[level-membership-for-endocrinology-diabetes-and-metabolism-category]CHAPTER 8
Proteinuria
CHAPTER OUTLINE
PROTEIN CONSERVATION BY THE KIDNEYS
The theory of molecular sieving
Tubular reabsorption of proteins
NORMAL URINARY PROTEIN CONTENT
Determinants of urine protein excretion
Proteinuria in staging and prognosis of chronic kidney disease
Glomerular proteinuria and nephrotic syndrome
Proteinuria of prerenal origin
MICROALBUMINURIA AS A MARKER OF RISK
Microalbuminuria and risk of diabetic complications
Microalbuminuria as a risk factor in other inflammatory processes
INTRODUCTION
For over 150 years, proteinuria has been recognized as one of the cardinal signs of renal disease and it continues to be used for the diagnosis and management of patients with nephropathy. However, the use of sensitive immunoassay methods to measure urine albumin has demonstrated pathologically significant proteinuria well below the detection limit of chemical methods. Since the discovery, several decades ago, that low-level albuminuria (microalbuminuria) identifies diabetic patients with incipient nephropathy, further large-scale studies have revealed that microalbuminuria is also a risk factor for macro- and microvascular disease, in both diabetic and non-diabetic populations. Studies over the last ten years have confirmed that microalbuminuria is not only associated with cardiovascular disease but also with any acute inflammatory condition, and is a reflection of systemic vascular endothelial dysfunction. Of particular importance, is the finding that, in some conditions, microalbuminuria is reversible with interventions that protect the vascular system. Thus modern assessment of proteinuria across the full pathological range has a role not just in the diagnosis and management of primary renal disease, but also as a marker of endothelial dysfunction in a variety of non-renal conditions.
PROTEIN CONSERVATION BY THE KIDNEYS
The kidneys receive approximately 25% of the resting cardiac output, which represents approximately 1.2 L/min of blood or 650 mL/min of plasma. The kidneys’ capacity to conserve protein can be judged from a simple calculation. Every 24 h, approximately 930 L of plasma containing about 70 g/L of protein pass through the kidneys, representing 65 kg of protein, of which < 100 mg (0.00015%) appear in the urine.
The filtration process is dependent on adequate renal blood flow, which is preserved by an autoregulatory system, despite variations in blood pressure (see p. 127). This mechanism allows vasodilatation as perfusion pressure falls and vasoconstriction as pressure rises. The mediators of this process include prostaglandins, kinins and atrial peptides (vasodilators), and angiotensin II, α-adrenergic hormones, thromboxane A2, noradrenaline (norepinephrine) and vasopressin (vasoconstrictors). In addition, renal arterioles respond within seconds to changes in vessel wall tension; thus, when renal perfusion pressure rises, vessel wall tone increases and, conversely, when renal perfusion pressure falls abruptly, there is a compensatory decrease in vessel wall tone. This phenomenon is called the myogenic reflex and helps to maintain a constant renal blood flow across a range of perfusion pressures.
A minimum intraglomerular pressure, derived from the pumping action of the heart, is required to overcome the two main opposing forces to filtration: the colloidal oncotic pressure and the hydrostatic pressure in the Bowman space. When the renal perfusion pressure falls below 50–60 mmHg, further vasodilatation does not occur and renal blood flow declines in proportion to the reduction in renal perfusion pressure. These mechanisms maintain renal blood flow, and hence glomerular filtration, independently of the normal fluctuations of blood pressure. However, recent studies suggest that chronic hypertension impairs the renal autoregulatory mechanism, which may contribute to hypertension-associated renal damage and proteinuria.
The glomerular capillary wall
The glomerular membrane consists of a modified capillary wall comprising endothelium, an acellular basement membrane and an outer specialized epithelial cell layer (Fig. 8.1). The endothelial cells are thin and fenestrated with 50–100 nm pores. The basement membrane, comprising the lamina rara interna, lamina densa and lamina rara externa, is around 300–350 nm thick and is best considered as a gel-like structure containing 3–5 nm long fibrils but no detectable pores. The numerous foot processes of the epithelial cells (also called podocytes) interdigitate and envelop the outer surface of the glomerular membrane. The foot processes are separated by slit diaphragms about 55 nm in width, which form the final barrier to plasma proteins.
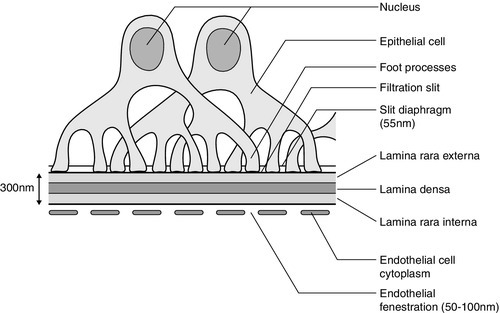
The whole of the glomerular membrane carries a fixed net negative charge, which is partly owing to a glycosialoprotein coat covering both endothelium and epithelium. The charge increases in density from the lamina rara interna towards the lamina rara externa, with the greatest density at the slit diaphragms of the epithelium.
The glomerular capillaries act as high pressure filters, allowing water and low molecular weight solutes to pass through freely and almost completely retaining plasma proteins. The concentration gradient of proteins across the glomerular membrane produces the colloidal oncotic pressure that must be overcome for filtration to take place.
The theory of molecular sieving
The glomerular membrane selectively allows passage of water and low molecular weight solutes into the tubule and restricts passage of larger molecular weight plasma proteins, based on a combination of molecular size, shape and charge. The pore size of the fenestrae of endothelial cells (50–100 nm) is too great to provide a major restriction to the passage of many proteins, which pass through to the glomerular membrane. Like the glomerular membrane, most plasma proteins carry a net negative charge, resulting in their electrochemical retention by the kidneys. The capillary side of the glomerular basement membrane (lamina rara interna) has a charge density of 35–45 mEq/L, so that electrochemical effects alone reduce the concentration of albumin to 5–10% of that in plasma. Thus, the lamina rara interna of the basement membrane is the first impediment to confront charged molecules, with the lamina densa providing the most effective size barrier to macromolecules. Some macromolecules accumulate at the slit diaphragms of the epithelium where the net negative charge is greatest, and there is evidence that some molecules are pinocytosed by the podocytes.
Based on their molecular radius, shape and charge, different proteins penetrate the glomerular membrane to a variable extent, for example, albumin (radius 3.6 nm, isoelectric point 4.7) is restricted at the lamina rara interna, while lactoperoxidase (radius 3.8 nm, isoelectric point 8.0) may reach the slit pores of the epithelial cells.
Despite the protein-retaining properties of the glomerular membrane, some protein does pass into the proximal tubular fluid. In healthy adults, the albumin content of glomerular filtrate is probably about 15 mg/L which, with a glomerular filtration rate (GFR) of 160 L/ 24 h, results in 2–3 g of albumin being presented to the renal tubules each day, most of which is reabsorbed.
Tubular reabsorption of proteins
The amount of protein reaching the proximal tubules is a function of the protein’s ability to cross the glomerular membrane and its plasma concentration. After reabsorption, proteins are catabolized in tubular cells and the amino acids, bound vitamins and trace metals returned to the plasma pool. For example, under normal circumstances the kidneys are responsible for about 10% of albumin catabolism, but this figure can rise to 60% when there is increased glomerular clearance of albumin such as occurs in nephrotic syndrome.
Normally, only a very small proportion of plasma proteins reaches the urine. This is primarily owing to retention of large molecular weight proteins by the glomeruli, and almost complete proximal tubular reabsorption and catabolism of any filtered lower molecular weight proteins that have passed through the glomerular membrane more easily. Thus, the renal clearance of plasma proteins represents a combination of these processes. The relative contributions of glomerular retention and tubular reabsorption to protein clearance by the kidney depend on molecular weight (Table 8.1).
TABLE 8.1
Relative clearances of plasma proteins
| Protein | Molecular weight (Da) | Clearance (%GFR) |
| IgG | 150 000 | 0.01 |
| Albumin | 69 000 | 0.02 |
| Amylase | 48 000 | 3 |
| Myoglobin | 17 800 | 75 |
| Lysozyme | 14 500 | 80 |
The likely mechanism of proximal tubular protein reabsorption is endocytosis at the tubular epithelial apical cell membrane mediated by two receptors (megalin and cubilin) and the cooperating protein amnionless. Mutations in one or other of these components are responsible for the tubular proteinuria of Imerslund–Grasbeck syndrome (selective vitamin B12 malabsorption) and Donnai–Barrow syndrome (a multisystem disorder affecting many organs that normally express megalin). Following reabsorption, proteins are transferred to lysosomes for degradation while the receptors are recycled from endosomes to the apical cell membrane. Mutations in the lysosomal and endosomal components are responsible for the tubular proteinuria in Dent disease and cystinosis; the molecular mechanism in Lowe syndrome is yet to be resolved (see Chapter 9).
Megalin and/or cubilin bind albumin and low molecular weight proteins but there is some evidence that, of the filtered proteins reaching the proximal tubule, those with lower molecular weights are reabsorbed by different mechanisms from those with higher molecular weights. For example, although increased amounts of low molecular weight proteins are excreted in tubular disease, such large increases are not seen in the urine of patients with massive glomerular proteinuria, when the reabsorptive mechanisms for large molecular weight proteins such as albumin must be saturated. Conversely, experimental induction of massive low molecular weight proteinuria through saturation of reabsorption with other low molecular weight proteins does not induce a similar increase in urine albumin.
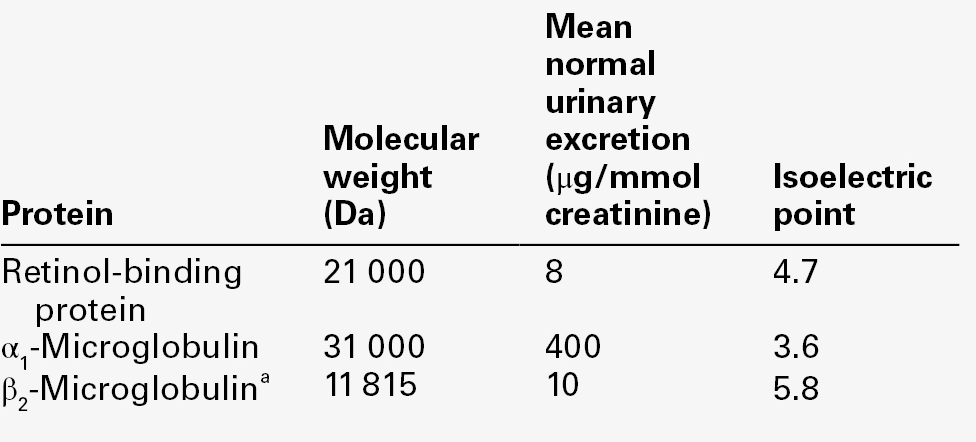
Low molecular weight proteins, such as α1-microglobulin and retinol-binding protein, that are readily filtered at the glomerulus and normally > 99.9% reabsorbed, can be used as sensitive markers of proximal tubular function, since even subtle tubular damage is associated with increased urinary excretion.
