Cardiovascular Emergencies
Edited by Anne-Maree Kelly
5.1 Chest pain
Steve Goodacre
Introduction
Chest pain is one of the most common presenting complaints in emergency medicine. It is also associated with life-threatening pathology, so it is arguably the most important complaint faced by emergency clinicians. It is certainly one of the most challenging. Failure to diagnose and manage appropriately patients with acute chest pain is a frequent cause of avoidable mortality and morbidity and is a leading cause of malpractice litigation. It is therefore not surprising that physicians often err on the side of caution, yet this can also have adverse consequences for the patient and society. Patient anxieties following unnecessary investigation are often unrecognized but may severely affect quality of life, while over-investigation and unnecessary hospital admission for chest pain waste millions of healthcare dollars each year.
Epidemiology
The incidence of acute chest pain presenting to the emergency department appears to be increasing. Awareness of the importance of early treatment for myocardial infarction has led to public information campaigns that increase emergency department attendances with chest pain. Meanwhile, general practitioners are increasingly being bypassed in favour of an emergency ambulance response. These changes in health service use have coincided in many developed countries with a decrease in the incidence of coronary heart disease. It therefore seems likely that patients presenting to the emergency department with acute chest pain have decreasing prevalence of acute coronary syndrome (ACS) and increasing prevalence of more benign conditions.
Differential diagnosis
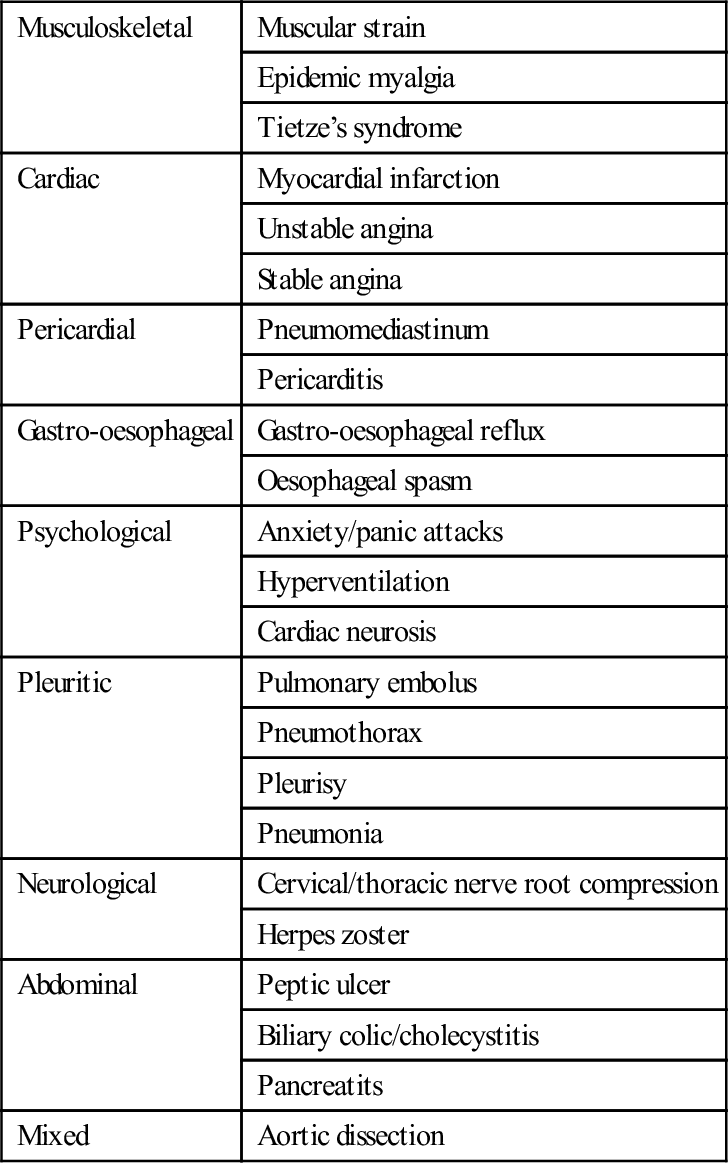
The main differential diagnoses are outlined in Table 5.1.1. The most common causes of acute chest pain are ACS (unstable angina or myocardial infarction), musculoskeletal pain, anxiety, gastro-oesophageal pain and non-specific chest pain. The most serious causes (in terms of threat to life) are ACS, pulmonary embolism and aortic dissection. Since ACS is both common and life threatening, it is inevitably the primary focus of assessment.
Table 5.1.1
| Musculoskeletal | Muscular strain |
| Epidemic myalgia | |
| Tietze’s syndrome | |
| Cardiac | Myocardial infarction |
| Unstable angina | |
| Stable angina | |
| Pericardial | Pneumomediastinum |
| Pericarditis | |
| Gastro-oesophageal | Gastro-oesophageal reflux |
| Oesophageal spasm | |
| Psychological | Anxiety/panic attacks |
| Hyperventilation | |
| Cardiac neurosis | |
| Pleuritic | Pulmonary embolus |
| Pneumothorax | |
| Pleurisy | |
| Pneumonia | |
| Neurological | Cervical/thoracic nerve root compression |
| Herpes zoster | |
| Abdominal | Peptic ulcer |
| Biliary colic/cholecystitis | |
| Pancreatits | |
| Mixed | Aortic dissection |
ACS is discussed in detail in Chapter 5.2, pulmonary embolus in Chapter 5.5 and aortic dissection in Chapter 5.10.
Musculoskeletal chest pain may be related to a precipitating episode, such as chest wall injury or physical over-exertion. Alternatively, it may be caused by inflammation in chest wall structures. Tietze’s syndrome (costochondritis) is most commonly seen in women and is characterized by tenderness of the costochondral cartilages. Epidemic myalgia (Bornholm disease) is due to inflammation of chest wall muscles and pleura occurring after viral infection, typically Coxsackie B. Herpes zoster produces severe pain along the distribution of a thoracic nerve and may be misdiagnosed as musculoskeletal pain if the patient presents before any rash or vesicles have developed.
Gastro-oesophageal pain occurs when gastric contents reflux into the oesophagus or when the oesophageal muscles spasm. Pneumomediastinum can occur spontaneously after vigorous exercise, vomiting or an asthma attack or may be associated with barotrauma from diving or inhalation during drug abuse. Pericarditis is most commonly caused by viral infection, but may be associated with systemic illness, such as uraemia or autoimmune disease, or follow myocardial infarction or cardiac surgery (Dressler syndrome).
Anxiety-related chest pain is a common and frequently unrecognized cause of acute chest pain. It may also coexist and be an important factor alongside other causes of chest pain. The patient with coronary heart disease and anxiety-related chest pain presents a particularly difficult diagnostic and management challenge. Anxiety may be related to a specific serious cause of chest pain and can be exacerbated by misguided efforts to provide reassurance through diagnostic testing. In extreme cases, this can lead to ‘cardiac neurosis’ in which the patient’s anxieties about cardiac disease cause more severe disruption to their daily activities and quality of life than would be expected from the pathology that worries them.
Pleurisy is typically caused by a viral infection and produces pain that is worse on inspiration. It may be differentiated from pulmonary embolus by the presence of systemic features and the absence of breathlessness or risk factors for thromboembolism, although investigation for pulmonary embolism is often required. Pneumonia and pneumothorax can also cause pleuritic pain but should be evident on chest radiography.
There are a number of serious abdominal complaints that may present as chest pain. These include biliary colic (acute biliary pain), cholecystitis, peptic ulcer disease and pancreatitis. Failure to take a careful history and examine the abdomen may lead to delayed diagnosis.
Finally, a substantial proportion of patients will be labelled entirely appropriately as ‘non-specific chest pain’ after emergency department evaluation. These patients have pain that simply cannot be categorized into a clear diagnostic group. It is more honest to accept this than apply an inaccurate diagnostic label.
Clinical features
Clinical assessment is primarily aimed at identifying patients with a significant risk of serious pathology who require further investigation and possibly inpatient care. The most common serious pathology is ACS, so clinical assessment is often focused upon associated features. Other serious conditions, such as pulmonary embolism and aortic dissection, should not be neglected.
ACS is classically associated with chest pain that is crushing, gripping or squeezing in nature and radiates to the left arm, but presenting features in the emergency department may be much more variable, particularly in patients with no past history of coronary heart disease and a non-diagnostic ECG. Table 5.1.2 shows the likelihood ratios of clinical features that may help to diagnose ACS. It is notable that pain radiating to the right arm or to both arms is a powerful predictor of ACS. Pain described as ‘burning’ or ‘like indigestion’ can be associated with ACS, as can pain occurring on exertion. So the diagnoses of gastro-oesophageal reflux or stable angina should be made with great caution. Pain that is sharp or associated with inspiration or movement is less likely to be cardiac, but these findings alone do not exclude ACS. Risk factors for coronary heart disease should be routinely recorded, although they may have surprisingly little diagnostic value. This is perhaps because patients are aware of these risk factors and take them into account when deciding whether or not to seek help for episodes of chest pain. In this respect, social and cultural factors may have an important influence upon patient’s interpretation of their symptoms and health-seeking behaviour.
Table 5.1.2
Likelihood ratios of clinical features useful for diagnosing acute myocardial infarction
| Useful for ruling in myocardial infarction | |
| Radiation to the right arm or shoulder | 4.7 |
| Radiation to both arms or shoulders | 4.1 |
| Described as burning or like indigestion | 2.8 |
| Association with exertion | 2.4 |
| Radiation to left arm | 2.3 |
| Associated with diaphoresis | 2.0 |
| Associated with nausea or vomiting | 1.9 |
| Worse than previous angina or similar to previous myocardial infarction | 1.8 |
| Described as pressure | 1.3 |
| Useful for ruling out myocardial infarction | |
| Described as pleuritic | 0.2 |
| Described as positional | 0.3 |
| Described as sharp | 0.3 |
| Reproducible by palpation | 0.3 |
| Inframammary location | 0.8 |
| Not associated with exertion | 0.8 |
Clinical examination is of limited diagnostic value and is mainly aimed at identifying non-cardiac causes of chest pain or complications of ACS, such as arrhythmia, heart failure or cardiogenic shock. Pain that can be reproduced by chest wall palpation is less likely to be cardiac, but this finding does not exclude the possibility of ACS. It is also important to determine specifically that chest wall palpation is reproducing the pain that led to presentation. Simply identifying chest wall tenderness has little value – everyone has a tender chest wall if you press hard enough!
Clinical assessment should not just focus upon ACS, but should aim positively to identify other causes. Pulmonary embolism is diagnostically challenging. Suspicion should be raised by chest pain that is clearly pleuritic in nature, haemoptysis, associated breathlessness, features of deep vein thrombosis or risk factors for venous thromboembolism (immobilization, malignancy, recent trauma or surgery, pregnancy, intravenous drug abuse or previous thromboembolism). Clinical examination may reveal tachycardia, tachypnoea or features of deep vein thrombosis (see Chapter 5.5). Aortic dissection is characterized by severe pain radiating to the back with associated diaphoresis. Neurological symptoms or signs, sometimes transient, are common. Clinical examination may reveal discrepancy between blood pressure in right and left arms (see Chapter 5.10).
Clinical assessment of chest pain should always include examination of the abdomen to identify tenderness, guarding, rebound tenderness or a positive Murphy’s sign.
Unnecessary investigation can be avoided if non-life-threatening pathology can be confidently diagnosed by clinical assessment. Pain that is reproduced by chest wall palpation in a patient at low risk of coronary heart disease and with no significant risk factors for pulmonary embolus can be confidently diagnosed as musculoskeletal. A positive diagnosis is particularly valuable for the patient who is primarily suffering from anxiety-related symptoms. In this case, pain is typically described as tightness around the chest and associated with a feeling of restricted breathing. Other features include palpitations (particularly awareness of the heartbeat), sweating, breathlessness, light-headedness, feelings of panic, or paraesthesia of the lips or fingertips.
Clinical investigations
The ECG is the most useful clinical investigation and should be performed on all patients presenting with acute non-traumatic chest pain. Table 5.1.3 shows the value of ECG features for diagnosing myocardial infarction. It is important to recognize that a normal ECG does not rule out myocardial infarction. ST-segment elevation or depression, new Q waves and new conduction defects are specific for acute myocardial infarction and predict adverse outcome. Patients with these features should be managed on a coronary care unit. Other changes associated with myocardial infarction are less helpful. T-wave changes are often non-specific and may be positional or due to numerous other causes. ECG changes in pulmonary embolism are also non-specific.
Table 5.1.3
Likelihood ratios of ECG features useful for diagnosing acute myocardial infarction
| New ST elevation>1 mm | 5.7–53.9 |
| New Q wave | 5.3–24.8 |
| Any ST-segment elevation | 11.2 |
| New conduction defect | 6.3 |
| New ST-segment depression | 3.0–5.2 |
| Any Q wave | 3.9 |
| Any ST-segment depression | 3.2 |
| T-wave peaking and/or inversion>1 mm | 3.1 |
| New T-wave inversion | 2.4–2.8 |
| Any conduction defect | 2.7 |
A standard 12-lead ECG may be augmented by serial ECG recording or continuous ST-segment monitoring. These may detect evolving ECG changes or dynamic ST-segment changes. However, these techniques may also identify non-specific false-positive changes, such as minor T-wave inversions, especially if they are used inappropriately in patients with a low risk of coronary heart disease. ST-segment monitoring was developed for the high-risk coronary care population; in low risk emergency department patients with chest pain, it has a very low yield of significant findings.
Like clinical examination, the chest radiograph is mainly intended to identify non-cardiac causes for chest pain, such as a pneumothorax or fractured rib and complications of myocardial infarction, such as left ventricular failure. Although it is often routinely ordered, it is not usually helpful.
Cardiac biomarkers are key investigations in acute chest pain and are a source of much heated debate. They are also a progressively developing technology, so this chapter will focus upon the principles that should guide their use.
Three key features determine the clinical value of a cardiac biomarker. Sensitivity tells us how good the marker is at identifying patients with disease, and thus how useful it is for ruling out myocardial ischaemia. Specificity tells us how good the marker is at identifying patients without disease, and thus how useful it is for ruling in myocardial ischaemia (i.e. a specific test that is positive suggests that the patient is very likely to have ischaemia). The prognostic value (often expressed as a relative risk) tells us how good the marker is at predicting future adverse events, such as death, myocardial infarction or life-threatening arrhythmia.
Intuitively, clinicians tend to be most concerned about sensitivity. If a marker lacks sensitivity then it may miss cases of myocardial infarction leading to potentially catastrophic discharge home without appropriate treatment. However, sensitivity and specificity are often related and may be influenced by the threshold of the marker used to determine a positive test. The lower the threshold used for a positive test, the higher the sensitivity and the lower the specificity. Many evaluations of new markers deliberately optimize sensitivity by selecting a low threshold and sacrificing specificity. This may be an acceptable trade-off in a high-risk population, but emergency department patients with no past history of coronary heart disease and a non-diagnostic ECG typically have a low prevalence of myocardial infarction (<10%). In these circumstances, a test with low specificity will generate many false-positive results requiring hospital admission and investigation, as well as unnecessary anxiety for the patient.
Furthermore, it should be remembered that even a random process can be made to appear sensitive by setting a low diagnostic threshold so that most of the results generated are deemed positive. For example, rolling a pair of dice to diagnose myocardial infarction will have 97% sensitivity if all results, except double six, are deemed to be positive. This is, of course, an extreme example, but biomarkers with diagnostic value that is little better than rolling dice have been promoted by studies that report high sensitivity while hiding away poor specificity. Sensitivity should always be reported with a corresponding specificity.
The prognostic value of a marker is arguably even more useful than its diagnostic parameters, particularly if the marker can predict high-risk patients who will benefit from treatment. If a prognostically useful marker is positive, then we know the patient has the potential to benefit from intervention; if it is negative then we know that, even if further investigation is required to identify the exact cause of their chest pain, they are unlikely to benefit from hospital admission and treatment.
Prognostic considerations explain changes in the definition of myocardial infarction. The original World Health Organisation (WHO) definition of myocardial infarction was based upon creatine kinase, a cardiac marker with limited sensitivity and specificity and only weak evidence of an association with adverse prognosis. Subsequent definitions of myocardial infarction, from the American Heart Association and European Society of Cardiology, used troponin as the biomarker based on research showing elevated troponin levels were associated with increased risk of adverse outcome. Furthermore, research has shown that informing clinicians that their patient has an elevated troponin level can alter patient management and reduce the risk of major adverse cardiac events over the following year. This is important evidence that measuring troponin provides patient benefit by reducing adverse outcomes.
Creatine kinase is released by damaged myocardium, but is also released by muscles and the liver and is measurable in the blood in the absence of pathology. Its MB isoenzyme (CK-MB) is more cardiac-specific but shares the same problems. Substantial myocardial damage is required to produce an elevated CK-MB, but CK-MB may also be elevated in the absence of myocardial injury. Its role in diagnosis is generally limited to situations where troponin testing is unavailable or likely to be unreliable.
There are two troponin assays, troponin I and troponin T, with little to choose between them in terms of diagnostic or prognostic performance. The diagnosis of myocardial infarction is based upon elevation of the troponin level above the 99th percentile upper reference limit of a normal reference population. This value will vary between assays. The precision of the assay is described by the coefficient of variation (CV) at the 99th percentile. There have been several generations of troponin assays with newer assays having higher sensitivity and better precision. High-sensitivity troponin assays have optimal precision (≤10% CV) at the 99th percentile and can detect myocardial infarction earlier after symptom onset than other assays. However, this appears to be at the cost of an increased number of low troponin rises that are of unclear diagnostic and prognostic significance.
Troponin elevation is not specific for myocardial infarction and low levels measured using a high sensitivity assay may not indicate significant pathology. Even substantial troponin elevations may not be due to ACS. Troponin can be elevated in pulmonary embolus, sepsis, renal failure, congestive cardiac failure and a number of other illnesses.
The use of troponin has tended to be limited by its lack of early sensitivity. It has been estimated that standard troponin assays take up to 12 hours after symptom onset to achieve optimal sensitivity, so if it is used too early after symptom onset it may produce a false-negative result. This has led to the widespread practice of delaying troponin measurement to at least 12 hours after symptom onset to achieve optimal sensitivity. This practice is problematic because most patients present a few hours after symptom onset, so enforcing a 12-hour delay will typically require hospital admission or use of observation facilities. If there is limited availability of such facilities, clinicians may feel under pressure to discharge the patient without any testing. Thus, a strategy intended to increase patient safety may paradoxically put patients at risk when applied to the real world. Newer assays allow a 6-hour testing strategy and recently developed high-sensitivity assays appear to have good early sensitivity and may be able to rule out myocardial infarction within a few hours of presentation to hospital. However, as mentioned above, this may be at the cost of limited specificity resulting in the generation of troponin elevations that have little diagnostic or prognostic significance. Thus, the choice of testing strategy involves a trade-off between sensitivity and specificity. It is important to know the test used in your institution and its performance characteristics in order to decide on an appropriate serial testing strategy.
Awareness of the limited early sensitivity of standard troponin assays has led to research into alternative biomarkers that can detect myocardial infarction in the initial hours after symptom onset. These biomarkers often have poor specificity so any improvement in sensitivity achieved by combining troponin with another biomarker involves a trade-off in terms of reduced specificity. The development of high-sensitivity troponin assays is likely to undermine the need to develop and use alternative early biomarkers. Future evaluations of alternative early biomarkers will need to include comparison with high-sensitivity troponin.
The most extensively evaluated early biomarkers to date are myoglobin, heart-type fatty acid binding protein (HFABP) and ischaemia modified albumen (IMA). Table 5.1.4 shows the sensitivity and specificity of these biomarkers at presentation to hospital, compared to troponin. None of the alternative biomarkers have sufficient sensitivity to rule out myocardial infarction at presentation. Adding HFABP or myoglobin to a contemporary troponin assay at presentation increases sensitivity at the expense of specificity, but it is not clear whether either biomarker can improve the early sensitivity of a high-sensitivity troponin assay. IMA appears to have insufficient specificity to play any useful role in diagnosis.
Table 5.1.4
Sensitivity and specificity of biomarkers for myocardial infarction at presentation to hospital
| Biomarker | Sensitivity (%) | Specificity (%) |
| Troponin I* | 77 | 93 |
| Troponin T* | 80 | 91 |
| Roche high sensitivity troponin T | 96 | 72 |
| ADVIA Centaur Ultra high sensitivity troponin I | 86 | 89 |
| Abbot Architect high sensitivity troponin I | 83 | 95 |
| Quantitative heart-type fatty acid binding protein | 81 | 80 |
| Qualitative heart-type fatty acid binding protein | 68 | 92 |
| Myoglobin | 62 | 83 |
| Ischaemia modified albumen | 77 | 39 |
The estimates are based on meta-analysis of data from heterogeneous studies and are therefore subject to substantial uncertainty. Goodacre S, Thokala P, Carroll C, Stevens J, Leaviss J, et al. Systematic review, meta-analysis and economic modelling of diagnostic strategies for suspected acute coronary syndrome. Health Technol Assess 2013;17(1) http://www.journalslibrary.nihr.ac.uk/hta/volume-17/issue-1 with permission.
*All assays combined, using 99th percentile as a diagnostic threshold.
Biomarkers are continually being developed and emergency physicians can expect to see headline-grabbing publications extolling their virtues. However, they should be wary before indiscriminately using new markers in their patients with chest pain. As described earlier, the emergency department population with chest pain are a heterogeneous population with a relatively low prevalence of ACS compared to the high-risk patients that usually comprise research study populations. Indiscriminate use of markers with limited specificity will lead to many false-positive results and consequent patient anxiety, unnecessary investigation and waste of resources.
Provocative cardiac testing, usually using an exercise treadmill, has been used in a number of emergency departments. Patients typically undergo a short period of observation and cardiac marker testing to rule out myocardial infarction before receiving an exercise treadmill test. Concerns about the safety of this procedure have been addressed by data from a number of centres, but it should be recognized that selection of low-risk patients plays a key role in ensuring safety.
Exercise treadmill testing has relatively poor sensitivity and specificity and cannot reliably rule in or rule out coronary artery disease. However, it is prognostically useful and predicts risk of adverse events over the months following attendance. It is therefore used to risk-stratify more than to diagnose. A patient with a negative treadmill test may have coronary artery disease but can be reassured that they are at low risk of adverse outcome. It is not clear whether the additional prognostic information provided by exercise treadmill testing justifies widespread use.
CT coronary angiography is developing an increasing role in the diagnostic evaluation of acute chest pain. Unlike exercise treadmill testing, CT coronary angiography detects coronary obstruction with a reasonable degree of accuracy. Sensitivity and specificity are both around 90% against a reference standard of invasive coronary angiography. However, coronary atheroma is a common finding and detecting atheroma, or even coronary obstruction, does not necessarily mean that this was the cause of the patient’s chest pain. Studies of the prognostic value of CT coronary angiography in patients with suspected ACS have been limited by selection of patients with low rates of adverse outcome and have, as yet, failed to show worthwhile prognostic value. This means that, although CT coronary angiography is a promising diagnostic test for acute chest pain, evidence is lacking that its use improves patient outcomes and its role in routine assessment remains unclear.
The combination of observation and cardiac marker testing to rule out myocardial infarction, followed by provocative cardiac testing to risk-stratify or CT coronary angiography to diagnose coronary artery obstruction, has been adopted in many hospitals in the form of a chest pain unit or pathway. These have a number of potential benefits for patients and health services and some evidence to suggest that they reduce the probability of admission, reduce the risk of discharge with ACS, improve patient satisfaction and quality of life and reduce health service costs. However, as an organizational intervention, the effect of the chest pain unit will depend upon local circumstances and may be influenced by staff attitudes, professional roles and local leadership. Furthermore, the presence of a chest pain unit may attract additional attendances with chest pain. Whether this represents identification of unmet demand or unnecessary work is a matter of opinion.
A number of clinical risk scores have been developed to risk-stratify patients with suspected ACS. The Goldman algorithm and acute cardiac ischaemia time insensitive predictive instrument (ACI-TIPI) were developed and validated on large cohorts of patients with chest pain in the 1980s and 1990s. The Goldman algorithm uses a series of questions about the patient’s age, clinical history and ECG findings to categorize patients into a low (<7%) or high (>7%) risk of myocardial infarction, based on the WHO definition used at the time. ACI-TIPI can be incorporated into a computerized ECG. The user enters the patient’s age, sex and whether chest or left arm pain is the primary symptom. The computer then uses these data and analysis of the ECG to generate a probability of acute cardiac ischaemia.
The thrombolysis in myocardial infarction (TIMI) score has been developed and validated as a predictor of adverse outcome in patients with diagnosed ACS (see Chapter 5.2). Studies have evaluated the TIMI score in emergency department patients with suspected ACS and shown that higher scores are associated with a higher risk of adverse outcome. This has led to the TIMI score being used to risk stratify patients with chest pain before a diagnosis of ACS has been confirmed. The ECG and cardiac biomarker components are the most powerful predictors among the elements of the TIMI score and a modified version of the TIMI score, with extra weighting for these elements, has been proposed for patients with acute chest pain. It is not clear whether either the original or modified TIMI score provides better prognostic information than that provided by ECG and biomarkers alone.
The Global Registry of Acute Coronary Events (GRACE) freedom-from-event score has recently been developed to predict adverse events in people with non-ST-elevation acute coronary syndrome and can identify up to 30% of the admitted population who are at low risk of death or any adverse in-hospital event. It is more complex to calculate than the TIMI score but bedside calculation can be easily facilitated by mobile technologies. The GRACE freedom-from-event score offers an alternative to the TIMI score for predicting adverse outcome but further validation in the wider chest pain population is required.
Treatment
Treatment of acute chest pain is directed at the specific cause. The treatment of ACS is outlined in Chapter 5.2, pulmonary embolus in Chapter 5.5 and aortic dissection in Chapter 5.10.
Musculoskeletal chest pain, whether due to muscular strain, chest wall injury, Tietze’s syndrome or epidemic myalgia, should be treated with simple analgesia and the patient advised to see their general practitioner if the pain persists beyond a few weeks. It is also worth considering whether anxiety may be exacerbating the patient’s symptoms.
Gastro-oesophageal pain can be treated acutely by antacids, although the diagnostic value of observing relief of pain with antacids (the so-called ‘GI cocktail’) is debatable. ACS can present as burning or indigestion-type pain and, pain being typically fluctuant, may ease coincidentally with administration of an antacid. Gastro-oesophageal pain should be diagnosed with caution and ideally only after ACS has been investigated and ruled out. In these circumstances, a course of treatment with a proton pump inhibitor is appropriate. Follow up will depend upon local practice along with the duration and severity of symptoms.
Anxiety-related symptoms range from simple chest wall muscular tension to panic attacks, hyperventilation syndrome and cardiac neurosis. Treatment should therefore be tailored to the patient’s individual needs. In many cases, anxiety will be an understandable reaction to concerns about heart disease or other serious pathology. The first step is therefore to provide clear and unequivocal reassurance. If diagnostic uncertainty makes this impossible then it may still be possible to provide reassurance by highlighting the excellent prognosis of patients with chest pain whose tests are negative. Information leaflets have been shown to reduce anxiety after emergency department attendance with chest pain. Patients with more severe symptoms may benefit from relaxation techniques, cognitive behavioural therapy or treatment with an antidepressant. These are best arranged through the patient’s general practitioner.
Managing anxiety in the emergency department patient is often complicated by difficulties in satisfactorily ruling out serious physical illness. A diagnosis of anxiety may be considered likely, but until cardiac testing is complete (perhaps even involving coronary angiography), the treating physician may be reluctant to discuss treatment of anxiety with the patient. This is inappropriate. If the patient has significant anxiety-related symptoms then this will adversely affect their quality of life and should be addressed regardless of whether they ultimately also need treatment for cardiac disease.
Non-specific chest pain obviously presents a challenge. With no clear diagnosis it is difficult to advise an appropriate treatment. However, patients can be advised that, although no clear diagnosis can be made, about half such patients presenting to the emergency department have no further episodes of pain over the following month. Those who do suffer further episodes of pain are unlikely to be troubled. Treatment is therefore unlikely to be required.
Finally, an acute episode of chest pain provides an opportunity to identify and manage cardiac risk factors at a time when the patient is likely to be most receptive to lifestyle advice. Smokers should be advised to use the episode as a stimulus to stop smoking and referral to a smoking cessation service arranged. General dietary and exercise advice may also be helpful. Blood pressure, blood glucose and lipid profile may be requested as part of clinical assessment, although any abnormalities identified should preferably be referred to the patient’s general practitioner, who will be best placed to provide overall cardiovascular risk assessment, intervention and long-term follow up.
Prognosis
Prognosis will also depend upon the underlying pathology and the prognoses of various causes of chest pain are discussed in the relevant chapters of this book. Patients with no obvious diagnosis after clinical assessment, ECG and troponin testing have an excellent prognosis. There is some evidence that patients who attend the emergency department with chest pain have a higher risk of adverse cardiac events than the general population, even if cardiac disease is ‘ruled out’ at initial presentation, but this risk is not high enough to warrant active intervention beyond ensuring that any cardiac risk factors identified have been addressed.
Likely developments over the next 5–10 years
Chest pain is responsible for a substantial and growing number of emergency medical admissions in many countries. This is placing a major burden upon healthcare systems. The value of hospital admission for low risk ACS and pulmonary embolus is being questioned and it is likely that there will be increasing efforts to manage acute chest pain without admission to hospital. These efforts may be successful for younger patients with no co-morbidities and a single potentially serious cause for their chest pain, but may be difficult to implement among the growing population of older patients with co-morbidities or multiple potentially serious causes for their pain.
High-sensitivity troponin assays are increasingly replacing other contemporary assays. This will lead to more positive results and concerns that some of these may represent false-positive results with little prognostic value. Emergency physicians may therefore need to be more circumspect in their use of troponin and avoid measuring troponin in patients with a very low risk of ACS in whom an elevated high-sensitivity troponin measurement just above the diagnostic threshold is likely to cause confusion rather than provide a clear diagnosis.
Point-of-care troponin testing has been available for some time but widespread adoption has been limited by a number of factors, including lack of usability and difficulties converting reduced turnaround times into reduced length of hospital stay. The development of hand-held point-of-care machines using finger-stick capillary blood samples could improve the usability of point-of-care testing and extend use to the pre-hospital setting.
CT coronary angiography is likely to be more widely used in the assessment of acute chest pain. In the absence of randomized trial evidence of effectiveness, it will not be clear whether patients are benefitting from increased use. Both high sensitivity troponin assays and CT coronary angiography have the potential to increase the diagnostic yield of positive tests in the investigation of acute chest pain leading to over-burdening of cardiology services.
Chest pain management is likely to be influenced by changes in health service policy, which are, in turn, likely to depend upon local social, political and economic factors. These changes will be variable and may be unpredictable. On one hand, public awareness of the medical significance of chest pain and policies aimed at increasing rapid access to care may lead to increased numbers of patients presenting with chest pain. On the other hand, reorganization of services and attempts to control costs may result in an opposite effect. Specifically, development of primary angioplasty services may lead to centralization of chest pain services and patients bypassing facilities that do not provide primary angioplasty.
Controversies
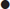
5.2 Acute coronary syndromes
Steve Goodacre and Anne-Maree Kelly
Introduction
Acute coronary syndrome (ACS) is the most common life-threatening condition in emergency medicine. Failure to identify and treat it promptly risks avoidable morbidity and mortality.
Aetiology, pathogenesis and pathology
ACS nearly always occurs as a consequence of atheroma in the coronary arteries, commonly known as coronary heart disease (CHD). Many people have coronary atheroma but are asymptomatic because it is not extensive enough to occlude coronary blood flow. Others have a degree of coronary occlusion that does not cause symptoms unless they exert themselves or if myocardial oxygen demand is increased by some other mechanism, such as anaemia. Cardiac chest pain that only occurs on exertion and is rapidly relieved by rest is known as stable angina and is not classified as an ACS.
ACS usually occurs when an atheromatous plaque ruptures or fissures. Haemorrhage may occur into the plaque or thrombus may accumulate over the fissure. The type of ACS that results depends on the extent of the rupture and degree of haemorrhage or thrombus formation. A gradually progressive occlusion will usually produce symptoms of unstable angina: progressive symptoms of myocardial ischaemia occurring on less exertion or at rest. A rapidly progressive occlusion may lead to myocardial infarction (MI), with severe pain at rest and the potential for serious complications, such as arrhythmia, heart failure, cardiogenic shock or sudden cardiac death.
If coronary occlusion is minor or transient, the consequent myocardial ischaemia will not lead to myocardial damage. If coronary occlusion is severe or prolonged then myocardial necrosis will occur.
Not all coronary artery occlusion is due to coronary atheroma. Prinzmetal angina describes a syndrome in which myocardial ischaemia is associated with coronary artery spasm and is characterized by transient ST-segment elevation on the electrocardiograph (ECG). Coronary angiography may show minor atheroma or normal coronary arteries. Uncommonly, coronary artery spasm may be severe enough to cause myocardial necrosis and an associated biomarker elevation.
Other rare causes of coronary artery occlusion include Kawasaki’s disease, in which occlusion is due to inflammation in the coronary artery and aortic dissection that involves the coronary arteries.
ACS may involve occlusion of one or more of the coronary arteries and the location of occlusion may determine the clinical presentation, ECG findings and likelihood of complications. The most common site for MI is the anterior or anteroseptal region. It usually results from occlusion of the left anterior descending artery. It has a worse prognosis than other types of MI and complications are more common. Sudden cardiac death may result from total occlusion of the left anterior descending artery, giving a lesion in this location the grim sobriquet of ‘widow-maker’. Lateral infarction is usually caused by occlusion of the circumflex artery or the diagonal branch of the left anterior descending artery. Inferior MI is usually caused by occlusion of the right coronary artery or the circumflex artery. It has a better prognosis than anterior infarction and ventricular dysfunction is less likely, although heart block due to involvement of the atrioventricular node is more common. Posterior infarction is usually due to occlusion of the right coronary artery or, less commonly, the circumflex artery in patients with dominance of the left coronary circulation. Posterior or inferior MI may result in right ventricular infarction leading to right ventricular failure.
ACS may be associated with a range of life-threatening complications, including arrhythmia, such as atrial fibrillation, ventricular tachycardia and ventricular fibrillation. Supraventricular tachycardias are not usually associated with ACS. Heart block may occur with infarcts affecting the nodal branch of the right coronary artery or septal infarcts. Infarction may lead to myocardial dysfunction, resulting in heart failure or cardiogenic shock. Uncommon complications include papillary muscle dysfunction and mitral regurgitation, ventricular septal defect or cardiac rupture. The probability of any of these complications increases with the severity of myocardial damage incurred.
Epidemiology
Coronary heart disease is the leading cause of death in the world. It is responsible for 6.8% of disability-adjusted life years (DALYs) lost through disease by men and 5.3% of DALYs lost by women. The global burden of CHD is expected to rise from 47 million DALYs in 1990 to 82 million in 2020. Most of this increased burden will be in developing countries. However, CHD mortality rates have dramatically decreased in many developed countries since the 1980s. Studies suggest that 50–75% of the fall in cardiac deaths can be attributed to population interventions, particularly those relating to smoking, hypertension and high cholesterol. The remaining 25–50% is due to treatments for patients with CHD, such as acute reperfusion therapies (including thrombolysis and percutaneous coronary intervention (PCI)), aspirin, angiotensin-converting enzyme inhibitors, statins and coronary artery bypass surgery.
The main risk factors for CHD are well established and include smoking, diabetes, hypertension, hyperlipidaemia and a family history of CHD at a young age, while obesity and lack of exercise may play a contributory role. Age and sex are also important. CHD prevalence increases with age and increases at an earlier age (40–50 years) in men than in women (over 60 years). Everyone over the age of 60 is effectively at risk of CHD. Conversely, a history of CHD presenting in a relative when they were aged over 60 should not be considered a significant risk factor.
Patients presenting to the emergency department (ED) with chest pain in general, and ACS specifically, show a diurnal variation with a peak of attendances during the morning, although many of these attendances relate to symptoms occurring overnight. Presentation is more common on a Monday, when cardiovascular mortality appears to be higher. Cardiovascular mortality also increases during the winter months, particularly in colder climates.
Prevention
Prevention of ACS is achieved principally by preventing underlying CHD, although secondary prevention of ACS in patients with established CHD can be attempted by ensuring appropriate treatment with daily low-dose aspirin, angiotensin-converting enzyme (ACE) inhibitor, β-blockers and lipid-lowering therapy.
Primary CHD prevention can take place by addressing the important coronary artery disease risk factors that are amenable to intervention. The most important modifiable risk factors at a population level are smoking, obesity and lack of exercise. These may be tackled by legislation and education and by economic and social policy. Diabetes, hypertension and hyperlipidaemia can be addressed at an individual level. It is increasingly recognized that the importance of any one risk factor depends on the presence of other risk factors, so cardiovascular risk is most appropriately assessed by a comprehensive assessment involving all risk factors, along with age and sex. Screening programmes should be based on overall cardiovascular risk assessment, rather than individual risk factors. Similarly, the decision to prescribe treatments for risk factors, particularly lipid-lowering therapy, should be based on overall cardiovascular risk.
This has implications for emergency medicine. It may be tempting to use the patient’s attendance at the ED to undertake opportunistic screening by, for example, measuring blood pressure, blood sugar or lipids, even though they will not influence management of the presenting complaint. This approach is inappropriate because it does not involve overall cardiovascular risk assessment. Furthermore, it may be considered unethical because the patient is effectively being screened (with potential implications for health insurance) without the opportunity to make an informed choice about whether they wish to receive screening. For these reasons, coronary risk assessment for primary prevention is best left to primary care physicians.
Although opportunistic screening in the ED is best avoided, opportunistic patient education about risk factors may be very salient, particularly if the patient has presented with symptoms that could be related to CHD. For example, an episode of chest pain, even if ultimately diagnosed as non-cardiac, may offer an ideal opportunity to promote smoking cessation.
Clinical features
Clinical assessment of suspected ACS is described in detail in Chapter 5.1. Chest pain is suggestive of MI if it radiates to either arm, both arms or shoulders; is described as burning, like indigestion, heavy, pressing or band-like; occurs on exertion; is associated with diaphoresis, nausea or vomiting; or is worse than previous angina or similar to previous MI. Chest pain is less likely to be MI if it is sharp, pleuritic, positional, reproduced by palpation, inframammary in location or not associated with exertion.
Clinical assessment of cardiac pain is required to determine whether it is due to stable angina or ACS. Stable angina is characterized by pain that is predictable, precipitated by exertion, relieved promptly by rest or glyceryl trinitrate (GTN) and is not becoming more frequent or severe. Unstable angina is caused by a dynamic narrowing of the coronary artery and is characterized by pain that may be unpredictable, may occur at rest or minimal exertion, may not be immediately relieved by rest or GTN or may be increasing in frequency or severity.
Patients with stable angina do not typically present to the ED. They are often used to their symptoms and will not seek medical help unless something unexpected happens. If a patient presents with apparently stable angina, the diagnosis should be considered carefully. It should be remembered that pain precipitated by exertion is known to be predictive of MI in ED patients.
Clinical examination is generally unhelpful in making the diagnosis of ACS, which should be based on clinical history and investigations. However, clinical examination is essential to identify complications of ACS and to rule out differential diagnoses. Heart failure may be identified by poor peripheral circulation, tachycardia, pulmonary crepitations, elevated jugular venous pressure and a third heart sound on cardiac auscultation. The additional finding of hypotension may suggest cardiogenic shock. A systolic murmur raises the possibility of papillary muscle rupture or ventricular septal defect secondary to MI, although pre-existing aortic or mitral valve disease are much more common.
Differential diagnosis
Alternative diagnoses and their differentiation from ACS are described in Chapter 5.1. The most potentially serious alternative diagnoses are pulmonary embolus and aortic dissection. These should be considered in any patient with suspected ACS who is diaphoretic, tachycardic, tachypnoeic, hypotensive or reports associated neurological symptoms (transient or persistent) but does not have definite ECG features of ACS.
Clinical investigations
The 12-lead ECG is the essential investigation to identify ACS requiring emergency reperfusion. It should be performed as soon as possible after arrival in any patient with any suspicion of ACS. Pre-hospital ECGs can be obtained by some emergency medical services and in some settings is used to prioritize patients and guide triage to high-dependency areas/cardiac catheter laboratories.
The critical decision is to determine whether there is evidence of ST-elevation MI (STEMI) or MI with new bundle branch block. If there is any doubt, senior or specialist advice should be sought immediately. Repeat ECG recording may be helpful if a senior clinician feels there is insufficient certainty to allow for an immediate decision but a high suspicion of evolving MI persists.
Identifying new bundle branch block presents a challenge, especially if previous medical records are not immediately available. In the past, the Sgarbossa criteria have been suggested as indicative of increased likelihood of MI. These criteria are ST elevation of 1 mm or more that is concordant with (in the same direction as) the QRS complex; ST depression of 1 mm or more in leads V1, V2 or V3 and ST-segment elevation of 5 mm or more that is discordant with (in the opposite direction to) the QRS complex. Recent research has questioned their utility, reporting low sensitivity for diagnosing MI. Other research has suggested that the presence of concordant ST changes is closely correlated with acute coronary occlusion but that the discordant criteria and left bundle branch block (LBBB) without Sgarbossa criteria are not. These findings have yet to be validated. In the absence of clear decision-making criteria, decisions regarding acute reperfusion will depend on specialist clinical judgement.
Other ECG changes may be useful in diagnosing AMI and are described in Chapter 5.1 and Table 5.1.3. Q waves typically follow ST elevation but may appear as early as 4 hours after symptom onset. Their presence does not therefore preclude early reperfusion. Tall, upright T waves (‘hyperacute’ T waves) may be present in the very early stages of infarction. Deep (>3 mm) inverted T waves suggest a subendocardial MI which would be confirmed by biomarker elevation. Similarly, patients with significant (>1 mm) ST depression have an increased risk of adverse outcome and are likely to have a troponin rise. Unfortunately, despite an association with increased risk of adverse outcome, neither ST depression nor deep T-wave inversion is associated with benefit from acute reperfusion therapy.
Other T-wave changes, such as small inversions (<3 mm), flat T waves and biphasic T waves, are common and non-specific. They may suggest ACS, but may also occur in patients with hypertension, patients who are hyperventilating and in the normal population.
In addition to changes directly suggesting ACS, the ECG should be inspected for any concurrent pathology or evidence of complications. Cardiac rate and rhythm and P-wave presence and morphology should be evaluated for evidence of arrhythmia or heart block. Tall R waves or S waves suggest ventricular strain or hypertrophy that may contribute to or be a consequence of ACS.
A subtle sign that can indicate ischaemia or ventricular dysfunction is poor anterior R-wave progression. Normally, R waves progressively increase in size across leads V1 to V4. Small R waves across these leads suggest pathology.
Repeated 12-lead ECG recording or continuous ST-segment monitoring can help to identify transient or dynamic ECG changes. The development of significant (>1 mm) ST deviation provides clear evidence of ischaemia, identifies high-risk patients and may facilitate rapid identification of patients requiring reperfusion. T-wave changes, by contrast, are non-specific and often arise as a result of hyperventilation or changes in patient position during monitoring. The incidence of significant ST changes decreases and the incidence of false-positive T-wave changes increases in patients with a lower likelihood of significant ACS. Therefore, repeated ECG recording and ST-segment monitoring should be reserved for high-risk patients.
A normal or non-diagnostic ECG does not rule out ACS or necessarily stratify the patient to a very low-risk group. In fact, the majority of patients admitted with ACS do not have diagnostic ECG changes. Serial ECG recordings and ST-segment monitoring do not substantially increase the negative predictive value of the ECG or provide very useful prognostic data. Negative ECG recording therefore has limited value.
Biochemical markers are discussed in detail in Chapter 5.1. Their role is to identify patients with probable ACS and to rule out ACS if negative. However, it should be remembered that a negative cardiac marker, even if highly sensitive and performed at an optimal time after the worst symptoms, does not rule out CHD. Patients with negative markers require risk stratification and further cardiac testing if CHD is considered a likely diagnosis, although further cardiac testing does not necessarily have to be undertaken at the initial hospital attendance.
Biochemical markers (particularly troponin) have a valuable prognostic role. Any patient with an elevated troponin is at increased risk of adverse outcome and has the potential to benefit from hospital admission, although the prognostic importance of small elevations of a high sensitivity assay is uncertain. If ACS is the likely cause of a troponin elevation, then the patient should be admitted under the care of a cardiologist. As a general rule, the higher the troponin level the greater the risk of adverse outcome. Patients with minor troponin elevations may be managed conservatively and possibly without ECG monitoring. A recent paper has reported the rate of significant ventricular arrhythmia in patients with troponin rise without ischaemic ECG changes as 0% (95% CI 0–2.3%). Those with substantial troponin elevations should be managed in a coronary care unit and considered for early percutaneous coronary intervention, even if they have no significant ECG changes.
Criteria for diagnosis
The term ACS covers a spectrum of disorders, including unstable angina, non-ST-elevation MI (NSTEMI) and STEMI. The diagnostic definition of MI has been a matter of intense debate in recent years and a consensus has gradually emerged. In contrast, the challenge of defining a diagnosis of ACS per se has been largely overlooked.
The original World Health Organization (WHO) diagnosis of MI is outlined in Table 5.2.1. It required an elevation of creatine kinase to more than twice the upper limit of the normal range. With the development of troponins, it became apparent that this definition failed to include a substantial number of patients with prognostically significant myocardial damage, as evidenced by a troponin rise. Therefore, the American Heart Association and European Society of Cardiology (AHA/ESC) developed a new definition of MI, outlined in Table 5.2.2, which required a rise in serum troponin above the 99th percentile of the values for a reference control group.
Table 5.2.1
WHO criteria for definite acute MI (1970)
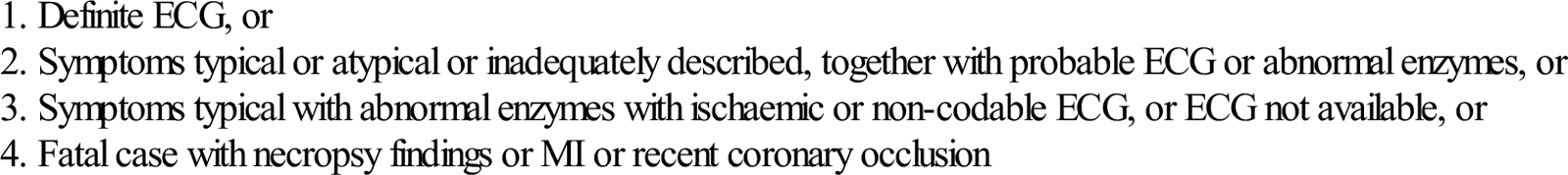
Table 5.2.2
The AHA/ESC criteria for MI (2012)
| Typical rise and fall of biochemical markers of myocardial necrosis with at least one of the following: |

