CHAPTER 219 Achondroplasia and Other Dwarfisms
Given our present understanding of the short-limb dysplasias, neurosurgical approaches to achondroplastic, thanatophoric, and hypochondroplastic patients are similar. Hydrocephalus and cervicomedullary compression are typically pediatric concerns, whereas spinal stenosis has traditionally been treated in adults; however, increased sensitivity to signs of spinal stenosis and improved surgical technology have allowed earlier treatment of clinically significant spinal compression. The initial symptoms frequently do not have strictly neurosurgical resolutions; thus, a comprehensive treatment plan involving a multidisciplinary team of physicians that includes a neurosurgeon, neurologist, pulmonary specialist, sleep specialist, geneticist, anesthesiologist, neuroradiologist, orthopedic surgeon, and otolaryngologist is useful.1–3 Because these patients are at risk for brainstem compression, comprehensive testing is directed toward the detection of central and obstructive apnea and cervicomedullary compression, all of which contribute to the risk for sudden death.
Several areas of research are leading to new initiatives in the management of skeletal dysplasias. First is the application of advanced diagnostic tools and imaging techniques.4 Second is an emphasis on outcome studies, a movement based on the well-supported assumption that different operative strategies can yield marked long-term differences in the patient’s health.4,5 Third, a better understanding of molecular biology has led to fresh ideas for targeted molecular treatment of achondroplasia, namely, those that inhibit the excess FGFR3 signals that are present in these individuals. A fourth area, the use of recombinant growth hormone to treat pediatric patients, is outside the scope of this chapter. Here, we describe our current approach to the evaluation and management of short-limb dysplasias and summarize the molecular biology, diagnostic findings, and outcome studies, mostly from our own institution.
Genetics and Epidemiology
Achondroplasia is an autosomal dominant disorder that results from a guanine (G)-to-adenine (A) mutation at base 1138 (G1138A) in the FGFR3 gene on chromosome 4 at 4p16.3.6–8 The gene codes for a tyrosine kinase receptor expressed in developing bones. The nucleotide (missense) mutation results in an amino acid glycine (Gly)-to-arginine (Arg) substitution at amino acid 380 (Gly380Arg) in the transmembrane domain of the protein. Penetrance of the Gly380Arg mutation is 100%; in other words, all individuals with the mutation will have achondroplasia. A few patients with achondroplasia have been demonstrated to instead have a Gly375Cys mutation of the FGFR3 gene.9 Achondroplasia appears to be genetically homogeneous. No significant racial difference has been detected in North America, Spain, Korea, Japan, China, or Sweden.10–15
The FGFR3 hypochondroplasia mutation pattern is more diverse than that for achondroplasia.14,16–18 In many patients, a C1620G or C1620A mutation results in an asparagine-to-lysine substitution at amino acid 540 (N450K) in the FGFR3 proximal tyrosine kinase domain. In a substantial number of patients who express a mild phenotype, the mutation has yet to be identified.16 The phenotypic diversity confounds estimates of incidence data. Accurate prenatal ultrasonographic diagnosis is rare.19 Before identification of the specific nucleotide mutations, some achondroplasia case reports probably included patients with hypochondroplasia. This overlap should be considered in efforts to compare current and past case series.
An arginine-to-cysteine (R248C) substitution in the extracellular domain of the receptor has been found in thanatophoric patients. Other mutations are likely because at least two phenotypes have been identified, thanatophoric dwarfism type I and type II. Less is known about this dysplasia because the mutation is almost always lethal neonatally.20
Compound mutations for achondroplasia and hypochondroplasia have been reported in children whose achondroplastic father or mother had the G380R mutation and the other parent had the hypochondroplasia N450K mutation.21,22 The phenotypic expression of the compound mutation appears to be more severe than that of achondroplasia.22
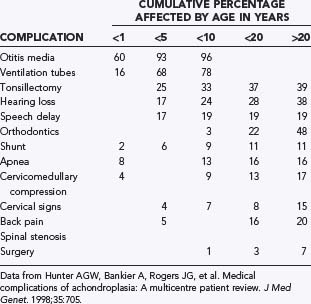
New mutations account for about 80% of children born with achondroplasia. In other words, most infants with FGFR3 mutations are born to parents without FGFR3 mutations. As in many autosomal dominant conditions, a positive correlation exists between advanced paternal age and the occurrence of new mutations. It was initially thought that factors influencing DNA replication or repair during spermatogenesis, but not during oogenesis, may predispose to occurrence of the G1138A mutation in the FGFR3 gene.23 However, more recent evidence suggests that sperm with the FGFR3 mutation have a selective advantage over sperm with a normal FGFR3 gene, thereby increasing the number of affected sperm with age.24–26 Offspring of couples who are both affected by achondroplasia have a 25% chance of inheriting both parental achondroplasia alleles, thereby resulting in homozygous achondroplasia, which is almost universally fatal within the first year of life.27 The skeletal features of achondroplasia are highly exaggerated in the homozygous condition: significantly shorter limbs, a smaller chest size, and a smaller foramen magnum. A brief summary of medical complications by age is presented in Table 219-1.
Approximately 150 skeletal dysplasias have been identified, a number of which are associated with neurological symptoms.28 Achondroplasia is the most common in clinical series. Although frequency estimates cluster between 1 in 10,000 and 1 in 35,000 live births, the true frequency must be recalculated because these data were generated before the FGFR3 mutations were identified.6,7,29,30
The FGFR3 gene encodes one of four tyrosine kinase receptors for fibroblast growth factor (FGFR1 to FGFR4) in mammals.31 Mutations of FGFR3 in achondroplasia have been shown to cause a gain of function, which correlates with the severity of the clinical phenotype.32,33 FGFR3 normally functions as an inhibitor of linear bone growth by acting negatively on both the proliferation and differentiation of growth plate chondrocytes.34,35 In achondroplasia, the normal function of FGFR3 is exaggerated. Numerous cellular mechanisms have been proposed that describe the increase in receptor tyrosine kinase activity that is common in all of the FGFR3 mutations in achondroplasia.32
Medical Complications
Most individuals with achondroplasia have normal intelligence. A cross-sectional survey (using self-reported 36-item short form health survey [SF-36] data) of the functional health status of adults with achondroplasia revealed that the mental component summary scores did not differ significantly from scores in the general population. In contrast, the physical component summary scores were significantly lower starting in the fourth decade of life.36 In children, motor milestones are delayed, partly because of generalized hypotonia and partly because of the mechanical disadvantage imposed by short limbs.1,27,37–39 Psychosocial problems arising from short stature include lack of acceptance by peers and the tendency for adults, including parents and teachers, to treat children with achondroplasia appropriately for their height rather than their age.40,41 Quality-of-life issues on which the patient’s perceived status is suboptimal require special attention during adolescence.42 Involvement in support groups with other families who have children of short stature can improve self-esteem and assist parents in guiding their achondroplastic children through the difficulties of growing up in a culture that often equates stature with status.