Glomerular diseases causing heavy proteinuria induce the release of a multitude of inflammatory and fibrogenic mediators, all of which cause tubulointerstitial fibrosis and renal scarring that contribute to renal impairment. Recent studies suggest that large proteins, such as albumin, which pass through the diseased glomerular membrane, cause tubular cell injury when they reach the proximal tubules. Reabsorption of excessive amounts of these proteins leads to overloading of lysosomes and activation of proximal tubular cells, which produce matrix metalloproteins, cytokines, leukocyte chemoattractants and vasoactive mediators, leading to interstitial inflammation and scarring. The mechanism of injury has been most studied for albumin and it appears likely that compounds attached to the protein, such as fatty acids, are major culprits. In minimal change glomerular disease with heavy proteinuria that is not associated with scarring, the albumin-fatty acid composition appears to be different from that in other glomerulopathies.
Tubular secretion of proteins
Proteins that are too large to be filtered by the normal glomerulus appear in urine either as a result of tubular secretion or from desquamation of tubular epithelial cells as part of the normal cellular turnover. Some large molecular weight proteins may also enter the urine during its postrenal passage along the urinary tract. Measurable activities of enzymes such as N-acetyl β-D-glucosaminidase (150 000 Da), γ-glutamyl transferase (120 000 Da) and lactate dehydrogenase (144 000 Da) can be found in normal urine.
The major urine protein exclusively of renal origin is uromodulin (previously known as Tamm–Horsfall glycoprotein), a heavily glycosylated protein secreted from the thick ascending limb of the loops of Henle. It has a molecular weight of 70 000 Da but is normally present in urine as a large non-covalently linked polymer. Since its isoelectric point is 3.3, it tends to precipitate as a gel during urinary acidification, forming casts by trapping whatever is in the vicinity such as albumin, red blood cells, tubular cells or cellular debris. Numerous mutations in the uromodulin gene have been identified in association with familial juvenile hyperuricaemic nephropathy and medullary cystic kidney disease type 2. These are probably different phenotypes of the same disease and are known collectively as uromodulin storage disease, owing to accumulation of the misfolded mutated protein in cells of the thick ascending limb of the loops of Henle. They are rare, primary tubulointerstitial disorders with autosomal dominant inheritance characterized by chronic progressive kidney disease, hyperuricaemia and gout, with minimal proteinuria.
The normal biological function of uromodulin remains unclear but it probably has a protective role in trapping potentially damaging material in the urinary space, allowing excretion as casts. Microscopic differentiation of the type of casts present in urine can give useful information about pathological processes within the kidney (see Chapter 7). Experimental studies suggest that uromodulin may have a role in protecting against stone formation. Its capacity to bind type-1 fimbriated E. coli has led to the suggestion that its excretion has evolved as a defence against urinary tract infections.
NORMAL URINARY PROTEIN CONTENT
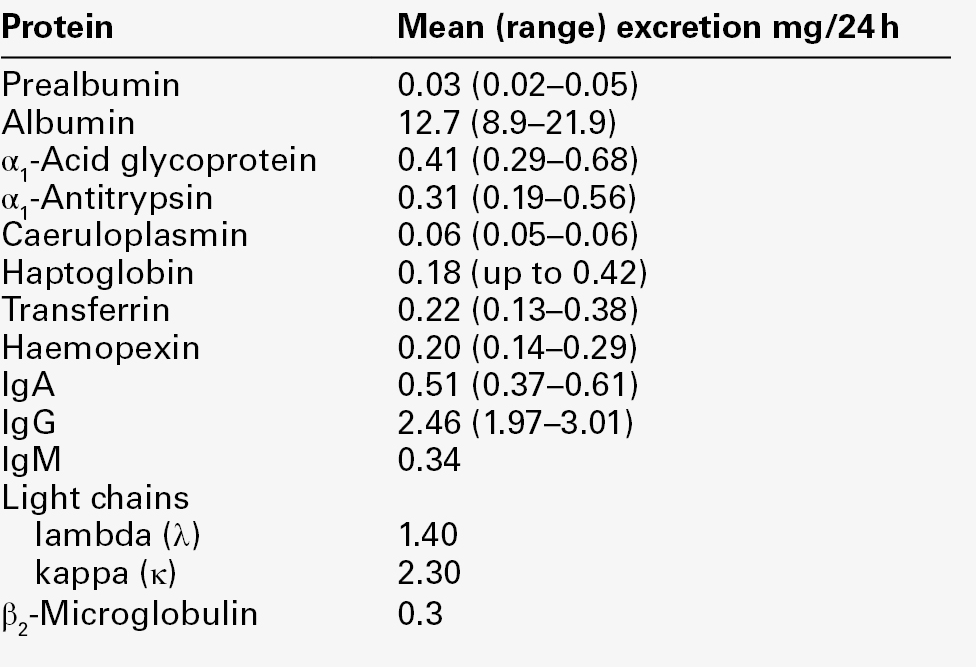
A normal adult excretes about 300 mg/24 h of non-dialysable material, of which < 140 mg/24 h is protein, but published reference ranges for total urinary protein excretion vary considerably with the analytical method used. Plasma proteins represent only some 25 mg/24 h of total urinary protein (Table 8.2), of which about half is albumin: the remaining proteins are of renal origin, uromodulin being the major contributor (70 mg/24 h). For these reasons, and because low-level albuminuria (microalbuminuria) has prognostic value both for renal and non-renal diseases, urine albumin immunoassays should be used to assess glomerular proteinuria when protein excretion rates are low.
Determinants of urine protein excretion
Age, sex and diurnal variation
In neonates, albumin excretion tends to be higher than in older children and adults: this has been attributed to greater permeability of the neonatal glomerulus. Total urine protein tends to fall after birth and rises with increasing age, reaching adult excretion rates by puberty. However, the urine protein/creatinine concentration ratio remains constant from 3 to 15 years of age, since urine creatinine excretion rises with increasing body mass. In children aged 4–16 years, albumin excretion rate, corrected for body surface area, rises with age and is slightly higher in females. Daytime excretion rates are higher than during the night and the sex difference disappears in overnight collections. There appears to be no sex difference in urine albumin excretion in adults, but, when expressed as a ratio to creatinine, the reference range is slightly higher in females owing to their lower muscle mass and hence lower creatinine excretion.
Posture
In both adults and children, ambulatory urine protein excretion is higher than it is overnight or during recumbency, with two- to ten-fold differences being reported for urinary albumin. Orthostatic proteinuria in otherwise healthy subjects has been the subject of controversy for some time. The discussion has been complicated by the variety and differing sensitivities of the protein assays used.
Proteinuria of < 1 g/24 h has been described in 0.6–9% of healthy young adults, in the absence of urinary red cells, white cells or casts, and can be divided into ‘constant’ and ‘postural’ based on its persistence after recumbency. Renal biopsies of patients with postural proteinuria reveal that 8% have unequivocal evidence of well-defined disease and 45% have subtle alterations in glomerular structure. However, more recent studies, using non-invasive Doppler ultrasound to compare recumbent and orthostatic blood flow in the left renal veins of young people with orthostatic proteinuria, have revealed that in > 50% of patients there is reduced blood flow to the left renal vein during standing owing to entrapment of the left renal vein.
The medical management of isolated or postural proteinuria in an otherwise healthy patient tends to be conservative, with annual assessment of proteinuria and renal function; biopsy is reserved for the rare patient who has evidence of progressive renal impairment.
Exercise and diet
Exercise-induced proteinuria was discovered over a century ago in soldiers after marches or drills. Five- to 100-fold increases in the excretion of proteins such as albumin, transferrin and immunoglobulins have been observed following 26-mile marathon runs, with smaller increases after less strenuous activities. The pattern of exercise-induced proteinuria is generally glomerular, although mixed glomerular and tubular proteinuria has also been described, which persists for over 3 h after exercise. The reason for exercise-induced proteinuria is unclear, but some degree of renal ischaemia owing to redistribution of blood during exercise has been suggested as a possible mechanism.
A large protein meal is associated with an increased urine albumin excretion, which appears to be secondary to an associated increase in GFR.
The lowest and most reproducible estimates of urine protein excretion are obtained from an early morning urine specimen after overnight recumbency.
Pregnancy
During normal pregnancy, the urinary albumin excretion rate generally remains within the non-pregnant range, although there is some evidence for a small increase in albumin excretion during the third trimester, which may be related to increased glomerular permeability and/or GFR. Total urine protein excretion increases owing to decreased renal tubular protein reabsorption.
Hypertension in pregnancy is associated with significant maternal and fetal morbidity and mortality. The reliable detection of significant proteinuria is most important in women with new-onset hypertension during pregnancy because it distinguishes between those pregnancies with pre-eclampsia and those with gestational hypertension, the former often requiring admission to hospital owing to the severity of potential complications. In the UK, NICE guidelines for the routine antenatal care of healthy pregnant women recommend blood pressure and urine protein measurement at each antenatal visit; 660 000 women each year will have at least 7–10 such checks.
Gestational hypertension is defined as new hypertension occurring after 20 weeks of pregnancy, but without significant proteinuria. In this group, routine urine protein measurement may be performed using an automated reagent-strip reading device (more reliable than a manual reading) or by a laboratory method. If a reagent strip reading is 1 + or greater, the proteinuria should be quantitated by a laboratory measure in a spot or 24 h urine sample. Women admitted with pre-eclampsia (new hypertension presenting after 20 weeks with significant proteinuria) do not need to have repeated measures of urine protein since there is no strong evidence linking the degree of proteinuria with adverse outcome. Significant proteinuria is defined as > 300 mg/24 h or > 30 mg/mmol creatinine in a random sample. There are insufficient studies of urine albumin excretion to be able to define cut-offs equivalent to those defining significant proteinuria in gestational hypertension or pre-eclampsia.
PROTEINURIA IN KIDNEY DISEASE
Richard Bright, in 1836, is generally credited with noting the association between proteinuria and kidney disease. Proteinuria remains the most frequent clinical finding and quantitation of proteinuria is valuable for diagnosing, monitoring and assessing the prognosis of kidney disease. Normally, total urine protein excretion is < 150 mg/24 h in adults and < 140 mg/m2/24 h in children, depending on the methods employed: normal concentrations are often undetectable by chemical methods. However, sensitive immunoassay of specific proteins has extended the detection limits to urine protein concentrations within the reference range. Measurements of low concentrations of specific proteins such as albumin, predominantly reflecting glomerular function, and α1-microglobulin or retinol-binding protein, reflecting tubular reabsorptive function, are now used as very sensitive and early markers of primary renal disease (e.g. glomerulonephritis) and secondary renal disease (e.g. in diabetes mellitus or hypertension). Thus, proteinuria can now be regarded as a continuum that extends from the measurable amount of protein normally excreted in urine up to the 1000-fold increases found in nephrotic syndrome.
