78
The underlying mechanisms contributing to the hypercoagulable state in malignant disease have been well studied and are multifactorial. Venous compression secondary to tumor growth, cancer-associated thrombocytosis, immobility, indwelling central lines, and chemotherapy or radiation therapy are risk factors that increase the possibility of VTE.79 However, the systemic prothrombotic response seen in malignant disease is mediated by cytokines, inhibitors of fibrinolysis, and procoagulants.80 Tumor cells can directly initiate hemostasis through constitutive expression of tissue factor. Tissue factor is not normally expressed on resting vascular endothelium but rather induced by chemical mediators during times of inflammation or vessel damage to bind factors VII and VIIa. This complex of tissue factor and factor VII activates factors X and XI through proteolysis, leading to the generation of thrombin.81 Another mediator in VTE associated with malignant disease is cancer procoagulant. The role of cancer procoagulant in coagulation is limited to its association with malignant disease as it has not been identified in normal healthy tissue. Cancer procoagulant serves as a direct activator of factor X independent of the presence of factor VIIa. Although cancer procoagulant has not been studied as intensely as tissue factor, the characterization of cancer procoagulant and hypercoagulability in acute myelogenous leukemia has been well published.82,83 The platelet adhesion molecules glycoprotein Ib and glycoprotein IIb/IIIa have also been identified on tumor cells. Such expression allows platelet activation and aggregation.84
The prothrombotic cytokines, such as vascular endothelial growth factor, tumor necrosis factor-α, and interleukin-1, mediate their actions through induction of tissue factor on vascular endothelium, monocytes, and leukocytes.81,82 In addition, interleukin-1 and tumor necrosis factor-α downregulate the expression of thrombomodulin, the receptor for thrombin, on the endothelial surface. This results in a decrease in thrombin-thrombomodulin complexes, the activating complex of protein C, a natural anticoagulant.81,82 Furthermore, interleukin-1 and tumor necrosis factor-α stimulate the production of plasminogen activator inhibitor-1 (PAI-1), the main physiologic inhibitor of fibrinolysis.85
As many as 90% of patients with cancer have abnormal coagulation parameters, including increased levels of coagulation factors, elevated fibrinogen or fibrin degradation products, and thrombocytosis.86 Elevated fibrinogen and thrombocytosis are the most common abnormalities, perhaps reflecting an overcompensated form of intravascular coagulation.86 Levels of the coagulation inhibitors antithrombin, protein C, and protein S also may be reduced in malignant disease.87
Markers of activated coagulation are elevated in the majority of patients with solid tumors and leukemia.86 Fibrinopeptide A levels reflect tumor activity, decreasing or increasing in response to treatment or progression of disease, suggesting that tumor growth and thrombin generation are intimately related.86 Furthermore, these levels may fail to normalize after administration of heparin to patients with cancer and DVT, perhaps explaining why these DVTs may be refractory to anticoagulants.86
VTE is also associated with the treatment of some cancers. DVT complicates 29% of general surgical procedures for malignant disease.87 Preoperative activation of the coagulation system, as reflected by elevated thrombin-antithrombin complex values, is associated with a 7.5-fold increased risk of postoperative DVT.86 Some chemotherapeutic regimens also predispose to DVT, and thrombotic complications may be as common as the more widely recognized infectious complications.88 VTE has been reported in up to 6% of patients undergoing treatment for non-Hodgkin’s lymphoma and in 17.5% of those receiving therapy for breast cancer.89,90 Among patients with stage II breast cancer, thrombosis was significantly more common in those randomly assigned to 36 weeks of chemotherapy (8.8%) than in those receiving only 12 weeks of treatment (4.9%).88 Potential thrombogenic mechanisms associated with chemotherapy include direct endothelial toxicity, induction of a hypercoagulable state, reduced fibrinolytic activity, tumor cell lysis, and use of central venous catheters.89,90 Some intravenous chemotherapeutic agents are associated with activation of coagulation and increased markers of thrombin generation, a response that is blocked by pretreatment with heparin.91
Surgery
The high incidence of postoperative DVT as well as the availability of easily repeatable, noninvasive diagnostic tests has allowed a greater understanding of the risk factors associated with surgery than in other conditions. Surgery constitutes a spectrum of risk that is influenced by age of the patient, coexistent thrombotic risk factors, type of procedure, extent of surgical trauma, length of procedure, and duration of postoperative immobilization.92 The type of surgical procedure is particularly important.47 The overall incidence of DVT is approximately 19% in patients undergoing general surgical operations and 24% in those having elective neurosurgical procedures; among those undergoing surgery for hip fracture, hip arthroplasty, and knee arthroplasty, it is 48%, 51%, and 61%, respectively.92 On the basis of these data, patients can be classified as being at low, moderate, or high risk for thromboembolic complications, as shown in Table 49-2.
Table 49-2
Risk of Postoperative Deep Venous Thrombosis
| Category | Characteristics |
| Low | Age <40 y, no other risk factors, uncomplicated abdominal/thoracic surgery |
| Age >40 y, no other risk factors, minor elective abdominal/thoracic surgery <30 min | |
| Moderate | Age >40 y, abdominal/thoracic surgery >30 min |
| High | History of recent thromboembolism |
| Abdominal or pelvic procedure for malignancy | |
| Major lower extremity orthopedic procedure |
From Hull RD, et al: Prophylaxis of venous thromboembolism. Chest 89(Suppl):374S, 1986.
Approximately half of postoperative lower extremity thrombi develop in the operating room; the remainder occur during the next 3 to 5 days.93 However, the risk for development of DVT does not end uniformly at hospital discharge. In one study, 51% of the thromboembolic events occurred after discharge from gynecologic surgical procedures.94 Similarly, up to 25% of patients undergoing abdominal surgery have DVT within 6 weeks of discharge.95 Heit et al94 found a nearly 22-fold higher risk of DVT and PE among patients who were hospitalized after previous surgery.
All components of Virchow’s triad may be present in the surgical patient—perioperative immobilization, transient changes in coagulation and fibrinolysis, and potential for gross venous injury. Immobilization is associated with a reduction in venous outflow and capacitance during the early postoperative period.96 Surgery is also accompanied by a transient, low-level hypercoagulable state, presumably mediated by the release of tissue factor, which is marked by a rise in thrombin activation markers shortly after the procedure begins.96 The thrombogenic potential of different surgical procedures appears to differ, with greater rises in thrombin activation markers during hip arthroplasty than after laparotomy.97 Increased levels of PAI-1 are also associated with a decrease in fibrinolytic activity on the first postoperative day, the “postoperative fibrinolytic shutdown.”98 This relationship between impaired fibrinolysis and postoperative DVT may be particularly important69; preoperative and early postoperative elevations in PAI-1 correlate with the development of thrombosis in orthopedic patients.98
Trauma
The prevalence of DVT among autopsied trauma casualties has been reported to be as high as 65%, comparable to the 58% incidence among injured patients in modern venographic series.13 Substantially lower DVT rates, ranging from 4% to 20%,99 have been noted in series employing duplex ultrasonography, although many patients studied were receiving prophylaxis and the limitations of ultrasound in screening of asymptomatic patients are well recognized. Recent trauma was associated with a nearly 13-fold increase in risk.94
Although the risk of DVT may be less than 20% in some injured patients,13 certain subgroups are at particularly high risk. Age (OR, 1.05 for each 1-year increment), blood transfusion (OR, 1.74), surgery (OR, 2.30), fracture of the femur or tibia (OR, 4.82), and spinal cord injury (OR, 8.59) have been significantly associated with the development of DVT in this population.13 Other reported risk factors are a hospital stay longer than 7 days,100 increased Injury Severity Score,101,102 Trauma Injury Severity Score of 85 or less,100 pelvic fractures,103 major venous injury,104 presence of femoral venous lines,105 duration of immobilization,106 and prolongation of the partial thromboplastin time.107
As with postoperative DVT, several pathophysiologic elements may be responsible for the high incidence of DVT in trauma patients. Immobilization by skeletal fixation, paralysis, and critical illness are associated with venous stasis, whereas mechanical injury is important after direct venous trauma and central venous cannulation. Less well appreciated is the hypercoagulable state after depletion of coagulation inhibitors and components of the fibrinolytic system. Fibrinopeptide A levels rise after injury,108 consistent with activation of coagulation, whereas fibrinolytic activity has been found to increase initially and then to decrease.109,110
Inherited Thrombophilia
Prothrombin Gene Mutation.
The prothrombin G20210A gene mutation was demonstrated when 28 families with a documented history of VTE were noted to have an 18% incidence of a transition from guanine to adenine at the 20210 nucleotide on the prothrombin gene. A comparison to healthy controls revealed only a 1% incidence of the same mutation.111 In this same study, Poort also reported that in 87% of individuals with such a mutation, the prothrombin levels were greater than 115% of normal. Thus, elevated levels of prothrombin, the precursor for thrombin, are associated with an increased risk of VTE. A large multicenter study revealed the incidence of this mutation in the general European population to be 0.7% to 4%.112 This mutation is rare in individuals of Asian or African descent. Additional investigations have shown that in those with a spontaneous DVT, the incidence of the mutation may range from 7% to 16%, depending on the patient population studied.113 The risk of VTE in the presence of the prothrombin G20210A mutation is threefold higher in heterozygotes than in those with the wild type, and the presence of homozygosity further increases this risk. The prothrombin G20210A gene mutation is frequently coinherited with factor V Leiden mutation in approximately 1% to 10% of symptomatic carriers.114,115
Factor V Leiden Mutation.
Resistance to activated protein C, characterized by the failure of exogenous activated protein C to prolong the activated partial thromboplastin time, was initially described by Dahlbäck in 1993.116 Subsequent studies from the Leiden University hospital in the Netherlands revealed this laboratory phenotype to be the result of a mutation in the gene for factor V of the coagulation cascade in 95% of instances. The transition mutation causes replacement of arginine with glutamine at position 506, such that factor Va is resistant to cleavage by activated protein C and is therefore inactivated at a decreased rate.117 Thus, more factor Va is available to participate in the generation of thrombin. In addition, after cleavage by activated protein C, the degradation product of factor V in combination with protein S acts as a cofactor for activated protein C, increasing the degradation of factor VIIIa as well as additional factor Va.118 The factor V Leiden mutation is inherited in an autosomal dominant manner and is 10 times more common than other heritable defects.119 The factor V Leiden mutation is present in 5% to 8% of European whites, with prevalence of up to 15% in some areas of Greece, Sweden, and Lebanon.120,121 In a study of 4047 Americans, the carrier frequencies of the mutation were 5.3% for whites, 2.2% for Hispanic Americans, 1.25% for Native Americans, 1.2% for African Americans, and 0.45% for Asian Americans. Carrier frequencies were similar in white men and women in this study.122 Factor V Leiden mutation has been reported in 20% to 50% of first-time VTE events; thus it is the most common cause of inherited thrombophilias.123 The relative risk of DVT is increased sevenfold in those who are heterozygous for the mutation. However, in individuals who are homozygous for the mutation, the relative risk is increased 80-fold. In addition, homozygous patients develop DVT at a younger age than do those who are heterozygous.124
The etiology of activated protein C resistance is not limited to factor V Leiden mutations. Increased plasma levels of factor VIII, the presence of antiphospholipid antibodies, older age, pregnancy, and the use of estrogens are well-known causes of increased activated protein C resistance.125
Protein C.
Protein C is a vitamin K–dependent serine protease that inhibits the coagulation system by inactivating factors Va and VIIIa, the two cofactors necessary for activation of factor X and thrombin generation.126 Once it is activated by the thrombin-thrombomodulin complex, activated protein C can be enhanced by protein S.127,128 Either an acquired or inherited reduction of protein C activity increases thrombotic risk. Heterozygous protein C deficiency is inherited in an autosomal dominant fashion; this deficiency may be classified as type I, characterized by a reduction in both antigenic and functional levels, or as type II, in which antigen levels are normal and functional activity is decreased.126 In a study of 10,000 healthy blood donors, the observed prevalence of inherited protein C deficiency was 1.45 per 1000.129 Furthermore, a study of 2132 patients with VTE found that 3.2% had protein C deficiency.130 It is estimated that individuals who are heterozygous for protein C deficiency have a sevenfold higher risk for development of VTE over baseline.131 Other than VTE, protein C deficiency is also associated with neonatal purpura fulminans and warfarin skin necrosis.
Neonatal purpura fulminans can result from either a homozygous or a double heterozygous genotype. It is characterized by microvascular thrombosis in the dermis, leading to severe ecchymoses followed by perivascular hemorrhage, extensive venous and arterial thrombosis, necrosis, laboratory findings consistent with disseminated intravascular coagulation, and extremely low levels of protein C.132 Warfarin-induced skin necrosis may occur in individuals with protein C deficiency during the first several days of warfarin therapy. Warfarin initiation can reduce protein C anticoagulant activity to 50% within 1 day.133 With the exception of factor VII, other procoagulant vitamin K–dependant clotting factors decline at a slower rate, inducing a transient hypercoagulable state. The clinical manifestations of warfarin-induced skin necrosis are similar to those seen in neonatal purpura fulminans.134 Acquired deficiency of protein C may also be seen in acute thrombotic events, liver disease, renal disease, disseminated intravascular coagulation, hemolytic-uremic syndrome, chemotherapy, thrombotic thrombocytopenic purpura, and acute infections.135
Protein S.
Protein S is a vitamin K–dependent cofactor for the protein C–mediated inactivation of factors Va and VIIIa. In addition, protein S can directly inhibit the clotting pathway through interactions with factors Va and Xa. Protein S is synthesized by hepatocytes and megakaryocytes and circulates bound to C4b-binding protein or as a free form; only the free form is able to interact with protein C.136 Protein S deficiency is autosomal dominant and is more common than protein C deficiency. The incidence of protein S–associated VTE is 5% to 7%; the prevalence in the general population is 0.13%.137–138 It is estimated that individuals who are heterozygous for protein S deficiency have an 8.5-fold higher relative risk for development of VTE.139
There are more than 130 types of protein S mutations, which can fall into three general categories. Type I deficiency is quantitative, type II deficiency is qualitative, and type III deficiency is caused by mutations that increase the affinity of protein S for C4b-binding protein.125 The clinical presentation of patients with heterozygous protein S deficiency is similar to that of patients with protein C deficiency: an increased incidence of VTE and warfarin-induced skin necrosis.140 Homozygous individuals are rare but will present with neonatal purpura fulminans. Acquired protein S deficiency has been reported in disseminated intravascular coagulation, diabetes mellitus, pregnancy, oral contraceptive use, nephritic syndrome, liver disease, and essential thrombocythemia.126
Antithrombin.
Antithrombin, a vitamin K–independent glycoprotein, inhibits thrombin as well as factors Xa, IXa, XIa, and XIIa. The action of antithrombin is accelerated by heparin.141 Antithrombin deficiency is inherited in an autosomal dominant manner. The majority of patients are heterozygous; homozygotes usually die in utero. Type I deficiency is a quantitative defect, whereas type II is a qualitative deficiency.125 The incidence of antithrombin deficiency–associated VTE has been reported at 0.5% to 3%, with a prevalence in the general population of 0.2%.130,141 However, it is estimated that heterozygotes for this deficiency have a 20-fold higher risk for development of VTE over that of the general population.142 The clinical presentations include thrombosis of the iliofemoral veins, upper limb deep veins, mesenteric veins, vena cava, renal veins, and retinal veins as well as cerebral venous thrombosis and Budd-Chiari syndrome.125,143
Hyperhomocysteinemia.
High plasma levels of homocysteine have been associated with an increased risk for both venous and arterial thrombosis.144 The metabolic breakdown of homocysteine involves N5-methyltetrahydrofolate reductase (MTHFR). Individuals who are homozygous for a substitution of cytosine by thymine on the MTHFR gene at position 677 (C677T) and have inadequate dietary intake may achieve moderately increased homocysteine levels.145 Despite this association, data supporting the MTHFR C677T mutation as an independent risk factor for VTE are lacking, but a new study is helping to provide mounting evidence for the importance of hyperhomocysteinemia in VTE.146
Other Inherited Thrombophilias.
Other inherited thrombophilias, such as dysfibrinogenemia and elevated clotting factors, are less well studied. Inherited defects of fibrinogen are detected in less than 1% of those presenting with first-time VTE.147 In such defects, fibrinogen may be unable to bind thrombin, or the fibrin itself is unable to be lysed by plasmin. Clinically, the majority of these patients are asymptomatic, yet 25% may present with bleeding and an additional 20% with thrombosis.148 Elevated levels of factors VIII, IX, and XI have been demonstrated to be an independent risk factor for VTE.149–151 Individuals with elevations of factors IX and XI above the 90th percentile in healthy controls have an odds ratio of 2.3 and 2.2, respectively, for DVT compared with those with levels below the 90th percentile.149,150 Elevated factor VIII levels have been reported in up to 25% of individuals presenting with VTE.152
Others have noted residual risk for thrombosis not explained by known inherited thrombophilic disorders. This additional risk is indirect evidence that more inherited thrombophilias are yet to be identified.153
Pregnancy
The incidence of VTE in the pregnant population is 6 to 10 times greater than in matched controls,154 leading to approximately 10% of all maternal deaths.155 By clinical evaluation, VTE has been reported at a rate of 1.3% to 7% during pregnancy and 6.1% to 23% in the postpartum period.156 However, studies that employed venography, Doppler ultrasonography, or ventilation-perfusion scans for evaluation of clinically suspected thromboembolism have suggested an incidence of 0.029% to 0.055% in this population.157 The risk of thrombosis appears to be two to three times greater during the puerperium, with the highest incidence found after cesarean section.158 Interestingly, when VTE has been objectively documented, the occurrence of thrombosis is equally distributed throughout all trimesters.159
DVT in pregnancy has been attributed to impaired venous outflow due to uterine compression, and 97% of reported thromboses have been isolated to the left leg.159 Furthermore, pregnancy is associated with a transient hypercoagulable state due to increases in the levels of fibrinogen, von Willebrand factor, and factors II, VII, VIII, and X. Compounding this, acquired functional resistance to activated protein C is also seen during pregnancy.159,160 Similarly, protein S levels are decreased by 50% to 60% early during pregnancy, with free protein S levels comparable to those in hereditary heterozygous protein S deficiency.161,162 The fibrinolytic system is also altered in pregnancy; levels of tissue plasminogen activator are decreased, and plasminogen activator inhibitors 1 and 2 are increased.163
Both retrospective and prospective studies have demonstrated that between 30% and 50% of women with a pregnancy-associated VTE also have an inherited thrombophilia. This high incidence has led to the recommendation of screening for thrombophilia in those pregnant patients with a personal or family history of VTE.164,165 The risk of puerperal DVT also increases with maternal age, suppression of lactation, hypertension, and assisted delivery but not with the number of pregnancies.166
Oral Contraceptives and Hormonal Therapy
As suggested by case reports in the early 1960s, further studies have now established the use of oral contraceptives as an independent risk factor for the development of DVT. These studies noted odds ratios of 3.8 to 11.0 for thrombosis167; an unweighted summary relative risk among 18 controlled studies was 2.9.168 Approximately one quarter of idiopathic thromboembolic events among women of childbearing age have been attributed to oral contraceptives. The risk of hospital admission for a thromboembolic event, including cerebral thrombosis, has been estimated to be 0.4 to 0.6 per 1000 for oral contraceptive users, compared with 0.05 to 0.06 for nonusers.169 Early studies also suggested that thromboembolism is responsible for approximately 2% of deaths in young women, with contraceptive-associated mortality rates of 1.3 and 3.4 per 100,000 among women aged 20 to 34 years and 35 to 44 years, respectively.170 The increased risk of thromboembolism appears to diminish soon after oral contraceptives are discontinued and is independent of the duration of use.171
Risk is correspondingly higher when oral contraceptive use is combined with other factors, such as surgery172 and inherited inhibitor deficiencies.173 The factor V Leiden mutation may be particularly important in this regard; resistance to activated protein C has been reported in up to 30% of patients with contraceptive-associated thromboembolism.174 The use of third-generation oral contraceptives may act synergistically with the factor V Leiden mutation, raising thromboembolic risk 30-fold to 50-fold.175–177
The risk of VTE among users of third-generation oral contraceptives may be up to eight times that of young women who do not use oral contraceptives. However, the higher risk associated with third-generation progestins has been questioned on the basis of the possible confounding effects of age and other prescribing biases.178
Estrogenic compounds also increase the risk of VTE when they are used for lactation suppression,166 in treatment of carcinoma of the prostate, and as postmenopausal replacement therapy.179 Although estrogen doses used for postmenopausal replacement therapy are approximately one sixth those in oral contraceptives, some data support an increased thromboembolic risk at these doses as well. Several studies show a twofold to fourfold higher risk of VTE among women taking hormone replacement therapy.179–183 This increased risk is greatest during the first year of treatment.180,183 However, given the relative infrequency of thromboembolism, this risk represents only one or two additional cases of thromboembolism per year in every 10,000 women in this age group.