Reproductive difficulties have not been conclusively documented, but reduced fertility, frequent fibroid cysts, and early menopause have been reported. The decreased reproductive rates in achondroplastic individuals may have been due in part to the social stigma present in those with reduced height in finding potential mates. However, with the establishment of organizations for those with reduced height, such as Little People of America, these individuals are now more likely to marry and have children.32 Women with achondroplasia must deliver their infants by cesarean section because of cephalopelvic disproportion,43 and the administration of spinal anesthesia is strongly discouraged.44
Obstructive sleep apnea, or sleep-disordered breathing secondary to a small upper airway, is common. Tonsillectomy and adenoidectomy decrease the degree of upper airway obstruction in most children. The majority do not have significant obstructive or central apnea, but a considerable minority are severely affected.45–48 The cause of different patterns of sleep disorders may be related to localized alterations in chondrocranial development.46 Many infants sleep with their necks in a hyperextended position, which functionally increases the size of the upper airway. Although the hyperextended neck position relieves intermittent obstruction, it can also exacerbate the neurological sequelae of cervicomedullary compression related to a small foramen magnum. Abnormal respiratory sinus arrhythmias may be present.49 A small thoracic cage can result in restrictive pulmonary disease in infancy. Respiration may be further compromised by aspiration secondary to gastroesophageal reflux, swallowing dysfunction, or both, and result in recurrent pneumonia. Anesthesia can be given safely to children, with special consideration for limited neck extension and the use of appropriately sized endotracheal tubes.47
A relatively high rate (about 3%) of jugular bulb dehiscence—complete absence of the roof over the jugular bulb—was identified in a series of 126 achondroplastic children. This increased incidence may account for unexplained hearing loss, tinnitus, and self-audible bruits in these children and poses a risk for difficult-to-control bleeding at myringotomy.50
Evaluation and Diagnosis
Cervicomedullary Compression
Clinical Findings and Pathology
Cervicomedullary compression stems primarily from a reduction in the diameter of the foramen magnum in both the sagittal and coronal dimensions, a reduction that is sometimes more than 5 SD less than normal.51–56 Cervicomedullary compression warrants early and aggressive treatment because it results in high cervical myelopathy and increases the risk for sudden death by central respiratory failure.57–60 A prospective evaluation of achondroplastic infants found radiographic evidence of craniocervical stenosis in 58% of the patients studied, and a diagnosis of cervicomedullary compression was made in 35%.61 Although these figures are derived from a selected population and are therefore higher than for the general population, they are a strong argument for careful evaluation and treatment of achondroplastic children. A retrospective study found increased mortality (in comparison to population standards) in achondroplastic children younger than 4 years, with sudden death from brainstem compression identified as the cause of half of the deaths. The same study also found a 7.5% risk for sudden death in the first year of life.27
Chronic medullary and upper cervical cord compression may exist as a neurologically asymptomatic lesion and exhibit neither signs of root compression in the arms nor symptoms of cranial nerve impairment. Nonetheless, microcystic histopathologic changes, cervical syringomyelia, necrosis, and gliosis have been reported in autopsies of achondroplastic children who died unexpectedly. Presumably, lesions of this type interrupt the neural respiratory pathways from the nucleus tractus solitarii to the phrenic nerve nucleus, thereby arresting the muscles of inspiration and resulting in sudden death in some cases. We therefore consider infants with a history of sleep apnea or other severe respiratory or neurological abnormalities to be at increased risk for respiratory complications resulting from occult cervicomedullary compression. Some authors have recommended performing sleep and imaging studies on all children with achondroplasia.62 We believe that a careful history and neurological examination should precede costlier and more uncomfortable diagnostics. A composite profile of patients with cervicomedullary compression includes upper or lower extremity paresis, apnea or cyanosis, hyperreflexia or hypertonia, and delay in motor milestones beyond achondroplastic standards. These patients can present a striking contrast to the usual floppy, hypotonic achondroplastic infant.63
Evaluation
Once a high-risk patient with respiratory or neurological symptoms or signs has been identified, we advise comprehensive testing. Parents should be carefully interviewed about the health and medical history of their child, with an emphasis on respiratory symptoms. A general physical examination should be performed, including chest measurements. Respiratory evaluation should include evaluation of blood pH, a chest radiograph, and overnight polysomnography. Electrocardiograms and echocardiograms should be performed to see whether there is cardiac evidence of chronic oxygen deprivation during sleep. A neurological examination for signs of brainstem compression, such as hyperreflexia, hypertonia, paresis, asymmetry of movement or strength, or abnormal plantar responses, is essential. Brainstem auditory evoked potential and upper extremity somatosensory evoked potential evaluation should be considered as an adjunct, especially in patients with normal results on neurological examination. Imaging studies of the craniocervical junction are necessary, particularly magnetic resonance imaging (MRI) in the sagittal plane. We also strongly recommend cardiac-gated MRI flow studies of cerebrospinal fluid (CSF) at the foramen magnum. There has been some evidence that dynamic MRI with the head in flexed and extended positions may increase the sensitivity for positional pathology in CSF dynamics.64
Indications for Surgery
Concerns have been expressed about the indications for surgical decompression of the foramen magnum in this population.65–67 Radiographic studies during the first years of life show some degree of compression at the foramen magnum level. MRI evidence of spinal cord compression, such as indentation or narrowing of the upper cervical cord, is a common finding that is usually graded as “marked” or “severe” in the MRI report.65 In one report, myelomalacia was observed in 13 of 30 achondroplastic children.55 Yet other than the routinely observed generalized hypotonia seen in the achondroplastic population, the majority of these children are asymptomatic and outgrow their developmental delays.66,68–70 Cervicomedullary decompression (CMD) as a standard routine prophylactic measure is therefore not warranted.
Several groups have published guidelines for surgical intervention.68,69,71 Our indications are based on lower limb symptoms, polysomnography, and MRI flow studies. The underlying principle must be to identify patients who are at risk for neurological damage or sudden death. We recommend that patients with cervicomedullary compression be identified and treated prophylactically, before abrupt and irreversible changes occur. For the purpose of diagnosis, we define clinically significant cervicomedullary compression to be (1) neurological evidence of upper cervical myelopathy or chronic brainstem compression (apnea, lower cranial nerve dysfunction, swallowing difficulties); (2) evidence of stenosis on imaging studies, including the absence of flow above and below the foramen magnum; and (3) frequently an otherwise unexplained respiratory or developmental abnormality.
