[level-membership-for-pediatrics-category]
Chapter 268 Acquired Immunodeficiency Syndrome (Human Immunodeficiency Virus)
Etiology
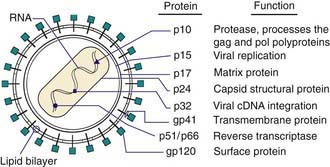
HIV-1 and HIV-2 are members of the Retroviridae family and belong to the Lentivirus genus, which includes cytopathic viruses causing diverse diseases in several animal species. The HIV-1 genome contains 2 copies of single-stranded RNA that is 9.2 kb in size. At both ends of the genome there are identical regions, called long terminal repeats, which contain the regulation and expression genes of HIV. The remainder of the genome includes 3 major sections: the GAG region, which encodes the viral core proteins (p24, p17, p9, and p6, which are derived from the precursor p55); the POL region, which encodes the viral enzymes (i.e., reverse transcriptase [p51], protease [p10], and integrase [p32]); and the ENV region, which encodes the viral envelope proteins (gp120 and gp41, which are derived from the precursor gp160). Other regulatory proteins, such as tat (p14), rev (p19), nef (p27), vpr (p15), vif (p23), vpu in HIV-1 (P16), and vpx in HIV-2 (P15) are involved in transactivation, viral messenger RNA expression, viral replication, induction of cell cycle arrest, promotion of nuclear import of viral reverse transcription complexes, downregulation of CD4 receptors and class I major histocompatibility complex, proviral DNA synthesis, and virus release and infectivity (Fig. 268-1).
Epidemiology
Transmission
Breast-feeding is the least common route of vertical transmission in industrialized nations but responsible for as many as 40% of perinatal infections in resource-limited countries. Both free and cell-associated viruses have been detected in breast milk from HIV-infected mothers. The risk for transmission through breast-feeding in chronically infected women is approximately 9-16% but 29-53% in women who acquire HIV postnatally, suggesting that the viremia experienced by the mother during primary infection at least triples the risk for transmission. It seems reasonable for women to substitute infant formula for breast milk if they are known to be HIV-infected or are at risk for ongoing sexual or parenteral exposure to HIV. However, the WHO recommends that in developing countries where other diseases (diarrhea, pneumonia, malnutrition) substantially contribute to a high infant mortality rate, the benefit of breast-feeding outweighs the risk for HIV transmission, and HIV-infected women in developing countries should breast-feed their infants for at least the 1st 6 mo of life (see later section on prevention).
Pathogenesis
HIV infection affects most of the immune system and disrupts its homeostasis (Fig. 268-2). In most cases, the initial infection is caused by low amounts of a single virus. Therefore, disease may be prevented by prophylactic drug(s) or vaccine. When the mucosa serves as the portal of entry for HIV, the 1st cells to be affected are the dendritic cells. These cells collect and process antigens introduced from the periphery and transport them to the lymphoid tissue. HIV does not infect the dendritic cell but binds to its DC-SIGN surface molecule, allowing the virus to survive until it reaches the lymphatic tissue. In the lymphatic tissue (e.g., lamina propria, lymph nodes), the virus selectively binds to cells expressing CD4 molecules on their surface, primarily helper T lymphocytes (CD4+ T cells) and cells of the monocyte-macrophage lineage. Other cells bearing CD4, such as microglia, astrocytes, oligodendroglia, and placental tissue containing villous Hofbauer cells, may also be infected by HIV. Additional factors (co-receptors) are necessary for HIV fusion and entry into cells. These factors include the chemokines CXCR4 (fusion) and CCR5. Other chemokines (CCR1, CCR3) may be necessary for the fusion of certain HIV strains. Several host genetic determinants affect the susceptibility to HIV infection, the progression of disease, and the response to treatment. These genetic variants vary in different populations. A deletion in the CCR5 gene that is protective against HIV infection (CCR5Δ32) is relatively common in whites but is rare in blacks. Several other genes that regulate chemokine receptors, ligands, histocompatibility complex, and cytokines have also been found to influence the outcome of HIV infection. Usually, CD4+ lymphocytes migrate to the lymphatic tissue in response to viral antigens and then become activated and proliferate, making them highly susceptible to HIV infection. This antigen-driven migration and accumulation of CD4 cells within the lymphoid tissue may contribute to the generalized lymphadenopathy characteristic of the acute retroviral syndrome in adults and adolescents. HIV preferentially infects the very cells that respond to it (HIV-specific memory CD4 cells), accounting for the progressive loss of these cells and the subsequent loss of control of HIV replication. The continued destruction of memory CD4+ cells in the gastrointestinal tract leads to reduced integrity of the gastrointestinal epithelium followed by leakage of bacterial particles into the blood and increased inflammatory response, which cause further CD4+ cell loss. When HIV replication reaches a threshold (usually within 3-6 wk from the time of infection), a burst of plasma viremia occurs. This intense viremia causes flu or mononucleosis-like symptoms (fever, rash, lymphadenopathy, arthralgia) in 50-70% of infected adults. With establishment of a cellular and humoral immune response within 2-4 mo, the viral load in the blood declines substantially, and patients enter a phase characterized by a lack of symptoms and a return of CD4 cells to only moderately decreased levels.