Estrogen in pharmacologic doses is associated with alterations in the coagulation system that may contribute to this thrombotic tendency. Such alterations include decreases in PAI-1184 and increases in blood viscosity, fibrinogen, plasma levels of factors VII and X, and platelet adhesion and aggregation.185,186 An associated prothrombotic state is implied by rises in markers of activated coagulation occurring in conjunction with elevations of circulating factor VIIa and decreases in antithrombin and protein S inhibitor activity.185,187 The extent to which antithrombin and protein S are depressed is significantly less with lower estrogen preparations.188
Blood Group
There also appears to be a relationship between VTE risk and the ABO blood groups, with a higher prevalence of blood type A and correspondingly lower prevalence of blood type O groups.189,190 In reviewing the literature, Mourant et al191 found the relative incidence of type A to be 1.41 times higher among patients with thromboembolism than among controls. The effect of blood type was greater in young women who were taking oral contraceptives or were pregnant; the relative incidence of type A among patients with thromboembolism was 3.12 in those taking oral contraceptives and 1.85 in those who were pregnant. A relationship between soluble endothelial cell markers and ABO blood group is known to exist, with significantly lower levels of von Willebrand factor among those with type O blood.192
Geography and Ethnicity
There are also geographic differences in the frequency of VTE; the incidence of postoperative DVT in Europe has been noted to be nearly twice that in North America.87,92 Higher rates of thromboembolism have also been noted in the central United States compared with either coast.193 Autopsy series suggest that although the prevalence of thromboembolism is identical among American black and white patients, it is significantly higher than in a matched Ugandan black population.194 A similar autopsy series noted the prevalence of thromboembolism to be 40.6% in Boston and 13.9% in Kyushu, Japan.195
Unfortunately, regional variations in underlying medical and surgical conditions as well as in prophylactic measures and diagnostic methods may confound any apparent differences in the incidence of thromboembolism among different ethnic and geographic groups. Nevertheless, it is certainly conceivable that differences between ethnic groups might arise from either genetic or environmental factors. Such differences seem likely on the basis of recognized geographic differences in the spectrum of mutations leading to congenital anticoagulant deficiencies.196 Such theoretical concerns are also supported by geographic variability in the incidence of the factor V Leiden mutation. The factor V Leiden allele has a prevalence of 4.4% in Europeans, corresponding to a carrier rate of 8.8%, but the allele has not been identified in Southeast Asian or African populations.121
Central Venous Catheters
The use of central venous cannulation for hemodynamic monitoring, infusion catheters, and pacemakers has been associated with a rising frequency of DVT. This is particularly true for upper extremity thrombi, as many as 65% of which are related to central venous cannulation.197 Although the incidence of symptomatic thrombosis may be low, studies employing objective surveillance have reported thrombosis to occur with a mean incidence of 28% after subclavian cannulation.198 This risk also extends to femoral venous catheters; ipsilateral thrombosis develops in 12% of patients undergoing placement of large-bore catheters for trauma resuscitation.199
Experimental studies in rats suggest that the catheter itself, associated vascular injury, and stasis contribute to the development of early perigraft thrombus in virtually all cases.200 Catheter material appears to be an important determinant of thromboembolism201; polytetrafluoroethylene (Teflon) and heparin-bonded catheters are associated with thrombosis in less than 10% of cases.202 Other determinants of thrombosis are catheter diameter, number of venipuncture attempts, duration of catheter placement, and composition of the infusate.198 Heit et al203 found that current or recent placement of a central venous catheter and previous placement of a transvenous pacemaker were strongly associated with upper extremity DVT, with more than a fivefold higher risk even after adjustment for comorbid conditions.203
Inflammatory Bowel Disease
Clinical series have reported VTE to complicate inflammatory bowel disease in 1.2% to 7.1% of cases.204,205 Crohn’s disease has incidence rates of 31.4 per 10,000 person-years and 10.3 per 10,000 person-years for DVT and PE, respectively. Ulcerative colitis also has a high incidence rate of 30.0 per 10,000 and 19.8 per 10,000 person-years for DVT and PE, respectively. Such thromboses frequently occur among young patients, are more common with active disease, and may affect unusual sites, such as the cerebral veins.204,205 Greater extent of colonic disease in ulcerative colitis portends a higher risk of VTE. Most cases are not associated with inherited inhibitor deficiencies, antiphospholipid antibodies, or factor V Leiden mutation. The significance of thrombocytosis and elevations of factor V, factor VIII, and fibrinogen during active episodes of inflammatory bowel disease is unclear because all are acute phase reactants.204,205
Other coagulation abnormalities, including depressed antithrombin values, elevated PAI-1 values, and presence of anticardiolipin antibodies, have been inconsistently reported.206 However, fibrinopeptide A elevations in inflammatory bowel disease suggest that active inflammation is associated with activation of coagulation, possibly mediated by endotoxin-induced monocyte activation.204–206
Systemic Lupus Erythematosus
A syndrome of arterial and venous thrombosis, recurrent abortion, thrombocytopenia, and neurologic disease may complicate systemic lupus erythematosus (SLE) when it is accompanied by the presence of antiphospholipid antibodies.207 Lupus anticoagulant and anticardiolipin antibodies may be seen in association with SLE, with other autoimmune disorders, with nonautoimmune disorders (such as syphilis and acute infection), with drugs (including chlorpromazine, procainamide, and hydralazine), and with older age.208
Lupus anticoagulant is present in 34% of patients with SLE and anticardiolipin antibodies in 44%, in comparison with 2% and 0% to 7.5%, respectively, of the general population.208 Among patients with SLE, those with lupus anticoagulant are at a sixfold higher risk for VTE, whereas those with anticardiolipin antibodies are at a twofold greater risk.209 The incidence of arterial or venous thrombosis is 25% in patients with lupus anticoagulant and 28% in patients with anticardiolipin antibodies.208
Varicose Veins and Superficial Thrombophlebitis
Varicose veins are also included as a risk factor for acute DVT, frequently only as a marker of previous venous disease.210 Most studies evaluating thrombotic risk have been performed in inpatients with other major risk factors for DVT. Such studies have inconsistently supported varicose veins as a risk factor in postoperative DVT and DVT after stroke or myocardial infarction.210–212 The importance of varicose veins in otherwise healthy outpatients with DVT has been questioned by some researchers213; varicose veins were not identified as an independent risk factor in young women,214 although some studies of postmenopausal women have reported varicose veins or superficial thrombophlebitis to be associated with odds ratios of 3.6 to 6.9 for the development of thromboembolism.180,183
Heit et al215 found, however, that varicose veins were independent predictors of DVT. They also found that age is an important factor in these patients, reporting a higher correlation between DVT and varicosity in young patients. For example, 45-year-old patients with varicose veins had a fourfold higher risk of VTE, 60-year-old patients had a twofold greater risk, and 75-year-old patients had no increase in risk. This group also found that patients with previous superficial vein thrombosis were more than four times more likely to have DVT or PE.215
Iliac Vein Compression
The association of VTE and anatomic anomalies or syndromes represents a congenital risk factor responsible for DVT in both the upper and lower extremities. Left iliac vein compression by the right iliac artery and fifth lumbar artery was first described in cadavers by May and Thurner, who hypothesized that chronic pulsation of the right iliac artery and mechanical obstruction led to intimal hypertrophy of the vein wall and subsequent venous obstruction.216 In actuality, it was Virchow more than a century earlier who had initially observed that iliofemoral vein thrombosis was five times more likely to occur in the left leg than in the right leg.217 Whereas left lower extremity venous hypertension, with or without left iliofemoral DVT, has come to be known as May-Thurner syndrome, it is important to recognize, if only for historical nomenclature, that a similar syndrome was described by Cockett in 1965 (Cockett’s syndrome) and took popularity over the term May-Thurner syndrome in Europe. However, it was Cockett who associated the acute phase of iliofemoral DVT secondary to compression of the iliac vein with the long-term consequence of chronic venous insufficiency. In addition, it was Cockett who noted that surgical intervention for the purpose of alleviating the skin ulcers and varicosities associated with iliac vein compression was ineffective unless the underlying disease process was identified.218
May-Thurner syndrome is more common in young to middle-aged women, especially after multiple pregnancies. The presenting symptom is often acute onset of left leg pain and swelling secondary to thrombosis. Atypically, patients may present with symptoms of chronic venous insufficiency that are unresponsive to compression stockings, leg elevation, and a calf exercise program.219 Early 20th century postmortem dissections revealed a 22% to 32% incidence of left iliac vein compression.220 Modern computed tomography imaging has revealed that in an asymptomatic population, the average amount of compression on the left iliac vein was 35%, and 24% of this population demonstrated greater than 50% compression.220 Whereas individuals in such series were asymptomatic, the incidence of symptomatic left common iliac vein compression presenting with either edema or DVT is estimated to be between 37% and 61%.221,222 Interestingly, the presence of an abdominal aortic aneurysm was associated with a significantly less amount of compression on the left common iliac vein by the right common iliac artery secondary to a higher prevalence of tortuosity in that artery.223
Upper Extremity Compression
Upper extremity DVT is less common than lower extremity DVT, accounting for only 1% to 2% of all DVT in the absence of central venous catheterization.224 Primary upper extremity DVT, or spontaneous thrombosis of the axillary-subclavian vein, accounts for up to 24% of all upper extremity DVT.225 Primary axillary-subclavian vein thrombosis was first described by Paget in 1875.226 However, it was von Schroetter who noted in 1884 that the spontaneous thrombosis of the axillary-subclavian vein was precipitated by a repetitive physical activity of the upper extremity and that extrinsic compression of the vein occurs by the musculoskeletal components of the thoracic outlet.227 Subsequently, axillary-subclavian vein thrombosis has come to be known as Paget-Schroetter syndrome. Activities such as throwing, swimming, and weightlifting can cause microtrauma of the venous intima that initiates thrombus formation.228 This syndrome, also called effort thrombosis, tends to affect young, healthy adults who participate in sports or whose professions require repetitive arm movements, such as painting. The presentation is often associated with spontaneous swelling of the arm, cyanotic discoloration, distention of the subcutaneous veins in the arm, fatigue, heaviness, or pain with use. The affected limb has been shown to be the dominant arm in 90% of cases.229 As this patient population tends to be healthy and often young, the diagnosis is often overlooked, and long-term outcomes of axillary-subclavian vein thrombosis can lead to thrombus progression, PE, and the postthrombotic syndrome.230 Postthrombotic syndrome of the upper extremity has been demonstrated to occur 7% to 46% of the time after DVT; it is associated with an increased functional disability of the affected limb and an overall decreased quality of life.231
Popliteal Vein Entrapment
Although arterial entrapment syndrome is well described in the literature, popliteal vein entrapment has been reported to occur either alone or with the artery in 10% of cases.232 Anatomic anomalies of the medial head of the gastrocnemius have been associated with popliteal vascular entrapment. However, when venous entrapment occurs in a setting without arterial entrapment, both the medial and the lateral heads of the gastrocnemius have been shown to contribute.233 Bone tumors and hypertrophied fibrous fascia have also been associated with isolated venous involvement.234,235 Traditionally, popliteal artery entrapment has been more associated with the male gender; venous entrapment has been reported to occur 70% of the time in women.236 The presentation is often that of a young adult with signs of chronic venous disease including leg swelling, varicosities, skin changes, and DVT.
Inferior Vena Cava Anomalies
Congenital hypoplasia or absence of the inferior vena cava (IVC) presents another anatomic risk factor for VTE. The incidence of IVC anomalies is estimated at 0.07% to 8.7% of the population; however, the incidence of IVC anomalies discovered in individuals presenting with a DVT is 5.1% to 6.7%.237–239 Anomalous vena cava development occurs in the sixth to eighth week of gestation. Often, such anomalies are compensated for by an enlarged vena azygos system as well as by an extensive system of deep venous collaterals that allow venous return from the lower extremities. Despite collateral flow, an absent or hypoplastic vena cava predisposes patients to venous stasis and thrombosis. Patients presenting with DVT and underlying vena cava anomalies are younger than those with DVT and no underlying IVC anomaly.239 Furthermore, two small case series reported the occurrence of bilateral DVT to be greater than 60% when IVC anomalies are involved.238 Unfortunately, IVC anomalies as well as venous thrombosis above the inguinal ligament are easily missed as B-mode ultrasound does not always allow accurate examination of abdominal veins.240 In individuals younger than 30 years with an apparent idiopathic bilateral DVT, further investigation may be warranted for anomalies of the IVC.237–239 IVC-related thrombosis can be predisposed by malignant disease, external compression, and antiphospholipid syndrome in addition to IVC anomalies, and workup for each of these is important.241 Furthermore, symptomatic PE occurs more frequently in those with IVC thrombosis, so some have recommended additional prophylactic measures, including filters, in these patients.
Other Risk Factors
Traditional risk factors for VTE have included obesity and cardiac disease; however, the evidence supporting these risk factors remains equivocal. Among postmenopausal women, a body mass index of greater than 25 to 30 kg/m2 has been associated with a significantly increased risk of VTE.181,183 Some investigators have reported obesity to be associated with a twofold greater risk for postoperative DVT, although multivariate analyses by others have not shown obesity to constitute an independent risk.210 Obesity (and past tobacco smoking) was not an independent risk factor of DVT in the Olmsted County study215 and has not been proved to be a risk factor for the development of DVT after stroke.212 Obesity is a risk factor, however, for recurrent DVT.242
Systemic hypercoagulability, congestive heart failure, and enforced bed rest may predispose patients who are hospitalized with acute myocardial infarction to DVT. The incidence of DVT in this population has been reported to be 20% to 40%.92,211,240 Some investigators have noted the incidence of DVT to be higher among patients in whom myocardial infarction was confirmed (34%) than in those in whom the diagnosis was excluded (7%). However, other researchers240 have found a high incidence of 125I-fibrinogen scan–detected thrombosis among patients with myocardial infarction (38%) and among those with other severe illnesses but without confirmed infarction (62%). The prevalence of PE among autopsied patients has also not differed substantially from that in other inpatients.193,194
Although myocardial infarction may be in question as a risk factor, those older than 60 years with congestive heart failure have a DVT rate of 54%.243 However, the evidence supporting congestive heart failure as an independent risk factor for DVT is also conflicting. A variety of thromboembolic complications account for nearly half the deaths among patients who did not undergo anticoagulation after hospitalization for congestive heart failure, but congestive heart failure has not been identified as an independent risk factor for postoperative DVT. The balance of evidence suggests that severely ill medical patients are at significant risk for VTE,244 although it is difficult to define the additional risk associated with cardiac disease.
Pathophysiology
Virchow’s Triad
As postulated by Virchow, three factors are important in the development of venous thrombosis: (1) abnormalities of blood flow, (2) abnormalities of blood, and (3) vascular injury. These tenets have subsequently been refined, and it currently appears that flow abnormalities determine the localization of venous thrombi, that abnormalities of blood may include aberrations of both the coagulation and fibrinolytic systems, and that biologic injury to the venous endothelium is potentially more important than gross trauma. It also is clear, however, that the origin of DVT is frequently multifactorial, with components of Virchow’s triad assuming variable importance in individual patients. In the early 1970s, Gwendylen Stewart theorized a relationship between thrombosis and inflammation. This relationship appears to play an important role in the thrombogenic process and the evolution of the thrombus.245
Mechanical venous injury plays a role in thrombosis associated with direct venous trauma,246 hip arthroplasty,247 and central venous catheters.200,247 Central venous cannulation is largely responsible for the increasing incidence of upper extremity thrombosis, whereas similar venous injury may be responsible for the fact that 57% of thrombi occurring after hip arthroplasty arise in the femoral vein rather than the usual site in the calf.248 However, the importance of mechanical venous injury in other situations is questionable. For example, venous injury cannot account for the observations that thromboses in trauma patients are more commonly bilateral (77%) than unilateral (23%) and may be as common in an injured limb as in an uninjured limb.246 Neither do mechanical crush injuries in animal models usually cause thrombosis, even when followed by stasis.249,250 Overt endothelial injury probably is neither necessary nor sufficient to cause thrombosis in the absence of other factors.250
Endothelium and Hemostasis
The endothelium provides a vasodilatory and local fibrinolytic environment. Thus, coagulation, platelet adhesion, platelet activation, inflammation, and leukocyte activation are inhibited. Mechanisms that maintain this environment include production of thrombomodulin with subsequent protein C activation; expression by endothelium of heparan sulfate and dermatan sulfate, both which can accelerate antithrombin and heparin cofactor activity; expression of tissue factor pathway inhibitor; and production of tissue plasminogen activator and urokinase-type plasminogen activator. In addition, the endothelium produces nitric oxide, prostacyclin, and interleukin-10, all of which inhibit adhesion and activation of leukocytes and promote vasodilatation.251
With homeostatic disturbances of the endothelium, prothrombotic and proinflammatory states occur. Vasoconstriction is facilitated by platelet-activating factor and endothelin-1; thrombosis is facilitated by production of von Willebrand factor, tissue factor, PAI-1, and activated factor V.251 Endothelial cells and activated platelets also upregulate cell adhesion molecules P-selectin and E-selectin, promoting leukocyte interactions. This sets the stage for the inflammatory contribution to the thrombotic process.
Inflammation and Thrombosis
Inflammation and thrombosis are interrelated.252 With inflammation, there is an increase in tissue factor, platelet reactivity, fibrinogen, phosphatidylserine on membrane surfaces, and PAI-1 (inhibiting fibrinolysis) and a decrease in thrombomodulin (and thus a decrease in the activity of protein C).253
Cell adhesion molecules allow leukocyte transmigration, and selectins are intimately involved in this process. Selectins are the first glycoproteins to be upregulated on activated platelets and endothelial cells.252 In the vein wall, P-selectin is upregulated first, followed by E-selectin.254 Clinically, in addition to the role of P-selectin in the pathogenesis of DVT, soluble P-selectin levels in combination with the Wells score have proved to aid in the diagnosis of DVT with a specificity of 96% and a positive predictive value nearing 100%.255
Venous stasis and ischemia result in P-selectin upregulation; this localizes microparticles, which are prothrombotic, to the area of evolving thrombosis.256–258 The receptor to P-selectin, P-selectin glycoprotein ligand-1, is expressed on leukocytes and their derived microparticles. Microparticles coexpressing tissue factor have been found to accumulate in growing thrombi in a P-selectin:P-selectin glycoprotein ligand-1–dependent fashion. The interaction between P-selectin and its receptor stimulates the production of thrombogenic microparticles from leukocytes, particularly monocytes, along with platelets and endothelial cells.259,260 These prothrombotic microparticles express tissue factor and possess a phosphatidylserine-rich anionic surface that may facilitate assembly of coagulation cascade complexes.261 The microparticles are then concentrated back in the area of thrombosis formation, leading to thrombus amplification.
