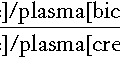CHAPTER 5
Hydrogen ion homoeostasis and tissue oxygenation and their disorders
CHAPTER OUTLINE
THE PHYSIOLOGICAL ROLE OF HYDROGEN IONS
THE ASSESSMENT OF ACID–BASE STATUS
DISORDERS OF HYDROGEN ION HOMOEOSTASIS
The interpretation of acid–base data
Mixed disorders of hydrogen ion homoeostasis
The role of haemoglobin in oxygen transport
The effects of pulmonary disease on oxygen uptake into blood
INTRODUCTION
Disorders of both hydrogen ion homoeostasis and tissue oxygenation will be discussed in this chapter. Abnormalities of hydrogen ion homoeostasis occur frequently in respiratory disorders, as a result of changes in the rate of excretion of carbon dioxide. Such disorders can also affect oxygenation, and impaired tissue oxygenation is an important potential cause of acidosis. Furthermore, hydrogen ion concentration, carbon dioxide and oxygen are measured using related technologies, usually with the same instrument.
The first part of this chapter deals with hydrogen ion homoeostasis and its disorders (colloquially often referred to as ‘acid–base balance’ and ‘acid–base disorders’, respectively), while the second part deals with the mechanism whereby oxygen is made available to the tissues, disorders in which tissue oxygenation is impaired and how tissue oxygenation is measured.
THE PHYSIOLOGICAL ROLE OF HYDROGEN IONS
Hydrogen ions are ubiquitous in the body and maintenance of appropriate concentrations is critical to normal function. The gradient of hydrogen ion concentration between the inner and outer mitochondrial membrane drives oxidative phosphorylation; changes in hydrogen ion concentration can affect the surface charge and physical conformation of proteins and thus their function, and hydrogen ion concentration determines the degree of ionization of weak acids and bases and can thus affect the disposition of such substances, among which are many with important physiological and pharmacological functions.
The hydrogen ion concentration of the blood is normally controlled within narrow limits, in health rarely exceeding 46 nmol/L or falling below 35 nmol/L. This regulation is achieved in spite of the continuous production of hydrogen ions as a result of the normal processes of metabolism. Intracellular hydrogen ion concentration is, in general, higher; in the cytosol being slightly so while in lysosomes it is several orders of magnitude higher. However, the interior of mitochondria is slightly alkaline.
Definitions
An increase in the hydrogen ion concentration of the blood is termed acidaemia and a decrease, alkalaemia. The term ‘acidosis’ strictly describes a pathological disturbance that can result in acidaemia, but may not necessarily do so because of the simultaneous existence of another disturbance (possibly the result of a physiological compensatory process) that has an opposing effect. Similarly, alkalaemia is not always present in alkalosis. These distinctions, although made much of by some authors, often only introduce confusion and will not be pursued in this chapter.
Strictly speaking, it is the activity of hydrogen ions and not their concentration that is relevant, and devices for measuring hydrogen ions respond to their activity. Activity and concentration are only the same in ideal solutions, which biological fluids are not, but the distinction can be ignored for practical purposes.
Hydrogen ion concentration can also be expressed in terms of pH. The pH of a solution is the logarithm (base 10) of the reciprocal of hydrogen ion concentration (or minus the logarithm of hydrogen ion concentration). Thus, a solution with hydrogen ion concentration of 100 nmol/L (100 × 10− 9 mol/L) has a pH of 7.00 (log10 1/100 × 10− 9). pH does not have units; it varies in a reciprocal and nonlinear fashion with hydrogen ion concentration. The reference range for the pH of blood, corresponding to the range for hydrogen ion concentration given above, is 7.36–7.42. It is necessary, in discussing hydrogen ion homoeostasis and its disorders, to consider the production and disposal of hydrogen ions; it is therefore logical to discuss the effects of changes in these processes on hydrogen ion concentration rather than on a derived unit. Furthermore, as will be seen, the analysis of disorders of hydrogen ion homoeostasis is facilitated considerably if direct measurements are used.
In health, the rates of formation of hydrogen ions and of their consumption and excretion are in balance, but disturbances of hydrogen ion homoeostasis occur frequently and in a wide variety of disease states. They are classified traditionally as respiratory or non-respiratory in origin, according to whether the primary abnormality is the result of an excess or deficiency of carbon dioxide (respiratory) or of bicarbonate (non-respiratory). Non-respiratory disturbances are often referred to as metabolic. As will be seen, however, a respiratory disorder that causes hypoxia may produce an acid–base abnormality whose characteristics are non-respiratory, or even combine respiratory and non-respiratory features. Nevertheless, the distinction is a useful one and is of considerable value in analysing disorders of hydrogen ion homoeostasis.
HYDROGEN ION HOMOEOSTASIS
Buffering
What follows is a brief, simplified account of the essential features of buffering, sufficient for an appreciation of the physiology of hydrogen ion homoeostasis.
In essence, buffer systems limit the extent to which the hydrogen ion concentration actually changes in the face of any tendency to change. A buffer system (or buffer pair) consists of a weak (that is, only partly dissociated) acid and its conjugate base (that is, the anion that combines with a hydrogen ion to form the acid). If the acid is HB and the conjugate base is B−, the relevant reactions are:
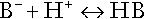
and
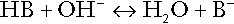
Bicarbonate
An example of central importance to physiology is the carbonic acid–bicarbonate system. Carbonic acid is a weak acid, which partly dissociates to bicarbonate and hydrogen ion:
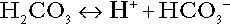
If an amount of hydrogen ions were to be added to a solution containing this buffer pair, some would combine with bicarbonate to form carbonic acid, thus limiting the increase in hydrogen ion concentration that otherwise would be expected to occur, but at the expense of consuming bicarbonate. Removal of hydrogen ions (or addition of hydroxyl, which, by combining with hydrogen ions to form water, has an identical effect) would cause dissociation of carbonic acid, producing hydrogen ions (and bicarbonate) and thus limiting the fall in hydrogen ion concentration that otherwise would be expected to occur.
It will be apparent that if a buffer is to function effectively in the face of equal tendencies for hydrogen ion concentration to increase and decrease, the concentrations of the acid and conjugate base should be equal, for example for the acid HB, [HB] = [B−]. Since at equilibrium:
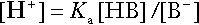
(where Ka is the equilibrium constant), it follows that a buffer will be most effective when the hydrogen ion concentration to be defended is numerically similar to the equilibrium constant. It should also be appreciated that hydrogen ion concentration, [H+], depends not on the absolute values for the concentrations of acid and conjugate base, but on the ratio of their concentrations. On the other hand, the buffer capacity, that is, the extent to which the buffer can absorb hydrogen ions, clearly will depend upon the absolute values of these concentrations.
For bicarbonate, it follows from the general equation (Eqn. 4) that:
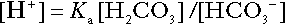
The concentration of carbonic acid cannot readily be determined, but it is directly proportional to the concentration of carbon dioxide [CO2], since carbonic acid is formed by the hydration of carbon dioxide:
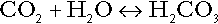
Thus, Equation 5 can be rewritten in the form:
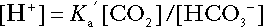
where Ka′ is a constant numerically incorporating the equilibrium constants for the reactions represented by Equations 3 and 5. The value of Ka′ is approximately 800 nmol/L and, at physiological extracellular hydrogen ion concentration (40 nmol/L), the numerical value of the molar ratio 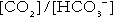 is approximately 0.05, suggesting that this system should be a poor buffer, particularly against a tendency for hydrogen ion concentration to fall. However, the body is a net producer of acid, that is, the tendency is for hydrogen ion concentration to rise. More importantly, carbon dioxide, generated by the buffering of hydrogen ion by bicarbonate and the subsequent dissociation of carbonic acid, can be removed by the lungs, keeping the carbon dioxide concentration constant. As a result, the effective buffering capacity of the carbonic acid–bicarbonate system is greatly increased, and it is a vitally important physiological buffer, particularly in the extracellular fluid.
is approximately 0.05, suggesting that this system should be a poor buffer, particularly against a tendency for hydrogen ion concentration to fall. However, the body is a net producer of acid, that is, the tendency is for hydrogen ion concentration to rise. More importantly, carbon dioxide, generated by the buffering of hydrogen ion by bicarbonate and the subsequent dissociation of carbonic acid, can be removed by the lungs, keeping the carbon dioxide concentration constant. As a result, the effective buffering capacity of the carbonic acid–bicarbonate system is greatly increased, and it is a vitally important physiological buffer, particularly in the extracellular fluid.
In practice, carbon dioxide concentration cannot be readily determined, but it is related to the partial pressure, PCO2, such that [CO2] = 0.225 × PCO2 when PCO2 is measured in kilopascals (kPa).
Phosphate
The monohydrogen and dihydrogen phosphate ions (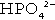 and
and 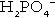 ) form a buffer pair with Ka being approximately 160 nmol/L. Although the Ka appears relatively favourable for buffering at physiological hydrogen ion concentrations, the concentration of phosphate in the extracellular fluid is too low for it to be of significance in this respect. Phosphate is, however, an important buffer in the urine, where its concentration is much greater.
) form a buffer pair with Ka being approximately 160 nmol/L. Although the Ka appears relatively favourable for buffering at physiological hydrogen ion concentrations, the concentration of phosphate in the extracellular fluid is too low for it to be of significance in this respect. Phosphate is, however, an important buffer in the urine, where its concentration is much greater.
Haemoglobin
All proteins can buffer hydrogen ions to some extent by virtue of their content of polar amino acid residues. Haemoglobin (Hb) is an important buffer. The reaction can be represented as:
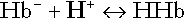
although each haemoglobin molecule is capable of buffering a number of hydrogen ions. It is relevant to note that haemoglobin is a more effective buffer for hydrogen ions when it is in the deoxygenated rather than the oxygenated form, and that oxygen release is facilitated by the buffering (the Bohr effect).
The efficacy of haemoglobin as a buffer is enhanced by the presence in erythrocytes of the enzyme carbonate dehydratase, which catalyses the hydration of carbon dioxide (Eqn. 6). Despite the fact that they are responsible for the majority of oxygen transport in the blood, erythrocytes obtain their energy anaerobically, by glycolysis, and thus do not generate carbon dioxide. In the capillary beds of tissues, carbon dioxide produced by aerobic metabolism readily diffuses down the concentration gradient into erythrocytes, where it is hydrated to form carbonic acid, which dissociates to form bicarbonate and hydrogen ions (Fig. 5.1). The latter are buffered by haemoglobin while bicarbonate diffuses out of the cells in exchange for chloride, so that the products of the dissociation of carbonic acid are removed, allowing further dissociation to occur and thus, by a mass action effect, stimulating its formation. In the lungs, alveolar PCO2 is lower than venous PCO2 and the process reverses, carbon dioxide being generated and excreted, while the release of hydrogen ions from haemoglobin favours oxygen uptake.
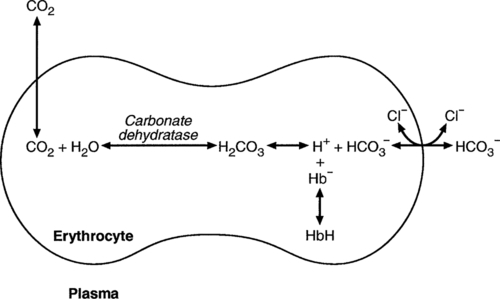
FIGURE 5.1 Transport and buffering of carbon dioxide in erythrocytes. Carbon dioxide is converted into carbonic acid in erythrocytes; this dissociates to form bicarbonate, which diffuses into the plasma in exchange for chloride and hydrogen ions, which are buffered by haemoglobin. In the alveoli, the reverse process liberates carbon dioxide.
Thus, in addition to transporting carbon dioxide, the conversion of carbon dioxide to bicarbonate serves to minimize the potential change in the value of the ratio 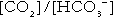 (and hence in hydrogen ion concentration) between arterial and venous blood. However, although erythrocytes are a potential source of bicarbonate, they can only generate bicarbonate if the hydrogen ion produced simultaneously can be buffered. There is clearly a limit to the extent to which this can occur, imposed by the buffering capacity of haemoglobin, so that this mechanism can have only a limited role in the correction of an acidosis. The way in which such correction is achieved is considered in a later section.
(and hence in hydrogen ion concentration) between arterial and venous blood. However, although erythrocytes are a potential source of bicarbonate, they can only generate bicarbonate if the hydrogen ion produced simultaneously can be buffered. There is clearly a limit to the extent to which this can occur, imposed by the buffering capacity of haemoglobin, so that this mechanism can have only a limited role in the correction of an acidosis. The way in which such correction is achieved is considered in a later section.
Other proteins
Although plasma proteins buffer hydrogen ions, their molar concentrations are lower than that of haemoglobin and their buffering capacity is less. In contrast, intracellular and tissue proteins make an important contribution to buffering. In chronic acidosis, they may contribute up to one-third of total buffer capacity. Buffering by bone is particularly important.
Ammonia
It is often stated that ammonia is an important buffer in the urine because it can combine with hydrogen ions to form ammonium ions. This cannot be true. Ammonium is a very weak acid, with an equilibrium constant approximately 100 times lower than physiological hydrogen ion concentration, so that when ammonia is produced in the body, it is immediately and almost completely converted to ammonium ions. The urinary excretion of ammonium is of relevance to hydrogen ion homoeostasis because it represents a route for the disposal of ammonium which, unlike urea synthesis, does not result in the generation of hydrogen ions (see p. 70).
Hydrogen ion turnover
The fact that, on a normal diet, the daily excretion of hydrogen ions by the kidneys (the only physiologically important route of excretion) is 40–80 mmol tends to distract from the fact that there is a massive endogenous turnover of hydrogen ions. In the resting adult, intermediary metabolism accounts for a hydrogen ion turnover of 2500–3000 mmol/24 h (Table 5.1).

Normally, the rates of hydrogen ion formation and utilization during intermediary metabolism are in overall balance, although any discrepancy can have a major effect on hydrogen ion concentration. But even this turnover of hydrogen ions appears insignificant in comparison with that which is associated with the turnover of adenine nucleotides – the movement of hydrogen ions that takes place across the mitochondrial membrane during oxidative phosphorylation and the synthesis and hydrolysis of ATP. This has been estimated at 500 mol/24 h. The potential for a disturbance in these processes to cause an acidosis is clearly colossal, although, in health, the rates of adenine nucleotide reduction and oxidation and of ATP formation and utilization are equal, so that these processes have no net effect on hydrogen ion homoeostasis. This may not, however, be true in disease.
As a result of oxidative metabolism, carbon dioxide is produced and excreted by the lungs. The rates of formation and excretion are normally equal, but carbon dioxide can combine with water to form carbonic acid, and the daily production of carbon dioxide in a resting adult is a potential source of approximately 15–20 mol of hydrogen ions. As has been alluded to, disorders affecting the excretion of carbon dioxide are an important cause of abnormalities of hydrogen ion homoeostasis.
Tendencies for hydrogen ion concentration to change can be limited to some extent by buffering, but this process can only offer a temporary solution to an imbalance between the rates of hydrogen ion production and disposal, because the body’s buffers have a limited capacity. Physiological processes often bring about a partial reversal of a change in hydrogen ion concentration (see Compensation, below), but ultimate correction of any disturbance requires equalization of the rates of production and disposal of hydrogen ion.