Conventionally, proteinuria has been classified into glomerular proteinuria; tubular proteinuria; nephrogenic proteinuria (e.g. uromodulin, basement membrane and tubular proteins); proteinuria of prerenal origin (e.g. overflow proteinurias such as light chain disease, myoglobinuria, haemoglobinuria, lysozyme in leukaemia and amylase in pancreatitis), and postrenal proteinuria owing to obstruction of the urinary tract or inflammation such as occurs in urinary tract infection. This is a convenient way of differentiating the principal sites of the renal abnormality, but is an over-simplification because, for example, glomerular disease leading to large amounts of plasma proteins being presented to the renal tubules leads to inflammatory changes within the tubules and renal scarring. Nor does this classification lend itself easily to the multifactorial causes of proteinuria in some disorders, e.g. HIV-associated glomerular and tubulointerstitial disease that may be modulated by co-infection with other viruses and drug-induced interstitial nephritis.
Proteinuria in staging and prognosis of chronic kidney disease
Numerous epidemiological studies have demonstrated the association of proteinuria with poorer prognosis in people in the general population and across all stages of chronic kidney disease (CKD): any renal disease is more likely to progress, there is an increased risk of developing acute kidney injury, and both all-cause and cardiovascular mortality are increased. These outcomes hold true, whether proteinuria is assessed by dipstick testing or by formal laboratory measurement of either total protein or albumin, in timed urine collections or in random samples. The quantitation of proteinuria (in the absence of a symptomatic urinary tract infection and preferably using the first morning urine) is an essential component of CKD staging, where the suffix ‘p’ is used to denote its presence. The decision limit is an albumin/creatinine ratio (ACR) > 30 mg/mmol (~ 300 mg/24 h) or urine protein/creatinine ratio (PCR) > 50 mg/mmol (~ 0.5 g/24 h), although the continuum of risk, albeit lower, extends into the reference range. The presence of proteinuria in CKD is sufficient indication to initiate blockade of the renin–angiotensin–aldosterone system (RAS) with angiotensin-converting-enzyme inhibitors (ACEI) or angiotensin-II receptor blockers (ARB). At higher excretions (ACR > 70 mg/mmol or PCR > 100 mg/mmol), RAS blockade should be titrated to the highest tolerable levels and referral of the patient to specialist care considered.
Glomerular proteinuria and nephrotic syndrome
In the normal adult, the renal tubules reabsorb about 2–3 g of filtered albumin every 24 h. Thus, even total failure of this process cannot explain albuminuria of > 3.0 g/24 h; such losses are usually secondary to increased glomerular permeability associated with glomerular damage.
Nephrotic syndrome can be defined as proteinuria severe enough to cause hypoalbuminaemia and oedema. The degree of proteinuria varies but is generally > 3.5 g/24 h and is accompanied by a plasma albumin < 25 g/L. However, it should be remembered that the amount of protein in the urine may decrease as the plasma protein concentration or the GFR falls. Causes of nephrotic syndrome are listed in Table 8.3. In addition to the nephrotic syndrome, glomerular proteinuria is a feature of several other syndromes of nephron injury, and the severity of proteinuria taken together with other clinical findings can allow useful diagnostic classification (Table 8.4).
TABLE 8.3
Causes of nephrotic syndrome
| Children (%) | Adults (%) | |
| Primary renal disease | ||
| Minimal change (steroid responsive) disease | 80 | 25 |
| Focal segmental glomerulosclerosis | 7 | 9 |
| Membranous nephropathy | 1 | 22 |
| Membranoproliferative glomerulonephritis | 10 | 26 |
| Glomerular disease in systemic conditions | ||
| Amyloidosis | – | 7 |
| Diabetic nephropathy | – | 3 |
| Systemic lupus erythematosus | – | 8 |
| Henoch–Schönlein purpura | 2 | < 1 |

Mechanisms underlying glomerular proteinuria
Glomerular injury can occur as a result of either primary or secondary kidney disease and there is no single pathogenetic pathway that will embrace all the possible mechanisms. The term glomerulonephritis is generally reserved for immunologically mediated diseases and excludes other conditions associated with glomerular damage such as diabetes mellitus or amyloidosis.
Glomerulonephritis can be subdivided immunologically into conditions mediated by antibodies against antigens that are either extrinsic or intrinsic to the kidney. Extrinsic antigens include microorganisms causing infections such as bacterial endocarditis or streptococcal infections, and DNA, as in systemic lupus erythematosus (SLE). Antibodies may also develop to an intrinsic structural component, such as the glomerular basement membrane, leading to immune complex formation. Whatever the original mechanism, antigen–antibody complexes become trapped in the glomerulus, activating both the classical and alternative complement pathways and leading to the release of anaphylatoxic components such as C3a and C5a. Anaphylatoxins, together with locally released kinins, prostaglandins and leukotrienes, attract polymorphonuclear neutrophils to the basement membrane, where they release lysosomal enzymes, leading to membrane disruption and glomerular proteinuria.
Examination of renal biopsies, by light and electron microscopy, reveals a gradation of morphological changes with increasing glomerular injury. Loss of anionic charge associated with fusion of the epithelial foot processes can produce a massive but selective proteinuria, while changes in the basement membrane tend to be associated with increasingly non-selective proteinuria (see later).
Minimal change disease
Light microscopy of renal biopsies shows little or no abnormality in this condition, and no immunoglobulin or complement components are seen on immunofluorescence. Electron microscopy shows fusion of epithelial cell foot processes, and the mechanism of proteinuria probably involves loss of the fixed negative charge on the glomerular basement membrane. Although the pathogenesis of minimal change disease has not been completely clarified, clinical and experimental observations suggest that T cell dysfunction plays a role.
In the developed world, minimal change disease, frequently of unknown aetiology, accounts for 90% of nephrotic syndrome in 2–6-year-olds and about 20% in adults. In children, there is massive selective proteinuria, which is predominantly albumin with little or no immunoglobulin. This form of nephrotic syndrome usually responds to a short course of prednisolone (steroid-responsive nephrotic syndrome) and a renal biopsy is unnecessary to confirm the diagnosis. The absence of serological evidence for complement activation gives additional confirmation. In adults, the diagnosis is more difficult and a renal biopsy is generally needed before treatment is started. Minimal change disease has a good long-term prognosis with sustained remission and preserved renal function as almost all patients are responsive to treatment.
Membranous nephropathy
This is the most frequent cause of nephrotic syndrome in adults. The common variant is idiopathic membranous nephropathy, in which 80% of patients have plasma antibodies to the phospholipase A2 receptor. These antibodies are rarely found in the secondary forms associated with SLE, hepatitis B virus and cancer. Light microscopy shows thickening of the glomerular basement membrane that is more marked in the later stages of the disease. Immunofluorescence microscopy always shows IgG deposits, but when other immunoglobulin classes (e.g. IgA, IgM) are present, the condition is more likely to be lupus nephritis. Progressive thickening of the basement membrane, demonstrated by electron microscopy, is the most frequent morphological finding in nephrotic syndrome in adults.
Over 80% of patients present with massive proteinuria that is only moderately or poorly selective, while the remaining 20% have asymptomatic proteinuria or microscopic haematuria. The course of the disease is variable, with about one-third of patients going into spontaneous remission and another third progressing to established renal failure. Risk factors for a poor prognosis include severe proteinuria, hypertension, older age, male gender and reduced GFR. Currently, treatment is aimed at lowering blood pressure to < 130/80 mmHg with an ACEI or ARB, which usually reduces proteinuria over six months. If proteinuria persists, immunosuppressive treatment is generally started; steroids alone are often ineffective but there remains debate over which additional agents are most effective: these include ciclosporin, chlorambucil and cyclophosphamide.
Membranoproliferative glomerulonephritis
This is characterized by mesangial proliferation and expansion and is not a specific disease but a pattern of injury. As with all glomerulonephritides, many different diseases can produce this pattern of immunologically mediated glomerular damage, including bacterial endocarditis, group A streptococcal infections, hepatitis C virus, monoclonal immunoglobulin deposition disease and SLE. The histological pattern tends to be similar and immunofluorescence microscopy frequently shows IgA, IgG and C3 deposits; circulating complement concentrations are frequently reduced with high concentrations of immune complexes present in some conditions.
Patients with membranoproliferative glomerulonephritis have proteinuria and microscopic haematuria; approximately 50% of those with nephrotic syndrome present with an unselective proteinuria. Management involves identification and treatment of the underlying cause and the use of immunosuppressive agents.
Focal segmental glomerulosclerosis
Hyaline material is deposited in the subendothelial spaces of affected capillary loops of some, but not all, glomeruli, which is why the terms focal and segmental are used. Patients present with mild proteinuria, recurrent haematuria or nephrotic syndrome. The aetiology is not known, although there is some evidence for the presence of a soluble permeability factor and some patients have SLE. Currently, adults are treated with corticosteroids for at least 4–6 months. Efficacy of treatment is judged by monitoring proteinuria; 20–30% will respond with a decrease or cessation of proteinuria. The prognosis in these patients is good, although they may suffer a relapse. Patients whose proteinuria fails to respond to steroids generally go on to develop CKD. Optimal management of this group is currently controversial, but regimens include use of lower-dose corticosteroids with the addition of ciclosporin for 12 months or more.
IgA nephropathy and Henoch–Schönlein purpura
IgA nephropathy is the commonest primary glomerular disease in the developed world; Henoch–Schönlein purpura appears to be a systemic form of the disease occurring primarily in children. A deficiency in glycosylation enzymes leads to the production of galactose-deficient IgA1. This leaves novel glycan residues exposed, to which autoantibodies develop, leading to immune complex deposition. Nephrotic syndrome is uncommon in IgA nephropathy and proteinuria rarely exceeds 5 g/24 h. Treatment involves immunosuppression and antihypertensive therapy with an ACEI or ARB to reduce proteinuria. In Henoch–Schönlein purpura, 50% of children have renal involvement, of whom 20% have nephrotic range proteinuria and associated poorer prognosis.
Urine protein selectivity and classification of glomerulonephritis
The concept of ‘selectivity’ was based on the assumption that there may be differential filtration of large molecular weight proteins among patients with glomerular disease. Protein selectivity is based on a comparison of the relative clearance of IgG (150 000 Da) and transferrin (69 000 Da) calculated as follows:

where:
[IgG]u = urine IgG concentration
[Trans]p = plasma transferrin concentration
[IgG]p = plasma IgG concentration
[Trans]u = urine transferrin concentration.
Where the selectivity index is > 0.5, the proteinuria is said to be ‘non-selective’, 0.3–0.5 ‘moderately selective’ and < 0.2 ‘highly selective’.
Patients with minimal change disease usually show highly selective proteinuria, while patients with membranous nephropathy and membranoproliferative glomerulonephritis do not. Patients with highly selective proteinuria tend to respond well to steroid therapy in contrast to those with non-selective proteinuria, although this is too variable to be useful to determine treatment.
In correlating glomerular disease severity with the type and magnitude of proteinuria, the assumption is made that all nephrons behave in a similar way. However, histological evidence suggests that glomerular disease can be focal with some glomeruli apparently being spared. But if only one nephron out of every 10 000 allowed unrestricted passage of large molecular weight plasma proteins, this would result in an increase in excretion of large molecular weight proteins from 18 to 800 mg/24 h, resulting in an unselective proteinuria, apparently indicative of more serious disease.