Microparticles normalize tail bleeding times in hemophilic mice,260 and human pericardium–derived microparticles increase thrombosis in a rodent venous stasis model.262 In addition to the importance of microparticle-derived tissue factor, tissue factor exposure by vein injury also exposes the tissue factor abundant in the outer portion of the vein wall.263 In DVT, microparticles are found to be elevated.264 PAI-1 is stored in the alpha granules of platelets.265 On activation, microparticles shed from platelets express PAI-1. As these microparticles are co-localized to the growing thrombus, they not only are prothrombotic but also inhibit fibrinolysis.266
The role of neutrophils in the pathogenesis of DVT has recently become more prominent. Neutrophils release a meshlike structure of DNA, histones, and antimicrobial particles designed to trap bacteria, termed neutrophil extracellular traps.267 These neutrophil extracellular traps have been found to interact with fibrin, von Willebrand factor, and coagulation factors and to act as scaffolding for the growing thrombus.268,269
Plasminogen System
Physiologic clot formation is balanced by a constant process of thrombolysis to prevent pathologic intravascular thrombosis. Plasmin is the main fibrinolytic enzyme with substrates fibrin and fibrinogen and other coagulation factors. Plasmin also interferes with platelet adhesion mediated by von Willebrand factor.270 Plasminogen activation provides localized proteolytic activity.271,272 The primary inhibitor of plasminogen activators is PAI-1. PAI-1 is secreted in an active form from the liver and endothelium and is stabilized by binding to vitronectin. The role of PAI-1 in venous thrombogenesis is contradictory in the literature.273,274 It is plausible that elevated levels of PAI-1 could suppress fibrinolysis and thus increase thrombosis.
Thrombus Resolution and Vein Wall Remodeling
Thrombus resolution involves profibrotic growth factors, collagen deposition, and matrix metalloproteinase expression and activation. Leukocytes invade the thrombus in a predictable sequence, with the neutrophil first.275 Neutrophils appear critical for early thrombus resolution by promoting fibrinolysis and collagenolysis.276 The monocyte appears to be more important for later DVT resolution. Monocyte influx correlates with elevated monocyte chemotactic protein-1 levels.277 Depletion of the CC chemokine receptor 2 is thought to be associated with late impairment of thrombus resolution.278
The changes in the vein wall after DVT mirror what is seen in the thrombus. Elastinolysis occurs early and is accompanied by elevation in activities of matrix metalloproteinases 2 and 9. However, early vein wall collagenolysis seems to occur within the first 7 days, representing an acute response to injury.279 Associated with the early biochemical injury from DVT is an elevation of a number of profibrotic mediators from the vein wall and thrombus including transforming growth factor-β, interleukin-13, and monocyte chemotactic protein-1. These mediators promote fibrosis and an increase in collagen I and III gene expression.280 The inhibition of inflammation decreases this vein wall fibrosis after venous thrombosis. P-selectin inhibition decreases many different mediators of vein wall fibrosis.257
Stasis
Regardless of etiology, most venous thrombi originate in areas of low blood flow, either in the soleal veins of the calf210 or behind valve pockets.281 Furthermore, many risk factors for acute DVT are associated with immobilization and slow venous flow. Several mechanisms exist to explain the role of stasis in thrombogenesis. In comparison with pulsatile flow, static streamline flow is associated with profound hypoxia at the depths of the venous valve cusps and may induce endothelial injury.282 The effects of hypoxia in cultured endothelial cells include stimulation of cytokine production and leukocyte adhesion molecule expression,283 perhaps accounting for the adhesion and migration of leukocytes observed in association with stasis.284 In addition, stasis allows the accumulation of activated coagulation factors and the consumption of inhibitors at sites prone to thrombosis. Stasis in the large veins may be particularly important because the low surface-to-volume ratio may prevent interaction with endothelial inhibitory pathways, particularly the endothelium-bound thrombomodulin–protein C system.285
Despite these observations, there is little evidence that stasis can activate coagulation, and in isolation, stasis appears to be inadequate to cause thrombosis alone.284 Experimentally, ligation of rabbit jugular veins for intervals of 10 to 60 minutes has consistently failed to cause thrombosis.285 From a clinical perspective, activation of coagulation during hip arthroplasty is associated with manipulation of the femoral component rather than simply occlusion of the femoral vein.97 Stasis might better be viewed as a permissive factor in venous thrombosis, localizing activated coagulation to thrombosis-prone sites.
Activation of Coagulation
Activation of coagulation appears to be critical in the pathogenesis of DVT. The coagulation cascade functions through serial activation of zymogens in the intrinsic and tissue factor pathways, with the ultimate generation of thrombin by the prothrombinase complex. Antithrombin and the thrombomodulin–protein C systems are the primary inhibitors of coagulation, whereas the fibrinolytic system serves to further limit fibrin deposition. Although the hemostatic system is continuously active, thrombus formation is ordinarily confined to sites of local injury by a precise balance between activators and inhibitors of coagulation and fibrinolysis. A prothrombotic state may result either from imbalances in the regulatory and inhibitory systems or from activation exceeding antithrombotic capacity.286 Activated coagulation can be successfully identified with certain byproducts of the coagulation cascade, including prothrombin fragment F1+2, generated by factor Xa in the cleavage of prothrombin to thrombin; fibrinopeptide A, formed in the thrombin-mediated conversion of fibrinogen to fibrin; and thrombin-antithrombin complex, formed by the combination of thrombin with its primary inhibitor.286,287 Increased levels of these markers have been seen with risk factors such as surgery, oral contraceptives, and malignant disease.288 The degradation of fibrin polymers by plasmin results in the creation of fragment E and two molecules of fragment D. Detection of D-dimer levels is another marker of clot formation and fibrinolysis. D-dimer has been found to accurately predict an ongoing risk of recurrent thrombolism.289
Just as the combination of stasis and injury may be insufficient to cause thrombosis alone,249 activated coagulation may be insufficient to provoke thrombosis. Ordinarily, activated coagulation factors are rapidly cleared from the circulation. Localized in regions of stasis, however, the coagulation cascade allows activated factors to rapidly amplify the thrombotic stimulus, leading to platelet aggregation and fibrin formation.249,285 Therefore, DVT appears to be multifactorial, requiring the convergence of several pathologic factors to produce a thrombotic event.290
Infusion of activated coagulation factors alone may not cause thrombosis, but infusion of tumor necrosis factor and antibody to protein C combined with low flow and subtle catheter injury consistently produces proximal venous thrombosis in a baboon model.291 Similarly, the thrombogenic potential of hip arthroplasty stems from the combination of injury to the femoral vein, venous occlusion, and measurable activation of coagulation during placement of the femoral component.97 The existence of a prothrombotic state before the precipitation of overt thrombosis is also well recognized.287,292 Increased thrombin activity may precede the development of a positive duplex leg scan result in surgical patients by several days,292 whereas thrombosis in patients with congenital thrombophilias is often triggered by events such as injury, surgery, and pregnancy.293
On this pathophysiologic background, the factors contributing to clinically important thrombosis have been most thoroughly evaluated in the valve pockets of the lower extremity veins. In flow models, primary and secondary vortices are produced beyond the valve cusps, which tend to trap red cells in a low-shear field near the apex of the cusp.294 The early nidus for thrombus formation may consist of red blood cell aggregates forming within these eddies. However, these aggregates appear to be transient unless stabilized by fibrin in the setting of locally activated coagulation. The events in the evolution of these thrombi are described by Sevitt295,296 in a series of detailed histologic studies.
Once early thrombi have formed in the valve pocket, they may become adherent to the endothelium near the apex of the valve cusp. These valve pocket thrombi appear to form on structurally normal endothelium and largely to spare the valve cusp. Laminated appositional growth may occur outward from the apex of the cusp, with propagation beyond the valve pocket depending on the relative balance between activated coagulation and thrombolysis. Once luminal flow is disturbed, prograde and retrograde propagation may occur.
Clinical symptoms develop only when a sufficient fraction of the venous outflow is occluded. If present, such symptoms appear 24 to 36 hours after the thrombus is detectable by 125I-fibrinogen scanning.93 Conversely, early thrombi may fail to propagate, with evidence of aborted thrombi appearing as endothelialized fibrin fragments within the valve pockets.295,296 A significant number of these early thrombi spontaneously resolve after serial 125I-fibrinogen scanning.22,297
Natural History
Thrombus Evolution as Determined by Noninvasive Studies
Unlike in animal models, in which there is a transient stimulus to thrombosis, the relative balance between organization, thrombolysis, propagation, and rethrombosis determines outcomes after human thrombosis. From a clinical perspective, the most important events after thrombosis are recanalization and recurrent thrombosis. However, the relative importance, frequency, and rate of these processes were not possible to define before noninvasive technology that permitted serial examinations of patients.
Impedance plethysmography was the first widely available noninvasive test permitting serial evaluations of outflow obstruction due to an acute DVT. Although this test could not distinguish between recanalization and the development of collateral venous outflow, results of such studies were found to normalize in 67% of patients by 3 months and in 92% of patients by 9 months.298 Venous duplex ultrasonography, with which individual venous segments can be observed over time, has further documented that recanalization does occur in most patients after acute DVT. In 21 patients monitored prospectively with duplex scanning, recanalization occurred in 44% of patients at 7 days and in 100% of patients by 90 days after the acute event.299 The percentage of initially involved segments that remained occluded decreased to a mean of 44% by 30 days and 14% by 90 days (Fig. 49-1).
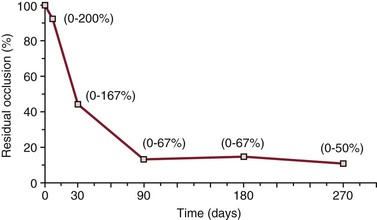
Figure 49-1 Mean percentage of initially occluded segments remaining occluded at follow-up among 21 patients with acute deep venous thrombosis (percentage residual occlusion = number of segments occluded at follow-up ÷ number of segments occluded at presentation). Numbers in parentheses are the ranges for individual cases, with numbers greater than 100% indicating extension of the initial thrombus. (From Killewich LA, et al: Spontaneous lysis of deep venous thrombi: rate and outcome. J Vasc Surg 9:89, 1989.)
van Ramshorst et al300 noted an exponential decrease in thrombus load during the first 6 months after femoropopliteal thrombosis. Most recanalization occurred within the first 6 weeks, with flow re-established in 87% of 23 occluded segments during follow-up. Killewich et al299 reported a linear decrease in thrombus burden; after 24 to 36 weeks, only 26% of the original thrombus remained. Using a somewhat different approach, Prandoni et al301 measured thrombus thickness under maximal compression and noted significant reduction in thrombus mass during the first 3 months after thrombosis. In aggregate, these studies confirm the histologic findings that recanalization begins early after an acute DVT, with the greatest reduction in thrombus load occurring within 3 months of the event. Complete thrombus resolution has been reported in 56% of patients monitored for 9 months.299,302 However, recanalization may continue for months to years after the acute event.301
Recurrent thrombosis appears to compete with recanalization early after an acute DVT. The importance of such events is well recognized clinically among patients with proximal DVT; recurrent thromboembolic events have been reported in 5.2% of patients who were treated with standard anticoagulation measures for 3 months compared with 47% of patients who were inadequately treated with a 3-month course of low-dose subcutaneous heparin.303,304 Proximal propagation may also complicate isolated calf vein thrombosis in up to 23% of untreated patients.305 Because of the risk of propagation and possible PE, new guidelines from the American College of Chest Physicians include the treatment of idiopathic calf vein DVT with 3 months of anticoagulation.306
Despite the significance of these observations, reports of serial noninvasive follow-up examinations suggest that these studies may underestimate the incidence of new thrombotic events. In the acute setting, Krupski et al307 found ultrasonographic evidence of proximal propagation in 38% of 24 patients being treated with intravenous heparin. In a larger series of 177 patients, most of whom were treated with standard anticoagulation measures, recurrent thrombotic events were observed in 52% of patients.308 New thrombi were observed in 6% of uninvolved contralateral extremities, whereas propagation of thrombi to new segments occurred in 30% of involved limbs and rethrombosis of a partially occluded or recanalized segment in 31% of extremities. Propagation in the ipsilateral limb tended to occur as an early event at a median of less than 40 days after presentation, whereas rethrombosis and extension to the contralateral limb tended to occur sporadically as late events.
Complications
Pulmonary Embolism
PE, with its attendant mortality, is the most devastating complication of acute DVT. In association with acute DVT, the majority of pulmonary emboli may be clinically silent. In patients presenting with symptomatic DVT, 50% to 80% have evidence of asymptomatic PE. Conversely, in those presenting with symptomatic PE, asymptomatic DVT can be demonstrated in approximately 80% of the cases.309 Approximately 90% of thromboemboli arise from the lower extremity veins, and inadequate treatment of proximal lower extremity venous thrombosis is associated with a 20% to 50% risk of clinically significant recurrent thromboembolism.309,310 The clinical significance of untreated PE was demonstrated by Barritt and Jordon in 1960 when they randomized patients with known PE either to receive or not to receive anticoagulant therapy. The group that did not receive anticoagulation demonstrated 50% incidence of recurrence, of which 25% was fatal.311 Symptomatic PE may also complicate 7% to 17% of proximal upper extremity thrombi.311–313
High-probability lung scan results may be obtained in 25% to 51% of patients with documented DVT in the absence of pulmonary symptoms, and some autopsy series have reported the combination of PE and DVT to be 1.8 times more common than proximal DVT alone.197 Modern imaging with computed tomography has revealed asymptomatic PE to be found in 1.5% of scans done for a reason other than suspected PE.314 In those with malignant disease undergoing staging, the incidence of asymptomatic PE found on computed tomography was 3.3%, whereas the overall incidence of VTE was found to be 6.3%.315 Symptomatic manifestation of PE may depend on the patient’s underlying cardiopulmonary reserve more than on the amount of the pulmonary circulation occluded.
The Rochester Epidemiology Project database of Olmsted County, Minnesota, demonstrated an overall average age- and sex-adjusted annual incidence of PE to be 66 per 100,000.316 In the United States, observational and autopsy data have suggested the incidence of symptomatic PE to be about 630,000, with 300,000 being fatal.253 The risk of death in patients with symptomatic PE is 18-fold higher than in patients presenting with DVT alone. For almost 25% of PE patients, the initial presentation is sudden death.203,215,316 In those patients who do not die suddenly, death occurs within 1 hour of presentation in 11% of patients with symptomatic PE and in 30% of those surviving 1 hour but in whom the diagnosis of PE is not made.317
Pulmonary Hypertension
The development of chronic thromboembolic pulmonary hypertension after PE was thought to have been a rare occurrence, affecting only 0.1% to 0.5% of individuals who survived the initial embolic event.318 However, more recent data suggest that chronic thromboembolic pulmonary hypertension occurs more often, with 3.8% of individuals developing the disorder after PE.319 The most common symptom is progressive exertional dyspnea, with worsening right ventricular failure, edema, chest pain, lightheadedness, and syncope developing as the disease progresses.318 However, many patients presenting with chronic thromboembolic pulmonary hypertension have no clear history of an acute thromboembolic event.320 The relationship of chronic thromboembolic pulmonary hypertension with PE needs to be recognized; medical management is associated with poor outcomes, and early surgical intervention with either pulmonary thromboembolectomy or pulmonary endarterectomy offers a curative approach.321
Postthrombotic Syndrome
Although less dramatic than PE, the postthrombotic syndrome is the most important late complication of acute DVT, and it is responsible for a greater degree of chronic socioeconomic morbidity. As many as 29% to 79% of patients may have some degree of long-term manifestations, such as pain, edema, heaviness, or hyperpigmentation. Severe manifestations occur in only 7% to 23% and ulceration in 4% to 6% of patients.322–327 Coon et al328 have estimated that severe postthrombotic changes are present in 5% of the U.S. population, yielding a prevalence of 6 to 7 million individuals with stasis changes and 400,000 to 500,000 with leg ulcers.
Severe manifestations of the postthrombotic syndrome are a consequence of ambulatory venous hypertension, which is determined by a combination of factors, including valvular reflux, persistent venous obstruction, and the anatomic distribution of these abnormalities.329 However, despite the importance of valvular reflux in postthrombotic sequelae, valvular destruction is not universally associated with acute DVT, and as many as one third of patients may remain free of chronic symptoms. Indeed, only 33% to 59% of initially thrombosed segments show evidence of reflux on duplex ultrasonography 1 year after the event.
These clinical observations are supported by the histologic evidence that thrombus organization rarely involves valve cusps. In most cases, the thrombus is separated from the valve cusp by a clear zone, which is postulated to arise from the local fibrinolytic activity of the valvular endothelium.330 This protective mechanism appears to fail in approximately 10% of cases in which thrombus adherence to the valve cusp is noted. Initial thrombus adherence is likely to account for the histologic findings in postthrombotic syndrome; approximately 50% of popliteal valves in such extremities demonstrate thrombus formation on the valve leaflets, usually on the surface facing the valve sinus, with associated loss of the endothelium and basement membrane.331
Newer data exist showing higher rates of postthrombotic syndrome and higher rates of recurrence in those with larger residual thrombus burden, which is further elucidated in the higher rates of postthrombotic syndrome in those who receive anticoagulation compared with those who had thrombolysis in the CaVenT study.332,333 Furthermore, several factors in the early natural history of a thrombotic event, including rate of recanalization, extent of reflux, anatomic distribution of reflux and obstruction, and recurrent thrombosis, do appear to influence the ultimate development of valvular incompetence and eventual postthrombotic syndrome. In addition, higher body mass index has been shown to be a risk factor for the postthrombotic syndrome.334 Depending on the segment involved, venous segments developing reflux have been noted to require 2.3 to 7.3 times longer for recanalization than segments in which valve function is preserved (Fig. 49-2).335 Recurrent thrombotic events are also detrimental to valve competence. Reflux may develop in 36% to 73% of segments with rethrombosis, a rate considerably higher than that in segments without rethrombosis (Fig. 49-3).308 Consistent with these observations is the finding of a sixfold higher risk of postthrombotic syndrome among patients with recurrent thrombosis.324
Figure 49-2 Median time (± interquartile range) from thrombosis to complete recanalization, stratified according to ultimate reflux status. Median times are 2.3 to 7.3 times longer among segments in which reflux developed, in all but the posterior tibial veins. CFV, Common femoral vein; GSV, greater saphenous vein; PFV, profunda femoris vein; PPV, popliteal vein; PTV, posterior tibial vein; SFD, superficial femoral vein; SFM, mid superficial femoral vein; SFP, proximal superficial femoral vein. (From Meissner MH, et al: Deep venous insufficiency: the relationship between lysis and subsequent reflux. J Vasc Surg 18:596, 1993.)
Figure 49-3 Reflux incidence in initially involved venous segments with and without subsequent rethrombosis. Numbers above bars indicate the number of segments in which reflux was observed divided by the number of segments in which reflux could be definitively assessed. Differences between segments with and without rethrombosis are statistically significant (*P < .005) for the mid superficial femoral vein (SFM), distal superficial femoral vein (SFD), and popliteal vein (PPV) segments. CFV, Common femoral vein; GSV, greater saphenous vein; PFV, profunda femoris vein; PTV, posterior tibial vein; SFP, proximal superficial femoral vein. (From Meissner MH, et al: Propagation, rethrombosis and new thrombus formation after acute deep venous thrombosis. J Vasc Surg 22:558, 1995.)
In a 25-year population-based retrospective cohort study, Mohr et al336 found that chronic venous insufficiency developed in 245 of 1527 patients with DVT or PE; 1-year, 5-year, 10-year, and 20-year cumulative incidence rates were 7.3%, 14.3%, 19.7%, and 26.8%, respectively. By 20 years, the cumulative incidence of venous ulcers was 3.7%. Thus, the incidence of venous stasis syndrome continues to rise for 20 years after VTE. Prandoni et al also demonstrated an increase in the incidence of the postthrombotic syndrome after acute DVT with a cumulative incidence of 22.8% after 2 years, 28.0% after 5 years, and 29.1% after 8 years.324 Furthermore, in patients 40 years or younger, Mohr et al demonstrated that venous stasis syndrome was 3.0-fold more likely in those with proximal DVT than in those with distal-only DVT.336
Mortality
DVT is frequently associated with comorbid medical conditions, and the rate of early death after a lower extremity thrombosis is substantial. Piccioli et al337 noted a 3-month mortality rate of 19%, with no deaths due to PE. Baglin et al338 reported a similar mortality (21%) during the first year after an acute DVT, and long-term follow-up at 3 and 5 years demonstrated mortality rates of 30% and 39%, respectively. The mortality rate after an acute DVT exceeds that in an age-matched population, and most deaths are related to malignant disease or cardiovascular disease. Higher short-term mortality rates have been seen in patients with upper extremity DVT, who tend to be more ill, with an increased prevalence of metastatic malignant disease. Six-month mortality may be as high as 48% among patients with upper extremity DVT compared with 13% in those with a lower extremity thrombosis.197
Calf Vein Thrombosis
Thrombosis isolated to the deep veins of the calf is often differentiated from that involving the proximal veins because of perceived differences in the associated rates of PE and postthrombotic complications. Noninvasive studies do suggest that such isolated thrombi recanalize more rapidly than proximal thrombi,335 with a mean 50% reduction in thrombus load by 1 month and complete recanalization within 1 year.339 The frequency of reflux in involved calf vein segments is also lower than in proximal venous segments.334 However, propagation to more proximal segments also occurs in 15% to 23% of limbs with thrombosis confined to the calf veins.340,341 In a review of the subject, Kearon342 found that in the absence of treatment, about one fourth to one third of symptomatic, isolated calf vein thrombosis extended to involve the proximal veins.