Hydrogen ion production
The processes involved in hydrogen ion production and utilization are summarized in Table 5.1. They comprise: processes involving carbon dioxide formation, reactions of intermediary metabolism and processes involving ‘fixed’ acids. In addition, and responsible for the bulk of hydrogen ion turnover, are the reactions involved in the complete oxidation of energy substrates. These processes are interlinked, but it is instructive to consider them separately.
Carbon dioxide
The role of carbon dioxide in relation to the formation of hydrogen ions has been mentioned above. Carbon dioxide, produced by oxidative metabolism, can become hydrated to carbonic acid, a weak acid that partly dissociates to hydrogen ion and bicarbonate:
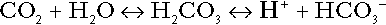
The equilibrium for this reaction strongly favours carbon dioxide and water, but in tissues containing carbonate dehydratase (e.g. tubular cells in the kidneys), the rate of formation of carbonic acid is increased and it can become an important source of bicarbonate and hydrogen ions.
Incomplete metabolism of glucose: glycolysis and lactate metabolism
The most familiar process of intermediary metabolism that results in the formation of hydrogen ions is anaerobic glycolysis, the metabolism of glucose to lactate. The overall equation for this reaction is:
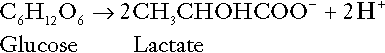
This process, which takes place particularly in skeletal muscle and erythrocytes, results in the formation of ~ 1.3 mol of hydrogen ions per 24 h in a 70 kg man at rest. The major route of disposal of lactate is glucose synthesis by gluconeogenesis in the liver and kidneys. The overall equation for this process is the reverse of that for glycolysis (most enzymes involved being common to both pathways, although some are unique). Gluconeogenesis thus consumes hydrogen ions. In health, lactate production and disposal are equal and so, too, are the production and disposal of hydrogen ions by these pathways. At rest, most of the lactate produced is converted back to glucose; during exercise, when lactate production is increased, 50% or more is completely oxidized instead. This process also consumes the hydrogen ions that are generated in its production and results in the formation of carbon dioxide and water:
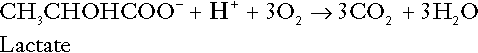
Thus, when lactate production and utilization are in balance, there is no net production of hydrogen ions. However, in disease states, an imbalance between these processes can be responsible for the development of acidosis.
The hydrogen ions generated in lactate production will be buffered principally by bicarbonate, so that Equation 10 could be written:
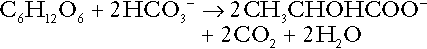
Clearly, the reverse reaction, gluconeogenesis, will regenerate the bicarbonate, and Equation 11 can be rewritten to show that this also occurs with complete oxidation of lactate:
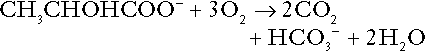
It is pertinent to point out that hyperlactataemia does not automatically indicate an acidosis. If it is a result of increased lactate production or decreased excretion, acidosis will only be present if the hydrogen ions produced simultaneously are not removed by excretion or metabolism. Intravenous fluids containing sodium lactate (e.g. Hartmann’s solution) are actually a source of alkali, because the lactate ions are metabolized to bicarbonate (Eqn. 13).
Incomplete metabolism of triglycerides: ketogenesis
The liberation of free fatty acids from triglyceride (triacylglycerol) in adipose tissue results in the generation of hydrogen ions. The process is exemplified by the equation:
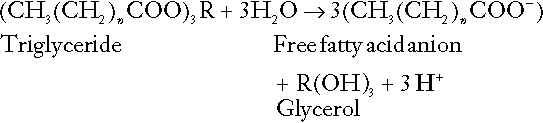
In the liver and adipose tissue, free fatty acids can be re-esterified to triacylglycerol, a process that consumes three hydrogen ions for each molecule of triacylglycerol synthesized. The further metabolism of free fatty acids to ketones in the liver (ketogenesis) also results in hydrogen ion production. An example of the reaction, starting from palmitate, is:
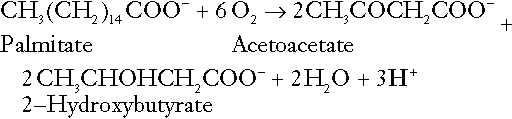
In health, this process may account for up to 0.4 mol hydrogen ion per 24 h, although in pathological states, its contribution may be much greater. However, ketoacids (strictly, oxoacids) are utilized as energy sources by skeletal muscle and other tissues. Their oxidation consumes hydrogen ions so that, in health, the overall rates of hydrogen ion production and disposal are equal:
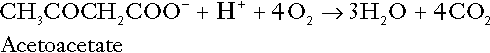
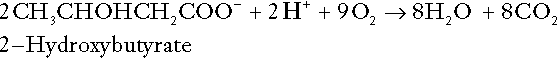
In disease, notably in diabetic ketoacidosis, excessive ketogenesis is an important cause of acidosis. Ketonuria may exacerbate the acidosis: ketoacid anions are a potential source of bicarbonate so that their loss in urine (in maximally acidic urine, about half the ketoacids are present in this form) effectively reduces the potential for bicarbonate generation.
Complete oxidation of glucose and triglycerides
Glucose can also be completely oxidized to carbon dioxide and water; indeed, this is the major route of glucose metabolism in the body.
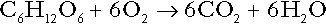
This is a complex process: oxidation is indirect, involving the transfer of hydrogen ions to adenine nucleotides, which are mainly oxidized by the mitochondrial electron transport system. Electrons are transferred to oxygen and combination with hydrogen ions produces water, while the energy released is transferred to ATP. But as Equation 18 indicates, the complete oxidation of glucose to carbon dioxide does not give rise to net formation of hydrogen ions: the products of its oxidation are carbon dioxide and water.
The same is true for the complete oxidation of triacylglycerols:
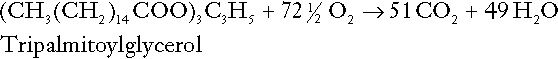
As with glucose oxidation, the process is far more complicated than appears from Equation 19, again involving the reduction of nucleotides, transfer of electrons to molecular oxygen, formation of water and trapping of energy in ATP, but the net production of hydrogen ions is zero.
Amino acid metabolism
Amino acid metabolism both produces and consumes hydrogen ions, according to the type of amino acid concerned. The metabolism of neutral amino acids eventually results in the formation of urea and carbon dioxide, for example:
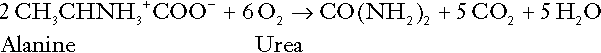
It is instructive, however, to look at this process in more detail. Most amino acids are metabolized by transamination in the liver to yield the corresponding oxoacid, the amino group being transferred to 2–oxoglutarate to form glutamate. Glutamate undergoes oxidative deamination, the amino group being converted to ammonium. The carbon skeletons of amino acids are in general glucogenic, although some are ketogenic. Ultimately, they will be completely metabolized. Omitting the transamination step, the intermediate stages are:
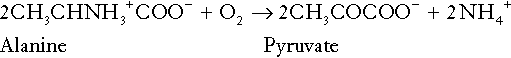
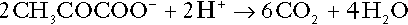
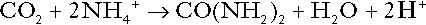
Thus, although urea synthesis generates hydrogen ions, these are utilized during the metabolism of the carbon skeleton so that the metabolism of neutral amino acids does not result in net generation of hydrogen ions provided that the nitrogen is converted into urea. If this does not occur, the metabolism of these amino acids consumes hydrogen ions. The relevance of this is discussed in a later section of this chapter.
The complete oxidation of a dibasic amino acid results in the generation of hydrogen ions, for example for lysine:
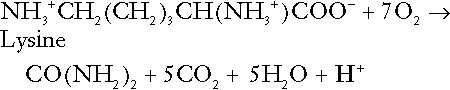
The complete oxidation of a dicarboxylic acid consumes hydrogen ions, for example for aspartate:
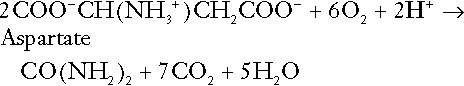
The complete oxidation of sulphur-containing amino acids (cysteine, methionine) generates hydrogen ions, for example for methionine:
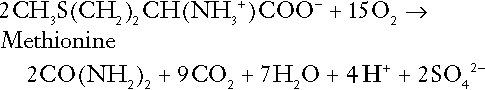
In each of these last three examples, it has been assumed that the end-product of the amino nitrogen is urea. If it is not, the effect on hydrogen ion metabolism may be different.
Overall, the amino acid composition of dietary protein and the manner of amino acid metabolism is such that, in health, there is a small net production of hydrogen ions. This is disposed of primarily by renal excretion.
Hydrogen ion excretion
Carbon dioxide
Carbon dioxide is excreted via the lungs. The respiratory control mechanisms are exquisitely sensitive to carbon dioxide so that, in health (and in the absence of any conscious effort to hypo- or hyperventilate), the rate of carbon dioxide elimination is made equal to the rate of production, and blood carbon dioxide concentration remains constant.
Hydrogen ions
Excess hydrogen ions are excreted in the urine and because, overall, the body is a net producer of acid, the urine is usually acidic. But before the urine can be acidified, there must be complete reabsorption of filtered bicarbonate.
Bicarbonate reabsorption
The glomerular filtrate contains bicarbonate at the same concentration as the plasma. Normally, the urine is virtually bicarbonate-free and, were the filtered bicarbonate not to be reabsorbed, the body’s bicarbonate pool, and thus buffering capacity, would rapidly be depleted. Generation of bicarbonate from carbon dioxide and water necessarily also involves the production of hydrogen ions.
The luminal membranes of renal tubular cells are relatively impermeable to bicarbonate, so that filtered bicarbonate cannot be reabsorbed directly. Instead, carbonic acid is generated from carbon dioxide and water in renal tubular cells (catalysed by carbonate dehydratase) and dissociates into hydrogen and bicarbonate ions; the former cross the luminal cell membranes in exchange for actively reabsorbed sodium and react with filtered bicarbonate, generating carbon dioxide. This can diffuse into tubular cells and, together with the carbon dioxide produced by aerobic metabolism, provides the substrate for the continued formation of carbonic acid. Although the equilibrium for the reaction favours carbon dioxide and water, the continual removal of hydrogen ions drives the enzyme-catalysed reaction in the direction of hydrogen and bicarbonate ions. The bicarbonate thus formed accompanies sodium as it is pumped across the basolateral membranes of tubular cells into the interstitial fluid. The net effect is the reabsorption of filtered bicarbonate ions, with an equivalent amount of sodium ions. In health, bicarbonate ‘reabsorption’ takes place almost entirely in the proximal renal tubules. This process is summarized in Figure 5.2.
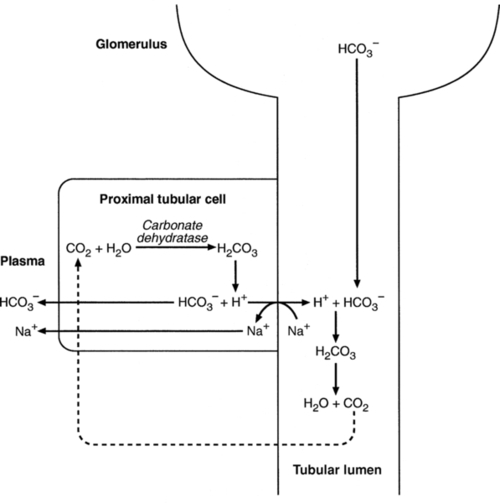
Acidification of the urine
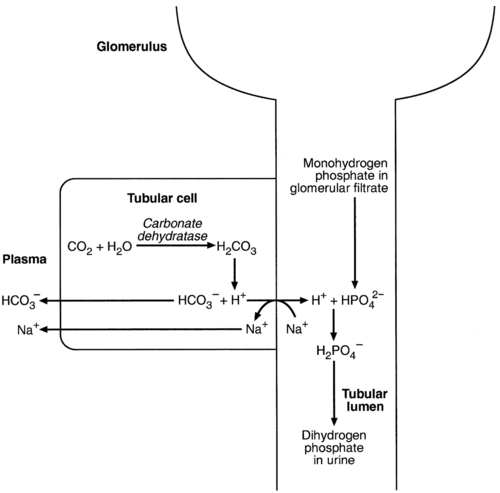
When bicarbonate reabsorption is complete, continued production of hydrogen ions by renal tubular cells and their movement into the tubular fluid will constitute net hydrogen ion excretion. Acidification of the urine is achieved by active secretion of hydrogen ions and hydrogen ion/potassium ion exchange by the α-intercalated cells of the distal tubules and proximal parts of the collecting ducts. There is, however, a limit to the acidity of urine that can be achieved. This is a pH of approximately 4.5, equivalent to a hydrogen ion concentration of 38 μmol/L. This represents a 1000-fold concentration gradient with respect to the extracellular fluid, but clearly, excretion of free hydrogen ions alone would be insufficient to remove the daily burden of acid produced by metabolic processes, which is measured in millimoles. Significant acid excretion is achieved by hydrogen ions being buffered by phosphate, titrating monohydrogen phosphate 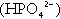 (the principal form in the plasma and thus the glomerular filtrate) to dihydrogen phosphate
(the principal form in the plasma and thus the glomerular filtrate) to dihydrogen phosphate 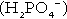 ions. It should be noted that, since the formation of hydrogen ions in renal tubular cells is accompanied by stoichiometric generation of bicarbonate ions, the excretion of hydrogen ions additionally results in the regeneration of bicarbonate ions and thus restores buffering capacity. This process is summarized in Figure 5.3.
ions. It should be noted that, since the formation of hydrogen ions in renal tubular cells is accompanied by stoichiometric generation of bicarbonate ions, the excretion of hydrogen ions additionally results in the regeneration of bicarbonate ions and thus restores buffering capacity. This process is summarized in Figure 5.3.
The role of urinary ammonium excretion
Although the urine contains ammonium and, indeed, the amount excreted increases considerably in states of chronic acidosis, this cannot, for the reasons indicated above, in itself, constitute net hydrogen ion excretion. Ammonium is produced in renal tubular cells by the action of the enzyme glutaminase on glutamine, the amide of glutamic acid (Eqn. 27), and the oxidative deamination of glutamate by glutamate dehydrogenase (Eqn. 28).
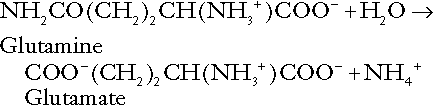
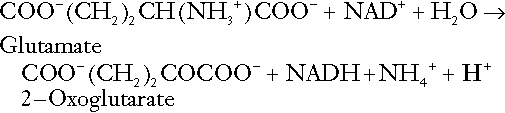
Glutamate is formed by transamination of 2–oxoglutarate with other amino acids, a process that does not involve either the production or utilization of hydrogen ions, and the equation for glutamine synthesis is the reverse of Equation 27. Thus, glutamine synthesis is a mechanism for the disposal of ammonium ions that does not (unlike urea synthesis) produce hydrogen ions. Although subsequent urinary ammonium excretion appears to be a means whereby hydrogen ions can be excreted in a buffered form, it does not represent direct excretion of hydrogen ions: rather, it is a process through which nitrogen can be excreted without the concomitant generation of hydrogen ions. As indicated in Equations 27 and 28, the production of ammonium from glutamine also yields 2–oxoglutarate: this is a substrate for gluconeogenesis, a process that consumes hydrogen ions.
