chapter 13
Abnormal Movements and Difficulty Walking Due to Central Nervous System Problems
In the previous chapter difficulty walking that is related to peripheral nervous system problems was discussed. The first half of this chapter deals with the more common central nervous system problems that result in difficulty walking; the second half discusses abnormal movements affecting the face, head and limbs, some of which lead to difficulty walking.
 ‘My legs are weak, Doc.’ When a patient states that their legs are weak, ask them to clarify exactly what they mean. Patients often use the term weakness as a non-specific way to say that something is wrong with their legs, and they do not necessarily mean that there is an actual loss of strength in their legs. Often, in fact, the legs are strong and the difficulty relates to other neurological and non-neurological problems.
‘My legs are weak, Doc.’ When a patient states that their legs are weak, ask them to clarify exactly what they mean. Patients often use the term weakness as a non-specific way to say that something is wrong with their legs, and they do not necessarily mean that there is an actual loss of strength in their legs. Often, in fact, the legs are strong and the difficulty relates to other neurological and non-neurological problems.DIFFICULTY WALKING
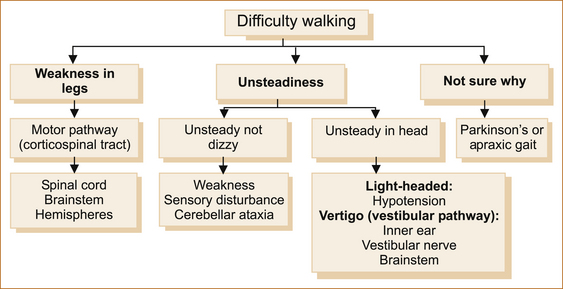
When a patient complains that they are have difficulty walking, simply ask if the difficulty relates to a sense of instability in the head or something wrong with their legs or if they are uncertain why they are having problems. Figure 13.1 shows the diagnostic possibilities in each of these three scenarios.
Difficulty walking related to weakness
The presence of weakness in the legs infers a problem involving the motor pathway, which extends from the motor cortex to the muscle.
Clues that the spinal cord is the site of the problem:
• Back or neck pain that coincides with the development of neurological symptoms in the lower limbs
• Alteration in sphincter function with constipation and/or urinary retention
• Altered sensation with a sensory level on the trunk (a strong indicator)
Clues that the brainstem is the site of the problem:
• Dysphagia (to a lesser extent)
• Ipsilateral facial sensory loss and contralateral loss to pain and temperature affecting the limbs (lateral brainstem involvement)
• A 12th, 6th or 3rd nerve palsy on the side opposite to the weakness points to the medulla, pons and midbrain, respectively (see Chapter 4, ‘The cranial nerves and understanding the brainstem’)
Clues that the cerebral hemisphere is the site of the problem:
• When there is upper motor neuron facial weakness in addition to the weakness in the arm and leg, the lesion must be above the mid pons on the contralateral side and, therefore, cannot be any lower in the brainstem or in the spinal cord. If areas in the hemispheres other than the motor pathway are affected, it may be possible to localise the lesion to the hemispheres, in particular the cerebral cortex.
• Dysphasia (dominant hemisphere lesions affecting the cortex)
• Visual field loss (hemianopia or quadrantanopia) if the visual pathways are affected
• Visual inattention if the parietal cortex is affected
• Cortical sensory signs (2-point discrimination, graphaesthesia, stereognosis or sensory inattention) (Other cortical phenomena are described in Chapter 5, ‘The cerebral hemispheres and cerebellum’.)
SPINAL CORD PROBLEMS
1. lesions external to the dura (traumatic spinal cord compression, prolapsed intervertebral discs, epidural abscess)
2. lesions beneath the dura but external to the spinal cord (neurofibroma, Chiari malformation)
3. intrinsic spinal cord problems (glioma, ependymoma, sarcoidosis, vasculitis, syphilis, arteriovenous malformation, spinal cord infarction and adrenomyeloneuropathy).
These are very rare conditions and are beyond the scope of this book.
The features that point to intrinsic cord pathology are:
• early development urinary or anal sphincter dysfunction
• early involvement of the spinothalamic tract
• suspended sensory loss, which is a classical feature of an intrinsic spinal cord problem where there is altered pain and temperature sensation in a pattern that resembles a cape with impaired sensation on the upper trunk but sparing the lower trunk and lower limbs.
• early sacral sensory loss occurs with extrinsic compression and sacral sparing is seen with intrinsic spinal cord lesions; the presence of radicular pain indicates lesions extrinsic to the dura.
Cervical spondylotic myelopathy (CSM): Cervical spondylotic myelopathy (CSM) is the commonest cause of spinal cord dysfunction in elderly patients. It is also the commonest cause of paraparesis or quadriparesis not related to trauma. Repeated occupational trauma, such as carrying axial loads, genetic predisposition and Down syndrome all predispose to an increased risk of cervical spondylosis.
Cervical spondylosis relates to degenerative changes initially in the cervical discs with subsequent subperiosteal bone formation, referred to as osteophytes. Cervical disc protrusion or extrusion, osteophytes and hypertrophy of the ligamentum flavum together result in spinal cord compression, and secondary spinal cord ischaemia can occur with severe compression. Rarely spondylolisthesis may occur and cause cord compression.
Cervical cord compression (see Figure 13.2) can be seen in younger patients with congenitally narrow spinal canals (10–13 mm). The spinal cord is stretched during flexion of the cervical spine and buckling of the ligamentum flavum occurs during extension of the cervical spine. Thus, repeated flexion and extension in the patient with significant canal narrowing may cause intermittent acute compression of the spinal cord, and it is thought that this may also account for the clinical deterioration seen in many cases of CSM [1].
