Chapter 44 Acute cerebrovascular complications
Cerebrovascular disease is common and its acute manifestations, known as stroke, produce considerable morbidity and mortality. Stroke is defined as an acute focal neurological deficit caused by cerebrovascular disease, which lasts for more than 24 hours or causes death before 24 hours. Transient ischaemic attack (TIA) also causes focal neurology, but this resolves within 24 hours. In the UK, stroke is responsible for 12% of all deaths and is the most common cause of physical disability in adults. The incidence in most developed countries is about 1–2/1000 population per year.1 The main causes of stroke are cerebral infarction as a consequence of thromboembolism and spontaneous intracranial haemorrhage (either intracerebral or subarachnoid haemorrhage (SAH)), causing about 85% and 15% of strokes, respectively. The main risk factors are increasing age, hypertension, ischaemic heart disease, atrial fibrillation, smoking, obesity, some oral contraceptives and raised cholesterol or haematocrit. The manifestations of stroke are:
PROGNOSIS IN ACUTE CEREBROVASCULAR DISEASE
Mortality after stroke averages 30% within a month, with more patients dying after SAH or intracerebral haemorrhage than after cerebral infarction, although survival to 1 year is slightly better in the haemorrhagic group. In all types of stroke about 30% of survivors remain disabled to the point of being dependent on others. Risk of stroke increases with age, so that it rises from 3/100 000 in the third and fourth decades to 300/100 000 in the eighth and ninth decades.2 Thus stroke is often accompanied by significant age-related medical comorbidity. In the past this may have been partially responsible for a relatively non-aggressive approach to the treatment of stroke patients, so that the gloomy prognosis of stroke becomes a self-fulfilling prophecy. The challenge for intensivists is to identify those patients who are most likely to survive and not to offer aggressive therapy to those who are not. It has been suggested that, by regarding stroke as a medical emergency, a ‘brain attack’ analogous to ‘heart attack’, and ensuring early intensive care support, outcome may be improved.3,4
CEREBRAL EMBOLISM
Embolism commonly occurs from thrombus or platelet aggregations overlying arterial atherosclerotic plaques, but 30% of cerebral emboli will arise from thrombus in the left atrium or ventricle of the heart. This is very likely in the presence of atrial fibrillation, left-sided valvular disease, recent myocardial infarction, chronic atrial enlargement or ventricular aneurysm. The presence of a patent foramen ovale or septal defects allows paradoxical embolism to occur. Iatrogenic air embolism may occur during cardiopulmonary bypass, cardiac catheterisation or cerebral angiography. Embolisation may also occur as a complication of attempted coil embolisation of cerebral aneurysms or arteriovenous malformations (AVMs) after SAH.
CLINICAL PRESENTATION
INVESTIGATIONS
ELECTROCARDIOGRAPHY
This may demonstrate atrial fibrillation or other arrhythmia, or recent myocardial infarct.
COMPUTED TOMOGRAPHY (CT) OR MAGNETIC RESONANCE IMAGING (MRI) SCANNING
The CT scan may be normal or show only minor loss of grey/white matter differentiation in the first 24 hours after ischaemic stroke but haemorrhage is seen as areas of increased attenuation within minutes. After a couple of weeks the CT appearances of an infarct or haemorrhage become very similar and it may be impossible to distinguish them if CT is delayed beyond this time. CT angiography will often demonstrate vascular abnormalities and vasospasm but multimodal MRI, a combination of diffusion and perfusion-weighted MRI and MR angiography (MRA), is much more sensitive in demonstrating small areas of ischaemia and targeting those patients most suitable for thrombolysis.5 Where cerebral infarction has occurred as a result of venous thrombosis, the best imaging technique is MRA. Other imaging techniques are appropriate to identify the source of stroke in specific areas. Any patient with a stroke or TIA in the internal carotid artery territory should have duplex Doppler ultrasonography which may demonstrate stenosis, occlusion or dissection of the internal carotid. Where trauma is an aetiological factor reconstruction CT bone window views.
MANAGEMENT
Ideally, treatment for stroke patients should be coordinated within a stroke unit, as there is a 28% reduction in mortality and disability at 3 months compared to patients treated on general medical wards.6 In general only those patients with a compromised airway due to depressed level of consciousness or life-threatening cardiorespiratory disturbances require admission to medical or neurosurgical ICUs. In either case, attention to basic resuscitation, involving stabilisation of airway, breathing and circulation, is self-evident.