In acidosis, hepatic glutamine synthesis is increased: in the kidneys, the formation of ammonium from glutamine, urinary ammonium excretion and gluconeogenesis are all increased. The net result is a decrease in hydrogen ion formation and an increase in bicarbonate generation, both of which tend to correct the acidosis. Renal ammonium excretion is illustrated in Figure 5.4.
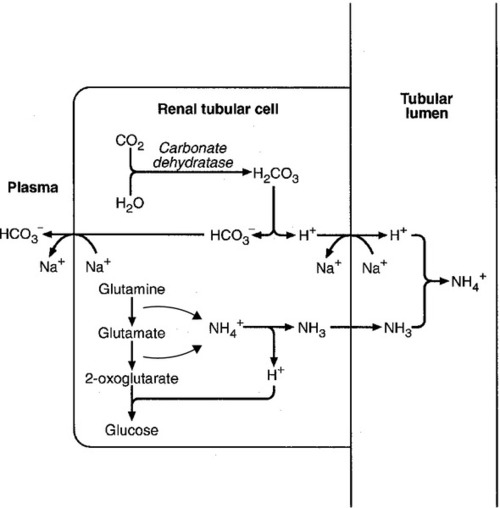
The luminal membranes of renal tubular cells are actually impermeable to ammonium ions, but are permeable to ammonia. Continued diffusion of small amounts of ammonia in equilibrium with ammonium within renal tubular cells into the tubular fluid, results in continued formation of ammonia from ammonium. In the lumens of nephrons, this ammonia is immediately converted back to ammonium ions. This process does not entail net excretion of hydrogen ions; indeed, the net result is the same as if ammonium ions were transported directly.
The role of the liver in hydrogen ion homoeostasis
Traditionally, the kidneys have been considered (together with the lungs) as the major organs responsible for hydrogen ion homoeostasis, but the liver also plays a role, although its extent remains controversial. While the kidneys are the only organs capable of excreting hydrogen ions from the body, the liver both generates and consumes hydrogen ions. As indicated above, it has a central role in the production (e.g. ketogenesis, ureagenesis) and utilization of hydrogen ions (e.g. gluconeogenesis), yet an abnormal hydrogen ion concentration is an uncommon finding in patients with liver failure unless this is severe or there is accompanying renal disease. But this should not be surprising, since it is well known that the liver has substantial reserve capacity, and functional impairment may only occur with massive liver damage. There is certainly evidence that acid–base status has an influence on hepatic urea synthesis and glutamine synthesis, and affects lactate and ketone metabolism. For example, acidosis tends to stimulate hepatic glutamine synthesis and lactate disposal, but inhibits ketogenesis.
Summary
Three organs are involved in hydrogen ion homoeostasis: the lungs, the kidneys and the liver. Extracellular hydrogen ion concentration depends on the ratio 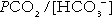 . The respiratory centre is exquisitely sensitive to arterial PCO2 and, given normal respiratory function, PCO2 is maintained within narrow limits by changes in the rate and depth of respiration. It has been calculated that cessation of carbon dioxide excretion would lead to potentially fatal acidosis within 30 min (although this is purely hypothetical, since the accompanying failure in oxygen supply would be fatal before this time).
. The respiratory centre is exquisitely sensitive to arterial PCO2 and, given normal respiratory function, PCO2 is maintained within narrow limits by changes in the rate and depth of respiration. It has been calculated that cessation of carbon dioxide excretion would lead to potentially fatal acidosis within 30 min (although this is purely hypothetical, since the accompanying failure in oxygen supply would be fatal before this time).
Nearly all the carbon dioxide produced each day is excreted through the lungs in the expired gas, although the liver is capable of disposing of a very small amount through the anaplerotic carboxylation of pyruvate to oxaloacetate. Although the liver can both generate and utilize hydrogen ions in metabolism, only the kidneys can excrete hydrogen ions from the body. In terms of the total turnover of hydrogen ions, the amount excreted this way is small. Nevertheless, it is vitally important, and a failure of renal hydrogen ion excretion frequently leads to acidosis, although even in acute kidney injury, if there are no additional causes of acidosis, it can take several days before the acidosis itself becomes severe.
Whilst any one of abnormal respiratory, hepatic or renal function may lead to the development of acidosis or alkalosis, changes in the others may ameliorate the effect on arterial hydrogen ion concentration. The compensatory hyperventilation seen in patients with non-respiratory acidosis is an example of this. Such compensatory processes are of vital importance in disturbances of hydrogen ion homoeostasis, and are discussed in relation to the specific conditions in which they occur. However, it is important to appreciate that, although they may restore an abnormal hydrogen ion concentration to, or at least towards, normal, they do not correct the underlying disturbance.
THE ASSESSMENT OF ACID–BASE STATUS
Clinical assessment
None of the clinical features of acidosis and alkalosis is specific to these disturbances and they may only be present when the disturbances are severe. The conditions that give rise to acidosis and alkalosis may have specific features, but, while clinical assessment is important in patients with disturbances of hydrogen ion homoeostasis, laboratory investigations are vital for their diagnosis, the assessment of their severity and for monitoring their progress.
Laboratory assessment
Hydrogen ion concentration and PCO2
Measurements of arterial blood hydrogen ion concentration (strictly, activity) and PaCO2 (the ‘a’ indicates arterial, and ‘A’ alveolar; in this chapter, for the sake of simplicity, the ‘a’ is omitted except where doing so could lead to ambiguity) are fundamental to the assessment of acid–base status. Analysers may express hydrogen ion concentration as pH, but the SI unit is concentration and the use of concentration greatly facilitates data interpretation.
Since the hydrogen ion concentration is determined by the ratio of PCO2 to bicarbonate concentration, bicarbonate concentration is not an independent variable and knowledge of it is not necessary for the characterization of acid–base disorders. Analysers calculate bicarbonate from PCO2, [H+] and Ka′, using the formula given in Equation 7. However, although Ka′ is supposedly a constant, there is evidence that it can vary unpredictably, particularly in severely ill patients.
True bicarbonate concentration is not readily measurable. Most laboratory measurements of ‘bicarbonate’ are in fact measurements of total carbon dioxide (TCO2), to which bicarbonate makes by far the greatest contribution, but which also measures dissolved carbon dioxide, carbonic acid and carbamino compounds (histidine residues in proteins that have combined with carbon dioxide). Together, all these other species contribute only about 10% to the TCO2. It should be noted that the TCO2 concentration of plasma and serum decreases in vitro, as carbon dioxide is lost to the atmosphere. Caution should thus be exercised in interpreting its value if there has been any significant delay between the times of the blood sample being taken and the plasma or serum being analysed. Arterial blood for analysis must be suitably anticoagulated; any gas bubbles must be expelled and the specimen protected from the atmosphere and either analysed immediately using a point of care instrument, or transported rapidly to the laboratory, chilled by placing the syringe in which it is collected in iced water.
Derived variables
Analysers that measure hydrogen ion concentration and PCO2 are generically termed ‘blood gas analysers’; they also measure PO2 and may provide other information of value in determining arterial oxygen content. They also frequently generate various derived terms, including ‘standard bicarbonate’, ‘base excess’ and ‘standard base excess’. These are calculated terms and add nothing to the characterization of disorders of hydrogen ion homoeostasis beyond what can be determined from consideration of [H+] and PCO2.
As will be seen, bicarbonate can be affected by both respiratory and non-respiratory disturbances. The standard bicarbonate is the bicarbonate concentration that would be expected in that blood sample were the PCO2 to be normal. It supposedly eliminates any contribution by a respiratory disturbance, so that an abnormal standard bicarbonate concentration is taken as indicating the presence of a non-respiratory disturbance of hydrogen ion homoeostasis. The base excess is the calculated amount of acid in millimoles that would have to be added to 1 L of the patient’s blood in vitro to restore the hydrogen ion concentration to normal in an alkalosis (the base deficit is the corresponding figure for alkali in an acidosis). The standard base excess extends the base excess to include the whole extracellular compartment. The calculation of these derived variables makes unwarranted assumptions, and the data themselves can confuse, rather than facilitate, the analysis of clinical disorders. The only measurement required to derive these variables in addition to PCO2 and [H+] is the haemoglobin concentration. Since, therefore, derived units do not incorporate any other independent measurements of acid–base status, it follows that they cannot provide any information that cannot be derived from the measured variables alone. Furthermore, although it is correct that abnormal values for standard bicarbonate and base excess indicate the presence of a non-respiratory component in an acid–base disorder, they do not distinguish between this being a primary disorder or a compensatory response to a primary respiratory disorder.
Base deficit is sometimes used to calculate the amount of bicarbonate that is required to correct an acidosis, but, in practice, if an acidosis is so severe that bicarbonate treatment is warranted, this should be given in small amounts and the effect assessed by frequent measurements of [H+] and PCO2.
Anion gap
The anion gap is the sum of the concentrations (in mmol/L) of the two major cations in plasma minus those of the two major anions (i.e. 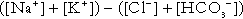 ). The total concentration of anionic charge in plasma must equal the concentration of cationic charge: the anion gap reflects mainly the anionic nature of most proteins in plasma at physiological hydrogen ion concentration, although phosphate and other anions make a small contribution. Its normal value is related to the concentrations of the species that determine it, and is ~ 15 mmol/L.
). The total concentration of anionic charge in plasma must equal the concentration of cationic charge: the anion gap reflects mainly the anionic nature of most proteins in plasma at physiological hydrogen ion concentration, although phosphate and other anions make a small contribution. Its normal value is related to the concentrations of the species that determine it, and is ~ 15 mmol/L.
Determination of the anion gap can be of help in determining the cause of a non-respiratory acidosis. When acidosis is due to loss of bicarbonate (e.g. renal tubular acidoses, loss of bicarbonate from the gut), there is increased renal chloride retention; the lost bicarbonate is replaced by chloride and the anion gap is unaffected. On the other hand, when acidosis is due to ingestion or excess generation of acids, the associated anions (e.g. lactate) replace bicarbonate as this is consumed by buffering, and the anion gap is increased. In practice, determination of the anion gap is only likely to be helpful if the cause of the acidosis is not already obvious. It certainly does not substitute for careful clinical assessment. For its calculation, plasma chloride concentration must be known, and in the UK, this often must be requested individually, rather than being available as part of a profile of tests. Also, the imprecision of the value of the anion gap, summating as it does the individual imprecisions of the measurements of the analytes that are required to calculate it, needs to be taken into account.
Other investigations
Other biochemical measurements may be of value in the assessment of disorders of hydrogen ion homoeostasis under certain conditions, for example in patients with diabetes, in neonates and in poisoned patients. These measurements are discussed in the sections that follow.
DISORDERS OF HYDROGEN ION HOMOEOSTASIS
Introduction
Although they differ in their pathogenesis, an understanding of the disorders of hydrogen ion homoeostasis is facilitated by considering them in a similar way. The processes involved are the generation of the disorder, buffering, physiological compensation and ultimate correction. In practice, there is often overlap between these.
Disorders of hydrogen ion homoeostasis may be simple, that is, involving only one type of disturbance, or mixed (two or more disturbances arising together). Unless rapidly corrected, simple disturbances typically generate secondary changes, which, in terms of bicarbonate concentrations and PCO2, can take on the characteristics of mixed disturbances.
Non-respiratory acidosis
This disorder can develop as a consequence of any, or a combination of, an increase in the rate of generation of hydrogen ions, a decrease in the rate of their utilization or excretion or a decrease in buffering capacity.
A list of some of the causes of non-respiratory acidosis is given in Box 5.1. Frequently, more than one cause may contribute to an acidosis in an individual patient. For example, patients with diabetic ketoacidosis are usually hypovolaemic and their renal function is impaired. Many of these conditions are discussed in detail in other chapters of this book; the reasons why they can cause acidosis should be evident from the first section of this chapter. Some conditions of particular interest are discussed later. Although many of the conditions causing acidosis have distinct clinical features, some of the features of acidosis, and of the body’s response to it, are common to them all. Non-respiratory acidosis can develop rapidly, particularly when it is due to increased production of hydrogen ions or loss of bicarbonate.
Compensatory responses in non-respiratory acidosis
Buffering
A tendency for hydrogen ion concentration to increase will be resisted by buffering; bicarbonate will be consumed and its concentration in the plasma will tend to fall. In the early stages of a condition that can cause acidosis, buffering, together with increased renal hydrogen ion excretion and bicarbonate regeneration, may prevent the development of a significant rise in hydrogen ion concentration. In chronic acidosis, buffering of hydrogen ions by tissue proteins also plays an important part in limiting the rise in hydrogen ion concentration.
Hyperventilation
Provided that respiratory function and its control are normal, acidosis stimulates ventilation through effects on aortic and carotid body chemoreceptors, and directly on the respiratory centre. Kussmaul breathing – deep, sighing respiration – is characteristic of non-respiratory acidosis. This not only allows excretion of carbon dioxide derived from the carbonic acid produced by buffering, but actually reduces the PCO2, which in turn tends to lower the hydrogen ion concentration towards normal. The muscular effort of hyperventilation itself generates carbon dioxide, and there is a lower limit to the PCO2 that can be obtained as a result of hyperventilation (approximately 1.4 –1.6 kPa). Many causes of non-respiratory acidosis are progressive conditions, and the acidosis may be so severe that respiratory compensation cannot normalize the hydrogen ion concentration. However, when the acidosis is only mild, a new steady state may be achieved in which hyperventilation maintains a hydrogen ion concentration that is only slightly higher than normal.
The increased ventilation, and thus respiratory compensation for non-respiratory acidosis, develops rapidly, so that the predicted changes of a ‘pure’, that is uncompensated, non-respiratory acidosis (elevated blood hydrogen ion concentration, decreased bicarbonate and normal PCO2) do not occur. However, compensation may take several hours to become maximal. This is because carbon dioxide equilibrates more rapidly across the blood–brain barrier than bicarbonate. Initial hyperventilation lowers cerebrospinal fluid (CSF) PCO2, and thus hydrogen ion concentration, and tends to counter the stimulation of respiration by peripheral chemoreceptors. Only as CSF bicarbonate concentration falls will its hydrogen ion concentration rise and augment the respiratory drive. The reverse phenomenon may be seen if rapid correction of an acidosis is attempted.
In patients with respiratory impairment, the efficacy of the compensatory process may be greatly reduced.