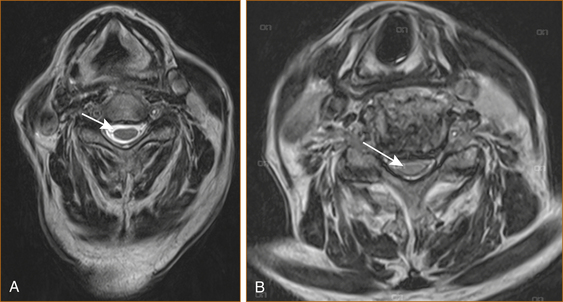
FIGURE 13.2 MRI scan of A normal cervical spine and B spinal cord compression due to cervical spondylosis
The natural course of CSM for any given individual is variable and precise prognostication is not possible. Once moderate signs and symptoms develop, however, patients are less likely to spontaneously improve. Worsening occurs more commonly in older patients whereas patients with mild disability are less likely to worsen [1].
CSM can present in a variety of ways.
1. The commonest presentation is with the insidious onset of difficulty walking related to weakness and/or stiffness (due to spasticity). Some patients will have neck pain from the cervical spondylosis; pain in the arm related to nerve root compression is less common.
2. Older patients in particular may present with weakness and wasting of the small muscles of the hands related to nerve root compression, and the signs of spinal cord compression are noted when they are examined.
3. A presentation that is much less common is the central cord syndrome resulting from a hyperextension injury in the setting of cervical canal stenosis where the weakness is greater in the upper than the lower limbs with or without a suspended sensory loss (affecting the shoulders and upper torso like a cape) and urinary retention.
4. Patients with cervical cord compression may experience an electric shock-like ensation radiating down their back or into their limbs with neck flexion, the so-called L’Hermitte phenomena commonly seen in patients with multiple sclerosis (MS).
 Some elderly patients assume that the gradual onset of difficulty walking is simply the result of advancing age and tend not to seek medical attention until the cord compression and limb weakness are severe.
Some elderly patients assume that the gradual onset of difficulty walking is simply the result of advancing age and tend not to seek medical attention until the cord compression and limb weakness are severe.Thoracic cord compression: Thoracic spinal cord compression most commonly occurs with metastatic malignancy, less commonly with a thoracic disc, epidural abscess or extrinsic spinal cord tumour such as a meningioma or a neurofibroma. There is an aphorism that ‘a thoracic cord lesion in a middle-aged to elderly female is a meningioma (benign tumour arising from the meninges) until proven otherwise’. Back pain is rare with a meningioma, more common but not a universal feature of thoracic discs, but increasingly severe back pain occurs for on average 8 weeks or longer in 80–95% of patients who subsequently develop malignant cord compression [2]. Patients who are not known to suffer from malignancy, but who develop increasingly severe midline (especially thoracic) back pain, should be investigated promptly in the hope of preventing malignant cord compression.
Although initially the pain is localised to the vertebra, subsequent nerve root compression can result in radicular pain. The back pain is often worse after lying down and this can be a vital clue. Rapidly developing weakness in the legs is the initial and dominant neurological symptom once malignant spinal cord compression occurs; sensory symptoms are less common and sphincter disturbance occurs late. A delay in the diagnosis of malignant cord compression is common with many patients only diagnosed after they lose their ability to walk [2, 3] and, unfortunately, once significant cord compression related to malignancy occurs the prognosis for recovery is poor.
In patients with preexisting malignancy, particularly breast and prostate, the development of back pain should raise the suspicion of possible secondary malignancy in the spinal column and prompt investigation (see Figure 13.3).
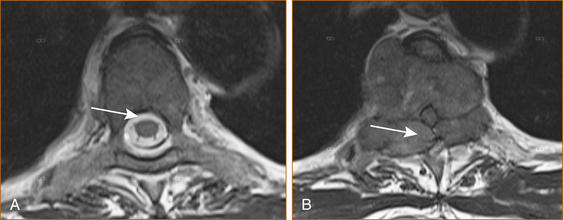
The following suggest spinal metastases:
• pain in the thoracic or cervical spine
• severe unremitting or progressive lumbar spinal pain
• spinal pain aggravated by straining
It has been recommended that patients with known malignancy should be advised to return urgently within 24 hours for review should they develop back pain in the midline [4].
Severe thoracic back pain followed by the rapid development of spinal cord compression can be the initial manifestation of malignancy in as many as one-third of patients with cancer of unknown primary origin, non-Hodgkin lymphoma, myeloma and lung cancer [2]. Metastatic disease less commonly affects the cervical or lumbar spine. Although almost any systemic cancer can metastasise to the spinal column, prostate, breast and lung are the most common [2].
Intrinsic spinal cord problems: Transverse myelitis is the commonest intrinsic cord lesion resulting in dysfunction in the lower limbs and difficulty walking in the younger patient while spinal cord ischaemia is seen in the elderly. Spinal cord ischaemia can develope abruptly or insidiously. Other intrinsic spinal cord problems such as tumours and the cavitating lesion referred to as syringomyelia are very rare and are beyond the scope of this book.
TRANSVERSE MYELITIS: Acute transverse myelitis (ATM) is a focal inflammatory disorder of the spinal cord, resulting in motor, sensory and autonomic dysfunction with many infectious and non-infectious causes [10].
The Transverse Myelitis Consortium Working Group [11] has established diagnostic criteria that require:
• bilateral signs and/or symptoms of spinal cord dysfunction affecting the motor, sensory and autonomic systems
• a clearly defined sensory level
• the exclusion of cord compression
• the demonstration of inflammation within the cerebrospinal fluid (CSF). If none of the inflammatory criteria is met at symptom onset, repeat MRI and lumbar puncture evaluation should be performed between 2 and 7 days following symptom onset.
The development of neurological symptoms and signs is often preceded by a febrile illness up to 4 weeks prior to onset [12], typically an upper respiratory tract infection. Patients present with an ascending sensory loss, with the development of a sensory level in the majority of patients with or without a paraparesis, paraplegia, quadriplegia and urinary retention [12, 13]. The symptoms may develop rapidly over as little as 4 hours or more slowly over several weeks.