Whereas clinical data suggest that the incidence of acute and long-term complications may be less than after proximal venous thrombosis, isolated calf vein thrombi are not entirely benign. Although prior embolization of more proximal thrombus cannot be excluded, concurrent PE has been noted in approximately 10% of patients with calf vein thrombosis when clinical suspicion is supplemented by objective tests343 and in up to 33% of patients in series employing routine ventilation-perfusion lung scans.344 Even if clinically significant emboli are presumed to arise only from proximal thrombus extension, a 20% rate of proximal propagation in untreated calf vein thrombosis theoretically would be associated with a 2% risk of fatal PE and a 5% to 10% risk of symptomatic recurrent thromboembolism.345
Although less common than after proximal thrombosis, postthrombotic symptoms may also complicate isolated calf vein thrombosis. Persistent symptoms of pain and swelling at 1 year have been noted in 23% of extremities with calf vein thrombosis compared with 54% of those with proximal thrombosis and 9% of uninvolved limbs (Fig. 49-4).339 In contrast, Prandoni et al325 found the rates of mild (21%) and severe (5%) postthrombotic sequelae 45 months after the acute event to be similar among patients with isolated calf and proximal venous thromboses. Other researchers have reported postthrombotic symptoms in 38% of patients after a mean follow-up of 3.4 years and 47% after 5 to 10 years. There has been some suggestion that calf vein thrombosis in named calf veins is different from thrombosis in the gastrocnemius and soleus veins and that such thrombi are associated with fewer significant clinical symptoms.346 However, a more recent study of muscular calf vein thrombosis in 128 patients revealed an 18.8% incidence of symptomatic recurrences during a mean follow-up of 26.7 months and a 7% incidence of PE at presentation, even in muscular calf vein thrombosis.347 This suggests that even muscular calf vein thrombosis should be treated with anticoagulation. Because of these findings and the potential complications of embolization and propagation, the American College of Chest Physicians 2012 guidelines include the recommendation for 3 months of anticoagulation after calf vein DVT.348
Figure 49-4 Prevalence of symptoms (edema, pain, hyperpigmentation, or ulceration) during follow-up among extremities with proximal deep venous thrombosis (DVT), extremities with isolated calf vein thrombosis (CVT), and uninvolved limbs contralateral to a calf vein thrombosis. The prevalence of symptoms in the three groups is significantly different at 12 months (P = .004). The table shows the total number of extremities within each group evaluated at each follow-up interval. (From Meissner MH, et al: Early outcome after isolated calf vein thrombosis. J Vasc Surg 26:749, 1997.)
Selected Key References
Coon WW, Willis PW 3rd, Keller JB. Venous thromboembolism and other venous disease in the Tecumseh community health study. Circulation. 1973;48:839–846.
Classic paper regarding the early estimates of the incidence and prevalence of VTE..
Esmon CT. Inflammation and thrombosis. J Thromb Haemost. 2003;1:1343–1348.
Extensive review on the procoagulant effects of inflammation..
Fuchs TA, Brill A, Wagner DD. Neutrophil extracellular trap (net) impact on deep vein thrombosis. Arterioscler Thromb Vasc Biol. 2012;32:1777–1783.
Review of the role of neutrophil extracellular traps..
Heit JA, Cohen AT, Anderson FJ. Estimated annual number of incident and recurrent, non-fatal venous thromboembolism (VTE) events in the US. Blood. 2005;106(11).
Data on the incidence and prevalence of VTE taken from the Rochester community..
Humphries J, McGuinness CL, Smith A, Waltham M, Poston R, Burnand KG. Monocyte chemotactic protein-1 (MCP-1) accelerates the organization and resolution of venous thrombi. J Vasc Surg. 1999;30:894–899.
This study identifies the role of monocytes and MCP-1 in VTE resolution..
Kearon C. Natural history of venous thromboembolism. Circulation. 2003;107(Suppl):I22–I30.
Meissner MH, Caps MT, Bergelin RO, Manzo RA, Strandness DE Jr. Propagation, rethrombosis and new thrombus formation after acute deep venous thrombosis. J Vasc Surg. 1995;22:558–567.
Careful serial ultrasound studies are used to determine the rate of thrombus extension or progression and rethrombosis..
Meissner MH, Manzo RA, Bergelin RO, Markel A, Strandness DE Jr. Deep venous insufficiency: the relationship between lysis and subsequent reflux. J Vasc Surg. 1993;18:596.
Prandoi P, Lensing A, Cogo A, Cuppini S, Villalta S, Carta M, Cattelan AM, Polistena P, Bernardi E, Prins MH. The long-term clinical course of acute deep venous thrombosis. Ann Intern Med. 1996;125:1.
Rosendaal FR. Thrombosis in the young: epidemiology and risk factors. A focus on venous thrombosis. Thromb Haemost. 1997;78:1–6.
Wakefield TW, Myers DD, Henke PK. Mechanisms of venous thrombosis and resolution. Arterioscler Thromb Vasc Biol. 2008;28:387–391.
The reference list can be found on the companion Expert Consult website at www.expertconsult.com.
References
1. Hull RD, et al. Prophylaxis of venous thromboembolism. An overview. Chest. 1986;89:374S–383S.
2. Biland L, et al. Varicose veins (VV) and chronic venous insufficiency (CVI). Medical and socio-economic aspects, Basle study. Acta Chir Scand Suppl. 1988;544:9–11.
3. Coon WW, et al. Venous thromboembolism and other venous disease in the Tecumseh community health study. Circulation. 1973;48:839–846.
4. Silva Mde C. Chronic venous insufficiency of the lower limbs and its socio-economic significance. Int Angiol. 1991;10:152–157.
5. Wakefield TW, et al. Call to action to prevent venous thromboembolism. J Vasc Surg. 2009;49:1620–1623.
6. Heit JA, et al. Estimated annual number of incident and recurrent, non-fatal venous thromboembolism (VTE) events in the US. Blood. 2005;106(11).
7. Heit JA, et al. Trends in the incidence of deep vein thrombosis and pulmonary embolism: a 35-year population based study. Blood. 2006;108:403a.
8. Heit JA. The epidemiology of venous thromboembolism in the community. Arterioscler Thromb Vasc Biol. 2008;28:370–372.
9. Keenan CR, et al. The effects of race/ethnicity and sex on the risk of venous thromboembolism. Curr Opin Pulm Med. 2007;13:377–383.
10. Cohen AT, et al. Venous thromboembolism (VTE) in Europe. The number of VTE events and associated morbidity and mortality. Thromb Haemost. 2007;98:756–764.
11. Geerts WH, et al. Prevention of venous thromboembolism: the Seventh ACCP Conference on Antithrombotic and Thrombolytic Therapy. Chest. 2004;126:338S–400S.
12. Anderson FA Jr, et al. Estimated annual numbers of US acute-care hospital patients at risk for venous thromboembolism. Am J Hematol. 2007;82:777–782.
13. Geerts WH, et al. A prospective study of venous thromboembolism after major trauma. N Engl J Med. 1994;331:1601–1606.
14. Cook DJ, et al. Prevalence, incidence, and risk factors for venous thromboembolism in medical-surgical intensive care unit patients. J Crit Care. 2005;20:309–313.
15. Muscedere JG, et al. Venous thromboembolism in critical illness in a community intensive care unit. J Crit Care. 2007;22:285–289.
16. Liebson CL, et al. Risk factors for venous thromboembolism in nursing home residents. Mayo Clin Proc. 2008;83:151–157.
17. Attia J, et al. Deep vein thrombosis and its prevention in critically ill adults. Arch Intern Med. 2001;161:1268–1279.
18. Azarbal A, et al. Duplex ultrasound screening detects high rates of deep vein thromboses in critically ill trauma patients. J Vasc Surg. 2011;54:743–747.
19. Heit JA, et al. Predictors of recurrence after deep vein thrombosis and pulmonary embolism: a population-based cohort study. Arch Intern Med. 2000;160:761–768.
20. Murin S, et al. Comparison of outcomes after hospitalization for deep venous thrombosis or pulmonary embolism. Thromb Haemost. 2002;88:407–414.
21. Lindblad B, et al. Incidence of venous thromboembolism verified by necropsy over 30 years. BMJ. 1991;302:709–711.
22. Kakkar VV, et al. Natural history of postoperative deep-vein thrombosis. Lancet. 1969;2:230–232.
23. Piccioli A, et al. Epidemiologic characteristics, management, and outcome of deep venous thrombosis in a tertiary-care hospital: the Brigham and Women’s Hospital DVT registry. Am Heart J. 1996;132:1010–1014.
24. Cogo A, et al. Acquired risk factors for deep-vein thrombosis in symptomatic outpatients. Arch Intern Med. 1994;154:164–168.
25. Oger E, et al. The value of a risk factor analysis in clinically suspected deep venous thrombosis. Respiration. 1997;64:326–330.
26. Anderson FA Jr, et al. The prevalence of risk factors for venous thromboembolism among hospital patients. Arch Intern Med. 1992;152:1660–1664.
27. Rosendaal FR. Thrombosis in the young: epidemiology and risk factors. A focus on venous thrombosis. Thromb Haemost. 1997;78:1–6.
28. Heit JA, et al. Risk factors for deep vein thrombosis and pulmonary embolism: a population-based case-control study. Arch Intern Med. 2000;160:809–815.
29. Heit JA, et al. The epidemiology of venous thromboembolism in the community. Thromb Haemost. 2001;86:452–463.
30. Nordstrom M, et al. A prospective study of the incidence of deep-vein thrombosis within a defined urban population. J Intern Med. 1992;232:155–160.
31. Hansson PO, et al. Deep vein thrombosis and pulmonary embolism in the general population. “The Study of Men Born in 1913”. Arch Intern Med. 1997;157:1665–1670.
32. Tardy B, et al. Effects of long travels in sitting position in elderly volunteers on biological markers of coagulation activation and fibrinolysis. Thromb Res. 1996;83:153–160.
33. Gibbs NM. Venous thrombosis of the lower limbs with particular reference to bed-rest. Br J Surg. 1957;45:209–236.
34. McLachlin AD, et al. Venous stasis in the lower extremities. Ann Surg. 1960;152:678–685.
35. Rohrer MJ, et al. A prospective study of the incidence of deep venous thrombosis in hospitalized children. J Vasc Surg. 1996;24:46–49.
36. Sandoval JA, et al. Incidence, risk factors, and treatment patterns for deep venous thrombosis in hospitalized children: an increasing population at risk. J Vasc Surg. 2008;47:837–843.
37. Buck JR, et al. Pulmonary embolism in children. J Pediatr Surg. 1981;16:385–391.
38. Coon WW. Epidemiology of venous thromboembolism. Ann Surg. 1977;186:149–164.
39. Wise RC, et al. Spontaneous, lower-extremity venous thrombosis in children. Am J Dis Child. 1973;126:766–769.
40. Leslie IJ, et al. A prospective study of deep vein thrombosis of the leg in children on halo-femoral traction. J Bone Joint Surg. 1981;63:168–170.
41. DeAngelis GA, et al. Prevalence of deep venous thrombosis in the lower extremities of children in the intensive care unit. Pediatr Radiol. 1996;26:821–824.
42. McBride WJ, et al. Pulmonary embolism in pediatric trauma patients. J Trauma. 1994;37:913–915.
43. Radecki RT, et al. Deep vein thrombosis in the disabled pediatric population. Arch Phys Med Rehabil. 1994;75:248–250.
44. Nguyen LT, et al. Spontaneous deep vein thrombosis in childhood and adolescence. J Pediatr Surg. 1986;21:640–643.
45. Bernstein D, et al. Pulmonary embolism in adolescents. Am J Dis Child. 1986;140:667–671.
46. DeAngelis GA, et al. Prevalence of deep venous thrombosis in the lower extremities of children in the intensive care unit. Pediatr Radiol. 1996;26:821–824.
47. Sigel B, et al. The epidemiology of lower extremity deep venous thrombosis in surgical patients. Ann Surg. 1974;179:278–290.
48. Warlow C, et al. Deep venous thrombosis of the legs after strokes. Part I—incidence and predisposing factors. Br Med J. 1976;1:1178–1181.
49. Cruickshank JM, et al. Air travel and thrombotic episodes: the economy class syndrome. Lancet. 1988;2:497–498.
50. Homans J. Thrombosis of the deep leg veins due to prolonged sitting. N Engl J Med. 1954;250:148–149.
51. Ledermann JA, et al. Acute pulmonary embolism following air travel. Postgrad Med J. 1983;59:104–105.
52. Thromboembolism and air travel. Lancet. 1988;2:797.
53. Sarvesvaran R. Sudden natural deaths associated with commercial air travel. Med Sci Law. 1986;26:35–38.
54. Symington I, et al. Pulmonary thromboembolism after travel. Br J Dis Chest. 1977;71:147.
55. Wessler S. Thrombosis in the presence of vascular stasis. Am J Med. 1962;33:648–666.
56. Landgraf H, et al. Economy class syndrome: rheology, fluid balance, and lower leg edema during a simulated 12-hour long distance flight. Aviat Space Environ Med. 1994;65:930–935.
58. Ferrari E, et al. Travel as a risk factor for venous thromboembolic disease: a case-control study. Chest. 1999;115:440–444.
59. Samama MM. An epidemiologic study of risk factors for deep vein thrombosis in medical outpatients: the Sirius study. Arch Intern Med. 2000;160:3415–3420.
60. Scurr JH, et al. Frequency and prevention of symptomless deep-vein thrombosis in long-haul flights: a randomised trial. Lancet. 2001;357:1485–1489.
61. Lapostolle F, et al. Severe pulmonary embolism associated with air travel. N Engl J Med. 2001;345:779–783.
62. Paganin F, et al. Venous thromboembolism in passengers following a 12-h flight: a case-control study. Aviat Space Environ Med. 2003;74:1277–1280.
63. Belcaro G, et al. Venous thromboembolism from air travel: the LONFLIT study. Angiology. 2001;52:369–374.
64. Hughes RJ, et al. Frequency of venous thromboembolism in low to moderate risk long distance air travellers: the New Zealand Air Traveller’s Thrombosis (NZATT) study. Lancet. 2003;362:2039–2044.
65. Sevitt S. The vascularisation of deep-vein thrombi and their fibrous residue: a post mortem angiographic study. J Pathol. 1973;111:1–11.
66. Simioni P, et al. The risk of recurrent venous thromboembolism in patients with an Arg506→Gln mutation in the gene for factor V (factor V Leiden). N Engl J Med. 1997;336:399–403.
67. De Stefano V, et al. The risk of recurrent deep venous thrombosis among heterozygous carriers of both factor V Leiden and the G20210A prothrombin mutation. N Engl J Med. 1999;341:801–806.
68. den Heijer M, et al. Is hyperhomocysteinaemia a risk factor for recurrent venous thrombosis? Lancet. 1995;345:882–885.
69. Prins MH, et al. A critical review of the evidence supporting a relationship between impaired fibrinolytic activity and venous thromboembolism. Arch Intern Med. 1991;151:1721–1731.
70. Bergqvist D, et al. Venous thromboembolism and cancer. Curr Probl Surg. 2007;44:157–216.
71. Shen VS, et al. Fatal pulmonary embolism in cancer patients: is heparin prophylaxis justified? South Med J. 1980;73:841–843.
72. Cornuz J, et al. Importance of findings on the initial evaluation for cancer in patients with symptomatic idiopathic deep venous thrombosis. Ann Intern Med. 1996;125:785–793.
73. Prandoni P, et al. Deep-vein thrombosis and the incidence of subsequent symptomatic cancer. N Engl J Med. 1992;327:1128–1133.
74. Monreal M, et al. Occult cancer in patients with deep venous thrombosis. A systematic approach. Cancer. 1991;67:541–545.
75. Griffin MR, et al. Deep venous thrombosis and pulmonary embolism. Risk of subsequent malignant neoplasms. Arch Intern Med. 1987;147:1907–1911.
76. Nordstrom M, et al. Deep venous thrombosis and occult malignancy: an epidemiological study. BMJ. 1994;308:891–894.
77. Sorensen HT, et al. The risk of a diagnosis of cancer after primary deep venous thrombosis or pulmonary embolism. N Engl J Med. 1998;338:1169–1173.
78. Khorana AA, et al. Frequency, risk factors, and trends for venous thromboembolism among hospitalized cancer patients. Cancer. 2007;110:2339–2346.
79. Winter PC. The pathogenesis of venous thromboembolism in cancer: emerging links with tumour biology. Hematol Oncol. 2006;24:126–133.
80. Osborne N, et al. Venous thromboembolism in cancer patients undergoing major surgery. Ann Surg Oncol. 2008;15:3567–3578.
81. Prandoni P, et al. Cancer and venous thromboembolism. Lancet Oncol. 2005;6:401–410.
82. Rickles FR, et al. Molecular basis for the relationship between thrombosis and cancer. Thromb Res. 2001;102:V215–V224.
83. Gordon SG, et al. An enzyme-linked immunosorbent assay for cancer procoagulant and its potential as a new tumor marker. Cancer Res. 1990;50:6229–6234.
84. Nijziel MR, et al. From Trousseau to angiogenesis: the link between the haemostatic system and cancer. Neth J Med. 2006;64:403–410.
85. Nachman RL, et al. Interleukin 1 induces endothelial cell synthesis of plasminogen activator inhibitor. J Exp Med. 1986;163:1595–1600.
86. Falanga A, et al. Hemostatic system activation in patients with lupus anticoagulant and essential thrombocythemia. Semin Thromb Hemost. 1994;20:324–327.
87. Clagett GP, et al. Prevention of venous thromboembolism in general surgical patients. Results of meta-analysis. Ann Surg. 1988;208:227–240.
88. Levine MN, et al. The thrombogenic effect of anticancer drug therapy in women with stage II breast cancer. N Engl J Med. 1988;318:404–407.
89. Doll DC, et al. Vascular toxicity associated with antineoplastic agents. J Clin Oncol. 1986;4:1405–1417.
90. Clarke C, et al. Thromboembolism. A complication of weekly chemotherapy in the treatment of non-Hodgkin’s lymphoma. Cancer. 1990;66:2027–2030.
91. Edwards RL, et al. Abnormalities of blood coagulation tests in patients with cancer. Am J Clin Pathol. 1987;88:596–602.
92. Clagett GP, et al. Prevention of venous thromboembolism. Chest. 1995;108:312S–334S.
93. Kakkar VV, et al. Deep vein thrombosis of the leg. Is there a “high risk” group? Am J Surg. 1970;120:527–530.
94. Heit JA, et al. Risk factors for deep vein thrombosis and pulmonary embolism: a population-based case-control study. Arch Intern Med. 2000;160:809–815.
95. Scurr JH, et al. Frequency and prevention of symptomless deep-vein thrombosis in long-haul flights: a randomised trial. Lancet. 2001;357:1485–1489.
96. Kambayashi J, et al. Activation of coagulation and fibrinolysis during surgery, analyzed by molecular markers. Thromb Res. 1990;60:157–167.
97. Sharrock NE, et al. The John Charnley Award. Thrombogenesis during total hip arthroplasty. Clin Orthop Relat Res. 1995;16–27.
