CHAPTER 41
Metabolic effects of tumours
Wassif S. Wassif; James E. East
CHAPTER OUTLINE
Multiple endocrine neoplasia type 1
Multiple endocrine neoplasia type 2
Other familial syndromes associated with multiple endocrine neoplasia
METABOLIC CHANGES IN MALIGNANCY
ENDOCRINE SEQUELAE OF TUMOURS AND THEIR TREATMENT
INTRODUCTION
Tumours may exert metabolic effects on the host via a wide range of mechanisms. Some of the metabolic derangements related to tumours are directly related to a hormone or other substance that the tumour secretes. Such hormone secretion can be appropriate to the cell line from which the tumour originates or may be inappropriate or ‘ectopic’. In the last decade, molecular genetic techniques have helped characterize tumours and explain how neoplastic tissues can secrete hormones that are not typically associated with them. Metabolic derangements can also arise non-specifically, in the absence of hormone secretion or immunological phenomena, for example owing to tumour burden compromising host metabolism or the effects on the host of rapid cell turnover. Autoimmune reactions evoked by tumour antigens may also give rise to systemic effects and are increasingly being implicated in the causation of various tumour-related or paraneoplastic, syndromes. The recognition and characterization of the antibodies to shared tumour and host antigens that play a prominent role in paraneoplastic syndromes have advanced their use in both diagnosis and treatment.
Scientific advances have rapidly opened up new possibilities for the management of tumours, even in their advanced stages, where formerly only observation and palliation was possible. Clinical biochemistry has a key role in both diagnosis and monitoring treatment.
NEUROENDOCRINE TUMOURS
Neuroendocrine cells can be identified by their production of a neurotransmitter, neuromodulator or neuropeptide hormone. Neuroendocrine tumours (NETs) are distinguished by their ability to secrete peptides causing characteristic endocrine syndromes. The gastroenteropancreatic system has diffuse neuroendocrine components and NETs most commonly arise from these tissues, more than 50% being carcinoid tumours.
Carcinoid tumours
Carcinoids are NETs that arise from enterochromaffin cells, principally in the intestine (~ 80% in the ileum), the main bronchi and, rarely, other tissues including the ovaries, thymus, pancreas and thyroid. Carcinoid tumours all show a similar pattern of neuroendocrine expression and may contain or secrete amines, peptides or prostaglandins. The carcinoid syndrome is the clinical result of systemic release of these substances, particularly serotonin.
Clinical presentation
The incidence of carcinoid tumours found at post-mortem has been estimated to be as high as 1 in 150, whereas carcinoid syndrome only occurs in approximately 1 in every 50 000 individuals, indicating that most tumours appear to be are non-secretory. The products of primary intestinal carcinoids are metabolized in the liver prior to release into the systemic circulation: the development of carcinoid syndrome in patients with a gastrointestinal tumour therefore indicates the presence of hepatic metastases. Carcinoid tumours at bronchial (~10% of the total) and other sites secrete products directly into the systemic circulation and can be associated with carcinoid syndrome without the presence of metastases. The main clinical features of carcinoid syndrome are flushing and diarrhoea (Box 41.1). The diarrhoea tends to be secretory in type and weight loss is common. Patients who flush repeatedly may, in time, develop a cyanotic telangiectatic appearance, which persists. Wheezing may occur, caused by transient bronchoconstriction, which is most commonly associated with bronchial carcinoids. Hypertension is not typically a feature of carcinoid syndrome: hypotension is more common. Carcinoid syndrome is also associated with fibrosis; the bowel, retroperitoneum (and hence ureters), lungs and heart can all be affected. Fibrosis is thought not to be linked directly to serotonin but to mitogenic growth factors that drive fibroblast proliferation. Tryptophan is a precursor of both serotonin and nicotinic acid. Thus, in carcinoid syndrome, excess production of serotonin may lead to nicotinamide deficiency, manifesting as pellagra with dermatitis of sun-exposed areas. Carcinoid crisis has been described in certain circumstances including induction of anaesthesia, liver biopsy and following combination chemotherapy. It is manifest as extreme flushing, explosive diarrhoea, labile blood pressure and cardiac rhythm irregularities, including asystole.
Metabolism of serotonin
The rate limiting step in the biosynthesis of serotonin is the hydroxylation, by tryptophan hydroxylase, of tryptophan to 5-hydroxytryptophan (5HTP); this is subsequently decarboxylated to yield 5-hydroxytryptamine (serotonin, 5HT) (see Fig. 41.1). 5-Hydroxytryptamine is stored in neurosecretory granules or secreted into the bloodstream. After secretion, some is taken up into platelets. Oxidative deamination, catalysed by monoamine oxidase and aldehyde dehydrogenase, inactivates 5HT to 5-hydroxyindoleacetic acid (5HIAA), which is excreted in the urine.
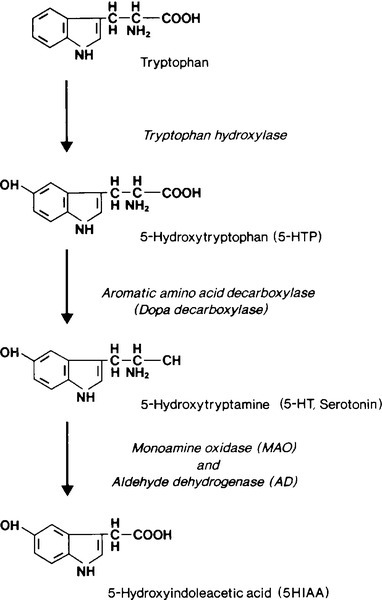
FIGURE 41.1 Pathway of synthesis and metabolism of 5-hydroxytryptamine.
The normal excretion of 5-HIAA is < 50 μmol/24 h. In patients with typical carcinoids, 99% of metabolized 5HT and 5HTP are excreted as 5HIAA. Urinary 5HIAA excretion is typically in the range 150–1500 μmol/24 h, generally exceeds 500 μmol/24 h and occasionally is as high as 3000 μmol/24 h. Carcinoid tumours of the colon and rectum do not contain the hydroxylase or decarboxylase enzymes and so do not form 5HTP or 5HT. A small number of patients have ‘atypical’ tumours. These patients excrete large amounts of 5HTP and 5HT in their urine. It is believed that these tumours, usually bronchial, are deficient in dopa decarboxylase and cannot convert 5HTP to 5HT so that the former is secreted into the bloodstream. Some of the 5HTP is converted to 5HT and subsequently to 5HIAA in extrarenal sites; some is decarboxylated in the kidneys and excreted into the urine as 5HT, and some escapes decarboxylation and is excreted directly into the urine. Thus, patients with atypical carcinoid tumours have a marked increase in 5HTP and 5HT excretion and only a moderate increase in 5HIAA excretion.