 Patients whose spinal cord symptoms reach maximal severity in < 4 hours from onset should be presumed to have an ischaemic aetiology [1].
Patients whose spinal cord symptoms reach maximal severity in < 4 hours from onset should be presumed to have an ischaemic aetiology [1].
In one study of acute transverse myelitis, a parainfectious cause was diagnosed in 38% of patients but the underlying infectious agent was identified in a minority of patients. In 36% of patients the aetiology remained uncertain and in 22% it was the first manifestation of possible MS. The MRI scan (see Figure 13.4) was abnormal in 96% of cases (i.e. a normal MRI is rare but does not exclude a diagnosis) [12].
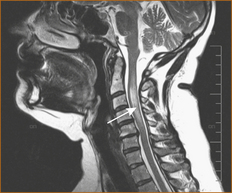
FIGURE 13.4 MRI scan of transverse myelitis
Acute non-compressive myelopathies can also be classified according to their aetiology [14]:
CONUS MEDULLARIS LESION: This chapter has discussed central nervous system causes of difficulty walking whereas the previous chapter discussed peripheral nervous system problems. There is one very rare syndrome that produces both upper and lower motor neuron signs in the lower limbs and this is a lesion at the level of the conus medullaris (the lower end of the spinal cord at the level of the 2nd lumbar vertebra) with or without the involvement of the cauda equina (the sheath of lumbosacral nerve that extends from the lower end of the spinal cord through the spinal canal exiting at the appropriate level and that looks like a horse’s tail, hence the name).
BRAINSTEM PROBLEMS CAUSING WEAKNESS IN THE LEG(S)
Weakness caused by brainstem problems is usually unilateral or bilateral weakness affecting the lower limbs with or without involvement of the upper limbs and, if above the mid pons, the face. It is the presence of symptoms and signs pointing to the brainstem (see Chapter 4, ‘The cranial nerves and understanding the brainstem’) that is the clue to the site of the pathology.
CEREBRAL HEMISPHERE PROBLEMS CAUSING WEAKNESS IN THE LEG(S)
Weakness related to cerebral hemisphere problems is almost invariably unilateral, although bilateral lower limb weakness can occur with involvement of the parafalcine region, typically with cerebral infarction in the distribution of the anterior cerebral artery. The presence of abulia (see Chapter 5, ‘The cerebral hemispheres and cerebellum’) is the clue that the lesion is affecting the distribution of the anterior cerebral artery.
Difficulty walking due to unsteadiness
UNSTEADINESS IN THE ABSENCE OF WEAKNESS OR DIZZINESS
Patients with marked impairment of proprioception in the lower limbs will be very unsteady on their feet, particularly in the dark and when they close their eyes, e.g. while having a shower or washing their hair. This is often referred to as a sensory ataxia. Vitamin B12 deficiency with subacute combined degeneration of the cord is probably the commonest cause now that syphilis has largely been eradicated. Very rarely, impairment of proprioception results from a dorsal root ganglionopathy or sensory neuronopathy. This occurs as a paraneoplastic phenomenon [15, 16], in patients with Sjögren’s syndrome [17] or pyridoxine abuse [18]. The sensory ganglionopathies are extremely rare and are mentioned here because the neurological problem may antedate the diagnosis of malignancy or indicate recurrence in a patient with known malignancy and should prompt a search for malignancy [16].
Truncal ataxia occurs with hypothyroidism [19] and alcoholism [20]. Patients with isolated truncal ataxia walk with a wide-based gait with little in the way of nystagmus or ataxia in the limbs.
The commonest cause of ataxia related to cerebellar disease would be cerebellar infarction. Two less common causes of cerebellar ataxia affecting the cerebellar hemispheres are the paraneoplastic cerebellar syndrome [21] and the hereditary spinocerebellar atrophies (SCA) [22]. The paraneoplastic cerebellar syndrome may be the initial presenting symptom of malignancy or develop after the diagnosis of malignancy is established. The clinical picture is one of very disabling ataxia affecting the limbs and trunk, together with dysarthria and nystagmus evolving rapidly over days to weeks. On the other hand, the hereditary spinocerebellar atrophies present with the insidious onset of ataxia affecting the limbs with or without nystagmus and dysarthria.
Difficulty walking not sure why
PARKINSON’S SYNDROME
• slowness of movement referred to as bradykinesia
• cogwheel rigidity (increased tone that has a sensation of the muscle giving way in little jerks).
The commonest cause of Parkinson’s syndrome is Parkinson’s disease, but drugs (such as phenothiazines, butyrophenones, metoclopramide, reserpine and tetrabenazine) may cause a reversible Parkinson’s syndrome, and toxins (such as manganese dust or carbon disulfide) and the recreational drug N-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP) cause an irreversible Parkinson’s syndrome [23] by destroying the dopamine neurons in the midbrain. The term ‘Parkinson’s disease’ is reserved for patients with the above clinical features who have the characteristic neuropathology of loss of pigmentation in the substantia nigra and the presence of the Lewy bodies.
Parkinson’s is largely a disease of the elderly, although in up to 10% of patients the onset is before the age of 50. The patient with Parkinson’s disease affecting both lower limbs walks with small shuffling steps where the sole of the shoe is often heard scraping along the ground. As the patient walks there is a lack of arm swing. They may also develop an involuntary sensation where they cannot stop themselves from walking with short accelerating steps, the so-called festinating gait. Occasionally, patients present with unilateral involvement mimicking a hemiparesis and are thought to have had a ‘stroke’. The insidious onset and the presence of tremor and cogwheel rigidity on examination should alert the clinician to the correct diagnosis.
