[level-membership-for-endocrinology-diabetes-and-metabolism-category]CHAPTER 7
The kidneys, renal function and kidney disease
David Makanjuola; Marta Lapsley
CHAPTER OUTLINE
Renal blood flow and its control
RENAL DISEASE AND ITS PRESENTATION
Manifestations of renal disease
Diseases affecting the kidneys
THE ASSESSMENT OF RENAL FUNCTION
Biochemical tests of renal function
ACUTE KIDNEY INJURY (ACUTE RENAL FAILURE)
Obstructive (postrenal) kidney injury
Acute kidney injury in the setting of chronic kidney disease
Metabolic consequences and management of acute kidney injury
Aetiology and pathogenesis of chronic kidney disease
Endocrine control of salt and water balance
ANATOMY
Gross anatomy
The kidneys are located against the posterior abdominal wall on either side of the vertebral column. They lie anterior to the diaphragm and various muscles; their anterior surfaces are covered by parietal peritoneum. The left kidney is posterior to the stomach, pancreas, spleen and descending colon, and the right to the liver, the second part of the duodenum and the ascending colon. Their superior poles are covered by the suprarenal (adrenal) glands. In the adult, the kidneys are about 11–13 cm long, 6 cm wide and 4 cm thick. They weigh approximately 150 g each, yet together receive about 25% of the resting cardiac output. This blood is supplied by the renal arteries, which are branches of the aorta. Venous drainage is into the renal veins, which drain into the inferior vena cava. The kidneys are innervated by sympathetic nerves arising from the sympathetic chains and by parasympathetic fibres arising from the vagus nerve.
Each kidney is covered by a poorly distensible capsule. This limits the swelling that can occur during acute inflammation, and in such circumstances there is an increase in tissue pressure that tends to decrease the glomerular filtration rate (GFR).
Congenital absence of one kidney occurs in approximately 1 in 2400 individuals; this is only of significance if renal surgery is contemplated for any reason. The remaining kidney usually undergoes compensatory hypertrophy. Another common (approximately 1 in 10 000) congenital anomaly is the single ‘horseshoe’ kidney, in which the lower poles of the potential two kidneys are conjoined; renal function is normal but, occasionally, ureteric obstruction may predispose to calculus formation and infection.
Microstructure
Each kidney contains approximately one million functional units, called nephrons. These are tubular structures, consisting of various histologically and functionally distinct elements. Each nephron has a single glomerulus, which is located in the outer part of the kidney (the cortex) and is responsible for the ultrafiltration of the blood. The remainder of the nephron consists of several contiguous tubular structures that progressively modify the composition of the ultrafiltrate before it is passed out as urine. These structures (Fig. 7.1) are:
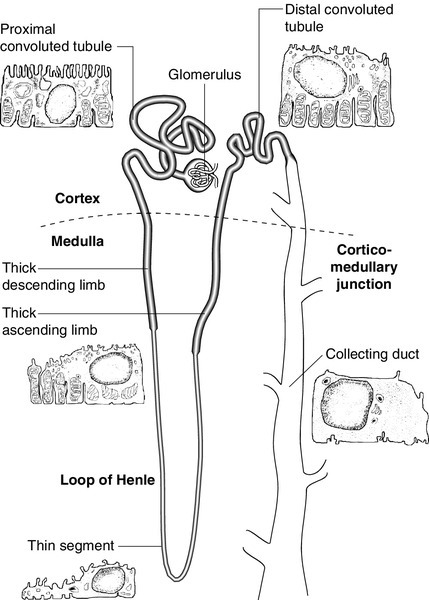
• the proximal convoluted tubule, also located in the cortex
• the loop of Henle, which has the configuration of a hairpin and extends into the deeper part of the kidney (the medulla), then doubles back on itself to return to the cortex
• the distal convoluted tubule, located in the cortex
• the collecting duct, which extends down through the medulla to the renal papillae, whence the urine drains into the renal pelvis. The renal pelvices are drained by the ureters, and the right and left ureters themselves drain into the urinary bladder, where urine is stored prior to voiding.
The glomerulus
Each glomerulus consists of a tuft of capillaries that protrudes into the dilated, blind end of the nephron (the Bowman capsule) (Fig. 7.2). The nephron is composed of epithelial cells, and these and the endothelial cells of the glomerular capillaries are separated by a basement membrane. This visceral basement membrane is continuous with a parietal basement membrane associated with the epithelial cells forming the outer part of the Bowman capsule, which are continuous with the epithelium of the proximal convoluted tubule. The microstructure of the glomerulus is described in detail in Chapter 8.
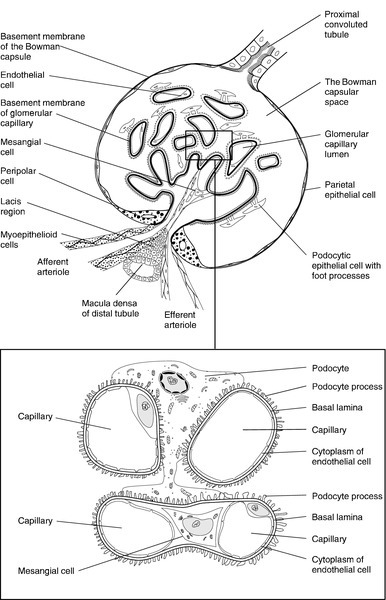
The glomerular filtrate is formed by the passage of fluid from the capillaries through fenestrations in the endothelial cells and through the basement membrane. This is covered on its epithelial aspect by the interdigitating foot processes of the epithelial cells, or podocytes, and the ultrafiltrate passes across a thin membrane (the epithelial slit diaphragm) into the filtration slits between the foot processes and then into the Bowman space, which is continuous with the lumen of the nephron. The selective retention of different constituents of the blood occurs at different stages. The endothelial fenestrations are too small to permit significant passage of erythrocytes, leukocytes or platelets; the major barrier to the filtration of proteins is mechanical and provided by the inner layer of the basement membrane, but the anionic surface layer also selectively retards anionic proteins. Finally, the foot processes and slit diaphragms also act as a mechanical barrier. The relative rate of progression of the constituents of the plasma into the Bowman space, therefore, depends on molecular mass, shape and charge. Glomeruli also contain mesangial cells. These stellate cells are contractile, are involved in the regulation of glomerular filtration and have phagocytic properties. The basement membrane does not extend between the mesangial cells and endothelial cells. This facilitates their ability to phagocytose large particles from the plasma. Their ability to ingest immune complexes plays an important part in the pathogenesis of certain forms of glomerular disease.
The proximal convoluted tubule
This structure is about 15 mm long and is composed of a single layer of inter-digitating epithelial cells united at their apices by tight junctions. The luminal surfaces of these cells have microvillous brush borders that provide the large surface area required for the absorptive function of the proximal tubule. The proximal tubule drains into a short straight segment directed towards the outer medulla and continuous with the descending limb of the loop of Henle.
The loop of Henle
The descending limb of the loop of Henle (Fig. 7.1) is composed of flat epithelial cells. The majority of the loops, whose glomeruli lie in the outer part of the cortex, are relatively short, but those with more deeply situated glomeruli have longer loops (up to 14 mm), which extend down into the medullary pyramids.
The thin descending limb turns back on itself, ascending towards the medulla, and the epithelial cells become cuboid, rich in mitochondria and have an invaginated luminal surface. The thick ascending limb extends towards the glomerulus of the same nephron and then leads into the distal convoluted tubule.
The distal convoluted tubule and collecting duct
The distal convoluted tubule is about 5 mm long and is made up of typical absorptive epithelial cells, although without a brush border. It passes close to the afferent and efferent arterioles of the same nephron. The tubular epithelial cells in this area have a special function in relation to the control of renin secretion and are known as the macula densa. Renin is secreted by myoepithelioid juxtaglomerular cells in the walls of the afferent arterioles. These cells, together with those of the macula densa and some other nearby cells (lacis cells), constitute the juxtaglomerular apparatus.
The distal convoluted tubule also contains intercalated (I) cells, responsible for the secretion of acid.
The collecting ducts are composed of I cells and also principal (P) cells, which become permeable to water under the influence of vasopressin. The ducts descend from the cortex down through the medulla, opening to the renal pelvis from the renal papillae.
Other specialized cells
In addition to the cells of the nephron and blood vessels, the kidneys contain medullary interstitial cells. These characteristically contain lipid droplets and secrete prostaglandins.
Blood vessels
Blood leaving glomerular capillary tufts passes not into venules, but into efferent arterioles. Those draining the cortical glomeruli divide into a capillary plexus that enmeshes the renal tubules. Those draining the juxtamedullary glomeruli form the vasa recta; these are bundles of vessels that plunge into the medulla to varying depths, forming capillary plexi that envelop the loops of Henle and then rejoin to form the ascending vasa recta, which eventually drain into the renal veins. The vasa recta form a countercurrent exchange system (see below), which is essential to the formation of both concentrated and dilute (with respect to plasma) urine.
RENAL FUNCTION
The principal functions of the kidneys are to excrete water-soluble (mostly nitrogenous) waste products and toxins from the body, and to control the volume and composition of the extracellular fluid (ECF). The kidneys also have important endocrine functions. It is perhaps surprising that the kidneys produce some 170 L of glomerular filtrate per 24 h, but only 1–2 L of urine. The bulk of the solvent and solute filtered is reabsorbed, but the control of the processes responsible gives considerable scope for the composition and volume of the urine to be adjusted to meet the physiological needs of the individual.
The physiology of the kidneys is well described in many standard physiology textbooks and will not be recounted in detail in this chapter. Rather, a brief summary is given and certain features that are pertinent to an understanding of the investigation of renal function and the mechanisms and consequences of renal impairment are emphasized.
Renal blood flow and its control
At rest, 20–25% of the cardiac output flows through the kidneys (1.1–1.3 L/min). The bulk of this blood perfuses the cortex; indeed, the flow rate to the medulla is actually less than to most other tissues.
The process of glomerular filtration is essential to renal function and is dependent on an adequate renal perfusion pressure. Autoregulatory mechanisms allow maintenance of renal blood flow and GFR within narrow limits in the face of a wide variety of external variables, including arterial pressure, venous pressure, ureteric hydrostatic pressure and plasma oncotic pressure. Renal blood flow and GFR are independent of mean arterial blood pressure over the range 80–200 mmHg. This is achieved by intrinsic, and humorally and neurally mediated, alterations in the tone of the renal blood vessels and, in particular, of the efferent glomerular arterioles.
Renal hypoperfusion stimulates the release of renin from juxtaglomerular cells. This enzyme converts circulating angiotensinogen to angiotensin I, which is in turn converted by angiotensin-converting-enzyme to angiotensin II, a powerful vasoconstrictor. This contributes to the maintenance of systemic blood pressure and, in the kidney, by causing efferent arteriolar vasoconstriction, helps to maintain the intraglomerular pressure despite a reduction in perfusion pressure. Various other mechanisms are involved in the maintenance of systemic blood pressure, but in the poorly perfused kidney, release of prostaglandins causes vasodilatation which, at least in mild hypotension, helps to maintain renal blood flow. A perfusion pressure of at least 50–60 mmHg is required to overcome the combined hydrostatic and oncotic pressures that oppose filtration; if mean arterial pressure falls below 80 mmHg, renal blood flow and GFR decline rapidly.
Glomerular function
Glomerular filtration is fundamental to the production of urine and to the homoeostatic functions of the kidney. Techniques for measuring the GFR, the amount of filtrate formed per unit time, are described in a later section (p. 131).
The glomerular filtration rate is determined by the balance of pressures across the filtration barrier in the glomerulus, and the physical nature and extent of the barrier itself. The forces include the difference between afferent and efferent glomerular arteriolar pressures (promoting filtration) opposed by the difference in osmotic pressures between the ultrafiltrate and the plasma, and the hydrostatic pressure in the Bowman space. The net filtration pressure is normally approximately 15 mmHg at the afferent end of the capillaries, falling to zero at the efferent ends. Factors influencing the GFR are summarized in Box 7.1.
A decrease in the area available for filtration can occur in many renal diseases in which there is glomerular damage. It can also result from contraction of mesangial cells mediated by agents such as angiotensin II, vasopressin, noradrenaline (norepinephrine), thromboxane A2 and prostaglandin F2; dopamine, natriuretic peptides and prostaglandin E2 have opposite effects. A reduction in filtration area may be an important physiological mechanism for reducing GFR and thus conserving fluid.
Tubular function
The proximal convoluted tubule
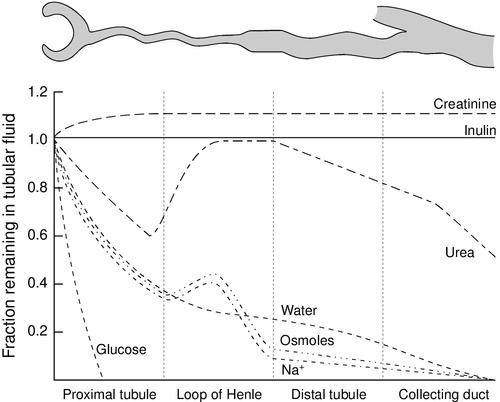
The proximal tubules are responsible for the active reabsorption of the bulk of filtered solute, accompanied by an iso-osmotic amount of water. Thus, virtually all the filtered glucose, amino acids, bicarbonate and potassium are absorbed here, together with some two-thirds of the filtered sodium. Absorption of solutes is an active, energy-requiring process and is isotonic, so that an equivalent amount of water is also absorbed and the fluid entering the descending limb of the loop of Henle is isotonic with plasma.
Transport occurs by way of ion channels, exchangers, co-transporters and pumps; filtered proteins are taken up into tubular cells by endocytosis and broken down into their constituent amino acids. Transport mechanisms have finite capacities and these may be exceeded in some circumstances. For example, glucose begins to be excreted in the urine if the plasma concentration exceeds about 10 mmol/L. Isolated or generalized disorders of renal tubular function result in the excretion of solutes normally reabsorbed in the proximal tubule even at normal plasma concentrations, as discussed further in Chapter 9.
The sodium content of the ECF is the primary determinant of ECF volume, the maintenance of which is critical to life. Given that about 26 000 mmol of sodium are filtered at the glomeruli each day (equivalent to some 8–9 times the total body sodium), it is clearly essential that most of the filtered sodium is reabsorbed. Indeed, the bulk of sodium reabsorption is obligatory. However, an increase in GFR (which potentially could lead to a massive increase in sodium excretion) results in increased proximal tubular sodium absorption and vice versa. This process, termed glomerulo-tubular balance, is thought to be mediated largely through changes in the oncotic pressure in the peri-tubular capillaries. This increases when the GFR increases, owing to the increase in the volume of water removed from the plasma. The re-absorption of several other solutes is affected similarly. Normal kidneys are capable of excreting between virtually zero and more than 400 mmol of sodium per 24 h. The fine control of sodium excretion is achieved in the distal convoluted tubule, where sodium reabsorption is stimulated by aldosterone.
Most of the filtered bicarbonate is absorbed in the proximal convoluted tubule, although indirectly. Hydrogen ions are generated in the tubular cells and are secreted into the tubular lumen (mainly in exchange for sodium) where they combine with the filtered bicarbonate ions. Bicarbonate ions are formed pari passu with hydrogen ions and are co-transported with sodium across the basolateral membranes of the tubular cells into the interstitial space (see Chapter 5).
Some substances are secreted into the urine from the proximal tubule. Examples include penicillins, creatinine, certain steroids and their glucuronides and derivatives of hippuric acid, for example p-aminohippuric acid (PAH). This property of PAH allows physiologists to use measurements of PAH clearance to determine renal plasma flow, but this is not an investigation that is used clinically.
The loop of Henle
This structure provides the countercurrent multiplier that generates the medullary hypertonicity that is essential for the regulation of water excretion and the production of concentrated urine.
Essentially, the process involves the active transport of solute (principally chloride and sodium ions) out of the thick ascending limb of the loop of Henle. This solute is not accompanied by water. As a result, the fluid within the lumen becomes hypotonic and the interstitial fluid surrounding the loop becomes hypertonic. Since water can diffuse out of the thin descending limb, but not the thick ascending limb, the net effect is that the fluid within the loop of Henle and the surrounding interstitial fluid become progressively hypertonic from the corticomedullary junction down into the medulla. There is further sodium and chloride absorption from the thick ascending limb. As water cannot diffuse out of the thick limb, the fluid entering the distal convoluted tubule is hypotonic with respect to plasma.
The anatomical disposition of the vasa recta allows them to act as a countercurrent exchanger, maintaining (though not actively contributing to) the osmotic gradient. The flows of water and solutes in the nephron are summarized in Figure 7.3. Whereas 60–70% of the filtered loads of solute and water have been reabsorbed from the glomerular filtrate at the beginning of the loop of Henle, a further 15% of water is removed during passage through this structure, so that only 10–20% of filtered water and somewhat less solute reach the distal convoluted tubule.
Tubuloglomerular feedback
There is a reciprocal relationship between the rate of flow of fluid through the ascending limb of the loop of Henle and the first part of the distal convoluted tubule of individual nephrons, and the rate of glomerular filtration through the same nephron. Thus, a decrease in flow rate increases filtration and vice versa. This process tends to result in a constant load of solute being presented to the distal convoluted tubule. Feedback is mediated through constriction and dilatation of the afferent arteriole by factors such as renal prostaglandins, adenosine and the renin–angiotensin system, in response to changes in chloride absorption in the macula densa. This mechanism becomes more sensitive when ECF volume is decreased and vice versa.
Tubuloglomerular feedback should not be confused with glomerulotubular balance, the change in proximal tubular sodium reabsorption in response to changes in GFR that has been discussed above and in Chapter 4.
The role of urea
Urea has an essential role in the generation of medullary hypertonicity. The thick ascending limb of the loop of Henle is permeable to urea (and sodium) but not to water. Movement of urea down its concentration gradient into the interstitium contributes significantly to medullary hypertonicity; the inner medullary portion of the collecting duct (see below) is also permeable to urea. The poor concentrating ability of the kidneys in the newborn is due to the decreased availability of urea to maintain medullary hypertonicity. In adults, renal concentrating power is dependent on an adequate protein intake (the source of urea) and is lower on a low protein diet.