Renal hydrogen ion excretion
Provided that renal dysfunction is not the cause of acidosis and renal function is not compromised, the urine hydrogen ion concentration rises to its maximum possible value. However, the capacity of the kidneys to excrete acid is limited: the fixed buffering capacity of the urine is approximately 2–3 times the normal acid load. In this process, filtered monohydrogen phosphate is titrated virtually completely to dihydrogen phosphate. In mild or transient acidosis, this may be sufficient to prevent a significant increase in blood hydrogen ion concentration occurring. If acidosis persists, however, there is an adaptive increase in the excretion of ammonium ions. Ammonium excretion may increase five-fold or more. As has been discussed above, although it has been widely considered that this represents the excretion of buffered hydrogen ions, it is more accurate to consider renal ammonium excretion as representing an alternative pathway for ammonium disposal that, unlike urea synthesis, does not involve the generation of hydrogen ions. Acidosis induces increased synthesis of glutaminase, the enzyme responsible for the formation of ammonium from glutamine. It is noteworthy in this respect that renal gluconeogenesis, which provides a pathway for the utilization of the 2–oxoglutarate derived from the carbon skeleton of glutamine, is also increased in acidosis.
Biochemical characteristics of non-respiratory acidosis
The cardinal features of a non-respiratory acidosis are an elevated blood hydrogen ion concentration and a decrease in bicarbonate. Hyperventilation results in a decrease in PCO2. The extent of the change in PCO2 predicted for a given decrease in bicarbonate is discussed later. Comparison of the observed change with the predicted change can indicate whether there is an additional component to the acid–base disturbance.
In addition to these changes, other consequences of acidosis, especially hyperkalaemia, may be present, together with any specific features of the cause of the acidosis.
Systemic effects of acidosis
Many of the systemic effects of acidosis are common to acidosis of whatever cause. The effects on the cardiovascular system, oxygen delivery to tissues, the nervous system, potassium homoeostasis and bone are of particular significance. Acidosis also has important effects on intermediary metabolism.
The cardiovascular system
Acidosis has a negative inotropic effect, although this is probably only of significance in severe acidosis. Acidosis causes arteriolar vasodilatation and constriction of peripheral veins, but in many forms of acidosis these responses are obscured by other influences on vasomotor tone.
Oxygen delivery to tissues
Acidosis causes a right shift in the oxyhaemoglobin dissociation curve (the Bohr effect, see p. 90) and this facilitates oxygen delivery to tissues. An increase in hydrogen ion concentration decreases erythrocyte 2,3-diphosphoglycerate (2,3-DPG) concentrations through effects on both synthesis and breakdown; this causes a left shift in the curve, but, whereas the Bohr effect is immediate, the fall in 2,3-DPG takes place over a matter of hours. The reverse is also true; so, if hydrogen ion concentration is restored to normal rapidly, oxygen delivery will be compromised until 2,3-DPG concentrations are restored to normal. This is a potential hazard if an attempt is made to correct an acidosis rapidly by the intravenous infusion of bicarbonate.
The nervous system
Patients with acidosis can demonstrate impaired consciousness of varying degrees of severity, but there is little correlation between this and the severity of the acidosis. In many patients with acidosis, other factors are operating that may affect CNS function, and changes in blood flow and oxygen delivery secondary to the acidosis may also be relevant.
Potassium homoeostasis
There is a well-known association between acidosis and hyperkalaemia. This is multifactorial: movement of potassium ions from the intracellular to the extracellular compartment used to be thought to be related to intracellular buffering of hydrogen ions, but is probably to a greater extent the result of a loss of intracellular potassium to the plasma for other reasons, including a decrease in ATPase activity. It is of practical importance to note that total body potassium stores are frequently depleted, despite the high plasma concentration, and treatment of the acidosis may cause hypokalaemia if potassium is not replaced.
Bone
In chronic non-respiratory acidosis, there is significant buffering of hydrogen ions by bone, accompanied by decalcification, leading to a negative calcium balance. This is one factor contributing to renal osteodystrophy, the bone disease of chronic kidney disease. In addition, acidosis tends to increase ionized calcium concentration and so the filtered load presented to the renal tubules; renal calcium reabsorption may be decreased and calcitriol synthesis decreased.
Other effects
Acute acidosis can cause a leukocytosis; chronic acidosis can have a detrimental effect on nitrogen balance and insulin resistance can be a feature of both acute and chronic acidosis.
Management of non-respiratory acidosis
The logical management of a non-respiratory acidosis is to treat the underlying cause(s) of the disturbance appropriately. However, this may not always be possible and then, if the acidosis itself is having a significantly adverse effect on the patient, it may be necessary to attempt to lower the blood hydrogen ion concentration by giving alkali. The potential advantages are the reversal of the effects discussed above, including the hyperventilation, which may be particularly distressing for a conscious patient. There are, however, significant disadvantages. As indicated above, rapid correction of an acidosis may have an adverse effect on oxygen delivery to tissues. When acidosis is associated with the presence of organic anions (e.g. lactate, acetoacetate), continued metabolism of these after correction of the acidosis (rather than pari passu with the correction, as would happen if the underlying cause were reversed) may, by consuming hydrogen ions, cause the blood hydrogen ion concentration to fall. This is a rebound phenomenon and not just a direct consequence of excessive alkali administration.
The alkali used most frequently when it is considered necessary to correct an acidosis is sodium bicarbonate. Buffering of hydrogen ions will result in the formation of carbon dioxide and blood PCO2 may rise. Since carbon dioxide equilibrates across the blood–brain barrier more quickly than bicarbonate, the resulting increase in CSF carbon dioxide may cause a paradoxical rise in CSF hydrogen ion concentration. This may perpetuate the hyperventilation, even though the peripheral stimulus has been reduced by restoration of blood hydrogen ion concentration to normal.
There is general agreement that, in otherwise uncomplicated cases of non-respiratory acidosis that have a reversible cause (e.g. ketoacidosis and some forms of lactic acidosis), bicarbonate administration should not be considered unless the arterial hydrogen ion concentration is unusually severe (e.g. > 100 nmol/L). In acute kidney injury, however, bicarbonate administration may have a rapid and beneficial effect on dangerous hyperkalaemia, while renal replacement treatment is being put in place. If intravenous bicarbonate is to be given, it should be given as a series of small quantities (e.g. 50 mmol) in isotonic solution (unless there is a danger of fluid overload, when a hypertonic solution may be used); after each infusion, the patient should be reassessed clinically and arterial [H+] and PCO2 measured.
The use of bicarbonate in the treatment of renal tubular acidosis (RTA) is discussed on p. 79.
Specific causes of non-respiratory acidosis
Ketoacidosis
Diabetic ketoacidosis is discussed in detail in Chapter 16. The primary abnormality is increased lipolysis and ketogenesis (production of acetoacetic and 2-hydroxybutyric (β-hydroxybutyric) acids), coupled with decreased utilization of these acids. In addition, dehydration may decrease the glomerular filtration rate (GFR) and impair renal hydrogen ion excretion. Some patients also have a lactic acidosis (see below). It is noteworthy that resolution of the acidosis and clinical improvement of conscious level can lag behind the correction of the hyperglycaemia and ketosis in this condition. As normovolaemia is restored by the administration of 0.9% saline, renal excretion of sodium together with ketoacid anions results in loss of a source of bicarbonate and there is a tendency to hyperchloraemic acidosis, which may not be so rapidly reversed.
Ketoacidosis can also occur in association with alcohol ingestion (see Chapter 40), typically in alcoholics one or more days after a drinking bout, so that ethanol may not be detectable in the blood at the time that acidosis becomes apparent. Ketoacidosis develops as a result of a combination of factors, which are summarized in Table 5.2. The term ketoacidosis is a relative misnomer when applied to this condition; the oxidation of ethanol to acetaldehyde and acetate increases the [NADH]/[NAD+] ratio and hence the ratio [2–hydroxybutyrate]/[acetoacetate], so that dipstick tests for urinary ketones (which do not detect 2–hydroxybutyrate) may be negative or only weakly positive.
TABLE 5.2
Factors involved in the pathogenesis of alcoholic ketoacidosis
| Factor | Mechanism(s) |
| Fasting | Decreased insulin and increased counter-regulatory hormones,a leading to: increased lipolysis increased ketogenesis decreased ketone utilization |
| Hypoglycaemia | Effects of fasting exacerbated |
| Dehydration | Increased counter-regulatory hormones Impaired renal ketone excretion Impaired insulin secretionb |
| Ethanol | Metabolism inhibits lipolysis but lipolysis is stimulated as ethanol is cleared from the blood |
a Counter-regulatory hormones include glucagon, adrenaline (epinephrine) and cortisol.
b Via stimulation of α-adrenergic receptors by adrenaline (epinephrine).
In both types of ketoacidosis, significant excretion of the organic acids can occur in urine; at the lowest attainable urinary hydrogen ion concentration, some 50% of 2–hydroxybutyrate, but only 10% of acetoacetate, is in the form of the undissociated acid. Although this is advantageous in that it represents a route of buffered hydrogen ion excretion, the fact that the bulk of acetoacetate and 50% of 2–hydroxybutyrate are excreted as organic cations has adverse consequences. It entails obligatory excretion of sodium and potassium as the accompanying anions, thus contributing to their deficits, and also removes substrate for the later regeneration of bicarbonate by oxidation of these anions. It is of interest that, in patients with ketoacidosis with poor renal function (in whom less excretion of organic anions will occur), the extent of the fall in plasma bicarbonate concentration at the time of admission tends to match the increase in the anion gap and in the plasma concentrations of organic acid anions; in contrast, in patients with good renal function, the anion gap is less and plasma chloride concentration is increased. A third, but rare, cause of ketoacidosis is severe starvation.
The non-enzymatic decarboxylation of acetoacetate to acetone consumes hydrogen ions, but adds to the burden of carbon dioxide to be excreted.
Other acid–base disturbances associated with alcohol
A range of other acid–base disturbances can occur in association with alcohol ingestion. In normal subjects, the severe hypoglycaemia that can occur when ethanol is taken after a period without food, can be accompanied by a mild lactic acidosis. In chronic alcoholics, severe vomiting may cause a non-respiratory alkalosis, and continued ethanol ingestion can cause a lactic acidosis.
In patients with cirrhosis, alcohol can precipitate hepatic encephalopathy, which is often associated with respiratory alkalosis, as a result of stimulation of the respiratory centre by ammonia and other nitrogenous toxins.
Lactic acidosis
Lactate is formed from pyruvate as the end-product of glycolysis. Normal venous plasma lactate concentration is 0.6–1.2 mmol/L, tending to the lower end of this range during fasting and to the higher end after meals. Concentrations of up to 10 mmol/L can occur during severe physical exercise, but they fall rapidly once exercise ceases. Lactate is generated by glycolysis, principally in skeletal muscle, brain, erythrocytes, the skin and the gut, and is disposed of by gluconeogenesis in the liver and kidneys, thereby providing an important source of glucose, and by complete oxidation.
Lactic acid is a strong acid and is virtually completely dissociated at normal physiological hydrogen ion concentrations. Thus, the generation of lactate is always accompanied by equimolar generation of hydrogen ions. Buffering or other compensatory processes may prevent a significant rise in hydrogen ion concentration so that hyperlactataemia is not always associated with acidosis, but if the lactate concentration is greater than about 5 mmol/L, the hydrogen ion concentration is usually elevated.
Lactic acidosis can occur as a result of either excessive lactate formation or decreased lactate disposal or a combination of both. It is conventionally divided into type A (hypoxic) lactic acidosis, and type B, in which hypoxia is not the primary event. Causes of type B lactic acidosis include exposure to drugs, toxins and other chemicals, severe liver disease and certain inherited metabolic diseases. Some of the more important causes of lactic acidosis are indicated in Box 5.2. This list is not meant to be exhaustive; many rare conditions that have been reported to cause lactic acidosis and some conditions that only occasionally cause it, have not been included.
Type A lactic acidosis is the commoner variety. It is due primarily to an increase in lactate formation, as a consequence of tissue hypoxia, resulting, for example, from cardiogenic or haemorrhagic shock. Approximately half the lactate is produced by the gut. Decreased disposal of lactate is also important. Hepatic uptake and metabolism of lactate may be decreased in acidosis and when perfusion is decreased. The effects of acidosis on the cardiovascular system (negative inotropism and vasoconstriction) may further impair tissue perfusion (so that in the gut, a vicious spiral may develop, whereby splanchnic vasoconstriction induced by acidosis itself contributes to the acidosis). Although an acidosis-induced right shift in the oxyhaemoglobin dissociation curve may promote oxygen delivery to tissues, type A lactic acidosis can become self-perpetuating unless vigorous measures are undertaken to reverse the disturbance and treat the underlying cause. The prognosis is poor and appears to be related directly to the blood lactate concentration, mortality exceeding 80% with a concentration > 9 mmol/L. The specific treatment of the acidosis, for example by the judicious administration of bicarbonate, has been described above. Measures to correct the underlying cause are more important.
The mechanisms of lactate accumulation in type B lactic acidosis vary. The classic descriptions of this condition relate to phenformin-induced lactic acidosis; phenformin is a biguanide formerly used for treating type 2 diabetes mellitus. It causes lactic acidosis primarily as a result of decreased utilization of lactate for gluconeogenesis, but increased production may also contribute. Lactic acidosis frequently leads to shock, and increased production may then become the predominant mechanism. Metformin, also a biguanide, has low risk of causing lactic acidosis except in patients with renal impairment, in whom it should not be used.
The inherited metabolic diseases shown in Box 5.2 are a useful paradigm to illustrate the mechanisms that can lead to the accumulation of lactate (Fig. 5.5). Two reactions in the glycolytic pathway are not simply reversible for gluconeogenesis. Deficiency of both fructose 1,6-diphosphatase and pyruvate carboxylase, the enzymes that catalyse these steps in gluconeogenesis, are associated with lactate accumulation, particularly when production is increased as, for example, during exercise. In glucose 6-phosphatase deficiency, glucose 6-phosphate released from glycogen cannot be converted to glucose and so is metabolized through the glycolytic pathway to lactate, particularly when glycogenolysis is stimulated, for example during fasting. Pyruvate dehydrogenase is responsible for converting pyruvate to acetyl coenzyme A, which subsequently enters the tricarboxylic acid cycle: its deficiency decreases pyruvate, and hence lactate, oxidation.
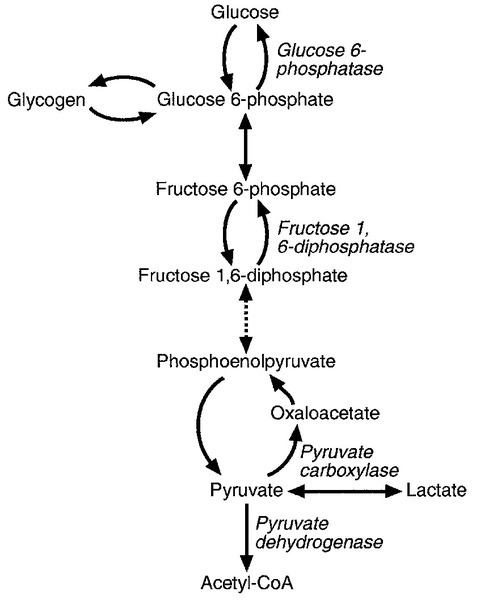
The association between alcohol and lactic acid, mentioned above, is not confined to ethanol. Methanol and ethylene glycol are metabolized by alcohol dehydrogenase and the reactions consume NAD+, which is then not available for the lactate dehydrogenase reaction. Particularly with ethylene glycol poisoning, other acids (e.g. glycolic and glyoxylic acids) also contribute to the acidosis (it should, however, be noted that glycolate may cause an apparent elevation of lactate concentration in lactate assays employing lactate oxidase).
