Chapter 15 Abnormalities of the Optic Nerve and Retina
Disorders of the optic nerve and retina are common causes of afferent visual loss in clinical neurology. The diagnosis of optic neuropathy should be considered when visual loss (affecting visual acuity, color vision, or visual field) is accompanied by abnormal optic disc appearance or a relative afferent pupillary defect (RAPD; see Chapter 17). The specific cause for an optic neuropathy often can be established on the basis of clinical history (i.e., character, progression of vision loss) and examination (i.e., pattern of visual field loss and optic disc appearance). Furthermore, optic neuropathies are classifiable by appearance of the optic disc: normal, swollen, or pale. Chapter 14 describes the various patterns of visual field loss and clinical history typically elicited in patients with specific optic nerve disorders. This chapter presents the differential diagnosis for optic neuropathies based on the optic disc appearance and discusses retinal disorders of particular interest in neurology. Chapter 36 discusses many of the entities described in this chapter in more detail.
Optic Nerve Anatomy and Physiology
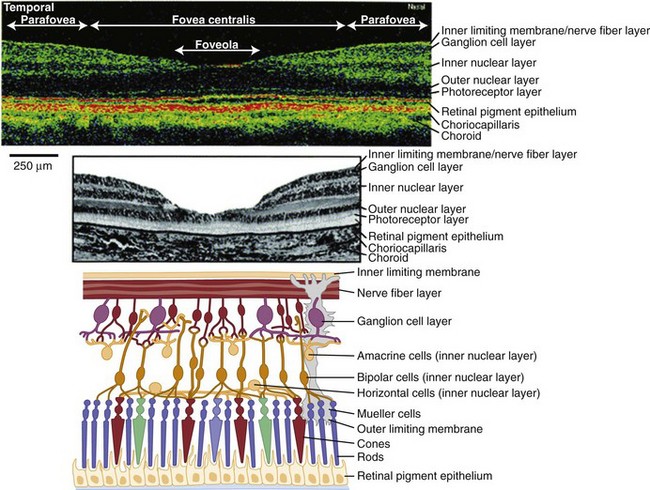
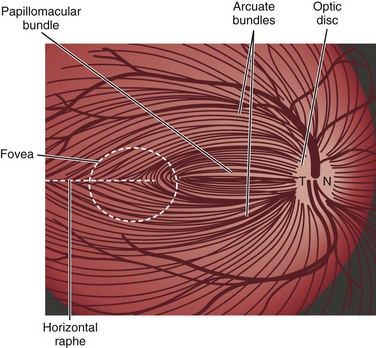
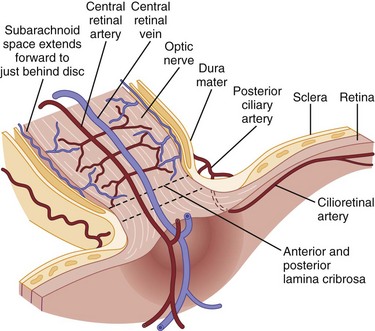
Light stimulates retinal photoreceptors whose signal reaches a ganglion cell after being modulated by bipolar, horizontal, and amacrine cells (Fig. 15.1). Two main types of retinal ganglion cells exist: parasol cells (which project to the magnocellular layer and are specialized for motion perception and coarse stereopsis) and midget cells (which project to the parvocellular layer and are specialized for high spatial resolution, color vision, and fine stereopsis). Temporal retinal fibers form arcuate bundles around the fovea, respecting the midline horizontal raphe, and then enter the optic disc superiorly and inferiorly (Fig. 15.2). Optic nerve fibers exit the globe at the scleral canal, where they receive physical support from the lamina cribrosa and receive metabolic support from intertwining astrocytes. Once nerve fibers pass the lamina cribrosa, they are supported by oligodendrocytes and become myelinated. After exiting the orbit, the nerve enters the optic canal within the lesser sphenoid wing. In this space, the nerve is particularly vulnerable to trauma or compressive lesions (Balcer, 2001; Sarkies, 2004).
(Adapted and reprinted with permission from Jaffe and Caprioli [2004] and www.webvision.med.utah.edu.)
Effective axonal transport is essential for maintenance of the ganglion cell axon’s structure and function. Orthograde transport (away from the ganglion cell body) occurs at two speeds: 400 mm per day for proteins and neurotransmitters packaged in vesicles, and 1 to 4 mm per day for structural elements of the cytoskeleton. Interference of axonal transport, for example from elevated intracranial pressure (ICP), ultimately damages axons of the optic nerve and causes atrophy (Hayreh, 1977).
The ophthalmic artery, arising from the internal carotid artery, provides blood supply to the eye via multiple short posterior ciliary arteries and the central retinal artery (Hayreh and Zimmerman, 2007) (Fig. 15.3). The short posterior ciliary arteries provide blood supply to the optic nerve head and the subretinal choroid. Each posterior ciliary artery supplies a variable segmental territory of the optic nerve head, and because anastomoses in this blood supply are scant, it can suffer watershed ischemia during hypoperfusion. Furthermore, the segmental blood supply underlies the sectoral disc swelling or atrophy that results from interrupted flow of a posterior ciliary artery and subsequent optic nerve infarction (Balcer, 2001; Fontal et al., 2007).
The Swollen Optic Disc
Unilateral Optic Disc Swelling
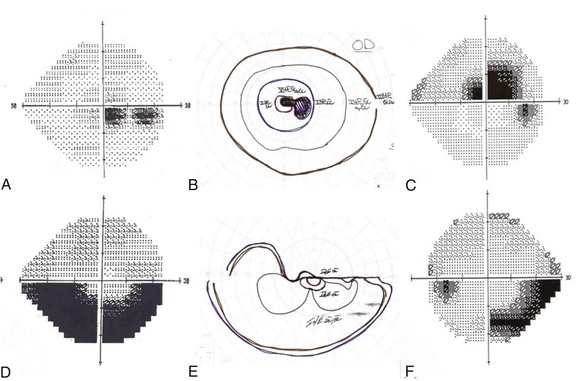
Despite suggestive patterns in the appearance of the optic nerve, it often is not possible to distinguish NAION, optic neuritis, and compressive optic neuropathies on this basis alone. Typically these diagnoses also rely on data from the clinical history and the pattern of the visual field deficit (Fig. 15.4). Vision loss generally is slowly progressive in patients with compressive lesions; it is rapidly progressive with subsequent improvement in those with optic neuritis; and it is maximal at onset with minimal improvement in patients with NAION. Both optic neuritis and compressive lesions generally produce central visual loss, whereas NAION typically produces a nerve fiber bundle–type field defect (originating from the physiological blind spot and respecting the horizontal meridian, owing to the arrangement of retinal ganglion cell axons traveling to the optic disc). However, considerable overlap exists in the patterns of visual field loss caused by the different forms of optic neuropathy.