In addition to glomerular filtration, renal clearances of plasma proteins also reflect tubular reabsorption. If the urine protein content were to represent the overall glomerular permeability, then it must be assumed that tubular reabsorption is entirely unselective. However, small changes in the relative proportion of filtered protein reabsorbed by the renal tubules can have a large effect on their urine concentrations. Despite these limitations, some recent studies have shown that in membranous nephropathy and focal segmental glomerulosclerosis, the selectivity index or IgG excretion predicts remission better than urine total protein. Similarly, progression to CKD is better predicted both by selectivity index or IgG excretion, and the tubular component of proteinuria (e.g. α1-microglobulin or retinol-binding protein) than by the total 24 h urine protein excretion. However, protein selectivity studies and monitoring of tubular proteinuria are not widely used in the routine diagnosis and management of patients with glomerular disorders.
Pathophysiological consequences of glomerular proteinuria
Hypoalbuminaemia
The severe hypoalbuminaemia found in nephrotic syndrome is not simply secondary to urinary losses exceeding hepatic synthesis, because in adults the maximum hepatic synthetic capacity for albumin is around 14 g/24 h, which in theory should replace urinary losses in all but the most severely proteinuric patients. In the nephrotic state, there is a decrease in the circulating albumin pool and hepatic synthesis is generally normal or increased. The loss of the fixed anionic charge on the glomerular membrane, which appears to be fundamental to the glomerular proteinuria of nephrotic syndrome, may also extend to the endothelium of all capillaries, leading to a generalized increase in capillary permeability to albumin.
Oedema and salt and water retention
Until recently, the explanation for the gross interstitial oedema associated with nephrotic syndrome was attributed solely to hypoalbuminaemia, the consequent reduction in plasma colloidal osmotic pressure being thought to lead to an increase in fluid loss from capillaries at their arteriolar ends and a reduction in tissue fluid reabsorption distally, as predicted from the operation of Starling forces. The resultant hypovolaemia was thought to stimulate the renin–aldosterone system and vasopressin release, leading to sodium and water retention. However, recent work suggests the explanation is more complex.
Normally, about 60% of total body albumin is in the interstitial space and 40% in the vascular space. Every hour, about 8 g of albumin cycle between these two fluid compartments. The gradual loss of plasma albumin from the circulation in nephrotic syndrome leads to a fall in plasma oncotic pressure (the osmotic pressure due to proteins), which in turn is accompanied by a decrease in tissue albumin and parallel fall in interstitial oncotic pressure. The balancing of falling oncotic pressure on either side of the capillary membrane protects against a dramatic fall in blood volume as a result of movement of fluid from the hypo-oncotic vascular space into the interstitium. However, the capacity of this protective mechanism is limited, and a decrease in blood volume can be expected if plasma oncotic pressure (normally 22–28 mmHg) falls below about 10 mmHg, or if the protein loss is very sudden. This is probably because the interstitial oncotic pressure is also maintained by the glycosaminoglycans matrix.
Thus, patients with nephrotic syndrome are not usually hypovolaemic and renal perfusion is generally normal. Despite this, there is intense renal sodium and water retention, which is not mediated by the renin–aldosterone system, and which exacerbates the interstitial oedema. The mechanism is not well understood, but may involve increased sodium reabsorption by epithelial sodium channels in the collecting ducts and decreased sensitivity to natriuretic peptides. In most patients, the oedema is managed by treatment with oral loop diuretics, most often furosemide. If this fails, treatment includes salt restriction, intravenous furosemide, additional thiazide or potassium-sparing diuretics and, occasionally, in severe cases, intravenous albumin.
Abnormalities of other plasma proteins
Patients with nephrotic syndrome have changes in the concentrations of circulating clotting factors, which may be responsible for an increased risk of thrombus formation including renal vein thrombosis, inferior vena cava thrombosis and pulmonary emboli. Urinary loss of the natural anticoagulant antithrombin III is probably involved, together with increased plasma concentrations of fibrinogen and factors V, VII, VIII and X, the reasons for which are unclear.
Urinary loss of vitamin D and vitamin D-binding globulin may lead to vitamin D deficiency with low plasma ionized calcium, secondary hyperparathyroidism and renal osteodystrophy. Some national guidelines recommend vitamin D supplementation, especially in children with nephrotic syndrome. Hypochromic microcytic anaemia, resistant to iron supplements, can occur as a result of urinary loss of transferrin. Patients with nephrotic syndrome may also lose immunoglobulins and complement components, with attendant increased risk of infections such as pneumococcal pneumonia and peritonitis.
Hyperlipidaemia
Many patients with nephrotic syndrome have massive increases in plasma cholesterol and triglyceride concentrations, which correlate inversely with the plasma concentration of albumin. Plasma very low density lipoprotein (VLDL) and low density lipoprotein (LDL) concentrations are increased, partly owing to reduced clearance and partly to increased hepatic synthesis, while high density lipoprotein (HDL) is reduced because of increased urinary loss. Very low density lipoprotein and LDL are apoB-containing lipoproteins, which are potentially atherogenic and have been linked to the pathogenetic processes that result in progressive glomerular and interstitial renal disease and increased cardiovascular risk. The activities of lecithin-cholesterol acyltransferase (LCAT) and the lipoprotein lipases are reduced in patients with nephrotic syndrome, yet increased urinary losses of these enzymes have not been demonstrated and the underlying mechanism for their reduced activity is not clear. Reduced LCAT activity may also reduce HDL production and hence, impair the patient’s ability to mobilize lipid from endothelium and peripheral tissues.
Hepatic synthesis of VLDL and LDL seems to be stimulated by the reduction in plasma oncotic pressure, since treatment with albumin or dextran infusions reduces hepatic lipoprotein synthesis. These abnormalities are summarized in Table 8.5. Nephrotic hyperlipidaemia is accompanied by an increased risk of cardiovascular complications and is usually treated aggressively with statins. The impact of this intervention on cardiovascular morbidity and mortality in patients with nephrotic syndrome has still to be demonstrated.
TABLE 8.5
Summary of lipoprotein and apolipoprotein abnormalities in nephrotic syndrome
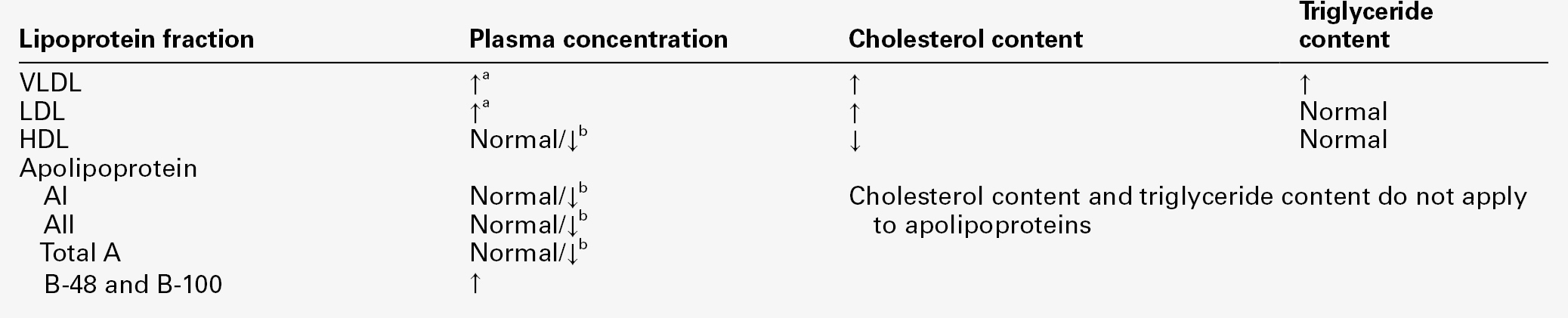
a Increased in more severe nephrotic states: inversely related to plasma albumin concentration.
b Depends on severity of nephrotic syndrome: lower concentrations occur in severe cases.
Tubular proteinuria
Unlike glomerular proteinuria, where protein excretion can reach 20 g/24 h and consists mainly of albumin, tubular proteinuria is generally < 1–2 g/24 h. In tubular proteinuria, although albumin remains a significant component, there is a relatively much greater increase in proteins of < 60 000 Da (Table 8.6).

Low molecular weight proteins are filtered rapidly by the glomerulus, and proteins of < 15 000 Da enter the Bowman space almost as easily as water and dissolved minerals. Under normal circumstances, the majority of filtered proteins and other solutes are reabsorbed by the proximal tubule, but in renal tubular disease, the reabsorption of protein together with water, ions, glucose and amino acids is impaired. The changing proportions of low and high molecular weight proteins in glomerular and tubular disease, shown in Table 8.6, can be explained by considering the renal handling of albumin (69 000 Da) and β2-microglobulin (11 815 Da).
Despite the large differences in plasma concentration of albumin (40 000 mg/L) and β2-microglobulin (2 mg/L), about 15 and 2 mg/L, respectively, appear in glomerular filtrate owing to differences in glomerular sieving. Assuming 160 L of filtrate are produced each day, 2400 mg of albumin and 320 mg of β2-microglobulin are filtered daily and would be excreted in the absence of tubular reabsorption. The maximum normal daily excretion of albumin is about 20 mg and of β2-microglobulin about 0.3 mg. Thus, in tubular disease, the increase in excretion of low molecular weight proteins is far greater than that found for larger proteins such as albumin.
Renal disorders associated with tubular proteinuria
In primary tubular proteinuria and interstitial nephritis, the renal interstitium and tubules are principally affected, rather than the glomeruli or renal blood vessels. However, recent studies have demonstrated that primary glomerular proteinuria leads to large amounts of plasma proteins being presented to the proximal renal tubules, absorption of which causes inflammation and secondary tubular damage. Thus, in addition to direct tubular toxic effects of conditions mediated by autoimmune or infective mechanisms, drugs, toxins, metabolic disorders or heavy metals, any pathology leading to glomerular proteinuria will also be associated with some degree of tubular dysfunction (Box 8.1).
Measurement of urine total protein or albumin is of limited value in differentiating between glomerular and tubular proteinuria, other than that the latter tends to be associated with proteinuria of < 1–2 g/24 h. Estimation of proteins that more specifically reflect tubular reabsorption is of more help in identifying primarily tubular disorders. In addition to tubular proteinuria, acute and chronic tubulointerstitial disease can lead to other tubular dysfunction with loss of water, sodium, potassium, calcium, glucose, phosphate, urate and amino acids. Bicarbonate wasting can lead to renal tubular acidosis (type 2). Tubular proteinuria, together with defective reabsorption described above, may form part of the Fanconi syndrome, which can be inherited or acquired (see Chapter 9).
Drug and heavy metal induced tubular damage
In general, the renal tubules are particularly susceptible to drug induced damage because the renal concentrating mechanism leads to high drug concentrations within the tubules. Aminoglycoside antibiotics bind to phospholipids and can disrupt cellular membranes. Analgesic drugs such as phenacetin or its metabolite acetaminophen (paracetamol) damage tubular cells because they are concentrated within the renal medulla, bind to cellular proteins and deplete stores of glutathione. Non-steroidal anti-inflammatory drugs (NSAIDs) inhibit the synthesis of renal vasodilatory prostaglandins, which can lead to unopposed vasoconstriction and ischaemia.