APRAXIA OF GAIT
Apraxia of gait is an inability to walk in the absence of weakness, sensory deficit, instability or incoordination. It is a perseveration of posture and an inability to perform the serial movements necessary for ambulation. In the initial stage there is difficulty in starting to walk or changing direction. Patients walk with small steps referred to as the ‘marche a petit pas’ (walks with little steps); unlike in Parkinson’s they do not shuffle but lift their feet off the ground. The features that help differentiate apraxia of gait from Parkinson’s disease are listed in Table 13.1.
TABLE 13.1
| Clinical feature | Parkinson’s | Frontal lobe apraxia |
| Resting tremor | Yes | No |
| Shuffles when walks | Yes | No |
| Lifts feet when walks with small steps | No | Yes |
| Swings arms when walking | No | Yes |
| Smiles spontaneously | No | Yes |
| Bradykinesia | Yes | Yes |
| Difficulty arising from chair | Yes | Yes |
| Grasp and palmo-mental reflexes | No∗ | Yes |
| Cogwheel rigidity(rigidity constant during testing) | Yes | No |
| Gegenhalten rigidity(rigidity increases with testing) | No | Yes |
∗May occur late in the course when cognitive decline is present.
To the inexperienced clinician these patients appear to have cogwheel rigidity but it is in fact Gegenhalten or ‘an involuntary, voluntary resistance to passive movement’. The way to differentiate between the cogwheel rigidity of Parkinson’s and the apparent cogwheel rigidity in frontal lobe disorders is that, in patients with Parkinson’s, the cogwheel rigidity is evident from the moment testing begins whereas, in patients with Gegenhalten, the increased tone that has the sensation of cogwheel rigidity is not present initially but, the more the clinician tests for increased tone, the greater the degree of resistance creating the impression of a cogwheel rigidity. If testing is momentarily interrupted by simply lowering the hand and wrist and then started again, the increased tone is not present initially but once again increases with further testing.
Communicating or ‘normal pressure’ hydrocephalus: Communicating hydrocephalus is relatively rare but important because it is treatable. The diagnosis is a largely a clinical one and is based on the triad of:
If there is an alteration in the rapidity with which a patient with Parkinson’s worsens, it is important to consider another cause. A common and readily reversible cause of a rapid deterioration in patients with Parkinson’s disease is a urinary tract infection. Worsening has also been described in patients with subdural haematoma [30]. Other
 In patients with chronic neurological diseases such as Parkinson′s, the rate of worsening tends to be fairly constant in the individual patient and, therefore, a rapid deterioration should alert the clinician to an alternative diagnosis.
In patients with chronic neurological diseases such as Parkinson′s, the rate of worsening tends to be fairly constant in the individual patient and, therefore, a rapid deterioration should alert the clinician to an alternative diagnosis.examples of this principle seen by this author include spinal cord compression and dermatomyositis, which lead to a more rapid decline in the patient’s ability to walk than had occurred with their Parkinson’s disease.1
ABNORMAL MOVEMENTS
Abnormal movements of the head, face and neck
Abnormal movements that affect the head, face and neck are:
Two uncommon causes of abnormal movements of the face and head, hemifacial spasm and spasmodic torticollis; the latter has been discussed in the section ‘Neck pain’ in Chapter 11, ‘Common neck, arm and upper back problems’.
HEAD TREMOR
Head tremor is usually a manifestation of benign essential/familial tremor or cerebellar disease; it is very rare in Parkinson’s disease [31]. Essential/familial tremor may also affect the mouth and at times the voice. Benign essential/familial tremor is discussed in more detail in the section on abnormal movements of the upper limbs where this form of tremor is more common.
TARDIVE DYSKINESIA
Tardive dyskinesia is a neurological syndrome first recognised in the 1950s [32] that consists of repetitive, involuntary, purposeless movements affecting the mouth, lips and tongue with tongue protrusion; lip smacking, puckering and pursing; facial grimacing and rapid eye blinking. The involuntary movements occur during most of the waking hours. Typically it relates to side effects from certain drugs with an increased incidence in the elderly, although it may occur in the absence of any drug therapy, particularly in elderly edentulous patients. Tardive dyskinesia may also affect the limbs and trunk where the abnormal movements are more athetoid (repetitive involuntary, slow, sinuous, writhing movements) in nature.
TOURETTE SYNDROME
Tourette syndrome is a condition predominantly seen in school-aged children [37]. The syndrome consists of tics that are sudden, brief, intermittent, involuntary or semi-voluntary movements (motor tics), such as blinking, nose twitching, head and limb jerking, mouth opening, torticollis, shoulder rotation and sustained eye closure (blepharospasm). More complex motor tics may occur, such as making obscene gestures (copropraxia) and imitating others’ gestures (echopraxia). Burping, retching, vomiting, fist shaking, trunk bending, jumping or kicking are also seen. Phonic or vocal tics can also occur and these may include simply sniffing, grunting, coughing, clearing the throat, barking, screaming, shouting obscenities (coprolalia) and repeating one’s own utterances (echolalia). One of the diagnostic characteristics is the ability of the patient to suppress their tics. The motor and phonic tics may persist during sleep. It is much more common in males and is often associated with attention-deficit/hyperactivity disorder or obsessive–compulsive disorder.
OCULOGYRIC CRISIS
This refers to the sudden involuntary contractions of some of the eye muscles that result in repetitive, conjugate ocular deviations usually, although not always, in an upward direction. The attack or crisis may last from seconds to minutes. Oculogyric crisis is most commonly seen following exposure to neuroleptic drugs. The incidence of oculogyric crises in patients treated with chronic neuroleptic therapy may be as high as 10%.
Abnormal movements of the limbs
• benign essential or familial tremor
• the tremor of Parkinson’s disease
Some abnormal movements occur only with activity, others occur at rest and the patient is unable to keep still (see Figure 13.5). Most are abolished by sleep, but myoclonus and hemiballismus can occur while the patient is asleep.