AIRWAY AND BREATHING
Patients with Glasgow Coma Scores (GCS) of 8 or less or those with absent gag will require intubation to preserve their airway and prevent aspiration. Where this requirement is likely to be prolonged, early tracheostomy should be considered. Adequate oxygenation and ventilation should be confirmed by arterial blood gas analysis, and supplemental oxygen prescribed if there is any evidence of hypoxia. If hypercarbia occurs then ventilatory support to achieve normocarbia is necessary to prevent exacerbation of cerebral oedema. Ventilatory support in such cases has been shown to result in improved prognosis.7
CIRCULATORY SUPPORT
A large number of stroke patients will have raised blood pressure on admission, presumably as an attempt by the vasomotor centre to improve cerebral perfusion. Hypertensive patients may have impaired autoregulation and regional cerebral perfusion may be very dependent on blood pressure.8 The patient’s clinical condition and neurological status should determine treatment rather than an arbitrary level of blood pressure. Control of even very high blood pressure (220/120 mmHg; 29.3/16.0 kPa) is not without risk and may result in progression of ischaemic stroke, so reduction should be monitored closely.9 It would seem reasonable on physiological grounds to avoid drugs that cause cerebral vasodilatation in that they may aggravate cerebral oedema, although there is no hard evidence for this. Animal experiments have suggested that haemodilution could improve blood flow by reducing whole blood viscosity but a recent multicentre study has failed to identify any clinical benefit.10 Cardiac output should be maintained and any underlying cardiac pathology such as failure, infarction and atrial fibrillation treated appropriately.
METABOLIC SUPPORT
Both hypo- and hyperglycaemia have been shown to worsen prognosis after acute stroke, therefore blood sugar levels should be maintained in the normal range.11 In the long term, nutritional support must not be neglected and early enteral feeding instituted by nasogastric intubation. In the longer term, particularly where bulbar function is reduced, percutaneous endoscopic gastrostomy is necessary.
ANTICOAGULATION
In theory, the use of anticoagulation reduces the propagation of thrombus and should prevent further embolism. In practice, the reduction in risk of further thromboembolic stroke is offset by a similar number of patients dying from cerebral or systemic haemorrhage as a result of anticoagulation.12,13 Anticoagulation can only be recommended in individuals where there is a high risk of recurrence, such as in those patients with prosthetic heart valves, atrial fibrillation with thrombus or those with thrombophilic disorders. A CT scan must be obtained prior to commencing therapy to exclude haemorrhage, and careful monitoring used. In patients with large infarcts there is always the risk of haemorrhage (haemorrhagic conversion) into the infarct and early heparinisation is best avoided.
THROMBOLYSIS
Systemic thrombolysis with streptokinase carries a very high risk of cerebral haemorrhage and should not be used.14 There is some evidence that intravenous recombinant tissue plasminogen activator (alteplase) has a better safety/efficacy profile. The National Institute of Neurological Disorders and Stroke (NINDS) trial showed a significant improvement in those patients given alteplase rather than placebo within 3 hours of acute stroke.15 This benefit was not found to be statistically significant in two European trials16,17 and the ATLANTIS trial showed no significant benefit at 90 days in the alteplase group together with an increased risk of intracranial haemorrhage.18 The 2006 Cochrane Library database states that thrombolysis appears to result in a significant net reduction in the proportion of patients dead or dependent in activities of daily living. However, there appears to be an increase in deaths within the first 7–10 days, symptomatic intracranial haemorrhage and deaths at follow-up at 3–6 months. The data from trials using intravenous recombinant tissue plasminogen activator, from which there is the most evidence on thrombolytic therapy so far, suggest that it may be associated with less hazard and more benefit. There is heterogeneity between the published trials for some outcomes so that the optimum criteria to identify patients most likely to benefit and least likely to be harmed, the latest time window, the agent, dose and route of administration are all unclear. Nevertheless, the data are promising and may justify the use of thrombolytic therapy with intravenous recombinant tissue plasminogen activator in experienced centres in highly selected patients where a licence exists. However, the data do not support the widespread use of thrombolytic therapy in routine clinical practice at this time, but suggest that further trials are needed to identify which patients are most likely to benefit from treatment and the environment in which it may best be given.19
INTRACEREBRAL HAEMORRHAGE
INVESTIGATIONS
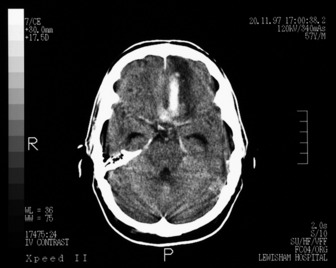
The general investigations are essentially those listed previously for ischaemic stroke, since it is difficult to distinguish between the two in the early stages. In addition, CT and/or MRI should be performed at the earliest opportunity. Lumbar puncture may be performed to exclude infection if mycotic aneurysm is suspected, but only after CT has excluded raised intracranial pressure or non-communicating hydrocephalus. Digital subtraction angiography will enable localisation of the source of the haemorrhage and will facilitate any surgical intervention for aneurysm or AVM (see Figures 42.7, 44.1 and 44.2).