98. Kluft C, et al. The postoperative fibrinolytic shutdown: a rapidly reverting acute phase pattern for the fast-acting inhibitor of tissue-type plasminogen activator after trauma. Scand J Clin Lab Invest. 1985;45:605–610.
99. Burns GA, et al. Prospective ultrasound evaluation of venous thrombosis in high-risk trauma patients. J Trauma. 1993;35:405–408.
100. Knudson MM, et al. Prevention of venous thromboembolism in trauma patients. J Trauma. 1994;37:480–487.
101. Napolitano LM, et al. Asymptomatic deep venous thrombosis in the trauma patient: is an aggressive screening protocol justified? J Trauma. 1995;39:651–657.
102. Hill SL, et al. Deep venous thrombosis in the trauma patient. Am Surg. 1994;60:405–408.
103. Knudson MM, et al. Thromboembolism following multiple trauma. J Trauma. 1992;32:2–11.
104. Sue LP, et al. Iliofemoral venous injuries: an indication for prophylactic caval filter placement. J Trauma. 1995;39:693–695.
105. Knudson MM, et al. Use of low molecular weight heparin in preventing thromboembolism in trauma patients. J Trauma. 1996;41:446–459.
106. Knudson MM, et al. Prevention of venous thromboembolism in trauma patients. J Trauma. 1994;37:480–487.
107. Burns GA, et al. Prospective ultrasound evaluation of venous thrombosis in high-risk trauma patients. J Trauma. 1993;35:405–408.
108. Gando S, et al. Posttrauma coagulation and fibrinolysis. Crit Care Med. 1992;20:594–600.
109. Garcia Frade LJ, et al. Changes in fibrinolysis in the intensive care patient. Thromb Res. 1987;47:593–599.
110. Kapsch DN, et al. Fibrinolytic response to trauma. Surgery. 1984;95:473–478.
111. Poort SR, et al. A common genetic variation in the 3′-untranslated region of the prothrombin gene is associated with elevated plasma prothrombin levels and an increase in venous thrombosis. Blood. 1996;88:3698–3703.
112. Rosendaal FR, et al. Geographic distribution of the 20210 G to A prothrombin variant. Thromb Haemost. 1998;79:706–708.
114. Makris M, et al. Co-inheritance of the 20210A allele of the prothrombin gene increases the risk of thrombosis in subjects with familial thrombophilia. Thromb Haemost. 1997;78:1426–1429.
115. Leroyer C, et al. Prevalence of 20210 A allele of the prothrombin gene in venous thromboembolism patients. Thromb Haemost. 1998;80:49–51.
116. Dahlbäck B, et al. Familial thrombophilia due to a previously unrecognized mechanism characterized by poor anticoagulant response to activated protein C: prediction of a cofactor to activated protein C. Proc Natl Acad Sci U S A. 1993;90:1004–1008.
117. Bertina RM, et al. Mutation in blood coagulation factor V associated with resistance to activated protein C. Nature. 1994;369:64–67.
118. Thorelli E, et al. Cleavage of factor V at Arg 506 by activated protein C and the expression of anticoagulant activity of factor V. Blood. 1999;93:2552–2558.
119. Koster T, et al. Activated protein C resistance in venous thrombosis. Lancet. 1994;343:541.
120. Irani-Hakime N, et al. The prevalence of factor V R506Q mutation–Leiden among apparently healthy Lebanese. Am J Hematol. 2000;65:45–49.
121. Rees DC, et al. World distribution of factor V leiden. Lancet. 1995;346:1133–1134.
122. Ridker PM, et al. Ethnic distribution of factor V Leiden in 4047 men and women. Implications for venous thromboembolism screening. JAMA. 1997;277:1305–1307.
123. Ho WK, et al. Risk of recurrent venous thromboembolism in patients with common thrombophilia: a systematic review. Arch Intern Med. 2006;166:729–736.
124. Rosendaal FR, et al. High risk of thrombosis in patients homozygous for factor V leiden (activated protein C resistance). Blood. 1995;85:1504–1508.
125. Franchini M, et al. Inherited thrombophilia. Crit Rev Clin Lab Sci. 2006;43:249–290.
126. Bick RL. Prothrombin G20210A mutation, antithrombin, heparin cofactor II, protein C, and protein S defects. Hematol Oncol Clin North Am. 2003;17:9–36.
127. Walker FJ. Protein S and the regulation of activated protein C. Semin Thromb Hemost. 1984;10:131–138.
128. Esmon CT, et al. Protein C activation. Semin Thromb Hemost. 1984;10:122–130.
129. Tait RC, et al. Prevalence of protein C deficiency in the healthy population. Thromb Haemost. 1995;73:87–93.
130. Mateo J, et al. Laboratory evaluation and clinical characteristics of 2,132 consecutive unselected patients with venous thromboembolism—results of the Spanish Multicentric Study on Thrombophilia (EMET-Study). Thromb Haemost. 1997;77:444–451.
131. Koster T, et al. Protein C deficiency in a controlled series of unselected outpatients: an infrequent but clear risk factor for venous thrombosis (Leiden Thrombophilia Study). Blood. 1995;85:2756–2761.
132. Marlar RA, et al. Neonatal purpura fulminans due to homozygous protein C or protein S deficiencies. Semin Thromb Hemost. 1990;16:299–309.
133. Vigano D’Angelo S, et al. Relationship between protein C antigen and anticoagulant activity during oral anticoagulation and in selected disease states. J Clin Invest. 1986;77:416–425.
134. McGehee WG, et al. Coumarin necrosis associated with hereditary protein C deficiency. Ann Intern Med. 1984;101:59–60.
135. Fourrier F, et al. Meningococcemia and purpura fulminans in adults: acute deficiencies of proteins C and S and early treatment with antithrombin III concentrates. Intensive Care Med. 1990;16:121–124.
136. Buchanan GS, et al. The inherited thrombophilias: genetics, epidemiology, and laboratory evaluation. Best Pract Res Clin Obstet Gynaecol. 2003;17:397–411.
137. Franco RF, et al. Genetic risk factors of venous thrombosis. Hum Genet. 2001;109:369–384.
138. Gladson CL, et al. The frequency of type I heterozygous protein S and protein C deficiency in 141 unrelated young patients with venous thrombosis. Thromb Haemost. 1988;59:18–22.
139. Martinelli I, et al. Different risks of thrombosis in four coagulation defects associated with inherited thrombophilia: a study of 150 families. Blood. 1998;92:2353–2358.
140. Sallah S, et al. Recurrent warfarin-induced skin necrosis in kindreds with protein S deficiency. Haemostasis. 1998;28:25–30.
141. Schafer AI, et al. Thrombotic disorders: diagnosis and treatment. Hematology Am Soc Hematol Educ Program. 2003;520–539.
142. Rosendaal FR. Risk factors for venous thrombotic disease. Thromb Haemost. 1999;82:610–619.
143. Das M, et al. Antithrombin III deficiency: an etiology of Budd-Chiari syndrome. Surgery. 1985;97:242–246.
144. den Heijer M, et al. Hyperhomocysteinemia as a risk factor for deep-vein thrombosis. N Engl J Med. 1996;334:759–762.
145. Lee R, et al. Hyperhomocysteinemia and thrombosis. Hematol Oncol Clin North Am. 2003;17:85–102.
146. Tonkin A, et al. Effects of fenofibrate on cardiovascular events in patients with diabetes, with and without prior cardiovascular disease: the Fenofibrate Intervention and Event Lowering in Diabetes (FIELD) study. Am Heart J. 2012;163:508–514.
147. Crowther MA, et al. Congenital thrombophilic states associated with venous thrombosis: a qualitative overview and proposed classification system. Ann Intern Med. 2003;138:128–134.
148. Roberts HR, et al. The dysfibrinogenaemias. Br J Haematol. 2001;114:249–257.
149. van Hylckama Vlieg A, et al. High levels of factor IX increase the risk of venous thrombosis. Blood. 2000;95:3678–3682.
150. Meijers JC, et al. High levels of coagulation factor XI as a risk factor for venous thrombosis. N Engl J Med. 2000;342:696–701.
151. Rosendaal FR. High levels of factor VIII and venous thrombosis. Thromb Haemost. 2000;83:1–2.
152. O’Donnell J, et al. Elevated plasma factor VIII levels—a novel risk factor for venous thromboembolism. Clin Lab. 2001;47:1–6.
153. Couturaud F, et al. Factors that predict risk of thrombosis in relatives of patients with unprovoked venous thromboembolism. Chest. 2009;136:1537–1545.
154. Kujovich JL. Hormones and pregnancy: thromboembolic risks for women. Br J Haematol. 2004;126:443–454.
155. James AH. Prevention and management of venous thromboembolism in pregnancy. Am J Med. 2007;120:S26–S34.
156. Friederich PW, et al. Frequency of pregnancy-related venous thromboembolism in anticoagulant factor–deficient women: implications for prophylaxis. Ann Intern Med. 1996;125:955–960.
157. Rutherford S, et al. Thromboembolic disease associated with pregnancy: an 11-year review. Am J Obstet Gynecol. 1991;164S:286.
158. Bates SM, et al. Use of antithrombotic agents during pregnancy: the Seventh ACCP Conference on Antithrombotic and Thrombolytic Therapy. Chest. 2004;126:644.
159. Ginsberg JS, et al. Venous thrombosis during pregnancy: leg and trimester of presentation. Thromb Haemost. 1992;67:519–520.
160. Eldor A. Thrombophilia and its treatment in pregnancy. J Thromb Thrombolysis. 2001;12:23–30.
161. Malm J, et al. Changes in the plasma levels of vitamin K–dependent proteins C and S and of C4b-binding protein during pregnancy and oral contraception. Br J Haematol. 1988;68:437–443.
162. Alving BM, et al. Recent advances in understanding clotting and evaluating patients with recurrent thrombosis. Am J Obstet Gynecol. 1992;167:1184–1191.
163. Brenner B. Haemostatic changes in pregnancy. Thromb Res. 2004;114:409–414.
164. Harvey D, et al. Factor V Leiden: association with venous thromboembolism in pregnancy and screening issues. Br J Biomed Sci. 2004;61:157–164.
165. McColl MD, et al. Risk factors for pregnancy associated venous thromboembolism. Thromb Haemost. 1997;78:1183–1188.
167. Prandoni P, et al. Symptomatic deep-vein thrombosis and the post-thrombotic syndrome. Haematologica. 1995;80:42–48.
168. Koster T, et al. Oral contraceptives and venous thromboembolism: a quantitative discussion of the uncertainties. J Intern Med. 1995;238:31–37.
169. Raskob G. Calf-vein thrombosis. Hull RD, et al. Venous thromboembolism: an evidence-based atlas. Futura: Armonk, NY; 1996:307.
170. Inman WH, et al. Investigation of deaths from pulmonary, coronary, and cerebral thrombosis and embolism in women of child-bearing age. Br Med J. 1968;2:193–199.
171. Rees DC, et al. World distribution of factor V Leiden. Lancet. 1995;346:1133–1134.
172. Greene GR, et al. Oral contraceptive use in patients with thromboembolism following surgery, trauma, or infection. Am J Public Health. 1972;62:680–685.
173. Pabinger I, et al. Thrombotic risk of women with hereditary antithrombin III–, protein C– and protein S–deficiency taking oral contraceptive medication. The GTH Study Group on Natural Inhibitors. Thromb Haemost. 1994;71:548–552.
174. Hellgren M, et al. Resistance to activated protein C as a basis for venous thromboembolism associated with pregnancy and oral contraceptives. Am J Obstet Gynecol. 1995;173:210–213.
175. Bloemenkamp KW, et al. Enhancement by factor V Leiden mutation of risk of deep-vein thrombosis associated with oral contraceptives containing a third-generation progestagen. Lancet. 1995;346:1593–1596.
176. Helmerhorst FM, et al. Oral contraceptives and thrombotic disease: risk of venous thromboembolism. Thromb Haemost. 1997;78:327–333.
177. Vandenbroucke JP, et al. Third-generation oral contraceptive and deep venous thrombosis: from epidemiologic controversy to new insight in coagulation. Am J Obstet Gynecol. 1997;177:887–891.
178. Farmer RD, et al. Population-based study of risk of venous thromboembolism associated with various oral contraceptives. Lancet. 1997;349:83–88.
179. Grodstein F, et al. Prospective study of exogenous hormones and risk of pulmonary embolism in women. Lancet. 1996;348:983–987.
180. Varas-Lorenzo C, et al. Hormone replacement therapy and the risk of hospitalization for venous thromboembolism: a population-based study in southern Europe. Am J Epidemiol. 1998;147:387–390.
181. Jick H, et al. Risk of hospital admission for idiopathic venous thromboembolism among users of postmenopausal oestrogens. Lancet. 1996;348:981–983.
182. Daly E, et al. Case-control study of venous thromboembolism risk in users of hormone replacement therapy. Lancet. 1996;348:1027.
183. Perez Gutthann S, et al. Hormone replacement therapy and risk of venous thromboembolism: population based case-control study. BMJ. 1997;314:796–800.
184. Scarabin PY, et al. Changes in haemostatic variables induced by oral contraceptives containing 50 micrograms or 30 micrograms oestrogen: absence of dose-dependent effect on PAI-1 activity. Thromb Haemost. 1995;74:928–932.
185. Quehenberger P, et al. Increased levels of activated factor VII and decreased plasma protein S activity and circulating thrombomodulin during use of oral contraceptives. Thromb Haemost. 1996;76:729–734.
186. Von Kaulla E, et al. Antithrombin 3 depression and thrombin generation acceleration in women taking oral contraceptives. Am J Obstet Gynecol. 1971;109:868–873.
187. Winkler UH, et al. Ethinylestradiol 20 versus 30 micrograms combined with 150 micrograms desogestrel: a large comparative study of the effects of two low-dose oral contraceptives on the hemostatic system. Gynecol Endocrinol. 1996;10:265–271.
188. Blann AD, et al. The influence of age, gender and ABO blood group on soluble endothelial cell markers and adhesion molecules. Br J Haematol. 1996;92:498–500.
189. Nordstrom M, et al. A prospective study of the incidence of deep-vein thrombosis within a defined urban population. J Intern Med. 1992;232:155–160.
190. Wautrecht JC, et al. The role of ABO blood groups in the incidence of deep vein thrombosis. Thromb Haemost. 1998;79:688–689.
191. Mourant AE, et al. Blood-groups and blood-clotting. Lancet. 1971;1:223–227.
192. Blann AD, et al. The influence of age, gender and ABO blood group on soluble endothelial cell markers and adhesion molecules. Br J Haematol. 1996;92:498–500.
193. Kniffin WD, et al. The epidemiology of diagnosed pulmonary embolism and deep venous thrombosis in the elderly. Arch Intern Med. 1994;154:861–866.
194. Thomas WA, et al. Incidence of myocardial infarction correlated with venous and pulmonary thrombosis and embolism. A geographic study based on autopsies in Uganda, East Africa and St. Louis, U.S.A. Am J Cardiol. 1960;5:41–47.
195. Gore I, et al. Myocardial infarction and thromboembolism: a comparative study in Boston and in Kyushu, Japan. Arch Intern Med. 1964;113:323–330.
196. Aiach M, et al. A review of mutations causing deficiencies of antithrombin, protein C and protein S. Thromb Haemost. 1995;74:81–89.
197. Hingorani A, et al. Upper extremity deep venous thrombosis and its impact on morbidity and mortality rates in a hospital-based population. J Vasc Surg. 1997;26:853–860.
198. Horattas MC, et al. Changing concepts of deep venous thrombosis of the upper extremity—report of a series and review of the literature. Surgery. 1988;104:561–567.
199. Meredith JW, et al. Femoral catheters and deep venous thrombosis: a prospective evaluation with venous duplex sonography. J Trauma. 1993;35:187–190.
200. Xiang DZ, et al. Composition and formation of the sleeve enveloping a central venous catheter. J Vasc Surg. 1998;28:260–271.
201. Monreal M, et al. Pulmonary embolism in patients with upper extremity DVT associated to venous central lines—a prospective study. Thromb Haemost. 1994;72:548–550.
202. Efsing HO, et al. Thromboembolic complications from central venous catheters: a comparison of three catheter materials. World J Surg. 1983;7:419–423.
203. Heit JA, et al. Predictors of survival after deep vein thrombosis and pulmonary embolism: a population-based, cohort study. Arch Intern Med. 1999;159:445–453.
204. Koenigs KP, et al. Thrombosis in inflammatory bowel disease. J Clin Gastroenterol. 1987;9:627–631.
205. Jackson LM, et al. Thrombosis in inflammatory bowel disease: clinical setting, procoagulant profile and factor V leiden. QJM. 1997;90:183–188.
206. Vecchi M, et al. Risk of thromboembolic complications in patients with inflammatory bowel disease. Study of hemostasis measurements. Int J Clin Lab Res. 1991;21:165–170.
207. Hughes GR, et al. The anticardiolipin syndrome. J Rheumatol. 1986;13:486–489.
208. Love PE, et al. Antiphospholipid antibodies: anticardiolipin and the lupus anticoagulant in systemic lupus erythematosus (SLE) and in non-SLE disorders. Prevalence and clinical significance. Ann Intern Med. 1990;112:682–698.
209. Wahl DG, et al. Risk for venous thrombosis related to antiphospholipid antibodies in systemic lupus erythematosus—a meta-analysis. Lupus. 1997;6:467–473.
210. Nicolaides AN, et al. The origin of deep vein thrombosis: a venographic study. Br J Radiol. 1971;44:653–663.
211. Maurer BJ, et al. Frequency of venous thrombosis after myocardial infarction. Lancet. 1971;2:1385–1387.
212. Warlow C, et al. Deep venous thrombosis of the legs after strokes: part 2—natural history. Br Med J. 1976;1:1181–1183.
213. Campbell B. Thrombosis, phlebitis, and varicose veins. BMJ. 1996;312:198–199.
214. Vessey MP, et al. Oral contraceptives and thromboembolic disease. Br Med J. 1968;2:696.
215. Heit JA, et al. The epidemiology of venous thromboembolism in the community. Thromb Haemost. 2001;86:452–463.
216. May R, et al. The cause of the predominantly sinistral occurrence of thrombosis of the pelvic veins. Angiology. 1957;8:419–427.
217. Virchow R. Über die Erweiterung kleiner Defässe. Arch Pathol Anat. 1851;3:427.
218. Cockett FB, et al. The iliac compression syndrome. Br J Surg. 1965;52:816–821.
219. Knipp BS, et al. Factors associated with outcome after interventional treatment of symptomatic iliac vein compression syndrome. J Vasc Surg. 2007;46:743–749.
220. Kibbe MR, et al. Iliac vein compression in an asymptomatic patient population. J Vasc Surg. 2004;39:937–943.
221. Wolpert LM, et al. Magnetic resonance venography in the diagnosis and management of May-Thurner syndrome. Vasc Endovascular Surg. 2002;36:51–57.
222. Chung JW, et al. Acute iliofemoral deep vein thrombosis: evaluation of underlying anatomic abnormalities by spiral CT venography. J Vasc Interv Radiol. 2004;15:249–256.
223. Moreland NC, et al. Decreased incidence of left common iliac vein compression in patients with abdominal aortic aneurysms. J Vasc Surg. 2006;44:595–600.
224. Joffe HV, et al. Upper-extremity deep vein thrombosis. Circulation. 2002;106:1874–1880.
225. Shebel ND, et al. Effort thrombosis (Paget-Schroetter syndrome) in active young adults: current concepts in diagnosis and treatment. J Vasc Nurs. 2006;24:116–126.
226. Paget J. Clinical lectures and essays. Longmans Green: London; 1875.
227. von Schroetter L. Handbuch der allgemeinen Pathologie und Therapie. Nothnagel: Berlin; 1884.
228. Oktar GL, et al. Paget-Schroetter syndrome. Hong Kong Med J. 2007;13:243–245.
229. Melby SJ, et al. Comprehensive surgical management of the competitive athlete with effort thrombosis of the subclavian vein (Paget-Schroetter syndrome). J Vasc Surg. 2008;47:809–820.