The AHA/ESC definition has been widely adopted, despite a number of concerns and criticisms. Patients with ACS who fulfil this definition have a higher risk of adverse outcome than those who do not. However, patients with MI according to the AHA/ESC criteria alone have a lower risk of adverse outcome than those who fulfil both the AHA/ESC and WHO criteria. This has led to problems in maintaining consistent care over time and some experts have suggested identifying a threshold level for troponin (e.g. troponin T>1 ng/mL) above which clinically important MI should be diagnosed. This controversy is unlikely to be resolved in the near future, particularly as newer and more sensitive biochemical markers are developed. However, the most important issue to recognize is that any detectable troponin is associated with a potentially increased risk of adverse outcome and, the higher the troponin level, the higher that risk.
MI can be usefully defined as STEMI or NSTEMI on the basis of the ECG. If there is evidence of significant ST elevation on any ECG (>2 mm in two consecutive chest leads, or>1 mm in two consecutive limb leads) then the patient has STEMI. These patients are likely to benefit from early reperfusion therapy. Patients without these changes but with evidence of myonecrosis based on cardiac markers are defined as having NSTEMI and do not benefit from reperfusion with thrombolytics, although PCI may be beneficial. NSTEMI and ACS without criteria for MI may be categorized together as non-ST-elevation ACS.
AHA/ECS have introduced a classification system for MI based on underlying pathophyisological mechanisms. Of most relevance to emergency medicine practice are type 1 and type 2 MI. Type 1 MI is defined as spontaneous MI related to atherosclerotic plaque rupture, ulceration, fissuring, erosion or dissection with resulting intraluminal thrombus in one or more of the coronary arteries leading to decreased myocardial blood flow or distal platelet emboli with ensuing myocyte necrosis. Type 2 MI is defined as instances of myocardial injury with necrosis where a condition other than coronary artery disease (CAD) contributes to an imbalance between myocardial oxygen supply and/or demand, e.g. coronary endothelial dysfunction, coronary artery spasm, coronary embolism, tachy-/brady-arrhythmias, anaemia, respiratory failure, hypotension and hypertension with or without left ventricular hypertrophy (LVH).
Treatment
Some treatments are indicated for all ACS, whereas others have specific application to STEMI, NSTEMI and other ACS.
Treatments for all ACS
Analgesia
Glyceryl trinitrate (GTN) and intravenous (IV) morphine are the analgesic agents of choice. Sublingual GTN may be appropriate if pain is mild to moderate, but severe pain usually requires titrated IV morphine. Doses of up to 20 mg, in small increments, are sometimes required. If IV morphine fails to control pain and the clinical condition is suitable, IV GTN by infusion at a rate titrated to effect (20–200 μg/min) is indicated. If this is insufficient to control pain and the patient is tachycardic, control of rate with small increments of IV β-blocker may be beneficial. It is important to note that ongoing severe pain, particularly in the absence of ECG changes, should raise concerns about an alternative diagnosis, such as aortic dissection.
Aspirin
Aspirin 300 mg should be administered unless already given (e.g. by emergency services or general practitioner) or contraindicated. The principal contraindication to aspirin is known allergy. A previous history of gastritis or indigestion is not a contraindication to the use of aspirin in ACS.
Oxygen
Recent analyses have raised questions about the role of routine oxygen therapy in the treatment for ACS. There is also a lack of evidence of benefit. Until further research clarifies the risk–benefit of supplemental oxygen therapy, its routine use is not recommended. Oxygen therapy is indicated for patients with hypoxia (oxygen saturation<93%) and those with evidence of shock to correct tissue hypoxia.
STEMI
Reperfusion
Patients with STEMI who present within 12 hours of symptom onset should have a reperfusion strategy implemented emergently. Reperfusion can be obtained by fibrinolytic therapy, PCI, or rarely, with emergency coronary artery bypass grafting. The choice of reperfusion therapy will depend on time from symptom onset, availability of PCI, delay to fibrinolysis, contraindications to fibrinolysis, location and size of the infarct and the presence or absence of cardiogenic shock.
PCI is the best available treatment if provided promptly. It is generally accepted that a delay of 90 minutes between presentation and balloon inflation is the maximum desirable. If this is not possible, fibrinolysis should be used. For patients presenting very early (symptom duration less than 1 hour), fibrinolytic therapy is highly effective, so the maximum tolerable delay to PCI is 1 hour from presentation. For patients aged less than 75 years with cardiogenic shock, PCI markedly improves outcomes.
Fibrinolytic agents include streptokinase and tissue fibrin-specific agents, such as alteplase and tenectaplase. Available evidence suggests that fibrin-specific agents reduce mortality compared to streptokinase, despite an increased risk of intracranial bleeding. Note that streptokinase should not be given to patients who have been previously exposed to it (more than 5 days ago) due to antibody formation. There is also some evidence that it may be less effective in populations with high levels of exposure to streptococcal skin infections, such as Aboriginal and Torres Strait Islander peoples. Contraindications to fibrinolytic therapy are shown in Table 5.2.3. All patients receiving fibrinolyic therapy should be transferred to a PCI-capable centre for further assessment and treatment. In cases of failed fibrinolyisis or if there is evidence of re-occlusion/re-infarction (e.g. recurrent ST elevation), this transfer should be emergent.
Table 5.2.3
Contraindications to fibrinolytic therapy in STEMI (ECS, 2011)
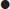