Even in those patients whose tumours produce predominantly 5HTP, urinary 5HIAA constitutes 50–60% of total urinary 5-hydroxyindoles and increased values are found in almost all patients.
Laboratory investigation
The most sensitive indicator of the turnover of serotonin and its metabolites is measurement of 5HIAA in a 24 h urine sample. A number of dietary substances can interfere in the measurement of 5HIAA and should be avoided for three days prior to and during the collection of the urine sample (see Box 41.2). The test is not completely specific and a negative result is to be expected in patients with non-secretory tumours and those of the hindgut that do not produce 5HT.
Chromogranin A (CgA) is a 48 kDa protein distributed in dense core granules of neuroendocrine cells, the highest plasma concentrations being found in patients with carcinoid tumours. It has been reported to have a sensitivity of 75–85% and a specificity of 84–95% in the diagnosis of carcinoid syndrome. It is not dependent upon serotonin secretion and is therefore particularly useful in patients with non-secretory or atypical carcinoids and as its concentration reflects tumour burden, it can be used as a marker for assessing response to treatment. False positive results can be seen in hepatic and renal failure, atrophic gastritis, proton pump inhibitor therapy, and inflammatory bowel disease. Prostatic cancers, which may contain a significant neuroendocrine component, myeloma, exercise, trauma and hypertension may also increase CgA concentrations.
Diagnostic imaging
In isolated carcinoid tumours, where surgical excision is a management option, anatomical localization may be useful. Conventional imaging modalities include computed tomography (CT), magnetic resonance imaging (MRI) and positron emission spectroscopy (PET). A more specific option is the use of somatostatin receptor scintography, e.g. using 111In-DTPA-pentetreotide (Octreoscan™), which binds to somatostatin receptors on the tumour.
Treatment
Circumscribed tumours may be removable by surgical resection. For patients with significant hepatic metastatic disease, radiofrequency ablation or hepatic embolization, alone or in combination with intra-arterial chemotherapy (chemoembolization), while not curative, may provide significant palliation. Somatostatin analogues have been used effectively in the management of the symptoms of carcinoid syndrome.
MULTIPLE ENDOCRINE NEOPLASIA
Multiple endocrine neoplasia (MEN) syndromes are disorders characterized by pathological hyperfunction of two or more endocrine organs. They have been classified into two distinct disorders: MEN type 1 (MEN1) and MEN type 2 (MEN2) with MEN2 further subdivided into three main variants (see Box 41.3).
Two distinct genetic defects contribute to tumourigenesis in MEN syndromes: in MEN1, inactivation (loss of function) of a tumour suppressor gene is thought to be responsible, while MEN2 is caused by overactivation (gain of function or overexpression) of a proto-oncogene. A tumour suppressor gene restrains cell proliferation and tumours are stimulated by inactivating mutations or deletions in such a gene, which render the gene product either absent or nonfunctional. In contrast, a proto-oncogene (the normal unmutated version of an oncogene) can be converted to an oncogene, which can cause cell proliferations or deregulated cell growth when overexpressed.
Multiple endocrine neoplasia type 1
Multiple endocrine neoplasia type 1 (MEN1) typically becomes manifest after the first decade of life, with most men and women developing symptoms in the fourth and third decades, respectively. Typically, MEN1 tumours appear two decades earlier than isolated endocrine tumours.
Parathyroid disease
Primary hyperparathyroidism (HPT) is the most frequent endocrinopathy in MEN1 and is the most common reason for the disease to come to the attention of physicians. It occurs in 90% of affected individuals between 20 and 25 years of age, rising to nearly 100% by the age of 50 years. However, MEN1 itself is rare and accounts for only 2–4% of patients with HPT. There is no sex difference in MEN1 prevalence compared with a ratio of 3:1 females:males in sporadic hyperparathyroidism. Hyperparathyroidism is often found during the second decade of life during screening of immediate family members of an index patient with proven MEN1. The pathological features are those of diffuse or asymmetrical hyperplasia, with all four glands being involved (although perhaps not all to the same degree) or with multiple adenomas. The investigation and management of HPT is discussed in Chapter 6.
Gastroenteropancreatic neuroendocrine tumours
Gastroenteropancreatic (GEP) neuroendocrine tumours are the second most common tumours in MEN1, with some 60% of patients being affected. They usually present between the second and fifth decades, unless diagnosed earlier by screening. Approximately half of the tumours are gastrinomas. The Zollinger–Ellison syndrome (ZES) is severe, intractable, multiple and recurrent peptic ulcer disease caused by a gastrin secreting tumour, which can be located in the duodenum or pancreas. The majority of gastrinomas associated with MEN1 are in the duodenum, where they are often small (< 0.5 cm in diameter) and multiple. Since gastrin secreting cells are not normally found in the duodenum or pancreas, gastrinomas in these sites should be regarded as ectopic and potentially malignant, irrespective of their histological grade. The clinical syndrome does not differ from that seen with non-MEN1 gastrinomas. The extreme hypersecretion of gastric acid may be associated with inactivation of pancreatic lipase, resulting in fat malabsorption and steatorrhoea. Hyperparathyroidism in MEN1 can exacerbate hypergastrinaemia. The diagnosis can be made by demonstrating increased gastric acid secretion with simultaneously elevated plasma gastrin concentrations. Secretin infusion (2 U/kg) may cause augmented gastrin production in those patients whose basal values are equivocal. Excellent treatments are now available for the medical management of peptic ulcer disease using histamine H2 receptor antagonists or proton pump inhibitors.
Other GEP NETs include insulinomas, vasoactive intestinal peptide secreting tumours (VIPomas) and glucagonomas. Their clinical features are discussed further in Chapter 12.
Chromogranin A is useful, particularly as a marker of midgut tumours, in the investigation of GEP NETs. Cocaine- and amfetamine-regulated transcript (CART), is a 116 amino acid peptide widely distributed in nervous and endocrine tissues and its physiological roles may include regulation of feeding and response to psychological stress. It is produced by various islet cell tumours and is of particular use as a marker in the investigation of NETs of pancreatic origin. Measurement of a screening panel of gut hormones may also be useful.
Pituitary tumours
The true prevalence of anterior pituitary tumours in MEN1 is unclear. Between 30% and 50% of patients with MEN1 develop anterior pituitary tumours. Whilst some are non-functional, approximately 60% secrete prolactin, 25% secrete growth hormone and 5% secrete ACTH. Occasionally, excess secretion of GH and cortisol in MEN1 is the result of ectopic secretion of GHRH and ACTH, respectively, e.g. by a pancreatic islet or carcinoid tumour. It is important to identify such patients in order to ensure appropriate therapy.