230. Lee AYY, et al. Venous thrombosis of the upper extremities. Curr Treat Options Cardiovasc Med. 2001;3:207–214.
231. Elman EE, et al. The post-thrombotic syndrome after upper extremity deep venous thrombosis in adults: a systematic review. Thromb Res. 2006;117:609–614.
232. Murray A, et al. Popliteal artery entrapment syndrome. Br J Surg. 1991;78:1414–1419.
233. Liu PT, et al. Popliteal vascular entrapment syndrome caused by a rare anomalous slip of the lateral head of the gastrocnemius muscle. Skeletal Radiol. 2005;34:359–363.
234. Leconte D, et al. An uncommon syndrome: the trapped popliteal vein. Apropos of a case. J Chir (Paris). 1986;123:358–361.
235. Scarborough J, et al. Stenosis of the popliteal vein caused by an osteochondroma of the distal femur: a case report. Diagn Imaging. 1979;48:167–170.
236. Gerkin TM, et al. Popliteal vein entrapment presenting as deep venous thrombosis and chronic venous insufficiency. J Vasc Surg. 1993;18:760–766.
237. Chee YL, et al. Inferior vena cava malformation as a risk factor for deep venous thrombosis in the young. Br J Haematol. 2001;114:878–880.
238. Ruggeri M, et al. Congenital absence of the inferior vena cava: a rare risk factor for idiopathic deep-vein thrombosis. Lancet. 2001;357:441.
239. Obernosterer A, et al. Anomalies of the inferior vena cava in patients with iliac venous thrombosis. Ann Intern Med. 2002;136:37–41.
240. Lensing AW, et al. Detection of deep-vein thrombosis by real-time B-mode ultrasonography. N Engl J Med. 1989;320:342–345.
241. Linnemann B, et al. Etiology and VTE risk factor distribution in patients with inferior vena cava thrombosis. Thromb Res. 2008;123:72–78.
242. Nicolaides AN, et al. Myocardial infarction and deep-vein thrombosis. Br Med J. 1971;1:432–434.
243. Kotilainen M, et al. Leg vein thrombosis diagnosed by 125I-fibrinogen test after acute myocardial infarction. Ann Clin Res. 1973;5:365–368.
244. Simmons AV, et al. Deep venous thrombosis after myocardial infarction. Predisposing factors. Br Heart J. 1973;35:623–625.
245. Stewart GJ. Neutrophils and deep venous thrombosis. Haemostasis. 1993;23(Suppl 1):127–140.
246. Dennis JW, et al. Reassessing the role of arteriograms in the management of posterior knee dislocations. J Trauma. 1993;35:692–695.
247. Sevitt S, et al. Venous thrombosis and pulmonary embolism. A clinico-pathological study in injured and burned patients. Br J Surg. 1961;48:475–489.
248. Stamatakis JD, et al. Femoral vein thrombosis and total hip replacement. Br Med J. 1977;2:223–225.
249. Aronson DL, et al. Experimental studies on venous thrombosis: effect of coagulants, procoagulants and vessel contusion. Thromb Haemost. 1985;54:866–870.
250. Thomas DP, et al. The relationship between vessel wall injury and venous thrombosis: an experimental study. Br J Haematol. 1985;59:449–457.
251. Becker BF, et al. Endothelial function and hemostasis. Z Kardiol. 2000;89:160–167.
252. Wakefield TW, et al. Mechanisms of venous thrombosis and resolution. Arterioscler Thromb Vasc Biol. 2008;28:387–391.
253. Esmon CT. Inflammation and thrombosis. J Thromb Haemost. 2003;1:1343–1348.
254. Myers D Jr, et al. Selectins influence thrombosis in a mouse model of experimental deep venous thrombosis. J Surg Res. 2002;108:212–221.
255. Ay C, et al. High concentrations of soluble P-selectin are associated with risk of venous thromboembolism and the P-selectin Thr715 variant. Clin Chem. 2007;53:1235–1243.
256. Myers DD, et al. P-selectin and leukocyte microparticles are associated with venous thrombogenesis. J Vasc Surg. 2003;38:1075–1089.
257. Myers DD, et al. Treatment with an oral small molecule inhibitor of P selectin (PSI-697) decreases vein wall injury in a rat stenosis model of venous thrombosis. J Vasc Surg. 2006;44:625–632.
258. Myers DD, et al. Inflammation-dependent thrombosis. Front Biosci. 2005;10:2750–2757.
259. Andre P, et al. Pro-coagulant state resulting from high levels of soluble P-selectin in blood. Proc Natl Acad Sci U S A. 2000;97:13835–13840.
260. Hrachovinova I, et al. Interaction of P-selectin and PSGL-1 generates microparticles that correct hemostasis in a mouse model of hemophilia A. Nat Med. 2003;9:1020–1025.
261. Jy W, et al. Platelet factor 3 in plasma fractions: its relation to microparticle size and thromboses. Thromb Res. 1995;80:471–482.
262. Biro E, et al. Human cell–derived microparticles promote thrombus formation in vivo in a tissue factor–dependent manner. J Thromb Haemost. 2003;1:2561–2568.
263. Day SM, et al. Macrovascular thrombosis is driven by tissue factor derived primarily from the blood vessel wall. Blood. 2005;105:192–198.
264. Rectenwald JE, et al. D-dimer, P-selectin, and microparticles: novel markers to predict deep venous thrombosis. A pilot study. Thromb Haemost. 2005;94:1312–1317.
265. Booth NA, et al. Plasminogen activator inhibitor (PAI-1) in plasma and platelets. Br J Haematol. 1988;70:327–333.
266. Podor TJ, et al. Vimentin exposed on activated platelets and platelet microparticles localizes vitronectin and plasminogen activator inhibitor complexes on their surface. J Biol Chem. 2002;277:7529–7539.
267. Amulic B, et al. Neutrophil extracellular traps. Curr Biol. 2011;21:R297–R298.
268. Fuchs TA, et al. Neutrophil extracellular trap (net) impact on deep vein thrombosis. Arterioscler Thromb Vasc Biol. 2012;32:1777–1783.
269. Brill A, et al. Neutrophil extracellular traps promote deep vein thrombosis in mice. J Thromb Haemost. 2012;10:136–144.
270. Adelman B, et al. Plasmin effect on platelet glycoprotein Ib–von Willebrand factor interactions. Blood. 1985;65:32–40.
271. Dano K, et al. Plasminogen activators, tissue degradation, and cancer. Adv Cancer Res. 1985;44:139–266.
273. Crowther MA, et al. Fibrinolytic variables in patients with recurrent venous thrombosis: a prospective cohort study. Thromb Haemost. 2001;85:390–394.
274. Schulman S, et al. The significance of hypofibrinolysis for the risk of recurrence of venous thromboembolism. Duration of Anticoagulation (DURAC) Trial Study Group. Thromb Haemost. 1996;75:607–611.
275. Wakefield TW, et al. Venous thrombosis–associated inflammation and attenuation with neutralizing antibodies to cytokines and adhesion molecules. Arterioscler Thromb Vasc Biol. 1995;15:258–268.
276. Varma MR, et al. Neutropenia impairs venous thrombosis resolution in the rat. J Vasc Surg. 2003;38:1090–1098.
277. Humphries J, et al. Monocyte chemotactic protein-1 (MCP-1) accelerates the organization and resolution of venous thrombi. J Vasc Surg. 1999;30:894–899.
278. Henke PK, et al. Targeted deletion of CCR2 impairs deep vein thrombosis resolution in a mouse model. J Immunol. 2006;177:3388–3397.
279. Henke PK, et al. Fibrotic injury after experimental deep vein thrombosis is determined by the mechanism of thrombogenesis. Thromb Haemost. 2007;98:1045–1055.
280. Deatrick KB, et al. Vein wall remodeling after deep vein thrombosis involves matrix metalloproteinases and late fibrosis in a mouse model. J Vasc Surg. 2005;42:140–148.
281. Sevitt S. Organization of valve pocket thrombi and the anomalies of double thrombi and valve cusp involvement. Br J Surg. 1974;61:641–649.
282. Hamer JD, et al. The PO2 in venous valve pockets: its possible bearing on thrombogenesis. Br J Surg. 1981;68:166–170.
283. Shreeniwas R, et al. Hypoxia-mediated induction of endothelial cell interleukin-1 alpha. An autocrine mechanism promoting expression of leukocyte adhesion molecules on the vessel surface. J Clin Invest. 1992;90:2333–2339.
284. Thomas DP, et al. The effect of stasis on the venous endothelium: an ultrastructural study. Br J Haematol. 1983;55:113–122.
285. Thomas D. Venous thrombogenesis. Br Med Bull. 1994;50:803–812.
286. Amiral J, et al. Thromboembolic diseases: biochemical mechanisms and new possibilities of biological diagnosis. Semin Thromb Hemost. 1996;22(Suppl 1):41–48.
287. Bauer KA, et al. The pathophysiology of the prethrombotic state in humans: insights gained from studies using markers of hemostatic system activation. Blood. 1987;70:343–350.
288. Wakefield TW. Venous thrombosis. Bope ET, et al. Conn’s current therapy 2011. WB Saunders: Philadelphia; 2011.
289. Palareti G, et al. D-dimer testing to determine the duration of anticoagulation therapy. N Engl J Med. 2006;355:1780–1789.
290. Rosendaal FR. Thrombosis in the young: epidemiology and risk factors. A focus on venous thrombosis. Thromb Haemost. 1997;78:1–6.
291. Wakefield TW, et al. Inflammatory and procoagulant mediator interactions in an experimental baboon model of venous thrombosis. Thromb Haemost. 1993;69:164–172.
292. Owen J, et al. Thrombin and plasmin activity and platelet activation in the development of venous thrombosis. Blood. 1983;61:476–482.
293. Ridker PM, et al. Age-specific incidence rates of venous thromboembolism among heterozygous carriers of factor V Leiden mutation. Ann Intern Med. 1997;126:528–531.
294. Karino T, et al. Flow through a venous valve and its implication for thrombus formation. Thromb Res. 1984;36:245–257.
295. Sevitt S. Organization of valve pocket thrombi and the anomalies of double thrombi and valve cusp involvement. Br J Surg. 1974;61:641–649.
296. Sevitt S. The structure and growth of valve-pocket thrombi in femoral veins. J Clin Pathol. 1974;27:517–528.
297. Doouss TW. The clinical significance of venous thrombosis of the calf. Br J Surg. 1976;63:377–378.
298. Huisman MV, et al. Utility of impedance plethysmography in the diagnosis of recurrent deep-vein thrombosis. Arch Intern Med. 1988;148:681–683.
299. Killewich LA, et al. Regression of proximal deep venous thrombosis is associated with fibrinolytic enhancement. J Vasc Surg. 1997;26:861–868.
300. van Ramshorst B, et al. Thrombus regression in deep venous thrombosis. Quantification of spontaneous thrombolysis with duplex scanning. Circulation. 1992;86:414–419.
301. Prandoni P, et al. A simple ultrasound approach for detection of recurrent proximal-vein thrombosis. Circulation. 1993;88:1730–1735.
302. Killewich LA, et al. Spontaneous lysis of deep venous thrombi: rate and outcome. J Vasc Surg. 1989;9:89–97.
303. Hull R, et al. Warfarin sodium versus low-dose heparin in the long-term treatment of venous thrombosis. N Engl J Med. 1979;301:855–858.
304. Hull RD, et al. Continuous intravenous heparin compared with intermittent subcutaneous heparin in the initial treatment of proximal-vein thrombosis. N Engl J Med. 1986;315:1109–1114.
305. Philbrick JT, et al. Calf deep venous thrombosis. A wolf in sheep’s clothing? Arch Intern Med. 1988;148:2131–2138.
306. Kearon C, et al. Antithrombotic therapy for VTE disease: Antithrombotic Therapy and Prevention of Thrombosis, 9th ed: American College of Chest Physicians Evidence-Based Clinical Practice Guidelines. Chest. 2012;141:e419S–494S.
307. Krupski WC, et al. Propagation of deep venous thrombosis identified by duplex ultrasonography. J Vasc Surg. 1990;12:467–474.
308. Meissner MH, et al. Propagation, rethrombosis and new thrombus formation after acute deep venous thrombosis. J Vasc Surg. 1995;22:558–567.
309. Buller HR, et al. Treatment of venous thromboembolism. J Thromb Haemost. 2005;3:1554–1560.
310. Girolami A, et al. The pathogenesis of venous thromboembolism. Haematologica. 1995;80:25–35.
311. Barritt DW, et al. Anticoagulant drugs in the treatment of pulmonary embolism. A controlled trial. Lancet. 1960;1:1309–1312.
312. Campbell CB, et al. Axillary, subclavian, and brachiocephalic vein obstruction. Surgery. 1977;82:816–826.
313. Donayre CE, et al. Pathogenesis determines late morbidity of axillosubclavian vein thrombosis. Am J Surg. 1986;152:179–184.
314. Gosselin MV, et al. Unsuspected pulmonary embolism: prospective detection on routine helical CT scans. Radiology. 1998;208:209–215.
315. Cronin CG, et al. Prevalence and significance of asymptomatic venous thromboembolic disease found on oncologic staging CT. AJR Am J Roentgenol. 2007;189:162–170.
316. Heit JA. The epidemiology of venous thromboembolism in the community. Arterioscler Thromb Vasc Biol. 2008;28:370–372.
317. Dalen JE, et al. Natural history of pulmonary embolism. Prog Cardiovasc Dis. 1975;17:259–270.
318. Piovella F, et al. Chronic thromboembolic pulmonary hypertension. Semin Thromb Hemost. 2006;32:848–855.
319. Pengo V, et al. Incidence of chronic thromboembolic pulmonary hypertension after pulmonary embolism. N Engl J Med. 2004;350:2257–2264.
320. McNeil K, et al. Chronic thromboembolic pulmonary hypertension (CTEPH). Heart. 2007;93:1152–1158.
321. Auger WR, et al. Chronic thromboembolic pulmonary hypertension. Cardiol Clin. 2004;22:453–466.
322. Monreal M, et al. Venographic assessment of deep vein thrombosis and risk of developing post-thrombotic syndrome: a prospective study. J Intern Med. 1993;233:233–238.
323. Lindner DJ, et al. Long-term hemodynamic and clinical sequelae of lower extremity deep vein thrombosis. J Vasc Surg. 1986;4:436–442.
324. Prandoni P, et al. The long-term clinical course of acute deep venous thrombosis. Ann Intern Med. 1996;125:1–7.
325. Prandoni P, et al. Symptomatic deep-vein thrombosis and the post-thrombotic syndrome. Haematologica. 1995;80:42–48.
326. Strandness DE, et al. Long-term sequelae of acute venous thrombosis. JAMA. 1983;250:1289–1292.
327. Meissner MH, et al. Acute venous disease: venous thrombosis and venous trauma. J Vasc Surg. 2007;46(Suppl S):25S–53S.
329. Johnson BF, et al. Relationship between changes in the deep venous system and the development of the postthrombotic syndrome after an acute episode of lower limb deep vein thrombosis: a one- to six-year follow-up. J Vasc Surg. 1995;21:307–312.
330. Glas-Greenwalt P, et al. Localization of tissue plasminogen activator in relation to morphologic changes in human saphenous veins used as coronary artery bypass autografts. Ann Surg. 1975;181:431–441.
331. Budd TW, et al. Histopathology of veins and venous valves of patients with venous insufficiency syndrome: ultrastructure. J Med. 1990;21:181–199.
332. Carrier M, et al. Residual vein obstruction to predict the risk of recurrent venous thromboembolism in patients with deep vein thrombosis: a systematic review and meta-analysis. J Thromb Haemost. 2011;9:1119–1125.
333. Enden T, et al. Long-term outcome after additional catheter-directed thrombolysis versus standard treatment for acute iliofemoral deep vein thrombosis (the CaVenT study): a randomised controlled trial. Lancet. 2012;379:31–38.
334. Ageno W, et al. Body mass index is associated with the development of the post-thrombotic syndrome. Thromb Haemost. 2003;89:305–309.
335. Meissner MH, et al. Deep venous insufficiency: the relationship between lysis and subsequent reflux. J Vasc Surg. 1993;18:596–605.
336. Mohr DN, et al. The venous stasis syndrome after deep venous thrombosis or pulmonary embolism: a population-based study. Mayo Clin Proc. 2000;75:1249–1256.
337. Piccioli A, et al. Epidemiologic characteristics, management, and outcome of deep venous thrombosis in a tertiary-care hospital: The Brigham and Women’s Hospital DVT registry. Am Heart J. 1996;132:1010–1014.
338. Baglin T, et al. Incidence of recurrent venous thromboembolism in relation to clinical and thrombophilic risk factors: prospective cohort study. Lancet. 2003;362:523–526.
339. Meissner MH, et al. Determinants of chronic venous disease after acute deep venous thrombosis. J Vasc Surg. 1998;28:826–833.
340. Lohr JM, et al. Lower extremity calf thrombosis: to treat or not to treat? J Vasc Surg. 1991;14:618–623.
341. Lagerstedt CI, et al. Need for long-term anticoagulant treatment in symptomatic calf-vein thrombosis. Lancet. 1985;2:515–518.
342. Kearon C. Natural history of venous thromboembolism. Circulation. 2003;107:22–30.
343. Passman MA, et al. Pulmonary embolism is associated with the combination of isolated calf vein thrombosis and respiratory symptoms. J Vasc Surg. 1997;25:39–45.
344. Kistner RL, et al. Incidence of pulmonary embolism in the course of thrombophlebitis of the lower extremities. Am J Surg. 1972;124:169–176.
345. Raskob G. Calf-vein thrombosis. Hull RD, et al. Venous thromboembolism. Futura: Armonk; 1996.
346. McLafferty RB, et al. Late clinical and hemodynamic sequelae of isolated calf vein thrombosis. J Vasc Surg. 1998;27:50–56.
347. Gillet JL, et al. Short-term and mid-term outcome of isolated symptomatic muscular calf vein thrombosis. J Vasc Surg. 2007;46:513–519.
348. Bates SM, et al. Diagnosis of DVT: Antithrombotic Therapy and Prevention of Thrombosis, 9th ed: American College of Chest Physicians Evidence-Based Clinical Practice Guidelines. Chest. 2012;141:e351S–418S.
[/level-membership-for-surgery-category][not-level-membership-for-surgery-category]Chapter 49
Acute Deep Venous Thrombosis
Pathophysiology and Natural History
Jordan P. Knepper, Thomas W. Wakefield
The complications of acute venous thromboembolism (VTE), including deep venous thrombosis (DVT), pulmonary embolism (PE), and the postthrombotic syndrome, are the most common preventable cause of hospital death1 and a source of substantial long-term morbidity.2–4 The impact on health is so great, the Surgeon General of the United States issued a Call to Action to aid in combating VTE.5 An understanding of the epidemiology, pathophysiology, and natural history of VTE is essential in guiding prophylaxis, diagnosis, and treatment. In addition, recognizing underlying risk factors and the multifactorial nature of VTE may aid in the identification of situations likely to provoke thrombosis in both high-risk individuals and those with unexplained thromboembolism. Furthermore, understanding of the natural history of VTE is important to define the relative risks and benefits of anticoagulation as well as the duration of treatment in individual patients.
Intimate to the pathophysiology is the triad initially described by Rudolf Virchow (stasis, endothelial damage or injury, and hypercoagulability). However, an improved understanding of the coagulation and fibrinolytic systems as well as of the role of the vascular endothelium and the inflammatory response in thrombosis and hemostasis has led to the identification of new prethrombotic conditions and also a more thorough appreciation of previously recognized risk factors. It now appears that many venous thrombi arise from the convergence of several risk factors on a background of an imbalance between coagulation and fibrinolysis. Similar factors define the balance between recanalization of the venous lumen and recurrent thrombotic events that may be important determinants of long-term outcome after an episode of acute venous thrombosis.