Pre-hospital fibrinolysis should be considered when delay to PCI exceeds 90 minutes and transfer times to a fibrinolysis-capable facility exceed 30 minutes.
The evolution of primary PCI has resulted in some systems identifying STEMI prior to hospital arrival and bypassing the ED, instead taking the patient directly to the catheterization laboratory. Even in these systems, it is important that emergency physicians remain knowledgeable and skilled in the diagnosis and management of STEMI, as a substantial proportion of cases self-present to the ED and pre-hospital diagnosis can sometimes be inaccurate.
P2Y12 receptor inhibitors
Patients undergoing primary PCI for reperfusion for STEMI should receive an antiplatelet agent. The choice of agent will depend on balancing the risk of recurrent ischaemic events and bleeding risk in individual patients. If clopidogrel is used it should be given as a high-dose clopidogrel regimen (600 mg oral bolus and 150 mg daily for 7 days, then 75 mg/day for at least 12 months). Potent oral antiplatelet agents (prasugrel and ticagrelor) are alternatives to clopidogrel for subgroups at high risk of recurrent ischaemic events (e.g. those with diabetes, stent thrombosis, recurrent events on clopidogrel or a high burden of disease on angiography). Prasugrel is contraindicated in patients with prior stroke/transient ischaemic attack. Neither prasugrel or ticagrelor should be used in patients with a previous haemorrhagic stroke or in patients with a moderate-to-severe liver disease. For patients undergoing fibrinolysis, 300 mg clopidogrel is recommended.
Antithrombin therapy
Antithrombin therapy should be used in conjunction with PCI and fibrin-specific fibrinolytic agents. The use of antithrombin therapy with Streptokinase is optional. Low molecular weight heparin (e.g. enoxaparin) or unfractionated heparin can be used. There are data suggesting that enoxaparin may provide benefit over unfractionated heparin and dosing and administration are easier leading to recommendations listing it as the preferred agent. If unfractionated heparin is used it should be administered according to local dosing and monitoring guidelines taking into account whether there is concomitant use of glycoprotein IIb/IIIa inhibitors (GP inhibitors) and whether PCI or fibrinolysis was the reperfusion strategy used.
Bivalirudin (a direct thrombin inhibitor) has shown similar efficacy to the heparins with less bleeding. Its place in the management of ACS is evolving.
Glycoprotein IIb/IIIa inhibitors
The role of GP inhibitors is evolving and data are conflicting and complex. Current recommendations are that the decision to use GP inhibitors is a specialist cardiologist decision and that there is no benefit in initiating it before the catheterization laboratory. An exception is that use before the catheterization laboratory may be considered in high-risk patients undergoing transfer for primary PCI in consultation with the receiving cardiologist. Abciximab is the preferred agent.
Non-STEMI
P2Y12 receptor inhibitors
In addition to aspirin, patients should receive P2Y12 receptor inhibitor agents, e.g. clopidogrel 300 mg loading dose and 75 mg/day. This should be withheld if emergency coronary bypass surgery is planned. There are limited data regarding newer antiplatelet agents (prasugrel and tricagrelor) in non-STEMI. In particular, data regarding prasugrel are limited to patients undergoing PCI. Both these agents have increased bleeding risk compared to clopidogrel. The decision to use them in preference to clopidogrel will require careful balancing of the bleeding risk and the risk of recurrent ischaemic events and is best left to the treating cardiologist.
Antithrombin therapy
Subcutaneous low molecular weight or unfractionated heparin should be given until angiography or for 48–72 hours. The dose of low molecular weight heparin should be reduced if there is renal impairment. The dose of heparin is as above.
Glycoprotein IIb/IIIa inhibitors
Current evidence supports selective use of GP inhibitors. Indications may include ongoing ischaemia despite antiplatelet and antithrombin therapy, diabetes and troponin elevation.
β-Blockers
Initiation of a β-blocker is recommended unless contraindicated.
Invasive management
Patients with NSTEMI should have early coronary angiography (ideally within 48 hours), unless they have severe co-morbidities.
Disposition
Disposition depends on the type of ACS. Patients with STEMI and NSTEMI require admission to hospital for further care. Those with STEMI should be admitted to a monitored bed in a cardiac care unit because of the small but significant risk of life-threatening arrhythmia. It has been the practice also to admit patients with NSTEMI to monitored beds, but this is being challenged on the basis that there are subgroups within this classification at very low risk of adverse events. Patients with ACS without ECG changes or cardiac marker elevations require a period of assessment in the ED/chest pain unit and disposition will depend on the risk identified during that process (see Chapter 5.1).
Complications
Arrhythmias and conduction disturbances (see Chapter 5.4)
Pericarditis (see Chapter 5.6)
Acute left ventricular failure and cardiogenic shock
Most MIs are accompanied by some degree of left ventricular failure, which may range in severity from asymptomatic to pulmonary oedema or cardiogenic shock. Mortality depends in part on the degree of left ventricular failure, with cardiogenic shock having a reported mortality of approximately 80%.
Management includes maintaining adequate oxygenation, correcting electrolyte imbalances and optimizing ventricular filling pressures. Patients with pulmonary oedema may require non-invasive ventilatory support (see Chapter 5.3), but this need not be routine as most cases respond to medical therapy. If there is hypotension or other evidence of inadequate perfusion in the presence of adequate intravascular volume, inotropes should be initiated early and aggressively. PCI has been shown markedly to improve outcome for patients with STEMI accompanied by cardiogenic shock. Left ventricular assist devices may bridge to recovery, cardiac surgery or transplantation in selected patients.
Thromboembolism
Thrombus can form on areas of hypokinetic myocardium due to relative stasis and the prothrombotic effects of local inflammatory changes. It is more common with large anterior infarctions with left ventricular aneurysm formation, where the incidence has been reported to be up to 10%. Echocardiography is used to confirm the presence of thrombus. Systemic anticoagulation is required to prevent embolic complications.
Mechanical defects




Prognosis
The prognosis of ACS varies substantially between patients, depending on age, co-morbidities, risk factors, severity of coronary occlusion and myocardial necrosis and the presence of complications. Treatment of ACS should be guided by prognosis: the worse the prognosis, the greater the potential impact of treatment. Prognostic scoring therefore has an important role to play in the management of ACS.
The thrombolysis in myocardial infarction (TIMI) score has been developed and validated as a predictor of adverse outcome (mortality, life-threatening arrhythmia or subsequent myocardial infarction) in ACS. Patients are ascribed a score between zero (lowest risk) and seven (highest risk) by scoring one point for each factor listed in Table 5.2.4. Higher scores are associated with a higher risk of adverse outcome, as shown in Table 5.2.5. The TIMI score is simple to calculate and applicable to a wide range of patients. It can be used to predict which patients will benefit from early invasive management. Patients with a TIMI score of 3 or more appear to benefit from early invasive treatment, whereas those with a TIMI score of 2 or less do not. Patients with a TIMI score of 3 or more with ACS should therefore be considered high risk and receive early coronary angiography.
Table 5.2.4
Age≥65 years
Previous coronary artery stenosis>50%
Three or more risk factors for coronary heart disease
ST-segment deviation
Aspirin use in the preceding 7 days
Two or more anginal events in last 24 hours
Elevated cardiac biomarkers
Table 5.2.5
TIMI score and risk of death or MI at 14 days
| TIMI score | Risk of death or MI at 14 days (%) |
| 0 or 1 | 3 |
| 2 | 3 |
| 3 | 4 |
| 4 | 6 |
| 5 | 11 |
| 6 or 7 | 19 |
An alternative to the TIMI score is the Global Registry of Acute Coronary Events (GRACE) score, which uses the components outlined in Table 5.2.6. Each component is weighted to give an estimate of the probability of in-hospital and 6-month death or MI. The weighting process makes the GRACE score a little more difficult to calculate in the clinical setting, but allows for a more precise estimate of prognosis. Both web-based calculators and apps for hand-held devices are readily available.
Table 5.2.6
Age
Heart rate
Systolic blood pressure
Creatinine
Killip class
Cardiac arrest at presentation
ST-segment deviation
Elevated cardiac markers
It is worth noting that ST-segment deviation on the ECG and elevated cardiac markers feature in both the TIMI and GRACE scores and are powerful predictors of adverse events following ACS.
Risk-stratification determines subsequent management of patients with ACS. Although this process is typically undertaken by cardiologists, it is worth emergency physicians understanding the process. If patients have a high risk of subsequent adverse events (e.g. 6-month mortality risk exceeding 3%), then early invasive coronary angiography is often recommended. It should also be considered for patients with unstable symptoms even if their estimated risk is lower. Patients whose symptoms are controlled and have a low risk of adverse events can be offered conservative treatment and cardiac testing for ischaemia, such as exercise treadmill testing. CT coronary angiography is increasingly being offered as an alternative to testing for ischaemia and as a step in the diagnostic pathway between risk stratification and invasive coronary angiography.
Likely developments over the next 5–10 years