Hyperlactataemia is common in patients with severe acute hepatic failure, but in many such patients the predominant acid–base disturbance is not acidosis, but alkalosis, either respiratory (as a result of direct stimulation of the respiratory centre by toxins) or non-respiratory, thought to be caused by a decrease in ureagenesis such that the consumption of hydrogen ions during the oxidation of the carbon skeletons of amino acids is not balanced by hydrogen ion formation.
The naturally occurring isomer of lactic acid in man is the L-isomer. Lactic acidosis due to accumulation of the D-isomer has been reported in patients with blind intestinal loops and short bowel syndrome, resulting from overproduction by the altered gut flora. The diagnosis may be missed, since D-lactate is not measured by the usual assays based on lactate dehydrogenase.
Dilutional (expansion) acidosis
Excessive infusion of isotonic saline solution can cause a mild acidosis. Expansion of the extracellular fluid (ECF) volume leads to a dilutional decrease in bicarbonate concentration, but, more importantly, volume expansion decreases renal bicarbonate reabsorption. This form of acidosis is self-correcting when ECF volume is allowed to return to normal.
Acidosis in renal disease
Acidosis is common in patients with renal disease. In acute kidney injury, acidosis is frequently multifactorial. Patients are often shocked and may have increased acid production, while the failure of urine production prevents renal acid excretion. In chronic kidney disease, the urine can usually be maximally acidified – that is, the hydrogen ion concentration can be raised (the pH lowered) to the same extent as can occur in normal individuals (exceptions include renal disease particularly affecting the medulla, for example chronic pyelonephritis, in which acidification may be impaired). In the early stages, the development of hyperphosphataemia results in an increase in the amount of phosphate filtered in each nephron, and phosphate reabsorption is decreased secondarily to increased secretion of parathyroid hormone. This may allow the excretion of a normal load of hydrogen ion buffered by phosphate. However, although individual nephrons may secrete a normal or even increased amount of ammonium, overall ammonium excretion decreases early in chronic renal disease and, as a consequence, systemic acidosis develops. Nevertheless, severe acidosis is unusual in chronic kidney disease, with plasma bicarbonate concentration rarely falling below 10–12 mmol/L. This is a result of extensive buffering of hydrogen ions by bone, a factor of importance in the pathogenesis of renal osteodystrophy. Uraemic acidosis is an example of a type of acidosis in which alkali treatment is beneficial, though usually with oral calcium carbonate rather than sodium bicarbonate in order to reduce the risk of sodium overload and also bind phosphate.
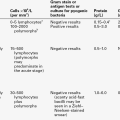
Impairment of renal tubular function is an important cause of acidosis. Three distinct syndromes of renal tubular acidosis (RTA) have been described: type 1 or distal RTA (sometimes referred to as ‘classic’ RTA); type 2 or proximal RTA, and type 4 or hyperkalaemic RTA. Both type 1 and type 4 owe their pathogenesis to defective mechanisms in the distal tubules: they are sometimes called hypokalaemic and hyperkalaemic distal RTA, respectively. Their principal characteristics are summarized in Table 5.3.
TABLE 5.3
Some causes and features of renal tubular acidosis

a Lower urinary pH may be achieved in ‘rate’ defects, see text for details.
Normal urinary acidification requires the generation of nearly a 1000-fold gradient in hydrogen ion concentration between the blood and the lumen of the nephron, and type 1 RTA can be due to a failure to maintain this gradient because secreted hydrogen ions diffuse back into the tubular cells or due to defective hydrogen ion secretion. It can be inherited as an isolated defect (autosomal dominant) or be acquired. Unusually, for a systemic acidosis, there is associated potassium wasting, leading to hypokalaemia, partly because more potassium is excreted to maintain electrochemical neutrality and partly because the impairment of hydrogen ion excretion impairs sodium reabsorption and may lead to secondary aldosteronism. Buffering of acid in bone contributes to hypercalciuria and there is a risk of nephrocalcinosis. Type 1 RTA is typically treated with modest amounts of oral sodium bicarbonate (typically 2–3 mmol/kg body weight/day; cf. type 2 RTA, below); potassium supplements, citrate and thiazide diuretics (which increase proximal tubular sodium – and thus bicarbonate – reabsorption by inducing volume contraction) may also be helpful. The principal aim of treatment is to allow normal growth in children and to correct the bone disease and reduce the risk of renal damage in adults.
Type 2 RTA is less common than type 1. It is due to a failure of proximal tubular bicarbonate reabsorption, which leads to bicarbonaturia. The fall in plasma bicarbonate concentration causes a systemic acidosis, but as the amount of filtered bicarbonate falls, the amount presented to the proximal tubules for reabsorption falls, and bicarbonate may be completely reabsorbed. Since the distal acidification mechanism is normal, excretion of an acidic urine is possible, albeit only at the expense of a systemic acidosis. Plasma bicarbonate concentration is typically 15–20 mmol/L. Type 2 RTA is usually associated with other abnormalities of proximal tubular function, for example glycosuria, amino aciduria, hypercitraturia and phosphaturia; it can be a feature of inherited diseases (classically, the Fanconi syndrome caused by cystinosis) or be acquired. Plasma potassium concentration is usually normal or only slightly decreased. Hypercalciuria is not a feature of type 2 RTA. However, metabolic bone disease (rickets in children, osteopenia in adults) occurs frequently. It is, in part, a direct consequence of the acidosis; defective synthesis of calcitriol and hypophosphataemia may also be contributory factors. Correction of the acidosis in type 2 RTA requires the ingestion of much larger quantities (typically 5–15 mmol/kg body weight/day) of bicarbonate than are required in RTA type 1. Appropriate treatment is particularly important in children, to optimize growth (the condition is in any case rare in adults). Bicarbonate replacement leads to bicarbonaturia and potassium wasting: potassium replacement is also required.
Renal tubular acidosis type 4 can be subdivided into two types. So-called ‘rate defects’ (which occur more frequently) are associated with decreased secretion of aldosterone, resulting from either primary adrenal disease or renal disease in which renin secretion is impaired (e.g. diabetic nephropathy, tubulointerstitial nephropathy). They can also be seen in syndromes of aldosterone resistance, in patients being treated with spironolactone and in patients treated with non-steroidal anti-inflammatory drugs (in whom it is probably as a result of a decrease in renin secretion secondary to decreased renal prostaglandin synthesis). The acidosis is due, in part, to reduced hydrogen ion secretion secondary to decreased sodium reabsorption, and usually responds to treatment with small doses of a synthetic mineralocorticoid together with bicarbonate, diuretics or ion exchange resins to correct the hyperkalaemia. However, there is also decreased ammonium excretion, and the fact that this, and the acidosis, may be corrected if the plasma potassium concentration is lowered without giving aldosterone suggests that the hyperkalaemia itself contributes to the acidosis. The other subtype comprises ‘voltage defects’, in which the secretion of hydrogen and potassium ions in the cortical collecting ducts is reduced as a result of a decrease in the usual negative intratubular electrical potential. This can occur with structural damage to the nephrons (e.g. sickle cell nephropathy), drugs (lithium, triamterene and amiloride, among others) and a decrease in the supply of sodium to the distal nephron as a result of avid proximal absorption (e.g. in hepatic cirrhosis). The urine pH can be reduced below 5.5 in rate defects, but not in voltage defects. In contrast to type 2 RTA, where acidosis is often severe, the acidosis in both subtypes of 4 RTA is often mild (plasma bicarbonate concentration typically 18–21 mmol/L), and the hyperkalaemia is often more remarkable.
If a diagnosis of renal tubular acidosis is not clear from the clinical findings and the simple laboratory measurements outlined above, tests of urinary acidification (for distal RTA) or bicarbonate reabsorption (for proximal RTA) may be required (see Chapter 9). Investigations that may be of value in the diagnosis of type 4 RTA include assessment of adrenal function and demonstration of a failure of plasma renin and aldosterone concentrations to respond to oral furosemide.
Respiratory acidosis
This is a consequence of carbon dioxide retention. In health, the rate and depth of respiration are adjusted precisely, so that the rate of excretion matches the rate of formation. Carbon dioxide excretion is a complex process, involving the transport of carbon dioxide in the blood to the pulmonary capillaries, diffusion into the alveoli and ventilation. Ventilation is controlled by the medullary respiratory centre, which receives input from peripheral and central chemoreceptors. Carbon dioxide retention can occur as a consequence of malfunction of either the excretory mechanism or its control. Some of the causes of respiratory acidosis are indicated in Box 5.3.
Compensatory responses in respiratory acidosis
Buffering
The role of erythrocytes in converting carbon dioxide to bicarbonate and buffering the hydrogen ions produced has been discussed above. This process is extremely effective: in health, the arteriovenous difference in hydrogen ion concentration is only ~ 3 nmol/L. It is salutary to examine the efficacy of this process in more detail. In the absence of any buffering, were an increase in PCO2 sufficient to cause an increase in bicarbonate concentration of only 1 mmol/L to occur (acutely, an increase of little more than 1 kPa is required to do this), it would be expected that hydrogen ion concentration would increase by the same amount, that is, 1 mmol/L. The fact that the increase is only a matter of a few nanomoles is almost entirely due to buffering by haemoglobin. Buffering of hydrogen ions by haemoglobin takes place rapidly, but the amount of haemoglobin available to buffer hydrogen ions is limited. The maximum increase in plasma bicarbonate concentration that occurs in acute respiratory acidosis is of the order of 4 mmol/L. In chronic carbon dioxide retention, buffering by other intracellular buffers occurs, and, as discussed below, increased renal ammonium excretion plays an important (albeit indirect) part in controlling the hydrogen ion concentration. Buffering of hydrogen ions by bone occurs to a lesser extent in chronic respiratory acidosis than in metabolic acidosis.
Hyperventilation
Although the increase in PCO2 will stimulate the respiratory centre, the underlying disease will mean that the respiratory apparatus is unable to respond adequately to this stimulus. Therapeutic measures to improve respiratory function may lower PCO2, but in chronic carbon dioxide retention, this may have undesirable consequences, as is discussed in the section on treatment, below.
Renal hydrogen ion excretion
In sustained carbon dioxide retention, renal bicarbonate reabsorption is maximal and phosphate is excreted almost entirely in the dihydrogen form. There is also a marked increase in urinary ammonium excretion. This has the effect of compensating for the increase in hydrogen ion formation from carbon dioxide, and may even restore blood hydrogen ion concentration to normal. It is accompanied by a further increase in plasma bicarbonate concentration (in addition to that generated directly by the erythrocyte mechanism). Although this is usually considered to be a consequence of increased renal hydrogen ion excretion (since bicarbonate is generated pari passu with hydrogen ions in renal tubular cells), it is probably also partly a result of diversion of ammonium from ureagenesis, requiring decreased buffering by bicarbonate of hydrogen ions produced during this process. Practically, it is important to appreciate that this compensation evolves over several days of carbon dioxide retention. If an attempt is made to reduce PCO2 rapidly, for example by artificial ventilation, temporary persistence of the compensatory process may result in the development of an alkalosis (Post-hypercapnic alkalosis, see p. 83).
There is a limit to which changes in renal acid excretion and in ammonium metabolism can compensate for an increase in PCO2; if this rises above 8 kPa, arterial hydrogen ion concentration will always be increased.
Biochemical characteristics of respiratory acidosis
The cardinal features of respiratory acidosis are an increased blood PCO2 and a high, or high–normal, hydrogen ion concentration; bicarbonate concentration is increased. The hydrogen ion and bicarbonate concentrations for any PCO2 depend on the extent of the compensatory increase in renal hydrogen ion and ammonium excretion (see above). In an acute disturbance, the increase in bicarbonate is only of the order of 2–4 mmol/L, even with massive increases in PCO2, but in compensated disturbances the increase is much greater.
Systemic effects of respiratory acidosis
In patients with respiratory acidosis, the manifestations of the underlying disorder and of hypoxaemia, if present, usually dominate the clinical findings, but effects due to acidosis and to hypercapnia may also be present. Hypoxaemia causes breathlessness, cyanosis and drowsiness. The consequences of acidosis have been discussed above. The effects of hypercapnia are seen predominantly in the central nervous and cardiovascular systems.
The neurological effects of hypercapnia cover a spectrum from anxiety and confusion through to impaired consciousness and coma. Particularly in chronic carbon dioxide retention, headache, papilloedema, extensor plantar responses and myoclonus may occur. Most of these effects are due to the increased cerebral blood flow that is a consequence of the vasodilatory action of carbon dioxide.
Systemic vasodilatation also occurs, but the cardiac output is increased so that blood pressure is usually maintained. The skin is warm and arterial pulses are bounding. The acidosis may cause venous constriction and chronic hypoxaemia may cause pulmonary hypertension and cor pulmonale, rendering the patient very susceptible to pulmonary oedema should intravenous fluids be given injudiciously.
Management
The logical management of respiratory acidosis is to treat the underlying cause and thus restore PCO2 to normal. This may not be possible and, in chronic carbon dioxide retention, if compensatory processes have restored the blood hydrogen ion concentration to normal or near normal, it may not be necessary. In practice, the management of respiratory disorders is usually dictated by the necessity to maintain an adequate arterial PO2. As mentioned above, rapid correction of an elevated PCO2 in a patient with chronic carbon dioxide retention is potentially dangerous. The compensatory changes can persist for several hours, or even a few days, and may cause the patient to become alkalotic.
This demonstrates the fact that the compensatory process in a respiratory acidosis can be regarded as the physiological generation of a non-respiratory alkalosis, although the patient’s hydrogen ion concentration does not fall below normal, that is, the patient does not become frankly alkalotic, as long as the PCO2 remains elevated.
Non-respiratory alkalosis
This disorder can develop because of excessive loss of hydrogen ions, decreased generation of hydrogen ions or exogenous alkali administration. Some of the causes are indicated in Box 5.4 and some of the more important of these are discussed later.
Non-respiratory alkalosis is characterized by an increase in plasma bicarbonate concentration. Since this bicarbonate is filtered at the glomerulus and, therefore, available for urinary excretion, the persistence of a non-respiratory alkalosis implies that it is being perpetuated by inappropriate reabsorption of filtered bicarbonate. Indeed, in healthy subjects, it is very difficult to produce a sustained alkalosis by the administration of, for example, sodium bicarbonate, whether orally or intravenously, because the excess is excreted in the urine. In considering the body’s responses to non-respiratory alkalosis, it is therefore necessary to take into account not only the underlying cause, but also the factors responsible for its perpetuation. Three such factors appear to be important:
• extracellular volume contraction
• potassium deficiency
• mineralocorticoid excess.
These are discussed further below.
Compensation for non-respiratory alkalosis
Buffering
A fall in blood hydrogen ion concentration results in the release of buffered hydrogen ions and, in consequence, the blood bicarbonate concentration increases. The excess bicarbonate could be excreted by the kidneys but, as mentioned above, this process is impeded in sustained alkalosis.