Optic Neuritis
Typical optic neuritis is an inflammatory optic neuropathy caused by demyelinating disease (Balcer, 2006) (Fig. 15.5). Visual loss in the affected eye typically occurs rapidly over several hours to a few days. Decreased color vision and contrast sensitivity are highly characteristic (Baier et al., 2005; Trobe et al., 1996). In addition, pain with eye movements precedes the vision loss in approximately 90% of cases (Optic Neuritis Study Group, 1991). The pain typically lasts 3 to 5 days; if it persists for longer than 7 days, optic neuritis should be considered less likely, and further workup should be pursued. Visual field defects commonly are present; they can be either diffuse or discrete scotomas and are nonspecific. Fundus examination reveals mild disc swelling in approximately one-third of affected eyes, which is considerably less prominent than the disc swelling associated with papilledema (Balcer, 2006; Beck, 1998) (Fig. 15.6). In the majority of patients, the fundus appearance is normal.
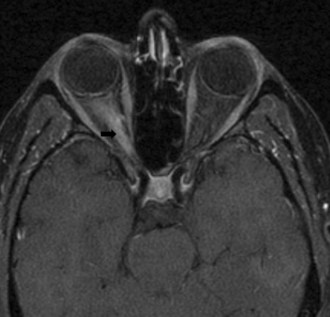
The prognosis for recovery of vision generally is good but is in relation to the severity of the initial deficit. Recovery typically begins within 1 month. The likelihood of progression of optic neuritis to multiple sclerosis (MS) is best predicted by brain magnetic resonance imaging (MRI) at the time of diagnosis. In the Optic Neuritis Treatment Trial, the risk of developing MS within 15 years was 72% among patients with one or more characteristic brain lesions, whereas it was 25% if the MRI was normal (Optic Neuritis Study Group, 2008). With features that are atypical for optic neuritis, however (e.g., painless visual loss, severe disc edema, disc or peripapillary hemorrhages, macular exudate), the risk of developing MS is significantly lower (Beck et al., 2003).
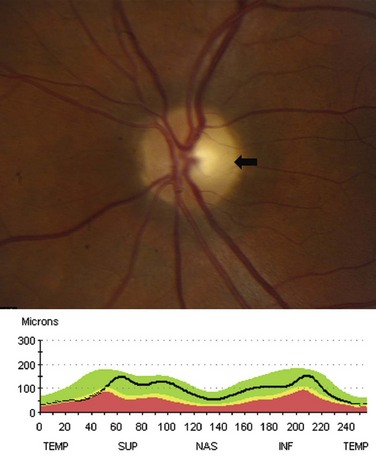
Following an episode of optic neuritis, the optic nerve often demonstrates pallor, suggesting that axonal loss has accompanied the episode of demyelination (Fig. 15.7). Optical coherence tomography (OCT) is a noninvasive imaging method that quantifies the atrophy of the nerve fiber layer (Balcer, 2006). It provides a reliable structural marker that complements clinical assessments of visual function.
Neuromyelitis optica (NMO), or Devic disease, is characterized by necrotizing demyelinating lesions of bilateral optic nerves and the spinal cord (Wingerchuk, 2006). It is believed to be a humorally mediated disease distinct from MS. The spinal lesion characteristic of NMO often extends contiguously over three or more vertebral segments. A serum antibody, neuromyelitis optica immunoglobulin G (NMO-IgG), which targets the autoantigen aquaporin 4 (AQP4), may be a useful marker in diagnosing the condition, although the exact specificity remains unknown (Jarius et al., 2008). Treatment with rituximab, a chemotherapeutic monoclonal antibody, may be of particular benefit in this group of patients (Wingerchuk, 2006).
Treating optic neuritis with high-dose intravenous (IV) corticosteroids reduces the risk of developing MS over the following 2 years (Beck et al., 1992). In the long term, however, this acute treatment is unlikely to affect the likelihood of progression to MS. In addition, IV corticosteroid treatment may hasten visual recovery, particularly for visual fields and contrast sensitivity, but does not significantly affect long-term visual outcomes. Because low-dose oral corticosteroids may be associated with an increased risk of recurrence of optic neuritis, this therapy should be avoided (Beck et al., 1992). In addition to IV corticosteroids, recent studies support the early use of immunomodulating treatments for high-risk patients to reduce the likelihood of progression to MS within 2 to 5 years (Balcer, 2006).
Ischemic Optic Neuropathy
GCA typically affects the extracranial medium- to large-caliber arteries, because they possess elastic lamina, which is the initial site of inflammation in this disorder (Salvarani et al., 2008). The condition is associated with polymyalgia rheumatica, consisting of proximal muscle ache, arthralgia, and stiffness, as well as with jaw claudication, fever, malaise, and scalp tenderness. The diagnosis is suggested by an elevated erythrocyte sedimentation rate and C-reactive protein and is confirmed by evidence of giant cells and endovascular inflammation on temporal artery biopsy. Acute vision loss is the presenting symptom in 7% to 60% of cases and is generally more severe than in NAION. In approximately 25% of cases, vision is limited to hand motion perception or worse (Balcer et al., 2003). In suspected cases, treatment with corticosteroids should not be delayed until a biopsy is obtained. Intraveous corticosteroids may help delay the progression of visual loss and decrease the likelihood of fellow eye involvement. The prognosis for recovery in the affected eye, however, is poor despite treatment (Hall and Balcer, 2004).
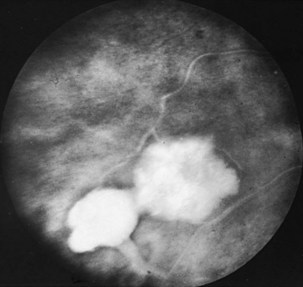
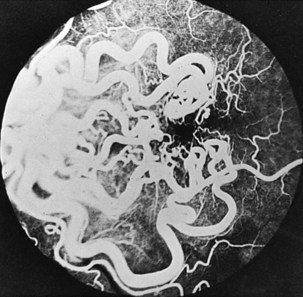
The optic disc in AAION typically has a chalky-white edematous appearance, and disc hemorrhages are likely to be present (Fig. 15.8). Coexisting retinal ischemia with cotton-wool spots is very typical for AAION. Fluorescein angiography reveals choroidal hypoperfusion (Fig. 15.9). Occasionally, GCA can be limited to the retro-orbital nerve and present without disc swelling; this situation is termed arteritic posterior ischemic optic neuropathy (PION).
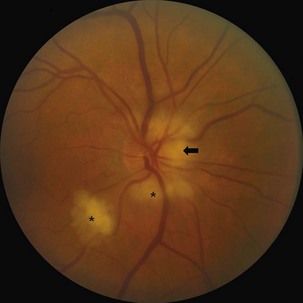
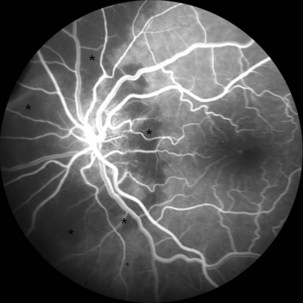
Nonarteritic AION is the most common cause of unilateral optic nerve swelling in adults older than 50 and is commonly associated with vascular risk factors such as diabetes or hypertension (Fontal et al., 2007). Other risk factors include a crowded optic nerve head and nocturnal hypotension, possibly precipitated by antihypertensive therapy (Arnold, 2003; Mathews, 2005). Swelling of a crowded optic nerve within the scleral canal may provoke a cycle of further vascular compression, ischemia, and swelling.