Hunter and coworkers conducted a multicenter review of 193 patients with chondrodysplasias. The study reported data on rates of medical complications at specific age intervals (see Table 219-1). At age 4 the rate of cervicomedullary compression was 6.8%. The authors emphasized the important role of surgery, primarily because progressive neurological symptoms continue into adulthood. Ultimately, about 17% of the patients in the series underwent CMD.72
Hydrocephalus
Clinical Findings and Pathology
Hydrocephalus in an achondroplastic patient is probably secondary to deformation of the cranial base. Constriction of the basal foramina, particularly the jugular foramina, is thought to reduce venous drainage and potentially raise intracranial venous pressure. Investigators have demonstrated a correlation between the degree of venous narrowing at the jugular foramina and the degree of hydrocephalus in achondroplaasia.73 In theory, absorption of CSF into the sagittal venous sinus is thus reduced and results in hydrocephalus.56 However, identifying patients at high risk for hydrocephalus is currently not possible. Hydrocephalus may resolve in some patients with continued growth of the skull base during puberty.
It is easy to suspect hydrocephalus in a patient with achondroplasia, given that macrocrania is a morphologic hallmark of the disease. Concerns about hydrocephalus may also arise because of the enlarged ventricles and the delayed acquisition of gross motor skills. Although hydrocephalus is associated with enlarged ventricles in the achondroplastic population, it generally resolves through growth and maturation of the cranial bones.74 An achondroplastic child typically displays transient hypotonia, but the papilledema that is expected with symptomatic hydrocephalus is rare. Radiographically, mild to moderate ventricular enlargement, prominent cortical sulci, and an increased frontal subarachnoid space are apparent. Hydrocephalus severe enough to require shunting is often discovered after craniocervical decompression, when CSF leaks often complicate wound healing.
Evaluation
A degree of abnormal CSF dynamics is common in achondroplasia. We believe that routine surveillance of achondroplastic infants for hydrocephalus is best limited to noninvasive measures that are easily incorporated into the pediatric examination and should not require referral to special centers and complex diagnostic procedures. Longitudinally evaluating head circumference and tracking the acquisition of developmental milestones, with comparison to published standards specific for achondroplasia,75,76 are usually sufficient. Imaging studies can be reserved for patients whose head circumference crosses percentiles on the achondroplastic chart or for those who exhibit unexplained neurological signs or developmental retardation.
Indications for Surgery
Stenosis of the jugular foramina contributes to the altered CSF dynamics in achondroplasia.56 Jugular foramen decompression is an option.77 However, we believe that this option should be used only in children with severe jugular stenosis and debilitating hydrocephalus in whom conventional ventriculoperitoneal shunting is contraindicated. Given the high percentage of complications, shunting is best reserved for those in whom the symptoms are severe and threatening.54 CSF pressure profiles can be determined with an external transducer attached to an open fontanelle, with an epidural pressure monitor, or with an intraventricular catheter in older patients. If no critical pressure elevations are detected during a 48-hour period, shunt placement is not required, ventriculomegaly notwithstanding. However, the presence of severe clinical stigmata for hydrocephalus obviates such demonstrations.
Spinal Stenosis
Clinical Findings and Pathology
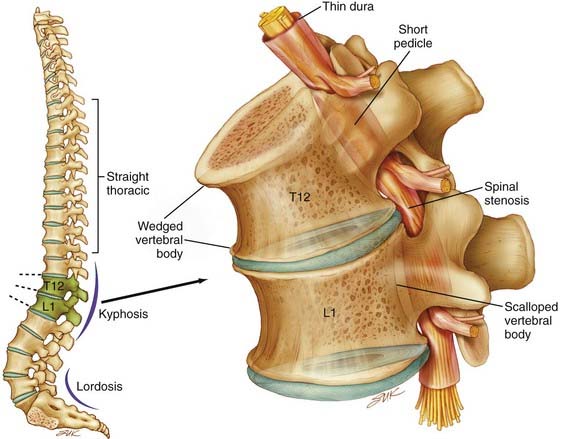
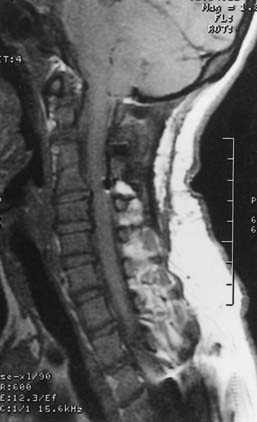
Spinal stenosis is the most common complication of achondroplasia and usually becomes symptomatic in the third decade or later. The anatomy of the achondroplastic spine is distinctive in several respects, all of which contribute to spinal cord compromise and nerve root compression. The superior and inferior articular facets of the vertebral bodies are susceptible to hypertrophy, which results in a “mushroom” shape that is clearly evident on MRI. Abbreviated and thickened pedicles of the vertebral arches result from premature fusion. Intervertebral disks tend to bulge prominently, thus further aggravating neural encroachment by the enlarged vertebral body articular surfaces. The interpediculate distance decreases in the lumbar region of the spine, which results in a canal that tapers caudally, the opposite of normal (the canal normally widens caudally). The overall picture is one of dramatic stenosis in every dimension of the spine (Fig. 219-1).
Although the problems related to hydrocephalus and cervicomedullary compression are frequently identified in infancy and childhood, neurological problems below the foramen magnum are traditionally thought to be manifested in late adolescence and adulthood. However, in our most recent surgical series of 44 pediatric achondroplastic patients who underwent spinal decompressive surgery, over half were younger than 12 years.78 Because the achondroplastic spinal canal tends to have severe congenital constriction, more intensive early screening might reveal substantial numbers of young achondroplastic patients with occult symptoms of spinal stenosis. Estimates of the incidence of symptomatic spinal stenosis range from 37% to 89%, thus suggesting that a significant proportion will eventually have this problem.72 Thorough urologic and neurological testing plays a useful part in the prospective evaluation for occult stenosis. Interestingly, in our pediatric series of achondroplastic spinal stenosis, 27 of 44 (61%) patients who underwent laminectomies for decompression had previously exhibited signs of cervicomedullary compression. In these patients the average age at the time of CMD was 3.5 years, whereas the average age at the time of spinal decompressive surgery was 11.5 years.78
In management of the achondroplastic spine, it is possible to distinguish between the neurosurgical and orthopedic aspects of this disease. The hypotonia that an achondroplastic infant typically exhibits suggests that muscular tone may be insufficient for adequate protection of pediatric skeletal structures in weight-bearing postures. Achondroplastic children are in fact developmentally delayed in supporting their heads independently, as well as in upright sitting and walking. In our opinion, parents should not encourage early sitting because of the potential for aggravation of thoracolumbar kyphosis in this posture. Sitting and standing postures affect the curvature of the spine, and in achondroplastic children, muscular weakness, short vertebral pedicles, and lax spinal ligaments complicate these mechanics. Attention has also been drawn to the dynamic effect of a small chest and a globulus abdomen in the progressive development of kyphosis.79 Moreover, delayed standing predisposes to the development of a gibbus, with wedging of one or more vertebral elements. These wedged deformities are both debilitating and preventable. Because surgical repair has risks, effort is well spent on prevention. Orthopedic bracing is used prophylactically when the formation of a wedged gibbus seems likely. Parents should also be urged to not use any infant carriers, strollers, or baby seats that exaggerate the thoracolumbar kyphosis.