Epidemiology
Incidence
The incidence of recurrent fatal and nonfatal VTE is estimated to exceed 900,000 cases annually in the United States alone.6 A 35-year population-based study using the Rochester Epidemiology Project database of Olmsted County, Minnesota, demonstrated an overall average age- and sex-adjusted annual VTE incidence of 122 per 100,000 person-years (DVT, 56 per 100,000; PE, 66 per 100,000).7 This study also demonstrated higher age-adjusted rates among men than among women (134 vs 115 per 100,000 respectively). First-time VTE is approximated to occur in about 250,000 U.S. white individuals annually.8 Compared with other racial populations, whites have a lower incidence of VTE than African Americans (104 vs 141 per 100,000) and a higher incidence of VTE than Hispanics and Asian/Pacific Islanders combined (104 vs 21 per 100,000).9 However, the problem of VTE is not just isolated to the United States; it is a global issue. The estimates of VTE across the European Union were 684,019 cases of DVT and 434,723 cases of PE, with 543,454 VTE-related fatalities.10
Populations Affected
Although estimated incidences of VTE in the community and hospital have been well published, those individuals who are at risk for DVT are less well studied. One large epidemiologic study addressed this issue using the Seventh American College of Chest Physicians Conference on Antithrombotic and Thrombolytic Therapy guidelines for VTE prevention.11 Of the estimated 38 million patient discharges in 2003, 31% were considered to be at risk for VTE because of a major surgery or a medical illness.12
Similarly, the incidence of VTE varies with the population studied, the use of thromboprophylaxis, the intensity of screening, and the accuracy of the diagnostic test employed. For example, individuals with acute spinal cord injury who were screened systematically with venography demonstrated DVT at a rate of 81%.13 However, medical-surgical intensive care unit (ICU) patients who received thromboprophylaxis had a DVT rate reported at 10% to 18% compared with a rate of 25% to 32% for those who were not given DVT prophylaxis.14 Interestingly, the risk of VTE in the critically ill is not limited to the time actually spent in the ICU. A single-center study noted that of the VTEs diagnosed in the critically ill, 64% were diagnosed with a VTE after discharge from the ICU; prolonged immobility after discharge from the ICU may have contributed to the high rate of DVT.15 Similarly, prolonged immobility contributes to increased rates of VTE in another population of patients, nursing home residents.16 In summary, it appears that medical-surgical ICU patients are at lower risk for DVT compared with acute spinal cord injury, trauma, or neurosurgery patients; at a comparable risk to that of patients who have had major orthopedic surgery; and at higher risk than that of medical-surgical ward patients.14,17 Furthermore, a more recent study noted a high (15.2%) rate of DVT in critically ill trauma patients within the first week that did not vary regardless of whether prophylaxis was used.18
Recurrence
VTE is a devastating acute disease process, but it may be manifested chronically. Nearly 30% of patients have a recurrence in a 10-year time span.19 Recurrences are more likely with the same event type as that of the incident event, that is to say, those who initially developed a PE are more likely to develop another PE instead of a DVT.20
Mortality
The severity of PE is shown in 30-year autopsy studies, which demonstrated a 26% incidence of PE in hospitalized patients, of which 9% were fatal. This translates to a 1% incidence of PE and a 0.36% incidence of death from PE in all hospitalized patients per year.21
Risk Factors
DVT occurring in the setting of a recognized risk factor is often defined as a secondary event; in the absence of risk factors, it is termed primary or idiopathic.22 Known risk factors for DVT are listed in Table 49-1.
Table 49-1
Risk Factors for Acute Deep Venous Thrombosis and Pulmonary Embolism
| Risk Factor | Odds Ratio | 95% Confidence Interval |
| Hospitalization | ||
| With recent surgery | 21.72 | 9.44-49.93 |
| Without recent surgery | 7.98 | 4.49-14.18 |
| Trauma | 12.69 | 4.06-39.66 |
| Malignant neoplasm | ||
| With chemotherapy | 6.53 | 2.11-20.23 |
| Without chemotherapy | 4.05 | 1.93-8.52 |
| Prior central venous catheter or pacemaker | 5.55 | 1.57-19.58 |
| Prior superficial vein thrombosis | 4.32 | 1.76-10.61 |
| Neurologic disease with extremity paresis | 3.04 | 1.25-7.38 |
| Varicose veins | ||
| Age 45 y | 4.19 | 1.56-11.30 |
| Age 60 y | 1.93 | 1.03-3.61 |
| Age 75 y | 0.88 | 0.55-1.43 |
| Congestive heart failure | ||
| Thromboembolism not categorized as a cause of death at postmortem examination | 9.64 | 2.44-38.10 |
| Thromboembolism categorized as a cause of death at postmortem examination | 1.36 | 0.69-2.68 |
Modified from Heit JA, et al: Risk factors for deep vein thrombosis and pulmonary embolism: a population-based case-control study. Arch Intern Med 160:809, 2000.
The high incidence of acute DVT in hospitalized patients, the availability of objective diagnostic tests, and the existence of clinical trials evaluating prophylactic measures have helped to identify high-risk groups in this population. In contrast, the risk factors for acute DVT are less well defined in population-based studies. Nonetheless, substantive differences have been noted in the distribution of risk factors between inpatients and outpatients.23 Malignant disease, surgery, and trauma within the previous 3 months remain significant risk factors for outpatient thrombosis, whereas the frequency of surgery and malignant disease is higher among inpatients with DVT.24,25 Approximately 47% of outpatients with a documented DVT have one or more recognized risk factors,24 and the incidence of VTE increases with the number of risk factors.26 Although some investigators have found the risk of acute DVT to be significantly higher only for those with three or more risk factors,25 others have reported the relative risk to increase from 2.4 in those with one risk factor to more than 20 in patients who have three or more risk factors.27 Heit et al28,29 identified several risk factors for VTE, including increasing age, male gender, surgery, trauma, hospital or nursing home confinement, malignant disease, neurologic disease with extremity paresis, central venous catheter or transvenous pacemaker, prior superficial vein thrombosis, and varicose veins. Among women, additional risk factors include pregnancy, oral contraceptives, and hormone replacement therapy. Independent predictors for recurrent DVT, which occurs in 30% of people within 10 years, include increasing age, obesity, malignant neoplasm, and extremity paresis.
Age
VTE occurs in all ages, although a higher incidence has consistently been associated with advanced age. Some investigators have identified cutoff points at which the risk of DVT is raised, whereas others have reported a relative increase with advancing age. In a community-based study of phlebographically documented DVT, the yearly incidence of DVT was noted to increase progressively from almost 0 in childhood to 7.65 cases per 1000 in men and 8.22 cases per 1000 in women older than 80 years.30 The incidence of DVT increased 30-fold from those aged 30 years to those older than 80 years. Rosendaal27 similarly noted an incidence of 0.006 per 1000 children younger than 14 years, which rose to 0.7 per 1000 among adults 40 to 54 years old. Furthermore, Hansson et al31 found the prevalence of objectively documented thromboembolic events among men to increase from 0.5% at the age of 50 years to 3.8% at the age of 80 years.
The influence of age on the incidence of VTE is likely to be multifactorial. The number of thrombotic risk factors increases with age, three or more risk factors being present in only 3% of hospitalized patients younger than 40 years but in 30% of those 40 years and older.26 Interestingly, it also appears that the number of risk factors required to precipitate thrombosis decreases with age.27 This may be related to an acquired prethrombotic state associated with aging as higher levels of thrombin activation markers are found among older people.32 Advanced age also has been associated with anatomic changes in the soleal veins and more pronounced stasis in the venous valve pockets.33,34 We have also noted biologic changes with aging in our research laboratory, such as elevations in P-selectin, tissue factor, and procoagulant microparticles, supporting the effect of age on venous thrombogenesis.
Venous diseases, including VTE, are usually regarded as rare in young children,3 with an incidence of 0.006 per 1000 children younger than 14 years,27 whereas the incidence in hospitalized children younger than 18 years has been estimated to be 0.05%.35 Early mobilization and discharge may partially explain the lower incidence in children.35 However, the diagnosis is often not considered in pediatric patients, and few studies have systematically evaluated children for DVT. Over time, the number of recognized cases in children hospitalized has increased from 0.3 to 28.8 per 10,000 from 1992 to 2005.36
VTE in children is almost always associated with recognized thrombotic risk factors,27,37–39 and multiple risk factors are often required to precipitate thrombosis.27 Venous thrombosis is more common in those children hospitalized in the ICU, those with spinal cord injuries, and those undergoing prolonged orthopedic immobilization, although the incidence is substantially lower than in corresponding adult populations. DVT may occur in as many as 3.7% of pediatric patients immobilized in halo-femoral traction for preoperative treatment of scoliosis,40 4% of children hospitalized in the ICU,41 and 10% of children with spinal cord injuries.42,43 Symptomatic postoperative DVT is regarded as unusual in children, although there are few data from studies employing routine surveillance,44 and autopsy-identified PE is approximately four times more frequent in pediatric patients who have undergone surgery than in the general pediatric medical population.37 Other thrombotic risk factors in hospitalized children are local infection and trauma, immobilization,39 inherited hypercoagulable states,27 oral contraceptive use,45 lower limb paresis,40 and use of femoral venous catheters.46 In addition, the risk factors for inpatient DVT in children include catheters and concomitant severe respiratory, oncologic, and infectious diseases; outpatient DVT is often associated with a prior DVT and thrombophilia.36
Immobilization
Many of the clinical presentations of thromboembolism support immobilization as a risk factor for VTE. In addition, stasis in the soleal veins and behind the valve cusps is exacerbated by advancing age and inactivity of the calf muscle pump,34 both of which are associated with an increased risk of DVT. The prevalence of lower extremity DVT in autopsy studies also parallels the duration of bed rest, with an increase during the first 3 days of confinement and a rapid rise to high levels after 2 weeks. DVT was found in 15% of patients dying after 0 to 7 days of bed rest, in comparison with 79% to 94% of those dying after 2 to 12 weeks.33 Preoperative immobilization is also associated with a twofold increase in risk of postoperative DVT,47 and DVT among stroke patients is significantly more common in paralyzed or paretic extremities (53% of limbs) than in nonparalyzed limbs (7%).48 Patients with neurologic disease and extremity paresis or paralysis have a threefold higher risk for DVT and PE that appears to be independent of hospital confinement.28
Travel
Immobilization as a thrombotic risk factor extends to include prolonged travel, particularly the “economy class syndrome,” which occurs in people who have sat in a cramped position during extended aircraft flights.49 Several case series have reported the occurrence of PE in relation to extended travel,49–54 although none has rigorously examined the prevalence relative to that of the general population, and few have thoroughly reported the presence of other risk factors. A high prevalence of preexisting venous disease and other thrombotic risk factors in this group of patients has sometimes been noted.32,54 The question of prolonged travel as a risk factor is moderated by observations that extreme duration of venous stasis alone may fail to produce thrombosis55 and that no consistent rheologic or prothrombotic changes have been demonstrated during prolonged travel.32,56 However, PE is the second leading cause of travel-related death, accounting for 18% of 61 deaths, suggesting that a relationship cannot be excluded.53 After a consensus meeting, the World Health Organization published the following conclusions57:
• An association probably exists between air travel and venous thrombosis.
• Similar links may exist for other forms of travel.
• The available evidence does not permit an estimation of actual risk.
More evidence of a connection between travel and DVT and PE has accumulated. In a case-control study, Ferrari et al58 found that long-distance travel increased the risk of DVT with an odds ratio (OR) of 4.0, and Samama59 made similar observations (OR, 2.3). Scurr et al60 found a 10% risk of calf DVT in patients who traveled without compression stockings. Lapostolle et al61 observed that during an 86-month period, 56 of 135.3 million airline passengers had severe PE. The frequency among those who traveled more than 5000 km was 150 times as high as the frequency among those who traveled less than 5000 km. In another case-control study, Paganin et al62 observed a high incidence of VTE in patients with risk factors for DVT who traveled long distances. In particular, history of previous VTE (OR, 63.3), recent trauma (OR, 13.6), presence of varicose veins (OR, 10), obesity (OR, 9.6), immobility during flight (OR, 9.3), and cardiac disease (OR, 8.9) increased the risk of DVT. These investigators concluded that low mobility during flight was a striking modifiable risk factor for development of PE and that travelers with risk factors should increase their mobility.63
Hughes et al64 noted that the frequency of VTE is increased even in travelers otherwise at low to moderate risk. In a prospective study that involved 878 long-distance air travelers, the frequency of VTE was 1.0% (9 of 878; 95% confidence interval, 0.5-1.9), which included 4 cases of PE and 5 of DVT. Two of the patients traveled in business class, five used aspirin, and four wore compression stockings.
History of Venous Thromboembolism
Approximately 23% to 26% of patients presenting with acute DVT have a previous history of thrombosis,23,30 and histologic studies confirm that acute thrombi are often associated with fibrous remnants of previous thrombi in the same or nearby veins.65 Depending on sex and age, population-based studies have demonstrated that recurrent thromboembolism occurs in 2% to 9% of people with a previous episode of thromboembolism.3
The risk of recurrent thromboembolism is higher among patients with idiopathic DVT.66 In addition, primary hypercoagulability appears to have a significant role in many recurrences. Simioni et al66 reported the cumulative incidence of recurrent thrombosis among patients who are heterozygous for the factor V Leiden mutation to be 40% at 8 years of follow-up, 2.4-fold higher than in patients without the mutation, although the importance of heterozygous factor V Leiden to recurrent thrombosis has been questioned.67 den Heijer et al68 estimated that 17% of recurrent thromboembolic events may be due to hyperhomocysteinemia. A similar relationship between impaired fibrinolysis and recurrent DVT has been suggested by several investigators, although the methodologic validity of these findings has been questioned.69
Malignant Disease
Approximately 20% of all first-time VTE events are associated with malignant disease.70 An estimated 1 in 200 individuals with malignant disease will develop either DVT or PE, a fourfold higher risk than in those without malignant disease.28 Considering all-cause mortality of in-hospital death for cancer patients, one in seven will die of PE.71 Unfortunately, the discovery of an occult malignant neoplasm associated with an otherwise first-time idiopathic VTE is as high as 12% to 17% in some series.72,73 In another 5% to 11% of patients, malignant disease appears within 1 to 2 years of presentation for DVT.72–74 Related to this, several series have documented a significantly higher risk of malignant disease in patients with idiopathic DVT.72,75 Among such patients, 7.6% have been noted to have a malignant neoplasm during follow-up, with an odds ratio of 2.3 in comparison with those with secondary thrombosis.73 The incidence of occult malignant disease diagnosed within 6 to 12 months of an idiopathic DVT is 2.2 to 5.3 times higher than that expected from general population estimates.76,77 Furthermore, in individuals with recurrent idiopathic DVT, a malignant neoplasm can be identified 17% of the time.73 A correlation with location of tumor reveals that the highest rates of VTE are associated with pancreatic malignant neoplasms, followed by kidney, ovary, lung, and stomach malignant neoplasms.78
The underlying mechanisms contributing to the hypercoagulable state in malignant disease have been well studied and are multifactorial. Venous compression secondary to tumor growth, cancer-associated thrombocytosis, immobility, indwelling central lines, and chemotherapy or radiation therapy are risk factors that increase the possibility of VTE.79 However, the systemic prothrombotic response seen in malignant disease is mediated by cytokines, inhibitors of fibrinolysis, and procoagulants.80 Tumor cells can directly initiate hemostasis through constitutive expression of tissue factor. Tissue factor is not normally expressed on resting vascular endothelium but rather induced by chemical mediators during times of inflammation or vessel damage to bind factors VII and VIIa. This complex of tissue factor and factor VII activates factors X and XI through proteolysis, leading to the generation of thrombin.81 Another mediator in VTE associated with malignant disease is cancer procoagulant. The role of cancer procoagulant in coagulation is limited to its association with malignant disease as it has not been identified in normal healthy tissue. Cancer procoagulant serves as a direct activator of factor X independent of the presence of factor VIIa. Although cancer procoagulant has not been studied as intensely as tissue factor, the characterization of cancer procoagulant and hypercoagulability in acute myelogenous leukemia has been well published.82,83 The platelet adhesion molecules glycoprotein Ib and glycoprotein IIb/IIIa have also been identified on tumor cells. Such expression allows platelet activation and aggregation.84
The prothrombotic cytokines, such as vascular endothelial growth factor, tumor necrosis factor-α, and interleukin-1, mediate their actions through induction of tissue factor on vascular endothelium, monocytes, and leukocytes.81,82 In addition, interleukin-1 and tumor necrosis factor-α downregulate the expression of thrombomodulin, the receptor for thrombin, on the endothelial surface. This results in a decrease in thrombin-thrombomodulin complexes, the activating complex of protein C, a natural anticoagulant.81,82 Furthermore, interleukin-1 and tumor necrosis factor-α stimulate the production of plasminogen activator inhibitor-1 (PAI-1), the main physiologic inhibitor of fibrinolysis.85
As many as 90% of patients with cancer have abnormal coagulation parameters, including increased levels of coagulation factors, elevated fibrinogen or fibrin degradation products, and thrombocytosis.86 Elevated fibrinogen and thrombocytosis are the most common abnormalities, perhaps reflecting an overcompensated form of intravascular coagulation.86 Levels of the coagulation inhibitors antithrombin, protein C, and protein S also may be reduced in malignant disease.87
Markers of activated coagulation are elevated in the majority of patients with solid tumors and leukemia.86 Fibrinopeptide A levels reflect tumor activity, decreasing or increasing in response to treatment or progression of disease, suggesting that tumor growth and thrombin generation are intimately related.86 Furthermore, these levels may fail to normalize after administration of heparin to patients with cancer and DVT, perhaps explaining why these DVTs may be refractory to anticoagulants.86
VTE is also associated with the treatment of some cancers. DVT complicates 29% of general surgical procedures for malignant disease.87 Preoperative activation of the coagulation system, as reflected by elevated thrombin-antithrombin complex values, is associated with a 7.5-fold increased risk of postoperative DVT.86 Some chemotherapeutic regimens also predispose to DVT, and thrombotic complications may be as common as the more widely recognized infectious complications.88 VTE has been reported in up to 6% of patients undergoing treatment for non-Hodgkin’s lymphoma and in 17.5% of those receiving therapy for breast cancer.89,90 Among patients with stage II breast cancer, thrombosis was significantly more common in those randomly assigned to 36 weeks of chemotherapy (8.8%) than in those receiving only 12 weeks of treatment (4.9%).88 Potential thrombogenic mechanisms associated with chemotherapy include direct endothelial toxicity, induction of a hypercoagulable state, reduced fibrinolytic activity, tumor cell lysis, and use of central venous catheters.89,90 Some intravenous chemotherapeutic agents are associated with activation of coagulation and increased markers of thrombin generation, a response that is blocked by pretreatment with heparin.91
Surgery
The high incidence of postoperative DVT as well as the availability of easily repeatable, noninvasive diagnostic tests has allowed a greater understanding of the risk factors associated with surgery than in other conditions. Surgery constitutes a spectrum of risk that is influenced by age of the patient, coexistent thrombotic risk factors, type of procedure, extent of surgical trauma, length of procedure, and duration of postoperative immobilization.92 The type of surgical procedure is particularly important.47 The overall incidence of DVT is approximately 19% in patients undergoing general surgical operations and 24% in those having elective neurosurgical procedures; among those undergoing surgery for hip fracture, hip arthroplasty, and knee arthroplasty, it is 48%, 51%, and 61%, respectively.92 On the basis of these data, patients can be classified as being at low, moderate, or high risk for thromboembolic complications, as shown in Table 49-2.
Table 49-2
Risk of Postoperative Deep Venous Thrombosis
| Category | Characteristics |
| Low | Age <40 y, no other risk factors, uncomplicated abdominal/thoracic surgery |
| Age >40 y, no other risk factors, minor elective abdominal/thoracic surgery <30 min | |
| Moderate | Age >40 y, abdominal/thoracic surgery >30 min |
| High | History of recent thromboembolism |
| Abdominal or pelvic procedure for malignancy | |
| Major lower extremity orthopedic procedure |
From Hull RD, et al: Prophylaxis of venous thromboembolism. Chest 89(Suppl):374S, 1986.