MANAGEMENT
The general management principles are identical to those for ischaemic stroke. There is, of course, no place for anticoagulation or thrombolysis, and reversal of any coagulation defect either primary or secondary to therapeutic anticoagulation must be undertaken as a matter of urgency. A full coagulation screen must be performed and the administration of vitamin K, fresh frozen plasma, cryoprecipitate, etc. directed by the results. Where intraventricular extension has occurred the insertion of an EVD may increase conscious level, particularly in the presence of secondary hydrocephalus. The EVD level should be set such that the cerebrospinal fluid (CSF) drains at around 10 mmHg. The normal production of CSF should produce an hourly output and a sudden fall in output to zero should alert staff to the possibility that the drain has blocked. This is particularly likely if the CSF is heavily blood-stained. The meniscus of the CSF within the drain tubing should be examined for transmitted vascular pulsation or the level of the drain temporally lowered by a few centimetres to see if drainage occurs. If the drain is blocked, secondary hydrocephalus will recur. Because of the risk of introducing infection and causing a ventriculitis, the drain must be unblocked in a sterile manner by the neurosurgeons. Operative decompression of the haematoma should only be undertaken in neurosurgical centres and safe transfer must be assured if this is considered. This is a controversial treatment as the most recently published trial shows that patients with spontaneous supratentorial intracerebral haemorrhage in neurosurgical units show no overall benefit from early surgery when compared with initial conservative treatment.20 Subgroup analysis showed that those patients with intracerebral haemorrhage less than 1 cm from the cortical surface benefited from early surgery. Patients presenting with a GCS of less than 8/15 had an almost universally poor outcome. Not all intracerebral haematomata are amenable to surgery, and the CT scans should be reviewed by the Neurosurgical Unit, preferably prior to transfer by digital image link. Ideally clot evacuation within 6 hours maximum should be the goal.
The management of hypertension following spontaneous intracerebral haemorrhage may be difficult as too high a blood pressure may provoke further bleeding whereas too low a blood pressure may result in ischaemia. The Stroke Council of the American Heart Association has recommended that in patients with chronic hypotension the mean arterial pressure should not exceed 130 mmHg and that, if systolic blood pressure falls to 90 mmHg, then vasopressors should be used.21 The administration of mannitol prior to transfer should be discussed with the Neurosurgical Unit. There is no place for steroids, and hyperventilation to PaCO2 of 30 mmHg (4 kPa) or less to control raised intracranial pressure will have detrimental effects on cerebral blood flow in other areas of the brain.
SUBARACHNOID HAEMORRHAGE
SAH refers to bleeding which occurs principally into the subarachnoid space and not into the brain parenchyma. The incidence of SAH is around 6/100 000: the apparent decrease, compared with earlier studies, is due to more frequent use of CT scanning which allows exclusion of other types of haemorrhage. Risk factors are the same as for stroke, but SAH patients are usually younger, peaking in the sixth decade, with a female-to-male ratio of 1.6:1. Black people have twice the risk for SAH as whites. Between 5 and 20% of patients with SAH have a positive family history, with first-degree relatives having a three- to sevenfold risk, whereas second-degree relatives have the same degree of risk as the general population. Specific inheritable disorders are rare and account for only a minority of all patients with SAH.22 The only modifiable risk factors for SAH are smoking, heavy drinking and hypertension, which increase the risk odds ratio by 2 or 3.23 Overall mortality is 50%, of which 15% die before reaching hospital, with up to 30% of survivors having residual deficit producing dependency.