In an adult, compromise can result from abnormalities such as hyperlordosis, minor disk bulging, hypertrophic osteoarthritis, or ligamentous hypertrophy. The presence of thoracolumbar kyphosis is also positively correlated with symptomatic spinal stenosis.79 Although low back pain is a common complaint in achondroplastic patients, symptomatic neurogenic claudication can develop in those with severe stenosis. Prolonged walking produces first paresthesias and later weakness of the lower extremities, which is usually bilateral. These symptoms are promptly relieved by resting, squatting, or leaning forward, which straightens the lordosis and increases the transverse diameter of the lumbosacral canal.80 With progressive stenosis, the distance walked before claudication ensues decreases, thus making this symptom a useful clinical parameter.
Operative Management
At our institution we use a common high-speed drill technique for both craniocervical and spinal decompression. Although laminectomy is a widely practiced spinal procedure, we believe that modifications that address several of its shortcomings, including inadequate decompression and secondary spinal instability, are necessary for its use in achondroplastic patients.81,82
Cervicomedullary Compression
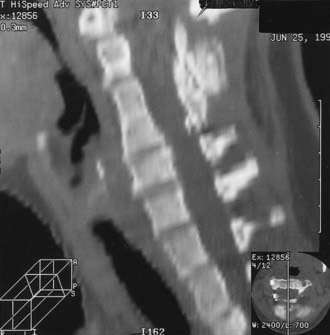
Craniocervical surgical decompression for cervicomedullary compression in children with achondroplasia has been used at several centers and generally yields good results.57,83 Decompression of the cervicomedullary junction has been shown to bring about dramatic and sustained improvement in neurological and respiratory function when it is combined with other therapy as needed for respiratory compromise.61,63 The surgeon must understand the anatomic difficulties presented by achondroplastic patients (Fig. 219-2). Clinical evaluation, moreover, is frequently difficult because achondroplastic patients can have respiratory difficulties for many reasons, some of which are unrelated to the neurological compromise. Long-term follow-up data that would allow definitive assessment of craniocervical decompression have also been lacking.65 As with any surgical procedure, detailed prior consultation must be conducted with the parents to inform them of the potential risks and expected benefits for their achondroplastic child.
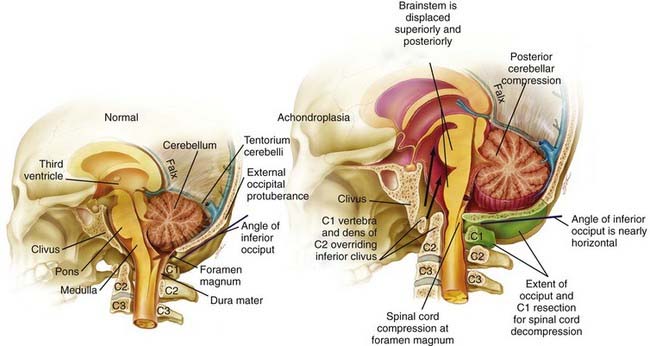
 A midline suboccipital incision is made for the decompression, and the ligaments and musculature are dissected subperiosteally to expose the occiput and the spinous processes and laminae of C1 and C2. The arch of C1 is then removed with a high-speed drill and small curets (Video 219-1). One frequently sees a thick, fibrous band or pannus above the level of C1, which should be left in place during dissection of the bone to protect the underlying dura and cord from incidental injury. Compression of the cervical cord occasionally necessitates removal of the arch of C2 or extension of the decompression even farther in a caudal direction. The posterior rim of the foramen magnum is thinned gradually with a high-speed drill and removed with small, straight and angled curets. Invariably, the bone of the foramen magnum is thickened, oriented more horizontally than usual, and severely indenting the underlying dura. Once decompression of the bone is complete, the fibrous pannus is removed as well, which often reveals a transverse dural channel that offers dramatic evidence of the extent of the dural constriction; consequently, adequate attention must be paid to the soft tissue aspects of the decompression. If a duraplasty is being performed, the dura is opened in the midline along the area of constriction. Adequate cord pulsations and CSF flow can be confirmed, and a dural patch graft can be performed with paraspinous fascia, pericranium, or commercially available human cadaveric dura. A watertight seal is confirmed, and the wound is copiously irrigated and closed in several layers; it is not drained, however, to avoid potentiating the development of a CSF fistula. In our early experience, CSF leaks developed in a significant number of patients despite efforts in making the closure watertight and required a ventriculoperitoneal shunt for definitive treatment. Therefore, it has been our recent practice to not routinely perform a duraplasty as part of the decompression. Instead, after removal of the bone and fibrous tissue, we use intraoperative ultrasonography to assess for residual compression of the cervicomedullary junction and to visualize the CSF spaces. If ultrasonography reveals adequate decompression and ample CSF spaces, we do not open the dura for a duraplasty. In our recent experience, only 2 of 43 patients underwent duraplasty, and both cases were complicated by postoperative CSF leaks.83
A midline suboccipital incision is made for the decompression, and the ligaments and musculature are dissected subperiosteally to expose the occiput and the spinous processes and laminae of C1 and C2. The arch of C1 is then removed with a high-speed drill and small curets (Video 219-1). One frequently sees a thick, fibrous band or pannus above the level of C1, which should be left in place during dissection of the bone to protect the underlying dura and cord from incidental injury. Compression of the cervical cord occasionally necessitates removal of the arch of C2 or extension of the decompression even farther in a caudal direction. The posterior rim of the foramen magnum is thinned gradually with a high-speed drill and removed with small, straight and angled curets. Invariably, the bone of the foramen magnum is thickened, oriented more horizontally than usual, and severely indenting the underlying dura. Once decompression of the bone is complete, the fibrous pannus is removed as well, which often reveals a transverse dural channel that offers dramatic evidence of the extent of the dural constriction; consequently, adequate attention must be paid to the soft tissue aspects of the decompression. If a duraplasty is being performed, the dura is opened in the midline along the area of constriction. Adequate cord pulsations and CSF flow can be confirmed, and a dural patch graft can be performed with paraspinous fascia, pericranium, or commercially available human cadaveric dura. A watertight seal is confirmed, and the wound is copiously irrigated and closed in several layers; it is not drained, however, to avoid potentiating the development of a CSF fistula. In our early experience, CSF leaks developed in a significant number of patients despite efforts in making the closure watertight and required a ventriculoperitoneal shunt for definitive treatment. Therefore, it has been our recent practice to not routinely perform a duraplasty as part of the decompression. Instead, after removal of the bone and fibrous tissue, we use intraoperative ultrasonography to assess for residual compression of the cervicomedullary junction and to visualize the CSF spaces. If ultrasonography reveals adequate decompression and ample CSF spaces, we do not open the dura for a duraplasty. In our recent experience, only 2 of 43 patients underwent duraplasty, and both cases were complicated by postoperative CSF leaks.83
Spinal Stenosis
Decompression of the achondroplastic spinal canal is difficult because of the extent and severity of the stenosis. The quantitative magnitude of this stenosis has been well documented.84 Moreover, poor postoperative results of spinal decompression were relatively common in achondroplastic patients.81,82,85 Before the era of computed tomography (CT) and MRI, the degree and extent of the spinal compression were often not appreciated with conventional myelography because of the lack of adequate diffusion of contrast medium. Insertion of bulky instruments under the laminae during the conventional techniques frequently traumatized neural tissue. Another source of poor results was postoperative instability resulting from overly wide laminectomies.