Clinical Manifestations
The HIV classification system is used to categorize the stage of pediatric disease by using 2 parameters: clinical status and degree of immunologic impairment (Table 268-1). Among the clinical categories, category A (mild symptoms) includes children with at least 2 mild symptoms such as lymphadenopathy, parotitis, hepatomegaly, splenomegaly, dermatitis, and recurrent or persistent sinusitis or otitis media (Table 268-2). Category B (moderate symptoms) includes children with LIP, oropharyngeal thrush persisting for >2 mo, recurrent or chronic diarrhea, persistent fever for >1 mo, hepatitis, recurrent (HSV) stomatitis, HSV esophagitis, HSV pneumonitis, disseminated varicella (i.e., with visceral involvement), cardiomegaly, or nephropathy (see Table 268-2). Category C (severe symptoms) includes children with opportunistic infections (e.g. esophageal or lower respiratory tract candidiasis, cryptosporidiosis (>1 mo), disseminated mycobacterial or cytomegalovirus infection, Pneumocystis pneumonia, or cerebral toxoplasmosis [onset >1 mo of age]), recurrent bacterial infections (sepsis, meningitis, pneumonia), encephalopathy, malignancies, and severe weight loss.
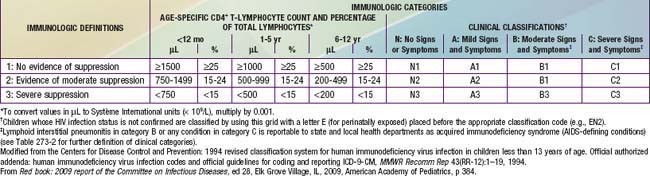
Table 268-2 CLINICAL CATEGORIES FOR CHILDREN YOUNGER THAN 13 YEARS OF AGE WITH HIV INFECTION
CATEGORY N: NOT SYMPTOMATIC
Children who have no signs or symptoms considered to be the result of HIV infection or have only 1 of the conditions listed in category A.
CATEGORY A: MILDLY SYMPTOMATIC
Children with 2 or more of the conditions listed but none of the conditions listed in categories B and C.
CATEGORY B: MODERATELY SYMPTOMATIC
Children who have symptomatic conditions other than those listed for category A or C that are attributed to HIV infection.
CATEGORY C: SEVERELY SYMPTOMATIC
Modified from the Centers for Disease Control and Prevention: 1994 revised classification system for human immunodeficiency virus infection in children less than 13 years of age. Official authorized addenda: human immunodeficiency virus infection codes and official guidelines for coding and reporting ICD-9-CM, MMWR Recomm Rep 43(RR-12):1–19, 1994.
The immune classification is based on the absolute CD4 lymphocyte count or the percentage of CD4 cells (see Table 268-1). Age adjustment of the absolute CD4 count is necessary because counts that are relatively high in normal infants decline steadily until 6 yr of age, when they reach adult norms. If there is a discrepancy between the CD4 count and percentage, the disease is classified into the more severe category.
Infections
Approximately 20% of AIDS-defining illnesses in children are recurrent bacterial infections caused primarily by encapsulated organisms such as Streptococcus pneumoniae and Salmonella (Table 268-3) due to disturbances in humoral immunity. Other pathogens, including Staphylococcus, Enterococcus, Pseudomonas aeruginosa, Haemophilus influenzae, and other gram-positive and gram-negative organisms, may also be seen. The most common serious infections in HIV-infected children are bacteremia, sepsis, and bacterial pneumonia, accounting for >50% of infections in these patients. Meningitis, urinary tract infections, deep-seated abscesses, and bone/joint infections occur less frequently. Milder recurrent infections, such as otitis media, sinusitis, and skin and soft tissue infections, are very common and may be chronic with atypical presentations.
Table 268-3 1993 REVISED CASE DEFINITION OF AIDS-DEFINING CONDITIONS FOR ADULTS AND ADOLESCENTS 13 YEARS OF AGE AND OLDER
Modified from the Centers for Disease Control and Prevention: 1993 revised classification system for HIV infection and expanded surveillance case definition for AIDS among adolescents and adults, MMWR Recomm Rep 41(RR-17):1–19, 1992.
Opportunistic infections (OIs) are generally seen in children with severe depression of the CD4 count. In adults, these infections usually represent reactivation of a latent infection acquired early in life. In contrast, young children generally have primary infection and often have a more fulminant course of disease reflecting the lack of prior immunity. This principle is best illustrated by Pneumocystis jirovecii (formerly Pneumocystis carinii) pneumonia, the most common opportunistic infection in the pediatric population (Chapter 236). The peak incidence of Pneumocystis pneumonia occurs at age 3-6 mo in the setting of undiagnosed perinatally acquired disease, with the highest mortality rate in children <1 yr of age. Aggressive approaches to treatment have improved the outcome substantially. While the overall incidence of opportunistic infections has markedly declined since the era of combination antiretroviral therapy (ART), OIs still occur in patients with severe immunodepletion as the result of unchecked viral replication, which often accompanies poor ART adherence.
Respiratory viruses such as respiratory syncytial virus (RSV) and adenovirus may present with prolonged symptoms and persistent viral shedding. In parallel with the increased prevalence of genital tract human papillomavirus (HPV) infection, cervical intraepithelial neoplasia (CIN) and anal intraepithelial neoplasia (AIN) also occur with increased frequency among HIV-1–infected adult women compared with HIV-seronegative women. The relative risk for CIN is 5-10 times higher for HIV-1 seropositive women. Multiple modalities are used to treat HPV infection (Chapter 258), although none is uniformly effective and the recurrence rate is high among HIV-1–infected persons.
Hematologic and Malignant Diseases
In contrast to the more frequent occurrence in adults, malignant diseases have been reported infrequently in HIV-infected children, representing only 2% of AIDS-defining illnesses. Non-Hodgkin lymphoma, primary CNS lymphoma, and leiomyosarcoma are the most commonly reported neoplasms among HIV-infected children. Epstein-Barr virus is associated with most lymphomas and with all leiomyosarcomas (Chapter 246). Kaposi sarcoma, which is caused by human herpesvirus 8, occurs frequently among HIV-infected adults but is exceedingly uncommon among HIV-infected children in resource rich countries (Chapter 249).