The distal convoluted tubule
This part of the nephron is responsible for the ‘fine tuning’ of urinary composition. Aldosterone stimulates sodium reabsorption, generating an electrochemical gradient that allows the secretion (and thus excretion) of potassium and hydrogen ions. Normal distal tubular function is essential to the maintenance of hydrogen ion homoeostasis.
The collecting duct
These structures extend from the ends of the distal convoluted tubules to the renal papillae. Their main function is to permit the reabsorption of solute-free water and thus regulate the osmolality of the urine. This is achieved through the action of vasopressin (antidiuretic hormone). The cells of the collecting ducts are normally impermeable to water. Under the influence of vasopressin, aquaporin-2 water channels are inserted into the tubular wall, such that they become permeable, allowing water to move out of the hypotonic fluid in the tubular lumen into the hypertonic interstitium, so producing concentrated urine. In the absence of vasopressin, no water is removed and the urine is hypotonic.
The extremes of achievable urine osmolality are 30–1400 mmol/kg. When maximally dilute, as much as 13% of the filtered water is excreted and the rate of urine production is about 16 mL/min; when maximally concentrated, < 0.5% of filtered water is excreted and urine production is as little as 500 mL/24 h.
Some solute is also absorbed in the collecting ducts. In the cortical portion, sodium reabsorption takes place in exchange for potassium and hydrogen ions, as in the distal convoluted tubules. The medullary segment is partially permeable to urea, which moves passively out into the interstitium and helps to maintain the high osmolality of the medulla.
Diuresis
While the maximal urine flow rate when a water load is being excreted is approximately 16 mL/min (assuming an average solute load), higher rates are attainable if there are increased quantities of non-reabsorbed solutes in the tubules. This osmotic diuresis is due to the direct osmotic effect of the solutes and to a secondary effect on sodium reabsorption. The retention of water in the lumen of the proximal tubules increases the concentration gradient against which sodium must be reabsorbed; the same factor limits sodium reabsorption in the thick ascending part of the loop of Henle, thus interfering with the generation of medullary hypertonicity. This is the explanation for the diuresis characteristic of hyperglycaemia.
RENAL DISEASE AND ITS PRESENTATION
Introduction
There are many specific renal diseases; these may present with clinical features clearly referable to the kidneys (e.g. a decrease or increase in urine output), but frequently have systemic manifestations (e.g. the ‘uraemic syndrome’ (see p. 142) and hypertension). The kidneys can also be involved in multisystem disease, for example diabetes mellitus, the connective tissue disorders and amyloidosis.
Manifestations of renal disease
There are relatively few cardinal manifestations of renal disease and none is specific to any one underlying disorder. They include anuria, oliguria and polyuria; respectively a complete absence, decreased production and excessive production of urine. Polyuria may come to notice only by virtue of causing excessive production of urine at night (nocturia). These are usually symptoms of disordered renal function, although anuria and oliguria can be secondary to obstruction of the urinary tract and polyuria can be due to extrarenal disease that impairs the ability of the kidneys to concentrate the urine.
Urinary frequency (the passing of urine more frequently than normal, but in normal total 24 h volumes) must be distinguished from polyuria; it is often related to irritation of the urinary tract, for example by infection, but can also be due to prostatic enlargement. Irritation of the urinary tract is frequently accompanied by dysuria – pain on micturition. Patients with renal disease may experience loin pain or have tenderness in the loins. Renal or ureteric colic, an intermittent pain of considerable severity, is characteristic of the passage of urinary calculi.
Patients with renal disease may present with systemic abnormalities, including hypertension, pyrexia, oedema and the ‘uraemic syndrome’. This latter term is used to describe the protean manifestations of renal failure. The urine may appear abnormal in renal disease; the causes are discussed in a later section (p. 130).
Finally, renal disease may be revealed for the first time by the finding of a biochemical abnormality, particularly an elevated plasma creatinine or urea concentration, or positive urine dip-stick tests for protein or blood.
Diseases affecting the kidneys
Although patients with renal disease frequently present with an obvious functional abnormality, for example increased plasma creatinine or proteinuria, these are not specific to any one renal disease.
A full discussion of the nature of individual renal diseases is beyond the scope of this chapter. Many renal diseases specifically affect one part of the kidney. Glomerulonephritis, which is often of immunological origin, can manifest by decreases in the GFR or changes in glomerular permeability (e.g. causing proteinuria). Glomerulonephritis may be of primary renal origin, or occur secondarily to an extrarenal disorder (see Chapter 8).
A considerable number of conditions, both inherited and acquired, limited to the kidneys and having extrarenal manifestations, can cause disorders of renal tubular function. They are discussed in detail in Chapter 9.
The kidneys can be affected by infection, tumours and infiltrative and degenerative disorders. Generalized disorders, particularly shock, hypertension, connective tissue diseases and diabetes, are also frequent causes of renal dysfunction. The kidneys are also susceptible to the effects of toxins, including many widely used drugs (e.g. the aminoglycoside antibiotics and some cytotoxic drugs), as well as industrial and other toxins, and exposure to such agents is another frequent cause of renal dysfunction.
The major part of this chapter is devoted to a discussion of generalized renal failure, both acute and chronic. These are potentially life-threatening conditions and the clinical biochemistry laboratory plays a major part in their diagnosis and management.
THE ASSESSMENT OF RENAL FUNCTION
Introduction
Many techniques are of value in the investigation of the kidneys and renal function. The major groupings of techniques are laboratory tests, particularly biochemical and immunological, radiological imaging and the histological examination of renal tissue usually obtained by percutaneous biopsy.
Imaging techniques: standard radiography, ultrasonography, computed tomography, magnetic resonance imaging, the use of radionuclides and duplex scanning, can provide valuable anatomical information. Such techniques can be used to diagnose, for example, structural abnormalities, tumours, hydronephrosis, polycystic disease and the scarring of pyelonephritis. Radionuclide techniques can also provide information concerning renal function, particularly divided function (the separate function of the two kidneys) and glomerular function. Duplex scanning is a good screening test to assess renal blood flow before proceeding to magnetic resonance angiography or renal angiography.
Useful immunological markers include the detection in serum of antiglomerular basement membrane antibodies (positive in Goodpasture syndrome), antineutrophil cytoplasmic antibodies (in vasculitis), antinuclear antibodies and double-stranded DNA antibodies (in systemic lupus erythematosus), complement components (in various types of glomerulonephritis) and antistreptolysin O antibodies (in post-streptococcal glomerulonephritis). Immunological techniques are also widely used in the histological examination of renal tissue. However, given the pivotal role of the kidneys in body fluid homoeostasis, it is not surprising that biochemical investigations that indicate derangements of homoeostasis are the most widely used tests of renal function. The remainder of this section covers such tests.
Biochemical tests of renal function
Urinalysis
The assessment of renal function should start with examination of the urine. Simple observations on the urine still contribute to the investigation of patients with suspected renal disease. To be valid, they must be made on a freshly passed sample.
Appearance
The normal colour of urine is due to urochrome pigments, and the deepness of the colouration, which can vary from a pale straw colour to deep amber, depends on urinary concentration. There are many causes of abnormal colouration of the urine, some of the more frequently encountered of which are indicated in Box 7.2. In some cases, the colour is pH dependent. The distinction between haemoglobin or myoglobin and other causes of a reddish-brown urine can simply be accomplished by testing the urine with a suitable reagent stick; both myoglobin and haemoglobin will give a positive reaction for ‘blood’.
Turbidity in a fresh sample suggests infection, but can also be due to the presence of fat in patients with the nephrotic syndrome. The presence of chyle in the urine may cause turbidity so severe that the urine appears milky. Anything more than a scant amount of foaming when the urine is shaken, suggests proteinuria. The most frequent abnormal odour is that of ammonia, owing to the presence of urea-splitting microbes, either because the patient has a urinary infection or, in stale samples, because of contamination.
Specific gravity and osmolality
Both of these give an indication of the concentration of the urine. When this needs to be known accurately, for example in tests of renal concentrating ability, measurements of osmolality are essential. A reagent stick assessment of specific gravity may sometimes help in the interpretation of a screening test for proteinuria, since false negative results can occur with very dilute urine and vice versa. However, these reagent sticks detect ionic species only, and underestimate the specific gravity if other substances, for example glucose, are present.
pH
Normal urine is acidic, except after meals. Measurements of urine pH are important in the investigation of renal stone formers and as part of a test of urinary acidification in the investigation of suspected renal tubular acidosis.
Glucose
Glycosuria can occur either because of an increase in blood glucose concentration or because of a low renal threshold for glucose, owing to impaired proximal tubular reabsorption. In the latter instance, the blood glucose concentration will be normal. Renal glycosuria is discussed further in Chapter 9.
Protein
Proteinuria is an important feature of renal disease. Normal urine protein excretion is < 200 mg/24 h and is not detectable with standard reagent sticks (e.g. ‘Albustix’). Pathological proteinuria may be:
• overflow (due to raised plasma concentrations of low molecular weight proteins)
• glomerular (due to increased glomerular permeability to protein)
• tubular (due to decreased tubular reabsorption of filtered protein or saturated reabsorption)
• secreted (derived from the epithelium of the urinary tract).
Further details of the causes, detection and investigation of proteinuria are discussed in Chapter 8 and will not be considered further here.
Urinary sediment
Microscopic examination of the sediment obtained by centrifuging a freshly passed sample of urine frequently reveals a few cells (e.g. erythrocytes, polymorphonuclear leukocytes and cells derived from the kidney and urinary tract), hyaline (or rarely granular) casts composed of uromodulin (Tamm–Horsfall protein), and sometimes fat droplets, mucus threads and pigment granules.
Increases in any of these may occur in renal diseases. Abnormal casts are also frequently present. These are accretions of uromodulin with other material, for example cells or cellular debris. Red cell casts imply haematuria from glomerular disease, and white cell casts, the presence of white cells within the renal tubules. In acute pyelonephritis, there is an increase in polymorphs and various types of cast, in contrast to lower urinary tract infection where polymorphs, but not casts, are increased. Microbiological examination in acute pyelonephritis will usually reveal bacteriuria. In acute glomerulonephritis, haematuria causes the urine to appear reddish-brown or smokey, and the sediment contains large numbers of red and white blood cells, and red cell, haemoglobin and other casts. In chronic glomerulonephritis, the amount of sediment is much less, but there is usually an excess of dysmorphic red blood cells, which suggests that red cells have passed through the glomerular membrane. Red cell and haemoglobin casts are sometimes present. Granular casts are seen in the urinary sediment in many renal diseases, but particularly in acute tubular necrosis; in this condition, casts containing tubular cells are also often present, but red cell casts are uncommon. Characteristic crystals may be seen when specific substances are present in excess or the pH of the urine promotes crystallization, for example, hexagonal cystine crystals, birefringent calcium oxalate crystals, calcium phosphate or uric acid crystals (see Chapter 9).
Other substances
Many other substances can be detected in the urine in appropriate circumstances, for example drugs or bilirubin. Their presence is due to the normal excretory function of the kidneys and is not a reflection of renal dysfunction, so they will not be considered further here.
Measurement of glomerular filtration rate
Glomerular filtration is essential to renal function, and investigations designed to measure the GFR are the most frequently performed tests of renal function. The factors that determine the GFR have been discussed above. Its measurement is based on the concept of clearance – the determination of the volume of plasma from which a substance is completely removed (the plasma is ‘cleared’ of the substance) by glomerular filtration during its passage through the kidney. Clearance is a theoretical concept – no substance is cleared completely from the plasma in this way – but can nevertheless be used as a valid measure of the GFR.
For the clearance of a substance to equal the GFR, it must be freely filtered by the glomeruli and eliminated from the body solely by this route (e.g. it must not be metabolized by the liver or secreted into or reabsorbed from the urine). The substance should be non-toxic and accurate measurement should be easily available in a routine laboratory. The clearance of a substance requires measurements of the plasma concentration and urinary excretion rate:
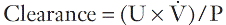
where U is the concentration of the substance in the urine, 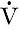 is the rate of formation of urine and P the plasma concentration of the substance. It should be noted that
is the rate of formation of urine and P the plasma concentration of the substance. It should be noted that 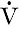 is a rate; it has the dimensions (volume/time) and its determination requires the collection and measurement of the total amount of urine formed over a known period of time. The units of the various quantities are usually adjusted so that the clearance is expressed as mL/min.
is a rate; it has the dimensions (volume/time) and its determination requires the collection and measurement of the total amount of urine formed over a known period of time. The units of the various quantities are usually adjusted so that the clearance is expressed as mL/min.
The critical properties of some of the substances used in clearance measurements to assess GFR are shown in Table 7.1.
TABLE 7.1
Properties of substances used to assess glomerular filtration rate

a The ideal substance would have all these properties. DTPA, diethylenetriaminepentaacetic acid.
Clearance can also be used to determine renal plasma flow, using a substance such as p-aminohippuric acid, which is almost completely cleared from the blood in a single passage through the kidneys by a combination of glomerular filtration and secretion.
Inulin clearance
Inulin is a plant polysaccharide that satisfies all the physiological criteria mentioned in the previous section. The measurement of inulin clearance remains the ‘gold standard’ for the estimation of GFR. However, it is a relatively complex procedure and, although test kits are commercially available, it is infrequently used in clinical practice. In essence, the test involves the injection of a bolus dose of inulin, followed by a maintenance infusion designed to produce a constant plasma concentration. Once this has been achieved, a series of timed urine samples is collected and blood is drawn for the measurement of inulin at the midpoints of the collection periods. The GFR is taken as the mean of the inulin clearances for each period.
Creatinine clearance
Creatinine is an endogenous substance, a normal product of muscle metabolism. Its rate of production is fairly constant from day-to-day, being determined by muscle bulk rather than by activity. Creatinine is removed from the body mainly by glomerular filtration and its clearance can be measured as an index of GFR, although in the UK, it is much less frequently used than previously.
Creatinine is actively secreted into the urine, so that its clearance tends to overestimate the GFR. This effect is of little significance at normal filtration rates, the ratio (creatinine clearance/inulin clearance) being between 1.1 and 1.2, but in advanced renal disease the contribution of active secretion to the total amount of creatinine excreted in the urine becomes high in relation to the amount filtered, and creatinine clearance may significantly overestimate the true GFR. This is despite the fact that in established renal failure, bacterial degradation of creatinine secreted into the gut may contribute approximately 2 mL/min to the total clearance.
Inaccuracies arising from methodological problems with creatinine measurement have largely been overcome, but the major cause of inaccuracy in the determination of GFR by creatinine clearance is the accurate measurement of urine volume. Traditionally, patients have been required to collect urine over a 24 h period. This is a convenient time, but requires the patient to void their urine completely at the beginning of the 24 h period and collect all the urine passed over the ensuing 24 h, making a final collection at the end of this period. The possibilities for mistakes resulting in an incomplete collection are considerable and even under ideal conditions, for example in a metabolic unit with highly motivated patients, the coefficient of variation for repeated measurements in the same individual is > 10%. The critical difference (the amount by which two estimates must differ to give a 95% probability that there has been a true change in GFR) is therefore > 33%. The accuracy of estimates of the GFR by measurement of creatinine clearance may be improved by making two or more consecutive 24 h urine collections, but this is often not practical even if it is acceptable to the patient.
There is, however, nothing special about the 24 h period. The use of shorter periods, although more convenient for the patient, may reduce accuracy through inadequate bladder emptying, but a good compromise is to make a collection overnight. As long as the bladder was emptied at the beginning of the test (before retiring) and when the final collection was made (on rising), the urine production rate can be calculated and substituted in the 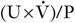 formula.
formula.
It is essential that a reliable method is employed to measure plasma and urine creatinine concentrations. Colorimetric assays tend to overestimate creatinine since they detect non-creatinine chromogens. But even when reliable methods are used, it should be appreciated that the creatinine clearance is based on four measurements: plasma and urine creatinine concentrations, urine volume and time. Each has an inherent inaccuracy and the overall analytical variance will be the sum of the individual variances.
Plasma creatinine concentration
The way in which creatinine is handled by the kidney, coupled with the fact that, in any one individual, the rate of production is relatively constant from day-to-day, means that the plasma creatinine concentration alone can be used as an index of renal function. But, although it is widely used, it has a number of disadvantages and these must be borne in mind when interpreting plasma creatinine concentrations. Perhaps the most important becomes apparent from considering the familiar 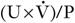 formula for deriving the GFR. Plasma creatinine concentration is inversely related to GFR, not directly related. This means that a plot of plasma creatinine concentration against GFR has the form of a hyperbola (Fig. 7.4). At any concentration of creatinine, halving the GFR will double the plasma creatinine concentration. But given the wide reference range for plasma creatinine concentration in the healthy population, this means that in an individual, the plasma creatinine concentration could be within the reference limits, yet the GFR be only one-half of normal.
formula for deriving the GFR. Plasma creatinine concentration is inversely related to GFR, not directly related. This means that a plot of plasma creatinine concentration against GFR has the form of a hyperbola (Fig. 7.4). At any concentration of creatinine, halving the GFR will double the plasma creatinine concentration. But given the wide reference range for plasma creatinine concentration in the healthy population, this means that in an individual, the plasma creatinine concentration could be within the reference limits, yet the GFR be only one-half of normal.
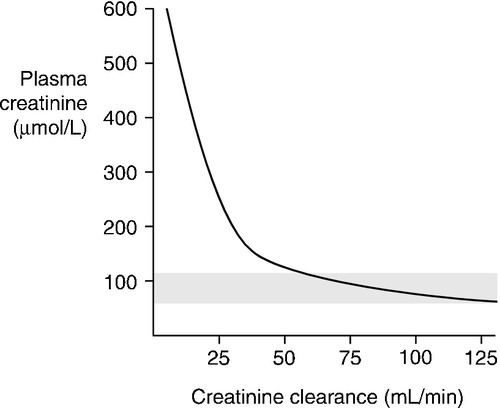
This is of critical importance to clinical practice, since in early renal disease (when it might be supposed that therapeutic intervention would be most beneficial), the plasma creatinine concentration may be ‘normal’ – that is, within the reference range – in spite of the GFR being reduced. Plasma creatinine concentration is thus a relatively insensitive index of mild renal functional impairment. At low values of GFR, however, it becomes extremely sensitive, although its accuracy declines because of the increased contribution of tubular secretion to overall renal excretion.