The investigation and management of pituitary tumours in patients with MEN1 is similar to that for other pituitary tumours and is discussed in detail in Chapter 18.
Foregut carcinoid tumours
Carcinoid tumours occur more frequently with MEN1 than in the general population; some 10% of MEN1 patients are affected. In contrast to sporadic carcinoid tumours, which are predominantly derived from the midgut and hindgut, MEN1 carcinoid tumours are usually derived primarily from the foregut. Foregut carcinoids rarely secrete serotonin, peptide hormones or calcitonin and are usually considered as clinically non-functional.
Adrenal tumours
Adrenal cortical lesions are common in MEN1: 20–40% of patients are affected, mostly with bilateral tumours. However, the majority are non-functional, clinically silent and rarely require treatment. Cushing and Conn syndromes are rare in MEN1.
Tumourigenesis in MEN1
MEN1 is the result of an inactivating mutation in the MEN-1 gene, a tumour suppressor gene located on chromosome 11q13. It consists of 10 exons and encodes a 610-amino acid nuclear protein termed menin. Menin interacts with diverse groups of transcription factors and coregulators, including JunD, suggesting a role in gene transcription, DNA replication and cell cycle control. Genetic mapping studies indicate somatic loss of heterozygosity in accord with the ‘two hit’ hypothesis. The first hit is a genetic mutation rendering the subject heterozygous for the MEN-1 mutant gene and predisposed to tumour development. A somatic inactivation of the unaffected allele then occurs (second hit) leading to the development of MEN1-associated tumours.
Diagnosis of MEN1
Occasionally, MEN1 may occur without a recognized family history. Some may be patients whose parents died before developing any manifestations of MEN1: a few may have MEN1 caused by two somatic mutations as described above. Sometimes it is difficult to make the distinction between sporadic tumours and MEN1, but an earlier onset and tumour multiplicity in the same organ increase the likelihood of MEN1. The dominant mode of inheritance, the occurrence of asymptomatic gene carriers and the risk of developing malignant disease make it essential to establish carrier status in all relatives whenever the diagnosis of MEN1 has been made in one family member.
Genetic screening
The identification of the MEN-1 gene (locus 11q13) has opened the possibility of genetic testing. However, over 1300 distinct germline mutations have been identified and there is a lack of genotype–phenotype correlation. Thus, genetic testing can be time consuming, arduous and expensive and a definitive result does not negate the requirement for further biochemical screening of carriers. However, for those found not to be carrying the mutant gene, there is no further need for surveillance or family screening.
Biochemical screening
Biochemical screening continues to play an important role in carrier ascertainment whenever genetic testing is not possible or has failed to detect germline mutations (see below) and is important for tumour surveillance in those known to have the affected gene.
The MEN1 syndrome rarely develops before the age of five or after the age of 70, and so screening should be performed every three years after the age of five until the age of 70 and at longer intervals thereafter. Over 95% of affected individuals will have been identified by the fourth decade. The measurement of serum calcium is a simple, cheap and reliable screening test; additional biochemical investigations include measurement of serum prolactin, gastrin (fasting) and CgA. Screening of patients with apparently sporadic pancreatic endocrine tumours for evidence of MEN1 is probably justified, especially in those with gastrinomas or insulinomas.
Surveillance of MEN1 patients and carriers
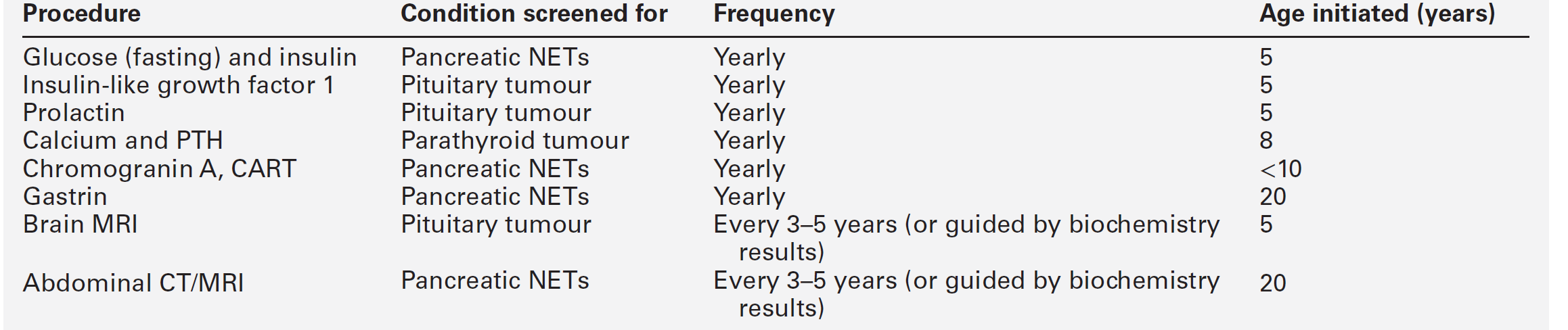
Once mutant MEN1 gene carriers have been identified by biochemical or genetic testing, they should be monitored rigorously to detect the development of new tumours or tumour recurrence. The age-related penetrance is virtually zero below the age of five years, rising to > 50% by 20 years, and > 95% by 40 years. Surveillance should include a regular history and examination focused on the clinical features of MEN1-associated tumours in combination with biochemical tests and imaging (Table 41.1). Although periodic screening for endocrine tumour expression in MEN1 appears to reduce morbidity and mortality as a result of early tumour recognition, this has not been proven.
Multiple endocrine neoplasia type 2
Multiple endocrine neoplasia type 2 (MEN2) involves two or more of medullary thyroid carcinoma (MTC), phaeochromocytoma and parathyroid tumours (see Box 41.3). Medullary thyroid cancer is a universal feature of MEN2 and is often the first tumour to present. MEN2A is the most common subtype, accounting for 80–90%. Patients with MEN2B, in addition to endocrine abnormalities, have a characteristic marfanoid habitus often with scoliosis and pes cavus. The presence of mucosal neuromas is characteristic (see Fig. 41.2). Some kindred appear to have a third form of MEN2, isolated familial MTC, but care should be taken that other features of MEN2 are not overlooked.