Diagnosis
Viral diagnostic assays, such as HIV DNA or RNA PCR or HIV culture, are considerably more useful in young infants, allowing a definitive diagnosis in most infected infants by 1-6 mo of age (Table 268-4). By 3-4 mo of age, the HIV culture and/or PCR identifies all infected infants. HIV DNA PCR is the preferred virologic assay in developed countries. Almost 40% of infected newborns have positive test results in the 1st 2 days of life, with >90% testing positive by 2 wk of age. Plasma HIV RNA assays, which detect viral replication, are as sensitive as the DNA PCR for early diagnosis. HIV culture has similar sensitivity to HIV DNA PCR; it is more technically complex and expensive, and results are often not available for several weeks compared with 2-3 days for PCR.
| TEST | COMMENT |
|---|---|
| HIV DNA PCR | Preferred test to diagnose HIV-1 subtype B infection in infants and children younger than 18 mo of age; highly sensitive and specific by 2 wk of age and available; performed on peripheral blood mononuclear cells. False negatives can occur in non-B subtype HIV-1 infections |
| HIV culture | Expensive, not easily available, requires up to 4 wk to do test; not recommended |
| HIV RNA PCR | Less sensitive than DNA PCR for routine testing of infants, because a negative result cannot be used to exclude HIV infection definitively. Some assays preferred test to identify non-B subtype HIV-1 infections. |
Ag, antigen; ICD, immune complex dissociated; PCR, polymerase chain reaction.
From Red book: 2009 report of the Committee on Infectious Diseases, ed 28, Elk Grove Village, IL, 2009, American Academy of Pediatrics, p 386.
Treatment
Combination Therapy
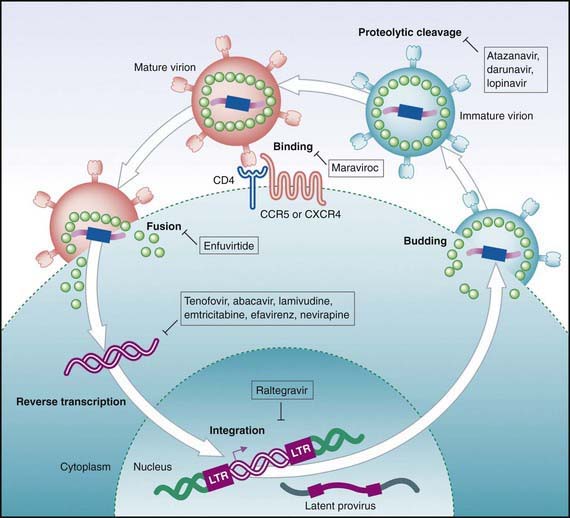
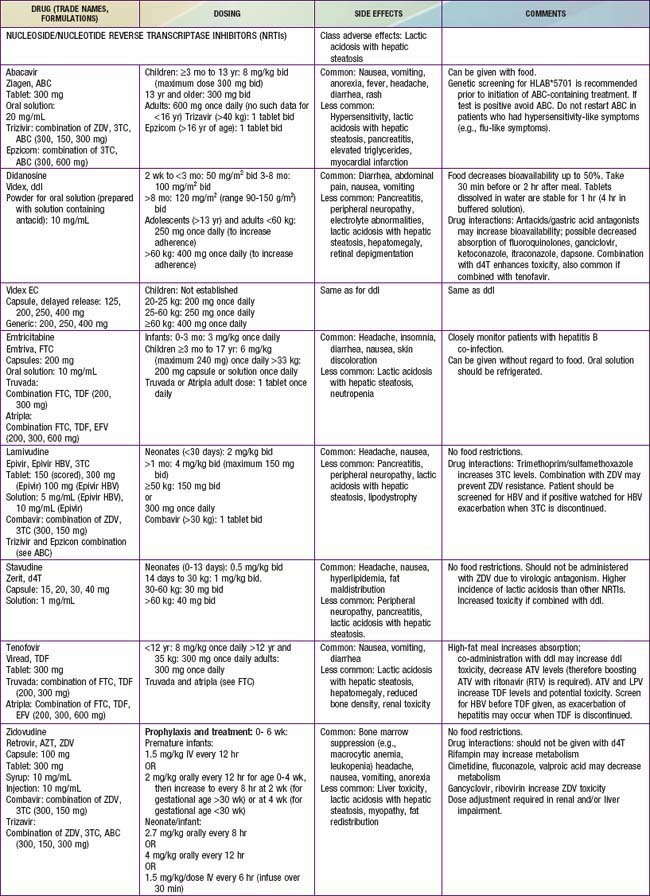
Antiretroviral drugs licensed as of 2010 are categorized by their mechanism of action, such as preventing viral entrance into CD4+ T cells, inhibiting the HIV reverse transcriptase or protease enzymes, or inhibiting integration of the virus into the human DNA. Within the reverse transcriptase inhibitors, a further subdivision can be made: nucleoside (or nucleotide) reverse transcriptase inhibitors (NRTIs) and non-nucleoside reverse transcriptase inhibitors (NNRTIs) (see Fig. 268-2). The NRTIs have a similar structure to the building blocks of DNA (e.g., thymidine, cytosine). When incorporated into DNA, they act like chain terminators and block further incorporation of nucleosides, preventing viral DNA synthesis. As of 2010, 7 NRTIs were licensed in the USA (Table 268-5). Among the NRTIs, thymidine analogs (e.g., stavudine [d4T], zidovudine [ZDV]) are found in higher concentrations in activated or dividing cells, and nonthymidine analogs (e.g., didanosine [ddI], lamivudine [3TC]) have more activity in resting cells. Activated cells are thought to produce >99% of the population of HIV virions. In contrast, resting cells account for <1% of the population but may serve as a reservoir for HIV. Suppression of replication in both populations is thought to be an important component of long-term viral control. NNRTIs (i.e., nevirapine, efavirenz, etravirine) act differently than the NRTIs. They attach to the reverse transcriptase and restrict its motility, reducing the activity of the enzyme. The protease inhibitors (PIs) are potent agents that act farther along the viral replicative cycle. As of 2010, 9 PIs were licensed in the USA, but only 5 of these agents (ritonavir, nelfinavir, fosamprenavir, tipranavir, and lopinavir) have pediatric formulations (e.g., liquid or powder) (see Table 268-5). The PIs bind to the site where the viral long polypeptides are cut to individual, mature, and functional core proteins that produce the infectious virions before they leave the cell. The virus entry into the cell is a complex process that involves cellular receptors and fusion. Several drugs have been developed to prevent this process. The fusion inhibitor, enfuvirtide, which binds to viral gp41, causes conformational changes that prevent fusion of the virus with the CD4+ cell and entry into the cell. Maraviroc is an example of a selective CCR5 co-receptor antagonist that blocks the attachment of the virus to this chemokine (an essential process in the viral binding and fusion to the CD4+ cells). Integrase inhibitors like raltegravir block the enzyme that catalyzes the incorporation of the viral genome into the host’s DNA.