A different viewpoint concerning the ideal strategy for spinal laminectomy recommends a wide decompression with foraminotomies and mandatory undermining of the facets.86 The rationale for this strategy is that compression is lateral as well as longitudinal in the achondroplastic spinal canal. Our experience, however, does not bear this out despite the hypothesized impact of small lateral recesses in the achondroplastic vertebral foramen. Moreover, the stabilization problems encountered with wide laminectomies can be more debilitating than the initial disease. In fact, there is no reason why every spinal level could not be subjected to the laminectomy we describe without the need for concomitant spinal stabilization. Therefore, in light of the results obtained at our institution with spinal decompression, we believe that a narrow, extensive laminectomy is the spinal decompression method of choice for achondroplastic patients. Some surgeons have adopted this operative technique for their nonachondroplastic patients as well. The goal is adequate decompression of neural elements and not simply enlargement of bony canals.
Thoracolumbar kyphosis is commonly present in achondroplastic children, with a prevalence as high as 87% in children between 1 and 2 years of age.87 Most of these patients, however, remain asymptomatic and may demonstrate spontaneous resolution by 3 years of age. Effective control of progressive thoracolumbar kyphosis of 30 degrees or greater with orthotic bracing has been demonstrated in the pediatric age group.88 However, decompression of the spine over areas of existing kyphosis warrants concern for progression as a result of removal of the posterior ligamentous tension band. Ain and colleagues demonstrated postlaminectomy kyphosis in 10 consecutive skeletally immature achondroplastic children who underwent decompressive laminectomy for spinal stenosis.89 They also demonstrated that achondroplastic children with thoracolumbar kyphosis can safely undergo instrumented fusion concomitant with laminectomies for neurological symptoms.90 Therefore, it is our current practice to recommend instrumented fusion in all skeletally immature achondroplastic children with thoracolumbar kyphosis at the intended levels of decompression.
Outcome
Craniocervical Decompression
We reviewed our experience in 43 children with achondroplasia who underwent CMD with a mean follow-up period of 62.5 months.83 There were no surgery-related deaths in these children. The most common complication was CSF leaks, which occurred in 7 patients from either the ventriculostomy site or the suboccipital incision. There were no instances of clinical deterioration immediately after the operation; however, 5 patients experienced worsening after a period of improvement. Imaging in these patients revealed recurrent stenosis at the foramen magnum, and all of them responded well to a revision decompression. The remainder of patients did not experience recurrence of clinical symptoms after the initial decompression during the extended follow-up of the study. We therefore concluded that CMD can be safely performed for cervicomedullary compression in achondroplastic patients with minimal morbidity, provided that the surgeon recognizes the intricacies of the anatomy in this population.
Spinal Restenosis in Achondroplasia
In a spine that has previously been decompressed, restenosis may occur because of accelerated facet hypertrophy, bony overgrowth, and scarring. This acceleration of facet hypertrophy may represent instability in the previously operated achondroplastic spine or some exaggerated response to normal motion that results from the genetic defect in this disease. There are many reports documenting the efficacy of decompressive therapy in the treatment of achondroplastic spinal stenosis. In several of these series, reoperation was often necessary for achondroplastic spinal restenosis.91
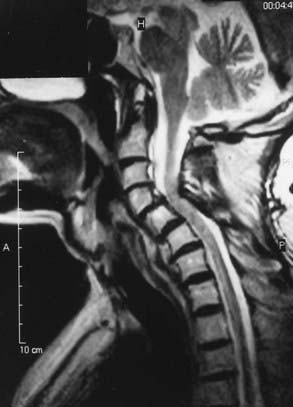
We reviewed our series of eight achondroplastic patients who underwent reoperation for spinal restenosis between 1994 and 1996. There were five men and three women with a mean age of 43 years. The most common neurological sign of recurrent stenosis was impaired motor function, which occurred in all eight (100%) patients. Seven (87.5%) of the patients had sensory dysfunction, four (50%) had neurogenic claudication, four (50%) had severe radicular pain, one (12.5%) had bladder incontinence (one also had bowel incontinence), and four (50%) had signs of myelopathy. Axial low back pain was present in all seven patients who had thoracolumbar stenosis. Two of the eight patients were seen because of abrupt deterioration of their neurological condition. All other patients experienced gradual deterioration over a mean interval of 8.9 months. All seven patients with thoracolumbar stenosis showed complete blockage on CT myelography; an incomplete block was observed in the patient with cervical stenosis. The most common cause of recurrent stenosis was facet hypertrophy in six (75%) patients (Fig. 219-3). Other causes included disk pathology in four (50%) patients, bony overgrowth in three (37.5%) patients, kyphosis in three (37.5%) patients, spur formation in two (25%) patients, and fusion construct (Figs. 219-4 and 219-5) in one (12.5%) patient. The mean interval between the most recent surgeries was 8.2 years; however, for surgery at the same level, the mean interval was 9.5 years. Complications included a dural tear and cerebellar hemorrhage in one patient and transient neurological worsening in another patient. One patient died 24 hours after surgery when acute respiratory insufficiency and fatal cardiac arrest developed after extubation. The patient had been placed in halo stabilization after a repeat cervical laminectomy and lateral mass fusion for cervical subluxation and progressive quadriparesis.
Acknowledgment
We would like to thank Ian Suk for his beautiful illustrations in Figures 219-1 and 219-2.
Ain MC, Browne JA. Spinal arthrodesis with instrumentation for thoracolumbar kyphosis in pediatric achondroplasia. Spine. 2004;29:2075.