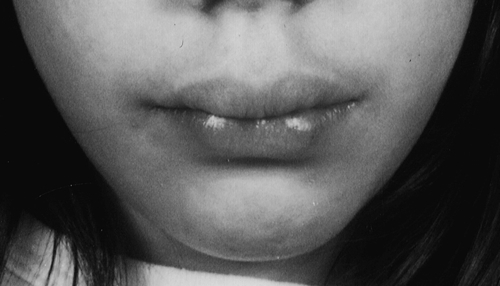
FIGURE 41.2 Typical features of a patient with MEN2B. (Photograph courtesy of Professor A G McGregor)
Germline activating mutations of the RET (rearranged during transfection) proto-oncogene, which is located on chromosome 10 (10q.11.2), have been identified in all three types of MEN2.
Diagnosis
RET germline mutation testing now forms the basis for carrier identification in MEN2 families. Medullary thyroid carcinomas secrete calcitonin, measurement of which serves as a useful tumour marker to assist in assessment of the response to therapy and for the detection of persistent or recurrent disease. Plasma carcinoembryonic antigen (CEA) concentration may be raised in medullary thyroid cancer and prove a useful additional tumour marker.
Before the establishment of DNA analysis for family screening for MEN2, measurement of calcitonin was often performed for this purpose, either on a basal sample or in response to stimulation with pentagastrin or calcium. Every effort should be made to identify gene carriers among the relatives of patients known to have MEN2 in order to offer prophylactic thyroidectomy against MTC. Strong genotype–phenotype correlation exists in MEN2, which can guide the timing of surgery and the screening protocol for optimal detection of parathyroid tumours and phaeochromocytomas.
Imaging
Ultrasonography of the thyroid may be useful. However, when invasion to surrounding structures or distant metastases are suspected, other imaging modalities such as MRI or CT scanning are necessary. 99mTechnetium-sestamibi scan is a more sensitive technique for the localization of metastases within the neck and chest. Scintigraphic imaging using 123I- or 131I-anti-CEA monoclonal antibodies is a promising development for the detection of occult disease and in selecting candidates for repeat neck exploration.
Treatment
If a mutation is found, surgical removal of the thyroid is recommended to prevent tumour development. Postoperatively, patients should be treated with suppressive doses of L-thyroxine. Since calcitonin gene expression is activated in inflamed or septic tissue, calcitonin concentration may remain elevated for 3-6 months after thyroid surgery. It is therefore prudent to delay calcitonin measurement for three months after thyroidectomy. Early and prophylactic total thyroidectomy has probably lowered the mortality from MTC to < 5%, well below the cancer-associated mortality in MEN1. RET tyrosine kinase inhibitor drugs are currently being developed and may have a role in the treatment of MEN2-related malignancies in the future. Follow-up of MTC is by measurement of calcitonin and CEA.
Surveillance
Genotype–phenotype correlation in MEN2 may be useful in guiding strategies for surveillance. However, a suggested scheme is that after the age of eight years PTH and calcium should be measured for detection of parathyroid tumours and for detection of phaeochromocytoma plasma free metanephrines and/or urinary fractionated metanephrines should be measured, with imaging using MRI or CT every 3–5 years or if biochemistry is abnormal. Patients with familial MTC require only periodic monitoring with imaging if biochemistry is abnormal.
Other familial syndromes associated with multiple endocrine neoplasia
Patients presenting with an endocrine tumour should have a full family history taken, looking for evidence of an inherited syndrome. Single, unilateral and late onset tumours are more likely to be sporadic. In contrast, a familial cause is more is likely in a patient who presents early, particularly if there is a family history or if there are multiple tumours or associated cutaneous features.
A further type of MEN (MEN4) has been described caused by mutations in cyclin-dependent kinase inhibitor genes. The exact phenotype is, as yet, poorly defined but includes parathyroid, and sometimes pituitary, tumours and other endocrine features.
Phaeochromocytoma is predominately a sporadic disorder. However, in 25% of patients, phaeochromocytoma may be familial or associated with a number of autosomal dominant hereditary syndromes. Examples of such syndromes are neurofibromatosis type 1 (NF1), von Hippel–Lindau syndrome and paraganglioma syndrome 1, 3 and 4. The cardinal features of NF1 are neurofibromas and dermal café-au-lait skin pigmentation. Other features include skeletal manifestations, vascular stenoses and a variety of endocrine tumours including phaeochromocytoma, somatostatin-producing carcinoid tumours and medullary thyroid cancer. The causative gene encodes a GTP-activating protein, neurofibromin. Von Hippel–Lindau syndrome is a multisystem cancer syndrome associated with retinal and cerebral haemangioblastomas, renal cysts and renal cell carcinomas, phaeochromocytomas and islet cell tumours.
Paragangliomas (PGL) are tumours derived from the sympathetic and parasympathetic nervous system. About half are familial. The sympathetic-associated PGL arise from the adrenal medulla or from the sympathetic ganglia. They are generally functional and secrete excess catecholamines. Hereditary PGL are characterized by the development of highly vascularized, slowly growing non-chromaffin tumours arising in the parasympathetic ganglia. Parasympathetic-derived PGL usually develop in the head and neck, most commonly at the bifurcation of the carotid artery, and are usually non-functional. Intraabdominal and thoracic catecholamine-secreting PGL are currently being reffered to as phaeochromocytomas.
Paragangliomas syndromes have been genetically characterized as PGL 1, 3 and 4 and are caused by mutations in the succinate dehydrogenase (SDH) subunit D, C and B genes, respectively (SDHD, SDHC and SDHB); the fourth subunit coded by the SDHA gene is not associated with hereditary PGL.
METABOLIC CHANGES IN MALIGNANCY
Introduction
Metabolic changes may herald the clinical onset of malignant disease and are often the cause of considerable morbidity and mortality in patients with cancer. During the rapid growth phase of a neoplasm, the nutritional and metabolic state of the host may become modified in favour of the neoplasm. The tumour-related metabolic changes may be a direct consequence of the presence of the tumour, or be secondary to secretory products of the tumour. These may either be normal secretory products of the cells of origin or substances not normally produced by them. Such metabolic alterations may, on occasions, be useful in the diagnosis and management of malignancy, that is, they act as tumour markers (see Chapter 42). In this section, some of the metabolic consequences of malignant disease are reviewed.
Paraneoplastic syndromes
A paraneoplastic syndrome is one that is attributable to a neoplasm, not as a direct result of local or metastatic presence of the tumour cells, but owing to substances secreted from them or autoantibodies produced against them. Such disorders may broadly be divided into neurological, humoral and other paraneoplastic syndromes.