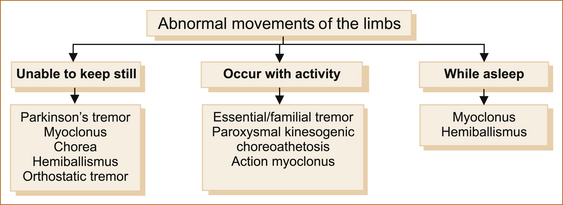
ABNORMAL MOVEMENTS WHERE THE PATIENT CANNOT KEEP STILL
Parkinson’s disease: Parkinson’s disease was first described by James Parkinson in 1817 [40]. Resting tremor abolished by movement is often the initial symptom although occasional patients with Parkinson’s disease may have a non-resting tremor. The associated features of rigidity and bradykinesia that are characteristic of Parkinson’s may be minimal or absent in the very early stages. Patients do not complain that their limbs are rigid or stiff; they tend to say that the muscles in the arms and legs ache and they do not work as well. With advanced disease there is increasing inability to undertake activities of daily living that results from the bradykinesia, although patients often assume it is simply the ageing process! Patients with Parkinson’s disease walk with a slightly stooped posture and do not swing their arms when they are walking.
Myoclonus: Myoclonus refers to sudden, shock-like, involuntary movements that can manifest in various patterns:
• focal, where a few adjacent muscles are involved
• multifocal, where many muscles jerk asynchronously
• generalised, where most of the muscles of the body are involved.
Myoclonic movements may be spontaneous or they may be activated by movement. Myoclonus is most commonly related to epilepsy and this has been discussed in Chapter 8, ‘Seizures and epilepsy’. Myoclonus might also occur after hypoxic brain damage either acutely, within 24 hours and while the patient is still comatose, or as a late complication. The myoclonus related to hypoxia is usually movement-induced, but it can be triggered by noise, touching the patient or tracheal suctioning. The acute form is associated with a poor prognosis. The chronic form develops days or weeks after the hypoxic insult and predominantly consists of an action myoclonus affecting the limbs. Violent flexion movements of the body, head and neck are precipitated by movement.
Propriospinal myoclonus (PSM) is a rare movement disorder characterised by myoclonic jerks arising in muscles corresponding to a single myotome and spreading rostrally and caudally to the other myotomes. PSM can be idiopathic, related to spinal cord lesions, drug use, malignancy or infection [45]. Patients present with myoclonic jerks involving abdominal wall muscles which worsen when lying down.
Chorea and athetosis: Chorea is the ceaseless irregular, rapid, uncontrolled complex body movements that look well coordinated and purposeful but are in fact involuntary. The term ‘chorea’ is derived from the Greek word ‘choreia’ or ‘khoreia’ for dancing. Chorea was thought to be suggestive of a grotesque dance. Chorea can affect the face, arms or legs. The abnormal movements are almost continuous.
Chorea is very uncommon; Huntington’s disease is now the main cause with Sydenham’s chorea largely disappearing along with rheumatic fever. A very rare but treatable cause of chorea is Wilson’s disease [47]. There are a number of even rarer inherited movement disorders that could result in dystonia, chorea or ataxia [48, 49, but are beyond the scope of this book.
HUNTINGTON’S DISEASE: Huntington’s disease [50] is a hereditary disorder with the genetic defect on the short arm of chromosome 4. The genetic defect leads to altered function of the ubiquitous protein, huntingtin, that culminates in neuronal loss in the caudate nucleus. The number of tri-nucleotide repeats (cysteine-adenine-guanidine; CAG) influences whether the disease shows incomplete or complete penetrance and also influences the age of onset, with juvenile-onset Huntington’s patients typically having more than 55 tri-nucleotide repeats.
Huntington’s disease is characterised by the insidious onset in middle age (35–44 years of age) of progressive cognitive decline associated with abnormal movements and psychiatric disturbances. The chorea may not be present at onset; often the initial symptoms are unsteadiness and a lack of coordination with the cognitive decline and psychiatric disturbances appearing later. Chorea is the typical movement disorder but often rigidity, bradykinesia and dystonia may predominate and be more disabling. Rarely does it develop before the age of 20 where it is referred to as juvenile Huntington’s disease. In juvenile Huntington’s disease, the symptoms are quite different – the patient often presents rigid and akinetic (absence or poverty of movement) – and the progression to disability is more rapid. The inheritance pattern of Huntington’s disease is autosomal dominant.
SYDENHAM’S CHOREA: Sydenham’s chorea was first described in 1686 by Thomas Sydenham in a work entitled ‘Schedula monitoria de novae febris ingress’, but it was not until 180 years later (in 1866) that the association with rheumatic fever was appreciated by Roger. It is a complication of rheumatic fever following infection with particular strains of streptococci (i.e. group A beta-hemolytic streptococci). It predominantly affects children although it can be seen in adults. It is much rarer these days since the virtual abolition of rheumatic fever.
The antecedent sore throat, polyarthritis and subcutaneous nodules the size of peas at joints such as the elbows and knees, together with the characteristic pink-red macular rash referred to as erythema marginatum, are the clues to the diagnosis. Sydenham’s chorea is a self-limiting illness that initially worsens over 2–4 weeks and then subsequently resolves spontaneously over 3–6 months, although some patients may have waxing and waning symptoms for up to 12 months [51].
ABNORMAL MOVEMENTS THAT OCCUR WITH ACTIVITY
Benign essential or familial tremor: Essential and familial essential tremor [53, 54] are the most common movement disorders in the elderly and the most common cause of an action tremor [55]. It can occur in patients in their teens.
Typically, the benign essential/familial tremor occurs when patients are either moving their arms or holding an object such as a book or a cup of tea where the cup rattles in the saucer and patients have to resort to half-filling their cup in order not to spill the tea. Eating becomes more difficult as the patient repeatedly spills food off the fork or soup out of the spoon. Although the tremor is bilateral, one side may be more severely affected than the other. The tremor sometimes leads to physical disability but progression to disability is usually very slow. Many patients are socially disabled as they are ‘too embarrassed’ to go out. Familial tremor is usually inherited as an autosomal dominant trait but with variable penetrance. Apart from the tremor, neurological examination is otherwise normal, in particular there is no rigidity or bradykinesia.