AETIOLOGY AND PATHOLOGY
The majority of cases of SAH are caused by ruptured saccular (berry) aneurysms (85%), the remainder being caused by non-aneurysmal perimesencephalic haemorrhage (10%) and rarer causes such as arterial dissection, cerebral or dural AVMs, mycotic aneurysm, pituitary apoplexy, vascular lesions at the top of the spinal cord and cocaine abuse. Saccular aneurysms are not congenital, almost never occur in neonates and young children and develop during later life. It is not known why some adults develop aneurysms at arterial bifurcations in the circle of Willis and some do not. It was thought that there was a congenital weakness in the tunica media, but gaps in the arterial muscle wall are equally as common in patients with or without aneurysms and once the aneurysm is formed, the weakness is found in the wall of the sac and not at its neck.24 The association with smoking, hypertension and heavy drinking would suggest that degenerative processes are involved. Sudden hypertension plays a role in causing rupture, as shown by SAH in patients taking crack cocaine or, rarely, with sulfenidil.
CLINICAL PRESENTATION
Classically, there is a ‘thunderclap’ headache developing in seconds, with half of patients describing its onset as instantaneous. This is followed by a period of depressed consciousness for less than an hour in 50% of patients, with focal neurology in about 30% of patients.25 About a fifth of patients recall similar headaches and these may have been due to ‘warning leaks’. The degree of depression of consciousness depends upon the site and extent of the haemorrhage. Meningism – neck stiffness, photophobia, vomiting and a positive Kernig’s sign – is common in those patients with higher GCS. The clinical severity of SAH is often described by a grade, the most widely used being that described by the World Federation of Neurological Surgeons (WFNS),26 which is summarised in Table 44.1.
Table 44.1 Clinical neurological classification of subarachnoid haemorrhage
| Grade | Signs | |
|---|---|---|
| I | Conscious patient with or without meningism | |
| II | Drowsy patient with no significant neurological deficit | |
| III | Drowsy patient with neurological deficit – probably intracerebral clot | |
| IV | Deteriorating patient with major neurological deficit (because of large intracerebral clot) | |
| V | Moribund patient with extensor rigidity and failing vital centres | |
| WFNS grade | GCS | Motor deficit |
|---|---|---|
| I | 15 | Absent |
| II | 14–13 | Absent |
| III | 14–13 | Present |
| IV | 12–7 | Present or absent |
| V | 3–6 | Present or absent |
WFNS, World Federation of Neurological Surgeons; GCS, Glasgow Coma Score.
COMPLICATIONS
ACUTE HYDROCEPHALUS
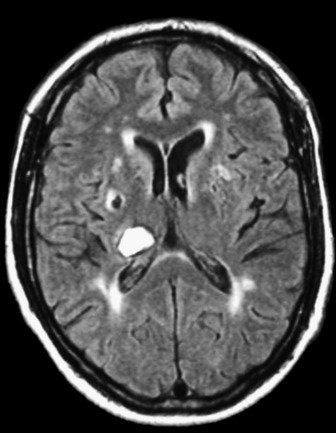
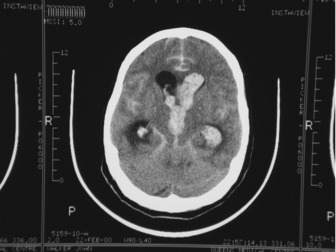
This may occur within the first 24 hours postictus and is often characterised by a one-point drop in the GCS, sluggish pupillary responses and bilateral downward deviation of the eyes (‘sunset eyes’). If these signs occur, CT scan should be repeated and, if hydrocephalus is confirmed, or there is a large amount of intraventricular blood, then a ventricular drain may be inserted. This is not without risk as it may provoke more bleeding and introduce infection. Only observational studies exist and these are contradictory so no firm recommendation can be made (Figure 44.3).