Arsenic, chromium, cadmium, copper, bismuth, mercury and lead have all been described as causing renal injury. Heavy metals react with membrane-bound sulphydryl groups, altering membrane permeability and causing cellular damage. Inhalation of mercury vapour appears to cause glomerular proteinuria, while ingestion of mercury salts can produce combined glomerular and tubular proteinuria. Acute lead intoxication in children can cause tubular proteinuria, although patients with clinically apparent lead poisoning sometimes have normal protein excretion. Cadmium is taken up by renal tubules as a metallothionein complex that is more toxic than cadmium alone. Increased excretion of β2-microglobulin was first described in cadmium workers, and markers of tubular proteinuria are now used to monitor occupationally exposed subjects.
Methods of assessing tubular damage
Renal tubular function can be assessed in a number of ways, such as by tests of reabsorption of glucose, phosphate, bicarbonate and amino acids. Discussion here will be limited to the use of urine proteins as markers of tubular damage by measurement of either high molecular weight proteins, such as enzymes or brush border antigens released by the damaged tubules, or of low molecular weight proteins filtered at the glomerulus and normally reabsorbed by the proximal renal tubule.
High molecular weight protein markers of renal tubular damage
Several enzymes not normally filtered by the glomeruli are released by damaged tubular cells and can be detected in the urine. They include lactate dehydrogenase, γ-glutamyl transferase and alkaline phosphatase. Their use as markers of tubular damage has been limited because of their instability in urine and the presence of urinary enzyme inhibitors.
The enzyme most widely employed for monitoring tubular damage is N-acetyl β-D-glucosaminidase (NAG), a lysosomal enzyme of 150 000 Da, which is found in high concentrations in the cells of the proximal tubule. Two isoenzymes of NAG are found in urine, one acidic and one basic: the acidic form is found in normal urine and both are excreted in patients with renal disease. Its lack of specificity limits its utility but NAG may be of value in monitoring inherited tubulopathies and in assessing tubular damage during cancer chemotherapy or treatment with antiretrovirals. Conditions associated with increased NAG excretion are given in Box 8.2.
Low molecular weight protein markers of renal tubular disease
Ideally, a low molecular weight urine protein that can be used to monitor tubular damage should have the following characteristics:
• it should have a constant plasma concentration
• be freely filtered by the glomerulus
• its tubular reabsorption should be near saturation so that small reductions in tubular function result in significant urinary excretion
• its tubular reabsorption should be unaffected by coexisting glomerular proteinuria
• it should be stable in urine
• it should be easily measurable.
Since the plasma concentration of low molecular weight proteins filtered by the glomeruli rises as the GFR falls, the tubules of those nephrons still functioning are presented with an increased low molecular weight protein load that causes overflow into the urine and confounds the interpretation of results. The characteristics of some proteins used as tubular markers are given in Table 8.7.
Lysozyme (muramidase) was the first low molecular weight marker of tubular function. Urine lysozyme excretion is increased in patients with Fanconi syndrome, but is often within normal limits in patients with nephrotic syndrome. Plasma lysozyme concentration is increased in inflammatory conditions and in some leukaemias, which limits its value as a tubular marker.
Increased excretion of β2-microglobulin was first identified in patients with cadmium poisoning and with Wilson disease. This protein is associated with the heavy chain of human leukocyte antigen surface protein on cells and is released during normal cellular turnover. Increased plasma concentrations are found in patients with liver disease and some malignancies such as myeloma and B cell lymphomas. However, β2-microglobulin is unstable in acidic urine (pH < 5.5) owing to degradation by neutrophil elastase. This necessitates strict control of urine pH after collection, although little can be done to prevent in vivo degradation in the bladder before voiding other than oral administration of bicarbonate to alkalinize the urine.
Retinol-binding protein (21 000 Da) forms an 85 000 Da complex with vitamin A and prealbumin in plasma and hence only a small proportion is free to be filtered by the glomeruli. Plasma concentration falls in inflammatory disease and in vitamin A deficiency. Plasma α1-microglobulin (31 000 Da) concentrations are increased in neoplastic diseases, but are reduced in liver disease in parallel with plasma albumin. Unlike β2-microglobulin, retinol-binding protein and α1-microglobulin are relatively stable in urine and are the most commonly measured markers of tubular proteinuria.
Proteinuria of prerenal origin
Proteinuria of prerenal origin has been defined as the occurrence in the urine of abnormal amounts of protein filtered by the glomeruli in the absence of any glomerular or tubular abnormality. The term is usually applied to ‘overflow’ proteinuria such as Bence Jones proteinuria, haemoglobinuria and myoglobinuria, where plasma concentrations of these proteins are increased. This definition has limitations because the very presence of abnormally high concentrations of protein at the glomerulus and in the tubular lumen can cause glomerular and tubular abnormalities. Furthermore, it is becoming apparent that non-renal conditions are associated with proteinuria that disappears when the condition resolves.
Myoglobinuria and haemoglobinuria
Myoglobin is found predominantly in skeletal and cardiac muscle cells. Any disease that causes rapid destruction of striated muscle (rhabdomyolysis) results in the release of myoglobin and other muscle proteins into the circulation. Having a molecular weight of 17 000 Da, myoglobin is filtered rapidly by the glomerulus; it has a renal threshold of about 15 mg/L.
Box 8.3 illustrates the diversity of causes of rhabdomyolysis, although a common factor appears to be the damaging effects on muscle cells of a failure to meet increased energy demands. Rhabdomyolysis is associated with acute kidney injury, but a causal relationship is not clear. Although myoglobin casts are found in rhabdomyolysis, infusion of pure myoglobin does not in general cause kidney damage, suggesting that there are other factors that lead to the kidney injury. Purine derivatives released from damaged muscle cells are rapidly converted to uric acid, leading to very high plasma urate concentrations and intrarenal deposition. Acidosis as a result of the initiating cause of rhabdomyolysis, for example hypovolaemic shock, or from the release of organic acids from muscle cells, may potentiate the degradation of myoglobin to globin and ferrihaemate within the tubules. Ferrihaemate is toxic to tubular epithelial cells. Desquamation adds to the tubular obstruction.
Intravascular haemolysis releases free haemoglobin, which binds to circulating proteins such as haptoglobin. Since haemoglobin itself is 68 000 Da and its plasma protein-complexed form is even larger, haemoglobinuria does not occur until haemolysis is severe and plasma haemoglobin concentration exceeds 1 g/L. Pure haemoglobin is not nephrotoxic, and acidosis and dehydration are important factors determining the nephrotoxicity of severe intravascular haemolytic episodes. Haemoglobin casts are found, and ferrihaemate released from degraded haemoglobin may also cause tubular epithelial cell damage.
Both myoglobin and haemoglobin possess peroxidase activity, which gives positive results for blood with reagent sticks. Measurement of urine myoglobin and haemoglobin is rarely necessary because other biochemical features give better clues to the diagnosis (Table 8.8).
TABLE 8.8
Biochemical features associated with myoglobinuria and haemoglobinuria
| Plasma | Myoglobinuria | Haemoglobinuria |
| Creatine kinase | ↑ ↑ ↑ | Normal |
| Visible haemolysis | None | Present |
| Calcium | ↓ | Normal |
| Haptoglobin | Normal | ↓ |
| Urate | ↑ ↑ ↑ | Normal |
| Creatinine | ↑a | Normal |
a Plasma creatinine concentration disproportionately increased compared to GFR.
Paraproteinaemias and Bence Jones proteinuria
Bence Jones proteinuria, the presence in the urine of immunoglobulin light chains, is a frequent finding in patients with multiple myeloma and may occur with other B cell malignancies (Table 8.9). This topic is considered in detail in Chapter 30.

There is an association between the presence of Bence Jones protein and the development of renal dysfunction in patients with multiple myeloma. Free light chains (molecular weight 22–24 000 Da) are normally cleared from the circulation by glomerular filtration followed by reabsorption and catabolism in the proximal tubules. Given the normal production rate of 500 mg/day and a reabsorptive capacity of 10–30 g/day, the production rate must increase substantially to cause overflow into the urine in the absence of renal dysfunction. Some light chains are toxic to the proximal tubules (a property that appears to relate to the amino acid sequence in the variable domain but is incompletely understood) and probably contribute to the development of Fanconi syndrome, renal tubular acidosis and renal failure in patients with multiple myeloma. Patients with light chain proteinuria show increased excretion of lysozyme, retinol-binding protein, β2-microglobulin and albumin (with β2-microglobulin excretion being higher in λ light chain proteinuria), suggesting that these proteins compete for common reabsorptive mechanisms. Light chains can complex with uromodulin, producing typical myeloma casts, especially when patients are dehydrated and/or acidotic. Partial degradation of light chains can cause polymerization and the formation of damaging amyloid deposits in the glomerular and tubular basement membranes. The specific detection and quantitation of urine paraproteins and Bence Jones protein requires electrophoresis, immunofixation and urine total protein analysis although specific immunoassays for urine free light chains are now available.
MICROALBUMINURIA AS A MARKER OF RISK
The term ‘microalbuminuria’ was coined to describe an increase in urine albumin that is detectable by sensitive immunoassays but is below the detection limit of chemical urine protein methods and dye-binding stick tests. Microalbuminuria is defined as an albumin excretion rate of 20–200 μg/min (30–300 mg/24 h or 3–30 mg/mmol creatinine). Diabetic patients with microalbuminuria are at increased risk of developing dipstick positive proteinuria and CKD. Early identification allows aggressive treatment to improve glycaemic control, blood pressure and plasma lipids and has been shown to improve outcome. More recently, large scale studies have shown that screening for microalbuminuria in non-diabetic as well as diabetic populations identifies patients at increased cardiovascular risk. Furthermore, microalbuminuria has been found to be a predictor of outcome in critically ill patients following insults such as major surgery, trauma or sepsis. The link between microalbuminuria and outcome in such apparently diverse groups of patients appears to be that microalbuminuria reflects the systemic microvascular endothelial dysfunction that, in various forms, is common to all these conditions.
Microalbuminuria and risk of diabetic complications
Diabetes mellitus is the commonest cause of established renal failure. It can affect the kidneys in a number of ways leading to glomerular disease, obstruction, tubular disease and a predisposition to infection, but it is diabetic glomerular disease that is the most important, involving about one third of patients. In the early stages of diabetic renal disease, there is hyperfiltration with an increased GFR, which may be associated with microalbuminuria. With adequate treatment, the GFR falls towards normal and microalbuminuria disappears. However, microalbuminuria returns during periods of poor glycaemic control and becomes persistent with established disease, usually after 5–20 years of diabetes. The risk of developing diabetic nephropathy can be reduced by improving glycaemic control and by treating hypertension aggressively. Once albuminuria reaches > 0.5 g/24 h or more, improved glycaemic control appears to have little effect on further progression.