By targeting different points in the viral life cycle and stages of cell activation and by delivering drug to all tissue sites, maximal viral suppression may be feasible. Combinations of 3 drugs, a thymidine analog NRTI (ZDV) and a nonthymidine analog NRTI (3TC) to suppress replication in both active and resting cells and a protease inhibitor (lopinavir/ritonavir or nelfinavir) or an NNRTI (efavirenz) have been shown to produce prolonged viral suppression. Less potent combinations such as triple NRTIs (abacavir, zidovudine, lamivudine) may be considered in special situations when there are concerns about significant drug interactions or adherence to a complex drug regimen. Combination treatment increases the rate of toxicities (see Table 268-5), and complex drug-drug interactions exist among many of the antiretroviral drugs. Many protease inhibitor drugs are inducers or inhibitors of the cytochrome P450 system and are therefore likely to have serious interactions with multiple drug classes, including nonsedating antihistamines and psychotropic, vasoconstrictor, antimycobacterial, cardiovascular, analgesic, and gastrointestinal drugs (cisapride). Whenever new medications are added to an antiretroviral treatment regimen, especially a protease inhibitor–containing regimen, a pharmacist and/or HIV specialist should be consulted to address possible drug interactions. The inhibitory effect of ritonavir (a protease inhibitor) on the cytochrome P450 system has been exploited, and small doses of the drug are added to several other protease inhibitors (lopinavir, tipranavir, atazanavir) to slow their metabolism by the P450 system and to improve their pharmacokinetic profile. This strategy provides more effective drug levels with less toxicity and less frequent dosing.
Initiation of Therapy
HIV-infected children with symptoms (clinical category A, B, or C) or with evidence of immune dysfunction (immune category 2 or 3) should be treated with antiretroviral therapy, regardless of age or viral load (see Tables 268-1 and 268-2). Children <1 yr of age are at high risk for disease progression, and immunologic and virologic tests to identify those likely to develop rapidly progressive disease are less predictive than in older children. Therefore, such infants should be treated with antiretroviral agents as soon as the diagnosis of HIV infection has been confirmed, regardless of clinical or immunologic status or viral load. Data suggest that HIV-infected infants who are treated before the age of 3 mo control their HIV infection better than infants whose antiretroviral therapy started later than 3 mo of age. Some of these infants even become HIV seronegative and lose their HIV specific immune response.
Supportive Care
Infants born to HIV-infected mothers should receive ZDV prophylaxis for 4-6 weeks +/− additional ART to prevent transmission. Guidelines for such prophylaxis are updated at least yearly and can be accessed at http://www.aidsinfo.nih.gov/default.aspx. A complete blood count, differential leukocyte count, and platelet count should be performed at 4 wk of age to monitor ZDV toxicity. If the child is found to be HIV-infected or if the HIV status is not clear, these tests should be continued every 1-3 mo to assess the hematologic effect of the disease or its treatment (prophylactic TMP-SMZ and antiretroviral therapy). If the child is found to be HIV infected, baseline laboratory assessment (e.g., CD4 count, HIV RNA, CBC, chemistries) should be done. Viral load and CD4 lymphocyte counts should be performed at 1 and 3 mo of age and should be repeated every 3 mo. The frequency of the test should be increased (every 4-6 wk) if the CD4 lymphocyte count or percentage declines rapidly.
All HIV-exposed and infected children should receive standard pediatric immunizations. In general, live oral polio vaccine should not be given (Fig. 268-3). The risk and benefits of rotavirus vaccination should be considered in infants born to HIV-infected mothers. Because <1% of these infants in developed countries will develop HIV infection, the vaccine should be given. In other situations, the considerable attenuation of the vaccine’s strains should be taken into account and unless the infant has clinical symptoms of AIDS or CD4 <15%, vaccination seems to be appropriate. Other live bacterial vaccines (e.g., bacillus Calmette-Guérin [BCG]) should be avoided due to the high incidence of BCG-related disease in HIV-infected infants. Varicella and measles-mumps-rubella (MMR) vaccines are recommended for children who are not severely immunosuppressed (i.e., CD4 cell percentage ≥15%), but neither varicella nor MMR vaccines should be given to severely immunocompromised children (i.e., CD4 cell <15%). Of note, prior immunizations do not always provide protection, as evidenced by outbreaks of measles and pertussis in immunized HIV-infected children. Durability of vaccine-induced titers is often short, especially if vaccines are administered when the child’s CD4 cell is <15%, and re-immunization when the CD4 count has increased (i.e., >15%) may be indicated.