REBLEEDING
This may occur within the first few hours after admission and 15% of patients may deteriorate from their admission status.26 They may require urgent intubation and resuscitation but not all rebleeds are unsurvivable and such deterioration should be treated. The chance of rebleeding is dependent on the site of the aneurysm, presence of clot, degree of vasospasm, age and sex of the patient. A preliminary report of the Co-operative Aneurysm Study gave the rebleeding rate in the first 2 weeks after the initial SAH was reported as 19% cumulative and then approximately 1.5% per day for the next 13 days in 1983.27,28
CEREBRAL VASOSPASM
This is the term used to describe narrowing of the cerebral blood vessels in response to SAH seen on angiography. It occurs in up to 70% of patients, but not all of these patients will have symptoms.29 Use of transcranial Doppler (TCD) to estimate middle cerebral artery blood velocity has shown that a velocity of more than 120 cm/s correlates with angiographic evidence of vasospasm. This technology allows diagnosis in the ICU and provides a means of monitoring the success of treatment to reduce vasospasm, which is undertaken to reduce the severity of delayed neurological deficit secondary to vasospasm. The problem is that not all patients who have angiographic vasospasm or high Doppler velocities have symptoms. If there is evidence of a depressed level of consciousness in the absence of rebleeding, hydrocephalus or metabolic disturbances, but there is evidence of vasospasm on TCD or angiogram, then it would seem appropriate to initiate treatment to reduce the vasospasm. If vasopasm occurs at the time of angiography or coiling, then intravascular vasodilators such as papaverine have been used.
PARENCHYMAL HAEMATOMA
This may occur in up to 30% of SAH following aneurysm rupture and has a much worse prognosis than SAH alone.29 If there is mass effect with compressive symptoms then evacuation of haematoma and simultaneous clipping of the aneurysm may improve outcome.
MEDICAL COMPLICATIONS
During the placebo-controlled study of nicardipine in WFNS grade I and II SAH patients, there was a 40% incidence of at least one life-threatening medical complication in the placebo group.30 The mortality due to medical complications was almost the same as that due to the combined effects of the initial bleed, rebleeds and vasospasm. The types of medical complication seen are shown in Table 44.2.
Table 44.2 Types of medical complication seen in patients with subarachnoid haemorrhage
| Medical complication | Incidence |
|---|---|
| Arrhythmias | 35% |
| Liver dysfunction | 24% |
| Neurogenic pulmonary oedema | 23% |
| Pneumonia | 22% |
| ARDS and atelectasis | 20% |
| Renal dysfunction | 5% |
ARDS, acute respiratory distress syndrome.
INVESTIGATIONS
The general investigations for stroke should be performed and early CT imaging is mandatory. Blood appears characteristically hyperdense on CT and the pattern of haemorrhage may enable localisation of the arterial territory involved. Very rarely, a false-positive diagnosis may be made if there is severe generalised oedema resulting in venous congestion in the subarachnoid space. Small amounts of blood may not be detected and the incidence of false-negative reports is around 2%.31 It may be difficult to distinguish between post-traumatic SAH and primary aneurysmal SAH, which precipitates a fall in the level of consciousness that provokes an accident or fall.32 MR scanning is particularly effective for localising the bleed after 48 hours when extravasated blood is denatured, and provides a good signal on MRI.33
Angiography via arterial catheterisation is still the most commonly used investigation for localising the aneurysm or other vascular abnormality prior to surgery. It is generally performed on patients who remain, or become, conscious after SAH. It is not without risk and aneurysms may rupture during the procedure and a meta-analysis has shown a complication rate of 1.8%.34 Other methods under investigation include CT angiography and MR angiography, and it is likely that these techniques may replace catheter angiography for diagnosis, although intra-arterial catheterisation would still be needed for endovascular therapy.35,36
Transcranial Doppler studies may be useful in detecting vasospasm or those patients in whom autoregulation is impaired.37 The technique is dependent on there being a ‘window’ of thin temporal bone allowing isonation of the Doppler signal along the middle cerebral artery. It is very user-dependent and 15% of patients do not have an adequate bone window.
BLOOD PRESSURE CONTROL
Elevation of blood pressure is commonly seen after SAH and there are no precise data on what constitutes an unacceptably high pressure that is likely to cause rebleeding. Equally, there are no precise data on a minimum level of pressure below which infarction is likely to occur, since this will depend on the patient’s normal pressure, degree of cerebral oedema and the presence or absence of intact autoregulation. One observational study has demonstrated reduced rebleeding but higher rates of infarction in newly treated compared with untreated post-SAH hypertensive patients.38 If blood pressure exceeds 200 mmHg (26.6 kPa) systolic or 100 mmHg (13.3 kPa) diastolic, then empirically it would seem wise to reduce this pressure in SAH patients who have unclipped aneurysms. β-adrenergic blockers or calcium antagonists are the most widely used agents, since drugs producing cerebral vasodilation may increase intracranial pressure.