Controversies






5.3 Assessment and management of acute pulmonary oedema
David Lightfoot
Introduction
Acute pulmonary oedema (APO) occurs mainly in elderly patients and, if severe, is associated with a very poor long-term prognosis (1-year mortality approaching 40%). It is a pathophysiological state characterized by fluid-filled alveolar spaces, with impaired alveolar gas exchange and reduced lung compliance. Acute dyspnoea, hypoxia and increased work of breathing are the resultant symptoms and signs. APO occurs when increased pulmonary capillary pressure, reduced plasma oncotic pressure or pulmonary capillary permeability changes lead to plasma leaving the capillaries and building up in the pulmonary interstitium. When this occurs at such a rate that lymphatic drainage from the lung cannot keep up, flooding of the alveoli results.
Aetiology and pathophysiology
The causes of APO can be divided into cardiogenic (the commonest cause in ED patients) and non-cardiogenic. In cardiogenic APO, an acute reduction in cardiac output associated with an increase in systemic vascular resistance (SVR) leads to back-pressure on the pulmonary vasculature, with resultant increased pulmonary capillary pressure. Once established, APO can lead to a downward spiral where decreasing oxygenation and increasing pulmonary vascular resistance (with its resultant increased right ventricular end-diastolic pressure) worsens left ventricular dysfunction and worsens pulmonary oedema. In most cases, the patient has a maldistribution of fluid rather than being fluid overloaded. They may, in fact, have a whole-body fluid deficit. This understanding has led to a change in the management of this condition, from the use of large doses of diuretics to a focus on vasodilators and non-invasive ventilation that reduce SVR and improve cardiac output. Some of the causes of cardiogenic pulmonary oedema are listed in Table 5.3.1.
Table 5.3.1
Causes of cardiogenic pulmonary oedema
Acute valvular dysfunction
Anaemia
Atrial fibrillation and other arrhythmias
Dietary, physical or emotional excess
Fluid overload – may be iatrogenic
Medication adverse effect
Medication non-compliance
Myocardial ischaemia/infarction
Myocarditis
Post-cardioversion
Pulmonary embolus
Severe hypertension
Worsening congestive cardiac failure
In non-cardiogenic APO, the mechanism is thought to be increased pulmonary vascular permeability, brought about by an insult, leading to alveolar flooding. Injury to alveolar cells will also reduce their ability to clear this oedema fluid from the alveolar space (this may also play some role in cardiogenic APO). Some of the causes of non-cardiogenic pulmonary oedema are listed in Table 5.3.2.
Table 5.3.2
Causes of non-cardiogenic pulmonary oedema
Airway obstruction
Aspiration
Asthma
Disseminated intravascular coagulopathy (DIC)
Eclampsia
Head injury, intracerebral haemorrhage, hyperbaric oxygen treatment, inhalation injury
Lung re-expansion, e.g. after treatment of a pneumothorax
Lung reperfusion
Near drowning/cold water immersion
Opiates and opiate antagonists (naloxone and naltrexone)
Pancreatitis
Pulmonary embolism (thrombus, fat, amniotic fluid, other)
Rapid ascent to high altitude
Renal/hepatic failure
SCUBA diving
Sepsis
Shock
Toxins
Trauma
Clinical assessment
History
As with all emergencies, clinical assessment and management should take place in parallel. There is usually a history of sudden-onset severe dyspnoea. A focused history concentrating on the recent occurrence of chest pain, a past history of ischaemic heart disease or congestive heart failure or an other causative factor is sought. Details of current medication and compliance are also important.
Examination
Patients are usually pale or cyanosed, sweaty (sometimes profusely) and frightened. They strive to maintain an upright position at all costs and may be unable to sit still. They may cough up pink or white frothy sputum, adding to their feeling of drowning. The respiratory rate is high, with use of the accessory muscles of respiration, and breathing is often noisy. Oxygen saturation is severely reduced, reflecting hypoxia. Most patients are hypertensive or normotensive. Hypotension indicates cardiogenic shock and a very poor prognosis. There may also be a raised jugular venous pressure (JVP), third heart sound or gallop rhythm and signs of right heart strain. Signs of chronic heart failure should also be sought, as well as murmurs that may hint at the cause. The chest may be dull to percussion and fine crepitations, which are often extensive, will be heard on auscultation. Importantly, there may be other adventitial lung sounds, including wheeze – so-called ‘cardiac asthma’.
Clinical investigations
Electrocardiogram
An ECG is required, looking for acute ischaemia, especially evidence of ST-elevation myocardial infarction (STEMI), which should be immediately treated with appropriate reperfusion strategies.
Imaging
A chest X-ray will show cardiac size (usually enlarged) and help differentiate APO from airways disease. The chest X-ray findings of pulmonary oedema reflect the changes in fluid distribution. Initially, blood is diverted to the upper lobe veins, which become more prominent than normal. As the oedema worsens, interstitial oedema results in basilar and hilar infiltrates, which are hazy and more confluent than patchy, and interlobular oedema is seen as Kerley B lines. There is loss of vascular delineation. In severe APO, widespread changes representing alveolar oedema appear. There may also be pleural effusions if the interstitial pressure exceeds pleural pressure. Changes associated with the underlying cause can also be seen, e.g. cardiomegaly and pleural effusions in cardiogenic APO. It is important to note that the X-ray changes may not be bilateral and may mimic consolidation from other causes, e.g. pneumonia.
Ultrasound scanning (USS) in the emergency department (ED) can be a useful adjunct to physical examination. During ultrasound of the chest, homogeneous vertical comet tails or B-lines, which originate from the pleural line and continue to the bottom of the screen, characterize pulmonary oedema. These are ultrasound resonance artefacts generated by oedematous interlobular septa and are initially seen in the lung bases but, as oedema worsens, extend upwards to the apices. USS can be used to help differentiate the cause of a patient’s dyspnoea. In acute lung injury or acute respiratory distress syndrome (ARDS), the B-lines are non-homogeneous with areas of sparing. In patients with airways disease, horizontal A-lines may be seen, with an absence of B-lines. The B-lines will resolve as pulmonary oedema improves and may be used to follow response to therapy. Findings of jugular venous distension on ultrasound exam are also consistent with APO. Ultrasound scanning of the heart (echocardiography) may also be useful in determining the cause of the APO (e.g. wall motion abnormalities in acute myocardial infarction, right ventricular strain in pulmonary embolus and acute valvular dysfunction).
Blood tests
Blood tests include haemoglobin, electrolytes, cardiac biomarkers or other cause specific bloods as indicated, e.g. lipase.
Brain or B-type natriuretic peptide (BNP) or N-terminal pro-brain natriuretic peptide (NT-proBNP) levels may be useful in distinguishing APO from other causes of acute dyspnoea. BNP is the active part of a hormone (pro-BNP) secreted by ventricular myocytes in response to stretch. Its physiological function is to oppose the effects of the activated renin–angiotensin–aldosterone system. NT-proBNP is the inactive part cleaved from pro-BNP and has a longer plasma half-life than BNP. Its levels are also age dependent meaning ‘rule in’ cut points must be age-qualified. Plasma levels rise during acute heart failure and APO. In the setting of acute dyspnoea, levels of BNP<100 pg/mL (or NT-proBNP<300 pg/mL) make a diagnosis of acute heart failure and APO less likely. Conversely, when the BNP levels are>500 pg/mL (or NT-proBNP>450 pg/mL (age<50),>900 pg/mL (age 50–75) and>1800 pg/mL (age>75)), acute heart failure is more likely. Unfortunately, intermediate levels are difficult to interpret and other diagnoses should be sought prior to making a diagnosis of APO. That said, it has been estimated that by combining the intermediate natriuretic peptide marker levels with historical and exam findings consistent with APO, a diagnostic probability of heart failure is around 75%. Diagnostic accuracy may also be improved by combining USS with NP levels or using predictive modelling. False-positive levels may result from such conditions as pulmonary embolism, sepsis and renal failure, as well as advancing age. Positive levels may also occur in patients with chronic heart failure, but with a different cause of their acute ED presentation of dyspnoea. In addition, false-negative results may occur in the early phases of APO, owing to the delay prior to secretion of natriuretic peptides, as well as in obese patients and those with valvular or pericardial heart disease. These aspects, as well as the relatively high cost of the test, have limited its uptake in EDs in some countries. Currently, its main utility in the Australasian context involves following levels as a guide to therapeutic response, often in chronic heart failure patients. There is potential for using natriuretic peptide level changes within an ED presentation to guide disposition decisions (including discharge of non-APO heart failure patients), but this has not yet been studied prospectively.
Oximetry
Oximetry (in some cases supplemented with arterial blood gases) will reflect severity and help monitor the patient’s response to therapy. Rarely, in more severe cases, invasive monitoring may be useful.
Treatment
In all patients with APO, management strategies should provide supportive care to maximize cardiac output and oxygenation, followed by treatment of the underlying cause.
Treatment of the patient with non-cardiogenic pulmonary oedema consists of removing the patient from the causative environment, supportive therapies aimed at maintaining oxygenation, including non-invasive and invasive ventilation in severe cases, and treating the underlying cause. These patients have lung injury and therefore low volume; low pressure lung protective ventilation regimens should be followed.
Most patients with APO in the ED have a cardiogenic aetiology. Therapy varies according to haemodynamic parameters.
Normotensive or hypertensive patients
The mainstays of treatment are reduction of preload and afterload with nitrates and optimization of oxygenation, often with non-invasive ventilatory support. The patient should be managed sitting up. This posture reduces ventilation–perfusion mismatch and helps with the work of breathing.
Nitrates
Nitrates are the mainstay of pharmacological therapy of APO. They act to increase cyclic guanosine monophosphate (cGMP) in smooth muscle cells, leading to relaxation. In lower doses, this predominantly causes venodilatation and preload reduction. At higher doses, the arterioles are also affected, leading to afterload and blood pressure reduction. In addition, coronary artery dilatation leads to increased coronary blood flow. Myocardial work and oxygen demands are reduced and oxygen delivery is improved. Nitrates are therefore the ideal agents for treating APO by reversing pathophysiological processes that underlie it. Their use is limited by their hypotensive effect and by the tachyphylaxis that occurs with prolonged use. Therefore, they should be titrated against the patient’s haemodynamics and require careful monitoring. Nitrates are contraindicated in those patients who have taken sildenafil or similar agents within the previous 24 hours, owing to profound vasodilatation and hypotension. They should also be used with caution in patients with fixed cardiac output (e.g. those with severe aortic stenosis or hypertrophic obstructive cardiomyopathy). Although nitrates may also be used topically or sublingually, in the patient with APO, the IV route is preferred, as dosing can be titrated to effect and therapy ceased promptly if the patient becomes hypotensive. Topical or sublingual therapy is often used as a temporizing measure until IV access can be secured. The peak effect of IV nitrates occurs after 5 minutes.
The usual dosing regimen is to begin the infusion at 5–10 μg/min and increase the rate by 10–20 μg/min every 3 minutes, titrated to clinical effect and limited by falling blood pressure. If a patient has not responded to nitrate by a dose of 200 μg/min, they are unlikely to respond to further dose increases. Some studies have looked at using higher-dose bolus IV nitrates and have shown good efficacy and safety, with improved results over low-dose nitrates, frusemide and non-invasive ventilation. These studies, however, have limitations of small patient numbers, retrospectivity or non-randomization and these dosing regimens are not currently widely used.
Sodium nitroprusside
Sodium nitroprusside is a potent vasodilator with rapid action on both the venous and arterial systems. It has beneficial actions in APO by reducing both preload and afterload. It can lead to rapid improvements in haemodynamic parameters and cardiac output without the renal complications of other agents. Due to its swift and sometimes profound hypotensive action, invasive blood pressure monitoring is recommended. Rebound vasoconstriction can occur if the drug is stopped abruptly, so it should be slowly weaned. In addition, longer duration of use (>72 hours) has been associated with cyanide toxicity, especially in patients with renal or hepatic disease, in whom it should be used with extreme caution. Nitroprusside is a valid vasodilator alternative to nitrates and can be used when nitrates are contraindicated in non-hypotensive patients.
Angiotensin-converting enzyme inhibitors (ACEIs)
ACEIs have a clear role in the treatment of chronic heart failure. Their use in APO is more controversial. ACEIs effectively reduce afterload and, in cardiogenic pulmonary oedema, can also improve pulmonary capillary wedge pressure and cardiac output. In a small prospective study, when added to standard therapy, they produced a more rapid improvement in haemodynamic parameters and symptoms than placebo. Their use in pulmonary oedema was also associated with reduced intubation rates and ICU length of stay. Unfortunately, there are no large randomized trials looking at the use of ACEIs in APO patients.
ACEIs can also produce prolonged first dose hypotension and can worsen renal function, especially when used in combination with diuretics. Therefore, these agents should be used with caution, especially in patients who have renal impairment or are hypotensive. At present, it is not recommended they be initiated as first-line therapy in APO patients, but should be commenced, with appropriate monitoring of blood pressure and renal function, once the patient has stabilized.
Frusemide
Frusemide has been the first-line treatment for patients with APO for many years. Its usefulness is due to venodilatatory properties that lead to reduced preload, as well as to its diuretic properties. The venodilatation occurs about 15 minutes after IV dosing, well before diuresis begins. Frusemide can, however, lead to increased peripheral vascular resistance via reflex sympathetic and renin–angiotensin system actions. As mentioned above, fluid overload is not usually a contributing factor in acute heart failure and so diuresis is not a necessary endpoint of therapy. The obvious exception is in patients with APO of iatrogenic origin after IV fluid therapy.
Although it is an established therapy, there are no controlled studies that show benefit from the use of frusemide in APO. At least two studies have shown that nitrates are more beneficial than frusemide in relation to haemodynamic and clinical outcomes. High-dose frusemide has been associated with worsening renal function, ICU admissions and poor outcomes in patients with acute heart failure. Nevertheless, a single dose of frusemide at 1–1.5 mg/kg is still commonly recommended in the initial management of this illness.
Morphine
In the past, morphine has been one of the major drugs used in the treatment of APO. It can cause vasodilatation and decreased sympathetic activation; however, it has significant adverse effects including respiratory and central nervous system depression, hypotension and decreased cardiac output. A small prospective study and a number of large retrospective studies have shown an association between morphine use in APO patients and increased rates of intubation, ICU admission and death. As there is no evidence showing benefit from the use of morphine in the treatment of APO and there is an increasing body of literature associating it with harm, morphine should not be used routinely in the treatment of patients with APO. It may be considered for the relief of chest pain that is resistant to nitrate therapy in patients who do not have an altered conscious state, respiratory depression or hypotension. If it is used, it should be in small, titrated IV doses with close observation.
Aspirin
The most common cause of APO in patients presenting to ED is myocardial ischaemia/infarction. Aspirin has been shown to reduce the risk of death and myocardial infarction in patients with myocardial ischaemia. Although it does not directly treat APO, when the cause is thought to be myocardial ischaemia, aspirin should be given.
Ventilatory support
Patients with APO should be given high-flow supplemental oxygen using an oxygen delivery system that can meet their minute volume needs, such as a Venturi system. They are hypoxic and, uncorrected, this will worsen APO through direct pulmonary vascular constriction and reduced myocardial oxygen delivery. Prolonged supranormal oxygenation, however, should be avoided, especially in those patients with underlying airways or other chronic lung disease. In these patients, oxygen should be used judiciously and weaned as soon as the patient is improving, aiming for low normal oxygen saturations.
Patients with a severely reduced level of consciousness, agonal respirations or respiratory arrest require endotracheal intubation and mechanical ventilatory support. This should be accomplished using rapid-sequence intubation.
The introduction of non-invasive ventilation (NIV) using continuous positive airway pressure (CPAP) or bi-level positive airway pressure (Bi-PAP) has allowed many patients to avoid endotracheal intubation. Initial CPAP pressures of 5–10 cm H2O are used and then titrated to effect. When using Bi-PAP, expiratory pressures are usually begun at 3–5 cm H2O, with the inspiratory pressure 5–8 cm higher. The benefits of these therapies are due to a number of effects. Oxygen concentration can be accurately controlled and higher percentages can be delivered than via a face-mask. By using CPAP, functional capacity is increased by alveolar recruitment, with a resultant increase in gas exchange area, improved pulmonary compliance and reduced work of breathing. The addition of inspiratory pressure support with Bi-PAP further reduces the work of breathing and may be more useful in hypercapnic or tiring patients. Cardiovascular effects result from positive intrathoracic pressures, with reduced venous return and reduced left ventricular transmural pressures. These preload and afterload effects improve cardiac output without increasing myocardial oxygen demand. In general, these therapies have few complications and are considered safe. Complications that have been reported include nasal bridge abrasions, patient intolerance, gastric distension and aspiration, pneumothorax and air embolism. The last three potentially serious adverse events are extremely rare and appear to occur in selected populations with other underlying disease processes (e.g. pneumothorax in patients with Pneumocystis carinii pneumonia).
A number of studies have compared CPAP and/or Bi-PAP both with each other and with conventional therapy in APO. Most studies have been small and a number of meta-analyses have analysed their combined data. When CPAP was compared to standard medical therapy, there were significant improvements in oxygenation, ventilation, respiratory rate and distress and heart rates, without significant adverse events. There were also significantly reduced rates of endotracheal intubation, intensive care unit length of stay and, more importantly, reduced mortality. There is also a clear reduction in the rate of intubation and ICU admission when using Bi-PAP compared to standard therapy. There also appears to be a trend towards a mortality benefit although, in most analyses, it does not reach significance. Bi-PAP is associated with a more rapid reduction in symptoms than CPAP and may be particularly useful in patients with coexisting airways disease and acute on chronic respiratory acidosis. In the earliest trials of Bi-PAP in APO, there appeared to be an unexplained increase in the rate of myocardial infarction among patients in the Bi-PAP groups. These trials involved very small numbers and had methodological issues. Subsequent trials and meta-analyses have not shown an increase in myocardial infarct rates among the Bi-PAP groups.
When CPAP and Bi-PAP were compared with each other, there was no significant difference in intubation, myocardial infarction or death rates. As there is no convincing evidence of benefit of Bi-PAP over CPAP and no proven mortality benefit of Bi-PAP over standard therapy, CPAP is currently the NIV method of choice in APO, unless the patient has underlying airways disease or significant acute on chronic respiratory acidosis.
New pharmacological agents
Levosimendan comes from a class of drugs known as the calcium sensitizers and is currently only available in Australasia via the special access scheme. It binds to troponin-C and stabilizes the molecule in its pro-contraction state, prolonging contraction. This occurs without impairment of diastolic relaxation and without increasing calcium concentration (with its concomitant risk of arrhythmia and cell death). Cardiac output increases and pulmonary capillary wedge pressures decrease without significantly increased oxygen demand. It also causes vasodilatation (venous and arteriolar) via potassium channel opening, leading to reduced preload and afterload. Adverse effects include tachyarrhythmias, hypotension, hypokalaemia and headache. QTc may also increase. A large randomized trial failed to show a mortality benefit when levosimendan was compared with dobutamine in patients with acute decompensated heart failure requiring inotropic support. Adverse events including hypotension, atrial fibrillation and hypokalaemia were also increased in the levosimendan group. As yet, there are no trials examining its use in patients with acute pulmonary oedema.
Nesiritide is currently available in the USA and some other regions, but not in Australia. It is a recombinant brain natriuretic peptide and acts to cause arterial (including coronary) and venous vasodilation and to suppress the renin–angiotensin–aldosterone and sympathetic nervous systems. Although its mechanism of action would suggest a benefit in patients with APO, a recent large randomized controlled study failed to show any benefit over standard care in symptom control, rehospitalization rate or death and demonstrated problems with hypotension in patients with acute decompensated heart failure. Its use is limited to patients not responding to or with contraindications to other therapies.
Nicorandil is a hybrid vasodilator that causes combined preload and afterload reduction via activated KATP channels and a nitrate-like effect. It also has a cardioprotective effect by activating mitochondrial KATP channels. When given as a bolus to heart failure patients, it decreases pulmonary capillary wedge pressure and improves cardiac index. It has been associated with decreased in-hospital mortality and readmission rates of patients in acute heart failure. There are no prospective trials and it has not been specifically studied among APO patients.
Adenosine A1 receptor antagonists are a class of drugs that can improve diuresis by inhibiting sodium-induced tubuloglomerular feedback (with resultant fall in renal blood flow and glomerular filtration rate) seen with loop diuretics. Initial studies of the first drug in this class, rolofylline, showed encouraging improvements in dyspnoea without worsening renal function in acute heart failure patients. However, when compared with placebo in a recent large randomized trial, the drug did not show benefits and there were increased adverse events, particularly seizures. It has not been studied in APO patients.
Serelaxin. Relaxin is a hormone released during pregnancy with actions that include nitric oxide production and inhibition of endothelin and angiotensin II. It produces systemic and renal vasodilatation and increased vascular compliance. In the RELAX-AHF trial, serelaxin, a synthetic form of human relaxin, was compared to placebo in addition to standard therapy in patients with acute heart failure. Serelaxin was associated with improved dyspnoea and signs of cardiac failure with less diuretic use, as well as decreased intensive care unit and hospital length of stay. There was no mortality or decreased rehospitalization rate at 60 days. As with all the newer acute heart failure drugs, there is only a small body of evidence concerning this medication and none looking specifically at APO.
Endothelin receptor antagonists. Endothelin is a potent vasoconstrictor and modulator of the sympathetic nervous and renin–angiotensin–aldosterone systems. Tezosentan, an endothelin receptor antagonist, was shown to increase cardiac index, reduce pulmonary capillary wedge and pulmonary arterial pressures and reduce systemic vascular resistance in patients with moderate to severe heart failure. Unfortunately, in large randomized trials (VERITAS 1 and 2), when compared with placebo and added to standard care, it showed no benefit in terms of dyspnoea, worsening heart failure or death.
Hypotensive patients
In general, patients with APO who are hypotensive are at the most severe end of the disease spectrum, defined as cardiogenic shock. They require both ventilatory and haemodynamic support. Endotracheal intubation using rapid-sequence intubation and ventilation maximizes oxygen delivery and minimizes oxygen utilization. A positive end-expiratory pressure of 5–10 cm H2O may be useful. These patients may have a fluid deficit and, therefore, cautious fluid bolus resuscitation should be titrated against haemodynamic parameters and clinical effect. Inotropic support is also required, with epinephrine (adrenaline) being the first-line agent. Cardiac output may be improved with dobutamine but can it lead to hypotension requiring the concomitant use of vasopressors. These drugs will increase cardiac output, but do so at the expense of increased myocardial oxygen demand and increased arrhythmogenicity. Invasive monitoring will be required in this group as it helps guide fluid and inotropic management. Some time may be bought by the use of invasive therapeutic manoeuvres, such as an intra-aortic balloon pump. This device reduces myocardial oxygen demand via afterload reduction and increases coronary flow through diastolic augmentation. Reversible causes should be treated, e.g. reperfusion for acute myocardial infarction or surgical correction of acute valvular dysfunction.