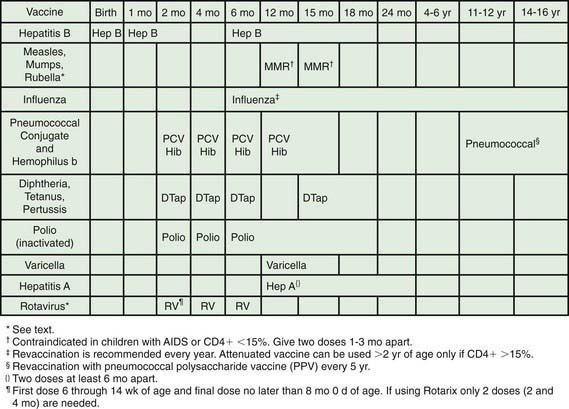
Prophylactic regimens are integral for the care of HIV-infected children. All infants between 4-6 wk and 1 yr of age who are proven to be HIV-infected should receive prophylaxis to prevent Pneumocystis jiroveci infection regardless of the CD4 count or percentage (see Tables 268-6 and 268-7). Infants exposed to HIV-infected mothers should receive the same prophylaxis until they are proven to be noninfected; however, prophylaxis does not have to be initiated if there is strong presumptive evidence of noninfection (i.e., non–breast-fed infant with 2 negative HIV PCR tests at >14 days and 4 wk of age, respectively). When the HIV-infected child is >1 yr of age, prophylaxis should be given according to the CD4 lymphocyte count (Table 268-6). The best prophylactic regimen is 150 mg/m2/day of trimethoprim component of TMP/SMZ given as 1-2 daily doses 3 days per wk. For severe adverse reactions to TMP/SMZ, alternative therapies include dapsone, atovaquone, or aerosolized pentamidine.
Table 268-6 RECOMMENDATIONS FOR PCP PROPHYLAXIS AND CD4 MONITORING FOR HIV-EXPOSED INFANTS AND HIV-INFECTED CHILDREN, BY AGE AND HIV INFECTION STATUS
| AGE/HIV INFECTION STATUS | PCP PROPHYLAXIS | CD4 MONITORING |
|---|---|---|
| Birth to 4-6 wk, HIV exposed | No prophylaxis | None |
| HIV infection reasonably excluded* | No prophylaxis | None |
| 4-6 wk to 4 mo, HIV exposed | Prophylaxis | 3 mo |
| 6 wk to 1 yr HIV-infected or indeterminate | Prophylaxis | 6, 9, and 12 mo |
| 1-5 yr, HIV infected | Prophylaxis if: | Every 3-4 mo† |
| CD4 <500 cells/µL or <15% | ||
| > 6 yr, HIV infected | Prophylaxis if: | Every 3-4 mo† |
| CD4 <200 cells/µL or <15%‡ |
The National Perinatal HIV Hotline (1-888-448-8765) provides consultation on all aspects of perinatal HIV care.
PCP, Pneumocystis carinii (some suggest jirovecii) pneumonia.
† More frequent monitoring (e.g., monthly) is recommended for children whose CD4 counts or percentages are approaching the threshold at which prophylaxis is recommended.
‡ Prophylaxis should be considered on a case-by-case basis for children who might otherwise be at risk for PCP, such as children with rapidly declining CD4 counts or percentages or children with category C conditions. Children who have had PCP should receive lifelong PCP prophylaxis.
Table 268-7 PROPHYLAXIS TO PREVENT FIRST EPISODE OF OPPORTUNISTIC INFECTIONS AMONG HIV-EXPOSED AND HIV-INFECTED INFANTS AND CHILDREN, USA*
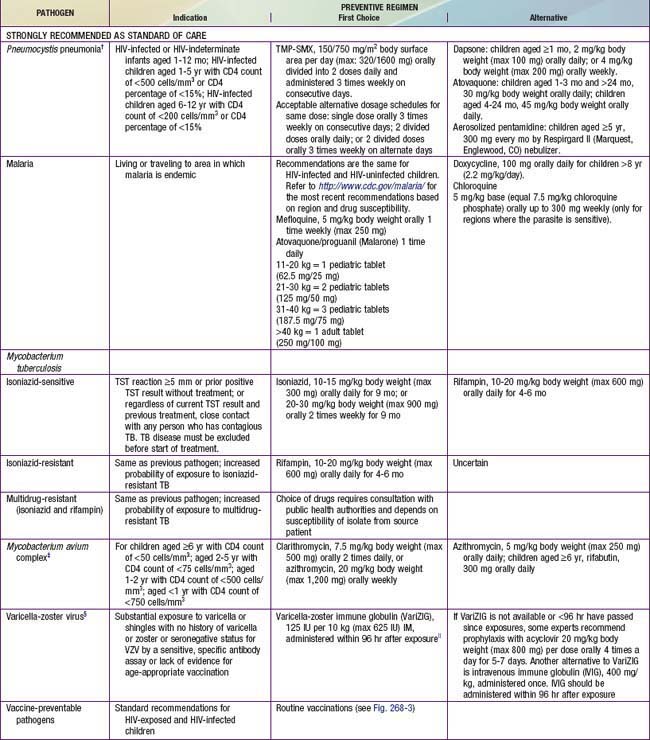
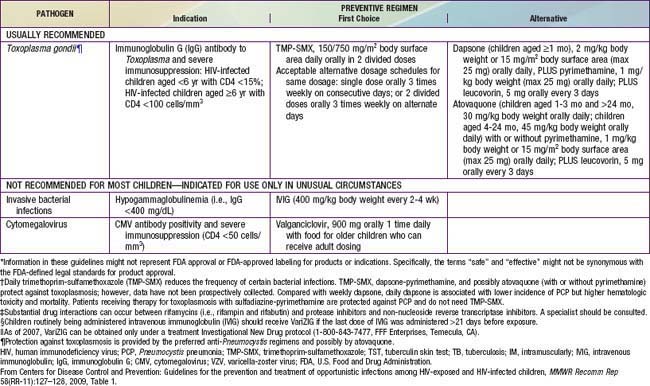
Prophylaxis against MAC should be offered to HIV-infected children with advanced immunosuppression (i.e., CD4 lymphocyte count <750 cells/mm3 in children <1 yr of age, <500 for children 1-2 yr of age, <75 cells/mm3 in children 2-5 yr of age, and <50 cells/mm3 in children >6 yr of age) (Table 268-7). The drugs of choice are azithromycin (20 mg/kg [maximum 1200 mg] once a week PO or 5 mg/kg [maximum 250 mg] once daily PO) or clarithromycin (7.5 mg/kg bid PO).