Ain MC, Shirley ED, Pirouzmanesh A, et al. Postlaminectomy kyphosis in the skeletally immature achondroplast. Spine. 2006;31:197.
Aryanpur J, Hurko O, Francomano C, et al. Craniocervical decompression for cervicomedullary compression in pediatric patients with achondroplasia. J Neurosurg. 1990;73:375.
Bagley CA, Pindrik JA, Bookland MJ, et al. Cervicomedullary decompression for foramen magnum stenosis in achondroplasia. J Neurosurg. 2006;104:166.
Deng C, Wynshaw-Boris A, Zhou F, et al. Fibroblast growth factor receptor 3 is a negative regulator of bone growth. Cell. 1996;84:911.
Erdincler P, Dashti R, Kaynar MY, et al. Hydrocephalus and chronically increased intracranial pressure in achondroplasia. Childs Nerv Syst. 1997;13:345.
Francomano CA. The genetic basis of dwarfism. N Engl J Med. 1995;332:58.
Goriely A, McVean GA, Rojmyr M, et al. Evidence for selective advantage of pathogenic FGFR2 mutations in the male germ line. Science. 2003;301:643.
Goriely A, McVean GA, van Pelt AM, et al. Gain-of-function amino acid substitutions drive positive selection of FGFR2 mutations in human spermatogonia. Proc Natl Acad Sci U S A. 2005;102:6051.
Horton WA, Hall JG, Hecht JT. Achondroplasia. Lancet. 2007;370:162.
Hunter AG, Bankier A, Rogers JG, et al. Medical complications of achondroplasia: a multicentre patient review. J Med Genet. 1998;35:705.
Lachman RS. Neurologic abnormalities in the skeletal dysplasias: a clinical and radiological perspective. Am J Med Genet. 1997;69:33.
Lemyre E, Azouz EM, Teebi AS, et al. Bone dysplasia series. Achondroplasia, hypochondroplasia and thanatophoric dysplasia: review and update. Can Assoc Radiol J. 1999;50:185.
Marin-Padilla M, Marin-Padilla TM. Developmental abnormalities of the occipital bone in human chondrodystrophies (achondroplasia and thanatophoric dwarfism). Birth Defects Orig Artic Ser. 1977;13:7.
McKusick VA, Amberger JS, Francomano CA. Progress in medical genetics: map-based gene discovery and the molecular pathology of skeletal dysplasias. Am J Med Genet. 1996;63:98.
Mogayzel PJJr, Carroll JL, Loughlin GM, et al. Sleep-disordered breathing in children with achondroplasia. J Pediatr. 1998;132:667.
Pauli RM, Breed A, Horton VK, et al. Prevention of fixed, angular kyphosis in achondroplasia. J Pediatr Orthop. 1997;17:726.
Pauli RM, Horton VK, Glinski LP, et al. Prospective assessment of risks for cervicomedullary-junction compression in infants with achondroplasia. Am J Hum Genet. 1995;56:732.
Reid CS, Pyeritz RE, Kopits SE, et al. Cervicomedullary compression in young patients with achondroplasia: value of comprehensive neurologic and respiratory evaluation. J Pediatr. 1987;110:522.
Ruiz-Garcia M, Tovar-Baudin A, Del Castillo-Ruiz V, et al. Early detection of neurological manifestations in achondroplasia. Childs Nerv Syst. 1997;13:208.
Sciubba DM, Noggle JC, Marupudi NI, et al. Spinal stenosis surgery in pediatric patients with achondroplasia. J Neurosurg. 2007;106:372.
Wassman ERJr, Rimoin DL. Cervicomedullary compression with achondroplasia. J Pediatr. 1988;113:411.
Yamada Y, Ito H, Otsubo Y, et al. Surgical management of cervicomedullary compression in achondroplasia. Childs Nerv Syst. 1996;12:737.
Zucconi M, Weber G, Castronovo V, et al. Sleep and upper airway obstruction in children with achondroplasia. J Pediatr. 1996;129:743.
1 Health supervision for children with achondroplasia. American Academy of Pediatrics Committee on Genetics. Pediatrics. 1995;95:443.
2 Carson B, Francomano C, Hurko O, et al. Management of achondroplasia and its neurosurgical complications. In: Schmidek H, editor. Operative Neurosurgical Techniques. Philadelphia: WB Saunders, 1999.
3 Carson B, Sponseller P, Guarnieri M. Congenital spine anomalies. In: Albright A, Pollack I, Adelson P, editors. Principals and Practices of Pediatric Neurosurgery. New York: Thieme, 1998.
4 Johnson MH, Smoker WR. Lesions of the craniovertebral junction. Neuroimaging Clin N Am. 1994;4:599.
5 Clancy CM, Eisenberg JM. Outcomes research: measuring the end results of health care. Science. 1998;282:245.
6 McKusick VA, Amberger JS, Francomano CA. Progress in medical genetics: map-based gene discovery and the molecular pathology of skeletal dysplasias. Am J Med Genet. 1996;63:98.
7 Francomano CA. The genetic basis of dwarfism. N Engl J Med. 1995;332:58.
8 Cohen MMJr. Achondroplasia, hypochondroplasia and thanatophoric dysplasia: clinically related skeletal dysplasias that are also related at the molecular level. Int J Oral Maxillofac Surg. 1998;27:451.
9 Superti-Furga A, Eich G, Bucher HU, et al. A glycine 375–to-cysteine substitution in the transmembrane domain of the fibroblast growth factor receptor-3 in a newborn with achondroplasia. Eur J Pediatr. 1995;154:215.
10 Tanaka H. Achondroplasia: recent advances in diagnosis and treatment. Acta Paediatr Jpn. 1997;39:514.
11 Climent C, Lorda-Sanchez I, Urioste M, et al. [Achondroplasia: molecular study of 28 patients.]. Med Clin (Barc). 1998;110:492.
12 Yang SW, Kitoh H, Yamada Y, et al. Mutation in the gene encoding the fibroblast growth factor receptor-3 in Korean children with achondroplasia. Acta Paediatr Jpn. 1998;40:324.
13 Zhao P, Ma H, Wang Y, et al. [Mutations of the fibroblast growth factor receptor 3 gene in achondroplasia.]. Zhonghua Yi Xue Yi Chuan Xue Za Zhi. 1999;16:16.
14 Ezquieta Zubicaray B, Iguacel AO, Varela Junquera JM, et al. [Gly380Arg and Asn540Lys mutations of fibroblast growth factor receptor 3 in achondroplasia and hypochondroplasia in the Spanish population.]. Med Clin (Barc). 1999;112:290.
15 Alderborn A, Anvret M, Gustavson KH, et al. Achondroplasia in Sweden caused by the G1138A mutation in FGFR3. Acta Paediatr. 1996;85:1506.