Neurological paraneoplastic syndromes
Various neurological paraneoplastic syndromes exist. Some have a classic presentation, e.g. Lambert–Eaton, syndrome, which resembles myasthenia gravis and is associated with small cell carcinoma of the bronchus. Some paraneoplastic syndromes appear to have an autoimmune basis; thus Lambert–Eaton syndrome is associated with the presence of anti-voltage gated calcium channel (VGCC antibodies). The majority of neurological paraneoplastic syndromes are thought to have an autoimmune basis, with an immune response being directed against antigens expressed by the tumour. The resulting antibodies then cross react with components of the nervous system. These antibodies can be detected in blood. In a patient with a neurological presentation, the presence of certain well characterized onconeural antibodies (anti-Ho, Yo, Ma2, CRMP-5, amphiphysin and Ri) enables the diagnosis of a paraneoplastic syndrome to be made whether or not a neoplasm has been detected. Neurological paraneoplastic syndromes and associated tumours and antibodies are discussed further in Chapter 36.
Humoral paraneoplastic syndromes
Humoral paraneoplastic syndromes arise from the secretion of substances from a tissue that does not normally produce them. The term ‘ectopic hormone production’ is used to refer to hormones secreted from sites that are not their physiological origin, in amounts sufficient to cause clinical effects. The ectopic secretion of such hormones tends not to be subject to the usual mechanisms of endocrine regulation and characteristic responses to dynamic function testing may be useful in diagnosis. The hormone itself may sometimes circulate in a molecular form distinct from that secreted from the eutopic source. Treatment of the tumour leads to resolution of the endocrine effects, but these reappear if the tumour recurs, accompanied by a demonstrable concentration of the hormone in serum. Some tumours are particularly associated with such ectopic hormone production, e.g. small cell lung cancer. Various theories have been developed in an attempt to explain the phenomenon of ectopic hormone production by tumours. One theory suggests that some fundamental change occurs at the genomic level that allows novel gene expression for that tissue. Thus, neoplastic change would cause certain genes to be switched on and others to be switched off. Another theory suggests that the neoplastic cell is derived from a stem cell that was capable of expressing the gene at an early developmental stage but is later suppressed, and that following neoplastic transformation, the cell undergoes dedifferentiation and regains some of its developmental properties. It has also been proposed that some widely expressed genes that are transcribed but not translated under normal conditions may, due to neoplastic transformation, be amplified and translated due to the action of different promoters.
Adrenocorticotropin
Ectopic secretion of adrenocorticotropin (ACTH) can occur with a variety of tumours, but is particularly associated with small cell carcinoma of the bronchus. Its clinical features and diagnosis are discussed in Chapter 18. The most common cause of ectopic ACTH production is the expression of the proopiomelanocortin (POMC) gene by tumour tissue. The POMC gene encodes for the 31-kDa precursor of ACTH, β-endorphin and melanocyte stimulating hormone. The principal source of POMC-derived peptides in the body is the pituitary gland; however, POMC immunoreactivity and POMC mRNA can be found in almost all tissues. The mRNA species produced in non-pituitary tissue is shorter than that expressed in pituitary tissue, which is thought to be due to the action of different promoters. One theory is that many tumours express the POMC gene, but what determines whether the tumour becomes an ectopic ACTH-secreting tumour is the switching of activity to the more conventional pituitary promoter. Excessive ACTH secretion can also be the result of ectopic secretion of corticotrophin-releasing hormone.
Vasopressin
The synthesis of vasopressin, resulting in the paraneoplastic syndrome of inappropriate anti-diuresis is particularly associated with small bronchial carcinoma, although can occur with other tumours (see Table 41.2). It may present acutely with severe symptomatic, hyponatraemia. This condition is discussed further in Chapter 4. Vaptans, agents which antagonize the action of vasopressin via blockade of the vasopressin receptor, have been used in management.
TABLE 41.2
Hormones arising from tumours derived from tissue not classically associated with their secretion
| Hormones | Associated tumour types |
| Adrenocorticotropic hormone, lipotrophin, melanocyte-stimulating hormone, endorphin, encephalin, other pro-opiomelanocortin fragments | Small cell carcinoma of bronchus Adenocarcinoma of bronchus Bronchial carcinoid tumour Thymic carcinoid tumour Medullary thyroid carcinoma Islet cell carcinoma of pancreas Phaeochromocytoma Gut, prostate, neurogenic and parotid tumours |
| Atrial natriuretic peptide | Small cell carcinoma of bronchus |
| Arginine vasopressin | Small cell carcinoma of bronchus Pancreatic carcinoma Breast carcinoma Thymic tumour |
| Calcitonin (clinically silent) | Small cell carcinoma of bronchus Bronchial carcinoid tumour |
| Calcitonin gene-related peptide | Small cell carcinoma of bronchus |
| Corticotrophin releasing hormone | Bronchial carcinoid tumour |
| Erythropoietin | Small cell carcinoma of bronchus Uterine fibroma Cerebellar haemangioblastoma Phaeochromocytoma Ovarian tumour |
| Gastrin releasing peptide | Small cell carcinoma of bronchus Medullary thyroid carcinoma |
| Glucagon | Non-β islet cell tumour Anaplastic lung carcinoma Renal adenocarcinoma |
| Growth hormone | Bronchial carcinoma Gastric carcinoma |
| Growth hormone releasing hormone | Pancreatic islet cell tumour Phaeochromocytoma |
| Human chorionic gonadotrophin | Renal and bladder carcinomas Breast carcinoma Prostatic carcinoma Melanoma Gynaecological carcinomas Head and neck carcinomas Lymphoma |
| Human placental lactogen | Lung and testicular tumours |
| Insulin-like growth factor 2 | Mesodermal and mesenchymal tumours |
| Neurotensin | Pancreatic endocrine tumours Carcinoid tumour |
| Parathyroid hormone-related peptide | Squamous cell carcinoma of head and neck, oesophagus cervix, bronchus Breast and ovarian carcinomas Carcinoma of pancreas Renal and bladder carcinomas Multiple myeloma Histiocytic lymphoma |
| Prolactin | Anaplastic lung carcinoma Small cell carcinoma of bronchus Renal adenocarcinoma |
| Renin | Renal adenocarcinoma Adenocarcinoma of pancreas Small cell carcinoma of bronchus Adenocarcinoma of lung Ovarian carcinoma Adrenocortical carcinoma |
| Somatostatin | Small cell carcinoma of bronchus Duodenal somatostatinoma Extra-adrenal paraganglionoma Phaeochromocytoma Medullary thyroid carcinoma |
| Vasoactive intestinal polypeptide | Bronchial carcinoid tumour Ganglioneuroblastoma Phaeochromocytoma Non-β islet cell tumour Small cell carcinoma of bronchus |
PTH-related peptide
Hypercalcaemia complicating malignancy is common. Mechanisms include prostaglandin-mediated bone resorption, osteoclast activating factors, e.g. tumour necrosis factors, and the action of PTH-related peptide (PTHrP), a protein with N-terminal amino acid homology with PTH. PTH-related peptide is not detected by most modern PTH assays which measure ‘intact’ PTH, hence the finding of a low PTH with a low phosphate in a hypercalcaemic patient is suggestive of hypercalcaemia secondary to PTHrP. Hypercalcaemia as a feature of malignancy is discussed further in Chapter 6.