Hypoventilation
In a systemic alkalosis, decreased stimulation of chemoreceptors would be expected to decrease the respiratory drive, leading to compensatory retention of carbon dioxide. However, any increase in PCO2 will tend to stimulate respiration and lessen the extent of the acute compensatory response. With the passage of time, this response may increase, because the central respiratory centre appears to become less sensitive to an increase in PCO2. However, hypoventilation will tend to decrease arterial PO2 and should the extent of this be so severe as to cause hypoxaemia, the stimulus of this will override any inhibitory effect of a low hydrogen ion concentration. Significant tissue hypoxia is, however, uncommon in the absence of respiratory disease: although alkalosis causes a left shift in the oxyhaemoglobin dissociation curve, chronically this is offset by a decrease in red cell 2,3-DPG, which reduces the affinity of haemoglobin for oxygen (see p. 90).
Renal bicarbonate excretion
As indicated above, persistence of a non-respiratory alkalosis implies continued and inappropriate renal bicarbonate reabsorption. This could be achieved by the combination of a fall in the glomerular filtration rate (GFR) with maintenance of normal rates of tubular bicarbonate reabsorption, or by enhanced tubular reabsorption with a normal GFR. In many patients with non-respiratory alkalosis, there is both a decrease in the GFR and increased bicarbonate reabsorption.
A decrease in ECF volume may lead to a decrease in the GFR. If this is associated with chloride deficiency, the requirement to maximize tubular sodium reabsorption may cause an obligatory increase in the reabsorption of filtered bicarbonate to maintain electrochemical neutrality, to the extent that the urine may become acidic. (Priority is given to the control of ECF volume over that of acid–base status.) In the majority of patients with non-respiratory alkalosis, correction of the alkalosis follows repletion of ECF volume by the infusion of an isotonic sodium chloride solution (hence ‘saline-responsive’ non-respiratory alkalosis).
Non-respiratory alkalosis is frequently associated with potassium deficiency, but the fact that, in many instances, the alkalosis can usually be corrected by volume expansion without replacement of potassium casts doubt on the precise role of potassium. However, potassium depletion can contribute to the maintenance of a non-respiratory alkalosis through an effect on bicarbonate reabsorption. Severe potassium depletion enhances proximal bicarbonate reabsorption. Distal tubular sodium reabsorption takes place in exchange for potassium and hydrogen ions; particularly when there is enhanced distal sodium reabsorption, potassium depletion may result in an increased secretion of hydrogen ions into the tubular fluid.
The third factor that can maintain a non-respiratory alkalosis is an increase in mineralocorticoid activity. This promotes distal tubular sodium reabsorption and results in increased excretion of both potassium and hydrogen ions. The effect is potentiated by potassium depletion, increased distal sodium delivery (as with diuretic treatment) and by the presence of non-resorbable anions, which accentuate the negativity of the luminal aspect of tubular cells. Increased mineralocorticoid secretion can occur secondarily to ECF volume contraction or be primary, as in Conn and Cushing syndromes. In these two conditions, ECF volume is expanded and the alkalosis, unusually, is not corrected by saline infusion.
Biochemical characteristics of non-respiratory alkalosis
The blood hydrogen ion concentration is low and the bicarbonate concentration increased; respiratory compensation may increase PCO2, but not to more than about 8 kPa. Hypokalaemia is almost always present. Paradoxically, for the reasons explained above, the urine may be acidic and urinary potassium excretion increased.
Systemic effects of alkalosis
In general, the effects of alkalosis are the opposite of those of acidosis. The effects on the cardiovascular system are, however, less in alkalosis and infrequently of clinical consequence. Alkalosis is rarely sustained over a long period and there is no evidence of any adverse effects on bone.
Chronic non-respiratory alkalosis is frequently associated with potassium depletion and hypokalaemia. In addition to any contribution from the causative condition (e.g. gastric secretions contain approximately 10 mmol/L potassium), this is related to increased distal tubular secretion of potassium as a consequence of decreased hydrogen ion secretion. Thus, alkalosis can cause potassium depletion and potassium depletion sustain alkalosis. If sodium depletion is also present, the stimulation of maximal distal tubular sodium reabsorption will also contribute to potassium depletion.
Neuromuscular hyperexcitability is present frequently in patients with acute respiratory alkalosis, manifest as paraesthesia, muscle cramps and tetany. It is unusual in non-respiratory alkalosis, except when the hydrogen ion concentration falls rapidly, as has been reported in patients with chronic respiratory acidosis treated with mechanical ventilation. Grand mal convulsions have been reported in such patients, and alkalosis can precipitate a fit in patients with epilepsy. This is, at least in part, a result of buffering of hydrogen ion by plasma proteins, particularly albumin, which decreases in alkalosis, leading to increased binding of calcium to protein, thus lowering the plasma ionized calcium concentration.
Management of non-respiratory alkalosis
Management should be directed towards treatment of the underlying cause of the alkalosis, when possible. As mentioned above, treatment can also be directed towards the correction of any factors tending to sustain the alkalosis. Most patients will respond to expansion of the ECF volume with isotonic saline. The demonstration of a low urinary chloride concentration reliably predicts those patients who will respond to this treatment. This is frequently combined with potassium replacement, although, in many instances, the alkalosis can be corrected by the administration of saline alone, even if there is potassium depletion (which may require correction in its own right).
Administration of saline is inappropriate (and potentially dangerous) in patients with saline-unresponsive causes of non-respiratory alkalosis. Management must be directed towards the underlying cause, for example removing the source of excessive mineralocorticoid secretion (or blockade of mineralocorticoid action) or replacement of potassium or magnesium, as appropriate.
Specific causes of non-respiratory alkalosis
Loss of gastric acid
The most severe non-respiratory alkalosis is seen in patients losing unbuffered acid from the stomach because of either gastric drainage or prolonged vomiting, particularly in association with pyloric stenosis, which prevents the concomitant loss of alkaline secretions from the proximal small intestine. The acid is hydrochloric acid so that patients become chloride depleted. The hydrogen ions are derived from carbonic acid, pari passu with bicarbonate. Initially, renal excretion of the excess bicarbonate may prevent the development of alkalosis, but with ECF volume contraction, the requirement to maximize renal sodium reabsorption in the face of hypochloraemia necessitates increased reabsorption of sodium with bicarbonate. Indeed, in severe cases, renal bicarbonate reabsorption may be complete in spite of the high plasma concentration, resulting in the excretion of an acidic urine. Potassium is lost in gastric fluid, but increased aldosterone secretion may result in significant loss of potassium in the urine as well, exacerbating the potassium depletion and the alkalosis. Thus all three of the factors mentioned above can be involved in the maintenance of the alkalosis. The alkalosis usually responds to re-expansion of the ECF volume with isotonic saline; isovolaemic infusion of saline prevents the development of alkalosis in patients whose gastric fluid is being drained (e.g. because of postoperative ileus).
Post-hypercapnic alkalosis
It has been pointed out that the rapid lowering of a chronically elevated PCO2 can result in the compensatory processes causing a frank non-respiratory alkalosis. If PCO2 falls to normal over a few days, bicarbonate is excreted and an alkalosis does not develop, but if this occurs in patients with contraction of the ECF volume, for example as a result of diuretic treatment, alkalosis may become apparent and persist. It can be treated by administration of isotonic saline, though with caution because these patients frequently have secondary cardiac disease.
Mineralocorticoid excess
Syndromes of actual or apparent mineralocorticoid excess are almost invariably associated with non-respiratory alkalosis, for reasons that have been outlined above. Extracellular fluid volume is increased and patients are usually hypertensive. Expansion of the ECF should oppose the effect of potassium depletion on proximal bicarbonate reabsorption, and it is likely that the alkalosis is maintained largely by increased distal reabsorption.
Miscellaneous
Several other conditions can cause non-respiratory alkalosis as a result of increased renal excretion of hydrogen ions, but are not associated with hypertension. They include Bartter and Gitelman syndromes, and magnesium and potassium depletion. Refeeding after starvation is also sometimes associated with transient non-respiratory alkalosis, the cause of which is unclear, but which may be perpetuated by concomitant hypovolaemia and avid renal sodium reabsorption.
Respiratory alkalosis
Respiratory alkalosis is a consequence of the rate of excretion of carbon dioxide exceeding the rate of production, leading to a decrease in PCO2. This is usually due to stimulation of the respiratory centre: the stimulus may be toxic, reflex, psychogenic or related to the presence of an intracranial lesion. The exception is mechanical ventilation, when normal respiratory control is overridden. It is a common abnormality in the critically ill. Some of the causes of respiratory alkalosis are indicated in Box 5.5.
Compensatory responses in respiratory alkalosis
Buffering
In acute respiratory alkalosis, the fall in PCO2 causes a decrease in hydrogen ion concentration and a slight fall in bicarbonate. Other buffers release hydrogen ions, so tending to counter the effect of the fall in PCO2; some of these hydrogen ions will combine with bicarbonate, causing its concentration in the blood to fall further. A new steady state can be attained rapidly and can persist for approximately six hours, after which the effect of changes in renal hydrogen ion metabolism become detectable.
Hypoventilation
Correction of a respiratory alkalosis is only possible if the rate of excretion of carbon dioxide can be restored to normal. The fact that an alkalosis develops (other than with mechanical ventilation) indicates that the inhibitory effect of the decrease in PCO2 on respiration is being overwhelmed by whatever stimulus is causing the hyperventilation.
Renal hydrogen ion excretion
If a low PCO2 persists for more than a few hours, decreased renal generation of bicarbonate (for which carbon dioxide is a substrate) will decrease urinary acidification and effect further compensation for the alkalosis, further lowering the plasma bicarbonate concentration.
Biochemical features of respiratory alkalosis
The cardinal feature of an acute respiratory alkalosis is a decrease in arterial PCO2, a decrease in hydrogen ion concentration and a small decrease in bicarbonate concentration, though not to less than about 18 mmol/L. In a chronic respiratory alkalosis, renal compensation may result in arterial hydrogen ion concentration being only marginally decreased, while the bicarbonate concentration falls further, but not to less than about 12 mmol/L. The finding of bicarbonate concentrations less than these values suggests the additional presence of a non-respiratory acidosis.
If the stimulus to hyperventilation is hypoxaemia, arterial hydrogen ion concentration may be affected predominantly by a resulting non-respiratory acidosis. The interpretation of measured acid–base parameters in mixed disorders of hydrogen ion homoeostasis is discussed in a later section.
Systemic effects of respiratory alkalosis
The manifestations of the underlying disorder often predominate in patients with respiratory alkalosis. In acute hypocapnia, cerebral vasoconstriction reduces cerebral blood flow, and light-headedness, confusion, impaired intellectual function, syncope and seizures may occur. In patients with sickle cell disease, hypocapnia has been recorded as causing strokes, presumably as a result of cerebral hypoxaemia engendered by the vasoconstriction. Perioral and peripheral paraesthesiae are common, in part at least, because of a fall in ionized calcium concentration. These features usually remit if the hypocapnia persists.
In contrast, cardiovascular changes can occur with both acute and chronic hypocapnia. They include an increase in heart rate, non-specific chest pain or even frank angina.
Mild hypokalaemia can occur with respiratory alkalosis; plasma phosphate concentration is often significantly reduced.
Management
Where possible, treatment should be directed at the underlying cause. In a psychogenically induced acute respiratory alkalosis, rapid symptomatic relief may be obtained by getting the patient to rebreathe from a paper bag. If the alkalosis is severe, and the nervous or cardiovascular features are giving cause for concern, it may occasionally be necessary to sedate the patient or prevent the hyperventilation by resorting to mechanical ventilation, always being sure that adequate oxygenation is maintained.
The interpretation of acid–base data
Many approaches to the diagnosis of disorders of hydrogen ion homoeostasis have been promulgated. These may involve the mathematical manipulation of data, or the plotting of data on a diagram that shows the ranges of the variables in the various disorders. Three points are worth emphasizing in this context. First, although the use of diagrams may facilitate the rapid diagnosis of an acid–base disturbance, it cannot add to the information that is available from an analysis of the data on which the diagrams are based. Second, diagnosis should be based on measured variables and not on secondarily derived data. Third, acid–base variables must always be assessed in their clinical context: this is particularly important in mixed disorders, in which results may be indistinguishable from those that can occur during the physiological compensation of single disorders.
The most logical acid–base diagram is thus a graph of hydrogen ion concentration against partial pressure of carbon dioxide, as shown in Figure 5.6. This shows the zones in which combinations of PCO2 and [H+] occur in the various disorders that have been described. If a pair of data for a patient falls outside one of these areas, it suggests that there is a mixed acid–base disorder. Such diagrams may be useful not only to help in defining the acid–base disorder in an individual patient, but also, when serial plots are made, to follow the response to treatment.
It will be apparent that diagnosis of the type of acid–base disorder requires only knowledge of PCO2 and [H+]; this must follow from the fact that, as shown by Equation 7, bicarbonate concentration depends on PCO2 and [H+] and cannot vary independently of them.
In Figure 5.6, the plot of [H+] against PCO2 is rectilinear in acute respiratory disorders. This fact makes it possible to diagnose acid–base disorders without recourse to diagrams. The slope of the plot is such that, in an acute respiratory disturbance (that is, before there has been time for significant compensation to occur), for every 1 kPa change in PCO2, [H+] would be expected to change by 5.5 nmol/L, an increase in PCO2 increasing [H+] and a decrease in PCO2 decreasing [H+] by this amount.
Thus, in a patient whose PCO2 is abnormal, it is possible to calculate whether the observed [H+] is what it would be expected to be if the respiratory disturbance were acute, that is, in the absence of any compensation. Suppose, for example, a patient has a PCO2 of 7.5 kPa and [H+] of 52 nmol/L – plotting these figures on Figure 5.6 suggests that the patient has an acute respiratory acidosis. But this can also be appreciated by calculation: assuming a normal PCO2 of 5.3 kPa and [H+] of 40 nmol/L, an acute increase in PCO2 to 7.5 kPa would be expected to increase [H+] to [40 + (5.5 × 2.2)] = 52 nmol/L, as is observed. Were the observed [H+] to be higher, it would indicate that the acidosis had a non-respiratory as well as a respiratory component. Were the observed [H+] to be lower than expected from the PCO2, it would indicate the presence of a non-respiratory alkalosis in addition to the respiratory acidosis.
In this latter instance, however, an alternative explanation could be that the respiratory acidosis had been partly compensated. As has been indicated, the compensatory processes in disturbances of hydrogen ion homoeostasis can be regarded as the generation of opposing disturbances. Indeed, in respiratory disturbances, in which the development of compensation lags behind the development of the primary disorder, the presence and nature of the compensation is made explicit if the primary abnormality is corrected rapidly, leaving the persisting compensatory process as the sole acid–base abnormality (see Post-hypercapnic alkalosis, above).
As previously mentioned, it is important to appreciate that examination of PCO2 and [H+], or of data derived from them, in isolation from clinical information and other, independent, laboratory data, may not permit differentiation between a compensated disorder of hydrogen ion homoeostasis or the simultaneous existence of two primary disorders having opposing effects on hydrogen ion concentration. In the descriptions of the major acid–base disorders, the maximum extent to which compensation can occur was indicated, and if data lie outside these limits then a mixed disturbance must be present; however, such a disturbance can be present even if the data lie within the limits of normal compensation.