Because of the frequent changes in these guidelines, physicians providing care to few HIV-exposed or infected children should periodically consult physicians with expertise in pediatric HIV infection as well as the U.S. Pediatric Guidelines for treatment of HI- infected children found at http://www.aidsinfo.nih.gov.
Prevention
Retrospective data suggest that even if a mother has received no antiretroviral therapy during gestation or delivery, the 6-wk component of the ZDV prophylactic regimen instituted for the newborn as soon as possible after delivery (preferably within 12-24 hr of birth) results in a significant reduction of transmission rate. Full-term infants and preterm infants should be treated with oral ZDV (see Table 268-5).
Although the most effective way to prevent postpartum transmission of HIV is to eliminate breast-feeding altogether and substitute replacement feeding, there is increasing evidence that early weaning may not be safe in very resource constrained settings, due to the high risk of malnutrition and diarrhea in formula-fed infants without a consistent source of clean water. The WHO has recommended exclusive breast-feeding (no additional solids or fluids other than water) for at least 6 mo in resource-limited settings, unless there is an acceptable, feasible, affordable, sustainable, and safe (AFASS) replacement feeding option. Multiple international studies have shown that there are 2 efficacious approaches to interrupt breast-feeding transmission. One option is to treat mothers with triple ART for the duration of breast-feeding and their infants with daily nevirapine from birth to 6 weeks of age; this is critical for mothers who require ART for their own health. The other option, only for women with relatively strong CD4 counts who do not require ART for their own health, is to discontinue maternal ART 1 wk after delivery and treat the infant with daily nevirapine from birth until all exposure to breast milk has ended. As more data become available, guidelines for prevention of mother to child transmission will be regularly updated at http://www.aidsinfo.nih.gov/default.aspx and the WHO website (http://www.who.int/en/index.html).
Aberg JA, Kaplan JE, Lubman H, et al. Primary care guidelines for the management of persons infected with human immunodeficiency virus: 2009 update by the HIV medicine association of the Infectious Diseases Society of America. Clin Infect Dis. 2009;49:651-681.
Beyrer C, Malinowska-Sempruch K, Kamarulzaman A, et al. Time to act: a call for comprehensive responses to HIV in people who use drugs. Lancet. 2010;376:551-562.
Bongaarts J, Pelletier F, Gerland P. How many more AIDS deaths? Lancet. 2010;375:103-104.
Centers for Disease Control and Prevention. Interim guidance: preexposure prophylaxis for the prevention of HIV infection in men who have sex with men. MMWR. 2011;60(3):65-68.
Centers for Disease Control and Prevention. Increase in newly diagnosed HIV infections among young black men who have sex with men—Milwaukee county, Wisconsin, 1999-2008. MMWR. 2011;60(4):99-102.
Centers for Disease Control and Prevention. Guidelines for the prevention and treatment of opportunistic infections among HIV-exposed and HIV-infected children. MMWR Recomm Rep. 2009;58(RR-11):1-166.
Centers for Disease Control and Prevention. Guidelines for prevention and treatment of opportunistic infections in HIV-infected adults and adolescents. MMWR Recomm Rep. 2009;58(RR-15):1-207.
Crothers K, Huang L, Goulet JL, et al. HIV infection and risk for incident pulmonary diseases in the combination antiretroviral therapy era. Am J Respir Crit Care Med. 2011;183:388-395.
Dollfus C, Le Chenadec J, Faye A, et al. Long-term outcomes in adolescents perinatally infected with HIV-1 and followed up since birth in the French perinatal cohort (EPF/ANRS CO10). Clin Infect Dis. 2010;51:214-224.
Donnell D, Boeten JM, Kiarie J, et al. Heterosexual HIV-1 transmission after initiation of antiretroviral therapy: a prospective cohort analysis. Lancet. 2010;375:2092-2098.
Grant RM, Lama JR, Anderson PL, et al. Preexposure chemoprophylaxis for HIV prevention in men who have sex with men. N Engl J Med. 2010;363:2587-2599.
Gray RH, Kigozi G, Serwadda D, et al. Male circumcision for HIV prevention in men in Rakai, Uganda: a randomized trial. Lancet. 2007;369:657-666.
Gupta RK, Gibb DM, Pillay D. Management of paediatric HIV-1 resistance. Curr Opin Infect Dis. 2009;22:256-263.
Havens PL, Mofenson LM, Committee on Pediatric AIDS. Evaluation and management of the infant exposed to HIV-1 in the United States. Pediatrics. 2009;123:175-187.