Type 1 diabetes usually presents acutely and microalbuminuria rarely occurs within five years of diagnosis, so annual monitoring of urine albumin is generally recommended only after five years’ disease duration. In contrast, type 2 diabetes will often have been present for many years prior to diagnosis, hence annual monitoring of urine albumin is recommended from the time of diagnosis. To reduce micro- and macrovascular risk, patients with persistent microalbuminuria (i.e. confirmed by two repeat measurements over 3–4 months) should be treated with an ACEI (or ARB), titrated to the maximum tolerable dose, to reduce intraglomerular pressure. Blood pressure should be maintained at < 130/80 mmHg by the use of additional antihypertensives if necessary. These measures, together with aggressive management of cardiovascular risk factors, aim to reduce the rate of progression of kidney disease and prevent cardiovascular events. However, it should be noted that a diminishing urine albumin excretion rate associated with treatment does not invariably translate into prevention of progression of renal disease as assessed by GFR.
Cardiovascular risk
Microalbuminuria is a marker for generalized vascular endothelial dysfunction. In the USA and the European Community, microalbuminuria is found in 6–10% of the general population. Numerous clinical studies in non-diabetic populations have found an association between microalbuminuria and cardiovascular risk factors, target organ damage and the presence of cardiovascular disease. Although microalbuminuria interacts with other cardiovascular risk factors, it is an independent predictor of cardiovascular disease. In large population studies, the presence of microalbuminuria has been shown to be associated with raised plasma C-reactive protein (CRP) concentrations (measured using high sensitivity assays), which is consistent with the view that microalbuminuria reflects systemic endothelial dysfunction mediated by inflammatory processes. Large cross-sectional prospective studies have shown an association between urine albumin excretion and both blood pressure and cardiovascular risk that extends from within the urine albumin reference range through microalbuminuria and to clinical proteinuria. In patients with primary hypertension, the prevalence of microalbuminuria is typically 4–6%. In subjects with mild hypertension and no cardiovascular complications, urine albumin excretion is determined by the haemodynamic load, whereas in subjects with more severe hypertension and associated target organ damage, the urinary albumin leak is probably the consequence of glomerular damage.
Treatment with an ACEI or ARB reduces urine albumin excretion and may prove to be a more targeted approach to reducing cardiovascular risk. Microalbuminuria can be considered not only as a risk factor for progressive renal damage, but as providing an integrated assessment of long-term damage to the cardiovascular system, and is therefore increasingly being used in cardiovascular risk assessment clinics. What remains to be shown is whether targeted treatment of microalbuminuria in the non-diabetic population reduces cardiovascular morbidity and mortality. However, there is general consensus that detection and quantitation of microalbuminuria is useful for the assessment of overall cardiovascular risk in hypertension, since it appears to be a cost-effective way of identifying patients at higher risk for whom additional preventive and therapeutic measures are advisable.
Microalbuminuria as a risk factor in other inflammatory processes
The emerging association between inflammation, vascular endothelial dysfunction and microalbuminuria has led to a large number of studies exploring the potential clinical value of this relationship. Interaction of activated leukocytes and the vascular endothelium is at the heart of the inflammatory process, and is normally under tight homoeostatic control, preventing the local inflammatory response from becoming a systemic, life-threatening process. Yet this homoeostasis can fail in patients with severe injuries, infection or multiple pathologies, resulting in systemic inflammatory response syndrome (SIRS). This is the underlying pathogenetic mechanism for the development of multiple organ failure, which is the major cause of death in the critically ill (see Chapter 20). Attempts to modulate the severe inflammatory response have been unsuccessful, partly because of the multiplicity of inflammatory pathways, such that blocking a single pathway is unlikely to be effective, and because such interventions must be given early in the evolution of SIRS to have any hope of success. Increased urine albumin excretion occurs within minutes of any acute inflammatory stimulus such as trauma, surgery, ischaemia reperfusion injury or infection, and normally returns to baseline within 6–8 h, depending on the nature and magnitude of the inflammatory insult. However, in patients who develop SIRS, microalbuminuria is sustained, reflecting systemic vascular endothelial dysfunction. Thus, serial monitoring of urine albumin during major inflammatory episodes has the potential to identify, within a few hours of onset, patients who require close monitoring in an intensive care unit, and who might be candidates for immune-modulating therapy. On the other hand, a return of urine albumin excretion to normal provides a marker of restoration of homoeostatic control of inflammation, which can be used as an indicator of successful recovery from the acute inflammatory insult.
CLINICAL INVESTIGATION OF PROTEINURIA
Urine dip-sticks
For many years, proteinuria has been identified by screening random urine specimens using a semiquantitative chemical stick test. These methods are based on the colour change of indicators such as bromophenol blue, which is buffered to pH 3.0 with citrate, and changes from yellow to blue when bound to albumin. Such stick tests for protein (that predominantly detect albumin) have a detection limit in the range 200–250 mg/L and are subject to errors, including false positives owing to alkaline pH from infection with urea-splitting organisms or contamination with antiseptics, and false negatives owing to very dilute urine and visual reading errors. With the growing awareness in diabetic medicine of the pathological significance of proteinuria at concentrations that are undetectable by conventional chemical sticks, specific semiquantitative immunoassay sticks for urine albumin have been developed that can detect albuminuria at ~ 30 mg/L.
Collection of urine
Measurement of urine protein or albumin as a concentration in milligrams or grams per litre makes no allowance for variations in urine flow rate. Formal correction can be made by collecting timed urine samples and expressing results in protein mass per unit time. However, this is both inconvenient for the patient and prone to urine collection errors because of misunderstanding of when the collection should start and finish. Numerous studies have demonstrated that expression of urine total protein or urine albumin as a ratio to creatinine concentration in an early morning sample is at least as reproducible as a timed urine, even if collected under ideal conditions, and is more convenient for the patient. There are now few indications for collecting timed urine specimens.
Urine protein measurement
Throughout this chapter, it has been emphasized that pathologically significant proteinuria extends from the upper reference limit for urine protein, which is in milligrams per litre, to tens of grams per litre, in nephrotic syndrome. Chemical methods used for measuring urine total protein, which are both accurate and precise, are generally adequate for monitoring patients with proteinuria > 1 g/L. However, there is a clear problem with using such techniques for quantitation of proteinuria in the mg/L range. First, external quality assessment programmes have highlighted the poor performance of urine total protein methods, particularly at low concentrations. This is partly related to the variable chromogenicity for the differing urine proteins of the different dyes used in total protein methods, resulting in different total urine protein reference ranges. Second, at low total protein concentrations, the relative contribution of proteins secreted into the urine by the kidneys, such as uromodulin, becomes more significant, thus masking within the total protein estimation small, but pathologically significant increases in urine albumin and other diagnostically important proteins (Fig. 8.2).
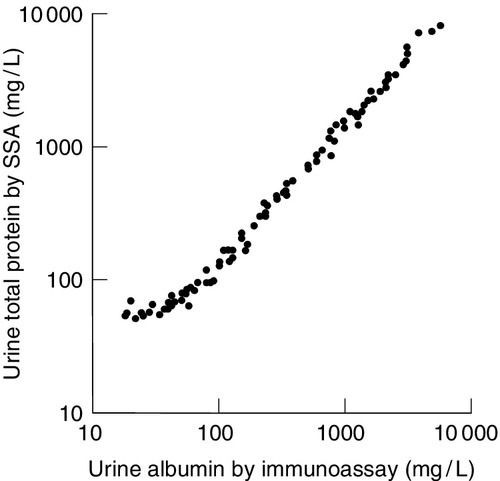
In the US National Kidney Foundation Guidelines, urine albumin is recommended as a sensitive marker for chronic kidney disease secondary to diabetes, glomerular disease and hypertension, and α1 and β2-microglobulins as sensitive markers of tubulointerstitial disease. For this reason, many laboratories now provide urine albumin as their first line test for assessing ‘proteinuria’, covering the range from normal values, through microalbuminuria to the albuminuric nephrotic range. However, at higher concentrations, the requirement for repeated dilutions of the sample for accurate measurement of albumin adds considerably to the cost. Above 0.5–1 g/L, some guidelines suggest that measurement of either urine albumin or total protein is acceptable for monitoring. In more specialized circumstances, such as screening for urinary light chains or monitoring of tubular dysfunction, specific protein immunoassays are the most reliable.
Stepwise investigation of proteinuria
Where proteinuria is confirmed, a stepwise approach to establishing the cause of proteinuria can be applied, as summarized in Figure 8.3. Particular regard should be given to evidence of oedema, diabetes mellitus, polycystic disease, hypertension, heart failure and other cardiovascular risk factors. A full clinical and laboratory assessment at this stage will identify the cause of proteinuria in the majority of patients. For example, proteinuria may be caused by protein leakage from inflamed epithelium during urinary tract infections. In other patients, proteinuria, especially microalbuminuria, may be associated with non-renal conditions, some of which are transient and disappear with the resolution of the associated condition (Box 8.4).
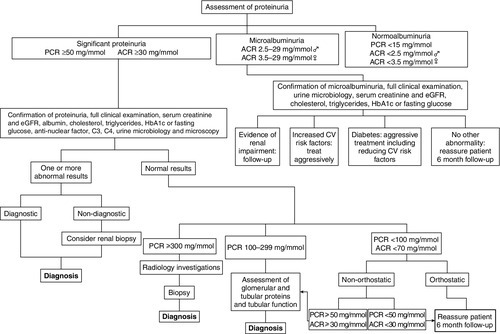
A third group of patients will demonstrate persistent asymptomatic proteinuria either in isolation or accompanied by other urinary abnormalities. Red cells can enter urine or filtrate at any point along the urinary tract, leading to haematuria. Proteinuria with haematuria or tubular cell, granular or leukocyte casts is likely to originate from the nephron. When red cell casts are also found, the nephron can be implicated with more confidence and, in general, this indicates more severe renal disease.
Studies have shown that ~ 70% of patients with asymptomatic non-orthostatic proteinuria have abnormal renal biopsies, while of those with orthostatic asymptomatic proteinuria, a much smaller proportion have abnormal biopsies. As a result, biopsy is now rarely performed in this latter group. The significance of the histological abnormalities is uncertain and the prognosis for both groups is excellent.
ACKNOWLEDGEMENT
I would like to thank Peter Gosling, who wrote this chapter for previous editions of the book.
Further reading
Although guidelines are available in print form, the latest versions incorporating updates and corrections are usually available only electronically (web addresses Accessed October 2013). Their titles are self-explanatory.
Kidney Disease Improving Global Outcomes. http://www.kdigo.org/home/guidelines.
NICE. http://www.nice.org.uk/nicemedia/live/12069/42116/42116.pdf.
NICE. http://www.nice.org.uk/nicemedia/live/11983/40803/40803.pdf.
NICE clinical guideline CG62: Antenatal care: routine care of the healthy pregnant woman (reviewed 2011), http://guidance.nice.org.uk/CG62.
NICE clinical guideline CG107: the management of hypertensive disorders during pregnancy. 2010. http://guidance.nice.org.uk/CG107.
NICE clinical guideline CG73: chronic kidney disease. 2008. http://guidance.nice.org.uk/CG73.
Renal Association. Clinical practice guideline on detection, monitoring and care of people with CKD. 5th ed. 2011. http://www.renal.org/Clinical/GuidelinesSection/Detection-Monitoring-and-Care-of-Patients-with-CKD.aspx.