Differentiating Parkinson’s from essential/familial tremor: Table 13.2 shows the differences between the tremor of Parkinson’s and that of familial essential tremor.
TABLE 13.2
The different features of the tremor of Parkinson’s disease and essential/familial tremor
| Clinical feature | Parkinson’s | Essential/familial |
| Affects the head and voice | No | Yes |
| Present at rest | Yes | No |
| Present when holding objects | No | Yes |
| Worse with walking | Yes | No |
| Influenced by alcohol | No | Yes |
| Frequency | 3–6 Hz | 5–12 Hz |
| Family history | Occasional | Often (familial) |
Cerebellar or intention tremor: Cerebellar tremor is also referred to as intention tremor. These patients present with tremor when using their arms. There is no tremor at rest, little or no tremor with the hands outstretched but obvious tremor when the patient is asked to perform finger-to-nose or heel-to-shin testing where instability is noticed as the patient stretches to reach the distant target. The oscillations of intention tremor are perpendicular to the direction of movement and usually of low frequency, less than 5 Hz.
ORTHOSTATIC TREMOR: Orthostatic tremor is a condition where the patient finds it impossible to stand still for any length of time without developing an increasingly severe sensation of instability (not dizziness) with their body developing a tremulous sensation. Patients rarely fall and usually hang on to something, sit or commence walking. Many patients avoid stopping while walking in order to avoid this sensation. The neurological examination is otherwise normal. Most cases are idiopathic [62], but it has been described in progressive supranuclear palsy [63] and following head injury [64].
Action myoclonus: Myoclonus is the one abnormal movement that occurs at rest and also persists in sleep. Action myoclonus refers to sudden arrhythmic muscular jerking induced by voluntary movement. The condition is usually associated with diffuse neuronal disease, such as post-hypoxic encephalopathy [68], uraemia and various forms of progressive myoclonic epilepsy such as the Ramsay–Hunt syndrome [69].
Paroxysmal kinesogenic choreoathetosis: Paroxysmal kinesogenic choreoathetosis (PKC) has its onset in childhood or early adulthood. The characteristic feature is discrete episodes of abnormal movements precipitated by sudden movement such as standing up from sitting or being startled. Chorea occurs as does hyperkinesias, dystonia, athetosis and ballism. The neurological exam is normal between the events. The attacks are brief (< 5 minutes) and may occur several times a day.
ABNORMAL MOVEMENTS THAT OCCUR DURING SLEEP
Hemiballismus: Hemiballismus (derived from the Greek word ‘ballismos’ that means jumping about or dancing) is considered a rare form of chorea and is almost continuous, violent, coordinated involuntary motor restlessness of half (very rarely bilateral, referred to as paraballism) of the body. The movements are usually continuous contorting movements and often rotatory in nature. It is most marked in the upper extremities and usually caused by a lesion involving the subthalamic nucleus of the opposite side of the brain, but it can arise from contralateral lesions in the cortex, basal ganglia and thalamus [72]. The commonest cause is an infarct, but it has been reported with demyelinating disease.
Rare but life-threatening movement disorders
These have also been referred to as movement disorder emergencies and are defined as any movement disorder that evolves over hours to days and include acute parkinsonism, dystonia, chorea, tics and myoclonus. The commonest is drug-induced (neuroleptics and antiemetics) parkinsonism, and cyanide, methanol, carbon monoxide, carbon disulfide, organophosphate pesticides and the designer drug MPTP are some of the toxins that may produce a severe encephalopathy with clinical features of parkinsonism. Only neuroleptic malignant syndrome and serotonin syndrome will be discussed here. For an excellent review of movement disorder emergencies see the review by Poston [39].
SEROTONIN SYNDROME
Serotonin syndrome [73] is caused mainly by the serotonin-specific reuptake inhibitors (sertraline, fluoxetine, paroxetine and fluvoxamine), clomipramine, ecstasy and the combination of monoamine oxidase inhibitors and meperidine that result in an increase in the biological activity of serotonin.
Serotonin syndrome consists of fever with confusion, hypomania, agitation, tachycardia, fever, sweating, shivering, tremor, diarrhoea, hypertension, incoordination, myoclonus, rigidity and hyperreflexia. The clinical features of serotonin syndrome and neuroleptic malignant syndrome overlap; it is the presence of myoclonus that distinguishes serotonin syndrome from neuroleptic malignant syndrome.
REFERENCES
1. Baron, E.M., Young, W.F. Cervical spondylotic myelopathy: A brief review of its pathophysiology, clinical course, and diagnosis. Neurosurgery. 2007;60(Supp1 1):S35–S41.
2. Prasad, D., Schiff, D. Malignant spinal-cord compression. Lancet Oncol. 2005;6(1):15–24.
3. Husband, D.J. Malignant spinal cord compression: Prospective study of delays in referral and treatment. BMJ. 1998;317(7150):18–21.
4. White, B.D., et al. Diagnosis and management of patients at risk of or with metastatic spinal cord compression: Summary of NICE guidance. BMJ. 337, 2008. [a2538].
5. Portenoy, R.K., et al. Identification of epidural neoplasm. Radiography and bone scintigraphy in the symptomatic and asymptomatic spine. Cancer. 1989;64(11):2207–2213.
6. Hyman, R.A., et al. 0.6 T MR imaging of the cervical spine: Multislice and multiecho techniques. Am J Neuroradiol. 1985;6(2):229–236.
7. Loblaw, D.A., et al. Systematic review of the diagnosis and management of malignant extradural spinal cord compression: The Cancer Care Ontario Practice Guidelines Initiative’s Neuro-Oncology Disease Site Group. J Clin Oncol. 2005;23(9):2028–2037.