Other factors that must be taken into account in assessing the significance of plasma creatinine concentrations include muscle mass, diet (a recent meal including meat, particularly if stewed, may cause a transient increase in plasma creatinine concentration) and the presence in the plasma of substances that interfere with the assay. Ketones, bilirubin, cephalosporins and spironolactone have all been implicated, depending on the method used, but especially in Jaffé-(picric acid) based assay systems. Enzymatic assays for creatinine are said to be more specific than older colorimetric methods, but they are still prone to interference by certain drugs such as acetylcysteine and phenindione.
The importance of muscle mass is exemplified by the changes that occur with age. In healthy individuals, GFR, and thus creatinine clearance, declines with increasing age from about the end of the 4th decade, at a rate of approximately 1 mL/min per year. However, plasma creatinine concentration does not normally rise with age; this is because of a decrease in production rate, thought to reflect the tendency for muscle mass to fall with age.
Although comparison of a measured plasma creatinine concentration with a reference range can be misleading, the fact that intraindividual variation is less than interindividual variation means that the detection of a change in creatinine concentration in an individual provides a more sensitive indication of a change in renal function. Nevertheless, the biological and analytical variance (even using the best assays) results in a critical difference of about 17% (20 μmol/L at a plasma creatinine concentration of 120 μmol/L).
In patients with advancing renal disease, the progressive loss of renal function with time can be expressed as a graph of reciprocal plasma creatinine concentration against time (since GFR is proportional to 1/[plasma creatinine]). A steady loss of renal function will cause this plot to be rectilinear (Fig. 7.5). Such plots are useful in patients with irreversibly declining renal function to help predict when renal replacement will become necessary, so that, for example appropriate means for vascular access for haemodialysis can be provided.
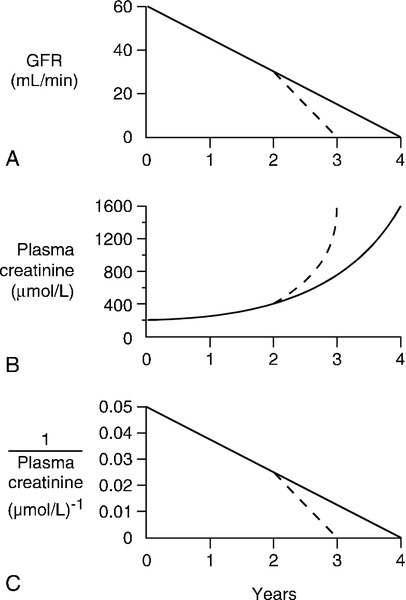
FIGURE 7.5 The progression of chronic kidney disease. Hypothetical curves to show (A) the decline in glomerular filtration rate (GFR); (B) the increase in plasma creatinine concentration and (C) the decrease in reciprocal plasma creatinine concentration. The broken lines show the changes that might be expected if the decline in function was to accelerate for any reason (see Box 7.6).
Given the problems associated with the measurement of plasma creatinine concentration and creatinine clearance, the practical application of these measurements to patients with, or suspected of having, renal disease is not straightforward. Since the rate of creatinine production is relatively constant in most individuals (the essential assumption being that lean body mass is constant), one approach is to measure the GFR (preferably by a reliable technique, see below) and the plasma creatinine concentration simultaneously and then to follow progress with serial plasma creatinine measurements.
Calculated creatinine clearance and estimated glomerular filtration rate
Given that the creatinine cleared by the kidney is almost entirely derived from muscle, and that muscle mass is related to body mass and to age, it is possible to derive formulae to estimate the creatinine clearance, and hence the GFR, from the plasma creatinine concentration. More than 25 different formulae have been suggested to correct for age, weight, gender and ethnic origin. The empirically derived equation of Cockcroft and Gault is one of the earliest and the best known of these. However, this and all similar formulae depend on measurements of plasma creatinine, and their accuracy can be impaired by all the factors that affect this. The early formulae, including that of Cockcroft and Gault, were derived using Jaffé based methods for the measurement of creatinine. Laboratories should take into account the relationship between their creatinine assay and the original assay used to derive any equation before advising clinicians on interpretation. None of the correction equations performs well in the physiological range of GFR, which limits their usefulness, for example in monitoring patients with diabetes or assessing potential kidney donors.
Cockcroft and Gault
The Cockcroft and Gault formula for males is:
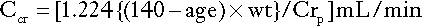
where Ccr is creatinine clearance, Crp is plasma creatinine concentration in μmol/L, age is measured in years and wt is body weight in kilograms. For females, Ccr is 15% less than for males, so that the figure 1.224 in the above formula is replaced by 1.04.
Modifications to the formula are required before it can be used in children or obese adults as it assumes that increasing weight is related to increasing muscle rather than fat mass. The formulae developed for children are particularly useful, since there are considerable practical difficulties in making quantitative urine collections in this age group. The commonest estimation used in the UK is the Schwartz formula:
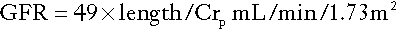
where length is measured in centimetres and plasma creatinine in μmol/L.
MDRD
The UK has adopted the system originally devised in the USA for deriving an estimate of GFR (eGFR), which can be used for population screening for kidney disease. This is based on the abbreviated MDRD (Modification of Diet in Renal Diseases) study equation. It is a useful estimate of GFR in adults up to a value of 60 mL/min per 1.73 m2 body surface area. The equation was developed from measurements in an independent sample of 1070 individuals. Over 90% of the estimates were within 30% of the measured GFR (using 125I-iothalamate), with only 2% having an error of more than 50%.
There are four variables (age, sex, race and creatinine concentration), but the equation only requires one biochemical value. The equation is being validated in other patient groups, including patients with type 1 diabetes without microalbuminuria, those with mild decreases in renal function or a normal GFR, and non-US populations, for example Indian and Chinese.
The original MDRD equation included an additional two variables (albumin and urea), but there is very little advantage in using it, as any small gain in accuracy is offset by the increase in variability from the extra measurements.
The abbreviated MDRD formula for Caucasians is:
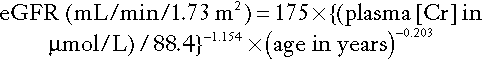
The factor 88.4 is required to convert μmol/L to mg/dL (the units for which the formula was derived). This equation applies to males; for females, the result is multiplied by 0.742; for African-Caribbeans, it is multiplied by 1.212.
The original abbreviated MDRD formula had 186 as the multiplier rather than 175; the change was made to align the formula with measurements of creatinine by methods whose calibration is traceable to the reference method (isotope dilution with mass spectrometry).
In a study of serum creatinine measurements in laboratories, the method group mean bias for a specimen with an assigned value of 79.7 μmol/L varied from − 5.25 to + 27.4 μmol/L. In order to standardize values for eGFR, UKNEQAS (UK National External Quality Assessment Service) has calculated additional corrections (slope and intercept adjusters) to align individual manufacturers’ methods to the reference method.
It should be noted that the MDRD equation for eGFR has not been validated for use in children, acute kidney injury, pregnancy, oedematous states, muscle wasting diseases, amputees or malnourished individuals. It is less accurate in obese than in lean individuals. When the eGFR is > 60 mL/min, many laboratories report the value as > 60 mL/min per 1.73 m2, owing to decreasing precision and accuracy at higher values of GFR. Values above 90 mL/min should never be reported as an absolute figure, because the correlation with measured GFR is poor.
CKD-EPI
The Chronic Kidney Disease Epidemiology Collaboration group produced a set of formulae based on pooled data from several studies, which correlate better with measured GFR than the original MDRD formula, especially at values > 60 mL/min per 1.73 m2. However, GFR is overestimated in certain groups of patients, especially those with a low body mass index. Different formulae are applied depending on the value of the serum creatinine. Further details are available on the websites of various renal organizations.
Definition of CKD using eGFR
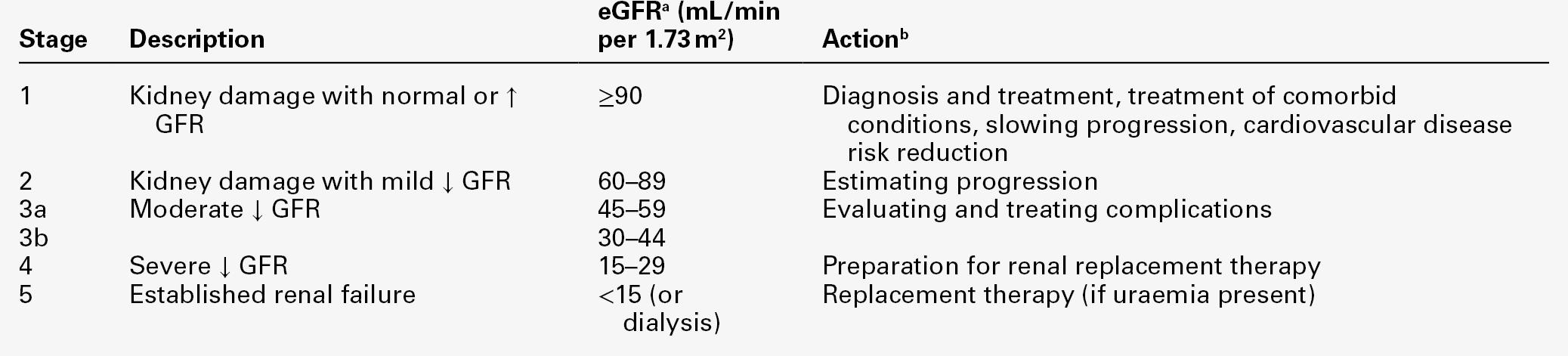
A persistent reduction in eGFR (< 60 mL/min) is defined as chronic kidney disease (CKD). However, patients with markers of kidney damage, like persistent albuminuria or abnormalities on imaging (such as polycystic kidneys) or on renal biopsy, are defined as having CKD even if their eGFR is normal.
The stage of CKD can be assigned based on the degree of renal GFR reduction, irrespective of diagnosis, according to the Kidney/Disease Outcomes Quality Initiative (K/DOQI) classification (see Table 7.2), and used to guide the appropriate management.
Cystatin C
This is a low molecular weight (13.4 kDa) protein, a cysteine protease inhibitor that is present on the surface of all nucleated cells and from which it is shed into the plasma and excreted by glomerular filtration. Its plasma concentration is much less dependent than that of creatinine on weight and height, muscle mass, age or sex, and many studies have suggested that its plasma concentration is a more sensitive index of mild renal impairment than that of creatinine. Estimates of GFR based on either cystatin C measurements alone or combined with creatinine, are more accurate than those based on creatinine alone. Plasma cystatin C concentration may be increased in malignancy, hyperthyroidism and by treatment with corticosteroids (a disadvantage in kidney transplant recipients), independently of the GFR. However, its greatest drawback is that it has to be measured by immunoassay, which is much more expensive than the usual colorimetric or enzymatic techniques used to measure creatinine. Although not widely available in routine laboratories in the UK, measurement may have a role in the detection of early renal impairment in patients with extremes of muscle bulk, such as body builders and small elderly women and in postpubertal teenage boys, for whom children’s reference ranges for creatinine are inappropriate.
Plasma urea concentration
Although of great historical importance and still widely used as a test of renal function, the measurement of plasma urea concentration suffers from many disadvantages for this purpose. Although the plasma urea concentration can provide useful information in some circumstances, particularly when it can be compared with the plasma creatinine concentration, the latter, in spite of the problems discussed above, is the more reliable test of renal function.
Urea is the end-product of the metabolism of many nitrogenous substances, particularly amino acids. It is freely filtered at the glomerulus, but its plasma concentration is dependent in part on its rate of formation, which can vary widely according to the overall rate of protein turnover and can also be affected by hepatic function. For example, plasma urea concentration frequently rises following a gastrointestinal bleed, owing to increased urea formation from the digestion of the blood in the gut, irrespective of any effect of the blood loss on GFR, while patients with combined hepatic and renal failure may have normal plasma urea concentrations because of decreased production despite decreased excretion. There is significant passive reabsorption of urea from the lumen of the nephron, and this increases at high plasma urea concentrations (as in renal failure) and if the rate of flow of fluid through the nephron is low (as when GFR is decreased by dehydration). The familiar observation that the plasma urea concentration tends to rise before the creatinine in patients with ‘prerenal’ renal impairment (see p. 137) reflects this latter fact.
Plasma β2-microglobulin
This protein, a component of the major histocompatibility (HLA) complex, is shed into the plasma at a constant rate and, having a molecular weight of only 11 815 Da, passes freely through the glomeruli. As a result, its plasma concentration is normally very low (< 2 mg/L), but it rises if renal function is impaired and can reach 40 mg/L. There is a direct relationship between plasma β2-microglobulin concentration and GFR, but its measurement has not been widely adopted for estimating the GFR for methodological reasons. Further, there are a number of conditions in which the production of β2-microglobulin is increased (e.g. lymphoid tumours and some inflammatory diseases) and these can result in increased plasma concentrations that do not reflect decreased clearance.
Isotopic techniques for measuring glomerular filtration rate
A number of radiolabelled compounds that are excreted entirely or largely by glomerular filtration have been used in tests designed to measure the GFR. These include 51Cr-labelled ethylenediaminetetraacetic acid (EDTA), 125I-iodothalamate and 99mTc-diethylenetriaminepentaacetic acid (DTPA). The Cr-EDTA-derived GFR is the accepted isotopic gold standard and is used when a very accurate estimate of GFR is required, for example in the work-up of potential kidney donors. In the simplest type of test, a single bolus injection is given and accurately timed blood samples are collected over 2 h (longer may be necessary in patients with renal impairment). The logarithm of plasma radioactivity is plotted against time and extrapolated back to zero time, to allow calculation of the notional initial volume of distribution. The GFR is then given by:
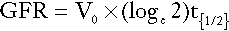
where Vo is the notional initial volume of distribution and t1/2 is the half-life for the decrease in plasma radioactivity.
Such methods compare reasonably well with the results of inulin clearance, but their accuracy is enhanced if classic clearance techniques are used. For this, the marker compound is given during a water diuresis and a series of measurements of plasma and urine radioactivity are made. The clearance formula is used to calculate individual clearances and the GFR is taken as the mean of these.
99mTc-DTPA is also used as a renal scanning agent (although the dose of radioactivity required is approximately ten times greater than for measurement of GFR). However, gamma camera scanning following the administration of this agent can be used to measure the individual contribution of each kidney to the overall GFR.
Other tests of renal function
Tests of proximal tubular function include the measurement of phosphate reabsorption (see Chapter 6) and the detection of aminoaciduria and glycosuria. The causes of glycosuria are discussed in Chapters 9 and 15. Amino aciduria can be due to increases in plasma amino acid concentration (‘overflow amino aciduria’), as occurs in several inherited disorders of amino acid metabolism, or to decreased renal tubular reabsorption of normally filtered amounts of amino acids (‘renal amino aciduria’). Renal amino aciduria can be due to isolated defects of amino acid transport or to generalized proximal tubular dysfunction, and is discussed in Chapter 9.
Tests of distal tubular function include formal tests of urinary concentration and dilution and of urinary acidification. These are discussed in Chapters 4 and 9. The assessment of urinary sodium excretion in relation to physiological requirements plays an important part in the investigation of renal function in certain circumstances, as discussed in the section on acute kidney injury (see later). In general, however, tests of renal tubular function are employed much less frequently than tests of glomerular function in the investigation of renal disease.
ACUTE KIDNEY INJURY (ACUTE RENAL FAILURE)
Introduction
Acute kidney injury (AKI) has now replaced the term acute renal failure. The new terminology enables healthcare professionals to consider the disease as a spectrum of injury. This spectrum extends from less severe forms of injury to more advanced injury requiring renal replacement therapy (RRT). Clinically, AKI is characterized by a rapid reduction in kidney function resulting in a failure to maintain fluid, electrolyte and acid–base homoeostasis. Acute kidney injury is potentially a life-threatening condition and, developing as it often does in patients who are already severely ill, it has a high mortality, despite the widespread availability of effective renal replacement therapy.
In many patients with AKI, there is oliguria (a urine flow rate of < 15 mL/h in the adult). However, this is not universally present. In particular, non-oliguric AKI may be seen in patients with burns, liver disease and drug-induced renal damage. Occasionally, there is no urine production at all (anuria). This can occur with particularly severe renal disease but is more frequently due to urinary tract obstruction (to which it is an important clue).
It is important to appreciate that a diagnosis of AKI on its own is incomplete and does not imply any particular cause. The principal clinical features of AKI are summarized in Table 7.3.
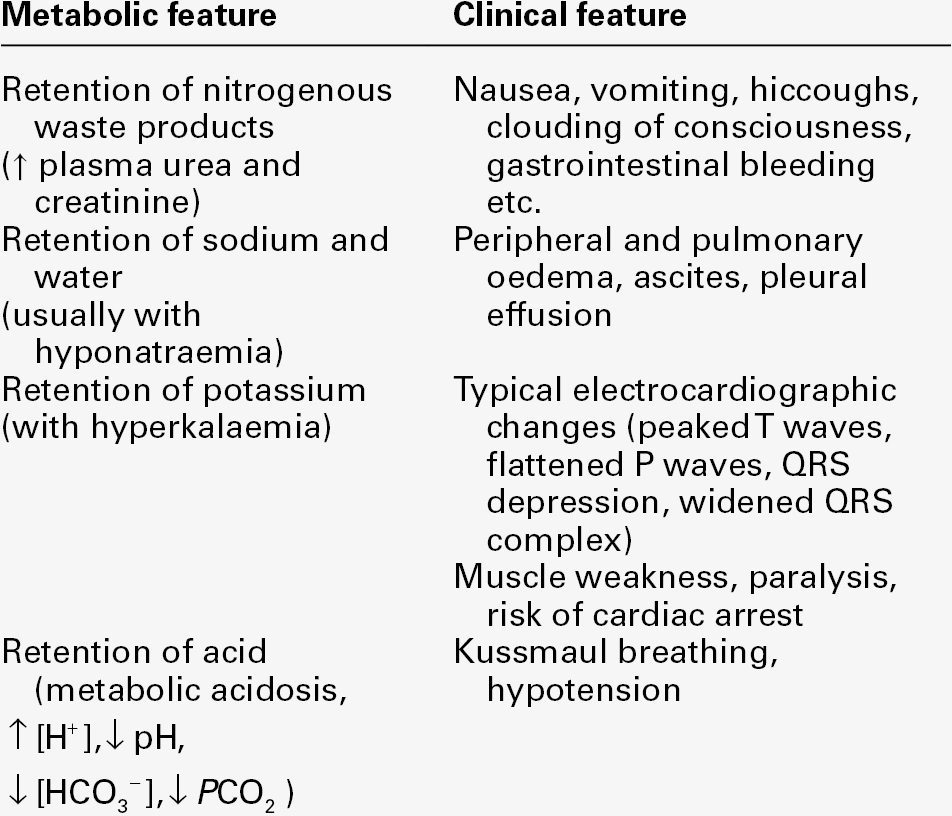
The use of the adjective ‘acute’ tends to emphasize the importance of the period of time over which the condition develops. While it is true that this time is usually only a few hours (in contrast to that for chronic kidney disease, CKD), a more fundamental difference between these two syndromes is that AKI is usually potentially reversible, whereas in CKD, there is progressive, irreversible loss of renal function over a period that can range from months to tens of years, leading eventually to established renal failure, a condition in which renal replacement (e.g. long-term dialysis or transplantation) is required for continued survival.