Tumour-induced osteomalacia
Tumour-induced osteomalacia (TIO) is a paraneoplastic syndrome resulting from the secretion of fibroblast growth factor 23 (FGF-23) by mesenchymal tumours. There may be a relatively long history of musculoskeletal symptoms such as bone pain and muscle weakness and, in children, rickets and growth failure may be observed. Classic biochemical features are normal plasma calcium and PTH concentrations and hypophosphataemia with renal phosphate wasting. Plasma 1,25-dihydroxyvitamin D concentration is low and FGF-23 elevated. The only effective treatment for TIO is resection of the tumour. Medical management is with phosphate supplements and calcitriol.
Other paraneoplastic syndromes and features of malignant disease
Haematological sequelae
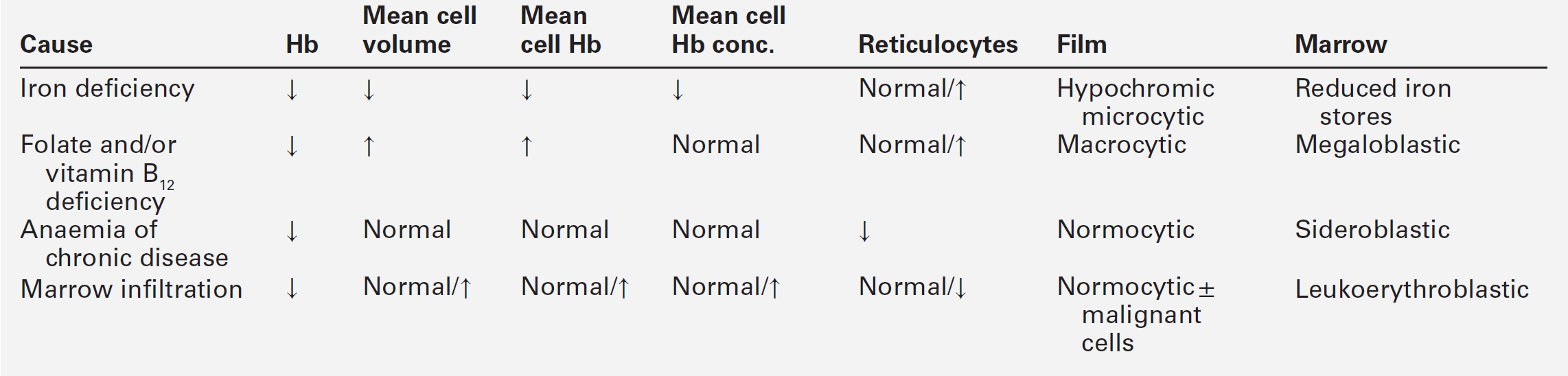
Anaemia is the most common haematological abnormality encountered in malignant disease. It may have various causes with differing laboratory findings (Table 41.3). Clinical and laboratory features are discussed further in Chapter 27; examples include anaemia of chronic disease, and folate deficiency resulting from altered metabolism or requirements due to the presence of the tumour. Autoimmune haemolytic anaemia can occur, most commonly in patients with B cell lymphoproliferative disorder, and is thought to be caused by an immune response evoked to antigens common to both the tumour cells and red blood cells. In contrast to idiopathic autoimmune haemolytic anaemia, response to corticosteroid therapy is uncommon. Pure red cell aplasia has been described mainly with thymomas, but also in association with adenocarcinomas, squamous cell carcinomas, anaplastic tumours and, rarely, with lymphoproliferative diseases. The mechanism is thought to be a T cell-mediated phenomenon. Microangiopathic haemolytic anaemia is a serious complication of certain malignancies and occurs when red cells are damaged by passage through blood vessels that have been distorted by either tumour or fibrin deposits. The blood film is characterized by the presence of schistocytes and polychromasia and there is biochemical evidence of haemolysis (increased plasma LDH activity, reduced haptoglobin concentration and haemosiderinuria). In contrast, some tumours, particularly of the kidneys, may be associated with erythrocytosis: this is thought to be the result of prostaglandin-mediated erythropoietin activity.
Hyperuricaemia
In certain patients with malignancy, predominantly those with leukaemia and lymphoma in whom there is a rapid turnover of cells, or massive cell lysis caused by cytotoxic agents (tumour lysis syndrome), hyperuricaemia can occur. Radiotherapy can produce a similar clinical scenario. Other features of the tumour lysis syndrome include hyperkalaemia, hyperphosphataemia and hypocalcaemia.
The increased filtration of urate and the increasing acidity and concentration of the tubular fluid leads to precipitation and renal obstruction. Adequate hydration is important prior to combination chemotherapy in those at risk and it is standard practice to administer allopurinol, a xanthine oxidase inhibitor, to reduce urate formation. Rasburicase (recombinant urate oxidase) catalyses the conversion of urate to allantoin, which is five to ten times more soluble than urate, and is helpful in reducing plasma urate in severely affected patients. Rasburicase should be avoided in patients with glucose 6-phosphate dehydrogenase deficiency as one of the major by-products of the enzymatic reaction is hydrogen peroxide, which may precipitate haemolysis. Preservation of samples at 4°C, or stabilization with perchloric acid, has been recommended prior to analysis of urate in patients receiving rasburicase, as degradation of uric acid can continue in collection tubes, resulting in a spuriously low urate concentration.
A number of other characteristic paraneoplastic syndromes exist. These include immune-mediated renal disease, thrombosis associated with antiphospholipid antibodies and finger clubbing and hypertrophic pulmonary osteoarthropathy attributed to the action of platelet-derived growth factor.
Cancer cachexia
Weight loss is common in cancer patients and in many, is the presenting feature. Cancer cachexia is a hypercatabolic state characterized by loss of body weight associated with a reduction in adipose tissue and muscle mass and loss of appetite. Weight loss can cause severe morbidity and decrease in quality of life and may be associated with an adverse prognosis and increased susceptibility to side-effects during treatment of the malignancy itself. The aetiology of the weight loss is complex: it appears to involve a combination of increased catabolism and decreased anabolism. The physical presence of the tumour may also contribute to weight loss, e.g. by causing obstruction in the gastrointestinal tract.