VASOSPASM
Angiographic demonstration of vasospasm may be seen in about 70% of SAH patients but only about 30% develop cerebral symptoms related to vasospasm. Transcranial Doppler-derived flow velocities in the middle cerebral arteries of more than 120 cm/s are accurate in predicting ischaemia.37 Symptoms tend to occur between 4 and 14 days postbleed, which is the period when cerebral blood flow is decreased after SAH.
One method of pre-empting vasospasm is the prescription of oral nimodipine at 60 mg given 4-hourly for 21 days, which has been shown to achieve a reduction in the risk of ischaemic stroke of 34%.39 Intravenous nimodipine should be used in the patients who are not absorbing, but it must be titrated against blood pressure to avoid hypotension. Other calcium antagonists, notably nicardipine and the experimental drug AT877, reduce vasospasm but do not improve outcome.
Low cerebral blood flow is known to worsen outcome and this resulted in the development of prophylactic hypertensive hypervolaemic haemodilution – so-called triple-H therapy.40 As originally described, the therapy involved the use of fluid loading to achieve haemodilution and vasopressor therapy to increase cerebral blood flow, and was combined with surgery within 24 hours if possible. The therapy was continued for 21 days and all patients remained neurologically stable or improved as a result, apparently, of the absence of vasospasm. Very few centres use the strict protocol as originally described, but fluid loading rather than fluid restriction is the norm and inotropes or vasopressors are used subsequently if neurological function decreases. Despite its widespread use, there remains no prospective randomised trial that demonstrates its utility.
SURGERY
Clipping of the aneurysm is the surgical treatment of choice, with wrapping, proximal ligation or bypass grafting being used if the aneurysm is inaccessible to Yasargil clipping. The timing of surgery remains debatable. Early surgery, within 3 days of the bleed, has the advantage of fewer deaths occurring from rebleeding but is technically difficult due to the friability of associated tissues. Delayed surgery, around 10–12 days postbleed, gives better operating conditions but allows some patients to suffer a rebleed. There is no type 1 evidence but an observational study suggested that there is no difference in outcome after early or late operation.41
ENDOVASCULAR COILING
The development of detachable microcoils made of platinum by Gugliemi in 1990 has resulted in endovascular embolisation of aneurysms or AVMs by interventional radiology.42 A microcatheter is passed from the femoral artery to the cerebral aneurysm and the coils positioned sequentially in the lumen of saccular aneurysms to occlude it. Rupture of the aneurysm or adjacent vessel occlusion, causing ischaemia, is the most frequent complication.43 The International Subarachnoid Aneurysm Trial (ISAT) of neurosurgical clipping versus endovascular coiling in 2143 patients with ruptured intracranial aneurysms – a randomised comparison of effects on survival, dependency, seizures, rebleeding, subgroups and aneurysm occlusion – has come down in favour of coiling over the open surgical technique at 1-year follow-up. In patients with ruptured intracranial aneurysms suitable for both treatments, endovascular coiling is more likely to result in independent survival at 1 year than neurosurgical clipping; the survival benefit continues for at least 7 years. The risk of late rebleeding is low, but is more common after endovascular coiling than after neurosurgical clipping.44 Additional complications of coiling include rupture during catheter placement in the aneurysm, coil embolisation and vasopasm. Not all aneurysms, particularly those with wide necks, multiple filling vessels or giant aneurysms, are suitable for coiling.
THERAPY OF MEDICAL COMPLICATIONS
This is obviously specific to the type of complication. Pneumonia may require continuous positive airways pressure or ventilatory support together with directed antimicrobial therapy; acute respiratory distress syndrome requires lung-protective/recruitment ventilatory strategies; and renal failure necessitates an appropriate means of renal replacement therapy. Arrhythmias require correction of trigger factors such as hypovolaemia and electrolyte or acid–base disturbances prior to the appropriate antiarrhythmic drug or DC cardioversion. Neurogenic pulmonary oedema may be associated with severe cardiogenic shock, which may require inotropic support or even temporary intra-aortic balloon counterpulsation. The cardiogenic shock is reversible and patients can make a good recovery despite the need for aggressive support.45
1 Wolfe CDA. The impact of stroke. Br Med Bull. 2000;56:275-276.
2 Bonita R. Epidemiology of stroke. Lancet. 1992;339:342-344.
3 Treib J, Grauer MT, Woessner R, et al. Treatment of stroke on an intensive stroke unit: a novel concept. Intens Care Med. 2000;26:1598-1611.