The approach to the interpretation of acid–base data described above is essentially physiological; it does not require the use of derived data which, as explained in an earlier section, do not provide additional, independent, information.
An alternative approach, which is essentially physicochemical in nature, is provided by the strong ion theory, originally developed by Stewart (1978). This was based on in vitro studies of the dissociation of water (and hence hydrogen ion concentration) in the presence of various solutes and carbon dioxide, and led to the conclusion that [H+] is determined by three independent variables. These are the ‘strong ion difference’ (SID, the sum of the concentrations of sodium, potassium, magnesium and calcium ions less than that of the sum of the concentrations of chloride, sulphate and organic acid anions); the total concentration of weak acids, i.e. phosphate and proteins (AT) and the partial pressure of carbon dioxide.
Strong ion difference is further differentiated into ‘apparent’ SID (SIDa) and ‘effective’ SID (SIDe). The former is provided by ([Na+] + [K+]) − [Cl−]); the latter is the sum of 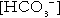 and non-bicarbonate buffers, that is, the anionic equivalences of albumin and phosphate. The difference between the two is the ‘strong ion gap’ (SIG). The normal value of SIG is zero. The various quantities are either measured or read from nomograms based on the solutions of a series of complex equations.
and non-bicarbonate buffers, that is, the anionic equivalences of albumin and phosphate. The difference between the two is the ‘strong ion gap’ (SIG). The normal value of SIG is zero. The various quantities are either measured or read from nomograms based on the solutions of a series of complex equations.
This approach defines additional primary acid–base disorders in addition to the four described above. There are two respiratory disorders (as with the physiological approach), but in non-respiratory acidosis, although SIDe is always low, this can occur with a high SIG (as in lactic acidosis) or a normal SIG (as with bicarbonate loss); in non-respiratory alkalosis, SIDe is high and SIG normal, but both acidosis and alkalosis can also be due to abnormal value of AT (hyperalbuminaemic acidosis and hypoalbuminaemic alkalosis, respectively). The contribution of differences in plasma protein concentration is not considered to be of significance in the physiological approach (and indeed, changes in plasma albumin concentration in vivo do not affect [H+]).
Although the strong ion theory has its proponents, it has not been widely adopted. It is considerably more complex than the physiological approach; it is derived entirely from observations made in vitro; it does not provide any quantitative data concerning the compensatory responses to primary disorders and, although there are some data suggesting that it better predicts outcome in critically ill patients than traditional approaches, most studies have failed to do this. (The interested reader will find further information and discussion of this topic in the Further reading section, below.)
Mixed disorders of hydrogen ion homoeostasis
Mixed acid–base disorders, that is, disorders with both respiratory and non-respiratory components, occur frequently. Some examples are shown in Box 5.6. They can be classified according to whether the component disturbances are additive or opposing in their effects on hydrogen ion concentration.
The presence of a mixed disorder can be inferred from any change in hydrogen ion concentration not being as predicted from the PCO2. In a mixed respiratory and non-respiratory acidosis, PCO2 will be elevated, but [H+] will be higher than would be predicted from the PCO2. In a mixed respiratory and non-respiratory alkalosis, PCO2 will be decreased, but [H+] will be lower than predicted. In each case, the plot of [H+] and PCO2 on the acid–base diagram (Fig. 5.6) will fall in the regions between those shown for the appropriate non-respiratory and acute respiratory disorders.
The diagnosis of mixed disorders in which the component disturbances have opposite effects on hydrogen ion concentration is less straightforward. As discussed above, the problem is that the changes in [H+] and PCO2 may be exactly the same as may be found as a result of physiological compensation. This stems from the fact that the compensatory process itself disturbs acid–base homoeostasis, albeit advantageously, tending as it does to restore [H+] towards normal. Usually, careful consideration of the clinical findings and other data that may be available will allow the correct diagnosis to be made. It should also be appreciated that the efficacy of compensatory processes is limited, particularly in non-respiratory disorders. Compensation restores the hydrogen ion concentration towards normal, but restoration to normal is only seen with mild chronic respiratory acidosis and chronic respiratory alkalosis. With more severe disturbances, compensation is incomplete. Overcompensation does not occur so that, for example, a patient with a slightly elevated [H+] and a low PCO2, must have a partially compensated non-respiratory acidosis and cannot have compensated respiratory alkalosis.
Since the causes of respiratory acidosis and alkalosis are retention and excessive excretion of carbon dioxide, respectively, these two disorders cannot coexist. However, several mechanisms can be responsible for the development of non-respiratory disorders, and the existence of a process producing an acidosis need not exclude the presence of one producing an alkalosis. Thus, a patient could have renal failure (a cause of acidosis) and be vomiting excessively (a cause of alkalosis). Whether the patient is actually acidotic or alkalotic will depend on which process predominates, but examination of [H+] and PCO2 alone will not reveal this type of mixed disturbance. It may only be inferred from clinical observations coupled with other biochemical data – particularly, for example, the anion gap – or become apparent when the predominant disorder is treated appropriately and apparent ‘over-swing’ occurs, for example the patient, having had a mild non-respiratory acidosis, becomes severely alkalotic.
Exceptionally, triple acid–base disorders can occur. Patients have been described with a severe non-respiratory alkalosis (usually due to prolonged vomiting), accompanied by a respiratory acidosis or alkalosis, in whom a high anion gap suggests a component of non-respiratory acidosis (e.g. a lactic acidosis). The [H+] (low) and PCO2 (also low) in such patients may suggest a partially compensated acute respiratory disturbance, but the clinical features will be inconsistent with this as the sole diagnosis.
In practice, because mixed acid–base disorders frequently develop in patients who are severely ill, the management of the acid–base disorder per se will usually be less important than that of the underlying illness. Indeed, the management of any acid–base disorder should include measures to correct the cause where possible, though, as has been indicated above, specific measures relating to the acidosis or alkalosis may also be necessary.
TISSUE OXYGENATION
Introduction
The process whereby atmospheric oxygen is made available to mitochondria, where the oxidation of carbon and hydrogen releases energy, is a complex one, depending on adequate alveolar ventilation and function, pulmonary and tissue blood flows, and the ability of the blood itself to take up oxygen in the alveoli and release it to tissues. Tissue oxygenation can be compromised by disease affecting any of these functions. Until relatively recently, the only readily available index of tissue oxygen supply was the arterial partial pressure of oxygen, PaO2. This measurement is still regarded as essential, but it has a number of limitations. It requires access to arterial blood, either by direct puncture or through an indwelling catheter; blood samples must be collected with considerable care and analysis performed without delay, but, perhaps most importantly, measurement of PaO2 provides incomplete information on oxygen transport. It is a measure of partial pressure, not of the oxygen content of blood nor the delivery of oxygen to tissues. However, although tissue oxygenation depends upon factors other than PaO2, maintenance of an adequate PaO2 is a prerequisite for normal tissue oxygenation. This is illustrated in the following sections, which discuss the transport of oxygen from the inspired gas to the tissues, where it is used for oxidative metabolism.
Pulmonary function
The lungs have two principal functions: to transfer oxygen from inspired gas to the blood, and to remove carbon dioxide from the blood to the expired gas. As discussed above, the latter is critical to hydrogen ion homoeostasis, and is compromised by hypoventilation. Hypoventilation invariably leads to decreased oxygen uptake into the blood, but, as will be seen, impaired oxygenation can occur in the absence of hypoventilation.
Alveolar ventilation
The partial pressure of oxygen in arterial blood, PaO2, depends on the alveolar oxygen tension (PaO2), which is in turn dependent on the fraction of the inspired gas comprising oxygen (FiO2), the arterial carbon dioxide tension (PACO2), the respiratory quotient (RQ), atmospheric barometric pressure (PB) and the partial pressure of water vapour (PH2O) such that:
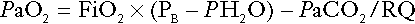
This is the alveolar gas equation. The respiratory quotient depends on the relative proportions of free fatty acids, carbohydrate and protein being used as energy substrates by the tissues, but it varies through only a small range, even in disease. Alveolar air is always saturated with water so that PH2O is constant. It follows that PAO2 can increase significantly as a result of either an increase in PB (requiring a hyperbaric chamber) or FiO2 (requiring the administration of oxygen) or a decrease in PACO2 (requiring an increase in ventilation). Only the last two are readily available to treat patients with a low PAO2. Inspection of Equation 29 will indicate that the effect of a change in PACO2 on PAO2 will be greater if FiO2 is normal than if it is raised; an increase in PACO2 (usually accepted as being equal to PaCO2) can significantly decrease PAO2 at normal values of FiO2, but it will have a lesser effect if the FiO2 is increased.
Oxygen uptake into blood
The continuous delivery of mixed venous blood of low oxygen content to the alveolar capillaries, and diffusion of oxygen down the concentration gradient from the alveolar space to the blood, result in a constant tendency for PAO2 to fall that is prevented by the delivery of oxygen to the alveoli by ventilation. In healthy young individuals, oxygen diffuses readily from alveoli into the plasma and PaO2 is usually only about 1 kPa less than PAO2, that is (breathing room air at sea level) about 13.3 kPa. The gradient increases with age and the approximate normal value is given by (PAO2 − PaO2) = (0.06 × age in years) kPa so that it may reach nearly 4 kPa in healthy 60-year-olds. Pulmonary disease that impairs diffusion is a potential cause of an increase in (PAO2 − PaO2) and hence a decrease in PaO2, but the nature of the alveoli is such that any impairment of diffusion must be considerable before it affects PaO2 at rest.
The maintenance of a normal PaO2 also requires a normal relationship between the perfusion of alveoli and their ventilation: the effects of a disturbance in this relationship (ventilation–perfusion imbalance) are further considered below.
In health, a small proportion of the blood reaching the lungs from the tissues bypasses the alveoli and does not take part in gas exchange (a right-to-left shunt). This is because the bronchial veins drain directly into the pulmonary veins, while some blood that perfuses the myocardium drains directly into the cavity of the left ventricle. Any increase in shunting due to a pathological process will tend to decrease PaO2.
The final factor that can affect PaO2 is the oxygen tension in the blood reaching the lungs, that is, mixed (‘mixed’ because this blood is derived from all the veins draining into the right side of the heart) venous PO2 or PvO2. If this is low, increased alveolar oxygen transport will be necessary to allow maximal PaO2. In health, increased tissue oxygen requirements (as, e.g. during exercise) result in a fall in PvO2 and the increased oxygen requirement is met by hyperventilation. Tissue oxygen requirements are often increased in disease, for example as a result of sepsis or the metabolic response to trauma, but, at the same time, the physiological responses leading to hyperventilation may be attenuated. Other factors that can contribute to a low PvO2 include decreased oxygen saturation, anaemia and a decrease in cardiac output, all of which, as will be discussed below, can decrease the delivery of oxygen to tissues.
In summary, a low PaO2, that is, hypoxaemia, can be caused by any of the following:
• hypoventilation
• decreased diffusion
• imbalance of ventilation and perfusion
• an increase in shunting
• a decrease in PvO2.
Hypoxaemia caused by hypoventilation or a decrease in PAO2 or PvO2 can be distinguished from the other causes by calculating the alveolar–arterial oxygen gradient (PAO2 − PaO2). PAO2 can be calculated from arterial blood gas measurements using the alveolar gas equation. The gradient is increased when hypoxaemia is a result of impaired diffusion, shunting or imbalance between ventilation and perfusion. In these conditions, PaCO2 is often normal. However, when, as in hypoxaemia caused by alveolar hypoventilation, PaCO2 is increased, (PAO2 − PaO2) is typically normal.
Hypoxaemia becomes recognizable clinically when it is sufficiently severe to cause central cyanosis. This requires a deoxygenated haemoglobin concentration of more than 50 g/L (saturation < 75% at normal haemoglobin concentrations). In patients with severe anaemia, the low haemoglobin may result in significant hypoxaemia being present without there being discernible cyanosis; conversely, patients with polycythaemia may have central cyanosis, despite a relatively normal oxygen saturation.
Peripheral cyanosis will always be present if there is central cyanosis, but peripheral cyanosis can occur in the absence of central cyanosis as a consequence of reduced peripheral circulation.
The role of haemoglobin in oxygen transport
Although the amount of oxygen present in physical solution in the blood is directly related to PaO2 (0.225 mL/L per kPa), only a small amount of oxygen is carried in this way (at normal PaO2, approximately 3 mL per litre of plasma). Oxygen is principally transported in the blood bound to haemoglobin. One gram of haemoglobin can bind 1.34 mL of oxygen when fully saturated. The normal PaO2 of arterial blood is approximately 13.3 kPa, at which haemoglobin is approximately 97% saturated. Total arterial oxygen content (CaO2) is given by the sum of the dissolved and haemoglobin-bound fractions:
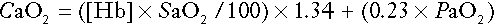
where [Hb] is the haemoglobin concentration (g/L) and SaO2 is the percentage saturation of haemoglobin with oxygen. At sea level, CaO2 is normally about 200 mL/L. SaO2 can be measured using a co-oximeter; these instruments also measure haemoglobin concentration and thus allow calculation of arterial oxygen content, which provides far more information than PaO2 alone. Modern blood gas analysers often incorporate a co-oximeter, and such instruments are widely used in intensive therapy units. Arterial oxygen content can also be measured non-invasively (transcutaneously) using a pulse oximeter. It is beyond the scope of this book to discuss in detail the analytical principles utilized in these instruments, but a summary is provided in a later section (see p. 91).
The familiar sigmoid relationship between Hb saturation and oxygen tension (Fig. 5.7) has a number of important clinical consequences. First, a considerable reduction in PaO2 below normal has little effect on the amount of oxygen carried in the blood. Saturation only falls below 90% when PaO2 falls below 8 kPa. If PaO2 falls further, however, oxygen saturation (and thus the amount of oxygen carried) decreases sharply. A further consequence is that, because haemoglobin is saturable, increasing PaO2 above that necessary to provide complete saturation has relatively little effect on arterial oxygen content, since only the small fraction present in solution is significantly increased.
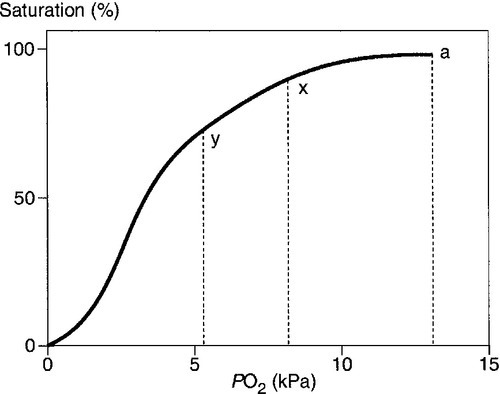
The effects of pulmonary disease on oxygen uptake into blood
The effects of alveolar hypoventilation and impaired diffusion on pulmonary function have been described above. Two other functional defects, shunting and ventilation–perfusion imbalance, can also have profound effects on the oxygenation of blood.