The regulation of appetite involves both central and peripheral mechanisms, which are integrated in the hypothalamus. There is no evidence that hypothalamic dysfunction per se is responsible for the cachexia of malignant disease. However, it has been suggested that malignant tissue causes anorexia by the secretion of biologically active molecules that depress feeding by interfering with central control mechanisms, e.g. serotonin from carcinoid tumours and bombesin from small cell bronchial cancers. Cytokines, particularly TNFα and IL-6, are thought to be important in the loss of appetite. They arise from tumour cells and as part of the host immune response; they are able to cross the blood–brain barrier and interact with mechanisms affecting appetite. Iatrogenic factors related to the treatment of cancer can also contribute to weight loss. Chemotherapy often causes nausea and vomiting, and radiotherapy can have short-term effects on the gut (e.g. mucositis) and cause long-term complications, e.g. enteritis that lead to malabsorption.
Changes in metabolism
Although reduced energy intake is common to both starvation and cancer cachexia, there are marked differences between the metabolic changes seen in the two states (see Table 41.4).
TABLE 41.4
Metabolic changes in cancer cachexia and starvation
| Starvation | Cachexia | |
| Acute phase response | No | Yes |
| Appetite | Increased | Decreased |
| Metabolic rate | Decreased | Increased |
| Skeletal muscle mass | Maintained | Decreased |
| Adipose tissue | Decreased | Decreased |
| Liver size | Decreased | Increased |
Many patients with cancer are mildly hypermetabolic with a resting energy expenditure greater than normal. However, only a fraction of the increased metabolic rate can be accounted for by the tumour tissue itself. Suggested mechanisms for cancer cachexia are shown in Figure 41.3.
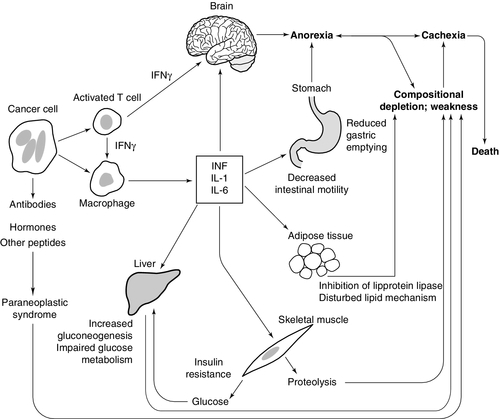
FIGURE 41.3 Possible mechanisms of cancer cachexia.
Many tumours produce energy via anaerobic glycolysis, which results in a net loss of ATP, there is partial uncoupling of oxidative phosphorylation and futile cycles may be activated leading to further energy wastage, e.g. nonesterified fatty acids released from adipose tissue are immediately re-esterified. Free fatty acids are oxidized, even when other energy sources are available, and there is increased protein turnover.
Marked derangements of carbohydrate metabolism are a feature of malignancy. They include abnormal glucose tolerance and hyperglycaemia, hypoglycaemia, lactic acidosis, increased glucose turnover and increased glucose transport into tumour cells. Tumour cells have an increased rate of glucose transport, which may be related to an increase in the number of membrane bound glucose transporters. The main energy source for many tumour cells is anaerobic metabolism of glucose, which results in the production of lactic acid. The lactic acid is then reconverted to glucose in the liver via the Cori cycle. This process also consumes energy, since glycolysis produces only two moles of ATP per mole of glucose, whereas gluconeogenesis consumes six moles of ATP. However, decreased hepatic capacity to utilize lactic acid may lead to its accumulation (lactic acidosis).
Glucose intolerance is a common feature of malignancy, affecting some 60% of cancer patients. Occasionally, frank hyperglycaemia is present. There is insulin resistance (at least with respect to effects on carbohydrate metabolism), although insulin receptor binding appears normal. Fasting insulin concentrations are also normal or slightly decreased but there is a decreased response to both endogenous and exogenous insulin. The picture is further complicated by other factors that may be present in cancer patients, i.e. starvation and malnutrition, sepsis, bed rest, dietary effects and drugs.
Abnormal lipid metabolism is a feature of some malignancies. Some tumours utilize lipids in preference to glucose as a major energy source, with mobilization of free fatty acids from the body’s fat stores and a consequent rise in their plasma concentration. Malignant cells may show abnormal lipid composition, particularly with respect to the cholesterol and phospholipid contents of their cell membranes and may synthesize unusual lipids. For example, hepatocellular carcinomas may produce 2-methyloleate. Loss of fat mass is facilitated by a tumour-produced lipid mobilizing factor. This appears to be a proteoglycan similar in structure to zinc α2-glycoprotein, which acts to sensitize adipose tissue to lipolytic stimuli by increasing production of cyclic AMP within adipocytes.
Patients with malignancy often have a wasting syndrome, where both adipose tissue and lean tissue are lost. The muscle wasting is due to increased protein catabolism as well as decreased synthesis. Proteolysis in cachexia is brought about via increased activity of a number of proteolytic pathways. The major mechanism for proteolysis in cancer cachexia is the ATP ubiquitin-dependent proteolytic pathway initiated in response to cytokines such as TNFα. Lysosomal cathepsins and the calcium/calpain pathway are also involved in proteolysis.
Treatment
Although giving nutrition support to patients with cancer cachexia would seem logical, there is little evidence that provision of energy to patients, via either enteral or parenteral routes, results in weight gain or significant improvement in outcome. Pharmacological management of cachexia has included use of appetite stimulants and drugs intended to counteract catabolism or stimulate anabolism. Corticosteroids have been used to stimulate appetite. Their effect is short lived and is not usually accompanied by weight gain. The progesterone analogue megestrol acetate has been shown to be more effective as an appetite stimulant. Various anabolic agents have been tried in cachexia. Growth hormone has proven efficacy in animal models but its use in clinical settings is associated with increased mortality, possibly owing to diversion of amino acids and energy to skeletal muscle away from the acute phase response. Examples of the use of drugs intended to treat cancer cachexia by modification of the immune response, include thalidomide and pentoxifylline, which both inhibit production of TNFα, and eicosapentaenoic acid, found in fish oil, which can reduce lipolysis produced by lipid mobilizing factor. None of these has been shown to be consistently effective.
ENDOCRINE SEQUELAE OF TUMOURS AND THEIR TREATMENT
With the development of successful therapies for specific malignancies, populations of treated patients are surviving for near-normal life spans. Long-term sequelae are becoming more evident as these survivors are being followed-up (Table 41.5). This section focuses on effects on somatic growth and reproductive capacity.