4 Wolfe C, Rudd A, Dennis M, et al. Taking acute stroke care seriously. Br Med J. 2001;323:5-6.
5 Jansen O, Schellinger P, Fiebach J, et al. Early recanalisation in acute ischaemic stroke scar tissue at risk defined by MRI. Lancet. 1999;353:2036-2037.
6 Stroke Trialists Collaboration. Collective systematic review of the randomised trials of organised inpatient (stroke unit) care after stroke. Br Med J. 1997;314:1151-1159.
7 Steiner T, Mendoza G, De Gorgia M, et al. Prognosis of stroke patients requiring mechanical ventilation in a neurological critical care unit. Stroke. 1997;28:711-715.
8 Fujii K, Satoshima S, Okada Y, et al. Cerebral blood flow and metabolism in normotensive and hypotensive patients with transient neurological deficits. Stroke. 1990;21:283-290.
9 Treib J, Haa BA, Stoll M, et al. Monitoring and management of antihypertensive therapy induced deterioration in acute ischaemia stroke. Am J Hypertens. 1996;9:513-514.
10 Aichner FT, Fazehar F, Bainin M, et al. Hypervolaemic hemodilution in acute ischaemic stroke. The Multicenter Austrian Hemodilution Stroke Trial (MAHST). Stroke. 1998;29:743-749.
11 Jorgensen HS, Nakayam H, Raaschon HO, et al. Effect of blood pressure and diabetes on stroke in progression. Lancet. 1994;344:156-159.
12 International Stroke Trial Collaborative Group. The International Stroke Trial (IST): a randomised trial of aspirin, subcutaneous heparin, both or neither among 19435 patients with acute ischaemic stroke. Lancet. 1997;349:1564-1565.
13 The Publications Committee for the Trial ORG 10172 in Acute Stroke Treatment (TOAST). Low molecular weight heparinoid ORG 10172 (Danaparoid) and outcome after acute ischaemic stroke. JAMA. 1998;279:1265-1272.
14 Multicentre Acute Stroke Trial – Europe Study Group. Thrombolytic therapy with streptokinase in acute ischaemic stroke. N Engl J Med. 1996;335:145-150.
15 National Institute of Neurological Disorders and Stroke rt-PA Stroke Study Group. Tissue plasminogen activator for acute ischaemic stroke. N Engl J Med. 1996;333:1-7.
16 Hacke W, Kaste M, Fieschi C, et al. Intravenous thrombolysis with recombinant tissue plasminogen activator for acute hemispheric stroke. The European Co-operative Acute Stroke Study (ECASS). JAMA. 1995;274:1017-1025.
17 Hacke W, Kaste M, Fieschi C, et al. Randomised double blind placebo-controlled trial of thrombolytic therapy with intravenous alteplase in acute ischaemic stroke (ECASS II). Lancet. 1998;352:1245-1251.
18 Clark WM, Wissman S, Albers GW, et al. Recombinant tissue type plasminogen activator (alteplase) for ischaemic stroke 3 to 5 hours after symptom onset. The ATLANTIS study: a randomised controlled trial. Alteplase thrombolysis for acute non-interventional therapy in ischaemic stroke. JAMA. 1999;282:2019-2026.
19 Wardlow JM, del Zoppo G, Yamaguchi T, et al. Thrombolysis for acute ischaemic stroke. Cochrane Database of Systematic Reviews. 2003;3:CD000213.
20 Siddique MS, Mendelow AD. Early surgery versus initial treatment in patients with spontaneous supratentorial haematomas in the International Surgical Trial in Cerebral Haemorrhage (STITCH): a randomised trial. Lancet. 2005;365:361-362.
21 Broderick JP, Adams HPJr, Barsan W, et al. Guidelines for the management of spontaneous intracerebral hemorrhage: a statement for healthcare professionals from a special writing group of the Stroke Council, American Heart Association. Stroke. 1999;30:905-915.
22 Van Gijn J, Rinkel GJE. Subarachnoid haemorrhage: diagnosis, causes and management. Brain. 2001;124:248-249.
23 Teunissen LL, Rinkel GJE, Algra A, et al. Risk factors for subarachnoid haemorrhage – a systematic review. Stroke. 1996;27:544-549.
24 Stehbens WE. Etiology of intracranial berry aneurysms. J Neurosurg. 1989;70:823-831.
25 Linn FH, Rinkel GJ, Algra A, et al. Headache characteristics in subarachnoid haemorrhage and benign thunderclap headache. J Neurol Neurosurg Psychiatry. 1998;65:791-793.