8. Sorensen, S., et al. Effect of high-dose dexamethasone in carcinomatous metastatic spinal cord compression treated with radiotherapy: A randomised trial. Eur J Cancer. 1994;30A(1):22–27.
9. Patchell, R.A., et al. Direct decompressive surgical resection in the treatment of spinal cord compression caused by metastatic cancer: A randomised trial. Lancet. 2005;366(9486):643–648.
10. al Deeb, S.M., et al. Acute transverse myelitis: A localized form of postinfectious encephalomyelitis. Brain. 1997;120(Pt 7):1115–1122.
11. Transverse Myelitis Consortium Working Group. Proposed diagnostic criteria and nosology of acute transverse myelitis. Neurology. 2002;59(4):499–505.
12. Harzheim, M., et al. Discriminatory features of acute transverse myelitis: A retrospective analysis of 45 patients. J Neurol Sci. 2004;217(2):217–223.
13. Misra, U.K., Kalita, J., Kumar, S. A clinical, MRI and neurophysiological study of acute transverse myelitis. J Neurol Sci. 1996;138(1–2):150–156.
14. de Seze, J., et al. Acute myelopathies: Clinical, laboratory and outcome profiles in 79 cases. Brain. 2001;124(Pt 8):1509–1521.
15. Croft, P. Neuromuscular syndromes associated with malignant disease. Br J Hosp Med. 1977;17(4):360–362. [356].
16. Rudnicki, S.A., Dalmau, J. Paraneoplastic syndromes of the peripheral nerves. Curr Opin Neurol. 2005;18(5):598–603.
17. Malinow, K., et al. Subacute sensory neuronopathy secondary to dorsal root ganglionitis in primary Sjögren’s syndrome. Ann Neurol. 1986;20(4):535–537.
18. Schaumburg, H., et al. Sensory neuropathy from pyridoxine abuse: A new megavitamin syndrome. N Engl J Med. 1983;309(8):445–448.
19. Jellinek, E.H., Kelly, R.E. Cerebellar syndrome in myxoedema. Lancet. 1960;2(7144):225–227.
20. Skillicorn, S.A. Presenile cerebellar ataxia in chronic alcoholics. Neurology. 1955;5(8):527–534.
21. Bariety, M., et al. [“Paraneoplastic” psychic and cerebellar syndrome reversible by radiotherapeutic treatment of its cause: “Anaplastic bronchial epithelioma”.]. Bull Mem Soc Med Hop Paris. 1960;76:650–661.
22. Richter, R. The hereditary nature of late cortical cerebellar atrophy. Trans Am Neurol Assoc. 73(73 Annual Meet), 1948. [85–78].
23. Davis, G.C., et al. Chronic parkinsonism secondary to intravenous injection of meperidine analogues. Psychiatry Res. 1979;1(3):249–254.
24. Messert, B., Baker, N.H. Syndrome of progressive spastic ataxia and apraxia associated with occult hydrocephalus. Neurology. 1966;16(5):440–452.
25. Dixon, G.R., et al. Use of cerebrospinal fluid flow rates measured by phase-contrast MR to predict outcome of ventriculoperitoneal shunting for idiopathic normal-pressure hydrocephalus. Mayo Clin Proc. 2002;77(6):509–514.
26. Panagiotopoulos, V., et al. The predictive value of external continuous lumbar drainage, with cerebrospinal fluid outflow controlled by medium pressure valve, in normal pressure hydrocephalus. Acta Neurochir (Wien). 2005;147(9):953–958. [discussion 958].
27. Pfisterer, W.K., et al. Continuous intraventricular pressure monitoring for diagnosis of normal-pressure hydrocephalus. Acta Neurochir (Wien). 2007;149(10):983–990. [discussion 990].
28. Wikkelso, C., et al. Normal pressure hydrocephalus. Predictive value of the cerebrospinal fluid tap-test. Acta Neurol Scand. 1986;73(6):566–573.
29. Bret, P., et al. [Clinical experience with the Sp[hy adjustable valve in the treatment of adult hydrocephalus: A series of 147 cases.]. Neurochirurgie. 1999;45(2):98–108. [discussion 108–109].
30. Wiest, R.G., Burgunder, J.M., Krauss, J.K. Chronic subdural haematomas and Parkinsonian syndromes. Acta Neurochir (Wien). 1999;141(7):753–757. [discussion 757–758].
31. Gan, J., et al. Possible Parkinson’s disease revealed by a pure head resting tremor. J Neurol Sci. 2009;279(1–2):121–123.
32. Schonecker, V.M. Ein eigentumliches syndrom in oralen Bereich bei Negaphenapplikation. Nervenarzt. 1957;28:35.
33. Tarsy, D., Baldessarini, R.J. Epidemiology of tardive dyskinesia: Is risk declining with modern antipsychotics? Mov Disord. 2006;21(5):589–598.
34. Kenney, C., Jankovic, J. Tetrabenazine in the treatment of hyperkinetic movement disorders. Expert Rev Neurother. 2006;6(1):7–17.
35. Sun, B., et al. Subthalamic nucleus stimulation for primary dystonia and tardive dystonia. Acta Neurochir Suppl. 2007;97(Pt 2):207–214.
36. Nisijima, K., Shioda, K., Iwamura, T. Neuroleptic malignant syndrome and serotonin syndrome. Prog Brain Res. 2007;162:81–104.
37. Jankovic, J. Tourette’s syndrome. N Engl J Med. 2001;345(16):1184–1192.
38. The Tourette Syndrome Classification Study Group. Definitions and classification of tic disorders. Arch Neurol. 1993;50(10):1013–1016.
39. Poston, K.L., Frucht, S.J. Movement disorder emergencies. J Neurol. 2008;255(Suppl 4):2–13.
40. Parkinson, J. An essay on the shaking palsy. London: Whittingham and Roland; 1817.
41. Polymeropoulos, M.H., et al. Mutation in the alpha-synuclein gene identified in families with Parkinson’s disease. Science. 1997;276(5321):2045–2047.