5.4 Arrhythmias
Marcus Eng Hock Ong, Swee Han Lim and Chi-Keong Ching
Introduction
Arrhythmia is the term used to describe an abnormal heart rhythm. The most common arrhythmias are atrial or ventricular ectopic beats. Tachycardia occurs when the heart rate is>100 beats per minute (bpm) and bradycardia is defined as a rate of<60 bpm. The management of cardiac arrhythmias depends on the presentation of the patient, haemodynamic stability, underlying heart disease (if any) and the exact type of the arrhythmia. Patients with asymptomatic stable arrhythmias in the absence of underlying heart disease usually do not require emergency treatment. However, patients with symptomatic arrhythmias, especially when associated with underlying heart disease, require more urgent therapy. The key objective in a patient with haemodynamic instability is restoration of adequate cardiac output to maintain cerebral perfusion as well as a stable rhythm, using interventions least likely to cause harm.
Pathophysiology and pathogenesis
An understanding of cardiac arrhythmia requires knowledge of the normal conduction system (Fig. 5.4.1). In the normal heart, electrical impulses start from the sinoatrial (SA) node and conduct via the atria to the atrioventricular (AV) node. The electrical impulses then conduct down the bundle of His to the right and left bundle branches and, subsequently, via the Purkinje fibres to the ventricular myocardium.
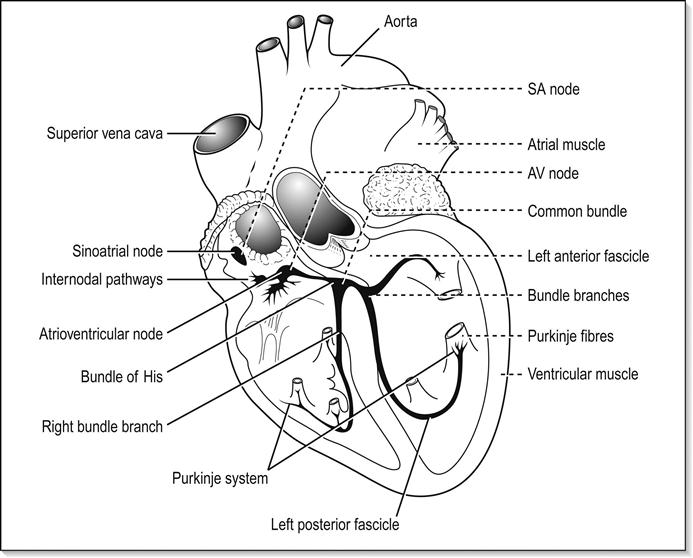
Different mechanisms, such as re-entry, enhanced automaticity and triggered activity, can result in arrhythmias. Generally, arrhythmias are thought of as abnormal impulse generation or abnormal impulse conduction. Abnormal impulse generation in the SA node, as in sick sinus syndrome, can result in failure of impulse formation. Abnormal impulse conduction in the AV node can result in failure of electrical conduction from the atrium to the ventricles, resulting in various degrees of AV block. Ectopic impulses in the atria result in atrial ectopics or atrial tachycardia. Accessory pathways between the atrium and ventricle can result in supraventricular tachycardia. Abnormalities in ventricular conduction can result in bundle branch blocks or a variety of intraventricular conduction abnormalities. The most dangerous arrhythmias arise from the ventricles, as these, especially in the presence of underlying structural heart disease, may be associated with sudden death.
Re-entry
Re-entry occurs when a closed loop of conducting tissue transmits an electrical impulse around the loop and stimulates atrial or ventricular electrical activity with each pass around the circuit. Atrial and ventricular fibrillation and flutter are examples of micro re-entry, whereas paroxysmal supraventricular tachycardia is an example of macro re-entry.
Principles of assessment and management
Patients with arrhythmias may present with symptoms due to the arrhythmia or may be asymptomatic and have the arrhythmia noticed during routine examination or investigations. All patients should initially be managed in an area where cardiac and other physiological monitoring is available.
The urgency of treatment is dictated by the patient’s clinical condition. For stable patients, the usual clinical process including history, physical examination and investigations (particularly ECG), is appropriate. For the unstable patient, urgent intervention is required, with restoration of a stable cardiac rhythm and cerebral perfusion being the priority. Clinical information, if promptly available, may be useful. For example, a patient with a history of renal failure or heart failure taking spironolactone should raise the suspicion that a wide complex tachycardia is due to hyperkalaemia.
Intravenous access should be obtained and blood drawn for investigations, such as full blood counts, electrolytes and cardiac markers (if indicated). A 12-lead ECG is essential and a chest X-ray may be helpful. Other specific investigations, such as serum digoxin level, thyroid function tests and theophylline levels, may sometimes be indicated, depending on the arrhythmia and the clinical context.
The management of arrhythmias should begin with attention to the airway, breathing and circulation. Management of cardiac arrest is discussed in Section 1.
Bradyarrhythmias
Bradycardia is defined as a heart rate of less than 60 bpm. It is important to take into account the patient’s underlying clinical state when treating bradyarrhythmias. ECG diagnosis of bradyarrhythmias can be simplified by the algorithm in Figure 5.4.2. Note that denervated transplanted hearts will not respond to atropine and so, if treatment is required, pacing, catecholamine infusion or both should be used.
Sinus bradycardia
Physiological sinus bradycardia may be associated with good physical conditioning (e.g. marathon runners), drug effects (e.g. β-blockers, calcium antagonists) and vagal stimulation (e.g. vomiting). More serious causes include acute inferior myocardial infarction, raised intracranial pressure, hypothermia and hypothyroidism.
Clinical features
There are often no signs or symptoms. Symptoms may be related to the underlying cause.
Clinical investigations



Treatment
Treat the underlying cause. If there is evidence of hypoperfusion, intravenous atropine 0.4–0.6 mg may be used while the cause is investigated. Physiological bradycardia does not require treatment. Disposition will depend on the cause.
Sick sinus syndrome (bradycardia–tachycardia syndrome)
This is most commonly found in elderly patients and results from fibrosis around the sinus node. It can also occur with congenital heart disease, rheumatic disorders, myocarditis, pericarditis, rheumatological disease, metastatic tumours, surgical damage, cardiomyopathies and ischaemic heart disease. It is a heterogeneous disorder that includes a wide variety of intermittent supraventricular tachycardias and bradyarrhythmias. Pathophysiologically, there is sinus bradycardia with intermittent failure of sinus node function, with the prolonged pause interrupted by a temporary escape rhythm. Drugs, such as β-blockers, digoxin and antiarrhythmics, as well as conditions, such as abdominal pain, thyrotoxicosis and hyperkalaemia, can exacerbate the condition.
Clinical features
Typical features are syncope, light-headedness, palpitations, dyspnoea, chest pain, collapse and cerebrovascular accidents.
Clinical investigations




Treatment
Unstable patients should be managed with atropine 0.4–0.6 mg IV as a bridge to pacing. If not effective, then consider adrenaline (epinephrine) infusion (2–10 μg/min) or dopamine (2–10 μg/kg/min) infusion if pacing is delayed. Transcutaneous pacing should be required if drugs are ineffective. Drug treatment for tachyarrhythmia risks aggravating pre-existing AV block or sinus arrest and should be avoided until pacemaker insertion. These patients eventually require a permanent pacemaker.
Heart block
First-degree AV block
In first-degree AV block, conduction of the atrial impulse to the ventricle is delayed. A P wave precedes each QRS complex, but the PR interval is more than 0.2 seconds. Causes include drug effects, vagal stimulation, inferior myocardial infarction and high vagal tone (especially in young patients). Rarely, it is a sign of myocarditis (e.g. rheumatic myocarditis), digoxin toxicity, idiopathic fibrosis or aortic valve disease.
There are no specific clinical features.
ECG features (Fig. 5.4.3) are:
 every P wave is followed by a QRS
every P wave is followed by a QRS
 PR interval is constant but>200 ms (five small squares on the ECG recorded at 25 mm/s).
PR interval is constant but>200 ms (five small squares on the ECG recorded at 25 mm/s).
Usually, no specific treatment is required. Unless associated with acute ischaemia, first-degree AV block is not itself an indication for hospital admission.
Second-degree AV block: Mobitz type I (Wenckebach)
In Mobitz type I AV block, conduction of the atrial impulses to the ventricles is intermittently blocked. This condition is due to impaired conduction in the AV node and so the atrial rate is greater than the ventricular rate. There is a progressive increase in the PR interval until a dropped QRS complex occurs. After the dropped QRS, AV conduction recovers, resulting in a normal PR interval and then the progressive increase in PR interval starts again. Anatomically, this block is above the bundle of His in the AV node. It is thought to be due to an increased impulse conduction interval at the AV node.
Causes include inferior myocardial infarction, digoxin toxicity and high vagal tone. The condition is nearly always benign and asymptomatic but, in the setting of acute ischaemia, may progress to complete heart block.
There are no specific clinical features.
ECG features (Fig. 5.4.4) are:



No treatment is required for stable patients. Atropine, dopamine/adrenaline infusion or cardiac pacing may be indicated in the haemodynamically unstable patient.
Second-degree Mobitz type ll AV block
Mobitz type II AV block is due to intermittent failure of conduction of atrial impulses to the ventricles. The PR interval remains constant, but there is regular, intermittent failure of P-wave conduction. This is usually due to impaired conduction in the bundle of His or bundle branches (i.e. it is infranodal). Advanced second-degree block is the block of two or more consecutive P waves. This condition may be seen with acute coronary syndrome involving the left coronary artery or, less commonly, idiopathic fissure of the bundle branches.
Although it may be asymptomatic, Mobitz type II AV block is more likely to be associated with stroke, Stoke–Adams attacks (syncope), a slow ventricular rate and sudden death.
ECG features (Fig. 5.4.5) are:

 atrial rhythm is regular (Ps plot through)
atrial rhythm is regular (Ps plot through)
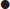 some P waves are not followed by a QRS (more Ps than QRS)
some P waves are not followed by a QRS (more Ps than QRS)
 PR interval may be within normal limits or prolonged, but is constant for each conducted QRS
PR interval may be within normal limits or prolonged, but is constant for each conducted QRS
 QRS complexes are dropped periodically. They may be narrow or widened.
QRS complexes are dropped periodically. They may be narrow or widened.
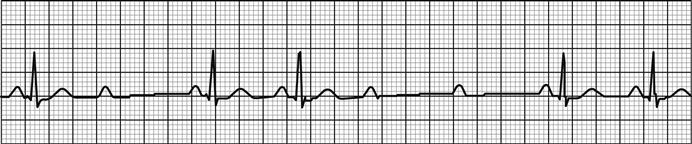
If the patient is haemodynamically unstable, give atropine 0.4–0.6 mg IV. If not effective, consider adrenaline (2–10 μg/min) or dopamine (2–10 μg/kg/min) infusion. Occasionally, pacing may be needed. Patients with this condition should be admitted, as it can deteriorate to complete heart block.
Third-degree (complete) AV block
In third-degree or complete AV block, conduction of the atrial impulse to the ventricles is completely blocked. Subsidiary pacemakers arise. If they are within the bundle of His, QRS complexes are narrow (Fig. 5.4.6). In contrast, if the block is infranodal, subsidiary pacemakers usually arise in the left or right bundle branches and the QRS complexes are wide (Fig. 5.4.7). The commonest cause of complete heart block is myocardial fibrosis. It is also seen in up to 8% of inferior myocardial infarctions, where it is often transient. Complete heart block is also associated with sick sinus syndrome, Mobitz type II block and transient second-degree block with new bundle branch or fascicular block.
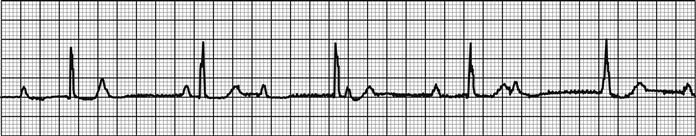
The patient may be asymptomatic, however, syncope or near syncope is common. Clinically, cannon ‘a’ waves may be seen in the neck veins and the first heart sound may vary in loudness.



Haemodynamically compromised patients should have measures taken to increase ventricular rate to a level that results in adequate perfusion. Judicious use of atropine 0.4–0.6 mg IV may be helpful. If this is unsuccessful, dopamine or adrenaline infusions, titrated to effect, may be effective. In ischaemic tissue, adrenaline is preferred, as coronary perfusion is better maintained. External pacing may be required if these measures are ineffective. Admission is required and permanent pacing is often necessary.
Never treat third-degree heart block with ventricular escape beats using lignocaine or any agent that suppresses ventricular escape rhythms as this will suppress the already slow heart rate, resulting in reduced cardiac output.
Tachyarrhythmias
There is a wide range of tachyarrhythmias. Immediate diagnosis and management may be considered on the basis of the width of the QRS complex and the regularity of the rhythm (Fig. 5.4.8).
Broad complex tachycardias
The differential diagnosis of a regular broad complex tachycardia includes ventricular tachycardia (VT) or supraventricular tachycardia (SVT) with aberrant conduction. Considerable research has been undertaken in an attempt to define ECG criteria that can reliably distinguish SVT from VT. Although relatively high sensitivities for some criteria have been reported, the high prevalence of VT in emergency department (ED) patients with broad complex tachycardia (approximately 80% in some studies) lowers the predictive value of those criteria. Features that increase the chance of a broad complex tachycardia being VT are shown in Table 5.4.1. It is usually safest to treat a broad complex tachycardia as VT unless there is very strong evidence to the contrary. In the older patient with underlying ischaemic heart disease, syncope and hypotension, VT would be the most likely diagnosis.
Table 5.4.1
Features that increase the chance of broad complex tachycardia being diagnosed as VT
| History | Clinical features | ECG features |
| Age>35 years | Cannon ‘a’ wave in JVP | AV dissociation |
| Smoker | Variable intensity of S1 | Fusion beats |
| Ischaemic heart disease | Unchanged intensity of S2 | Capture beats |
| Previous VT | QRS with>140 ms (<120 ms SVT) | Left axis variation >30° favours VT |
| Active angina | Concordance of QRS vectors in pericardial leads | QRS morphology in V1 |
Ventricular tachycardias
The proper identification of a ventricular arrhythmia is important in the evaluation of a patient. The management of ventricular arrhythmia depends on the correct identification of the rhythm, assessment of the risk–benefit ratio of antiarrhythmic drug therapy and an awareness of non-pharmacological modes of treatment. The presence or absence of heart disease and left ventricular function (ejection fraction) also influence the management approach. Risk increases with the severity of structural heart disease and left ventricular dysfunction.
Monomorphic ventricular tachycardia
Sustained VT is defined as a succession of ventricular impulses at a rate of>100 per minute and lasting more than 30 seconds or resulting in haemodynamic compromise. If the patient is haemodynamically stable, a 12-lead ECG should be recorded to characterize morphology.
Patients may be asymptomatic or complain of palpitations, dizziness or chest pain. Cannon ‘a’ waves may be seen in the neck veins. The patient may lose consciousness.
ECG features (Figs 5.4.9 and 5.4.10) are:


 rate>100 bpm: commonly 150–200 bpm
rate>100 bpm: commonly 150–200 bpm
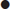 rhythm regular, although there is some beat-to-beat variability
rhythm regular, although there is some beat-to-beat variability
 constant QRS axis, often with marked left axis deviation or northwest axis
constant QRS axis, often with marked left axis deviation or northwest axis
 deep S wave with r/S ratio<1 in right bundle branch block (RBBB) morphology VT.
deep S wave with r/S ratio<1 in right bundle branch block (RBBB) morphology VT.
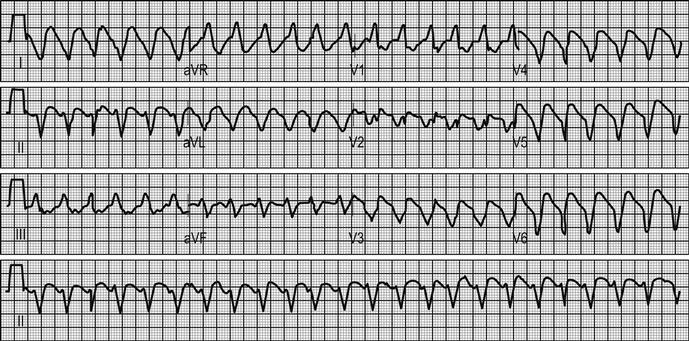
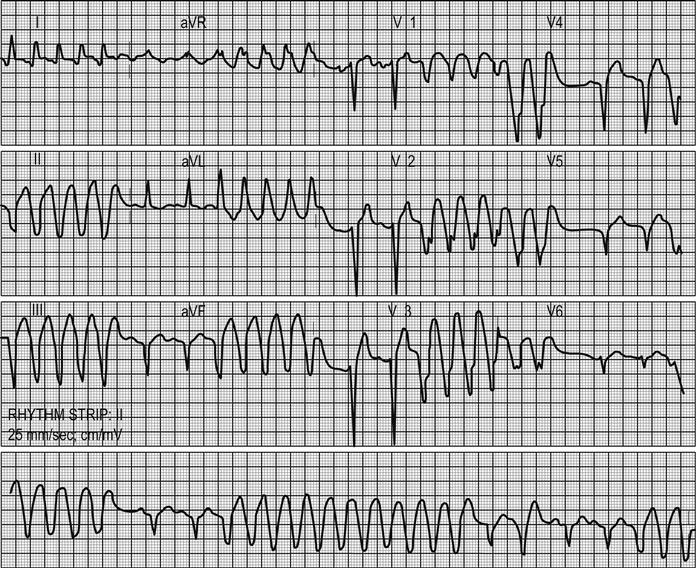
AV dissociation, captured beats or fusion beats, if present, are pathognomonic of VT. However, they are rarely seen because the rate of VT must be slow, usually<120 bpm for it to be easily visible.
Treatment
VT should be managed according to current AHA/ACC guidelines. Management of pulseless VT is addressed in Section 1 of this book. All patients with VT require oxygen therapy and IV access, at which time blood for electrolyte and cardiac marker analysis is obtained. Electrolyte imbalances, particularly of potassium, should be corrected.
An unstable patient requires emergency cardioversion, with sedation as required. Caution is needed, especially as these patients usually have low blood pressure. The first recommended DC shock should be 100 J (synchronized). If this does not convert the rhythm, it can be increased to 150 J and then 200 J. This can be further increased to 360 J if required. The equivalent biphasic energy should be used for biphasic defibrillators (e.g. escalating 70 J, 120 J, 150 J, 170 J). Shock-resistant VT may respond after administration of amiodarone. Lignocaine, magnesium and procainamide are considered second-line adjuncts to cardioversion as there is less evidence to support their efficacy. Nifekalant can also be considered, where available. An infusion of either amiodarone or lignocaine should be commenced after cardioversion. If the blood pressure is low, consider the use of inotropic support, for example dopamine infusion.
Stable patients may be treated with:



Sotolol 1 mg/kg is used as second-line agent. Following successful conversion, an infusion of the successful agent should be commenced for maintenance therapy. If pharmacological therapy is unsuccessful, cardioversion under sedation is indicated.
Polymorphic VT
VT with a continuously varying QRS morphology is called polymorphic VT. It is often associated with ischaemia and tends to be more electrically unstable than monomorphic VT.
Polymorphic VT includes a specific variant called torsades de pointes, which is associated with a prolonged QT. This is characterized by QRS peaks that twist around the baseline (Fig. 5.4.11) and this occurs in the presence of repolarization abnormalities. Causes are summarized in Table 5.4.2.
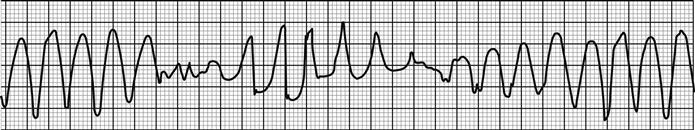
Table 5.4.2
Hypomagnesaemia
Hypocalcaemia
Class I and class II antiarrhythmic drugs
Phenothiazines
Tricyclic antidepressants
Congenital prolonged QT syndrome
Organophosphates
Complete heart block
Drug interaction of terfenidine with erythromycin
Syncope is the usual presenting symptom.