Shunting
Haemoglobin is only about 97% saturated in normal arterial blood. This is, in part, because of physiological shunting. In some conditions (e.g. lobar pneumonia, pulmonary oedema, adult respiratory distress syndrome), some alveoli become filled with fluid and do not take part in gas exchange, although they are still perfused with blood. Shunting is thereby increased and this leads to arterial hypoxaemia. Atelectasis (collapse of a lung or part of a lung so that it is not aerated) has the same effect. Under such circumstances, increasing PAO2 by increasing FiO2 has little effect on overall PaO2. This is because no gas exchange will be taking place in the alveoli that are acting as shunts, while haemoglobin in blood leaving normally functioning alveoli will already be fully saturated with oxygen. Increasing FiO2 will increase only the small proportion of oxygen held in physical solution in this blood, and this will have little effect on total arterial oxygen content.
Ventilation–perfusion imbalance
The fact that haemoglobin is not fully saturated in normal arterial blood is also related to an imbalance between alveolar ventilation and perfusion. At rest, ventilation is about 4.2 L/min and pulmonary blood flow is about 5.5 L/min, so that the overall ventilation–perfusion ratio (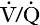 ratio) is approximately 0.8. However, this ratio is not uniform throughout the lungs, ranging between the approximate limits 0.5 and 3.0. Some alveoli are better ventilated than they are perfused (
ratio) is approximately 0.8. However, this ratio is not uniform throughout the lungs, ranging between the approximate limits 0.5 and 3.0. Some alveoli are better ventilated than they are perfused (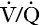 > 1), so that a proportion of ventilation is ‘wasted’ and the effect is an increase in ‘dead space’; in others,
> 1), so that a proportion of ventilation is ‘wasted’ and the effect is an increase in ‘dead space’; in others, 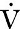 and
and 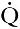 are in balance. In both cases, effective oxygenation of the blood takes place. However, in those alveoli in which perfusion exceeds ventilation (
are in balance. In both cases, effective oxygenation of the blood takes place. However, in those alveoli in which perfusion exceeds ventilation (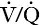 < 1), complete oxygenation of the blood is impossible. Because haemoglobin is saturable, oxygen transport in the well-ventilated and normally ventilated alveoli cannot compensate for decreased oxygenation of blood in poorly ventilated alveoli. Many pulmonary diseases, notably chronic obstructive pulmonary disease and interstitial pulmonary disease, give rise to an increase over the normal imbalance of ventilation and perfusion. However, in contrast to disease in which significant shunting occurs, increasing the FiO2 in such conditions, because it will increase oxygen transport in poorly ventilated alveoli, can increase overall PaO2.
< 1), complete oxygenation of the blood is impossible. Because haemoglobin is saturable, oxygen transport in the well-ventilated and normally ventilated alveoli cannot compensate for decreased oxygenation of blood in poorly ventilated alveoli. Many pulmonary diseases, notably chronic obstructive pulmonary disease and interstitial pulmonary disease, give rise to an increase over the normal imbalance of ventilation and perfusion. However, in contrast to disease in which significant shunting occurs, increasing the FiO2 in such conditions, because it will increase oxygen transport in poorly ventilated alveoli, can increase overall PaO2.
Differential effects of pulmonary disease on PaCO2 and PaO2
It will be instructive at this point to compare the effects of respiratory disease on PaCO2 and PaO2. Carbon dioxide is transported in blood effectively in physical solution (albeit mostly in the form of bicarbonate). In contrast to the oxyhaemoglobin dissociation curve, the curve relating the carbon dioxide content of blood to partial pressure is almost linear over the physiological range. As a result, a significant change in PaCO2 will always cause a significant change in the carbon dioxide content of blood. In contrast, as has been seen, considerable changes in PaO2 can occur with little effect on the blood’s oxygen content.
PaCO2 is determined by alveolar ventilation: in health, any increase in carbon dioxide formation (e.g. during exercise) can be matched by an increase in excretion and PaO2 is not affected. In contrast, in pulmonary disease, if PaCO2 is increased, then PaO2 will always be decreased. Thus, if hypoventilation causes hypercapnia, it will also cause hypoxaemia. However, hypercapnia is not always present in patients who are hypoxaemic. Carbon dioxide can diffuse between the blood and the alveolar air more readily than oxygen; defects in diffusion rarely cause hypercapnia, although they can cause hypoxaemia. Shunting causes a decrease in PaO2, but this will stimulate ventilation, increasing the excretion of carbon dioxide from ventilated alveoli and preventing a rise in PaCO2 or even causing hypocapnia. Only with extensive shunting will the PaCO2 be increased.
In pulmonary disease causing an increase in 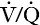 imbalance, there is a tendency for PaO2 to fall, for the reasons discussed above; this will stimulate respiration but, while the increased ventilation of well-perfused alveoli cannot compensate for the impaired oxygenation, it can increase carbon dioxide excretion and PaCO2 fall. With more severe disease, however, hypoxaemia will be accompanied by hypercapnia.
imbalance, there is a tendency for PaO2 to fall, for the reasons discussed above; this will stimulate respiration but, while the increased ventilation of well-perfused alveoli cannot compensate for the impaired oxygenation, it can increase carbon dioxide excretion and PaCO2 fall. With more severe disease, however, hypoxaemia will be accompanied by hypercapnia.
Respiratory failure, defined by PaO2 being < 8 kPa or PaCO2 being > 7 kPa, is thus divided into two types: type I, in which PaO2 is low and PaCO2 is normal or low (typically seen in condition such as pneumonia, pulmonary oedema and acute lung injury, where lung tissue is damaged) and type II, in which PaO2 is low but PaCO2 is high (typically seen in chronic obstructive pulmonary disease and conditions causing hypoventilation).
Oxygen transport to tissues
Oxygen delivery
The transport of oxygen to tissues depends not only on adequate transfer of oxygen from the inspired gas to the alveolar capillaries, but also on an adequate cardiac output. The total amount of oxygen delivered by the cardiopulmonary apparatus (DO2) is given by the product of oxygen content (CaO2) and cardiac output (CO):
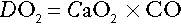
However, the amount of oxygen available to tissues will depend upon local perfusion and the affinity of haemoglobin for oxygen, which determines how readily oxygen can be released.
Oxygen uptake
Oxygen is taken up into tissues because their PO2 is lower than that of the blood, and oxygen diffuses down its concentration gradient. However, examination of the oxyhaemoglobin dissociation curve indicates that the amount of oxygen taken up will depend on the haemoglobin saturation (SO2) at a given PO2. Mixed venous oxygen saturation (SvO2) at rest is approximately 75%, corresponding to a PvO2 of 5.3 kPa and permitting the uptake of 46 mL of oxygen from each litre of blood. Resting cardiac output is approximately 5 L/min, and total tissue uptake of oxygen is about 250 mL/min at rest. However, if the oxyhaemoglobin curve were to shift to the left, less oxygen would be released from haemoglobin for the same fall in pO2, whereas a right shift would increase the availability of oxygen.
Such shifts occur physiologically. An increase in PCO2 or [H+] (both of which occur as blood traverses the capillary beds) causes a right shift, as does an increase in temperature. The position of the curve is also determined by the concentration of 2,3-DPG in erythrocytes. An increase in 2,3-DPG concentration (normally about 4 mmol/L of red cells) causes a right shift. This occurs in chronic hypoxia and thus facilitates oxygen uptake by tissues. These effects are illustrated in Figure 5.8. The position of the oxyhaemoglobin saturation curve can be defined by P50, the partial pressure of oxygen at which haemoglobin is 50% saturated. It is normally about 3.7 kPa.
Hypoxia
Tissue hypoxia can be due to a disturbance occurring at any stage in the delivery of oxygen to the cells where it is utilized (Table 5.4), to increased demand or to deficient oxygen uptake. The rational treatment of hypoxia obviously depends on knowing the cause.

Measurement of oxygen delivery to tissues
This requires determination of arterial oxygen content, preferably from measurements of haemoglobin and SaO2, and of cardiac output. SaO2 can be calculated from PaO2, using an assumed value for P50, but direct measurement, by oximetry, is more reliable. It may be possible to infer the adequacy of cardiac output from clinical observation, but it can be measured directly by the thermodilution method using a pulmonary artery (Swan–Ganz) catheter incorporating a thermistor.
Oximeters – instruments that measure the saturation of haemoglobin with oxygen – are of two types. Co-oximeters measure the optical absorbance of blood in vitro at multiple wavelengths; because oxyhaemoglobin and deoxygenated haemoglobin (and, indeed, carboxyhaemoglobin and methaemoglobin) have different absorbance characteristics, these instruments can be calibrated to quantitate these species separately, and thus to measure SaO2. Pulse oximeters are non-invasive. They have sensors that are usually placed on a thin part of the patient’s anatomy (e.g. a finger tip or ear lobe) that direct light of two wavelengths (660 and 940 nm) through the tissue to a detector that measures the relative absorbance and hence haemoglobin and oxyhaemoglobin. The influence of absorption by tissues and venous and capillary blood is minimized by measuring the changing absorption (hence pulse oximetry) that is due to haemoglobin in arterial blood alone. Reflectance pulse oximeters are also available, but are less widely used.
Pulse oximeters do not detect carboxyhaemoglobin and other haemoglobin variants and may thus give misleading results if these are present. For example, while the saturation may be normal (e.g. 97%), if 12% of the haemoglobin were to be in the form of carboxyhaemoglobin, the true percentage of oxyhaemoglobin would only be 85% of the total. Pulse co-oximeters that combine the two techniques and allow more reliable non-invasive measurement of SaO2 have been developed.
Most blood gas analysers provide an estimate of SaO2 based on measurements of [H+], PaO2 and empirical equations. This approach relies on a number of assumptions that may not be valid in individual patients, with the result that these estimates can vary significantly from measured values, particularly if abnormal haemoglobins (e.g. carboxy-, methaemoglobin) are present. Direct measurements are to be preferred. Blood gas analysers may also provide a value for arterial oxygen content, but as with saturation, these are only estimates, not true measurements.
Detection of tissue hypoxia
Although the accumulation of lactic acid is a consequence of severe tissue hypoxia, the development of hyperlactataemia is a relatively late phenomenon, and by the time it is detectable, hypoxic tissue damage may already have occurred. Furthermore, it can occur for other reasons, for example impaired hepatic function, strenuous muscle contraction (not only in exercise but also due to rigors or convulsions) and as a result of improved perfusion of previously poorly perfused tissue (‘washout’ phenomenon).
The measurement of mixed venous oxygen saturation (SvO2) is now widely used in the assessment of the critically ill. SvO2 can be measured either in vitro in a blood sample taken through a pulmonary artery catheter or in vivo using a catheter containing fibre-optic bundles, which transmit light of appropriate wavelength to the blood and transmit reflected light back to a measuring device. It reflects the difference between arterial oxygen supply and tissue consumption. SvO2 is normally of the order of 75%, but frequently falls to 50% or less when there is anaerobic metabolism, although such a fall can also be due to appropriately increased tissue uptake of oxygen or to a decrease in cardiac output. Because the measured SvO2 is effectively a mean value of venous blood from all tissues, it can be affected by changes in the relative distribution of blood between different tissues. Measurements of SvO2 can be misleading; in severe sepsis, reduced tissue oxygenation is sometimes associated with a high SvO2, as a result of impaired oxygen extraction and local arteriovenous shunting.
Oxygen delivery to tissues (approximately 16 mL/kg body weight/min at rest) normally exceeds demand, so that if delivery falls demand may still be met and consumption be independent of supply. However, if supply falls below a critical level (approximately 8 mL/kg per min), oxygen consumption becomes supply dependent (Fig. 5.9). If this occurs, it may be possible to improve oxygenation by the use of inotropes or red cell transfusion (to improve supply) or sedation (to reduce consumption).
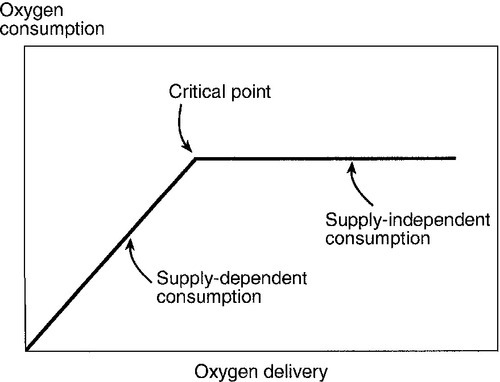
Management of respiratory failure
It is beyond the scope of this book to discuss this topic in detail. Appropriate measures, depending on the cause, may include treatment of any airways obstruction, infection or pulmonary oedema, increasing FiO2, improving alveolar hypoventilation to decrease PACO2, using inotropic drugs to stimulate cardiac output, increasing the oxygen-carrying capacity of the blood by red cell transfusion and the use of measures to increase tissue perfusion or reverse the effects of toxins. Hypophosphataemia is a sometimes unrecognized cause of respiratory muscle weakness, especially in the critically ill, as is malnutrition. In patients with chronic hypercapnia who are hypoxaemic, it has long been thought that increasing the FiO2 is potentially dangerous. The rationale for this notion is that acquired insensitivity of the respiratory centre to carbon dioxide can cause hypoxaemia to be providing the major stimulus to respiratory effort, and that its abolition would lead to respiratory arrest. However, hypoxaemia is a greater threat to life than hypercapnia, and this danger is now thought too have been exaggerated.
There are many techniques for improving alveolar hypoventilation, including the provision of invasive respiratory support through the use of a mechanical ventilators or even extracorporeal oxygenation techniques. Ventilators may be used in a variety of modalities, for example to take over completely a patient’s breathing (e.g. when there is paralysis of the respiratory muscles or in deep coma leading to absent or poor respiratory effort) or to provide assistance to inspiratory efforts initiated by the patient. However, mechanical ventilation, although a potentially life-saving technique in critical hypoxia, is not without its disadvantages, including impaired cardiac filling (and hence a decrease in cardiac output); too rapid a fall in PaCO2; depressing the normal respiratory drive; dilutional hyponatraemia due to inappropriate antidiuresis, and a risk of mechanical damage to the airways and lungs (which may already be damaged). Oxygen in high concentrations is potentially toxic, and the lowest concentration compatible with reversing and preventing hypoxia should be used.
Ventilatory support is not synonymous with invasive mechanical ventilation. Patients able to breathe spontaneously may be helped by the maintenance of continuous positive airways pressure (CPAP) through the use of tight-fitting nasal or facial masks. The increased pressure aids gas entry during inspiration and reduces airways collapse during expiration.
CONCLUSION
Disorders of hydrogen ion metabolism and of tissue oxygenation are frequently encountered in clinical practice and may coexist, often because of a common aetiology, but sometimes because of a direct effect of one upon the other. For both groups of disorders, a sound understanding of the physiological principles governing homoeostasis in normal individuals is essential for correct diagnosis and effective management. These principles are essentially simple, and pertain no matter how complex any particular disturbance may appear.
Further reading
Critical Care Medicine Tutorials. http://www.ccmtutorials.com/rs/oxygen/page11.htm; [Accessed 19.01.12].
A succinct explanation of the concept of delivery dependent oxygen consumption.