Effects on somatic growth
Somatic growth may be profoundly suppressed in a child affected with, for example, acute lymphoblastic leukaemia. This effect may be mediated by therapy aimed at destroying the malignant cells, including cranial irradiation with subsequent hypopituitarism, direct radiotherapy to bony structures that determine final height, for example the spine, or the administration of pharmacological doses of glucocorticoids and intensive chemotherapy.
The hypothalamus is more susceptible to radiation damage than the pituitary. Growth hormone secretion appears to be particularly vulnerable to the effects of radiation, and, while doses of 18–50 Gy may lead to decreased growth hormone secretion alone, higher doses (> 60 Gy) can cause panhypopituitarism through damage to both the hypothalamus and pituitary. Effects of chemotherapy and glucocorticoids can have significant effects on skeletal development, affecting both final height and bone mineral density. The effects of chemotherapeutic agents on chondrocytes, the extracellular matrix and osteocytes are wide ranging, but evidence for direct effects on chondrocytes’ proliferation and ossification is limited; however, disruption of growth plate activity will result in skeletal growth disturbance. Chemotherapy seems to enhance the growth-suppressing effects of radiotherapy when they are given in combination, possibly by enhancing the effects of radiation on the hypothalamo–pituitary axis. It has been hypothesized that chemotherapy effects may relate to suppression of IGF1 synthesis in the liver and/or its effects on growth plates.
For growth hormone to effect growth optimally, normal rhythms of secretion must be maintained. This will only occur if there is normal hypothalamo–pituitary function. The secretion of growth hormone may be affected directly as a result of radiation-induced damage to the hypothalamus or pituitary, or secondarily, for example, by depression, which can accompany any severe illness and is associated with impaired growth hormone secretion. Thyroid hormone and glucocorticoids are necessary for the efficient biological activity of growth hormone; the secretion of either may be decreased following radiotherapy to the hypothalamo–pituitary unit and lead indirectly to impairment of growth. Pharmacological doses of glucocorticoids suppress growth hormone secretion. Additionally, they cause a degree of functional hypogonadism (hypogonadotrophic hypogonadism). If gonadal steroidogenesis is suppressed, the pubertal growth spurt will not occur and final height will be compromised. Glucocorticoids and gonadal steroids ultimately bring about long bone epiphyseal fusion. Their pharmacological use may hasten this fusion and so prevent the attainment of the genetic potential for final height. Low-dose cranial irradiation can also result in early puberty with long bone fusion before attainment of final height.
Strategies to promote growth in these patients can be divided into two groups: either the use of growth hormone replacement in growth hormone deficient individuals, or the use of GnRH analogues to delay puberty in those in whom it is occurring early. Earlier and more aggressive use of growth hormone in those who are growth hormone deficient has resulted in improvements in final height, with safety data suggesting that this does not lead to an increased risk of tumour recurrence. Caution should be exercised, however, as longer term follow-up data are not yet available to determine whether there is an increase in secondary neoplasms. The use of GnRH analogues in combination with growth hormone therapy is more controversial: only some of the few studies suggest an improvement in final height.
Reproductive consequences of therapy
Disease itself, or radiotherapy or chemotherapy directed against populations of actively dividing cells, may influence reproductive potential. This effect may be mediated by a direct action on the germ cell population or be an indirect effect mediated by suppression of hypothalamic GnRH or pituitary gonadotrophin secretion. Specific guidelines are available that may help to predict doses of radiotherapy that may be tolerated, or drug combinations that are less likely to cause depletion of the germ cell pool. Thus, a chemotherapeutic regimen of adriamycin, bleomycin, vinblastine and dacarbazine (ABVD) is less likely to cause infertility than the alternative regimen of nitrogen mustard, oncovin, procarbazine and prednisone (MOPP) in the treatment of Hodgkin disease, a fact that may influence the choice in a young patient who is anxious to subsequently have a family. As the germ cell pool is greater in younger than older women, the risk of ovarian failure as a result of treatment increases with age. Use of GnRH analogues to suppress oocyte maturation via the hypothalamo–pituitary axis and to make the germinal epithelium quiescent (a premenarche state) has shown some success in protecting ovarian function against chemotherapy in human and animal studies. Other strategies that may be used include oophoropexy (an operation to relocate the ovaries anatomically so that they are shielded from the radiotherapy field) or sperm banking prior to therapy. Freezing ovarian tissue pre-chemotherapy with transplant post-chemotherapy has been shown to be successful at restoring function, including fertility, although this process carries with it the potential risk of the re-introduction of tumour cells.
Radiotherapy can affect the uterus, with a decrease in its volume and changes in its musculature and vascular supply. Embryos or oocytes can be frozen prior to therapy, but success rates for subsequent viable pregnancy are limited by the freeze–thaw process. Furthermore, the time factor involved in stimulating a population of oocytes for retrieval prior to commencement of therapy often militates against this, and ovarian stimulation itself has the potential to increase tumour growth in oestrogen-sensitive tumours.
Some tumours, such as prostatic or breast cancers, are actively stimulated by a particular endocrine milieu. Thus, androgens stimulate the growth of prostatic cancer, while oestrogens may encourage the growth of breast cancer. Direct modulation of the endocrine axis may, in itself, constitute therapy for these particular malignancies. Thus growth of prostatic cancer may be suppressed by orchidectomy or by suppression of the hypothalamo–pituitary–testicular axis by a depot injection of a GnRH analogue. This seeming paradox may be explained by the fact that chronic stimulation with GnRH is associated with downregulation of pituitary receptors and inhibition of gonadotrophin release. Alternatively, the target organ effect of androgens may be blocked by the administration of an anti-androgen such as cyproterone acetate or flutamide. In women, oestrogen effects may be opposed by the administration of an anti-oestrogen, such as tamoxifen, or the use of an aromatase inhibitor such as 4-hydroxyandrostenedione, which blocks the penultimate step in oestrogen biosynthesis.
These patients have specific concerns not just about their reproductive capacity, but also about the likelihood of transmission of the malignancy to their offspring or the risk of damage to their germ cells, making fetal malformation more likely than in the normal population. With the exception of the heritable syndromes, there is no evidence of an increased likelihood of malignancy or an increased risk of fetal malformation in progeny of treated patients at the present time.
CONCLUSION
Tumours can have profound metabolic effects, even when very small. These can be due to the physical presence of the tumour, to the products of its secretion (whether expected on the basis of its tissue of origin or not) or to the body’s response to the presence of the tumour. Metabolic derangements can also occur as a side-effect of treatment, whether this be surgical or with radiotherapy or chemotherapy.