Classification and causes
As has been discussed above, the primary determinants of the GFR are the hydrostatic pressure of blood in the glomerular capillaries, the intrinsic physical properties of the glomeruli and the hydrostatic pressure of fluid in the lumina of the nephrons. It follows that changes in any one or more of these may cause a decrease in GFR. Acute kidney injury has traditionally been classified into prerenal (due to impaired renal perfusion), intrinsic renal and postrenal (obstructive) causes (Box 7.3). This classification, though particularly useful in formulating an approach to diagnosis and management, has limitations; in any patient, more than one factor may be contributing to the development of renal failure. Furthermore, both ‘prerenal’ and ‘postrenal’ AKI can lead to the development of intrinsic renal damage. Also, intrinsic renal disease can have both detrimental effects on renal perfusion and lead to obstruction of urinary flow. Finally, the syndrome of acute kidney injury can develop in patients with CKD as when, for example, complete bladder outflow obstruction leads to retention of urine and AKI in a man who already has chronically impaired renal function as a result of partial obstruction due to prostatic hypertrophy.
Prerenal acute kidney injury
This type of injury is characterized, at least in its early stages, by a lack of structural renal damage, preservation of normal tubular function (e.g. concentrating ability, sodium reabsorption) and rapid reversibility provided that the underlying cause is managed appropriately. In essence, it is a consequence of reduced renal perfusion, secondary to either cardiovascular insufficiency (hypovolaemia and hypotension) or a derangement of intrarenal haemodynamics. Some causes are indicated in Box 7.3. In many hospitals, a significant proportion of cases of acute kidney injury follows trauma or surgery and may be exacerbated by the administration of nephrotoxic drugs or coexistent medical conditions.
In health, renal blood flow and GFR remain unchanged over a wide range of renal perfusion pressures, but a fall in perfusion pressure below a mean of about 80 mmHg leads to a reduction in renal blood flow and GFR.
Homoeostatic mechanisms involved in the defence of ECF volume include increased sodium reabsorption and increased water retention. The major stimulus to sodium reabsorption is through angiotensin II-induced secretion of aldosterone. Water reabsorption is stimulated by increased vasopressin secretion; stimuli to vasopressin secretion in this context may include hypovolaemia, angiotensin II and, if hypovolaemia is due to hypotonic fluid loss, increased plasma osmolality. In addition, vasoconstriction of efferent glomerular arterioles by angiotensin II increases the filtered fraction of the plasma, thus increasing the protein content (and oncotic pressure) of blood in these vessels and in the peritubular capillaries, so enhancing proximal tubular reabsorption of water.
Diagnosis
The accurate diagnosis of prerenal AKI is important, since rapid intervention may prevent progression to intrinsic AKI. In prerenal AKI, the plasma urea concentration is usually increased disproportionately to that of creatinine. As urine flow rate falls, the diffusion of urea from the lumen of the nephron into the interstitial fluid increases. An increase in urea may also be related to the increased catabolism that frequently is present as a result of the underlying disorder. In contrast, in intrinsic kidney injury, plasma urea and creatinine concentrations tend to rise in parallel.
Urinalysis may be informative and should be performed in all patients with AKI. As discussed on page 130, abnormalities apart from the presence of hyaline casts are uncommon in prerenal AKI, but are invariably present in intrinsic AKI. The fact that tubular function is intact in prerenal AKI underlies a number of diagnostic tests (see Table 7.4). In prerenal AKI, the homoeostatic responses to hypovolaemia described above should maximize renal sodium reabsorption and water retention. In consequence, the urine will tend to be concentrated and have a low sodium content, whereas if there is tubular damage, there will be a failure of this response, and the urine will be dilute and contain significant quantities of sodium. Although in general these theoretical conclusions are supported by clinical observations, considerable overlap may occur in the results obtained in patients with prerenal and established AKI. It must be emphasized that the tests are only useful if there is oliguria, and may be vitiated if the patient has been given a diuretic (which will increase sodium excretion, even in prerenal AKI), or mannitol or an X-ray contrast medium, both of which increase urine osmolality even in intrinsic AKI.

Although these tests are of help in management, the diagnosis of intrinsic AKI may only be made on the basis of an inexorably rising plasma creatinine concentration despite correction of any ‘prerenal’ factors that may have been identified.
Management
The management of prerenal AKI involves rapid restoration of euvolaemia, discontinuation of potentially nephrotoxic drugs and increasing tubular flow to prevent tubular obstruction. Delay in achieving euvolaemia increases the risk of progression to intrinsic kidney injury. Fluid replacement must be done with a view to achieving adequate cardiac output but avoiding fluid overload. In patients with shock, it may be necessary to infuse vasoactive substances to achieve an adequate blood pressure.
Intrinsic acute kidney injury
Intrinsic AKI can be a progression from a prerenal phase, but many conditions causing AKI do so without a prerenal component. Nephrotoxins, intrinsic renal disease (e.g. glomerulonephritis) and systemic disease affecting the kidneys (e.g. septicaemia) are all important causes of kidney injury (see Box 7.3).
The term ‘acute tubular necrosis’ is sometimes used synonymously with ‘intrinsic AKI’, but should not be. Ischaemic injury and many nephrotoxins cause acute tubular necrosis, but in other cases of renal injury, the brunt of the damage is borne by the glomeruli. Although the treatment in such patients will involve management of the kidney injury per se, the prognosis often depends on the underlying cause.
Diagnosis
The distinction between prerenal and intrinsic AKI has been discussed above. In addition, proteinuria is usually present in intrinsic injury. The urinary sediment frequently contains epithelial cells, both free and in casts, and in kidney injury due to glomerulonephritis, haematuria and red cell casts are present.
The plasma biochemical disturbances are similar in all types of kidney injury though without intervention, they become more severe if prerenal AKI progresses to intrinsic injury. The plasma urea and creatinine concentrations then tend to rise in parallel.
Special investigations that may be of value (but will not be required in all cases) include radiology (ultrasound, isotopic scanning, CT and MRI scans) and renal histology.
Ultrasound is helpful in the exclusion of urinary tract obstruction. It shows the size and cortical thickness of the kidneys (small kidneys imply chronicity). Asymmetry of renal size may suggest renovascular disease. This can be confirmed by duplex scanning. If poor images are obtained, blood flow can be determined by magnetic resonance angiography (MRA). This technique does not require the use of X-ray contrast media, but in patients with eGFR < 30 mL/min, the gadolinium which is used as a contrast agent for MRI scanning has been implicated in the development of a condition known as nephrogenic systemic fibrosis. There is no known cure for this condition. Computed tomography (CT) scanning is also useful in further definition of anatomical renal tract abnormalities. Contrast-enhanced CT scanning can, however, exacerbate renal impairment by causing contrast nephropathy. It should, therefore, be used with caution in patients with acute kidney injury.
Renal biopsy is not required if the cause of AKI is obvious (e.g. post-trauma), but will inform management if the development of kidney injury is unexpected and if intrinsic renal disease, for example glomerulonephritis, is suspected.
Acute tubular necrosis
Pathogenesis
While the reduction in GFR that occurs with renal hypoperfusion is readily explicable, the cause of its persistence after restoration of the circulation is a more complex issue, which is still not fully understood. Several mechanisms may be involved (Fig. 7.6). These include continued renal vasoconstriction due to the intrarenal release of vasoactive substances, such as endothelin and prostaglandins, and to angiotensin II (renin secretion may remain high secondary to decreased delivery of solute, especially sodium, to the macula densa); direct damage to the glomeruli resulting in decreased filtration, and physical obstruction of the lumina of nephrons by swollen tubular cells or tubular debris. Diffusion of fluid from the lumina of nephrons into the interstitial tissue through damaged tubular walls (stimulated by the difference in oncotic pressures), will tend to oppose the process of glomerular filtration.
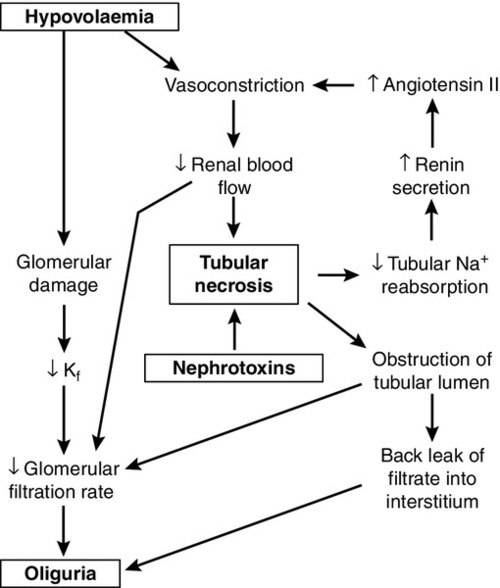
There is evidence from experimental models to support all these mechanisms. In all probability, all are involved in the pathogenesis of acute tubular necrosis, but the relative contributions of the various mechanisms are likely to differ in each case.
Whatever the cause, however, injury, whether nephrotoxic or ischaemic, appears to be central to the development of acute tubular necrosis, and the restoration of normal renal function requires not only restoration of glomerular filtration, but also regeneration of the tubular epithelium of nephrons.
Natural history
The natural history of acute tubular necrosis (ATN) can be divided into three phases: the initial, oliguric phase; a diuretic phase, as glomerular filtration begins to recover, but a combination of persisting tubular damage and a high osmotic load causes a diuresis, and a recovery phase. The time course is variable between a few days and several months. Although patients who survive usually recover normal renal function, a small number develop renal cortical necrosis, from which functional recovery is not possible.
Exceptions occur and some patients do not have an oliguric phase. The details of management of each phase are different although the principles – avoidance of complications and the provision of renal replacement therapy as required – are common to all patients with AKI.
The mortality of AKI has decreased considerably with the widespread availability of renal replacement treatment, but remains in the region of 50%. It is related not only to the severity of the kidney injury and the efficiency of treatment, but also to the underlying cause and any comorbidities, for example multiple trauma. Acute kidney injury frequently occurs in patients who are already severely ill. The prognosis is generally better in non-oliguric AKI than when oliguria is present.
Obstructive (postrenal) kidney injury
The possibility of an obstructive cause must be considered in any patient with AKI. The history and examination (e.g. previous symptoms suggestive of prostate disease, or a palpable bladder on clinical examination) may provide a clue. Ultrasonography may reveal dilatation of the ureters if there is ureteric obstruction.
Obstructive AKI may be due to urethral (e.g. benign prostatic hypertrophy), bilateral ureteric (e.g. calculi, retroperitoneal fibrosis) or intrarenal (e.g. light chain precipitation in myeloma) obstruction. Any of these conditions can also cause chronic kidney disease.
The procedures involved in the diagnosis of obstructive AKI often reveal the cause. Management is directed at restoring a normal flow of urine, for example by percutaneous nephrostomy, or removal or ultrasonic destruction of calculi. Relief of obstruction often leads to a temporary diuresis and natriuresis, and the urine output should be monitored closely to guide fluid replacement therapy.
Acute kidney injury in the setting of chronic kidney disease
Acute kidney injury can occur and precipitate presentation in patients with pre-existing (chronic) kidney disease. Clues to this possibility are indicated in Box 7.4. In practice, it is often necessary to manage the acute event and investigate the patient for chronic renal disease later.
Hepatorenal syndrome
Patients with liver disease can develop acute kidney injury. The causes may be common (e.g. paracetamol (acetaminophen) toxicity), or independent. Prerenal and intrinsic renal disease can occur. The hepatorenal syndrome is a unique form of AKI that can develop in patients with chronic liver failure. It is discussed in detail in Chapter 14. The hepatorenal syndrome can progress to acute tubular necrosis; this has a particularly poor prognosis and recovery usually only occurs if there is some improvement in hepatic function.
Metabolic consequences and management of acute kidney injury
The importance of prevention has already been emphasized. Many cases of AKI occur in patients at predictably high risk of developing the condition.
Accurate diagnosis is essential in the management of acute kidney injury. It is essential to consider the following possibilities:
• is there a prerenal component?
• is there any urinary tract obstruction?
• is there any intrinsic renal disease?
• is there a chronic component?
The approaches to answering these questions have been discussed above.
If renal function is not rapidly restored to normal despite appropriate management of any antecedent condition, the aim of treatment becomes the prevention of the consequences of the condition. There are two aspects to management: general measures and, if these alone are insufficient, renal replacement therapy. Specific measures to treat the underlying cause should, of course, be continued. All patients require rigorous clinical and laboratory monitoring throughout.
General management
Fluid and electrolyte balance
The prevention of fluid overload or electrolyte imbalance is crucial to the management of AKI. In calculating fluid balance, allowance must be made for endogenous water production of some 500 mL/24 h and insensible losses (approximately 0.5 mL/kg body weight/h, but significantly higher if there is pyrexia). Endogenous production derives from oxidative metabolism and will be increased if (as is usual) the patient is catabolic.
Objective assessment of extracellular fluid (ECF) volume, preferably using flow-based measurements (or, alternatively, central venous pressure), is valuable, and in many patients is essential. Sodium intake must be limited in oliguric patients and special note taken of any covert sodium administration if drugs are given as their sodium salts. In patients with non-oliguric AKI and during the diuretic phase of acute tubular necrosis, adequate fluid replacement is required and should be informed by continuous assessment of urinary volume.
Hyperkalaemia is a life-threatening complication of AKI and frequent monitoring of plasma potassium concentration is essential. Several factors contribute to the increase in plasma potassium concentration, including decreased renal excretion, acidosis and loss of intracellular potassium to the ECF as a result of catabolism. Potassium intake should be minimized; intravenous calcium chloride can be used to antagonize the effects of hyperkalaemia. An infusion of dextrose and insulin may temporarily reduce the plasma potassium concentration, but persistent significant hyperkalaemia is an indication for renal replacement treatment.
Other disturbances that may occur in AKI include hyperphosphataemia, hypocalcaemia, hyperuricaemia and hypermagnesaemia. Hypercalcaemia may develop, particularly in patients with AKI related to crush injuries, owing to release of calcium from damaged muscle and increased synthesis of calcitriol as falling phosphate concentrations decrease the inhibition of renal 1α-hydroxylase.
Acid–base balance
Since the kidneys are the only organs capable of excreting significant amounts of hydrogen ions, and the net daily acid load that has to be excreted is of the order of 40–80 mmol, patients with AKI are usually acidotic. Acid–base status may also be adversely affected by comorbid conditions, for example lactic acidosis secondary to sepsis.
Nutrition
Active nutritional management is a vital part of the management of patients with AKI, whether being treated conservatively or with renal replacement. The aim is to maintain nutritional status where it is adequate and to improve it where it is compromised, in order to limit catabolism, promote the healing of any wounds and enhance resistance to infection. The mainstay of nutritional management is to provide adequate energy substrates to obtain positive energy balance. Nitrogen requirements are variable and there are few reliable data to guide the amount of protein or amino acids that should be provided. In established renal failure, an increase in plasma urea concentration not associated with an increase in creatinine suggests excessive provision of nitrogen. It is important to provide adequate vitamins and trace elements.
If possible, the enteral route should be used to provide nutrients. In patients who cannot eat, or eat enough, proprietary liquid feeds may be given through a nasogastric tube. For patients with intestinal failure, and for those in whom enteral feeding is contraindicated for some other reason or in whom enteral feeding cannot, on its own, provide sufficient energy, parenteral feeding will be required.
The principles of nutritional management in AKI are no different from those in any other condition: an adequate regimen must be provided; a suitable route for its provision must be available, and the patient’s response must be carefully monitored (see Chapter 11). It should be appreciated that patients receiving renal replacement in general have increased requirements for nutrients, since there will be loss from the bloodstream into the dialysate.
There is no reason not to use lipid emulsions together with glucose solutions as an energy source in patients with AKI receiving parenteral feeding, unless there is a problem with lipid utilization; this will be apparent from simple inspection of the plasma, which will appear lipaemic.
Other measures
Infection is a frequent cause of death in patients with AKI, even if it has not precipitated the condition. It is essential to treat any infection early and adequately and to take every practicable step possible to prevent infection developing. When prescribing drugs, including antibiotics, care must be taken to avoid any that are potentially nephrotoxic. The dosages of any drugs that are excreted by the kidneys should be reviewed and, where appropriate, dosage guided by measurement of plasma concentrations.
Renal replacement treatment
Major indications for renal replacement include the following:
• fluid overload (e.g. pulmonary oedema)
• the need to ‘create space’ to allow provision of parenteral nutrition or other solutions in the oliguric patient
• severe hyperkalaemia (plasma potassium > 7.0 mmol/L)
• severe acidosis (arterial [H+] > 70 nmol/L, pH < 7.15 or plasma bicarbonate < 12 mmol/L).
Haemodialysis
This technique is discussed on page 149. It is usually performed intermittently, each treatment lasting approximately 4 h. Clearance rates of more than 30 mL/min can be achieved. Intermittent haemodialysis is an efficient technique, but there is a danger of ‘disequilibrium syndrome’ due to too rapid removal of metabolites leading to osmotic imbalance between body fluid compartments with consequent rapid shifts of fluid. Headache, vomiting and, occasionally, fits may occur.
Because excess fluid is removed intermittently, the provision of continuous parenteral feeding may be more difficult with haemodialysis than with the filtration techniques described in the following sections.