Although the clinical profile of NAION may occasionally overlap with the findings of optic neuritis (Rizzo et al., 1991), typical features of NAION include nerve fiber hemorrhages, altitudinal visual field loss, moderate to severe disc edema, and the absence of pain (Figs. 15.10 and 15.11). Because the optic nerve head is supplied by an end-arterial system of short posterior ciliary arteries and the circle of Zinn-Haller, sectoral ischemic disc swelling is common. NAION may follow ocular surgery, because an associated increase in intraocular pressure may compromise optic nerve head perfusion (Fontal et al., 2007).
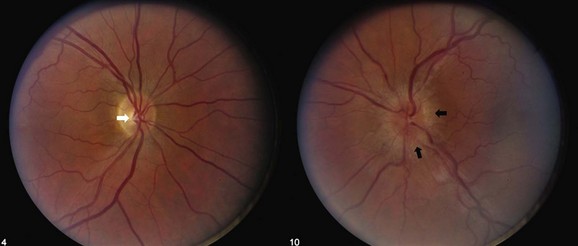
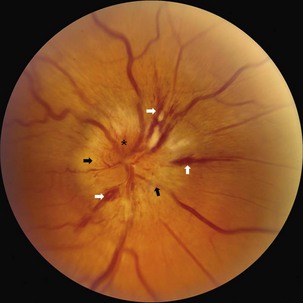
Many patients with NAION will have a stable deficit, although one minority may experience visual loss progressing over one month. Spontaneous improvement may occur in the first 6 months, although in many patients this reflects improved ability with eccentric fixation (Hayreh and Zimmerman, 2008). In 30% to 40% of patients, subsequent involvement of the fellow eye also occurs, and this rate is increased by the presence of vascular risk factors. When the second eye is affected in AION, optic atrophy has already developed in one eye, and acute disc edema occurs in the fellow eye; this clinical presentation is called the pseudo–Foster Kennedy syndrome. (A true Foster Kennedy syndrome is produced by optic atrophy due to compression, typically from an expanding tumor, and papilledema in the fellow eye secondary to increased ICP.) Occasionally, premonitory disc swelling in an asymptomatic eye will be noted, which may progress to frank visual loss or remit spontaneously (Hayreh and Zimmerman, 2007). Recurrence of NAION in an affected eye, however, is rare, possibly because optic nerve atrophy following the initial event decompresses the nerve. There does not appear to be a significantly higher rate of stroke in patients with nonarteritic ischemic optic neuropathy, suggesting that its pathophysiology may differ from simple vasoocclusion (Arnold and Levin, 2002).
Other Causes
Inflammatory conditions are an important cause of subacute optic neuropathy. Optic discs may appear swollen or normal, the latter indicating retrobulbar involvement. Optic nerve involvement is common in neurosarcoidosis, which can be accompanied by anterior uveitis or posterior segment vitritis (Prasad et al., 2008) (Fig. 15.12). Visual loss due to this condition is often steroid responsive. Optic neuropathy and retinal involvement may also occur with other inflammatory disorders, such as systemic lupus erythematosus and Sjögren disease (Fig. 15.13). Occasionally, optic nerve infiltration produces optic disc edema without affecting visual function, but more often there is a decrease in visual acuity and visual field loss.
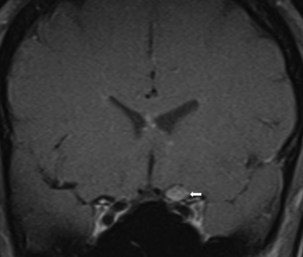
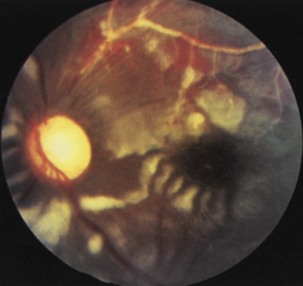
Fig. 15.13 Optic disc pallor and multiple cotton-wool spots in a patient with systemic lupus erythematosus.
Infectious conditions are another frequent cause of optic neuropathy (March and Lessell, 1996). Neuroretinitis, in which optic neuropathy coexists with characteristic peripapillary or macular exudates, should be distinguished from acute demyelinating optic neuritis (Fig. 15.14). The initial clinical presentation of these conditions may be similar, but the characteristic macular star of neuroretinitis will appear within 1 to 2 weeks, establishing the diagnosis. The distinction is critical because neuroretinitis has no association with an increased risk of MS and may be due to cat scratch disease (Bartonella henselae), syphilis (Treponema pallidum), or Lyme disease (Borrelia burgdorferi). In most cases, Bartonella infection is self-limited and does not require treatment, but in severe cases doxycycline may be effective (Balcer et al., 2003). Other infectious causes of optic neuropathy include human immunodeficiency virus (HIV) and opportunistic infections including toxoplasmosis, cytomegalovirus, and cryptococcosis. Paranasal sinusitis or mucocele may lead to either compressive or inflammatory optic neuropathy.
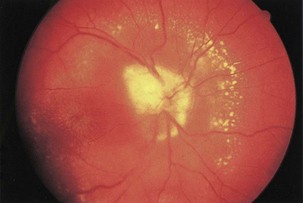
Paranasal sinus disease can cause a condition that mimics optic neuritis, with acute optic neuropathy and pain on eye movements, or can cause a progressive optic neuropathy resulting from compression (Rothstein et al., 1984). Consider optic neuropathy due to sinusitis and mucocele in patients who have clinical evidence of optic neuritis with seemingly atypical features, particularly elderly patients with severe sinus disease, a history of fevers, ophthalmoplegia, or progression of vision loss beyond 2 weeks.
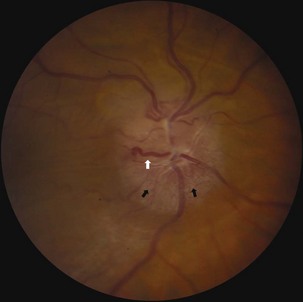
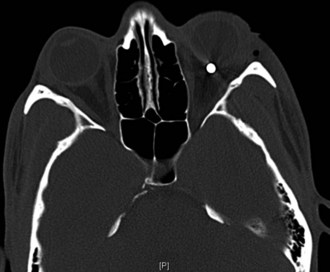
Several compressive mass lesions cause a progressive optic neuropathy. The optic disc swells in cases of intraorbital compression, but in cases of retro-orbital compression, disc swelling only occurs if ICP is elevated. Chronic disc edema due to compressive lesions may be accompanied by optociliary shunt vessels and glistening white bodies on the disc surface (pseudodrusen from extruded axoplasm) (Fig. 15.15). Important causes of compressive optic neuropathy include neoplasm (including optic nerve sheath or skull base meningioma, pituitary adenoma, and craniopharyngioma), sinus lesions, bony processes (such as fibrous dysplasia), enlarged extraocular muscles (as in Graves disease ophthalmopathy), or aneurysms (Fig. 15.16). Meningiomas of the optic nerve sheath occur primarily in women and can cause acuity loss associated with either disc swelling or atrophy.