Torsades de pointes may be sensitive to magnesium. A bolus of 2 g over 1–2 minutes followed by an infusion will usually cause reversion. Cardioversion is recommended if the patient is haemodynamically compromised, but torsades de pointes can be resistant. Accelerating the heart rate, thereby shortening ventricular repolarization (e.g. by overdrive pacing to a rate of 90–120 bpm or isoprenaline infusion titrated to similar effect) may be successful. Treatment of the underlying cause is essential as torsades de pointes is very difficult to control. Beta-blockers can also be considered for congenital prolonged QT syndrome.
Idiopathic ventricular tachycardia
Idiopathic ventricular tachycardia is a monomorphic VT that occurs in the absence of structural heart disease. It is often exercise dependent and is named according to its site of origin. The QRS morphology during tachycardia can indicate the site of origin.
Idiopathic ventricular tachycardia presents as one of two subclasses:


Right ventricular outflow tract ventricular tachycardia
Right ventricular outflow tract ventricular tachycardia has a typical LBBB inferior axis morphology. The aetiology is believed to be cyclic-AMP (cAMP)-mediated triggered activity.
It typically occurs in young patients and is slightly more common in females. There is usually no evidence of underlying structural heart disease.
 QRS is broad (>120 ms) with a left bundle branch inferior axis morphology
QRS is broad (>120 ms) with a left bundle branch inferior axis morphology
 AV dissociation is not usually seen as the tachycardia is often very rapid
AV dissociation is not usually seen as the tachycardia is often very rapid

It is usually responsive to β-blockers as it is catecholamine sensitive. It is important to exclude underlying structural heart disease, especially arrhythmogenic right ventricular cardiomyopathy.
Idiopathic left ventricular tachycardia (ILVT)
Idiopathic left ventricular tachycardia is a fascicular ventricular tachycardia. The pathophysiology is a re-entrant phenomenon in the posterior fascicle of the left bundle branch.
It occurs in young patients without structural heart disease.
On ECG (Fig. 5.4.12), QRS morphology is broad and shows a right bundle branch block pattern with left axis deviation. The duration of the QRS complex is 100–140 ms with an RS interval<80 ms. For ischaemic VT the duration of the QRS complex is usually>140 ms and the RS interval>100 ms. ECG may also show capture beats or fusion beats.
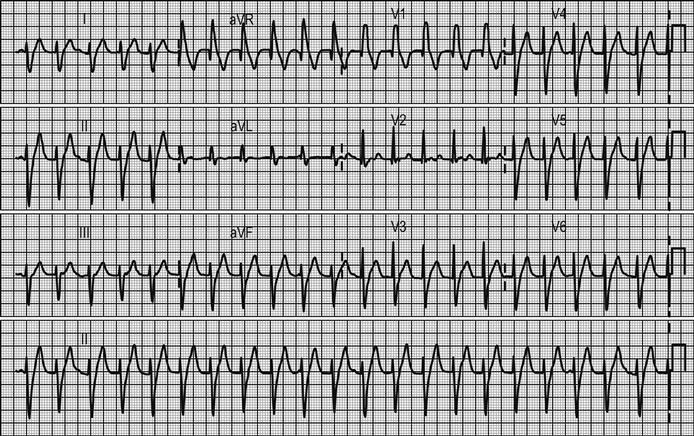
The drug of choice is intravenous verapamil. Amiodarone and sotalol have been reported to be equally effective. Vagal manoeuvres and intravenous adenosine are ineffective in converting this arrhythmia. Lignocaine is not effective for ILVT.
Pre-excited atrial fibrillation
Pre-excited atrial fibrillation (Wolff–Parkinson–White (WPW) AF) is a differential diagnosis for an irregular, wide complex tachycardia.
The patient is usually young (age<50 years) with a previous history of palpitations, rapid heart rate, syncope or documented history of WPW.
ECG features (Fig. 5.4.13) are:

 broad and bizarre QRS complex, signifying conduction down the aberrant pathway
broad and bizarre QRS complex, signifying conduction down the aberrant pathway
 occasionally, a narrow QRS can be seen, representing conduction through the AV node
occasionally, a narrow QRS can be seen, representing conduction through the AV node
 changing RR intervals; a QRS complex that changes frequently.
changing RR intervals; a QRS complex that changes frequently.
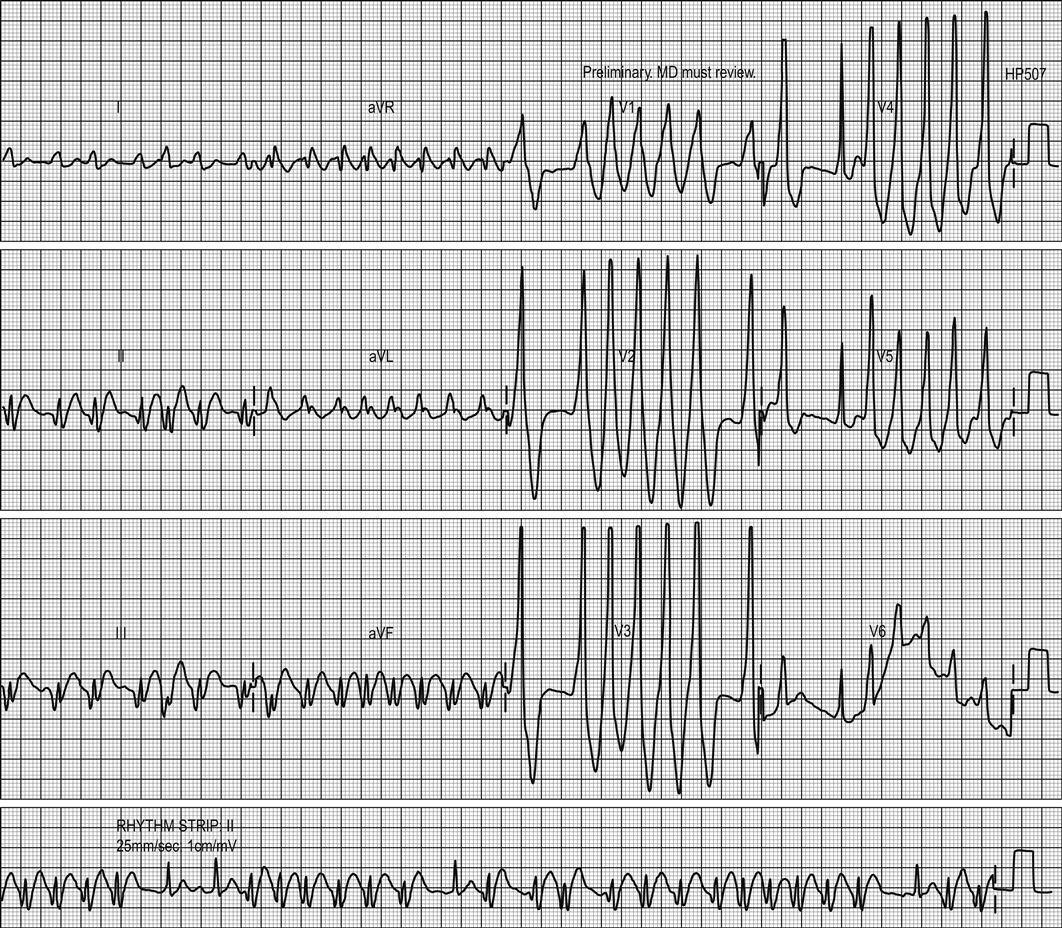
During sinus rhythm, the ECG of patients with WPW shows a PR interval<0.12 seconds, a slurred R-wave upstroke (δ wave) and a wide QRS>0.10 seconds (Fig. 5.4.14).
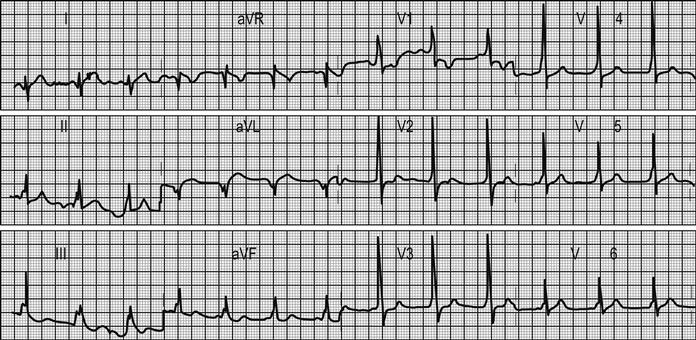
It is important to distinguish WPW AF from other wide complex tachyacardias. Certain subtypes of polymorphic VT, such as torsades de pointes, present with an undulating baseline. In contrast, WPW AF usually has a stable electrocardiographic baseline with no alteration in the polarity of the QRS complexes.
AF with aberrant conduction occurs when a patient with a pre-existing bundle branch block (or a rate-responsive bundle branch block) has a rapid ventricular response to AF. The ECG will show a wide complex tachycardia of irregular rate with stable beat-to-beat QRS configuration, contrasting with the variable beat-to-beat QRS configuration in WPW AF.
Haemodynamically unstable pre-excited AF is managed by immediate synchronized cardioversion. Haemodynamically stable pre-excited AF could be treated with:



Amiodarone administration modifies sinus and AV node properties with little, if any, effect on fast-channel tissues (i.e. accessory pathways) and it is not effective in treatment of WPW with AF.
Narrow complex tachycardias
Sinus tachycardia
Sinus tachycardia is defined as a heart rate>100 bpm. It not an arrhythmia per se, but rather an indication of an underlying disorder. Common causes include shock, hypoxia, cardiac failure, anaemia, drug effects, fever/infection, pain, anxiety, thyrotoxicosis and pregnancy.
Patients may complain of palpitations, but are often asymptomatic. Clinical features would be those of the underlying cause.




Management is focused on treatment of the underlying cause.
Paroxysmal supraventricular tachycardia
Paroxysmal supraventricular tachycardia (PSVT) originates from either an ectopic atrial focus or a re-entry circuit. Those caused by atrial flutter and fibrillation will be considered separately below.
Re-entry circuits are responsible for pre-excitation, which exists when the whole or part of the ventricular muscle is activated earlier than anticipated. The majority of re-entry circuits involve the AV node. Retrograde conduction may also involve an AV bypass tract. WPW syndrome is the most common of these. It is characterized by an electrically conductive muscle bridge (bundle of Kent) connecting atria and ventricle and bypassing the AV node. ECG when in sinus rhythm may show a PR interval of less than 0.12 s, a δ wave (slurred upstroke) and a wide QRS>0.10 s (see Fig. 5.4.14).
Patients may have palpitations, chest pain or syncope.
ECG features (Fig. 5.4.15) are:







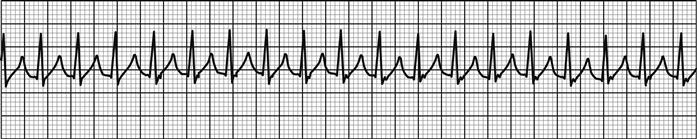
If the patient is haemodynamically unstable, cardioversion is indicated. Sedation is usually required. If the patient is mildly symptomatic but blood pressure is>100 mmHg, vagal manoeuvres may be tried. If vagal manoeuvres are unsuccessful or inappropriate, the choice of drug therapy lies between adenosine (escalating 6 mg, 12 mg and 18 mg rapid boluses into a large vein) or verapamil (continuous slow infusion of 1 mg/min to a maximum of 20 mg or 5 mg IV slowly, which may be repeated). Diltiazem infusion (2 mg/min to a maximum of 50 mg) is an alternative. There is scant evidence to show one approach is more effective than the other. Both have adverse effects. With adenosine, patients experience a transient sense of impending doom, chest discomfort and shortness of breath that can be very distressing. With verapamil, hypotension may occur, hence the cautious administration. Concurrent use of β-blockers may potentiate this. Flecainide (2 mg/kg over 30–45 minutes) would be considered third-line therapy. Patients who are resistant to chemical cardioversion may require electrical cardioversion.
Atrial flutter
Atrial flutter rarely occurs in the absence of underlying heart disease. Causes include ischaemic heart disease, acute myocardial infarction, congestive cardiac failure, pulmonary embolism, myocarditis, chest trauma and digoxin toxicity.
Patients may have palpitations and chest pain or more commonly are asymptomatic.
ECG features (Fig. 5.4.16) are:
 rate: atrial rate 250–350/min, flutter rate usually about 300/min
rate: atrial rate 250–350/min, flutter rate usually about 300/min

 rhythm: atrial rhythm regular; ventricular rhythm usually regular, but may be irregular
rhythm: atrial rhythm regular; ventricular rhythm usually regular, but may be irregular
 P waves: sawtoothed ‘flutter waves’. Best seen in II, III, and aVF
P waves: sawtoothed ‘flutter waves’. Best seen in II, III, and aVF

 QRS: usually<0.10 s, but may be widened if flutter waves are buried in the QRS complex.
QRS: usually<0.10 s, but may be widened if flutter waves are buried in the QRS complex.
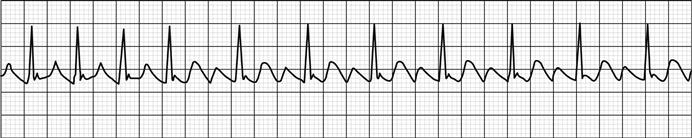
Treatment of the underlying illness is essential and will often result in spontaneous reversion. Haemodynamically unstable patients will usually respond to low-energy cardioversion (e.g. 50 J). Flecainide can be used for chemical reversion and verapamil for rate control, if required.
Atrial fibrillation
AF is the result of chaotic atrial depolarization from multiple areas of re-entry within the atria. Thus, there is a lack of coordinated atrial activity. AF is characterized by an irregularly irregular rhythm without discrete P waves and may be acute or chronic. Causes of AF seen in the ED are summarized in Table 5.4.3.
Table 5.4.3
Cardiac
Ischaemic heart disease
Pericarditis
Hypertension
Rheumatic heart disease
Pre-excitation syndromes
Cardiomyopathy
Atrial septal defect
Atrial myxoma
Postoperative
Non-cardiac
Electrolyte imbalances
Sepsis
Pulmonary embolism
Drug and alcohol intoxication
Chronic obstructive airways disease
Thyrotoxicosis
Lung cancer
Intrathoracic pathology



Clinical features
Patients with acute episodes of AF often experience palpitations, dyspnoea, dizziness or angina. Those with chronic AF often have no specific symptoms, especially if their heart rate is<100 bpm. Clinically, the pulse is irregularly irregular and S1 varies in intensity.
Clinical investigations
Investigations will be guided by the clinical context. Patients with acute-onset AF should have electrolyte studies and thyroid function tests as well as consideration of cardiac marker levels in appropriate clinical contexts. All patients with AF should have echocardiography, if not recently performed. This can be done as an outpatient procedure for those successfully treated in the ED.
ECG features (Fig. 5.4.17) are:
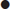
 chaotic irregular baseline – fibrillatory waves
chaotic irregular baseline – fibrillatory waves
 regularly irregular RR cycles – fast or slow AF
regularly irregular RR cycles – fast or slow AF
 wide QRS due to aberrance may occur intermittently (Ashman’s phenomenon).
wide QRS due to aberrance may occur intermittently (Ashman’s phenomenon).
Treatment
Emergency management depends on the chronicity of the condition, the ventricular response rate, haemodynamic stability, the presence of underling structural heart disease and any associated conditions.
The aim of treatment for patients with chronic AF is rate control and treatment of any associated illness. In patients with acute-onset AF, the choice lies between cardioversion in the ED (electrical or pharmacological), rate control and anticoagulation with delayed elective electrical cardioversion or rate control alone. The choice will be governed by the duration of the episode, the age of the patient, known structural heart disease, co-morbidities and previous attempts at cardioversion. There is no evidence that a rhythm control strategy is superior to a rate control strategy. That said, a rhythm-control strategy may be preferred for younger patients, those who are symptomatic, those presenting for the first time with lone AF and those with AF secondary to a treatable/correctable precipitant. A rate-control strategy may be preferred for patients aged over 65, those with coronary artery disease, those with contraindications to antiarrhythmic drugs, those unsuitable for cardioversion and those with congestive heart failure.
Treatment of AF in the ED is summarized in Figure 5.4.18. Haemodynamically stable patients where onset of AF is within 48 hours may be treated with either pharmacological or electrical cardioversion. Where AF is of a longer duration, a delayed elective cardioversion or rate-control-only strategy is indicated. There is a case in otherwise well patients with very recent-onset paroxysmal AF in the absence of structural heart disease or other underlying condition for a period of observation as up to 90% will revert spontaneously – usually within 24 hours.
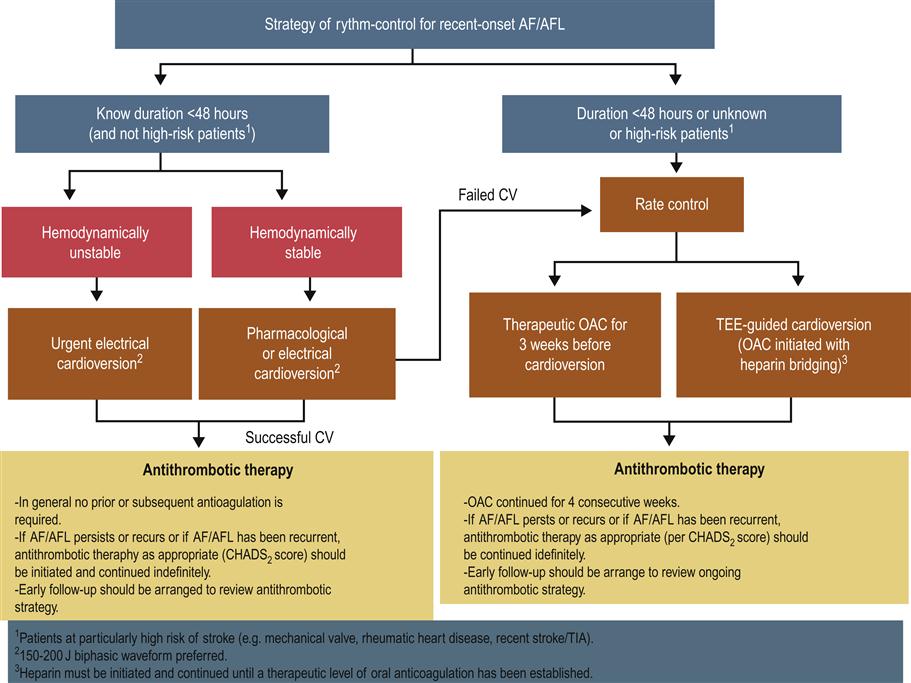
For pharmacological cardioversion in patients without structural heart disease, a class Ic agent, such as flecainide (2 mg/kg over 20–30 minutes) or propafenone, is recommended. Where structural heart disease is present, amiodarone (2–3 mg/kg over 5–10 minutes, repeated if necessary) is the drug of choice. Ibutilide, dofetilide, quinidine or procainamide (where available) may also be useful for cardioversion.
Rate control can be achieved with β-blockers (e.g. metoprolol 2.5–5 mg IV over 2 minutes, repeated if necessary to a maximum of 15 mg), verapamil (5–10 mg IV over 2–5 minutes), amiodarone or diltiazem.
In patients with life-threatening haemodynamic instability, emergency electrical cardioversion should be attempted, irrespective of the duration of the AF. In patients with WPW syndrome, flecainide may be used as an alternative to attempt pharmacological cardioversion. Atrioventricular node-blocking agents (such as diltiazem, verapamil or digoxin) should not be used.
In patients with known permanent AF where haemodynamic instability is caused mainly by a poorly controlled ventricular rate, a pharmacological rate-control strategy should be used. Beta-blockers or rate-limiting calcium antagonists are the agents of choice or, where these are contraindicated or ineffective, amiodarone should be used.
Multifocal atrial tachycardia
This rare arrhythmia is characterized by three or more atrial foci, a ventricular rate of more than 100 bpm and variable PP, PR and RR intervals. It is associated with chronic obstructive lung disease, hypoxia, electrolyte disturbance, pulmonary embolus and digoxin toxicity. Treatment is directed at improving the underlying condition. Specific treatment of the arrhythmia is rarely required and often not helpful.
Unifascicular blocks
A unifascicular block is a conduction block that affects one of the major infranodal conduction pathways: right bundle branch block (RBBB), left anterior fascicular block (LAFB) or left posterior fascicular block (LPFB). Conduction blocks can be caused by ischaemia, cardiomyopathies, valvular disease, myocarditis, surgery, congenital disease and degenerative diseases (Lenegre or Lev disease).
Left anterior fascicular block
There are no specific clinical features.


Usually no treatment needed. Treat the underlying cause.
Left posterior fascicular block
There are no specific clinical features.