Hazra R, Siberry GK, Mofenson LM. Growing up with HIV: children, adolescents, and young adults with perinatally acquired HIV infection. Ann Rev Med. 2010;61:169-185.
Johnston R. HIV cure: controversy, consensus, and a consortium. AIDS Res Human Retrovir. 2010;26:943-946.
Kumwenda NI, Hoover DR, Mofenson LM, et al. Extended antiretroviral prophylaxis to reduce breast-milk HIV-1 transmission. N Engl J Med. 2008;359:119-128.
Landovitz RJ, Currier JS. Postexposure prophylaxis for HIV infection. N Engl J Med. 2009;361:1768-1774.
Lockman S, Creek T. Acute maternal HIV infection during pregnancy and breast-feeding: substantial risk to infants. J Infect Dis. 2009;200:667-669.
Marhefka SL, Lyon M, Koenig LJ, et al. Emotional and behavioral problems and mental health service utilization of youth living with HIV acquired perinatally or later in life. AIDS Care. 2009;21:1447-1454.
Maron G, Gaur AH, Flynn PM. Antiretroviral therapy in HIV-infected infants and children. Pediatr Infect Dis J. 2010;29(4):360-363.
Mathers BM, Degenhardt L, Ali H, et al. HIV prevention, treatment, and care services for people who inject drugs: a systematic review of global, regional, and national coverage. Lancet. 2010;375:1014-1028.
Migueles SA, Connors M. Long-term nonprogressive disease among untreated HIV-infected individuals. JAMA. 2010;304:194-200.
Mofenson LM. Protecting the next generation—eliminating perinatal HIV-1 infection. N Engl J Med. 2010;362(24):2316-2318.
Panel on Antiretroviral Therapy and Medical Management of HIV-Infected Children: Guidelines for the use of antiretroviral agents in pediatric HIV infection. August 16, 2010 (PDF file), aidsinfo.nih.gov/ContentFiles/PediatricGuidelines.pdf. Accessed January 4, 2011
Panel on Treatment of HIV-Infected Pregnant Women and Prevention of Perinatal Transmission: Recommendations for use of antiretroviral drugs in pregnant HIV-1-infected women for maternal health and interventions to reduce perinatal HIV transmission in the United States. May 24, 2010 (PDF file), aidsinfo.nih.gov/ContentFiles/PerinatalGL.pdf. Accessed January 4, 2011
Public Health Service Task Force: Recommendations for use of antiretroviral drugs in pregnant HIV-1-infected women for maternal health and interventions to reduce perinatal HIV-1 transmission in the United States (website), www.aidsinfo.nih.gov/Guidelines/GuidelineDetail.aspx?GuidelineID=9. Accessed January 4, 2011
Richman DD, Margolis DM, Delaney M, et al. The challenge of finding a cure for HIV infection. Science. 2009;323:1304-1307.
Sidibé M, Buse K. Fomenting a prevention revolution for HIV. Lancet. 2010;375:533-534.
Silverman JG. Key to prevent HIV in women: reduce gender-based violence. Lancet. 2010;376:6-8.
UNAIDS: Global Report: UNAIDS report on the global AIDS epidemic 2010 (website), http://www.unaids.org/globalreport/Global_report.htm. Accessed January 4, 2011
Volberding PA, Deeks SG. Antiretroviral therapy and management of HIV infection. Lancet. 2010;376:49-60.
Volkow ND, Montaner J. Enhanced HIV testing, treatment, and support for HIV-infected substance users. JAMA. 2010;303(14):1423-1424.
Working Group on Antiretroviral Therapy and Medical Management of HIV-Infected Children: Guidelines for the use of antiretroviral agents in pediatric HIV infection (website), www.aidsinfo.nih.gov/Guidelines/GuidelineDetail.aspx?GuidelineID=8. Accessed September 20, 2010
World Health Organization: Antiretroviral drugs for treating pregnant women and preventing HIV infection in infants: recommendations for a public health approach. 2010 version (PDF file), whqlibdoc.who.int/publications/2010/9789241599818_eng.pdf. Accessed January 4, 2011
World Health Organization: Antiretroviral therapy for HIV infection in infants and children: towards universal access. Recommendations for a public health approach. 2010 revision (PDF file), whqlibdoc.who.int/publications/2010/9789241599801_eng.pdf. Accessed January 4, 2011
World Health Organization: HIV and infant feeding (website), www.who.int/nutrition/publications/hivaids/9789241595964/en/index.html. Accessed September 20, 2010
[/level-membership-for-pediatrics-category][not-level-membership-for-pediatrics-category]
Chapter 268 Acquired Immunodeficiency Syndrome (Human Immunodeficiency Virus)
Etiology
HIV-1 and HIV-2 are members of the Retroviridae family and belong to the Lentivirus genus, which includes cytopathic viruses causing diverse diseases in several animal species. The HIV-1 genome contains 2 copies of single-stranded RNA that is 9.2 kb in size. At both ends of the genome there are identical regions, called long terminal repeats, which contain the regulation and expression genes of HIV. The remainder of the genome includes 3 major sections: the GAG region, which encodes the viral core proteins (p24, p17, p9, and p6, which are derived from the precursor p55); the POL region, which encodes the viral enzymes (i.e., reverse transcriptase [p51], protease [p10], and integrase [p32]); and the ENV region, which encodes the viral envelope proteins (gp120 and gp41, which are derived from the precursor gp160). Other regulatory proteins, such as tat (p14), rev (p19), nef (p27), vpr (p15), vif (p23), vpu in HIV-1 (P16), and vpx in HIV-2 (P15) are involved in transactivation, viral messenger RNA expression, viral replication, induction of cell cycle arrest, promotion of nuclear import of viral reverse transcription complexes, downregulation of CD4 receptors and class I major histocompatibility complex, proviral DNA synthesis, and virus release and infectivity (Fig. 268-1).