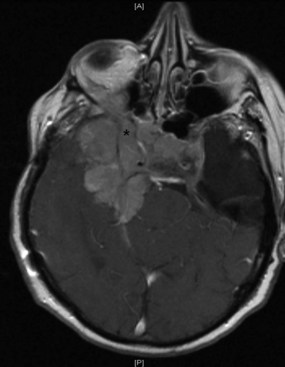
Primary optic nerve neoplasms include benign juvenile pilocytic glioma in children and (rarely) malignant glioblastoma in adults (Fig. 15.17). Juvenile pilocytic astrocytoma is often associated with neurofibromatosis type 1 and may be managed conservatively with frequent ophthalmological examination through adolescence (Listernick et al., 2007). When clinical or radiographic progression is detected, chemotherapy should be first-line therapy, followed by radiation and rarely surgery. Malignant optic nerve glioblastoma is much rarer, affects adults, and has a considerably worse prognosis (Spoor et al., 1980). Other neoplastic conditions include lymphoma, leukemia, carcinomatous meningitis, and optic nerve metastasis. Almost any form of carcinoma can metastasize to the optic nerve; breast and lung carcinomas are the most common.
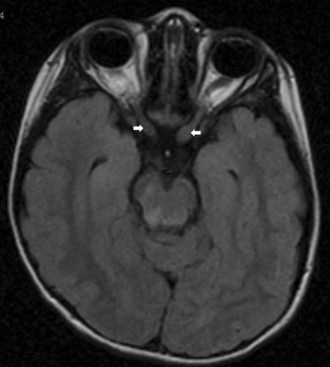
Optic neuropathy may occur as a delayed effect of radiation therapy. It can occur with or without disc edema and can sometimes be difficult to distinguish from tumor recurrence (Danesh-Meyer, 2008). Radiation optic neuropathy is suggested by exposure (typically 50-Gy dosage), characteristic 6- to 24-month time lag to symptoms, and accompanying radiation changes in proximal tissues. Progression occurs over weeks to months, and spontaneous recovery is rare. Corticosteroids may help by reducing edema in the affected optic nerve.
Visual loss in a patient with known or suspected cancer raises the possibility of a paraneoplastic optic neuropathy or retinopathy (Damek, 2005). In paraneoplastic optic neuropathy, evidence of other neurological dysfunction is common, and the antibody most commonly identified is directed toward collapsin response mediator protein 5 (CRMP5). Paraneoplastic retinopathies, on the other hand, include cancer-associated retinopathy (with antibodies to recoverin protein) and melanoma-associated retinopathy (with antibodies to rod ganglion cells).
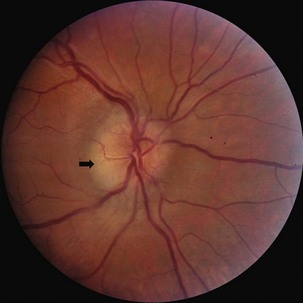
Leber hereditary optic neuropathy (LHON) is a subacute, sequential, maternally inherited optic nerve disorder in which 80% to 90% of affected persons are males in the second or third decade of life (see Chapter 14) (Man et al., 2002; Newman, 2005). Although true disc edema is not present, the optic disc may appear hyperemic and mildly swollen in the acute phase; nevertheless, fluorescein angiography should confirm the absence of capillary leakage. Circumpapillary telangiectatic vessels, frequently present in the peripapillary nerve fiber layer, are an important clue to the diagnosis (Fig. 15.18). These early funduscopic changes also may be noted in presymptomatic eyes. As the condition progresses, the discs become atrophic. Because fibers mediating the pupillary light reflex may be selectively spared, the light reflex may be preserved despite significant visual loss. Genetic diagnosis of LHON is based on the identification of related mitochondrial DNA mutations (see Chapter 63). Most patients have permanent vision loss, although a minority will experience some recovery of vision. The prognosis depends upon the specific mutation harbored; patients with mtDNA mutation T14484C are more likely to have spontaneous recovery than patients with mutations G11778A or G3460A (Newman, 2005). At present, no effective treatment for this condition is available (Chinnery and Griffiths, 2005).
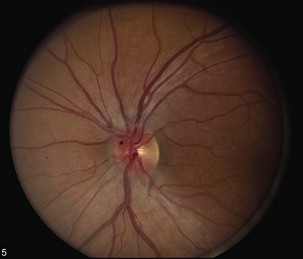
Direct traumatic optic neuropathy (TON) may include nerve avulsion or transection and is easily recognized by the relevant history of injury (Sarkies, 2004) (Fig. 15.19). Fundus examination may reveal extensive intraocular hemorrhages. On the other hand, posterior indirect traumatic optic neuropathy will present with visual loss in the absence of significant fundus abnormalities; it may result from shearing forces and subsequent edema within the optic canal. Up to half of these patients may improve spontaneously (Sarkies, 2004). There is weak evidence that corticosteroid therapy may be helpful within the first 8 hours, but no other medical or surgical interventions have proven effective (Yu Wai Man and Griffiths, 2005; Yu Wai Man and Griffiths, 2007).
Bilateral Optic Disc Swelling
Papilledema
The term papilledema refers specifically to optic disc swelling secondary to increased ICP. Disc swelling in papilledema results from blockage of axoplasmic flow in nerve fibers, increasing the volume of axoplasm in the optic disc (Hayreh, 1977). Although papilledema is typically bilateral, it can be asymmetrical, based on anatomical differences in the meningeal covering of the intracranial optic nerves that can lead to differences in transmitted pressure. On the basis of the chronicity and fundus appearance, papilledema can be divided into four stages: early, fully developed (acute), chronic, and atrophic. The acute phase of papilledema is strongly suggested by a mismatch between a markedly swollen disc and relatively spared optic nerve function, particularly central visual acuity. The most common visual field defects encountered in patients with early or acute papilledema are enlargement of the physiological blind spot, concentric constriction, and inferior nasal field loss. When acute papilledema is accompanied by decreased acuity (and often metamorphopsia), fluid typically extends within the retina to the macula itself.
In early papilledema, swelling is most prominent at the superior and inferior poles of the optic disc where the nerve fiber layer is thickest. With further development of papilledema, swelling encompasses the disc surface more uniformly, and the degree of disc elevation increases (Fig. 15.20). The retinal veins may distend slightly, and the disc may appear mildly hyperemic. These vascular changes result from nerve fiber swelling causing compression of capillaries and venules, leading to venous stasis and dilation, formation of microaneurysms, and finally disc and peripapillary splinter hemorrhages (see Fig. 15.20). Correspondingly, in fluorescein angiography, fluorescence may be absent during the retinal arterial phase as a result of delayed circulation caused by disc swelling. Dilated capillaries, microaneurysms, and flame-shaped hemorrhages may appear in the arteriovenous phase, and fluorescein may leak from dilated capillaries in the venous phase. Retinal cotton-wool spots may occur secondary to ischemia in the nerve fiber layer. Spontaneous venous pulsations usually are absent once the ICP exceeds 18 cm H2O. Although papilledema typically is bilateral, it can be asymmetrical because of differences in transmitted pressure related to anatomical variation in the meningeal covering of the intracranial and intracanalicular optic nerves.