Usually no treatment needed. Treat the underlying cause.
Right bundle branch block
RBBB can be a normal variant. Other causes include pulmonary embolism, right ventricular hypertrophy, ischaemic heart disease, congenital heart disease and cor pulmonale.
Clinical features will be those of the underlying cause.
ECG features (Fig. 5.4.19) are:
 rSR pattern, most noted in V1 and V2
rSR pattern, most noted in V1 and V2
 broad S wave in left ventricular leads
broad S wave in left ventricular leads
 QRS>120 ms in complete RBBB and≤120 ms with incomplete RBBB.
QRS>120 ms in complete RBBB and≤120 ms with incomplete RBBB.
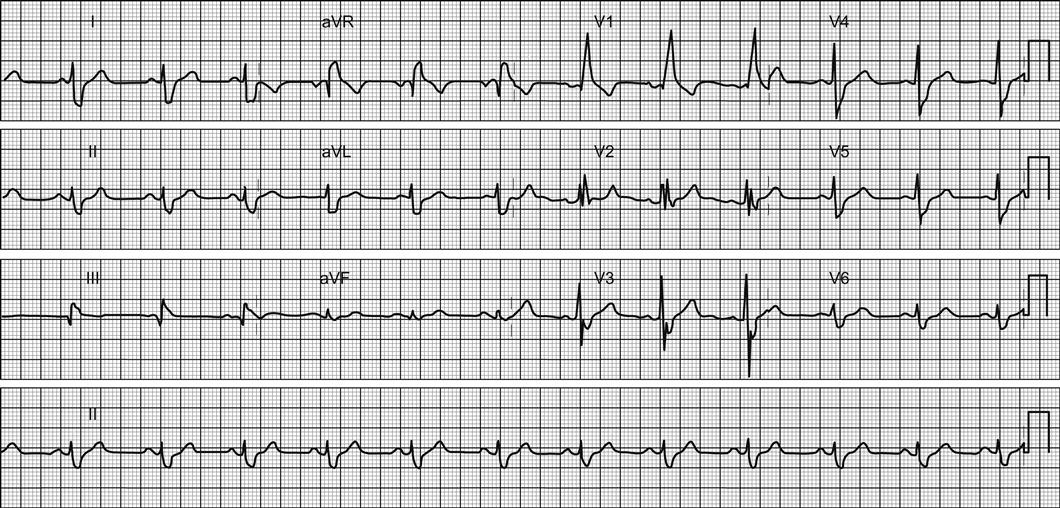
Treatment is of the underlying cause. If there is a new RBBB, the cause should be actively determined.
Left bundle branch block
LBBB is usually pathological. Causes include myocardial infarction, ischaemic heart disease, left ventricular hypertrophy, congenital heart disease and left ventricular strain.
Clinical features will be those of the underlying cause.
ECG features (Fig. 5.4.20) are:


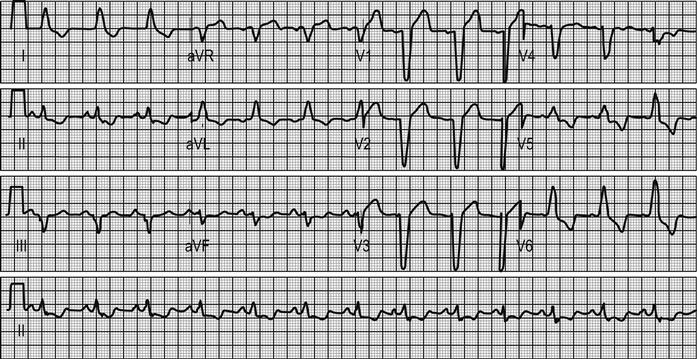
Treatment is of the underlying cause. If there is a new LBBB in the setting of myocardial ischaemia, management should be as for acute myocardial infarction.
Combination blocks
A bifascicular block is a conduction block that affects two of the major infranodal conduction pathways. This may be an LBBB or a combination of RBBB and LAFB or LPFB. Trifascicular block is a combination of conduction blocks of all three fascicles. Examples include:
 RBBB and LAFB with first-degree AV block
RBBB and LAFB with first-degree AV block
 RBBB and LPFB with first-degree AV block
RBBB and LPFB with first-degree AV block


Blocks may be permanent or transient. In the setting of an acute myocardial infarction, both bi- and trifascicular blocks may degenerate to complete heart block. Thus admission to a monitored bed is needed and pacemaker insertion considered.
Other disturbances of cardiac rhythm and conduction
Atrial ectopics
Atrial ectopics are mostly asymptomatic and may be precipitated by alcohol, nicotine and caffeine. They may be associated with AF, underlying heart disease or respiratory disease.
 complexes are usually earlier than normal (premature)
complexes are usually earlier than normal (premature)
 P-wave morphology different from sinus P. May be lost or deformed
P-wave morphology different from sinus P. May be lost or deformed
 PR interval may be short or long
PR interval may be short or long


Usually no treatment is necessary.
Junctional rhythm
A junctional rhythm is usually asymptomatic.





Usually no treatment needed.
Brugada syndrome
The Brugada syndrome is a syndrome with the ECG showing right bundle branch morphology and coved ST segment elevation in V1 and V2 with terminal T inversion (Fig. 5.4.21). The ECG pattern may be associated with sudden cardiac death from ventricular fibrillation. There may be a family history of sudden death. Occasionally, the ECG pattern is seen only during fever or after taking antiarrhythmic drugs, especially class Ic antiarrhythmics, such as flecainide or propafenone. Patients with the Brugada pattern should be referred for further evaluation and risk stratification.
Controversies




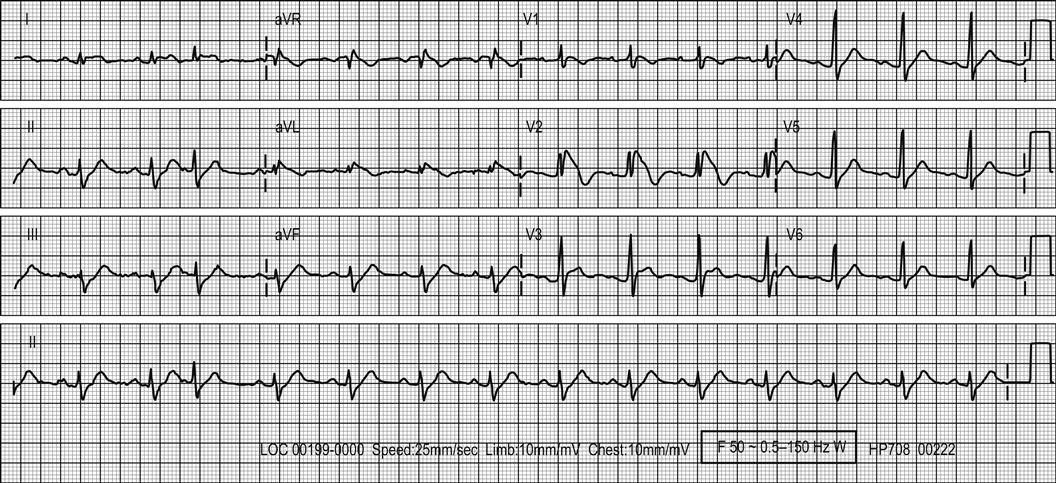
5.5 Pulmonary embolism
David Mountain
Introduction
Pulmonary embolus (PE) is the third most common cardiovascular disease. Historical data suggest that, left untreated, it has high mortality. Treating PE with anticoagulation reduces the overall in-hospital mortality to 4–12%. Of these, most (4–9%) are due to co-morbidity and 1.5–5% directly to PE.
The diagnosis and management of PE is often difficult. It requires careful clinical assessment and methodical application of aids to diagnosis. It relies on the estimation of probabilities rather than any truly definitive test. At all times, the possibility of alternative serious conditions causing the clinical presentation should be considered. All patients being considered for any diagnostic test for PE (including D-dimer assay) should have a documented risk assessment to ensure that use of those tests is safe and appropriate (see diagnostic risk assessment below). In general, if the risk of PE is below 5% (e.g. most low-risk emergency patients), then an alternative diagnosis should be sought first and investigation for PE pursued only if no alternative disease can be found. Indeed, in this population, excessive aggressive radiological investigation may find more false-positive venous thromboembolism (VTE) than real PE or deep venous thrombosis (DVT). The threshold for diagnosis is around 70–80% probability of PE, at which point the benefits of treatment outweigh the risks of non-treatment. These probabilities may vary in individuals if the risks of PE are high (e.g. pre-existing severe lung/heart disease) or bleeding risks are excessive (e.g. high risk of falls, recent major non-compressible bleeding).
Most institutions now use computed tomographic pulmonary angiography (CTPA) and/or ventilation/perfusion (V/Q) scan where definitive diagnosis is necessary. Most diagnostic pathways also include an initial D-dimer assay and venous ultrasound (or CT venography) in cases where there is clinical doubt after imaging. Some centres may also use echocardiography for risk stratification after diagnosing PE and, if available, it should be used for unstable cases. Magnetic resonance imaging (MRI) has been investigated in some centres but is not routinely used (Fig. 5.5.1).
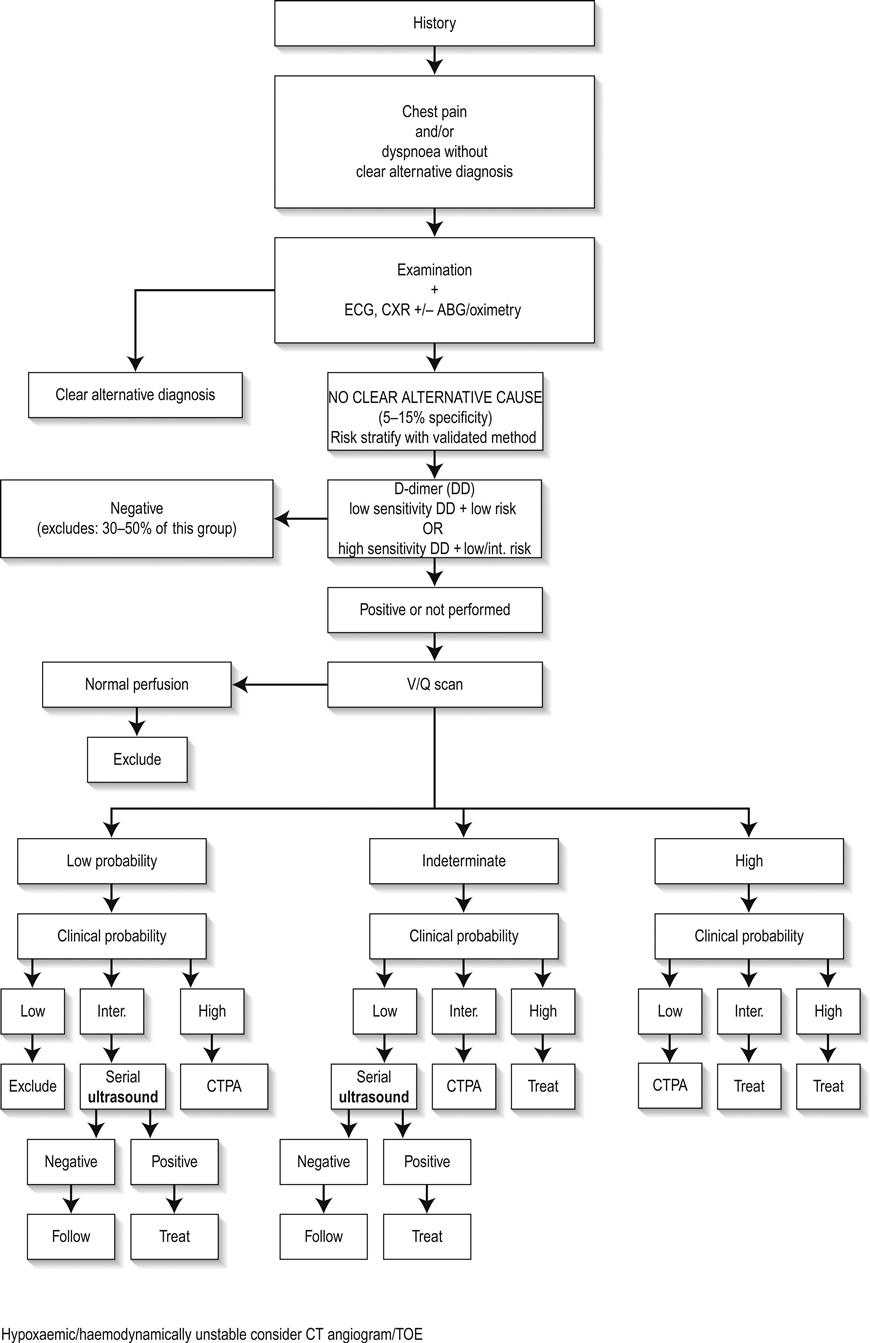
Aetiology, pathogenesis and pathology
Many risk factors and conditions are associated with PE, with major risks (e.g. cancer and surgery) clearly provocative. However, up to 30% of PEs, particularly those presenting to the emergency department (ED), are idiopathic. Most risk factors act via more than one of the processes in Virchow’s triad, e.g. vessel wall injury, venous stasis or hypercoagulable states. The major risk factors associated with secondary (provoked) PE are:
 surgery or trauma (particularly affecting the pelvis, lower limb or CNS) (15–30%)
surgery or trauma (particularly affecting the pelvis, lower limb or CNS) (15–30%)


 a past history of DVT/PE (particularly unprovoked and recent).
a past history of DVT/PE (particularly unprovoked and recent).
Other provoking factors important individually, but not common overall, are hypercoagulable states (e.g. antithrombin 111 deficiency, antiphospholipid syndrome, protein C and S deficiencies and hyperhomocysteinaemia) and many others both congenital and acquired. Other associations include increasing age (particularly age over 60), indwelling venous devices, hormone replacement therapy or contraception, obesity, pregnancy, some vasculitic diseases with venous involvement, smoking, use of non-steroidal anti-inflammatory drugs and long-haul air travel.
Prevention
The greatest single preventable cause of DVT is surgery. Low-dose preventative anticoagulation should be routine with low molecular weight heparins making this safer and more effective for most groups. Venous stasis as a result of illness-related bed rest, serious cardiopulmonary disease and (to a much lesser degree) travel are important avoidable causes of PE and early mobilization techniques are important for the hospitalized. Low-dose anticoagulation has been demonstrated to improve outcomes in medical patients admitted for bed rest. This should be started as early as possible. There is a role for emergency physicians and nurses to identify DVT risk and start thromboprophylaxis in high-risk ED patients.
Clinical Features
History
The history is the most important clue to a diagnosis of PE. Virtually all patients with PE will present with a history of either recent onset dyspnoea (particularly if rapid or recurrently episodic), chest pain (of any type) or both (sensitivity 97%, specificity 10%). Syncope either with respiratory symptoms or signs (even if transient) or in patients at high risk of VTE, is often a marker for severe PE. Symptoms of DVT should be sought in all patients. Haemoptysis has some predictive value but is an uncommon clinical feature.
Other symptoms (e.g. clear musculoskeletal symptomatology) are less important in the diagnosis of PE, but often help to suggest or exclude other causes in low-risk cases. Associated risk factors, divided into major and minor as described below, increase the probability of PE and should be documented and incorporated into clinical risk assessment. No single symptom or sign has the sensitivity or specificity to either establish or exclude the diagnosis.
Examination
Physical signs confirming PE are rare. That said, some increase the likelihood of a diagnosis of PE. A persistent unexplained tachycardia at rest increases the concern for PE and is part of many prediction rules. Leg (or arm) signs of DVT, particularly a swollen, oedematous leg with pain in the venous distribution, or significant thrombophlebitis, significantly increase the PE risk and mandates imaging for a complete work-up. Other features include tachypnoea (50–80%), cough (10–20%), mild fever (<38.5°C), wheeze and pleural effusion. They are not discriminatory for PE. Occasionally, elevated jugular venous pressure (JVP), a loud cardiac S(P)2 heart sound and/or pulmonary systolic murmur may be found and are markers of right ventricular strain.
Risk assessment for the diagnosis of PE
The pre-test probability (PTP) calculation used to decide on investigation strategies is based on history, examination and investigations, including chest X-ray, arterial blood gas analysis or oximetry, ECG and investigations for alternative diagnoses depending on presentation. For non-experts, this estimate is best made using validated scoring systems. The best validated and most widely disseminated is the Wells rule (Table 5.5.1). This is usually followed by a D-dimer test if PE has not been excluded on initial clinical review, particularly in low- or intermediate-risk patients. Other validated risk assessments are the Geneva score, modified (dichotomous) Wells score and estimation by senior experienced/expert clinicians.
Table 5.5.1
Wells clinical criteria for PE†
| Clinical signs of DVT | 3.0 |
| Pulse rate>100 (at rest) | 1.5 |
| Immobilized≥3 days | 1.5 |
| Surgery<4 weeks | 1.5 |
| Past history PE/DVT | 1.5 |
| Haemoptysis | 1.0 |
| Current/recent neoplasm | 1.0 |
| No alternative diagnosis more likely than PE | 3.0* |
| Score | |
| Low | <2 |
| Moderate | 2–6 |
| High | >6 |

*Including information from ECG, ABG, CXR and other tests for alternative diagnoses.
†A dichotomized (modified Wells) scale of≤4 (low) or>4 (high) is also validated and allows lower sensitivity D-dimers to exclude more patients. On line version at http://www.mdcalc.com/wells-criteria-for-pulmonary-embolism-pe/
Recently, the PERC rule (available on line at http://www.mdcalc.com/perc-rule-for-pulmonary-embolism/) developed by Kline et al. has been used in EDs to exclude some low-risk patients from any further investigations, including D-dimer, by making them very low risk (<2% chance of PE). It is only safe when applied to patients thought to be low risk either by another rule, e.g. Wells, or by an experienced treating clinician.
Investigations
The initial screening tests for PE are discussed below.
Chest X-ray
A normal chest X-ray (CXR) in the presence of significant hypoxia is suggestive of PE. However, the CXR is reportedly abnormal in 80–90% of patients diagnosed with PE. If definitely present, an enlarged descending pulmonary artery, pulmonary oligaemia or cut-off, a plump pulmonary artery and ‘Hampton’s hump’ (a semicircular opacity with the base abutting the pleural surface) are all quite specific for PE (70–90%). However, these signs may be subtle (e.g. often identified in retrospect) and have poor sensitivity. Pleural effusion, plate atelectasis, enlarged heart shadow and non-specific consolidation are seen commonly but do not occur more often in PE patients than in other diagnoses. The main role of CXR is the identification of alternative diagnoses. It also assists in deciding which radiological test is best to use between non-SPECT V/Q and computed tomography pulmonary angiogram (CTPA).
ECG
At least 21 potential features on ECG have been postulated as suggesting PE, but they are insensitive and most lack specificity. The most significant are tachycardias (particularly atrial), right bundle branch block (including incomplete or transient patterns), right axis deviation (particularly S1–3), T-wave inversion (especially deep V1–3) and the S1-Q3-T3 pattern. The more features that are present on the ECG, the more suggestive it is for right ventricular strain. These changes have been associated with poorer outcome in PE, particularly the rare S1-Q3-T3 pattern.
Arterial blood gas analysis (ABG)
In recent years, the role of ABG in PE has been challenged. A PaO2<80 mmHg without other cause makes PE much more likely, however, 12% of patients with PE have PaO2>80 mmHg. An abnormal A–a gradient marginally increases PE risk, but 20% of patients with PE have normal A–a gradients. In most patients, oximetry is sufficient to exclude significant hypoxaemia (<92%) and little is gained from routine ABG.
D-dimer tests
D-dimer tests have been available since the 1980s. They are well validated in emergency practice and recommended by current guidelines. They have only been shown to have reasonable specificity when used in ED populations.
The D-dimer test is sensitive but non-specific. Positive results occur with many other conditions. Their main role is to reduce the need for further investigations in a significant number of patients (20–60%). By identifying many low-risk patients who do not undergo further investigation, they increase the PE yield in the remaining investigated patients to over 5%, making further investigation of these patients worthwhile. D-dimers are only useful when negative, allowing PE to be excluded in low/intermediate-risk patients.
To be used safely and efficiently, it is important that the following steps are used:
 They should be used as part of a documented diagnostic process.
They should be used as part of a documented diagnostic process.