42. Miyasaki, J.M., et al. Practice parameter: Initiation of treatment for Parkinson’s disease: An evidence-based review: Report of the Quality Standards Subcommittee of the American Academy of Neurology. Neurology. 2002;58(1):11–17.
43. Parkinson Study Group. Pramipexole vs levodopa as initial treatment for Parkinson disease: A randomized controlled trial. JAMA. 2000;284(15):1931–1938.
44. Miyasaki, J.M., et al. Practice parameter: Evaluation and treatment of depression, psychosis, and dementia in Parkinson disease (an evidence-based review): Report of the Quality Standards Subcommittee of the American Academy of Neurology. Neurology. 2006;66(7):996–1002.
45. Roze, E., et al. Propriospinal myoclonus revisited: Clinical, neurophysiologic, and neuroradiologic findings. Neurology. 2009;72(15):1301–1309.
46. Venkatesan, A., Frucht, S. Movement disorders after resuscitation from cardiac arrest. Neurol Clin. 2006;24(1):123–132.
47. Taly, A.B., et al. Wilson disease: Description of 282 patients evaluated over 3 decades. Medicine (Baltimore). 2007;86(2):112–121.
48. Sharma, N., Standaert, D.G. Inherited movement disorders. Neurol Clin. 2002;20(3):759–778. [vii].
49. Jen, J.C., et al. Primary episodic ataxias: Diagnosis, pathogenesis and treatment. Brain. 2007;130(Pt 10):2484–2493.
50. Huntington, G. On chorea. The Medical and Surgical Reporter:. A Weekly Journal. 1872;26(15):317–321.
51. Gordon, N. Sydenham’s chorea, and its complications affecting the nervous system. Brain Dev. 2009;31(1):11–14.
52. Cox DW, Roberts E. Wilson’s disease. 2006. Gene Tests website. Available: http://www.ncbi.nlm.nih.gov/bookshelf/br.fcgi?book=gene&part=wilson#wilson (14 Dec 2009).
53. Critchley, M. Observations on essential (heredofamial) tremor. Brain. 1949;72(Pt 2):113–139.
54. Davis, C.H., Kunkle, E.C. Benign essential (heredofamilial) tremor. Trans Am Neurol Assoc. 1951;56:87–89.
55. Thanvi, B., Lo, N., Robinson, T. Essential tremor – the most common movement disorder in older people. Age Ageing. 2006;35(4):344–349.
56. Gulcher, J.R., et al. Mapping of a familial essential tremor gene, FET1, to chromosome 3q13. Nat Genet. 1997;17(1):84–87.
57. Higgins, J.J., Pho, L.T., Nee, L.E. A gene (ETM) for essential tremor maps to chromosome 2p22-p25. Mov Disord. 1997;12(6):859–864.
58. Zesiewicz, T.A., et al. Practice parameter: Therapies for essential tremor: Report of the Quality Standards Subcommittee of the American Academy of Neurology. Neurology. 2005;64(12):2008–2020.
59. Bhidayasiri, R. Differential diagnosis of common tremor syndromes. Postgrad Med J. 2005;81(962):756–762.
60. Nandi, D., Aziz, T.Z. Deep brain stimulation in the management of neuropathic pain and multiple sclerosis tremor. J Clin Neurophysiol. 2004;21(1):31–39.
61. Wishart, H.A., et al. Chronic deep brain stimulation for the treatment of tremor in multiple sclerosis: Review and case reports. J Neurol Neurosurg Psychiatry. 2003;74(10):1392–1397.
62. Gates, P.C. Orthostatic tremor (shaky legs syndrome). Clin Exp Neurol. 1993;30:66–71.
63. de Bie, R.M., Chen, R., Lang, A.E. Orthostatic tremor in progressive supranuclear palsy. Mov Disord. 2007;22(8):1192–1194.
64. Sanitate, S.S., Meerschaert, J.R. Orthostatic tremor: Delayed onset following head trauma. Arch Phys Med Rehabil. 1993;74(8):886–889.
65. Poersch, M. Orthostatic tremor: Combined treatment with primidone and clonazepam. Mov Disord. 1994;9(4):467.
66. Finkel, M.F. Pramipexole is a possible effective treatment for primary orthostatic tremor (shaky leg syndrome). Arch Neurol. 2000;57(10):1519–1520.
67. Guridi, J., et al. Successful thalamic deep brain stimulation for orthostatic tremor. Mov Disord. 2008;23(13):1808–1811.
68. Fahn, S. Posthypoxic action myoclonus: Literature review update. Adv Neurol. 1986;43:157–169.
69. Lance, J.W. Action myoclonus, Ramsay Hunt syndrome, and other cerebellar myoclonic syndromes. Adv Neurol. 1986;43:33–55.
70. Tomita, H., et al. Paroxysmal kinesigenic choreoathetosis locus maps to chromosome 16p11.2-q12.1. Am J Hum Genet. 1999;65(6):1688–1697.
71. Wein, T., et al. Exquisite sensitivity of paroxysmal kinesigenic choreoathetosis to carbamazepine. Neurology. 1996;47(4):1104–1106.
72. Dewey, R.B., Jr., Jankovic, J. Hemiballism-hemichorea: Clinical and pharmacologic findings in 21 patients. Arch Neurol. 1989;46(8):862–867.
73. Sternbach, H. The serotonin syndrome. Am J Psychiatry. 1991;148(6):705–713.
74. Lappin, R.I., Auchincloss, E.L. Treatment of the serotonin syndrome with cyproheptadine. N Engl J Med. 1994;331(15):1021–1022.
75. Graudins, A., Stearman, A., Chan, B. Treatment of the serotonin syndrome with cyproheptadine. J Emerg Med. 1998;16(4):615–619.