16 Ramaswami U, Rumsby G, Hindmarsh PC, et al. Genotype and phenotype in hypochondroplasia. J Pediatr. 1998;133:99.
17 Tsai FJ, Wu JY, Tsai CH, et al. Identification of a common N540K mutation in 8/18 Taiwanese hypochondroplasia patients: further evidence for genetic heterogeneity. Clin Genet. 1999;55:279.
18 Matsui Y, Yasui N, Kimura T, et al. Genotype phenotype correlation in achondroplasia and hypochondroplasia. J Bone Joint Surg Br. 1998;80:1052.
19 Lemyre E, Azouz EM, Teebi AS, et al. Bone dysplasia series. Achondroplasia, hypochondroplasia and thanatophoric dysplasia: review and update. Can Assoc Radiol J. 1999;50:185.
20 Baker KM, Olson DS, Harding CO, et al. Long-term survival in typical thanatophoric dysplasia type 1. Am J Med Genet. 1997;70:427.
21 Huggins MJ, Smith JR, Chun K, et al. Achondroplasia-hypochondroplasia complex in a newborn infant. Am J Med Genet. 1999;84:396.
22 Chitayat D, Fernandez B, Gardner A, et al. Compound heterozygosity for the achondroplasia-hypochondroplasia FGFR3 mutations: prenatal diagnosis and postnatal outcome. Am J Med Genet. 1999;84:401.
23 Wilkin DJ, Szabo JK, Cameron R, et al. Mutations in fibroblast growth-factor receptor 3 in sporadic cases of achondroplasia occur exclusively on the paternally derived chromosome. Am J Hum Genet. 1998;63:711.
24 Goriely A, McVean GA, Rojmyr M, et al. Evidence for selective advantage of pathogenic FGFR2 mutations in the male germ line. Science. 2003;301:643.
25 Goriely A, McVean GA, van Pelt AM, et al. Gain-of-function amino acid substitutions drive positive selection of FGFR2 mutations in human spermatogonia. Proc Natl Acad Sci U S A. 2005;102:6051.
26 Tiemann-Boege I, Navidi W, Grewal R, et al. The observed human sperm mutation frequency cannot explain the achondroplasia paternal age effect. Proc Natl Acad Sci U S A. 2002;99:14952.
27 Hecht JT, Francomano CA, Horton WA, et al. Mortality in achondroplasia. Am J Hum Genet. 1987;41:454.
28 Lachman RS. Neurologic abnormalities in the skeletal dysplasias: a clinical and radiological perspective. Am J Med Genet. 1997;69:33.
29 Gardner RJ. A new estimate of the achondroplasia mutation rate. Clin Genet. 1977;11:31.
30 Passos-Bueno MR, Wilcox WR, Jabs EW, et al. Clinical spectrum of fibroblast growth factor receptor mutations. Hum Mutat. 1999;14:115.
31 Ornitz DM, Marie PJ. FGF signaling pathways in endochondral and intramembranous bone development and human genetic disease. Genes Dev. 2002;16:1446.
32 Horton WA, Hall JG, Hecht JT. Achondroplasia. Lancet. 2007;370:162.
33 Naski MC, Wang Q, Xu J, et al. Graded activation of fibroblast growth factor receptor 3 by mutations causing achondroplasia and thanatophoric dysplasia. Nat Genet. 1996;13:233.
34 Colvin JS, Bohne BA, Harding GW, et al. Skeletal overgrowth and deafness in mice lacking fibroblast growth factor receptor 3. Nat Genet. 1996;12:390.
35 Deng C, Wynshaw-Boris A, Zhou F, et al. Fibroblast growth factor receptor 3 is a negative regulator of bone growth. Cell. 1996;84:911.
36 Mahomed NN, Spellmann M, Goldberg MJ. Functional health status of adults with achondroplasia. Am J Med Genet. 1998;78:30.
37 Thompson NM, Hecht JT, Bohan TP, et al. Neuroanatomic and neuropsychological outcome in school-age children with achondroplasia. Am J Med Genet. 1999;88:145.
38 Ruiz-Garcia M, Tovar-Baudin A, Del Castillo-Ruiz V, et al. Early detection of neurological manifestations in achondroplasia. Childs Nerv Syst. 1997;13:208.
39 Castiglia PT. Achondroplasia. J Pediatr Health Care. 1996;10:180.
40 Haverkamp F, Noeker M. hort stature in children–a questionnaire for parents’: a new instrument for growth disorder–specific psychosocial adaptation in children. Qual Life Res. 1998;7:447.
41 Fowler ES, Glinski LP, Reiser CA, et al. Biophysical bases for delayed and aberrant motor development in young children with achondroplasia. J Dev Behav Pediatr. 1997;18:143.
42 Apajasalo M, Sintonen H, Rautonen J, et al. Health-related quality of life of patients with genetic skeletal dysplasias. Eur J Pediatr. 1998;157:114.
43 Allanson JE, Hall JG. Obstetric and gynecologic problems in women with chondrodystrophies. Obstet Gynecol. 1986;67:74.
44 Berkowitz ID, Raja SN, Bender KS, et al. Dwarfs: pathophysiology and anesthetic implications. Anesthesiology. 1990;73:739.
45 Mogayzel PJJr, Carroll JL, Loughlin GM, et al. Sleep-disordered breathing in children with achondroplasia. J Pediatr. 1998;132:667.
46 Tasker RC, Dundas I, Laverty A, et al. Distinct patterns of respiratory difficulty in young children with achondroplasia: a clinical, sleep, and lung function study. Arch Dis Child. 1998;79:99.
47 Sisk EA, Heatley DG, Borowski BJ, et al. Obstructive sleep apnea in children with achondroplasia: surgical and anesthetic considerations. Otolaryngol Head Neck Surg. 1999;120:248.
48 Zucconi M, Weber G, Castronovo V, et al. Sleep and upper airway obstruction in children with achondroplasia. J Pediatr. 1996;129:743.
49 DiMario FJ, Bauer L, Baxter D. Respiratory sinus arrhythmia of brainstem lesions. J Child Neurol. 1999;14:229.
50 Pauli RM, Modaff P. Jugular bulb dehiscence in achondroplasia. Int J Pediatr Otorhinolaryngol. 1999;48:169.
51 Thomas IT, Frias JL, Williams JL, et al. Magnetic resonance imaging in the assessment of medullary compression in achondroplasia. Am J Dis Child. 1988;142:989.
52 Marin-Padilla M, Marin-Padilla TM. Developmental abnormalities of the occipital bone in human chondrodystrophies (achondroplasia and thanatophoric dwarfism). Birth Defects Orig Artic Ser. 1977;13:7.
53 Hecht JT, Nelson FW, Butler IJ, et al. Computerized tomography of the foramen magnum: achondroplastic values compared to normal standards. Am J Med Genet. 1985;20:355.