 The test should not be used if PE is not a realistic part of the differential diagnosis.
The test should not be used if PE is not a realistic part of the differential diagnosis.
Knowledge of the diagnostic performance of the D-dimer test being used is required to guide test application and interpretation. If a high-sensitivity test, such as VIDAS or one of the newer validated rapid latex tests, is available it is safe to use in low- and intermediate-ED risk groups (<30% chance of PE). Low-sensitivity tests, such as Simpli-Red or most latex agglutination tests, are only appropriate for use in low-risk patients (<10% chance of PE).
Further investigation
If the D-dimer is positive and no alternative diagnosis has become clear, further investigation is indicated. Following screening tests, more definitive investigations, including CTPA or V/Q scan, in some cases supplemented by ultrasound (US) or CT venography, should be performed. V/Q or CTPA are the usual initial investigations. If both are available, the choice is driven by patient-related issues (discussed below with each modality) and local logistics. Pulmonary angiography (PA) is now rarely available as most radiology departments have replaced it with multislice scanners. In unstable patients, echocardiography is the preferred initial test, looking for severe right ventricular dilatation and overload. MRI scanning may be used occasionally for patients where V/Q and CTPA are contraindicated or unavailable.
Computed tomographic pulmonary angiography and venography (CTPA and CTV)
Multislice (16–128) CTPA is available in most centres and after-hours availability is generally better than V/Q scanning. CTPA is preferable to V/Q scan in most patients with pre-existing lung disease because of the likelihood of intermediate probability V/Q scans in these patients. CTPA appears accurate in diagnosing main, lobar and most segmental vessel emboli. Sensitivity for subsegmental emboli is low, but the prevalence (reported to be 6–30%) and clinical significance of these PE is not clear. CTPA with 32–256 slices increases sensitivity for smaller PE, reported as over 90% in some studies and with good specificity (93–99%). However, the PIOPED2 study suggested sensitivity as low as 85% for PE, although when combined with CT venography (CTV), sensitivity improved to 90%. Large numbers of patients have been followed up after a negative CTPA, plus leg imaging if appropriate, with recurrent PE/death only seen in 1–2% and 0.2–0.5% respectively. This prognostic performance is similar to that reported for negative PA or normal V/Q scans.
Many centres now use CTPA alone to exclude significant PE. This is probably a safe strategy in all but high-risk patients without an alternative diagnosis. High-risk patients with negative CTPA should have leg imaging to exclude an embolic source (Fig. 5.5.2).
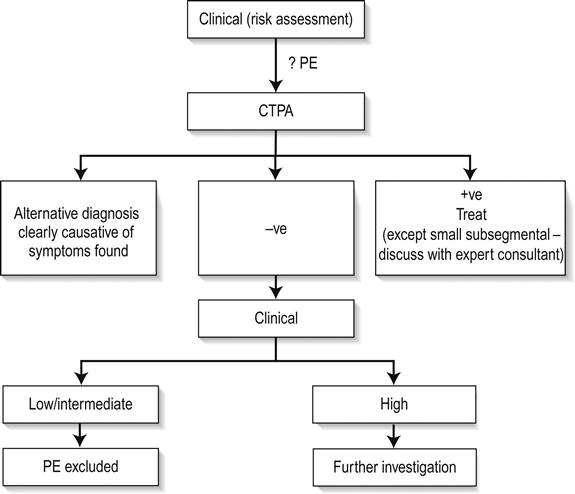
A major proposed advantage of CTPA is that other thoracic causes of chest pain may be identified. In some series, abnormal findings are seen in up to 60% of scans, with acute serious conditions in 20–30%. However, it is important to note that many of these findings may be incidental but then lead to prolonged further work-ups and additional radiation. Additionally, as scanners become quicker, data on right ventricular size, shape and function may help PE risk stratification.
In some centres, CTV, using the dye run-off from CTPA, images the venous system (legs to heart) instead of separate ultrasound testing. Almost all the additional yield (3–5% additional PE/DVT diagnosed in some studies) is from leg veins. Radiation doses to gonadal areas from pelvic/abdominal scanning should be avoided.
CTPA has some significant problems in clinical practice. In some studies, up to 15% of scans are technically inadequate. It requires significant dye loads, so is unsuitable for patients with renal dysfunction or contrast/iodine allergies. The radiation dose is high. In males, scans are generally to radio-insensitive tissues but, in women with proliferative breast tissues, high radiation doses (2–4 Gy per breast) confer significant increased lifetime breast cancer risk. V/Q scanning and US of the legs should be preferred in all young (including pregnant) female patients. The dose of radiation to the fetus is minimal in both CTPA and V/Q scanning if proper precautions are taken.
Ventilation–perfusion scan (V/Q scan)
V/Q scanning is usually readily available, has a low complication rate, moderate radiation exposure and can be used in patients with renal dysfunction and dye allergies. Problems are that potentially unwell patients may spend a long time in often distant nuclear medicine departments and many centres do not provide 24/7 scanning. Additionally, critics complain that the majority of V/Q scans are non-diagnostic (>50% in the PIOPED study if not risk stratified), requiring additional testing to rule PE in or out adequately. Because patients with obvious CXR abnormalities (e.g. major collapse, pleural effusions, etc.) or major lung disease almost always have indeterminate V/Q scans, they should have CTPA if available.
Two major studies (PIOPED and McMaster) have defined the probabilities of various V/Q scan results and their combination with clinical risk assessment. In most hospitals, scans were defined as normal, low, intermediate or high probability according to the number±size of unperfused lung segments, matched against ventilation. Further management depends on combining clinical risk and V/Q results as discussed below:


Much debate continues on how to manage patients with low clinical pre-test probability and intermediate V/Q. In PIOPED, this combination had rates of PE of 16%. However, studies have shown that with serial, or even single negative lower limb studies excluding DVT, recurrent PE or death rates are acceptably low. Patients with dichotomous risk versus V/Q results (low vs high) or intermediate/intermediate results need further alternative investigation with CTPA and/or leg vein imaging.
V/Q single-photon emission computed tomography (V/Q SPECT)
V/Q SPECT is a new technique combining V/Q scanning with computed tomographic scanning (circular arrays) allowing three-dimensional imaging of the lungs. This improves the clarity of images, gives better anatomical delineation of perfusion defects against known vascular domains and can be combined with low dose CT to look at patients with abnormal CXR or known abnormalities. This allows those units that have familiarity with this technique to give a definitive diagnosis or exclusion of PE in 95% of cases.
There is good evidence that V/Q SPECT gives better information and results than planar V/Q. On this basis, it would be an acceptable alternative to CTPA. Small numbers of outcomes studies suggest that there is good negative predictive value. To date, direct comparisons with CTPA have been limited so the true performance between the two tests is still unclear. Early evidence suggests that V/Q SPECT is probably slightly more sensitive but less specific. The specificity issues are probably reduced by combining V/Q SPECT with a low dose thorax CT to look for lung abnormalities that may explain poor perfusion, particularly in those with known lung disease or with abnormal CXR. Caution should be applied when the result is not concordant with the clinical picture (e.g. low-risk patient with multiple perfusion defects, etc.) and cross-over examinations to a CTPA/US may be advisable. The same is true for single subsegmental perfusion defects. V/Q SPECT is the preferred technique where CTPA is relatively contraindicated or where inadequate CTPA studies have occurred (10–25%). Further studies directly comparing V/Q SPECT with CTPA are awaited.
Magnetic resonance imaging
Magnetic resonance imaging (MRI) techniques are improving rapidly. Although sensitivity for central emboli is>90% with high specificity, its diagnostic performance is much poorer even for PE in large segmental vessels. There is little place currently for MRI in the acute setting unless CT/VQ are unavailable/contraindicated.
Echocardiography – transthoracic or transoesophageal (TTE/TOE)
Echocardiography is a rapid, accurate method of diagnosing massive PE in patients with instability. It helps exclude other causes of hypotension and raised venous pressure, such as cardiac tamponade and major valve or myocardial dysfunction. In massive PE, it can demonstrate right heart distension/dysfunction and sometimes central pulmonary artery or atrial clot. Importantly, it can be performed in resuscitation areas and used to see if initiation of thrombolytic treatment for unstable patients is indicated. It is insensitive for peripheral emboli and is not used to diagnose non-massive PE. However, in haemodynamically stable patients, echocardiography provides prognostic information. If right heart strain is seen, there is a much higher risk of poor outcomes (5–15% vs 0–2% mortality). If available, this information assists management decision making after diagnosis of PE.
Pulmonary angiography
Pulmonary angiography has been the ‘gold standard’ in diagnosing PE with very good sensitivity (98%) and specificity (97–100%) in all but the smallest PE. Because of significant technical, logistical and clinical difficulties and a 0.3% mortality and 3% complication rate, it is now rarely used.
Further investigation of isolated sub-segmental clots
Whenever isolated subsegmental clots are reported on imaging, further tests to exclude false positives are recommended. These include:
 leg imaging if not already performed
leg imaging if not already performed
 an alternative imaging test to confirm the original result if available/feasible
an alternative imaging test to confirm the original result if available/feasible

It is important to remember that false positives are common in patients with these isolated small subsegmental findings no matter what imaging is used.
Treatment
Risk stratification
Patients diagnosed with PE should be risk stratified for short-term/in-hospital prognosis. There are significant differences in both ongoing therapeutic options and the monitoring required for PE patients with different prognoses. Features that determine prognosis are overt haemodynamic instability (including syncope) or prolonged respiratory failure, investigations suggesting right ventricular overload/strain or severe V/Q mismatch and severe underlying co-morbidities.
Haemodynamic or respiratory instability should be clinically obvious, although the use of venous/arterial gases to expose unexpected lactic acidosis/hypoxia, if there is clinical concern, is reasonable. In patients without obvious instability, prognosis may be determined by considering:



Other features that raise concerns for patients are a demonstration of a massive clot load (e.g. massive DVT, centrally located emboli), recurrent PE on adequate therapy and serious cardiopulmonary co-morbidity.
Prognostic scores
Combinations of various clinical features and/or investigations have been studied to try to define low-risk groups for possible early or immediate discharge. The two major systems are the PESI (http://www.mdcalc.com/pulmonary-embolism-severity-index-pesi/) and Geneva prognostic scores, both of which have been prospectively validated. The PESI score was also validated in a large randomized trial confirming safety in selecting patients with PE for home therapy.
General measures
Almost all patients with diagnosed PE need to have a combination of supportive care, including oxygen therapy, thromboprophylaxis, analgesia and careful observation for an initial period while early prognosis is determined and heparin or fondoparinux plus warfarin therapy are initiated (unless there is an absolute contraindication). Thought should be given to taking a thrombophilia screen before heparin is started when there is an unprovoked PE or if the PE is recurrent and thrombophilia has not been investigated previously.
Heparin therapy
Heparin therapy can be given either as intravenous unfractionated heparin (UFH) or low molecular weight heparin (LMWH). LMWH therapy has been shown to be at least as effective as UFH (clinically and by cost) and is probably safer than UFH for DVT and PE. The evidence for this in PE is weaker than for DVT, but the equivalence of the therapies is accepted by evidence-based care bodies such as the Cochrane Collaboration and NICE and most accept an overall benefit from LMWH compared to UFH. Dosing for enoxaparin is usually 1.5 mg/kg daily or 1 mg bd, but all LMWHs probably provide adequate therapy. Dosing must be reduced for those with poor renal function (reduced dose and frequency) and the morbidly obese (e.g. 100 kg maximum lean weight) to avoid accumulation or excessive dosing.
LMWH is particularly useful if early discharge from inpatient units or even home therapy is being contemplated. Home therapy is now widely used in North America in low-risk PE and, in well-selected cases, it seems to be safe. It is recommended in some current guidelines but relies on good risk stratification. Currently, the best validated way to predict low risk is the PESI score (see above).
Warfarin therapy
Warfarin should be started at the same time as initial heparin therapy and maintained for at least 3 months. Duration of treatment should probably be longer in those at high risk (e.g. cancer patients, unprovoked large PE, etc.). The risk of anticoagulation causing major bleeding may be as high as 10% over 6 months in very high-risk patients (2–4% in well-controlled patients), with 1–3% intracranial haemorrhage rates in some registries and mortality from bleeding of 0.1–1% pa. Risk scores for assessing who is at high risk of bleeding are not validated in the PE population and have limited evidence of discrimination. Clearly, elderly, frail, hypertensive, falling, recently bleeding, alcoholic or severely co-morbid patients are at much higher risk.
Caval interruption techniques
The use of caval interruption techniques should be considered in cases of recurrent PE despite coagulation, or where there is bleeding precluding anticoagulation. It has also been recommended for massive PE or massive leg DVT with PE. Fatal PE usually occurs as a result of further clot progressing along the inferior vena cava (IVC). Percutaneous IVC umbrellas may be inserted relatively easily and prevent further deterioration.
Patients at high risk of deterioration
Patients without cardiorespiratory instability but with features of RV strain or major PE, such as a history of syncope/collapse, ongoing hypoxia or tachycardia, elevated troponins or BNP, large clot loads, RV strain on echocardiography or clinician concern, should undergo close observation and/or continuous monitoring in a high-dependency or critical care area. There is a high rate of deterioration or death in these patients, with up to 25% requiring inotropes, intubation or thrombolysis for instability and mortality rates of up 10%. A management plan should have been decided upon before transfer out of the ED, preferably with criteria for initiation of thrombolysis/thrombectomy or catheter endarterectomy.
Unstable patients
Patients presenting with overt shock, rapidly deteriorating respiratory failure or a history of recovery from cardiorespiratory arrest should be resuscitated, stabilized and, in most cases, given thrombolysis (or alternative treatments if thrombolysis is contraindicated; see below). Severe hypoxaemia may require endotracheal intubation and ventilation. Haemodynamic instability requires gentle intravascular fluid loading with 250–500 mL boluses of crystalloids totalling no more than 1 L unless dehydration or hypovolaemia are clearly coexistent with PE. The reason for this is that, in massive PE, the right ventricle is already pressure overloaded and failing and excessive fluids will further overstretch a failing ventricle (Starling’s law).
Persistent hypotension will require inotropic support. There is little evidence to support the use of norepinephrine (noradrenaline) over epinephrine (adrenaline) as the inotrope of choice. Patients requiring inotropes should not be treated with isoprenaline as this results in vasodilatation, reduced peripheral resistance and increased cardiac output, without improving coronary perfusion.
Thrombolysis
The widespread use of thrombolytic therapy for coronary disease led to a reappraisal of thrombolytics in PE. There is definite evidence of reduced pulmonary artery pressures and improved right ventricular function after thrombolysis, which may persist following the acute episode. One small, randomized trial in shocked patients showed a clear mortality benefit for thrombolysis. In addition, some meta-analyses suggest that in the sickest patients with PE there is probably a mortality benefit. Very few clinicians would withhold thrombolytics for massive (hypotensive/unstable) PE. There is widespread use of thrombolytics in Europe for moderate-sized PE causing right ventricular strain, with some registry evidence of improved mortality or reduced complications. However, there is much less acceptance in the USA and Australia for this indication as the evidence is weak and thrombolysis is actively discouraged in haemodynamically stable, non-deteriorating patients.
Tissue plasminogen activator (rTPA) appears to be the easiest and quickest to give and to have the fewest side effects (excepting intracranial haemorrhage [ICH]) compared to urokinase and streptokinase. rTPA has been used as an infusion of 100 mg over 2 hours. Bolus reteplase (two doses of 10 units separated by half an hour) or tenectaplase (weight-based infusion) should be just as effective, although they have not been properly studied in PE and do not have TGA approval for this indication.
Thrombolysis is associated with a major bleeding episode in up to 20% of patients, with ICH rates as high as 4% and death from bleeding in 0.3–2%.
Embolectomy
Patients with persistent haemodynamic instability or hypoxia with major contraindications to thrombolysis should be considered for thoracotomy and/or embolectomy. Patients in this category are not necessarily at hospitals with facilities for cardiopulmonary bypass and, therefore, alternative therapies have been developed. The use of mechanical clot disruption (catheter embolectomy) for massive PE has been reported in case studies, but controlled studies are difficult to design because of the infrequency of the event and the emergency nature of massive PE. Unlike other surgical techniques, the expertise and equipment for this procedure are readily available in most, larger hospitals. Pulmonary embolectomy without cardiac bypass has been used as a last resort for haemodynamically unstable patients, with a reported survival of more than 50%. Following cardiac arrest, survival rates are much lower, although survivors have been reported.
There is no evidence that mechanical removal of clot results in better outcome than does thrombolysis; in fact, it could be worse. In the pre-arrest or arrested patient, the transfer to cardiopulmonary bypass may buy additional time. In general, hospitals should decide on thrombolysis versus thrombectomy as their preferred management of unstable PE to avoid confusion and unnecessary delays to management in shocked patients.
Prognosis
Prognosis is largely dependent on coexistent illness and the size and eventual position of the initial PE. Patients with cardiorespiratory arrest or shock have mortality rates of 25–50% even with thrombolysis/thrombectomy. Right ventricular strain in haemodynamically stable patients has a mortality rate of 5–15%. In addition, this group is at risk of developing chronic thromboembolism and persistent pulmonary hypertension. Patients without significant co-morbidity or signs of severity have very low rates of poor outcomes. Even with anticoagulation, hospital mortality may still be high (2.5–10%), although much of this mortality is due to co-morbidity. Recurrent PE occurs in about 25% of patients by 8 years.
Disposition
Patients with haemodynamic instability, recovery post-arrest, those with significant persistent hypoxia (<92%) and those with evidence of right ventricular strain should be admitted to an ICU or high-dependency unit. Most patients with stable PE can be admitted directly to the ward. Early discharge on LMWH (or Fondoparinux) should be considered for compliant patients with smaller PEs, good home circumstances and a low risk of complications.







5.6 Pericarditis, cardiac tamponade and myocarditis
Jason Harney, Anne-Maree Kelly and James Hayes
Pericarditis
Introduction
Pericarditis may be acute, subacute or chronic. It is defined as inflammation of the pericardium. It should be noted, however, that the condition is better described as perimyocarditis. In the majority of cases, there are variable degrees of associated ‘epimyocarditis’, which has important clinical implications. The causes of pericarditis are listed in Table 5.6.1.
Table 5.6.1
| Idiopathic (about 25%) | Most of these are probably viral |
| Malignancy (about 25%) | Primary, e.g. sarcoma and mesotheliomas |
| Secondary, e.g. haematological, breast, lung and melanoma | |
| Infective | Viral, e.g. Coxsackie B, mumps, Epstein–Barr virus, influenza, HIV |
| Bacterial, e.g. staphylococcal, streptococcal, Gram-negatives and TB | |
| Mycotic, e.g. histoplasmosis | |
| Autoimmune/connective tissue | Rheumatoid arthritis, systemic lupus erythematosus (SLE), sarcoidosis, scleroderma, Stevens–Johnson syndrome, inflammatory bowel disease |
| Trauma | Blunt or penetrating |
| Post-pericardiotomy syndrome | |
| Radiation injury | |
| Myocardial infarction associated | Acute: days to weeks following transmural myocardial infarction |
| Dressler’s syndrome: weeks to months following myocardial infarction | |
| Drugs | SLE-type syndromes, e.g. hydralazine |
| Hypersensitivity syndromes, e.g. penicillin | |
| Systemic illnesses | Uraemia |
| Myxoedema | |
| Other | Dissecting aneurysm |

Clinical features
History
Idiopathic or viral types may have a history of a recent viral illness and the history should be directed towards the known causative pathologies. The pain is usually retrosternal, sometimes with radiation to the trapezius muscle ridges, but not generally to the arms. It may also be pleuritic in nature, and worse with movement and respiration. It is typically worse when lying supine and better when sitting up and leaning forward. True dyspnoea is not a feature unless there are secondary complications, such as cardiac tamponade or chronic constrictive pericarditis. Dyspnoea may also be due to the underlying disease process causing pericarditis. Respiration may however be shallow because of pain.
Examination
With viral or idiopathic types, fever may be present. Sinus tachycardia is common. A pericardial friction rub may be heard, caused by rubbing between parietal and visceral pericardial layers or between parietal pericardium and lung pleura. A rub may therefore be heard even with the presence of a large effusion. It may be audible anywhere over the precordium, but is best heard with the diaphragm of the stethoscope over the lower left sternal edge, where the least amount of lung tissue intervenes between the pericardium and the chest wall, with the patient leaning forward in full expiration. The rub has a superficial scratching or ‘Velcro-like’ quality. Rubs may be difficult to detect, as they can be transient and migratory. The patient should be examined for any signs of a complicating cardiac tamponade. A search should also be made for any signs of an underlying causative condition.
Temperature above 38°C is uncommon and may indicate purulent (i.e. bacterial) pericarditis, a much more serious condition.
High-risk features
The following high-risk clinical features should raise suspicion of serious underlying pathology, such as tuberculosis or other bacterial infection, malignancy or autoimmune disease:









Clinical investigation
Blood tests





Chest X-ray
Chest X-ray does not confirm the diagnosis of pericarditis but may rule out other causes of pleuritic chest pain. It may also find evidence of a complicating pericardial effusion or evidence of causative pathology, such as malignancy.
ECG
The ECG is the most important investigation and will show abnormalities in 90% of patients with acute pericarditis. ECG changes are the result of the associated epimyocarditis. The pericardium is electrically neutral and does not produce ECG changes. Therefore, in the occasional ‘pure’ case of pericarditis, the ECG will be normal. It may follow the typical evolution of changes, but in a sizeable minority will not.
The typical pattern follows four stages:

 diffuse concave upwards ST elevation. This may occur in all leads apart from AVR and often VI (Fig. 5.6.1)
diffuse concave upwards ST elevation. This may occur in all leads apart from AVR and often VI (Fig. 5.6.1)

 Stage 2: the PR and ST segments normalize, which can lead to a transiently normal ECG.
Stage 2: the PR and ST segments normalize, which can lead to a transiently normal ECG.


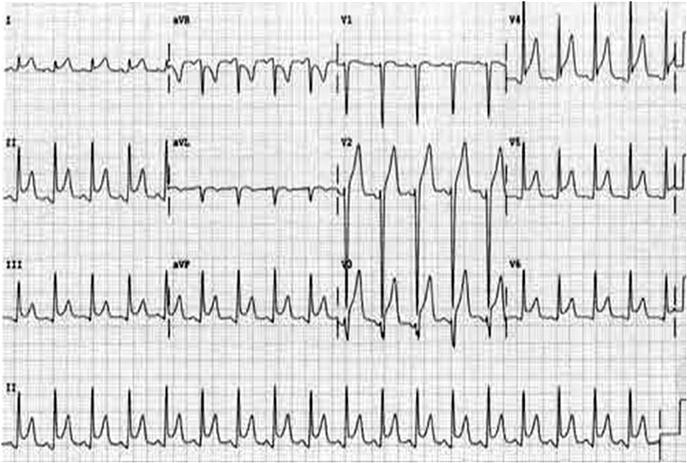




Echocardiography
This may give indirect evidence for pericarditis by showing the presence of an effusion or a thickened pericardium. High-quality echocardiograms are able to distinguish bloody from serous effusions. Transoesophageal echocardiography (TOE) is better at measuring thickness of the pericardium than transthoracic echocardiography (TTE). A normal echocardiogram does not rule out a diagnosis of pericarditis.
Computed tomography (CT) scan/magnetic resonance imaging (MRI)
CT and MRI have the advantages of a larger field of view and excellent imaging of anatomy that is not possible with echocardiography. They also have high soft-tissue contrast. In most patients, they provide excellent images of the pericardium, including thickness, the presence of effusions and any pericardial lesions.
Pericardiocentesis and biopsy
Rarely required, pericardiocentesis and biopsy may be indicated in selected cases. Indications include suspected serious underlying pathology, such as a bacterial, tuberculous or neoplastic pericarditis, when cardiac tamponade is present or in chronic or recurrent cases for diagnostic purposes.