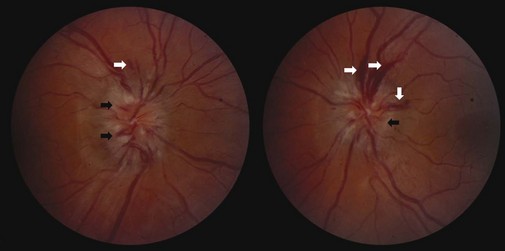
The disc appearance changes as papilledema becomes chronic, usually after weeks to months. The nerve fiber layer may appear pale and take on a gliotic appearance as a result of optic atrophy and astrocytic proliferation (Fig. 15.21). Hemorrhages are less prominent (and often have resolved completely). The disc takes on a “champagne cork” appearance in which small, glistening white bodies (pseudodrusen) result from extruded axoplasm after prolonged stasis. Shunt vessels due to compensatory dilation of preexisting communications between the retinal and ciliary circulation begin to appear.
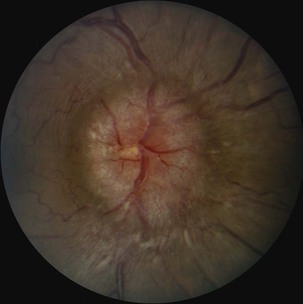
Pseudotumor cerebri, or idiopathic intracranial hypertension, can lead to disc swelling and progressive visual loss (Friedman, 2004). The condition is most common in obese women, but modest weight gain (by 5%-15%) even in nonobese women is a risk factor for disease. Additional risk factors are the use of tetracycline derivatives or vitamin A, and when present, these agents should be discontinued. Weight loss can be imperative in the management of pseudotumor cerebri. In the short term, treatment with acetazolamide can improve symptoms and reduce optic disc swelling. In refractory cases, optic nerve sheath fenestration or cerebrospinal fluid (CSF) shunting may be indicated.
Malignant Hypertension
A marked elevation in blood pressure may produce bilateral optic disc swelling indistinguishable from papilledema (Hayreh, 1977). Peripapillary cotton-wool spots also are a prominent feature in patients with malignant hypertension. Encephalopathic signs are common but not always present. Disc edema tends to develop at a lower blood pressure in patients with renal failure than in those without renal disease.
Diabetic Papillopathy
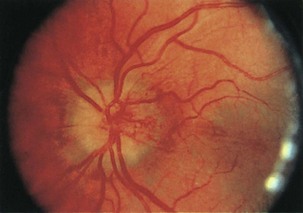
Diabetic papillopathy is a rare cause of bilateral (or sometimes unilateral) disc swelling in patients with type 1 diabetes (Barbera et al., 1996). This entity is distinct from typical NAION in that there is often bilateral, simultaneous optic nerve involvement. Often visual loss is minimal, with the exception of an enlarged physiological blind spot. Disc edema may be accompanied by marked capillary telangiectasias overlying the disc surface (Fig. 15.22). Neuroimaging and lumbar puncture may be necessary to distinguish this condition from papilledema. The pathogenesis is unclear but may relate to a mild impairment of blood flow causing disc swelling without infarction of the optic nerve head (as in the case of premonitory NAION). In many cases, the optic disc edema resolves without residual visual deficit.
Pseudopapilledema
In patients with pseudopapilledema, visible optic disc drusen (hyaline bodies) may be present. Even when disc drusen are not apparent, the distinction between true disc swelling and pseudopapilledema can frequently be made on the basis of fundus examination findings (Table 15.1). The most important distinguishing feature is the clarity of the peripapillary nerve fiber layer. In patients with true disc edema, the nerve fiber layer is hazy, obscuring the underlying retinal vessels, whereas in pseudopapilledema, this layer can remain distinct. In addition, the presence of spontaneous venous pulsations (SVPs) supports the diagnosis of pseudopapilledema, although SVP can be absent in pseudopapilledema as well. Although subretinal hemorrhages may be present in patients with pseudopapilledema (particularly in the setting of optic disc drusen), splinter hemorrhages are characteristic of true papilledema. Finally, fluorescein angiography will show leakage from vessels in papilledema, which is not seen in pseudopapilledema (Davis and Jay, 2003).
Table 15.1 Differentiation of Early Papilledema and Pseudopapilledema
| Feature | Papilledema | Pseudopapilledema |
|---|---|---|
| Disc color | Hyperemic | Pink, yellowish pink |
| Disc margins | Indistinct early at superior and inferior poles, later entire margin | Irregularly blurred, may be lumpy |
| Vessels | Normal distribution, slight fullness; spontaneous venous pulsations absent | Emanate from center, frequent anomalous pattern, ± spontaneous venous pulsations |
| Nerve fiber layer | Dull as a result of edema, which may obscure blood vessels | No edema; may glisten with circumpapillary halo of feathery light reflections |
| Hemorrhages | Splinter | Subretinal, retinal, vitreous |
Reprinted with permission from Beck, R.W., Smith, C.H., 1988. Neuro-Ophthalmology: A Problem-Oriented Approach. Little, Brown, Boston.
Optic Disc Drusen
Optic disc drusen, which constitute a common cause of pseudopapilledema, refer to calcium deposits within the optic nerve head. Although etiology is unclear, drusen may result from axonal degeneration from altered axoplasmic flow, particularly in the setting of a small optic canal (Auw-Haedrich et al., 2002). In children, disc drusen tend to be buried, whereas in adults, they often are visible on the disc surface (Figs. 15.23 and 15.24). The progression from buried to surface drusen in individual patients has been well documented. The prevalence of optic disc drusen is approximately 2% within the general population, and they can be bilateral in two-thirds of cases. Optic disc drusen are much more common in caucasian patients than in African Americans and may be genetic, inherited in an autosomal dominant pattern with incomplete penetrance.
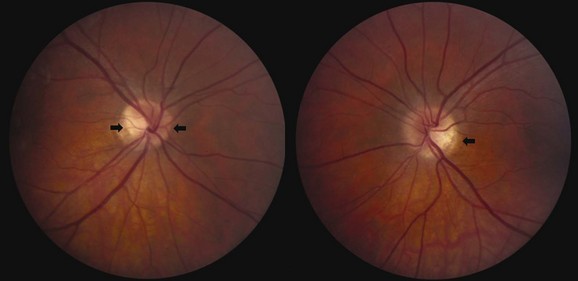
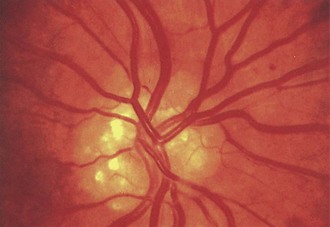
Fig. 15.24 Optic disc drusen in a 50-year-old man.