26 Drake CG, Hunt WE, Kassell NF, et al. Report of the World Federation of Neurological Surgeons Committee on a universal subarachnoid haemorrhage grading scale. J Neurosurg. 1996;84:985-986.
27 Fujii Y, Takeuchi S, Sasaki O, et al. Ultra-early rebleeding in spontaneous subarachnoid haemorrhage. J Neurosurg. 1996;84:35-42.
28 Kassell NF, Torner JC. Aneurysmal rebleeding: a preliminary report from the Cooperative Aneurysm Study. Neurosurgery. 1983;13:479-481.
29 Weir B, MacDonald L. Cerebral vasospasm. Clin Neurosurg. 1992;40:40-45.
30 Hauerberg J, Eskesen V, Rosenova J. The prognostic significance of intracerebral haematoma as shown on CT scanning after subarachnoid haemorrhage. Br J Neurosurg. 1994;8:333-339.
31 Solenski NJ, Haley EC, Kassell NF, et al. Medical complications of aneurysmal subarachnoid haemorrhage: a report of the multicenter cooperative aneurysm study. Crit Care Med. 1995;25:1007-1017.
32 Van der Wee N, Rinkel GJ, Hasan D, et al. Detection of subarachnoid haemorrhage on early CT: is lumbar puncture still needed after a negative scan? J Neurol Neurosurg Psychiatry. 1995;58:357-359.
33 Vos PE, Zwienenberg M, O’Hannion KL, et al. Subarachnoid haemorrhage following rupture of an ophthalamic artery aneurysm presenting as traumatic brain injury. Clin Neurol Neurosurg. 2000;102:29-32.
34 Noguchi K, Ogawa T, Seto H, et al. Sub-acute and chronic subarachnoid haemorrhage: diagnosis with fluid attenuated inversion – recovery MR imaging. Radiology. 1997;203:257-262.
35 Cloft HJ, Joseph GJ, Dion JE. Risk of cerebral angiography in patients with subarachnoid haemorrhage, cerebral aneurysm and arteriovenous malformation: a meta-analysis. Stroke. 1999;30:317-320.
36 Wardlaw JM, White PM. The detection and management of unruptured intracranial aneurysms. Brain. 2000;123:205-221.
37 Hashimoto H, Iida J, Hironaka Y, et al. Use of spiral CT angiography in patients with subarachnoid haemorrhage in whom subtraction angiography did not reveal cerebral aneurysms. J Neurosurg. 2000;92:278-283.
38 Vora YY, Suarez-Almazor M, Steinke DE, et al. Role of transcranial Doppler in the diagnosis of cerebral vasospasm after subarachnoid haemorrhage. Neurosurgery. 1999;44:1237-1247.
39 Wijdicks EF, Vermeulen M, Murray GD, et al. The effects of treating hypertension following aneurysmal subarachnoid haemorrhage. Clin Neurol Neurosurg. 1990;92:111-117.
40 Pickard JD, Murray GD, Illingworth R, et al. Effect of oral nimodipine on cerebral infarction and outcome after subarachnoid haemorrhage: British Aneurysm Nimodipine Trial (BRANT). Br Med J. 1989;298:636-642.
41 Origatano TC, Wascher TM, Reichman OU, et al. Sustained increased cerebral blood flow with prophylactic hypertensive hemodilution (‘triple-H’ therapy) after subarachnoid haemorrhage. Neurosurgery. 1990;27:729-740.
42 Kassell NF, Torner JC, Jane JA, et al. The International Cooperative Study on the Timing of Aneurysm Surgery. Part 2: surgical results. J Neurosurg. 1990;73:18-36.
43 Guglielmi G, Vinuela F, Duckwriter G, et al. Endovascular treatment of posterior circulation aneurysms by electrothrombosis using electric-ally detachable coils. J Neurosurg. 1992;77:515-524.
44 Molyneux AJ, Kerr RSC, Yu LM, et al. The International Sub Arachnoid Aneurysm Trial (ISAT) of neurosurgical clipping versus endovascular coiling in 2143 patients with ruptured intracranial aneurysms: a randomised comparison of effects on survival, dependency, seizures, rebleeding, subgroups and aneurysm occlusion. Lancet. 2005;366:809-817.
45 Parr MJ, Finfer SR, Morgan MK. Reversible cardiogenic shock complicating subarachnoid haemorrhage. Br Med J. 1996;313:681-683.