Continuous venovenous haemofiltration (CVVH)
This technique (see p. 150) allows clearance rates of approximately 10 mL/min. In haemofiltration, large volumes of fluid are removed, but over a longer period of time, thus minimizing the risks of circulatory imbalance. There is, therefore, ample scope for the provision of parenteral nutrition when required, although other fluid replacement may usually be required in addition.
Continuous venovenous haemodiafiltration (CVVHDF)
In this technique, the filter is also perfused by dialysis solution. This combines the advantages of continuous filtration (particularly the ease of removal of fluid) with those of dialysis (higher clearances). Correction of metabolic imbalances, for example hyperkalaemia, is more easily achieved with CVVHDF than CVVH.
Peritoneal dialysis
Peritoneal dialysis (see p. 150) is a simpler technique than haemodialysis or haemofiltration, and does not require access to the circulation. However, it is less efficient than haemodialysis (clearance rates approximately 20 mL/min) and so is less suitable for hypercatabolic patients. Considerable quantities of protein (up to 40 g) can be lost into the dialysate at each session and there is a risk of peritonitis. Nevertheless, if the requirements needed to set up haemodialysis or filtration are not immediately available, this technique is valuable in the management of patients with AKI.
CHRONIC KIDNEY DISEASE
Introduction
Chronic kidney disease is a syndrome of persistent renal impairment involving loss of both glomerular and tubular function such that the kidneys’ homoeostatic functions are compromised. It usually has a gradual onset and, in many cases, progresses inexorably to a critical state (established or end-stage renal failure) in which the patient’s continued survival requires the initiation of renal replacement treatment, either by some form of dialysis or by renal transplantation. Although there are many causes of CKD, the clinical features of the condition tend to be similar regardless of the cause, being in effect due to a decrease in the number of functioning nephrons. Despite this, it is, as discussed below, important to attempt to determine the cause, although this is not always possible.
Although CKD may be discovered early, before it has given rise to any clinical disturbance (e.g. by the routine assessment of renal function in patients at risk of developing CKD), it frequently presents late, with a GFR already < 20 mL/min. Some patients will also have clinical features of the ‘uraemic syndrome’. This term is used to describe the clinical features that can occur in patients with CKD, but is caused by the retention of many substances, of which urea itself is relatively unimportant, and by other (e.g. endocrine) abnormalities that arise as a result of renal dysfunction.
Aetiology and pathogenesis of chronic kidney disease
Some of the more frequent causes of CKD are indicated in Box 7.5. As alluded to above, it is not always possible to determine the cause; patients presenting with advanced uraemia frequently have small, shrunken kidneys (unless diabetes, amyloid or polycystic disease is the cause of the condition) and specific diagnostic features may not be discernible even in a biopsy. It is, however, important to try to determine the cause, particularly in patients with less advanced disease. This may reveal a condition that is susceptible to treatment, so that progression can be delayed or even halted. The cause will, to some extent, determine the prognosis and allow planning of the eventual need for renal replacement and, if transplantation is contemplated, of the likelihood of recurrent disease in the grafted kidney. Finally, the identification of a familial cause (e.g. polycystic disease) may usefully lead to screening of other members of the family.
Successful treatment of the underlying cause of CKD does not always arrest the progress of the condition. It appears that, once started, the loss of nephrons in CKD itself contributes to disease progression. This has important therapeutic implications and the possible mechanisms have been extensively investigated.
The progression of loss of renal function
If 80% of a rat’s renal mass is removed, the animal develops proteinuria, hypertension and progressive renal failure; glomerular sclerosis is demonstrable in the renal remnant. Before hypertension and histological changes develop, there is a massive increase in renal blood flow, owing to a decrease in afferent arteriolar resistance. This leads to an increase in capillary hydrostatic pressure, increased capillary permeability and passage of macromolecules, including plasma proteins and lipoproteins, through the capillary wall. Some of the protein appears as proteinuria, but some is scavenged by mesangial cells, and it is thought that overloading of these phagocytic cells may cause functional derangement and cellular proliferation and contribute to the glomerular sclerosis. Increased numbers of macrophages may liberate growth factors, also stimulating cellular proliferation, and direct endothelial damage may lead to platelet activation and intraglomerular thrombosis, with consequent fibrosis and liberation of platelet-derived growth factor and other stimulants of cellular proliferation.
Systemic hypertension is common in chronic kidney disease (though less so if the underlying pathology is in the medulla, e.g. reflux nephropathy, than in the cortex, e.g. glomerulonephritis), owing to salt and water retention and inappropriate release of renin and, thus, formation of angiotensin I. This hypertension itself has a harmful effect on renal function, by exacerbating the processes described above and by causing arteriolar damage and ischaemic glomerular sclerosis.
Chronic kidney disease causes hyperphosphataemia, and the phosphate retention can contribute to further renal damage. Dyslipidaemia is a frequent finding in chronic renal disease, and the accumulation of lipids in the glomeruli is also thought to contribute to the progression of renal disease and suggests another possible therapeutic manoeuvre that may retard this process.
In addition to these factors, incidental conditions can contribute to the progression of renal failure in patients with renal disease (see Box 7.6). As the homoeostatic capacity of the kidneys decreases, so the patient becomes more susceptible to what might otherwise be easily tolerated events.
The uraemic syndrome
The clinical syndrome associated with CKD is protean in its manifestations. None is specific to the condition, but the overall clinical disturbance is characteristic. Disorders of the cardiovascular, neurological, skeletal, gastrointestinal, endocrinological, haematological and immunological systems are of particular clinical importance, but any system can be affected. The clinical features are summarized in Box 7.7.
These changes result to a considerable extent from the effects of the retained ‘uraemic toxins’; no single substance is responsible, although some, for example parathyroid hormone, appear to be particularly important. Box 7.8 indicates some of the toxins that have been identified, but it should be emphasized that not only is it incomplete, but many uraemic toxins still await identification. In addition, deficiencies, for example of the hormones erythropoietin and calcitriol, contribute to the uraemic syndrome; while dialysis may relieve the clinical features of the syndrome by removing toxins, those related to deficiencies (e.g. anaemia) require treatment by specific supplementation.
Clinical features
A comprehensive discussion of the clinical manifestations of CKD is beyond the scope of this chapter. In the cardiovascular system, hypertension, congestive cardiac failure and accelerated atherosclerosis are all frequently encountered. The neurological involvement can include both the central nervous system (uraemic encephalopathy) and the peripheral nervous system. The skeletal features of CKD, which used to be called renal osteodystrophy, but is now called chronic kidney disease – mineral and bone disorder (CKD-MBD), comprise a complex amalgam of osteomalacia, secondary hyperparathyroidism, osteosclerosis and osteopenia; they largely stem from the impaired production of calcitriol (1,25-dihydroxycholecalciferol), the hormone derived from vitamin D. More recently, fibroblast growth factor 23 (FGF-23) and klotho, a transmembrane protein produced by the osteocyte, which is required for FGF-23 receptor activation, have been identified as being important in this area, but their exact roles are still under investigation. Renal bone disease is discussed in more detail in Chapter 31. Gastrointestinal problems include hiccoughs, anorexia and gastritis, which can all cause great distress to the patient; gastrointestinal bleeding can be life-threatening.
The anaemia of CKD is usually normochromic and normocytic (see p. 147); fragmented (‘burr’) cells may be present on the blood film. A multifactorial bleeding diathesis is often present, with disturbances of platelet function and of coagulation. Depression of both the humoral and cellular arms of the immune system are frequent in CKD and patients have increased susceptibility to infection, are more likely to develop cancers and – a possibly beneficial effect – may experience remission of immunologically mediated disease.
Metabolic disturbances in CKD
Retention of nitrogenous waste products
Patients with CKD invariably have high plasma concentrations of urea and creatinine, but both can be affected by extrarenal factors. Thus, plasma urea concentration can be significantly reduced, even in advanced disease, by restriction of dietary protein, or in patients with chronic liver disease, and a sudden increase may occur if the patient suffers a gastrointestinal haemorrhage. Patients with CKD often have muscle wasting, which results in decreased creatinine production.
Hyperuricaemia is usually present in patients with CKD, although the concentration does not often exceed 600 μmol/L and attacks of acute gout solely due to the CKD are rare.
Potassium metabolism
Potassium balance can often be maintained in CKD until the GFR falls to < 5 mL/min. This is because of an adaptive response whereby distal tubular potassium secretion is increased; the mechanisms include increased aldosterone secretion and increased sodium delivery to the distal nephron, but there is also a poorly understood direct effect on potassium excretion. Furthermore, colonic potassium excretion, which is not usually a major route of potassium loss, becomes quantitatively important in patients with CKD.
Hyperkalaemia tends to occur earlier in patients with hyporeninaemic hypoaldosteronism, for example secondary to diabetes, and if there is significant acidosis. Hyperkalaemia is a life-threatening complication of CKD and can be precipitated by the injudicious administration of potassium or a drug that interferes with potassium excretion, for example potassium-sparing diuretics, angiotensin-converting-enzyme (ACE) inhibitors or β-adrenergic blockers.
Acid–base metabolism
In health, the kidneys excrete some 40–80 mmol of hydrogen ions per 24 h, this being the net rate of production on a normal diet. In CKD, renal hydrogen ion excretion is impaired, and once the GFR falls to below ~ 30 mL/min, systemic acidosis is likely to develop. This is despite the fact that the pH of the urine can usually be reduced to the lowest level achievable in health. The causes of the acidosis are discussed in detail in Chapter 5, but in summary are primarily due to reduced phosphate excretion, which diminishes the buffering capacity of the urine, and reduced ammoniagenesis which, albeit indirectly, also decreases renal hydrogen ion excretion.
In addition, there is often a partial defect in the reabsorption of filtered bicarbonate, a consequence, in part, of the expanded ECF volume and, in part, of the increased secretion of parathyroid hormone (see below). Severe acidosis is, however, uncommon in CKD and, despite a continuing positive hydrogen ion balance, the blood hydrogen ion concentration tends to remain stable for weeks or even months at a time. This appears to be due to the extensive buffering of hydrogen ions that occurs in bone.
Although the usual acid–base disturbance in CKD is a non-respiratory acidosis, the ability of the kidneys to excrete excess bicarbonate is also decreased, and patients in whom there is excessive non-renal loss of acid, for example due to vomiting, may occasionally develop alkalosis.
Calcium, phosphate and magnesium metabolism and renal bone disease
These topics are reviewed in detail in Chapters 6 and 31. In essence, CKD leads to a progressive decrease in the synthesis of calcitriol from 25-hydroxycholecalciferol, as a result of both inhibition of the enzyme by retained phosphate (an effect that becomes apparent as the GFR falls to < 60–70 mL/min) and, in more advanced disease, a decrease in the amount of enzyme as the renal mass decreases. A lack of calcitriol leads to decreased absorption of calcium from the gut, exacerbated by a low dietary intake of calcium, common in patients with CKD. There is thus a tendency to hypocalcaemia which, in health, should be corrected by increased secretion of parathyroid hormone (PTH) and mobilization of calcium from bone. In CKD, although the plasma PTH concentration is characteristically increased, often to a very high level (secondary hyperparathyroidism), there is resistance to its action, probably because of the low calcitriol concentrations. In consequence, plasma calcium concentration is usually below normal.
It should be noted that the results of older assays for PTH may be misleading, owing to metabolism of the hormone and selective retention of the biologically inactive C-terminal fragment in CKD. Assays that measure only intact PTH are more reliable indicators of parathyroid activity.
As discussed in detail in Chapter 6, hypercalcaemia can occur in CKD if parathyroid autonomy develops or after successful renal transplantation (tertiary hyperparathyroidism).
Plasma phosphate concentrations are usually normal, but can be low in early CKD. Despite a decrease in the number of functioning nephrons, the fractional excretion of phosphate by each is increased due to the elevated PTH. In more advanced disease, further loss of nephrons leads to the characteristic phosphate retention.
Plasma magnesium concentrations tend to be increased in CKD. Although toxic concentrations do not occur in uncomplicated cases, problems have been encountered in patients given magnesium salts therapeutically, for example as antacids.
Protein metabolism
Patients with CKD tend to be mildly catabolic, although this process can be accelerated by the development of intercurrent illness. Although the limitation of protein intake is an important aspect of the conservative management of CKD (see p. 148), it must not be so severe that it becomes a contributory factor to the loss of body protein.
Endocrine disturbances in CKD
There is good reason to expect endocrine disturbances in renal disease since the kidneys are an important site of hormone production, action and clearance (Table 7.5). Quite apart from the effect of the kidneys themselves on the production, excretion and activity of various hormones, impairment of non-endocrine renal function can secondarily affect the endocrine system. For example, CKD causes significant alterations in the internal environment that may influence the secretory control of hormones, their transport, activation and degradation, the relative amounts of free and protein-bound hormones in plasma and the responsiveness of hormone receptors at the cellular or subcellular level. Thus, in addition to renal clearance, the extrarenal clearance of some hormones may be abnormal in CKD, for example impaired hepatic degradation of biologically active PTH or diminished breakdown of insulin by skeletal muscle. Also, the function of endocrine organs may be influenced by treatment such as dietary restriction, drug therapy and renal replacement treatment.
The major mechanisms whereby chronic kidney disease influences endocrine function are outlined in Table 7.6. When kidney disease becomes advanced, the plasma or tissue concentrations and/or the functions of most hormones are altered, resulting in a number of well-recognized endocrine abnormalities (Box 7.9).
TABLE 7.6
Mechanisms by which chronic kidney disease influences endocrine function
| Mechanism | Example |
| Damage to renal parenchyma | Reduced synthesis of hormones |
| Reduced clearance of hormones | |
| Reduced action of hormones | |
| Disturbance of fluid and electrolyte balance | Activation of homoeostatic mechanisms, e.g. renin–angiotensin system; PTH secretion in response to hypocalcaemia |
| Inappropriate secretion of hormones | Hypersecretion of hormones, e.g. prolactin |
| Appropriate homoeostatic feedback mechanisms | Secretion of LH in response to reduced testosterone |
| Impaired extrarenal catabolism | Accumulation of hormones, e.g. PTH, insulin |
| Reduced hormonal function | Presence of multimolecular forms with varied biological activity, e.g. glucagon and calcitonin |
| Accumulation of hormones with opposing actions | |
| Accumulation of metabolic inhibitors | |
| Renal loss of binding proteins | Disturbance of bound:free ratio |
| Target organ resistance to hormonal action | Abnormal receptor binding, e.g. 1,25-dihydoxyvitamin D |
| Abnormal postreceptor events, e.g. PTH, insulin | |
| Reduced clearance of drugs | May interfere with endocrine function |
| Specific drug therapy, e.g. steroids | May interfere with endocrine function or induce endocrine disease, e.g. Cushing syndrome |
| Malnutrition | May have profound effects on endocrine function, e.g. growth and reproductive function |
| Increased susceptibility to sepsis | May have profound effects on endocrine function |
Growth retardation
Short stature is common among children with advanced kidney disease; its pathogenesis is multifactorial (Box 7.10). From an endocrine point of view, growth is a complex process involving primarily growth hormone (GH), but it is also dependent upon normal adrenal, thyroid and sex hormone function as well as an adequate supply of nutrients. In CKD, plasma immunoreactive GH concentrations rise in proportion to the decline in creatinine clearance and probably also to protein malnutrition. This rise in GH is because of reduced clearance and consequently increased plasma half-life. Growth failure in the presence of raised plasma GH concentrations indicates a resistance to the actions of GH in peripheral tissues. The homoeostatic mechanisms regulating GH secretion also appear to be abnormal in CKD, with exaggerated GH responses to arginine, thyrotrophin releasing hormone (TRH) and insulin-induced hypoglycaemia.
The skeletal growth-promoting actions of GH are mediated by the insulin-like growth factors (IGFs). In CKD, the plasma concentrations of IGFs tend to be normal, but their bioavailability is reduced. This is probably because of increases in the plasma concentrations of their respective binding proteins.
Sexual dysfunction
Impaired sexual function is a frequent and distressing symptom in both male and female patients with advanced CKD. In men, the major problem is impotence, which persists despite dialysis. This may have a multifactorial aetiology, being either directly related to the CKD or to coexistent conditions (Box 7.11). However, in many patients the organic component of impotence can be directly related to a disorder of sex hormones (Box 7.12), and several mechanisms may contribute to the hormonal abnormalities found. For example, hyperprolactinaemia is secondary both to hypersecretion and a reduction in clearance. The former is thought to be caused by a decrease in the sensitivity of lactotrophs to dopamine; chromatographic analysis of the circulating prolactin has confirmed that the majority is intact prolactin and not fragments.
In addition to problems of potency, males with CKD are frequently infertile, with evidence of defective spermatogenesis. Testicular biopsy shows maturation arrest, thickening of tubular basement membrane and interstitial fibrosis, consistent with damage to seminiferous tubules. These observations lend support to the theory that sexual dysfunction in advanced CKD is due primarily to toxic effects of circulating ‘uraemic toxins’ on both Leydig and Sertoli cell function. The identity of these ‘toxins’ remains undetermined, although there is good evidence to suggest that both PTH and prolactin may play a role.
The course of progressive deterioration in testicular function is not markedly altered by either haemodialysis or peritoneal dialysis. The best treatment is renal transplantation, which restores plasma gonadotrophin and sex steroid concentrations to normal and may result in effective spermatogenesis. The immunosuppressive therapy does not appear to have any deleterious effects apart from suppression of adrenal androgen secretion by prednisolone, although there is some evidence that men treated with sirolimus based regimens have a lower sperm count than those who are not.
Other notable problems that occur in dialysed males include gynaecomastia and priapism. The former does not appear to be related to either prolactin concentrations or androgen:oestrogen ratios, and has been compared to the type of gynaecomastia that occurs in malnourished patients during refeeding. The cause of priapism is unknown, but may be related to hypovolaemia after dialysis, the use of heparin or treatment with androgens.
In children, delayed bone age and delayed puberty are found. In boys, Leydig cell function appears to be relatively normal, as shown by the normal testosterone:dihydrotestosterone ratios, suggesting that 5α-reductase activity is normal. As might be expected, this results in normal LH concentrations, but FSH concentrations are elevated, which appears to reflect damage to the germinal cell epithelium prior to the initiation of spermatogenesis. These observations suggest that Sertoli cell damage in renal impairment occurs early while Leydig cell damage either does not occur until late or only occurs in adult patients.