(Reprinted with permission from Beck, R.W., Smith, C.H., 1988. Neuro-Ophthalmology: A Problem-Oriented Approach. Little, Brown, Boston.)
Patients with optic disc drusen generally do not complain of visual symptoms, although rarely a patient may experience transitory visual obscurations similar to those described by patients with true papilledema. Although patients may be unaware of a visual field defect, such deficits are common, occurring in approximately 70% of eyes with visible disc drusen and in 35% of those with pseudopapilledema but no visible drusen (Auw-Haedrich et al., 2002). The scotoma probably results from nerve fiber layer thinning and axonal dysfunction caused by the drusen. The visual field defects, therefore, generally follow a nerve fiber bundle distribution, most commonly affecting the inferior nasal visual field. Enlargement of the blind spot and generalized field constriction also may occur. Progression of visual field defects in the setting of drusen is well documented. In addition, visual field loss in the setting of optic disc drusen occurs secondary to hemorrhage, superimposed ischemic optic neuropathy, or an associated retinal degeneration. Visual acuity loss associated with drusen is rare, however, and should prompt an evaluation for alternative causes.
Optic Neuropathies with Normal-Appearing Optic Discs
Unilateral Presentations
The most common causes of unilateral retrobulbar optic neuropathy are optic neuritis and compressive lesions. The time course of vision loss usually is helpful in distinguishing between these two entities. No definite way exists to differentiate these disorders on examination, but the detection of a superior temporal field defect in the fellow eye (a junctional scotoma) is highly suggestive of a compressive lesion affecting the anterior optic chiasm and the posterior optic nerve, involving the decussating fibers (termed Willebrand knee or genu). Posterior (retrobulbar) ischemic optic neuropathy (PION) may occur in patients with giant cell arteritis, other vasculitides, or severe blood loss (Chang and Miller, 2005; Hayreh, 2004). For practical purposes, no retrobulbar correlate to NAION exists.
Optic Neuropathies with Optic Atrophy
Glaucomatous optic neuropathy is easily distinguished from optic neuritis, since it occurs in the setting of elevated intraocular pressure and optic disc cupping (Fig. 15.25) (Jonas and Budde, 2000). However, angle closure glaucoma may present with painful acute visual loss, resembling the features of optic neuritis. Distinguishing characteristics include the severity of pain (which can be excruciating) and a red eye with an enlarged, nonreactive pupil. Normal-tension glaucoma is more difficult to recognize but will present with optic disc cupping and progressive field constriction, despite normal intraocular pressures; many of these patients have a fairly benign natural history (Anderson et al., 2001).
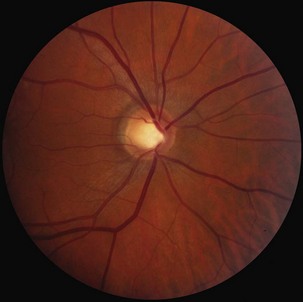
Dominantly inherited optic atrophy typically presents with insidious asymmetrical visual loss in childhood (Newman, 2005). These patients often have a striking disc appearance, with pallor and excavation of the temporal portion of the disc (Fig. 15.26). The disorder is due to mutations of the OPA1 gene, with autosomal inheritance and variable penetrance. The OPA gene product is believed to target the mitochondria and support membrane stability. Because over 90 different pathogenic OPA1 mutations have been described, a simple DNA test does not exist as it does for LHON.
Congenital Optic Disc Anomalies
Tilted Optic Disc
A tilted optic disc usually is easily recognizable on fundus examination. The disc may appear foreshortened on one side, and one portion may appear elevated, with the opposite end depressed (Fig. 15.27). Often the retinal vessels run in an oblique direction. Tilted optic discs are of neurological importance in that they usually are bilateral and may be associated with temporal field loss, thus mimicking a chiasmal syndrome. However, differentiation from chiasmal disease generally is possible because visual field defects in patients with tilted discs typically do not respect the vertical meridian.
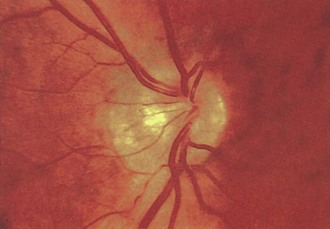
Optic Nerve Dysplasia
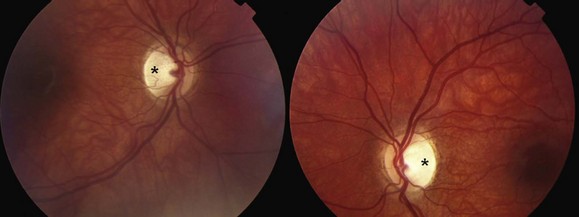
Of the several types of optic nerve dysplasia, optic nerve hypoplasia is the most common (Taylor, 2007). In this condition, the optic disc appears small and surrounded by choroid and retinal pigment changes that resemble a double ring (Fig. 15.28). The abnormality may be unilateral or bilateral. In most cases, no specific cause is identifiable. The frequency of optic nerve hypoplasia appears to be increased in children of mothers who had diabetes mellitus or ingested antiepileptic drugs, quinine, or lysergic acid diethylamide (LSD) during pregnancy. De Morsier syndrome (septo-optic dysplasia) is characterized by developmental abnormalities of structures sharing an embryological forebrain derivation, including bilateral optic nerve hypoplasia, absent septum pellucidum, and pituitary gland dysfunction (classic growth hormone deficiency) (Taylor, 2007). Optic nerve aplasia, or complete absence of the optic discs, is extremely rare.
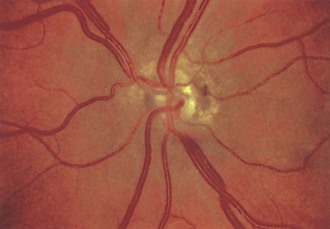
Fig. 15.28 Optic nerve hypoplasia.
(Reprinted with permission from Beck, R.W., Smith, C.H., 1988. Neuro-Ophthalmology: A Problem-Oriented Approach. Little, Brown, Boston.)
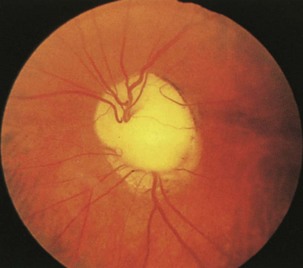
Optic nerve coloboma is more common than optic nerve hypoplasia and results from incomplete closure of the fetal fissure (Fig. 15.29). It may occur as an isolated finding or as part of a congenital syndrome including Aicardi syndrome and trisomy 13. Another type of congenital anomaly, the optic pit, is manifested as a small grayish area, usually located in the inferior temporal portion of the optic disc. In some optic nerve dysplasias, the disc appears enlarged. This is true of the so-called morning glory disc in which a large whitish concavity is surrounded by pigmentation that resembles a morning glory flower. This appearance occurs because defective closure of the embryonic fissure is followed by growth of glial tissue and vascular remnants.