Approximately half of postoperative lower extremity thrombi develop in the operating room; the remainder occur during the next 3 to 5 days.93 However, the risk for development of DVT does not end uniformly at hospital discharge. In one study, 51% of the thromboembolic events occurred after discharge from gynecologic surgical procedures.94 Similarly, up to 25% of patients undergoing abdominal surgery have DVT within 6 weeks of discharge.95 Heit et al94 found a nearly 22-fold higher risk of DVT and PE among patients who were hospitalized after previous surgery.
All components of Virchow’s triad may be present in the surgical patient—perioperative immobilization, transient changes in coagulation and fibrinolysis, and potential for gross venous injury. Immobilization is associated with a reduction in venous outflow and capacitance during the early postoperative period.96 Surgery is also accompanied by a transient, low-level hypercoagulable state, presumably mediated by the release of tissue factor, which is marked by a rise in thrombin activation markers shortly after the procedure begins.96 The thrombogenic potential of different surgical procedures appears to differ, with greater rises in thrombin activation markers during hip arthroplasty than after laparotomy.97 Increased levels of PAI-1 are also associated with a decrease in fibrinolytic activity on the first postoperative day, the “postoperative fibrinolytic shutdown.”98 This relationship between impaired fibrinolysis and postoperative DVT may be particularly important69; preoperative and early postoperative elevations in PAI-1 correlate with the development of thrombosis in orthopedic patients.98
Trauma
The prevalence of DVT among autopsied trauma casualties has been reported to be as high as 65%, comparable to the 58% incidence among injured patients in modern venographic series.13 Substantially lower DVT rates, ranging from 4% to 20%,99 have been noted in series employing duplex ultrasonography, although many patients studied were receiving prophylaxis and the limitations of ultrasound in screening of asymptomatic patients are well recognized. Recent trauma was associated with a nearly 13-fold increase in risk.94
Although the risk of DVT may be less than 20% in some injured patients,13 certain subgroups are at particularly high risk. Age (OR, 1.05 for each 1-year increment), blood transfusion (OR, 1.74), surgery (OR, 2.30), fracture of the femur or tibia (OR, 4.82), and spinal cord injury (OR, 8.59) have been significantly associated with the development of DVT in this population.13 Other reported risk factors are a hospital stay longer than 7 days,100 increased Injury Severity Score,101,102 Trauma Injury Severity Score of 85 or less,100 pelvic fractures,103 major venous injury,104 presence of femoral venous lines,105 duration of immobilization,106 and prolongation of the partial thromboplastin time.107
As with postoperative DVT, several pathophysiologic elements may be responsible for the high incidence of DVT in trauma patients. Immobilization by skeletal fixation, paralysis, and critical illness are associated with venous stasis, whereas mechanical injury is important after direct venous trauma and central venous cannulation. Less well appreciated is the hypercoagulable state after depletion of coagulation inhibitors and components of the fibrinolytic system. Fibrinopeptide A levels rise after injury,108 consistent with activation of coagulation, whereas fibrinolytic activity has been found to increase initially and then to decrease.109,110
Inherited Thrombophilia
Prothrombin Gene Mutation.
The prothrombin G20210A gene mutation was demonstrated when 28 families with a documented history of VTE were noted to have an 18% incidence of a transition from guanine to adenine at the 20210 nucleotide on the prothrombin gene. A comparison to healthy controls revealed only a 1% incidence of the same mutation.111 In this same study, Poort also reported that in 87% of individuals with such a mutation, the prothrombin levels were greater than 115% of normal. Thus, elevated levels of prothrombin, the precursor for thrombin, are associated with an increased risk of VTE. A large multicenter study revealed the incidence of this mutation in the general European population to be 0.7% to 4%.112 This mutation is rare in individuals of Asian or African descent. Additional investigations have shown that in those with a spontaneous DVT, the incidence of the mutation may range from 7% to 16%, depending on the patient population studied.113 The risk of VTE in the presence of the prothrombin G20210A mutation is threefold higher in heterozygotes than in those with the wild type, and the presence of homozygosity further increases this risk. The prothrombin G20210A gene mutation is frequently coinherited with factor V Leiden mutation in approximately 1% to 10% of symptomatic carriers.114,115
Factor V Leiden Mutation.
Resistance to activated protein C, characterized by the failure of exogenous activated protein C to prolong the activated partial thromboplastin time, was initially described by Dahlbäck in 1993.116 Subsequent studies from the Leiden University hospital in the Netherlands revealed this laboratory phenotype to be the result of a mutation in the gene for factor V of the coagulation cascade in 95% of instances. The transition mutation causes replacement of arginine with glutamine at position 506, such that factor Va is resistant to cleavage by activated protein C and is therefore inactivated at a decreased rate.117 Thus, more factor Va is available to participate in the generation of thrombin. In addition, after cleavage by activated protein C, the degradation product of factor V in combination with protein S acts as a cofactor for activated protein C, increasing the degradation of factor VIIIa as well as additional factor Va.118 The factor V Leiden mutation is inherited in an autosomal dominant manner and is 10 times more common than other heritable defects.119 The factor V Leiden mutation is present in 5% to 8% of European whites, with prevalence of up to 15% in some areas of Greece, Sweden, and Lebanon.120,121 In a study of 4047 Americans, the carrier frequencies of the mutation were 5.3% for whites, 2.2% for Hispanic Americans, 1.25% for Native Americans, 1.2% for African Americans, and 0.45% for Asian Americans. Carrier frequencies were similar in white men and women in this study.122 Factor V Leiden mutation has been reported in 20% to 50% of first-time VTE events; thus it is the most common cause of inherited thrombophilias.123 The relative risk of DVT is increased sevenfold in those who are heterozygous for the mutation. However, in individuals who are homozygous for the mutation, the relative risk is increased 80-fold. In addition, homozygous patients develop DVT at a younger age than do those who are heterozygous.124
The etiology of activated protein C resistance is not limited to factor V Leiden mutations. Increased plasma levels of factor VIII, the presence of antiphospholipid antibodies, older age, pregnancy, and the use of estrogens are well-known causes of increased activated protein C resistance.125
Protein C.
Protein C is a vitamin K–dependent serine protease that inhibits the coagulation system by inactivating factors Va and VIIIa, the two cofactors necessary for activation of factor X and thrombin generation.126 Once it is activated by the thrombin-thrombomodulin complex, activated protein C can be enhanced by protein S.127,128 Either an acquired or inherited reduction of protein C activity increases thrombotic risk. Heterozygous protein C deficiency is inherited in an autosomal dominant fashion; this deficiency may be classified as type I, characterized by a reduction in both antigenic and functional levels, or as type II, in which antigen levels are normal and functional activity is decreased.126 In a study of 10,000 healthy blood donors, the observed prevalence of inherited protein C deficiency was 1.45 per 1000.129 Furthermore, a study of 2132 patients with VTE found that 3.2% had protein C deficiency.130 It is estimated that individuals who are heterozygous for protein C deficiency have a sevenfold higher risk for development of VTE over baseline.131 Other than VTE, protein C deficiency is also associated with neonatal purpura fulminans and warfarin skin necrosis.
Neonatal purpura fulminans can result from either a homozygous or a double heterozygous genotype. It is characterized by microvascular thrombosis in the dermis, leading to severe ecchymoses followed by perivascular hemorrhage, extensive venous and arterial thrombosis, necrosis, laboratory findings consistent with disseminated intravascular coagulation, and extremely low levels of protein C.132 Warfarin-induced skin necrosis may occur in individuals with protein C deficiency during the first several days of warfarin therapy. Warfarin initiation can reduce protein C anticoagulant activity to 50% within 1 day.133 With the exception of factor VII, other procoagulant vitamin K–dependant clotting factors decline at a slower rate, inducing a transient hypercoagulable state. The clinical manifestations of warfarin-induced skin necrosis are similar to those seen in neonatal purpura fulminans.134 Acquired deficiency of protein C may also be seen in acute thrombotic events, liver disease, renal disease, disseminated intravascular coagulation, hemolytic-uremic syndrome, chemotherapy, thrombotic thrombocytopenic purpura, and acute infections.135
Protein S.
Protein S is a vitamin K–dependent cofactor for the protein C–mediated inactivation of factors Va and VIIIa. In addition, protein S can directly inhibit the clotting pathway through interactions with factors Va and Xa. Protein S is synthesized by hepatocytes and megakaryocytes and circulates bound to C4b-binding protein or as a free form; only the free form is able to interact with protein C.136 Protein S deficiency is autosomal dominant and is more common than protein C deficiency. The incidence of protein S–associated VTE is 5% to 7%; the prevalence in the general population is 0.13%.137–138 It is estimated that individuals who are heterozygous for protein S deficiency have an 8.5-fold higher relative risk for development of VTE.139
There are more than 130 types of protein S mutations, which can fall into three general categories. Type I deficiency is quantitative, type II deficiency is qualitative, and type III deficiency is caused by mutations that increase the affinity of protein S for C4b-binding protein.125 The clinical presentation of patients with heterozygous protein S deficiency is similar to that of patients with protein C deficiency: an increased incidence of VTE and warfarin-induced skin necrosis.140 Homozygous individuals are rare but will present with neonatal purpura fulminans. Acquired protein S deficiency has been reported in disseminated intravascular coagulation, diabetes mellitus, pregnancy, oral contraceptive use, nephritic syndrome, liver disease, and essential thrombocythemia.126
Antithrombin.
Antithrombin, a vitamin K–independent glycoprotein, inhibits thrombin as well as factors Xa, IXa, XIa, and XIIa. The action of antithrombin is accelerated by heparin.141 Antithrombin deficiency is inherited in an autosomal dominant manner. The majority of patients are heterozygous; homozygotes usually die in utero. Type I deficiency is a quantitative defect, whereas type II is a qualitative deficiency.125 The incidence of antithrombin deficiency–associated VTE has been reported at 0.5% to 3%, with a prevalence in the general population of 0.2%.130,141 However, it is estimated that heterozygotes for this deficiency have a 20-fold higher risk for development of VTE over that of the general population.142 The clinical presentations include thrombosis of the iliofemoral veins, upper limb deep veins, mesenteric veins, vena cava, renal veins, and retinal veins as well as cerebral venous thrombosis and Budd-Chiari syndrome.125,143
Hyperhomocysteinemia.
High plasma levels of homocysteine have been associated with an increased risk for both venous and arterial thrombosis.144 The metabolic breakdown of homocysteine involves N5-methyltetrahydrofolate reductase (MTHFR). Individuals who are homozygous for a substitution of cytosine by thymine on the MTHFR gene at position 677 (C677T) and have inadequate dietary intake may achieve moderately increased homocysteine levels.145 Despite this association, data supporting the MTHFR C677T mutation as an independent risk factor for VTE are lacking, but a new study is helping to provide mounting evidence for the importance of hyperhomocysteinemia in VTE.146
Other Inherited Thrombophilias.
Other inherited thrombophilias, such as dysfibrinogenemia and elevated clotting factors, are less well studied. Inherited defects of fibrinogen are detected in less than 1% of those presenting with first-time VTE.147 In such defects, fibrinogen may be unable to bind thrombin, or the fibrin itself is unable to be lysed by plasmin. Clinically, the majority of these patients are asymptomatic, yet 25% may present with bleeding and an additional 20% with thrombosis.148 Elevated levels of factors VIII, IX, and XI have been demonstrated to be an independent risk factor for VTE.149–151 Individuals with elevations of factors IX and XI above the 90th percentile in healthy controls have an odds ratio of 2.3 and 2.2, respectively, for DVT compared with those with levels below the 90th percentile.149,150 Elevated factor VIII levels have been reported in up to 25% of individuals presenting with VTE.152
Others have noted residual risk for thrombosis not explained by known inherited thrombophilic disorders. This additional risk is indirect evidence that more inherited thrombophilias are yet to be identified.153
Pregnancy
The incidence of VTE in the pregnant population is 6 to 10 times greater than in matched controls,154 leading to approximately 10% of all maternal deaths.155 By clinical evaluation, VTE has been reported at a rate of 1.3% to 7% during pregnancy and 6.1% to 23% in the postpartum period.156 However, studies that employed venography, Doppler ultrasonography, or ventilation-perfusion scans for evaluation of clinically suspected thromboembolism have suggested an incidence of 0.029% to 0.055% in this population.157 The risk of thrombosis appears to be two to three times greater during the puerperium, with the highest incidence found after cesarean section.158 Interestingly, when VTE has been objectively documented, the occurrence of thrombosis is equally distributed throughout all trimesters.159
DVT in pregnancy has been attributed to impaired venous outflow due to uterine compression, and 97% of reported thromboses have been isolated to the left leg.159 Furthermore, pregnancy is associated with a transient hypercoagulable state due to increases in the levels of fibrinogen, von Willebrand factor, and factors II, VII, VIII, and X. Compounding this, acquired functional resistance to activated protein C is also seen during pregnancy.159,160 Similarly, protein S levels are decreased by 50% to 60% early during pregnancy, with free protein S levels comparable to those in hereditary heterozygous protein S deficiency.161,162 The fibrinolytic system is also altered in pregnancy; levels of tissue plasminogen activator are decreased, and plasminogen activator inhibitors 1 and 2 are increased.163
Both retrospective and prospective studies have demonstrated that between 30% and 50% of women with a pregnancy-associated VTE also have an inherited thrombophilia. This high incidence has led to the recommendation of screening for thrombophilia in those pregnant patients with a personal or family history of VTE.164,165 The risk of puerperal DVT also increases with maternal age, suppression of lactation, hypertension, and assisted delivery but not with the number of pregnancies.166
Oral Contraceptives and Hormonal Therapy
As suggested by case reports in the early 1960s, further studies have now established the use of oral contraceptives as an independent risk factor for the development of DVT. These studies noted odds ratios of 3.8 to 11.0 for thrombosis167; an unweighted summary relative risk among 18 controlled studies was 2.9.168 Approximately one quarter of idiopathic thromboembolic events among women of childbearing age have been attributed to oral contraceptives. The risk of hospital admission for a thromboembolic event, including cerebral thrombosis, has been estimated to be 0.4 to 0.6 per 1000 for oral contraceptive users, compared with 0.05 to 0.06 for nonusers.169 Early studies also suggested that thromboembolism is responsible for approximately 2% of deaths in young women, with contraceptive-associated mortality rates of 1.3 and 3.4 per 100,000 among women aged 20 to 34 years and 35 to 44 years, respectively.170 The increased risk of thromboembolism appears to diminish soon after oral contraceptives are discontinued and is independent of the duration of use.171
Risk is correspondingly higher when oral contraceptive use is combined with other factors, such as surgery172 and inherited inhibitor deficiencies.173 The factor V Leiden mutation may be particularly important in this regard; resistance to activated protein C has been reported in up to 30% of patients with contraceptive-associated thromboembolism.174 The use of third-generation oral contraceptives may act synergistically with the factor V Leiden mutation, raising thromboembolic risk 30-fold to 50-fold.175–177
The risk of VTE among users of third-generation oral contraceptives may be up to eight times that of young women who do not use oral contraceptives. However, the higher risk associated with third-generation progestins has been questioned on the basis of the possible confounding effects of age and other prescribing biases.178
Estrogenic compounds also increase the risk of VTE when they are used for lactation suppression,166 in treatment of carcinoma of the prostate, and as postmenopausal replacement therapy.179 Although estrogen doses used for postmenopausal replacement therapy are approximately one sixth those in oral contraceptives, some data support an increased thromboembolic risk at these doses as well. Several studies show a twofold to fourfold higher risk of VTE among women taking hormone replacement therapy.179–183 This increased risk is greatest during the first year of treatment.180,183 However, given the relative infrequency of thromboembolism, this risk represents only one or two additional cases of thromboembolism per year in every 10,000 women in this age group.
Estrogen in pharmacologic doses is associated with alterations in the coagulation system that may contribute to this thrombotic tendency. Such alterations include decreases in PAI-1184 and increases in blood viscosity, fibrinogen, plasma levels of factors VII and X, and platelet adhesion and aggregation.185,186 An associated prothrombotic state is implied by rises in markers of activated coagulation occurring in conjunction with elevations of circulating factor VIIa and decreases in antithrombin and protein S inhibitor activity.185,187 The extent to which antithrombin and protein S are depressed is significantly less with lower estrogen preparations.188
Blood Group
There also appears to be a relationship between VTE risk and the ABO blood groups, with a higher prevalence of blood type A and correspondingly lower prevalence of blood type O groups.189,190 In reviewing the literature, Mourant et al191 found the relative incidence of type A to be 1.41 times higher among patients with thromboembolism than among controls. The effect of blood type was greater in young women who were taking oral contraceptives or were pregnant; the relative incidence of type A among patients with thromboembolism was 3.12 in those taking oral contraceptives and 1.85 in those who were pregnant. A relationship between soluble endothelial cell markers and ABO blood group is known to exist, with significantly lower levels of von Willebrand factor among those with type O blood.192
Geography and Ethnicity
There are also geographic differences in the frequency of VTE; the incidence of postoperative DVT in Europe has been noted to be nearly twice that in North America.87,92 Higher rates of thromboembolism have also been noted in the central United States compared with either coast.193 Autopsy series suggest that although the prevalence of thromboembolism is identical among American black and white patients, it is significantly higher than in a matched Ugandan black population.194 A similar autopsy series noted the prevalence of thromboembolism to be 40.6% in Boston and 13.9% in Kyushu, Japan.195
Unfortunately, regional variations in underlying medical and surgical conditions as well as in prophylactic measures and diagnostic methods may confound any apparent differences in the incidence of thromboembolism among different ethnic and geographic groups. Nevertheless, it is certainly conceivable that differences between ethnic groups might arise from either genetic or environmental factors. Such differences seem likely on the basis of recognized geographic differences in the spectrum of mutations leading to congenital anticoagulant deficiencies.196 Such theoretical concerns are also supported by geographic variability in the incidence of the factor V Leiden mutation. The factor V Leiden allele has a prevalence of 4.4% in Europeans, corresponding to a carrier rate of 8.8%, but the allele has not been identified in Southeast Asian or African populations.121
Central Venous Catheters
The use of central venous cannulation for hemodynamic monitoring, infusion catheters, and pacemakers has been associated with a rising frequency of DVT. This is particularly true for upper extremity thrombi, as many as 65% of which are related to central venous cannulation.197 Although the incidence of symptomatic thrombosis may be low, studies employing objective surveillance have reported thrombosis to occur with a mean incidence of 28% after subclavian cannulation.198 This risk also extends to femoral venous catheters; ipsilateral thrombosis develops in 12% of patients undergoing placement of large-bore catheters for trauma resuscitation.199
Experimental studies in rats suggest that the catheter itself, associated vascular injury, and stasis contribute to the development of early perigraft thrombus in virtually all cases.200 Catheter material appears to be an important determinant of thromboembolism201; polytetrafluoroethylene (Teflon) and heparin-bonded catheters are associated with thrombosis in less than 10% of cases.202 Other determinants of thrombosis are catheter diameter, number of venipuncture attempts, duration of catheter placement, and composition of the infusate.198 Heit et al203 found that current or recent placement of a central venous catheter and previous placement of a transvenous pacemaker were strongly associated with upper extremity DVT, with more than a fivefold higher risk even after adjustment for comorbid conditions.203
Inflammatory Bowel Disease
Clinical series have reported VTE to complicate inflammatory bowel disease in 1.2% to 7.1% of cases.204,205 Crohn’s disease has incidence rates of 31.4 per 10,000 person-years and 10.3 per 10,000 person-years for DVT and PE, respectively. Ulcerative colitis also has a high incidence rate of 30.0 per 10,000 and 19.8 per 10,000 person-years for DVT and PE, respectively. Such thromboses frequently occur among young patients, are more common with active disease, and may affect unusual sites, such as the cerebral veins.204,205 Greater extent of colonic disease in ulcerative colitis portends a higher risk of VTE. Most cases are not associated with inherited inhibitor deficiencies, antiphospholipid antibodies, or factor V Leiden mutation. The significance of thrombocytosis and elevations of factor V, factor VIII, and fibrinogen during active episodes of inflammatory bowel disease is unclear because all are acute phase reactants.204,205
Other coagulation abnormalities, including depressed antithrombin values, elevated PAI-1 values, and presence of anticardiolipin antibodies, have been inconsistently reported.206
[/not-level-membership-for-surgery-category]