In women with CKD, there is typically hypothalamic anovulation with amenorrhoea along with loss of libido and inability to reach orgasm. These women frequently exhibit hyperprolactinaemia with reduced oestrogen concentrations and elevated plasma concentrations of gonadotrophins and gonadotrophin releasing hormone (GnRH). Even in women who menstruate, the midcycle LH surge appears to be absent leading to anovulatory cycles and low progesterone concentrations, which in turn may cause dysfunctional bleeding. It is not surprising, therefore, that such patients are infertile. With treatment, however, premenopausal women may resume normal menstruation with ovulatory cycles and may even become pregnant. As with males, renal transplantation produces the most satisfactory results.
Thyroid abnormalities
Patients with CKD have several clinical disturbances that could potentially affect thyroid function. For example, they are chronically ill, often malnourished and display multiple hormonal and metabolic derangements. It is, therefore, not surprising that various abnormalities of thyroid function have been well documented (Box 7.13). These may complicate the diagnosis of coexistent thyroid disease (there is an increased prevalence of goitre and an increased incidence of primary hypothyroidism in CKD, particularly in women). The development of subclinical thyroid function test abnormalities has been explained by a mixture of defects (Box 7.14), which may also be observed in other chronic illnesses and malnutrition states (see Chapter 19). The high incidence of goitre in uraemic patients suggests that there is a circulating goitrogen in CKD. This does not appear to be either TSH or autoimmune antibodies, but there is circumstantial evidence to suggest that PTH may be involved since the incidence of thyroid nodules and goitres is greatly increased at post-mortem in patients with primary hyperparathyroidism.
With peritoneal dialysis (PD), plasma total T4 concentrations are not as low as with haemodialysis (HD), but T3 concentrations are lower. The low T4 concentrations in PD may relate to low concentrations of thyroxine binding globulin (TBG), while in haemodialysis there may be an inhibitor of binding present. Continuous ambulatory peritoneal dialysis (CAPD) appears to correct unbound, plasma free T4 concentrations better than HD; TSH is normal in both groups. Following successful transplantation, the plasma concentrations of thyroid hormones return to normal, while the TSH response to TRH may either be normal or decreased.
The diagnosis of thyroid disease in patients with CKD is difficult. A diagnosis of primary hypothyroidism can only be made with confidence if the plasma TSH is unequivocally elevated. In less clear-cut cases, measurement of free T4 may be helpful, but can be unreliable. The response to treatment should be monitored using plasma free T4 concentrations. The diagnosis of hyperthyroidism in renal impairment rests on the demonstration of an elevated plasma free T4 concentration in the presence of suppressed TSH, although hyperthyroidism is extremely rare in patients with this condition.
Anaemia
In patients with established renal failure, plasma erythropoietin concentrations are usually normal or even elevated. However, these concentrations are generally inappropriately low for the degree of anaemia and so there exists a state of relative erythropoietin deficiency, which contributes to the normochromic normocytic anaemia. Other factors include depression of the erythroid marrow by ‘uraemic toxins’, bleeding and haemodilution due to water retention. Following renal transplantation, the anaemia improves and the plasma erythropoietin concentrations return towards normal. However, very occasionally they remain elevated and erythrocytosis develops. This is due to some residual secretion of erythropoietin from the diseased kidneys.
Endocrine control of salt and water balance
Chronic kidney disease influences the activity of a number of renal and extrarenal hormones that control salt and water balance as well as blood pressure and volume. Renin release appears to be maintained by diseased kidneys under many circumstances. Thus, depending upon fluid and electrolyte balance, the plasma renin activity (PRA) in patients with renal impairment may be low, normal or high.
Plasma renin activities and 18-hydroxycorticosterone concentrations increase in patients on CAPD more than in those treated with HD. These increased concentrations have been attributed to the continuous fluid volume depletion induced by peritoneal ultrafiltration. Aldosterone concentrations are also elevated secondarily to the hyperkalaemia; they are more effectively reduced by HD than CAPD. Renal transplantation returns PRA and aldosterone towards normal. In addition, the normal physiological responses to sodium restriction, upright position, hypo- or hypervolaemia and angiotensin-converting-enzyme inhibitors appear to be preserved.
Plasma cortisol concentrations are at the upper limit of normal in CKD and respond normally during a tetracosactide test or insulin-induced hypoglycaemia, although the prolactin and GH responses are blunted during the latter, consistent with a degree of anterior pituitary dysfunction.
Plasma catecholamine concentrations are usually elevated in advanced CKD owing to a combination of several mechanisms, including reduced clearance, catabolism and neuronal uptake, and increased sympathetic efflux. After renal transplantation, normal plasma concentrations of noradrenaline (norepinephrine) with moderately elevated concentrations of adrenaline (epinephrine) have been found. Arginine vasopressin (AVP) is normally freely filtered by glomeruli and catabolized by tubules, so plasma AVP concentrations are elevated in CKD. The hormone is not significantly cleared by dialysis and remains elevated in response to the chronic fluid depletion. Following transplantation, both normal and elevated AVP concentrations have been reported. The plasma concentrations of natriuretic peptides (e.g. BNP, see Chapter 4) rise in direct relationship to the rise in plasma creatinine and represent a normal homoeostatic response to fluid overload. Once effective dialysis is established or renal transplantation performed, the concentrations return towards normal.
Impairment of the kidneys’ regulatory function results in defects in both the excretory and conservatory mechanisms for sodium and water (Fig. 7.7).
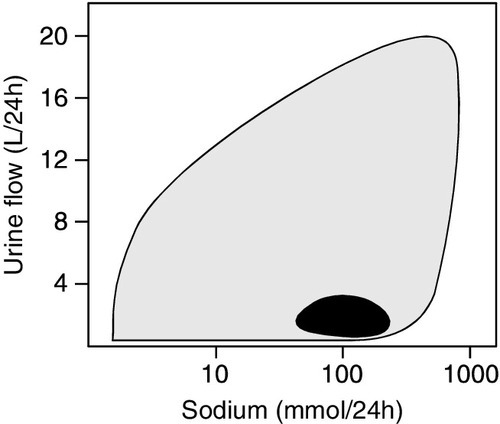
Thirst and nocturia are common features of chronic renal disease, reflecting the diminished capacity to concentrate the urine and loss of the normal diurnal variation in urine production. These effects are primarily due to the solute load causing osmotic diuresis in the remaining functional nephrons. Patients tend not to complain of polyuria; the low GFR sets a low limit on the maximum urine volume of not more than about 4 L/24 h. The capacity to dilute the urine may be preserved longer than that to concentrate it, but in advanced disease, neither dilution nor concentration can take place. The urine tends to have a fixed osmolality (~ 300 mmol/kg) and the patient is at great risk of both over- and under-hydration if fluid intake is not carefully regulated.
Early in the course of CKD, normal sodium balance may be maintained by increased excretion through the remaining functional nephrons. Particularly in patients with primarily tubular disorders, sodium conservation may be defective, leading to sodium depletion with clinical evidence of decreased ECF volume. Later, the tendency is to sodium retention, but this may not be apparent until the GFR falls below 10 mL/min and, even then, some patients continue to leak sodium. Since a fall in ECF volume can further impair renal function, careful attention to the maintenance of sodium balance is essential in the management of patients with CKD.
Carbohydrate metabolism and lipid metabolism
Impaired glucose tolerance secondary to insulin resistance is a feature of CKD. A number of mechanisms appear to contribute to the generation of this metabolic abnormality (Box 7.15), which is only partially corrected by dialysis and may persist following renal transplantation. However, this may well be related to the effect of steroid therapy since glucocorticoids are well known for their ability to cause hyperglycaemia. Some 70% of patients with CKD exhibit abnormalities of both lipids and lipoproteins, thought to be secondary to the hyperinsulinism. Typically, there is elevation of plasma triglycerides, very low density lipoprotein and intermediate density lipoprotein, but normal total cholesterol, with high density lipoprotein being decreased. The apoprotein and lipid content of the lipoproteins is also altered.
The major defects underlying these abnormalities appear to be low activities of tissue lipoprotein lipase, hepatic lipase and lecithin-cholesteryl acyltransferase. These abnormalities undoubtedly contribute to the increase in cardiovascular morbidity and mortality observed in patients with CKD.
Management
General management
This entails measures to evaluate and manage comorbid conditions, slow progression of kidney disease, reduce the risk of cardiovascular disease, prevent and treat complications of chronic kidney disease and to prepare for eventual renal replacement treatment. The measures specific to kidney disease are discussed further below. Management of comorbid conditions will not be discussed further here.
The staging of chronic kidney disease as defined by the chronic kidney disease K/DOQI classification (see Table 7.2) is used as the basis for the development of clinical management plans for individual patients.
Slowing the progression of kidney disease
If the cause of the CKD can be identified, it should of course be treated appropriately. Care must be taken to avoid conditions that may accelerate the decline in renal function, for example dehydration and infection (see Box 7.6).
Dietary measures are important both in an attempt to retard the progression of disease and in the avoidance of complications. Dietary management should ideally be supervised by a dietitian with special training in the management of renal disease. This person will not only be able to give advice on an appropriate diet, but should be able to design one that is acceptable to the patient and monitor compliance. The essentials of dietary management include maintenance of sodium, potassium and water balance, which usually entails restriction of sodium and potassium intake; reduction in phosphate intake, and dietary protein restriction (to about 0.8 g/kg per 24 h).
Experimental work suggests that judicious reduction in protein intake may decrease the progression of CKD, even when this is well established (plasma creatinine concentration > 400 μmol/L). Dietary protein restriction may protect against the progression of CKD by haemodynamically mediated reductions in intraglomerular pressure and by changes in cytokine expression and matrix synthesis. The haemodynamic effects of protein-induced hyperfiltration may be due to changes in hormones (such as glucagon and insulin-like growth factor-1), alterations in the renin–angiotensin system, and intrarenal effects, including tubuloglomerular feedback. The benefits of moderate dietary protein restriction (0.6–0.8 g/kg per day) on the progression of CKD in humans remain controversial. Current data suggest that, at best, a small reduction in the rate of decline of glomerular filtration rate (GFR) may be observed with a low protein diet.
When planning nutrition in patients being treated with dialysis, account must be taken of the loss of protein and amino acids into the dialysate. This occurs in patients both on peritoneal dialysis and haemodialysis, although the protein loss tends to be larger with the former and may be up to 40 g/24 h.
Supplementary vitamins may need to be prescribed both for patients treated conservatively and, particularly, those on dialysis. Careful clinical and biochemical monitoring of nutritional status is essential. Adequate energy intake must be maintained (at least 35 kcal/kg per 24 h) to meet energy requirements and prevent catabolism.
The need for careful attention to sodium and potassium intake should be obvious from earlier sections. There are experimental data to suggest that phosphate restriction is also desirable to prevent the development of renal metabolic bone disease, for which purpose the maintenance of plasma phosphate concentration within the reference range is the ideal. Although dietary protein restriction usually also entails dietary phosphate restriction, adequate control may be difficult to achieve. It is also important to avoid decreasing calcium intake, which may contribute to the development of bone disease. Oral aluminium salts can be given to bind phosphate in the gut and reduce its absorption; the risk of aluminium-induced neurological and bone toxicity that led to avoidance of their use may have been overstated, but if they are used, it is important to monitor the serum aluminium concentration regularly. Calcium containing phosphate binders such as calcium carbonate are widely used, but care must be taken to avoid inducing hypercalcaemia and there is a worry that they may increase the risk of vascular calcification. Newer, non-calcium-based phosphate binders are now available.
Dyslipidaemia is frequently present in patients with CKD. There are conflicting data concerning the effect of lipid lowering therapy with statins on progression of CKD. Some studies suggest that statins slow the rate of decline in renal function in patients with mild-to-moderate renal dysfunction, while others have found that they do not. All the data evaluating the effects of statin therapy on CKD progression were based on subset analyses of trials designed to evaluate the efficacy of statin therapy on cardiovascular disease. Thus, statin therapy cannot be recommended solely for renal protection.
Prevention of complications
It is essential to avoid dehydration. Particular care with fluid balance will be required if a patient has abnormal losses, for example because of vomiting, diarrhoea, or too vigorous treatment with diuretics. Blood pressure must be monitored regularly and any hypertension treated adequately. Any infections, but particularly those of the urinary tract, must be treated promptly, preferably with non-nephrotoxic drugs. Indeed, all medication must be chosen carefully to avoid nephrotoxicity.
The prevention of bone disease involves minimizing hyperphosphataemia (see above) and the administration of 1-hydroxylated metabolites of vitamin D, with or without calcium supplements. If hypercalcaemia occurs owing to tertiary hyperparathyroidism and patients are unfit for surgery, calcimimetics (e.g. cinacalcet) can be used. These drugs act directly on the calcium-sensing receptor in the chief cells of the parathyroid glands to reduce PTH synthesis and secretion.
Recombinant erythropoietin is available for the treatment of anaemia. It is effective and can greatly increase the well-being of patients with CKD, whether treated conservatively or by dialysis. It is, however, expensive and care is needed with patient selection and with monitoring the response to ensure efficient use of the drug. Iron stores must be replete for erythropoietin to work effectively.
Renal replacement treatment
By definition, the patient reaching end-stage renal failure requires renal replacement treatment in order to survive. In the face of such severe loss of renal function, the general measures outlined above will no longer be sufficient to maintain an internal environment compatible with the maintenance of vital functions. There are essentially two options: dialysis (peritoneal or haemodialysis; haemofiltration and haemodiafiltration are related techniques) and renal transplantation. These are not exclusive; most patients will need to undergo dialysis until transplantation becomes possible and, even after transplantation, some will require dialysis until adequate graft function is established or may need it again should the graft fail.
The optimum time to start a patient on dialysis will depend on several factors:
• the onset of major complications (pericarditis, neuropathy, encephalopathy, persistent hyperkalaemia)
• other life-threatening or intolerable features of uraemia.
Early initiation of maintenance dialysis is now preferred to a protracted period of dietary restriction. According to the K/DOQI guidelines (see Table 7.2) dialysis should be initiated electively in those whose eGFR is < 15 mL/min. Some patients in the UK are eligible for pre-emptive transplantation if dialysis is predicted to be required within six months (i.e. eGFR is predicted to fall to < 15 mL/min).
Haemodialysis
The dialyser consists of two contiguous compartments separated by a semi-permeable membrane. Blood flows through one compartment and dialysis fluid flows in the opposite direction through the other compartment. Waste products diffuse down the concentration gradient from blood to dialysate; water moves according to the relative osmolalities of plasma and dialysate, and other substances that can pass through the membrane (e.g. sodium, potassium) do so in relation to their own gradients (Fig. 7.8). The most frequently employed dialysers consist of an array of hollow fibres with semi-permeable walls; blood is pumped through the fibres, which are surrounded by dialysate.
The composition of the dialysate is similar to that of interstitial fluid, but with a lower potassium concentration and a higher glucose concentration (to make the fluid hypertonic and thus remove water).
Most patients dialyse for 12–15 h/week, spread over three sessions, at home, in a satellite unit or in hospital. It may be possible to relax the dietary restrictions required during conservative management somewhat after starting dialysis, but while more protein (e.g. 1 g/kg per 24 h) may be permitted, potassium, sodium, phosphate and water intake will still require restriction.
Clinical assessment provides an important guide to the adequacy of dialysis, but is inadequate on its own. Measurements of pre- and post-dialysis urea and creatinine concentrations also have limitations. Many alternative means of assessment have been studied, of which ‘urea kinetic modelling’ (UKM) is the most widely used. Measurements of plasma urea concentration are used to calculate the total urea clearance (K) (residual clearance plus clearance by the dialyser, in mL/min). This can be used to calculate the function ‘Kt/V’, where t is the dialysis time in minutes and V the volume of distribution of urea in mL (equal to the total body water or 65% of body weight). The ideal value of Kt/V is > 1.0. However, urea is not an ideal marker of renal function; estimates of V may underestimate true values and the desired clearance and actual clearance achieved may not be the same. Furthermore, it is not clear whether increasing benefit may result from the achievement of higher values of Kt/V. Thus, the use of Kt/V to guide dialysis therapy is not as straightforward as it might appear.
Haemofiltration
While in haemodialysis, waste products are removed by diffusion, in haemofiltration they are removed by convection. Fluid crosses the filtration membrane as a result of the pressure difference. Haemofiltration membranes are generally more permeable than those used in dialysis. This has the potential advantage that larger molecules (e.g. low molecular weight polypeptides) can be cleared from the plasma than is possible with haemodialysis.
There are systems that incorporate both diffusion and convection, that is, haemodiafiltration. In haemofiltration and haemodiafiltration, the volume ultrafiltered is replaced by intravenous infusion of pre-prepared replacement fluid from bags. Replacement fluid can also be prepared on line by the dialysis machine, for which the water quality has to be ultra-pure.
Peritoneal dialysis
In this technique, the semi-permeable membrane is the peritoneal membrane. Dialysate is instilled into the peritoneal cavity through a permanent silastic dialysis catheter, allowed to equilibrate and then removed. Continuous ambulatory peritoneal dialysis, in which the process is continuous with three or four fluid changes taking place over 24 h, is widely used.
Automated peritoneal dialysis (APD) is a term used to refer to all forms of PD using a machine to assist with the delivery and drainage of the fluid. Exchanges are typically carried out overnight, which leaves the patient free during the day.
Residual renal function can contribute as much as one-third of creatinine clearance; however, it declines with time so the dialysis prescription needs to be kept under review to ensure adequate clearance.
Renal transplantation
In addition to removing the requirement for repeated dialysis, successful transplantation removes the restrictions on the patient’s food and fluid intake. The patient will have to take immunosuppressive therapy for life, and, although this poses some risks (increased susceptibility to infection and incidence of lymphoproliferative disorders), they can be minimized by careful monitoring, including therapeutic monitoring of plasma immunosuppressive drug concentrations.
There are many immunosuppressive regimens. Briefly, they include a monoclonal antibody against the interleukin-2 (IL-2)-receptor (e.g. basiliximab or daclizumab) on induction, a calcineurin inhibitor (ciclosporin or tacrolimus), an antiproliferative agent (azathioprine or mycophenolate mofetil) and corticosteroids. Sirolimus is another immunosuppressive drug that is not nephrotoxic; calcineurin inhibitors are. Therapeutic monitoring of these drugs is essential (see Chapter 39). If there is graft dysfunction, toxic concentrations of calcineurin inhibitors are an important reversible cause.