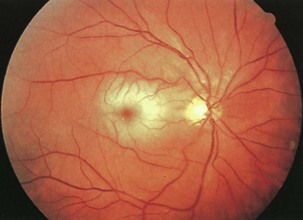
Retinal Disorders
Retinal Arterial Disease
Retinal arterial disease can manifest as a central retinal artery occlusion, branch retinal artery occlusion (CRAO/BRAO), or amaurosis fugax (transient monocular visual loss). Carotid artery atherosclerotic disease is the most common cause; cardiac valvular disease also must be considered. Evaluation and treatment for retinal arterial disease are similar to those for stroke and cerebrovascular disease in general because the annual risk of stroke or death in patients with visible retinal emboli can be increased 10-fold to 8.5% compared with controls. Acute retinal artery occlusion (CRAO/BRAO) is characterized by retinal whitening (edema) secondary to infarction. In CRAO, these findings usually are more prominent in the posterior pole than they are in the periphery (Fig. 15.30). A marked narrowing of the retinal arterioles often is noted. Because the fovea (the center of the macula) receives its blood supply from the choroid and there are no overlying retinal ganglion cells, this area retains its normal reddish-orange color, producing the characteristic cherry-red spot. The retinal edema usually subsides fairly rapidly over days to weeks. After resolution, the retinal appearance typically returns to normal, although the prognosis for visual recovery generally is poor.
When present, retinal emboli most often are located at arteriolar bifurcations (Fig. 15.31). Visualization of retinal emboli is more common in BRAO than in CRAO. They take on a glistening or whitish or yellowish appearance and may be located on or near the optic disc or in the retinal periphery. The three major types of retinal emboli are (1) cholesterol (Hollenhorst plaques, most commonly from the carotid artery), (2) platelet-fibrin (most commonly from the cardiac valves), and (3) calcific (from either a carotid or cardiac source). It is difficult to accurately distinguish among these on the basis of fundus examination alone. With impaired blood flow after a CRAO, a portion of a retinal arteriole may take on a whitish appearance. This represents not an embolus, but rather stagnant lipid in the blood or changes in the arteriole wall.
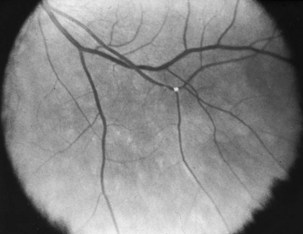
Fig. 15.31 Hollenhorst plaque at a retinal arteriole bifurcation.
(Reprinted with permission from Beck, R.W., Smith, C.H., 1988. Neuro-Ophthalmology: A Problem-Oriented Approach. Little, Brown, Boston.)
Branch Retinal Artery Occlusions and Encephalopathy (Susac Syndrome)
Branch retinal artery occlusions and encephalopathy (Susac syndrome) is a rare disorder characterized by multiple branch retinal artery occlusions and neurological dysfunction (Gross and Eliashar, 2005; Susac, 2004). Susac syndrome most commonly affects women between the ages of 20 and 40 years. A viral syndrome may precede the development of ocular and neurological signs. The most prominent neurological manifestations are impaired mentation and sensorineural hearing loss. Cerebrospinal fluid in patients with Susac syndrome shows a mild lymphocytic pleocytosis and elevated protein. Antinuclear antibody (ANA) testing and cerebral arteriography are generally normal, but brain MRI most often demonstrates multiple areas of high signal intensity that resemble demyelinating plaques on T2-weighted images.
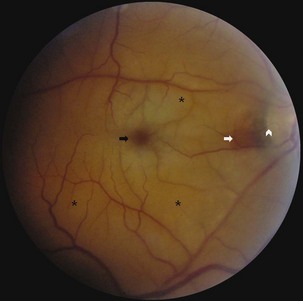
Retinal Vein Occlusion
Central or branch retinal vein occlusions rarely occur in patients younger than 50 years. The diagnosis is established clinically by the presence of characteristic retinal hemorrhages in the setting of acute vision loss. These occur diffusely in central retinal vein occlusion and focally in branch retinal vein occlusion (Fig. 15.32). Disc edema often is present and in some cases is the predominant fundus feature. In ischemic occlusion, treatment with panretinal photocoagulation can improve prognosis. No direct associations between retinal vein occlusion and carotid artery atherosclerotic disease are recognized. Patients require evaluation for vascular risk factors but generally do not require carotid imaging or ultrasound examination. In cases of bilateral retinal vein occlusion, evaluate the patient for hyperviscosity syndromes or hypercoagulable states.
Retinal Degenerations
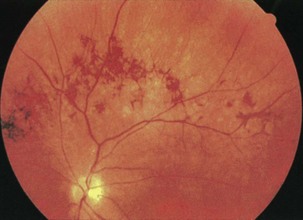
Among the many diseases of retinal degeneration, several are associated with neurological disease. The cause of retinitis pigmentosa (RP) is degeneration of the retinal rods and cones. Rods are predominantly affected early in the course of RP, impairing night vision. Visual field loss occurs first in the midperiphery and progresses to severe field constriction. Pigmentary retinal changes that look like bony spicules are the hallmark of RP (Fig. 15.33). In some cases, however, pigment changes are not prominent, and the visual field loss may be mistaken for a neurological disorder. Even without the characteristic bony spicule–type changes, the diagnosis of RP can be made on the basis of the retinal thinning, narrowing of retinal arterioles, and waxy optic disc pallor. RP may be associated with Kearns-Sayre syndrome, Cockayne syndrome, Refsum syndrome, Batten disease, inherited vitamin E deficiency, and spinocerebellar ataxia type 7.
Phakomatoses
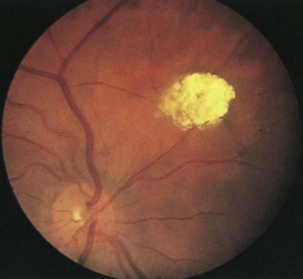
Retinal findings are common in phakomatoses that affect the nervous system, particularly tuberous sclerosis and von Hippel-Lindau disease. Neurological features of phakomatoses are described in Chapter 65. In tuberous sclerosis, retinal astrocytic hamartomas are characteristic (Fig. 15.34). These usually are multiple and may appear either as a fullness in the retinal nerve fiber layer or as a nodular refractile lesion (mulberry type). Von Hippel-Lindau disease is characterized by the presence of one or more retinal angiomas that appear as reddish masses with a feeding artery and a draining vein (Fig. 15.35). Treatment with photocoagulation or cryotherapy may be necessary. Wyburn-Mason disease is characterized by racemose arteriovenous malformations in the retina (Fig. 15.36).
Anderson D.R., Drance S.M., Schulzer M. Natural history of normal-tension glaucoma. Ophthalmology. 2001;108:247-253.
Arnold A.C. Pathogenesis of nonarteritic anterior ischemic optic neuropathy. J Neuroophthalmol. 2003;23:157-163.
Arnold A.C., Levin L.A. Treatment of ischemic optic neuropathy. Semin Ophthalmol. 2002;17:39-46.
Auw-Haedrich C., Staubach F., Witschel H. Optic disk drusen. Surv Ophthalmol. 2002;47:515-532.
Baier M.L., Cutter G.R., Rudick R.A., et al. Low-contrast letter acuity testing captures visual dysfunction in patients with multiple sclerosis. Neurology. 2005;64:992-995.