[/level-membership-for-endocrinology-diabetes-and-metabolism-category][not-level-membership-for-endocrinology-diabetes-and-metabolism-category]CHAPTER 8
Proteinuria
CHAPTER OUTLINE
PROTEIN CONSERVATION BY THE KIDNEYS
The theory of molecular sieving
Tubular reabsorption of proteins
NORMAL URINARY PROTEIN CONTENT
Determinants of urine protein excretion
Proteinuria in staging and prognosis of chronic kidney disease
Glomerular proteinuria and nephrotic syndrome
Proteinuria of prerenal origin
MICROALBUMINURIA AS A MARKER OF RISK
Microalbuminuria and risk of diabetic complications
Microalbuminuria as a risk factor in other inflammatory processes
INTRODUCTION
For over 150 years, proteinuria has been recognized as one of the cardinal signs of renal disease and it continues to be used for the diagnosis and management of patients with nephropathy. However, the use of sensitive immunoassay methods to measure urine albumin has demonstrated pathologically significant proteinuria well below the detection limit of chemical methods. Since the discovery, several decades ago, that low-level albuminuria (microalbuminuria) identifies diabetic patients with incipient nephropathy, further large-scale studies have revealed that microalbuminuria is also a risk factor for macro- and microvascular disease, in both diabetic and non-diabetic populations. Studies over the last ten years have confirmed that microalbuminuria is not only associated with cardiovascular disease but also with any acute inflammatory condition, and is a reflection of systemic vascular endothelial dysfunction. Of particular importance, is the finding that, in some conditions, microalbuminuria is reversible with interventions that protect the vascular system. Thus modern assessment of proteinuria across the full pathological range has a role not just in the diagnosis and management of primary renal disease, but also as a marker of endothelial dysfunction in a variety of non-renal conditions.
PROTEIN CONSERVATION BY THE KIDNEYS
The kidneys receive approximately 25% of the resting cardiac output, which represents approximately 1.2 L/min of blood or 650 mL/min of plasma. The kidneys’ capacity to conserve protein can be judged from a simple calculation. Every 24 h, approximately 930 L of plasma containing about 70 g/L of protein pass through the kidneys, representing 65 kg of protein, of which < 100 mg (0.00015%) appear in the urine.
The filtration process is dependent on adequate renal blood flow, which is preserved by an autoregulatory system, despite variations in blood pressure (see p. 127). This mechanism allows vasodilatation as perfusion pressure falls and vasoconstriction as pressure rises. The mediators of this process include prostaglandins, kinins and atrial peptides (vasodilators), and angiotensin II, α-adrenergic hormones, thromboxane A2, noradrenaline (norepinephrine) and vasopressin (vasoconstrictors). In addition, renal arterioles respond within seconds to changes in vessel wall tension; thus, when renal perfusion pressure rises, vessel wall tone increases and, conversely, when renal perfusion pressure falls abruptly, there is a compensatory decrease in vessel wall tone. This phenomenon is called the myogenic reflex and helps to maintain a constant renal blood flow across a range of perfusion pressures.
A minimum intraglomerular pressure, derived from the pumping action of the heart, is required to overcome the two main opposing forces to filtration: the colloidal oncotic pressure and the hydrostatic pressure in the Bowman space. When the renal perfusion pressure falls below 50–60 mmHg, further vasodilatation does not occur and renal blood flow declines in proportion to the reduction in renal perfusion pressure. These mechanisms maintain renal blood flow, and hence glomerular filtration, independently of the normal fluctuations of blood pressure. However, recent studies suggest that chronic hypertension impairs the renal autoregulatory mechanism, which may contribute to hypertension-associated renal damage and proteinuria.
The glomerular capillary wall
The glomerular membrane consists of a modified capillary wall comprising endothelium, an acellular basement membrane and an outer specialized epithelial cell layer (Fig. 8.1). The endothelial cells are thin and fenestrated with 50–100 nm pores. The basement membrane, comprising the lamina rara interna, lamina densa and lamina rara externa, is around 300–350 nm thick and is best considered as a gel-like structure containing 3–5 nm long fibrils but no detectable pores. The numerous foot processes of the epithelial cells (also called podocytes) interdigitate and envelop the outer surface of the glomerular membrane. The foot processes are separated by slit diaphragms about 55 nm in width, which form the final barrier to plasma proteins.
The whole of the glomerular membrane carries a fixed net negative charge, which is partly owing to a glycosialoprotein coat covering both endothelium and epithelium. The charge increases in density from the lamina rara interna towards the lamina rara externa, with the greatest density at the slit diaphragms of the epithelium.
The glomerular capillaries act as high pressure filters, allowing water and low molecular weight solutes to pass through freely and almost completely retaining plasma proteins. The concentration gradient of proteins across the glomerular membrane produces the colloidal oncotic pressure that must be overcome for filtration to take place.
The theory of molecular sieving
The glomerular membrane selectively allows passage of water and low molecular weight solutes into the tubule and restricts passage of larger molecular weight plasma proteins, based on a combination of molecular size, shape and charge. The pore size of the fenestrae of endothelial cells (50–100 nm) is too great to provide a major restriction to the passage of many proteins, which pass through to the glomerular membrane. Like the glomerular membrane, most plasma proteins carry a net negative charge, resulting in their electrochemical retention by the kidneys. The capillary side of the glomerular basement membrane (lamina rara interna) has a charge density of 35–45 mEq/L, so that electrochemical effects alone reduce the concentration of albumin to 5–10% of that in plasma. Thus, the lamina rara interna of the basement membrane is the first impediment to confront charged molecules, with the lamina densa providing the most effective size barrier to macromolecules. Some macromolecules accumulate at the slit diaphragms of the epithelium where the net negative charge is greatest, and there is evidence that some molecules are pinocytosed by the podocytes.
Based on their molecular radius, shape and charge, different proteins penetrate the glomerular membrane to a variable extent, for example, albumin (radius 3.6 nm, isoelectric point 4.7) is restricted at the lamina rara interna, while lactoperoxidase (radius 3.8 nm, isoelectric point 8.0) may reach the slit pores of the epithelial cells.
Despite the protein-retaining properties of the glomerular membrane, some protein does pass into the proximal tubular fluid. In healthy adults, the albumin content of glomerular filtrate is probably about 15 mg/L which, with a glomerular filtration rate (GFR) of 160 L/ 24 h, results in 2–3 g of albumin being presented to the renal tubules each day, most of which is reabsorbed.
Tubular reabsorption of proteins
The amount of protein reaching the proximal tubules is a function of the protein’s ability to cross the glomerular membrane and its plasma concentration. After reabsorption, proteins are catabolized in tubular cells and the amino acids, bound vitamins and trace metals returned to the plasma pool. For example, under normal circumstances the kidneys are responsible for about 10% of albumin catabolism, but this figure can rise to 60% when there is increased glomerular clearance of albumin such as occurs in nephrotic syndrome.
Normally, only a very small proportion of plasma proteins reaches the urine. This is primarily owing to retention of large molecular weight proteins by the glomeruli, and almost complete proximal tubular reabsorption and catabolism of any filtered lower molecular weight proteins that have passed through the glomerular membrane more easily. Thus, the renal clearance of plasma proteins represents a combination of these processes. The relative contributions of glomerular retention and tubular reabsorption to protein clearance by the kidney depend on molecular weight (Table 8.1).
TABLE 8.1
Relative clearances of plasma proteins
| Protein | Molecular weight (Da) | Clearance (%GFR) |
| IgG | 150 000 | 0.01 |
| Albumin | 69 000 | 0.02 |
| Amylase | 48 000 | 3 |
| Myoglobin | 17 800 | 75 |
| Lysozyme | 14 500 | 80 |
The likely mechanism of proximal tubular protein reabsorption is endocytosis at the tubular epithelial apical cell membrane mediated by two receptors (megalin and cubilin) and the cooperating protein amnionless. Mutations in one or other of these components are responsible for the tubular proteinuria of Imerslund–Grasbeck syndrome (selective vitamin B12 malabsorption) and Donnai–Barrow syndrome (a multisystem disorder affecting many organs that normally express megalin). Following reabsorption, proteins are transferred to lysosomes for degradation while the receptors are recycled from endosomes to the apical cell membrane. Mutations in the lysosomal and endosomal components are responsible for the tubular proteinuria in Dent disease and cystinosis; the molecular mechanism in Lowe syndrome is yet to be resolved (see Chapter 9).
Megalin and/or cubilin bind albumin and low molecular weight proteins but there is some evidence that, of the filtered proteins reaching the proximal tubule, those with lower molecular weights are reabsorbed by different mechanisms from those with higher molecular weights. For example, although increased amounts of low molecular weight proteins are excreted in tubular disease, such large increases are not seen in the urine of patients with massive glomerular proteinuria, when the reabsorptive mechanisms for large molecular weight proteins such as albumin must be saturated. Conversely, experimental induction of massive low molecular weight proteinuria through saturation of reabsorption with other low molecular weight proteins does not induce a similar increase in urine albumin.
Low molecular weight proteins, such as α1-microglobulin and retinol-binding protein, that are readily filtered at the glomerulus and normally > 99.9% reabsorbed, can be used as sensitive markers of proximal tubular function, since even subtle tubular damage is associated with increased urinary excretion.
Glomerular diseases causing heavy proteinuria induce the release of a multitude of inflammatory and fibrogenic mediators, all of which cause tubulointerstitial fibrosis and renal scarring that contribute to renal impairment. Recent studies suggest that large proteins, such as albumin, which pass through the diseased glomerular membrane, cause tubular cell injury when they reach the proximal tubules. Reabsorption of excessive amounts of these proteins leads to overloading of lysosomes and activation of proximal tubular cells, which produce matrix metalloproteins, cytokines, leukocyte chemoattractants and vasoactive mediators, leading to interstitial inflammation and scarring. The mechanism of injury has been most studied for albumin and it appears likely that compounds attached to the protein, such as fatty acids, are major culprits. In minimal change glomerular disease with heavy proteinuria that is not associated with scarring, the albumin-fatty acid composition appears to be different from that in other glomerulopathies.
Tubular secretion of proteins
Proteins that are too large to be filtered by the normal glomerulus appear in urine either as a result of tubular secretion or from desquamation of tubular epithelial cells as part of the normal cellular turnover. Some large molecular weight proteins may also enter the urine during its postrenal passage along the urinary tract. Measurable activities of enzymes such as N-acetyl β-D-glucosaminidase (150 000 Da), γ-glutamyl transferase (120 000 Da) and lactate dehydrogenase (144 000 Da) can be found in normal urine.