Many factors influence patient survival, but rates continue to improve, and one-year survival rates of > 95% for patients are now common. Typically, > 88% of deceased donor grafts and > 95% of live related grafts are still functioning at one year.
Following transplantation, careful monitoring is required to assess graft function, monitor the patient’s hydration and to assist the early diagnosis of rejection. Regular measurements are required of urine volume, plasma creatinine, sodium, potassium, bicarbonate, glucose, calcium, phosphate and albumin concentrations, liver function tests, haemoglobin and white blood cell and platelet counts, as well as therapeutic monitoring of immunosuppressive drugs.
CONCLUSION
The kidneys have an essential role in the regulation of the composition and volume of the body fluids and in the excretion of waste products of metabolism. They also have important endocrine functions. They can be affected by disease processes confined to the kidneys, but they are frequently affected by primarily extrarenal disease.
The informed use of biochemical tests plays an important part in the investigation of renal integrity and function, although many other types of investigation are also used in the management of patients with renal disease.
Renal impairment can be acute (when it is usually potentially reversible) or chronic (when it usually progresses inexorably to established (end-stage) renal failure). In both types, there are profound systemic consequences. Conservative measures are important in management, but renal replacement treatment, by some form of dialysis or haemofiltration, is often required. In established (end-stage) renal failure, death will ensue unless the patient either undergoes successful renal transplantation or receives long-term renal replacement treatment.
Note about terminology
The terms ‘acute kidney injury’ and ‘chronic kidney disease’ have now replaced ‘acute renal failure’ and ‘chronic renal failure’, respectively.
ACKNOWLEDGEMENT
We would like to acknowledge the contribution of Sui Phin Kon and William Marshall, who wrote the chapters for previous editions of the book.
Further reading
The Renal Association. http://www.renal.org
The website of The Renal Association, which has further information about eGFR and chronic kidney disease.
[/level-membership-for-endocrinology-diabetes-and-metabolism-category][not-level-membership-for-endocrinology-diabetes-and-metabolism-category]CHAPTER 7
The kidneys, renal function and kidney disease
David Makanjuola; Marta Lapsley
CHAPTER OUTLINE
Renal blood flow and its control
RENAL DISEASE AND ITS PRESENTATION
Manifestations of renal disease
Diseases affecting the kidneys
THE ASSESSMENT OF RENAL FUNCTION
Biochemical tests of renal function
ACUTE KIDNEY INJURY (ACUTE RENAL FAILURE)
Obstructive (postrenal) kidney injury
Acute kidney injury in the setting of chronic kidney disease
Metabolic consequences and management of acute kidney injury
Aetiology and pathogenesis of chronic kidney disease
Endocrine control of salt and water balance
ANATOMY
Gross anatomy
The kidneys are located against the posterior abdominal wall on either side of the vertebral column. They lie anterior to the diaphragm and various muscles; their anterior surfaces are covered by parietal peritoneum. The left kidney is posterior to the stomach, pancreas, spleen and descending colon, and the right to the liver, the second part of the duodenum and the ascending colon. Their superior poles are covered by the suprarenal (adrenal) glands. In the adult, the kidneys are about 11–13 cm long, 6 cm wide and 4 cm thick. They weigh approximately 150 g each, yet together receive about 25% of the resting cardiac output. This blood is supplied by the renal arteries, which are branches of the aorta. Venous drainage is into the renal veins, which drain into the inferior vena cava. The kidneys are innervated by sympathetic nerves arising from the sympathetic chains and by parasympathetic fibres arising from the vagus nerve.
Each kidney is covered by a poorly distensible capsule. This limits the swelling that can occur during acute inflammation, and in such circumstances there is an increase in tissue pressure that tends to decrease the glomerular filtration rate (GFR).
Congenital absence of one kidney occurs in approximately 1 in 2400 individuals; this is only of significance if renal surgery is contemplated for any reason. The remaining kidney usually undergoes compensatory hypertrophy. Another common (approximately 1 in 10 000) congenital anomaly is the single ‘horseshoe’ kidney, in which the lower poles of the potential two kidneys are conjoined; renal function is normal but, occasionally, ureteric obstruction may predispose to calculus formation and infection.
Microstructure
Each kidney contains approximately one million functional units, called nephrons. These are tubular structures, consisting of various histologically and functionally distinct elements. Each nephron has a single glomerulus, which is located in the outer part of the kidney (the cortex) and is responsible for the ultrafiltration of the blood. The remainder of the nephron consists of several contiguous tubular structures that progressively modify the composition of the ultrafiltrate before it is passed out as urine. These structures (Fig. 7.1) are:
• the proximal convoluted tubule, also located in the cortex
• the loop of Henle, which has the configuration of a hairpin and extends into the deeper part of the kidney (the medulla), then doubles back on itself to return to the cortex
• the distal convoluted tubule, located in the cortex
• the collecting duct, which extends down through the medulla to the renal papillae, whence the urine drains into the renal pelvis. The renal pelvices are drained by the ureters, and the right and left ureters themselves drain into the urinary bladder, where urine is stored prior to voiding.
The glomerulus
Each glomerulus consists of a tuft of capillaries that protrudes into the dilated, blind end of the nephron (the Bowman capsule) (Fig. 7.2). The nephron is composed of epithelial cells, and these and the endothelial cells of the glomerular capillaries are separated by a basement membrane. This visceral basement membrane is continuous with a parietal basement membrane associated with the epithelial cells forming the outer part of the Bowman capsule, which are continuous with the epithelium of the proximal convoluted tubule. The microstructure of the glomerulus is described in detail in Chapter 8.
The glomerular filtrate is formed by the passage of fluid from the capillaries through fenestrations in the endothelial cells and through the basement membrane. This is covered on its epithelial aspect by the interdigitating foot processes of the epithelial cells, or podocytes, and the ultrafiltrate passes across a thin membrane (the epithelial slit diaphragm) into the filtration slits between the foot processes and then into the Bowman space, which is continuous with the lumen of the nephron. The selective retention of different constituents of the blood occurs at different stages. The endothelial fenestrations are too small to permit significant passage of erythrocytes, leukocytes or platelets; the major barrier to the filtration of proteins is mechanical and provided by the inner layer of the basement membrane, but the anionic surface layer also selectively retards anionic proteins. Finally, the foot processes and slit diaphragms also act as a mechanical barrier. The relative rate of progression of the constituents of the plasma into the Bowman space, therefore, depends on molecular mass, shape and charge. Glomeruli also contain mesangial cells. These stellate cells are contractile, are involved in the regulation of glomerular filtration and have phagocytic properties. The basement membrane does not extend between the mesangial cells and endothelial cells. This facilitates their ability to phagocytose large particles from the plasma. Their ability to ingest immune complexes plays an important part in the pathogenesis of certain forms of glomerular disease.
The proximal convoluted tubule
This structure is about 15 mm long and is composed of a single layer of inter-digitating epithelial cells united at their apices by tight junctions. The luminal surfaces of these cells have microvillous brush borders that provide the large surface area required for the absorptive function of the proximal tubule. The proximal tubule drains into a short straight segment directed towards the outer medulla and continuous with the descending limb of the loop of Henle.
The loop of Henle
The descending limb of the loop of Henle (Fig. 7.1) is composed of flat epithelial cells. The majority of the loops, whose glomeruli lie in the outer part of the cortex, are relatively short, but those with more deeply situated glomeruli have longer loops (up to 14 mm), which extend down into the medullary pyramids.
The thin descending limb turns back on itself, ascending towards the medulla, and the epithelial cells become cuboid, rich in mitochondria and have an invaginated luminal surface. The thick ascending limb extends towards the glomerulus of the same nephron and then leads into the distal convoluted tubule.
The distal convoluted tubule and collecting duct
The distal convoluted tubule is about 5 mm long and is made up of typical absorptive epithelial cells, although without a brush border. It passes close to the afferent and efferent arterioles of the same nephron. The tubular epithelial cells in this area have a special function in relation to the control of renin secretion and are known as the macula densa. Renin is secreted by myoepithelioid juxtaglomerular cells in the walls of the afferent arterioles. These cells, together with those of the macula densa and some other nearby cells (lacis cells), constitute the juxtaglomerular apparatus.
The distal convoluted tubule also contains intercalated (I) cells, responsible for the secretion of acid.
The collecting ducts are composed of I cells and also principal (P) cells, which become permeable to water under the influence of vasopressin. The ducts descend from the cortex down through the medulla, opening to the renal pelvis from the renal papillae.
Other specialized cells
In addition to the cells of the nephron and blood vessels, the kidneys contain medullary interstitial cells. These characteristically contain lipid droplets and secrete prostaglandins.
Blood vessels
Blood leaving glomerular capillary tufts passes not into venules, but into efferent arterioles. Those draining the cortical glomeruli divide into a capillary plexus that enmeshes the renal tubules. Those draining the juxtamedullary glomeruli form the vasa recta; these are bundles of vessels that plunge into the medulla to varying depths, forming capillary plexi that envelop the loops of Henle and then rejoin to form the ascending vasa recta, which eventually drain into the renal veins. The vasa recta form a countercurrent exchange system (see below), which is essential to the formation of both concentrated and dilute (with respect to plasma) urine.
RENAL FUNCTION
The principal functions of the kidneys are to excrete water-soluble (mostly nitrogenous) waste products and toxins from the body, and to control the volume and composition of the extracellular fluid (ECF). The kidneys also have important endocrine functions. It is perhaps surprising that the kidneys produce some 170 L of glomerular filtrate per 24 h, but only 1–2 L of urine. The bulk of the solvent and solute filtered is reabsorbed, but the control of the processes responsible gives considerable scope for the composition and volume of the urine to be adjusted to meet the physiological needs of the individual.
The physiology of the kidneys is well described in many standard physiology textbooks and will not be recounted in detail in this chapter. Rather, a brief summary is given and certain features that are pertinent to an understanding of the investigation of renal function and the mechanisms and consequences of renal impairment are emphasized.
Renal blood flow and its control
At rest, 20–25% of the cardiac output flows through the kidneys (1.1–1.3 L/min). The bulk of this blood perfuses the cortex; indeed, the flow rate to the medulla is actually less than to most other tissues.
The process of glomerular filtration is essential to renal function and is dependent on an adequate renal perfusion pressure. Autoregulatory mechanisms allow maintenance of renal blood flow and GFR within narrow limits in the face of a wide variety of external variables, including arterial pressure, venous pressure, ureteric hydrostatic pressure and plasma oncotic pressure. Renal blood flow and GFR are independent of mean arterial blood pressure over the range 80–200 mmHg. This is achieved by intrinsic, and humorally and neurally mediated, alterations in the tone of the renal blood vessels and, in particular, of the efferent glomerular arterioles.
Renal hypoperfusion stimulates the release of renin from juxtaglomerular cells. This enzyme converts circulating angiotensinogen to angiotensin I, which is in turn converted by angiotensin-converting-enzyme to angiotensin II, a powerful vasoconstrictor. This contributes to the maintenance of systemic blood pressure and, in the kidney, by causing efferent arteriolar vasoconstriction, helps to maintain the intraglomerular pressure despite a reduction in perfusion pressure. Various other mechanisms are involved in the maintenance of systemic blood pressure, but in the poorly perfused kidney, release of prostaglandins causes vasodilatation which, at least in mild hypotension, helps to maintain renal blood flow. A perfusion pressure of at least 50–60 mmHg is required to overcome the combined hydrostatic and oncotic pressures that oppose filtration; if mean arterial pressure falls below 80 mmHg, renal blood flow and GFR decline rapidly.
Glomerular function
Glomerular filtration is fundamental to the production of urine and to the homoeostatic functions of the kidney. Techniques for measuring the GFR, the amount of filtrate formed per unit time, are described in a later section (p. 131).
The glomerular filtration rate is determined by the balance of pressures across the filtration barrier in the glomerulus, and the physical nature and extent of the barrier itself. The forces include the difference between afferent and efferent glomerular arteriolar pressures (promoting filtration) opposed by the difference in osmotic pressures between the ultrafiltrate and the plasma, and the hydrostatic pressure in the Bowman space. The net filtration pressure is normally approximately 15 mmHg at the afferent end of the capillaries, falling to zero at the efferent ends. Factors influencing the GFR are summarized in Box 7.1.
A decrease in the area available for filtration can occur in many renal diseases in which there is glomerular damage. It can also result from contraction of mesangial cells mediated by agents such as angiotensin II, vasopressin, noradrenaline (norepinephrine), thromboxane A2 and prostaglandin F2; dopamine, natriuretic peptides and prostaglandin E2 have opposite effects. A reduction in filtration area may be an important physiological mechanism for reducing GFR and thus conserving fluid.
Tubular function
The proximal convoluted tubule
The proximal tubules are responsible for the active reabsorption of the bulk of filtered solute, accompanied by an iso-osmotic amount of water. Thus, virtually all the filtered glucose, amino acids, bicarbonate and potassium are absorbed here, together with some two-thirds of the filtered sodium. Absorption of solutes is an active, energy-requiring process and is isotonic, so that an equivalent amount of water is also absorbed and the fluid entering the descending limb of the loop of Henle is isotonic with plasma.
Transport occurs by way of ion channels, exchangers, co-transporters and pumps; filtered proteins are taken up into tubular cells by endocytosis and broken down into their constituent amino acids. Transport mechanisms have finite capacities and these may be exceeded in some circumstances. For example, glucose begins to be excreted in the urine if the plasma concentration exceeds about 10 mmol/L. Isolated or generalized disorders of renal tubular function result in the excretion of solutes normally reabsorbed in the proximal tubule even at normal plasma concentrations, as discussed further in Chapter 9.
The sodium content of the ECF is the primary determinant of ECF volume, the maintenance of which is critical to life. Given that about 26 000 mmol of sodium are filtered at the glomeruli each day (equivalent to some 8–9 times the total body sodium), it is clearly essential that most of the filtered sodium is reabsorbed. Indeed, the bulk of sodium reabsorption is obligatory. However, an increase in GFR (which potentially could lead to a massive increase in sodium excretion) results in increased proximal tubular sodium absorption and vice versa. This process, termed glomerulo-tubular balance, is thought to be mediated largely through changes in the oncotic pressure in the peri-tubular capillaries. This increases when the GFR increases, owing to the increase in the volume of water removed from the plasma. The re-absorption of several other solutes is affected similarly. Normal kidneys are capable of excreting between virtually zero and more than 400 mmol of sodium per 24 h. The fine control of sodium excretion is achieved in the distal convoluted tubule, where sodium reabsorption is stimulated by aldosterone.
Most of the filtered bicarbonate is absorbed in the proximal convoluted tubule, although indirectly. Hydrogen ions are generated in the tubular cells and are secreted into the tubular lumen (mainly in exchange for sodium) where they combine with the filtered bicarbonate ions. Bicarbonate ions are formed pari passu with hydrogen ions and are co-transported with sodium across the basolateral membranes of the tubular cells into the interstitial space (see Chapter 5).
Some substances are secreted into the urine from the proximal tubule. Examples include penicillins, creatinine, certain steroids and their glucuronides and derivatives of hippuric acid, for example p-aminohippuric acid (PAH). This property of PAH allows physiologists to use measurements of PAH clearance to determine renal plasma flow, but this is not an investigation that is used clinically.
The loop of Henle
This structure provides the countercurrent multiplier that generates the medullary hypertonicity that is essential for the regulation of water excretion and the production of concentrated urine.
Essentially, the process involves the active transport of solute (principally chloride and sodium ions) out of the thick ascending limb of the loop of Henle. This solute is not accompanied by water. As a result, the fluid within the lumen becomes hypotonic and the interstitial fluid surrounding the loop becomes hypertonic. Since water can diffuse out of the thin descending limb, but not the thick ascending limb, the net effect is that the fluid within the loop of Henle and the surrounding interstitial fluid become progressively hypertonic from the corticomedullary junction down into the medulla. There is further sodium and chloride absorption from the thick ascending limb. As water cannot diffuse out of the thick limb, the fluid entering the distal convoluted tubule is hypotonic with respect to plasma.
The anatomical disposition of the vasa recta allows them to act as a countercurrent exchanger, maintaining (though not actively contributing to) the osmotic gradient. The flows of water and solutes in the nephron are summarized in Figure 7.3. Whereas 60–70% of the filtered loads of solute and water have been reabsorbed from the glomerular filtrate at the beginning of the loop of Henle, a further 15% of water is removed during passage through this structure, so that only 10–20% of filtered water and somewhat less solute reach the distal convoluted tubule.
Tubuloglomerular feedback
There is a reciprocal relationship between the rate of flow of fluid through the ascending limb of the loop of Henle and the first part of the distal convoluted tubule of individual nephrons, and the rate of glomerular filtration through the same nephron. Thus, a decrease in flow rate increases filtration and vice versa. This process tends to result in a constant load of solute being presented to the distal convoluted tubule. Feedback is mediated through constriction and dilatation of the afferent arteriole by factors such as renal prostaglandins, adenosine and the renin–angiotensin system, in response to changes in chloride absorption in the macula densa. This mechanism becomes more sensitive when ECF volume is decreased and vice versa.
Tubuloglomerular feedback should not be confused with glomerulotubular balance, the change in proximal tubular sodium reabsorption in response to changes in GFR that has been discussed above and in Chapter 4.
The role of urea
Urea has an essential role in the generation of medullary hypertonicity. The thick ascending limb of the loop of Henle is permeable to urea (and sodium) but not to water. Movement of urea down its concentration gradient into the interstitium contributes significantly to medullary hypertonicity; the inner medullary portion of the collecting duct (see below) is also permeable to urea. The poor concentrating ability of the kidneys in the newborn is due to the decreased availability of urea to maintain medullary hypertonicity. In adults, renal concentrating power is dependent on an adequate protein intake (the source of urea) and is lower on a low protein diet.
The distal convoluted tubule
This part of the nephron is responsible for the ‘fine tuning’ of urinary composition. Aldosterone stimulates sodium reabsorption, generating an electrochemical gradient that allows the secretion (and thus excretion) of potassium and hydrogen ions. Normal distal tubular function is essential to the maintenance of hydrogen ion homoeostasis.