Balcer L.J. Anatomic review and topographic diagnosis. Ophthalmol Clin North Am. 2001;14:1-21.
Balcer L.J. Clinical practice. Optic neuritis. N Engl J Med. 2006;354:1273-1280.
Balcer L.J., Beck R.W. Inflammatory optic neuropathies and neuroretinitis. Yanoff M., Duker M. Ophthalmology, second ed, St. Louis: Mosby, 2003.
Balcer L.J., Galetta S.L. Optic neuropathies. In: Noseworthy J.H., editor. Neurological Therapeutics: Principles and Practice. London: Martin Dunitz, 2003.
Barbera L.G., Weiss M.J., Hofeldt A.J. Diabetic retinopathy and diabetic papillopathy. Semin Neurol. 1996;16:179-185.
Beck R.W. Optic neuritis. Miller N.R., Newman N.J. Walsh and Hoyt’s Clinical Neuro-Ophthalmology, fifth ed, Baltimore: Williams & Wilkins, 1998.
Beck R.W., Cleary P.A., Anderson M.M., et al. A randomized, controlled trial of corticosteroids in the treatment of acute optic neuritis. The Optic Neuritis Study Group. N Engl J Med. 1992;326:581-588.
Beck R.W., Trobe J.D., Moke P.S., et al. High- and low-risk profiles for the development of multiple sclerosis within 10 years after optic neuritis: experience of the Optic Neuritis Treatment Trial. Arch Ophthalmol. 2003;121:944-949.
Chang S.H., Miller N.R. The incidence of vision loss due to perioperative ischemic optic neuropathy associated with spine surgery: the Johns Hopkins Hospital Experience. Spine. 2005;30:1299-1302.
Chinnery P.F., Griffiths P.G. Optic mitochondriopathies. Neurology. 2005;64:940-941.
Damek D.M. Paraneoplastic retinopathy/optic neuropathy. Curr Treat Options Neurol. 2005;7:57-67.
Danesh-Meyer H.V. Radiation-induced optic neuropathy. J Clin Neurosci. 2008;15:95-100.
Davis P.L., Jay W.M. Optic nerve head drusen. Semin Ophthalmol. 2003;18:222-242.
Fontal M.R., Kerrison J.B., Garcia R., et al. Ischemic optic neuropathy. Semin Neurol. 2007;27:221-232.
Friedman D.I. Pseudotumor cerebri. Neurol Clin. 2004;22:99-131.
Gross M., Eliashar R. Update on Susac’s syndrome. Curr Opin Neurol. 2005;18:311-314.
Hall J.K., Balcer L.J. Giant cell arteritis. Curr Treat Options Neurol. 2004;6:45-53.
Hayreh S.S. Optic disc edema in raised intracranial pressure. V. Pathogenesis. Arch Ophthalmol.. 1977;95:1553-1565.
Hayreh S.S. Posterior ischaemic optic neuropathy: clinical features, pathogenesis, and management. Eye. 2004;18:1188-1206.
Hayreh S.S., Zimmerman M.B. Incipient nonarteritic anterior ischemic optic neuropathy. Ophthalmology. 2007;114:1763-1772.
Hayreh S.S., Zimmerman M.B. Nonarteritic anterior ischemic optic neuropathy: natural history of visual outcome. Ophthalmology. 2008;115:298-305.
Jaffe G., Caprioli J. Optical coherence tomography to detect and manage retinal disease and glaucoma. Am J Ophthalmol. 2004;137(1):156-169.
Jarius S., Paul F., Franciotta D., et al. Mechanisms of disease: aquaporin-4 antibodies in neuromyelitis optica. Nat Clin Pract Neurol. 2008;4:202-214.
Jonas J.B., Budde W.M. Diagnosis and pathogenesis of glaucomatous optic neuropathy: morphological aspects. Prog Retin Eye Res. 2000;19:1-40.
Listernick R., Ferner R.E., Liu G.T., et al. Optic pathway gliomas in neurofibromatosis-1: controversies and recommendations. Ann Neurol. 2007;61:189-198.
Man P.Y., Turnbull D.M., Chinnery P.F. Leber hereditary optic neuropathy. J Med Genet. 2002;39:162-169.
March G.A.Jr., Lessell S. Infectious optic neuropathy. Int Ophthalmol Clin. 1996;36:197-205.
Mathews M.K. Nonarteritic anterior ischemic optic neuropathy. Curr Opin Ophthalmol. 2005;16:341-345.
Newman N.J. Hereditary optic neuropathies: from the mitochondria to the optic nerve. Am J Ophthalmol. 2005;140:517-523.
Optic Neuritis Study Group. The clinical profile of optic neuritis. Experience of the optic neuritis treatment trial. Arch Ophthalmol. 1991;109:1673-1678.
Optic Neuritis Study Group. Multiple sclerosis risk after optic neuritis: final optic neuritis treatment trial follow-up. Arch Neurol. 2008;65:727-732.
Patten J. Vision, the visual fields, and the olfactory nerve. In: Patten J., editor. Neurological Differential Diagnosis. London: Springer-Verlag, 2004.
Prasad S., Moss H.E., Lee E.B., et al. Clinical reasoning: a 42-year-old man with sequential monocular visual loss. Neurology. 2008;71:e43-49.
Prasad S., Volpe N.J., Balcer L.J. Approach to optic neuropathies: clinical update. Neurologist. 2010;16:23-34.
Rizzo J.F.3rd, Lessell S. Optic neuritis and ischemic optic neuropathy. Overlapping clinical profiles. Arch Ophthalmol. 1991;109:1668-1672.
Rothstein J., Maisel R.H., Berlinger N.T., et al. Relationship of optic neuritis to disease of the paranasal sinuses. Laryngoscope. 1984;94:1501-1508.
Salvarani C., Cantini F., Hunder G.G. Polymyalgia rheumatica and giant-cell arteritis. Lancet. 2008;372:234-245.
Sarkies N. Traumatic optic neuropathy. Eye. 2004;18:1122-1125.
Spoor T.C., Kennerdell J.S., Martinez A.J., et al. Malignant gliomas of the optic nerve pathways. Am J Ophthalmol. 1980;89:284-292.
Susac J.O. Susac’s syndrome. AJNR Am J Neuroradiol. 2004;25:351-352.
Taylor D. Developmental abnormalities of the optic nerve and chiasm. Eye. 2007;21:1271-1284.
Trobe J.D., Beck R.W., Moke P.S., et al. Contrast sensitivity and other vision tests in the Optic Neuritis Treatment Trial. Am J Ophthalmol. 1996;121:547-553.
Wingerchuk D.M. Neuromyelitis optica. Adv Neurol. 2006;98:319-333.
Yu Wai Man, P., Griffiths, P.G., 2005. Surgery for traumatic optic neuropathy. Cochrane Database Syst Rev CD005024.
Yu Wai Man, P., Griffiths, P.G., 2007. Steroids for traumatic optic neuropathy. Cochrane Database Syst Rev CD006032.