Epidemiology
Transmission
Breast-feeding is the least common route of vertical transmission in industrialized nations but responsible for as many as 40% of perinatal infections in resource-limited countries. Both free and cell-associated viruses have been detected in breast milk from HIV-infected mothers. The risk for transmission through breast-feeding in chronically infected women is approximately 9-16% but 29-53% in women who acquire HIV postnatally, suggesting that the viremia experienced by the mother during primary infection at least triples the risk for transmission. It seems reasonable for women to substitute infant formula for breast milk if they are known to be HIV-infected or are at risk for ongoing sexual or parenteral exposure to HIV. However, the WHO recommends that in developing countries where other diseases (diarrhea, pneumonia, malnutrition) substantially contribute to a high infant mortality rate, the benefit of breast-feeding outweighs the risk for HIV transmission, and HIV-infected women in developing countries should breast-feed their infants for at least the 1st 6 mo of life (see later section on prevention).
Pathogenesis
HIV infection affects most of the immune system and disrupts its homeostasis (Fig. 268-2). In most cases, the initial infection is caused by low amounts of a single virus. Therefore, disease may be prevented by prophylactic drug(s) or vaccine. When the mucosa serves as the portal of entry for HIV, the 1st cells to be affected are the dendritic cells. These cells collect and process antigens introduced from the periphery and transport them to the lymphoid tissue. HIV does not infect the dendritic cell but binds to its DC-SIGN surface molecule, allowing the virus to survive until it reaches the lymphatic tissue. In the lymphatic tissue (e.g., lamina propria, lymph nodes), the virus selectively binds to cells expressing CD4 molecules on their surface, primarily helper T lymphocytes (CD4+ T cells) and cells of the monocyte-macrophage lineage. Other cells bearing CD4, such as microglia, astrocytes, oligodendroglia, and placental tissue containing villous Hofbauer cells, may also be infected by HIV. Additional factors (co-receptors) are necessary for HIV fusion and entry into cells. These factors include the chemokines CXCR4 (fusion) and CCR5. Other chemokines (CCR1, CCR3) may be necessary for the fusion of certain HIV strains. Several host genetic determinants affect the susceptibility to HIV infection, the progression of disease, and the response to treatment. These genetic variants vary in different populations. A deletion in the CCR5 gene that is protective against HIV infection (CCR5Δ32) is relatively common in whites but is rare in blacks. Several other genes that regulate chemokine receptors, ligands, histocompatibility complex, and cytokines have also been found to influence the outcome of HIV infection. Usually, CD4+ lymphocytes migrate to the lymphatic tissue in response to viral antigens and then become activated and proliferate, making them highly susceptible to HIV infection. This antigen-driven migration and accumulation of CD4 cells within the lymphoid tissue may contribute to the generalized lymphadenopathy characteristic of the acute retroviral syndrome in adults and adolescents. HIV preferentially infects the very cells that respond to it (HIV-specific memory CD4 cells), accounting for the progressive loss of these cells and the subsequent loss of control of HIV replication. The continued destruction of memory CD4+ cells in the gastrointestinal tract leads to reduced integrity of the gastrointestinal epithelium followed by leakage of bacterial particles into the blood and increased inflammatory response, which cause further CD4+ cell loss. When HIV replication reaches a threshold (usually within 3-6 wk from the time of infection), a burst of plasma viremia occurs. This intense viremia causes flu or mononucleosis-like symptoms (fever, rash, lymphadenopathy, arthralgia) in 50-70% of infected adults. With establishment of a cellular and humoral immune response within 2-4 mo, the viral load in the blood declines substantially, and patients enter a phase characterized by a lack of symptoms and a return of CD4 cells to only moderately decreased levels.
Clinical Manifestations
The HIV classification system is used to categorize the stage of pediatric disease by using 2 parameters: clinical status and degree of immunologic impairment (Table 268-1). Among the clinical categories, category A (mild symptoms) includes children with at least 2 mild symptoms such as lymphadenopathy, parotitis, hepatomegaly, splenomegaly, dermatitis, and recurrent or persistent sinusitis or otitis media (Table 268-2). Category B (moderate symptoms) includes children with LIP, oropharyngeal thrush persisting for >2 mo, recurrent or chronic diarrhea, persistent fever for >1 mo, hepatitis, recurrent (HSV) stomatitis, HSV esophagitis, HSV pneumonitis, disseminated varicella (i.e., with visceral involvement), cardiomegaly, or nephropathy (see Table 268-2). Category C (severe symptoms) includes children with opportunistic infections (e.g. esophageal or lower respiratory tract candidiasis, cryptosporidiosis (>1 mo), disseminated mycobacterial or cytomegalovirus infection, Pneumocystis pneumonia, or cerebral toxoplasmosis [onset >1 mo of age]), recurrent bacterial infections (sepsis, meningitis, pneumonia), encephalopathy, malignancies, and severe weight loss.
Table 268-2 CLINICAL CATEGORIES FOR CHILDREN YOUNGER THAN 13 YEARS OF AGE WITH HIV INFECTION
CATEGORY N: NOT SYMPTOMATIC
Children who have no signs or symptoms considered to be the result of HIV infection or have only 1 of the conditions listed in category A.
CATEGORY A: MILDLY SYMPTOMATIC
Children with 2 or more of the conditions listed but none of the conditions listed in categories B and C.
CATEGORY B: MODERATELY SYMPTOMATIC
Children who have symptomatic conditions other than those listed for category A or C that are attributed to HIV infection.
CATEGORY C: SEVERELY SYMPTOMATIC
[/not-level-membership-for-pediatrics-category]