The collecting duct
These structures extend from the ends of the distal convoluted tubules to the renal papillae. Their main function is to permit the reabsorption of solute-free water and thus regulate the osmolality of the urine. This is achieved through the action of vasopressin (antidiuretic hormone). The cells of the collecting ducts are normally impermeable to water. Under the influence of vasopressin, aquaporin-2 water channels are inserted into the tubular wall, such that they become permeable, allowing water to move out of the hypotonic fluid in the tubular lumen into the hypertonic interstitium, so producing concentrated urine. In the absence of vasopressin, no water is removed and the urine is hypotonic.
The extremes of achievable urine osmolality are 30–1400 mmol/kg. When maximally dilute, as much as 13% of the filtered water is excreted and the rate of urine production is about 16 mL/min; when maximally concentrated, < 0.5% of filtered water is excreted and urine production is as little as 500 mL/24 h.
Some solute is also absorbed in the collecting ducts. In the cortical portion, sodium reabsorption takes place in exchange for potassium and hydrogen ions, as in the distal convoluted tubules. The medullary segment is partially permeable to urea, which moves passively out into the interstitium and helps to maintain the high osmolality of the medulla.
Diuresis
While the maximal urine flow rate when a water load is being excreted is approximately 16 mL/min (assuming an average solute load), higher rates are attainable if there are increased quantities of non-reabsorbed solutes in the tubules. This osmotic diuresis is due to the direct osmotic effect of the solutes and to a secondary effect on sodium reabsorption. The retention of water in the lumen of the proximal tubules increases the concentration gradient against which sodium must be reabsorbed; the same factor limits sodium reabsorption in the thick ascending part of the loop of Henle, thus interfering with the generation of medullary hypertonicity. This is the explanation for the diuresis characteristic of hyperglycaemia.
RENAL DISEASE AND ITS PRESENTATION
Introduction
There are many specific renal diseases; these may present with clinical features clearly referable to the kidneys (e.g. a decrease or increase in urine output), but frequently have systemic manifestations (e.g. the ‘uraemic syndrome’ (see p. 142) and hypertension). The kidneys can also be involved in multisystem disease, for example diabetes mellitus, the connective tissue disorders and amyloidosis.
Manifestations of renal disease
There are relatively few cardinal manifestations of renal disease and none is specific to any one underlying disorder. They include anuria, oliguria and polyuria; respectively a complete absence, decreased production and excessive production of urine. Polyuria may come to notice only by virtue of causing excessive production of urine at night (nocturia). These are usually symptoms of disordered renal function, although anuria and oliguria can be secondary to obstruction of the urinary tract and polyuria can be due to extrarenal disease that impairs the ability of the kidneys to concentrate the urine.
Urinary frequency (the passing of urine more frequently than normal, but in normal total 24 h volumes) must be distinguished from polyuria; it is often related to irritation of the urinary tract, for example by infection, but can also be due to prostatic enlargement. Irritation of the urinary tract is frequently accompanied by dysuria – pain on micturition. Patients with renal disease may experience loin pain or have tenderness in the loins. Renal or ureteric colic, an intermittent pain of considerable severity, is characteristic of the passage of urinary calculi.
Patients with renal disease may present with systemic abnormalities, including hypertension, pyrexia, oedema and the ‘uraemic syndrome’. This latter term is used to describe the protean manifestations of renal failure. The urine may appear abnormal in renal disease; the causes are discussed in a later section (p. 130).
Finally, renal disease may be revealed for the first time by the finding of a biochemical abnormality, particularly an elevated plasma creatinine or urea concentration, or positive urine dip-stick tests for protein or blood.
Diseases affecting the kidneys
Although patients with renal disease frequently present with an obvious functional abnormality, for example increased plasma creatinine or proteinuria, these are not specific to any one renal disease.
A full discussion of the nature of individual renal diseases is beyond the scope of this chapter. Many renal diseases specifically affect one part of the kidney. Glomerulonephritis, which is often of immunological origin, can manifest by decreases in the GFR or changes in glomerular permeability (e.g. causing proteinuria). Glomerulonephritis may be of primary renal origin, or occur secondarily to an extrarenal disorder (see Chapter 8).
A considerable number of conditions, both inherited and acquired, limited to the kidneys and having extrarenal manifestations, can cause disorders of renal tubular function. They are discussed in detail in Chapter 9.
The kidneys can be affected by infection, tumours and infiltrative and degenerative disorders. Generalized disorders, particularly shock, hypertension, connective tissue diseases and diabetes, are also frequent causes of renal dysfunction. The kidneys are also susceptible to the effects of toxins, including many widely used drugs (e.g. the aminoglycoside antibiotics and some cytotoxic drugs), as well as industrial and other toxins, and exposure to such agents is another frequent cause of renal dysfunction.
The major part of this chapter is devoted to a discussion of generalized renal failure, both acute and chronic. These are potentially life-threatening conditions and the clinical biochemistry laboratory plays a major part in their diagnosis and management.
THE ASSESSMENT OF RENAL FUNCTION
Introduction
Many techniques are of value in the investigation of the kidneys and renal function. The major groupings of techniques are laboratory tests, particularly biochemical and immunological, radiological imaging and the histological examination of renal tissue usually obtained by percutaneous biopsy.
Imaging techniques: standard radiography, ultrasonography, computed tomography, magnetic resonance imaging, the use of radionuclides and duplex scanning, can provide valuable anatomical information. Such techniques can be used to diagnose, for example, structural abnormalities, tumours, hydronephrosis, polycystic disease and the scarring of pyelonephritis. Radionuclide techniques can also provide information concerning renal function, particularly divided function (the separate function of the two kidneys) and glomerular function. Duplex scanning is a good screening test to assess renal blood flow before proceeding to magnetic resonance angiography or renal angiography.
Useful immunological markers include the detection in serum of antiglomerular basement membrane antibodies (positive in Goodpasture syndrome), antineutrophil cytoplasmic antibodies (in vasculitis), antinuclear antibodies and double-stranded DNA antibodies (in systemic lupus erythematosus), complement components (in various types of glomerulonephritis) and antistreptolysin O antibodies (in post-streptococcal glomerulonephritis). Immunological techniques are also widely used in the histological examination of renal tissue. However, given the pivotal role of the kidneys in body fluid homoeostasis, it is not surprising that biochemical investigations that indicate derangements of homoeostasis are the most widely used tests of renal function. The remainder of this section covers such tests.
Biochemical tests of renal function
Urinalysis
The assessment of renal function should start with examination of the urine. Simple observations on the urine still contribute to the investigation of patients with suspected renal disease. To be valid, they must be made on a freshly passed sample.
Appearance
The normal colour of urine is due to urochrome pigments, and the deepness of the colouration, which can vary from a pale straw colour to deep amber, depends on urinary concentration. There are many causes of abnormal colouration of the urine, some of the more frequently encountered of which are indicated in Box 7.2. In some cases, the colour is pH dependent. The distinction between haemoglobin or myoglobin and other causes of a reddish-brown urine can simply be accomplished by testing the urine with a suitable reagent stick; both myoglobin and haemoglobin will give a positive reaction for ‘blood’.
Turbidity in a fresh sample suggests infection, but can also be due to the presence of fat in patients with the nephrotic syndrome. The presence of chyle in the urine may cause turbidity so severe that the urine appears milky. Anything more than a scant amount of foaming when the urine is shaken, suggests proteinuria. The most frequent abnormal odour is that of ammonia, owing to the presence of urea-splitting microbes, either because the patient has a urinary infection or, in stale samples, because of contamination.
Specific gravity and osmolality
Both of these give an indication of the concentration of the urine. When this needs to be known accurately, for example in tests of renal concentrating ability, measurements of osmolality are essential. A reagent stick assessment of specific gravity may sometimes help in the interpretation of a screening test for proteinuria, since false negative results can occur with very dilute urine and vice versa. However, these reagent sticks detect ionic species only, and underestimate the specific gravity if other substances, for example glucose, are present.
pH
Normal urine is acidic, except after meals. Measurements of urine pH are important in the investigation of renal stone formers and as part of a test of urinary acidification in the investigation of suspected renal tubular acidosis.
Glucose
Glycosuria can occur either because of an increase in blood glucose concentration or because of a low renal threshold for glucose, owing to impaired proximal tubular reabsorption. In the latter instance, the blood glucose concentration will be normal. Renal glycosuria is discussed further in Chapter 9.
Protein
Proteinuria is an important feature of renal disease. Normal urine protein excretion is < 200 mg/24 h and is not detectable with standard reagent sticks (e.g. ‘Albustix’). Pathological proteinuria may be:
• overflow (due to raised plasma concentrations of low molecular weight proteins)
• glomerular (due to increased glomerular permeability to protein)
• tubular (due to decreased tubular reabsorption of filtered protein or saturated reabsorption)
• secreted (derived from the epithelium of the urinary tract).
Further details of the causes, detection and investigation of proteinuria are discussed in Chapter 8 and will not be considered further here.
Urinary sediment
Microscopic examination of the sediment obtained by centrifuging a freshly passed sample of urine frequently reveals a few cells (e.g. erythrocytes, polymorphonuclear leukocytes and cells derived from the kidney and urinary tract), hyaline (or rarely granular) casts composed of uromodulin (Tamm–Horsfall protein), and sometimes fat droplets, mucus threads and pigment granules.
Increases in any of these may occur in renal diseases. Abnormal casts are also frequently present. These are accretions of uromodulin with other material, for example cells or cellular debris. Red cell casts imply haematuria from glomerular disease, and white cell casts, the presence of white cells within the renal tubules. In acute pyelonephritis, there is an increase in polymorphs and various types of cast, in contrast to lower urinary tract infection where polymorphs, but not casts, are increased. Microbiological examination in acute pyelonephritis will usually reveal bacteriuria. In acute glomerulonephritis, haematuria causes the urine to appear reddish-brown or smokey, and the sediment contains large numbers of red and white blood cells, and red cell, haemoglobin and other casts. In chronic glomerulonephritis, the amount of sediment is much less, but there is usually an excess of dysmorphic red blood cells, which suggests that red cells have passed through the glomerular membrane. Red cell and haemoglobin casts are sometimes present. Granular casts are seen in the urinary sediment in many renal diseases, but particularly in acute tubular necrosis; in this condition, casts containing tubular cells are also often present, but red cell casts are uncommon. Characteristic crystals may be seen when specific substances are present in excess or the pH of the urine promotes crystallization, for example, hexagonal cystine crystals, birefringent calcium oxalate crystals, calcium phosphate or uric acid crystals (see Chapter 9).
Other substances
Many other substances can be detected in the urine in appropriate circumstances, for example drugs or bilirubin. Their presence is due to the normal excretory function of the kidneys and is not a reflection of renal dysfunction, so they will not be considered further here.
Measurement of glomerular filtration rate
Glomerular filtration is essential to renal function, and investigations designed to measure the GFR are the most frequently performed tests of renal function. The factors that determine the GFR have been discussed above. Its measurement is based on the concept of clearance – the determination of the volume of plasma from which a substance is completely removed (the plasma is ‘cleared’ of the substance) by glomerular filtration during its passage through the kidney. Clearance is a theoretical concept – no substance is cleared completely from the plasma in this way – but can nevertheless be used as a valid measure of the GFR.
For the clearance of a substance to equal the GFR, it must be freely filtered by the glomeruli and eliminated from the body solely by this route (e.g. it must not be metabolized by the liver or secreted into or reabsorbed from the urine). The substance should be non-toxic and accurate measurement should be easily available in a routine laboratory. The clearance of a substance requires measurements of the plasma concentration and urinary excretion rate:
where U is the concentration of the substance in the urine, is the rate of formation of urine and P the plasma concentration of the substance. It should be noted that is a rate; it has the dimensions (volume/time) and its determination requires the collection and measurement of the total amount of urine formed over a known period of time. The units of the various quantities are usually adjusted so that the clearance is expressed as mL/min.
The critical properties of some of the substances used in clearance measurements to assess GFR are shown in Table 7.1.
TABLE 7.1
Properties of substances used to assess glomerular filtration rate
a The ideal substance would have all these properties. DTPA, diethylenetriaminepentaacetic acid.
Clearance can also be used to determine renal plasma flow, using a substance such as p-aminohippuric acid, which is almost completely cleared from the blood in a single passage through the kidneys by a combination of glomerular filtration and secretion.
Inulin clearance
Inulin is a plant polysaccharide that satisfies all the physiological criteria mentioned in the previous section. The measurement of inulin clearance remains the ‘gold standard’ for the estimation of GFR. However, it is a relatively complex procedure and, although test kits are commercially available, it is infrequently used in clinical practice. In essence, the test involves the injection of a bolus dose of inulin, followed by a maintenance infusion designed to produce a constant plasma concentration. Once this has been achieved, a series of timed urine samples is collected and blood is drawn for the measurement of inulin at the midpoints of the collection periods. The GFR is taken as the mean of the inulin clearances for each period.
Creatinine clearance
Creatinine is an endogenous substance, a normal product of muscle metabolism. Its rate of production is fairly constant from day-to-day, being determined by muscle bulk rather than by activity. Creatinine is removed from the body mainly by glomerular filtration and its clearance can be measured as an index of GFR, although in the UK, it is much less frequently used than previously.
Creatinine is actively secreted into the urine, so that its clearance tends to overestimate the GFR. This effect is of little significance at normal filtration rates, the ratio (creatinine clearance/inulin clearance) being between 1.1 and 1.2, but in advanced renal disease the contribution of active secretion to the total amount of creatinine excreted in the urine becomes high in relation to the amount filtered, and creatinine clearance may significantly overestimate the true GFR. This is despite the fact that in established renal failure, bacterial degradation of creatinine secreted into the gut may contribute approximately 2 mL/min to the total clearance.
Inaccuracies arising from methodological problems with creatinine measurement have largely been overcome, but the major cause of inaccuracy in the determination of GFR by creatinine clearance is the accurate measurement of urine volume. Traditionally, patients have been required to collect urine over a 24 h period. This is a convenient time, but requires the patient to void their urine completely at the beginning of the 24 h period and collect all the urine passed over the ensuing 24 h, making a final collection at the end of this period. The possibilities for mistakes resulting in an incomplete collection are considerable and even under ideal conditions, for example in a metabolic unit with highly motivated patients, the coefficient of variation for repeated measurements in the same individual is > 10%. The critical difference (the amount by which two estimates must differ to give a 95% probability that there has been a true change in GFR) is therefore > 33%. The accuracy of estimates of the GFR by measurement of creatinine clearance may be improved by making two or more consecutive 24 h urine collections, but this is often not practical even if it is acceptable to the patient.
There is, however, nothing special about the 24 h period. The use of shorter periods, although more convenient for the patient, may reduce accuracy through inadequate bladder emptying, but a good compromise is to make a collection overnight. As long as the bladder was emptied at the beginning of the test (before retiring) and when the final collection was made (on rising), the urine production rate can be calculated and substituted in the formula.
It is essential that a reliable method is employed to measure plasma and urine creatinine concentrations. Colorimetric assays tend to overestimate creatinine since they detect non-creatinine chromogens. But even when reliable methods are used, it should be appreciated that the creatinine clearance is based on four measurements: plasma and urine creatinine concentrations, urine volume and time. Each has an inherent inaccuracy and the overall analytical variance will be the sum of the individual variances.
Plasma creatinine concentration
The way in which creatinine is handled by the kidney, coupled with the fact that, in any one individual, the rate of production is relatively constant from day-to-day, means that the plasma creatinine concentration alone can be used as an index of renal function. But, although it is widely used, it has a number of disadvantages and these must be borne in mind when interpreting plasma creatinine concentrations. Perhaps the most important becomes apparent from considering the familiar formula for deriving the GFR. Plasma creatinine concentration is inversely related to GFR, not directly related. This means that a plot of plasma creatinine concentration against GFR has the form of a hyperbola (Fig. 7.4). At any concentration of creatinine, halving the GFR will double the plasma creatinine concentration. But given the wide reference range for plasma creatinine concentration in the healthy population, this means that in an individual, the plasma creatinine concentration could be within the reference limits, yet the GFR be only one-half of normal.
This is of critical importance to clinical practice, since in early renal disease (when it might be supposed that therapeutic intervention would be most beneficial), the plasma creatinine concentration may be ‘normal’ – that is, within the reference range – in spite of the GFR being reduced. Plasma creatinine concentration is thus a relatively insensitive index of mild renal functional impairment. At low values of GFR, however, it becomes extremely sensitive, although its accuracy declines because of the increased contribution of tubular secretion to overall renal excretion.
Other factors that must be taken into account in assessing the significance of plasma creatinine concentrations include muscle mass, diet (a recent meal including meat, particularly if stewed, may cause a transient increase in plasma creatinine concentration) and the presence in the plasma of substances that interfere with the assay. Ketones, bilirubin, cephalosporins and spironolactone have all been implicated, depending on the method used, but especially in Jaffé-(picric acid) based assay systems. Enzymatic assays for creatinine are said to be more specific than older colorimetric methods, but they are still prone to interference by certain drugs such as acetylcysteine and phenindione.
The importance of muscle mass is exemplified by the changes that occur with age. In healthy individuals, GFR, and thus creatinine clearance, declines with increasing age from about the end of the 4th decade, at a rate of approximately 1 mL/min per year. However, plasma creatinine concentration does not normally rise with age; this is because of a decrease in production rate, thought to reflect the tendency for muscle mass to fall with age.
Although comparison of a measured plasma creatinine concentration with a reference range can be misleading, the fact that intraindividual variation is less than interindividual variation means that the detection of a change in creatinine concentration in an individual provides a more sensitive indication of a change in renal function. Nevertheless, the biological and analytical variance (even using the best assays) results in a critical difference of about 17% (20 μmol/L at a plasma creatinine concentration of 120 μmol/L).
In patients with advancing renal disease, the progressive loss of renal function with time can be expressed as a graph of reciprocal plasma creatinine concentration against time (since GFR is proportional to 1/[plasma creatinine]). A steady loss of renal function will cause this plot to be rectilinear (Fig. 7.5). Such plots are useful in patients with irreversibly declining renal function to help predict when renal replacement will become necessary, so that, for example appropriate means for vascular access for haemodialysis can be provided.
FIGURE 7.5 The progression of chronic kidney disease. Hypothetical curves to show (A) the decline in glomerular filtration rate (GFR); (B) the increase in plasma creatinine concentration and (C) the decrease in reciprocal plasma creatinine concentration. The broken lines show the changes that might be expected if the decline in function was to accelerate for any reason (see Box 7.6).
[/not-level-membership-for-endocrinology-diabetes-and-metabolism-category]