54 Hurko O, Pyeritz R, Uemastu S. Neurological considerations in achondroplasia. In: Nicoletti B, Kopits S, Ascani E, et al, editors. Human Achondroplasia. New York: Plenum Press; 1988:153.
55 Boor R, Fricke G, Bruhl K, et al. Abnormal subcortical somatosensory evoked potentials indicate high cervical myelopathy in achondroplasia. Eur J Pediatr. 1999;158:662.
56 Steinbok P, Hall J, Flodmark O. Hydrocephalus in achondroplasia: the possible role of intracranial venous hypertension. J Neurosurg. 1989;71:42.
57 Colamaria V, Mazza C, Beltramello A, et al. Irreversible respiratory failure in an achondroplastic child: the importance of an early cervicomedullary decompression, and a review of the literature. Brain Dev. 1991;13:270.
58 Mador MJ, Tobin MJ. Apneustic breathing. A characteristic feature of brainstem compression in achondroplasia? Chest. 1990;97:877.
59 Wang H, Rosenbaum AE, Reid CS, et al. Pediatric patients with achondroplasia: CT evaluation of the craniocervical junction. Radiology. 1987;164:515.
60 Benglis DM, Sandberg DI. Acute neurological deficit after minor trauma in an infant with achondroplasia and cervicomedullary compression. J Neurosurg. 2007;107:152.
61 Reid CS, Pyeritz RE, Kopits SE, et al. Cervicomedullary compression in young patients with achondroplasia: value of comprehensive neurologic and respiratory evaluation. J Pediatr. 1987;110:522.
62 Thomas IT, Frias JL. The prospective management of cervicomedullary compression in achondroplasia. Birth Defects Orig Artic Ser. 1989;25:83.
63 Aryanpur J, Hurko O, Francomano C, et al. Craniocervical decompression for cervicomedullary compression in pediatric patients with achondroplasia. J Neurosurg. 1990;73:375.
64 Danielpour M, Wilcox WR, Alanay Y, et al. Dynamic cervicomedullary cord compression and alterations in cerebrospinal fluid dynamics in children with achondroplasia. Report of four cases. J Neurosurg. 2007;107:504.
65 Rekate HL. Management of cervicomedullary compression. Childs Nerv Syst. 1997;13:359.
66 Wassman ERJr, Rimoin DL. Cervicomedullary compression with achondroplasia. J Pediatr. 1988;113:411.
67 Larsen PD, Snyder EW, Matsuo F, et al. Achondroplasia associated with obstructive sleep apnea. Arch Neurol. 1983;40:769.
68 Rimoin DL. Cervicomedullary junction compression in infants with achondroplasia: when to perform neurosurgical decompression. Am J Hum Genet. 1995;56:824.
69 Pauli RM, Horton VK, Glinski LP, et al. Prospective assessment of risks for cervicomedullary-junction compression in infants with achondroplasia. Am J Hum Genet. 1995;56:732.
70 Pauli RM. Surgical intervention in achondroplasia. Am J Hum Genet. 1995;56:1501.
71 Yamada Y, Ito H, Otsubo Y, et al. Surgical management of cervicomedullary compression in achondroplasia. Childs Nerv Syst. 1996;12:737.
72 Hunter AG, Bankier A, Rogers JG, et al. Medical complications of achondroplasia: a multicentre patient review. J Med Genet. 1998;35:705.
73 Moritani T, Aihara T, Oguma E, et al. Magnetic resonance venography of achondroplasia: correlation of venous narrowing at the jugular foramen with hydrocephalus. Clin Imaging. 2006;30:195.
74 Erdincler P, Dashti R, Kaynar MY, et al. Hydrocephalus and chronically increased intracranial pressure in achondroplasia. Childs Nerv Syst. 1997;13:345.
75 Horton WA, Rotter JI, Rimoin DL, et al. Standard growth curves for achondroplasia. J Pediatr. 1978;93:435.
76 Hunter AG, Hecht JT, Scott CIJr. Standard weight for height curves in achondroplasia. Am J Med Genet. 1996;62:255.
77 Lundar T, Bakke SJ, Nornes H. Hydrocephalus in an achondroplastic child treated by venous decompression at the jugular foramen. Case report. J Neurosurg. 1990;73:138.
78 Sciubba DM, Noggle JC, Marupudi NI, et al. Spinal stenosis surgery in pediatric patients with achondroplasia. J Neurosurg. 2007;106:372.
79 Kopits S. Thoracolumbar kyphosis and lumbosacral lordosis. In: Nicoletti B, Kopits S, Ascani E, et al, editors. Human Achondroplasia. New York: Plenum Press; 1988:241.
80 Siebens AA, Hungerford DS, Kirby NA. Curves of the achondroplastic spine: a new hypothesis. Johns Hopkins Med J. 1978;142:205.
81 Lutter LD, Langer LO. Neurological symptoms in achondroplastic dwarfs—surgical treatment. J Bone Joint Surg Am. 1977;59:87.
82 Morgan DF, Young RF. Spinal neurological complications of achondroplasia. Results of surgical treatment. J Neurosurg. 1980;52:463.
83 Bagley CA, Pindrik JA, Bookland MJ, et al. Cervicomedullary decompression for foramen magnum stenosis in achondroplasia. J Neurosurg. 2006;104:166.
84 Lutter LD, Longstein JE, Winter RB, et al. Anatomy of the achondroplastic lumbar canal. Clin Orthop Relat Res. 1977;126:139.
85 Shikata J, Yamamuro T, Iida H, et al. Surgical treatment of achondroplastic dwarfs with paraplegia. Surg Neurol. 1988;29:125.
86 Lonstein J. Treatment of kyphosis and lumbar stenosis in achondroplasia. In: Nicoletti B, Kopits S, Ascani E, et al, editors. Human Achondroplasia. New York: Plenum Press; 1988:283.
87 Kopits SE. Thoracolumbar kyphosis and lumbosacral hyperlordosis in achondroplastic children. Basic Life Sci. 1988;48:241.
88 Pauli RM, Breed A, Horton VK, et al. Prevention of fixed, angular kyphosis in achondroplasia. J Pediatr Orthop. 1997;17:726.
89 Ain MC, Shirley ED, Pirouzmanesh A, et al. Postlaminectomy kyphosis in the skeletally immature achondroplast. Spine. 2006;31:197.
90 Ain MC, Browne JA. Spinal arthrodesis with instrumentation for thoracolumbar kyphosis in pediatric achondroplasia. Spine. 2004;29:2075.
91 Ain MC, Elmaci I, Hurko O, et al. Reoperation for spinal restenosis in achondroplasia. J Spinal Disord. 2000;13:168.