The major urine protein exclusively of renal origin is uromodulin (previously known as Tamm–Horsfall glycoprotein), a heavily glycosylated protein secreted from the thick ascending limb of the loops of Henle. It has a molecular weight of 70 000 Da but is normally present in urine as a large non-covalently linked polymer. Since its isoelectric point is 3.3, it tends to precipitate as a gel during urinary acidification, forming casts by trapping whatever is in the vicinity such as albumin, red blood cells, tubular cells or cellular debris. Numerous mutations in the uromodulin gene have been identified in association with familial juvenile hyperuricaemic nephropathy and medullary cystic kidney disease type 2. These are probably different phenotypes of the same disease and are known collectively as uromodulin storage disease, owing to accumulation of the misfolded mutated protein in cells of the thick ascending limb of the loops of Henle. They are rare, primary tubulointerstitial disorders with autosomal dominant inheritance characterized by chronic progressive kidney disease, hyperuricaemia and gout, with minimal proteinuria.
The normal biological function of uromodulin remains unclear but it probably has a protective role in trapping potentially damaging material in the urinary space, allowing excretion as casts. Microscopic differentiation of the type of casts present in urine can give useful information about pathological processes within the kidney (see Chapter 7). Experimental studies suggest that uromodulin may have a role in protecting against stone formation. Its capacity to bind type-1 fimbriated E. coli has led to the suggestion that its excretion has evolved as a defence against urinary tract infections.
NORMAL URINARY PROTEIN CONTENT
A normal adult excretes about 300 mg/24 h of non-dialysable material, of which < 140 mg/24 h is protein, but published reference ranges for total urinary protein excretion vary considerably with the analytical method used. Plasma proteins represent only some 25 mg/24 h of total urinary protein (Table 8.2), of which about half is albumin: the remaining proteins are of renal origin, uromodulin being the major contributor (70 mg/24 h). For these reasons, and because low-level albuminuria (microalbuminuria) has prognostic value both for renal and non-renal diseases, urine albumin immunoassays should be used to assess glomerular proteinuria when protein excretion rates are low.
Determinants of urine protein excretion
Age, sex and diurnal variation
In neonates, albumin excretion tends to be higher than in older children and adults: this has been attributed to greater permeability of the neonatal glomerulus. Total urine protein tends to fall after birth and rises with increasing age, reaching adult excretion rates by puberty. However, the urine protein/creatinine concentration ratio remains constant from 3 to 15 years of age, since urine creatinine excretion rises with increasing body mass. In children aged 4–16 years, albumin excretion rate, corrected for body surface area, rises with age and is slightly higher in females. Daytime excretion rates are higher than during the night and the sex difference disappears in overnight collections. There appears to be no sex difference in urine albumin excretion in adults, but, when expressed as a ratio to creatinine, the reference range is slightly higher in females owing to their lower muscle mass and hence lower creatinine excretion.
Posture
In both adults and children, ambulatory urine protein excretion is higher than it is overnight or during recumbency, with two- to ten-fold differences being reported for urinary albumin. Orthostatic proteinuria in otherwise healthy subjects has been the subject of controversy for some time. The discussion has been complicated by the variety and differing sensitivities of the protein assays used.
Proteinuria of < 1 g/24 h has been described in 0.6–9% of healthy young adults, in the absence of urinary red cells, white cells or casts, and can be divided into ‘constant’ and ‘postural’ based on its persistence after recumbency. Renal biopsies of patients with postural proteinuria reveal that 8% have unequivocal evidence of well-defined disease and 45% have subtle alterations in glomerular structure. However, more recent studies, using non-invasive Doppler ultrasound to compare recumbent and orthostatic blood flow in the left renal veins of young people with orthostatic proteinuria, have revealed that in > 50% of patients there is reduced blood flow to the left renal vein during standing owing to entrapment of the left renal vein.
The medical management of isolated or postural proteinuria in an otherwise healthy patient tends to be conservative, with annual assessment of proteinuria and renal function; biopsy is reserved for the rare patient who has evidence of progressive renal impairment.
Exercise and diet
Exercise-induced proteinuria was discovered over a century ago in soldiers after marches or drills. Five- to 100-fold increases in the excretion of proteins such as albumin, transferrin and immunoglobulins have been observed following 26-mile marathon runs, with smaller increases after less strenuous activities. The pattern of exercise-induced proteinuria is generally glomerular, although mixed glomerular and tubular proteinuria has also been described, which persists for over 3 h after exercise. The reason for exercise-induced proteinuria is unclear, but some degree of renal ischaemia owing to redistribution of blood during exercise has been suggested as a possible mechanism.
A large protein meal is associated with an increased urine albumin excretion, which appears to be secondary to an associated increase in GFR.
The lowest and most reproducible estimates of urine protein excretion are obtained from an early morning urine specimen after overnight recumbency.
Pregnancy
During normal pregnancy, the urinary albumin excretion rate generally remains within the non-pregnant range, although there is some evidence for a small increase in albumin excretion during the third trimester, which may be related to increased glomerular permeability and/or GFR. Total urine protein excretion increases owing to decreased renal tubular protein reabsorption.
Hypertension in pregnancy is associated with significant maternal and fetal morbidity and mortality. The reliable detection of significant proteinuria is most important in women with new-onset hypertension during pregnancy because it distinguishes between those pregnancies with pre-eclampsia and those with gestational hypertension, the former often requiring admission to hospital owing to the severity of potential complications. In the UK, NICE guidelines for the routine antenatal care of healthy pregnant women recommend blood pressure and urine protein measurement at each antenatal visit; 660 000 women each year will have at least 7–10 such checks.
Gestational hypertension is defined as new hypertension occurring after 20 weeks of pregnancy, but without significant proteinuria. In this group, routine urine protein measurement may be performed using an automated reagent-strip reading device (more reliable than a manual reading) or by a laboratory method. If a reagent strip reading is 1 + or greater, the proteinuria should be quantitated by a laboratory measure in a spot or 24 h urine sample. Women admitted with pre-eclampsia (new hypertension presenting after 20 weeks with significant proteinuria) do not need to have repeated measures of urine protein since there is no strong evidence linking the degree of proteinuria with adverse outcome. Significant proteinuria is defined as > 300 mg/24 h or > 30 mg/mmol creatinine in a random sample. There are insufficient studies of urine albumin excretion to be able to define cut-offs equivalent to those defining significant proteinuria in gestational hypertension or pre-eclampsia.
PROTEINURIA IN KIDNEY DISEASE
Richard Bright, in 1836, is generally credited with noting the association between proteinuria and kidney disease. Proteinuria remains the most frequent clinical finding and quantitation of proteinuria is valuable for diagnosing, monitoring and assessing the prognosis of kidney disease. Normally, total urine protein excretion is < 150 mg/24 h in adults and < 140 mg/m2/24 h in children, depending on the methods employed: normal concentrations are often undetectable by chemical methods. However, sensitive immunoassay of specific proteins has extended the detection limits to urine protein concentrations within the reference range. Measurements of low concentrations of specific proteins such as albumin, predominantly reflecting glomerular function, and α1-microglobulin or retinol-binding protein, reflecting tubular reabsorptive function, are now used as very sensitive and early markers of primary renal disease (e.g. glomerulonephritis) and secondary renal disease (e.g. in diabetes mellitus or hypertension). Thus, proteinuria can now be regarded as a continuum that extends from the measurable amount of protein normally excreted in urine up to the 1000-fold increases found in nephrotic syndrome.
Conventionally, proteinuria has been classified into glomerular proteinuria; tubular proteinuria; nephrogenic proteinuria (e.g. uromodulin, basement membrane and tubular proteins); proteinuria of prerenal origin (e.g. overflow proteinurias such as light chain disease, myoglobinuria, haemoglobinuria, lysozyme in leukaemia and amylase in pancreatitis), and postrenal proteinuria owing to obstruction of the urinary tract or inflammation such as occurs in urinary tract infection. This is a convenient way of differentiating the principal sites of the renal abnormality, but is an over-simplification because, for example, glomerular disease leading to large amounts of plasma proteins being presented to the renal tubules leads to inflammatory changes within the tubules and renal scarring. Nor does this classification lend itself easily to the multifactorial causes of proteinuria in some disorders, e.g. HIV-associated glomerular and tubulointerstitial disease that may be modulated by co-infection with other viruses and drug-induced interstitial nephritis.
Proteinuria in staging and prognosis of chronic kidney disease
Numerous epidemiological studies have demonstrated the association of proteinuria with poorer prognosis in people in the general population and across all stages of chronic kidney disease (CKD): any renal disease is more likely to progress, there is an increased risk of developing acute kidney injury, and both all-cause and cardiovascular mortality are increased. These outcomes hold true, whether proteinuria is assessed by dipstick testing or by formal laboratory measurement of either total protein or albumin, in timed urine collections or in random samples. The quantitation of proteinuria (in the absence of a symptomatic urinary tract infection and preferably using the first morning urine) is an essential component of CKD staging, where the suffix ‘p’ is used to denote its presence. The decision limit is an albumin/creatinine ratio (ACR) > 30 mg/mmol (~ 300 mg/24 h) or urine protein/creatinine ratio (PCR) > 50 mg/mmol (~ 0.5 g/24 h), although the continuum of risk, albeit lower, extends into the reference range. The presence of proteinuria in CKD is sufficient indication to initiate blockade of the renin–angiotensin–aldosterone system (RAS) with angiotensin-converting-enzyme inhibitors (ACEI) or angiotensin-II receptor blockers (ARB). At higher excretions (ACR > 70 mg/mmol or PCR > 100 mg/mmol), RAS blockade should be titrated to the highest tolerable levels and referral of the patient to specialist care considered.
Glomerular proteinuria and nephrotic syndrome
In the normal adult, the renal tubules reabsorb about 2–3 g of filtered albumin every 24 h. Thus, even total failure of this process cannot explain albuminuria of > 3.0 g/24 h; such losses are usually secondary to increased glomerular permeability associated with glomerular damage.
Nephrotic syndrome can be defined as proteinuria severe enough to cause hypoalbuminaemia and oedema. The degree of proteinuria varies but is generally > 3.5 g/24 h and is accompanied by a plasma albumin < 25 g/L. However, it should be remembered that the amount of protein in the urine may decrease as the plasma protein concentration or the GFR falls. Causes of nephrotic syndrome are listed in Table 8.3. In addition to the nephrotic syndrome, glomerular proteinuria is a feature of several other syndromes of nephron injury, and the severity of proteinuria taken together with other clinical findings can allow useful diagnostic classification (Table 8.4).
TABLE 8.3
Causes of nephrotic syndrome
| Children (%) | Adults (%) | |
| Primary renal disease | ||
| Minimal change (steroid responsive) disease | 80 | 25 |
| Focal segmental glomerulosclerosis | 7 | 9 |
| Membranous nephropathy | 1 | 22 |
| Membranoproliferative glomerulonephritis | 10 | 26 |
| Glomerular disease in systemic conditions | ||
| Amyloidosis | – | 7 |
| Diabetic nephropathy | – | 3 |
| Systemic lupus erythematosus | – | 8 |
| Henoch–Schönlein purpura | 2 | < 1 |
Mechanisms underlying glomerular proteinuria
Glomerular injury can occur as a result of either primary or secondary kidney disease and there is no single pathogenetic pathway that will embrace all the possible mechanisms. The term glomerulonephritis is generally reserved for immunologically mediated diseases and excludes other conditions associated with glomerular damage such as diabetes mellitus or amyloidosis.
[/not-level-membership-for-endocrinology-diabetes-and-metabolism-category]
