CHAPTER 6
Calcium, phosphate and magnesium
Timothy Cundy; Andrew Grey; Ian R. Reid
CHAPTER OUTLINE
Regulation of calcium metabolism
Biochemical assessment of calcium metabolism
CALCIUM METABOLISM
Biological role of calcium
Calcium is a divalent cation with multiple roles in vertebrate physiology, which can be grouped as either structural or metabolic. Its structural role is in the skeleton where calcium deficiency leads to skeletal disease, either in the form of osteoporosis or osteomalacia. Its metabolic roles are more numerous.
The extracellular concentration of calcium ions influences the threshold for nerve action potentials, a high calcium raising the threshold and a low calcium having the opposite effect. The extracellular calcium concentration appears to alter the gating of sodium and potassium channels, possibly the result of calcium ions being attracted to and ‘screening’ the negative charge on the cell surface in the region of these channels.
Calcium has a key role in intracellular signalling. The depolarization of nerve or muscle cells, or the binding of a hormone or cytokine to its receptor on the surface of other types of cell, results in an increase in cytosolic calcium concentration via either one or both of two mechanisms: the influx of calcium through plasma membrane channels and the release of calcium from intracellular stores (e.g. the sarcoplasmic or endoplasmic reticulum). The latter pathway depends on receptor-activated hydrolysis of inositol phospholipids in the plasma membrane, leading to the release of inositol trisphosphate into the cytosol. This compound binds to a receptor on the endoplasmic reticulum resulting in the release of calcium into the cytosol. The regulation of a variety of cell functions can then follow, some enzymes being directly affected by the cytosolic calcium concentration (e.g. protein kinase C) and others indirectly by the calcium receptor protein, calmodulin. These pathways are integral to muscle contraction, neuroendocrine secretion, cell metabolism and growth.
Distribution of calcium
The total body calcium in a human adult is approximately 1 kg, 99% of which is contained in the skeleton. Approximately 1% of skeletal calcium is freely exchangeable with calcium in the extracellular fluid (ECF). Calcium ions diffuse freely throughout the extracellular space, where their concentration is approximately 1.2 mmol/L. The plasma ionized calcium concentration is the same, but the plasma total calcium is approximately two-fold higher because of protein binding of calcium. Calcium also forms complexes with phosphate, citrate and bicarbonate in plasma and interstitial fluid (Table 6.1). It is the ionized calcium concentration that is physiologically important and closely regulated.
TABLE 6.1
Distribution of plasma calcium
| Fraction | Total (%) |
| Ultrafilterable | 53 |
| Ionized | 47 |
| Complexed | 6 |
| Protein bound | 47 |
| Albumin | 37 |
| Globulin | 10 |
| Total (2.2–2.6 mmol/L) | 100 |
Calcium is principally an extracellular ion; cytosolic concentrations are of the order of 10− 4 to 10− 3 mmol/L. The low intracellular calcium concentration is necessary for calcium to function as an intracellular messenger. It is maintained by calcium pumps and exchangers on cell membranes. The endoplasmic reticulum and the mitochondria also have the capacity to remove calcium from the cytosol.
Calcium fluxes
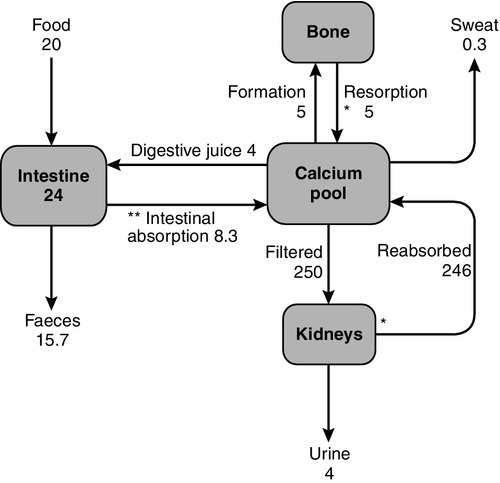
Three principal organs are involved in the body’s handling of calcium: the gastrointestinal tract, bone and the kidneys (Fig. 6.1).
Gastrointestinal tract
Intestinal absorption of calcium is mediated by two mechanisms. One is an active transcellular process, regulated by calcitriol (1,25-dihydroxyvitamin D; 1,25(OH)2D) in the duodenum. This involves uptake of calcium into the enterocyte by TRPV6 (membrane calcium channel transient receptor potential cation channel, subfamily V, member 6), followed by intracellular binding of calcium to the calcium-binding protein CaBP-9k, then energy-dependent transport of the calcium across the basolateral membrane via PMCA1b, an alternatively spliced transcript of plasma membrane Ca2 +-ATPase. 1,25-Dihydroxyvitamin D increases gene expression of TRPV6 and CaBP-9k, thus increasing calcium absorption. Calcium is also absorbed passively throughout the small intestine and possibly in the colon. Because the duodenum is relatively short compared to the rest of the small bowel, it may account for less than half of the calcium absorbed at normal dietary intakes, despite its greater absorptive capacity per unit length. At low calcium intakes, active absorption predominates, but this is saturable, and at higher intakes, the change in net absorbed calcium in relation to any change in intake is relatively small and mediated by passive paracellular absorption.
The gut not only absorbs calcium but also secretes it in the digestive juices. The unabsorbed portion of this secreted calcium (approximately 2–3 mmol/24 h) is known as the endogenous faecal calcium. Thus at very low intakes, the absorbed dietary calcium will be less than that lost from secretion in digestive juice and the net calcium absorption will be negative. Net calcium absorption is positive in healthy adults when their daily calcium intake is greater than 5 mmol.
The absorption of calcium is influenced by other dietary constituents. The presence of anions such as phosphate, oxalate (found in some fruit and vegetables) and phytate (found in some unrefined cereals) diminishes calcium solubility and thus net absorption. Gastrointestinal absorption of calcium tends to decline with age, but is increased during pregnancy and lactation. The principal regulator of intestinal absorption is 1,25(OH)2D, and so either deficiency or excess of this hormone is associated with parallel changes in calcium absorption.
The average calcium intake of healthy adults consuming a Western diet is about 20 mmol/day, of which 20–40% is absorbed. The principal source of calcium in the Western diet is dairy products. There are significant amounts of calcium in some green vegetables such as spinach, though the bioavailability is lower.
Kidneys
The ionized and complexed fractions of plasma calcium are filtered at the glomeruli, amounting to approximately 250 mmol/24 h. Of this, ~ 98% is reabsorbed, mostly in the proximal tubules. Calcium reabsorption in the proximal tubules is through a paracellular, therefore passive, process, which is closely linked with that of sodium and does not appear to be hormonally regulated. The fine-tuning of calcium excretion occurs in the distal parts of the nephrons where ~ 15% of the filtered load is reabsorbed. This section consists of the distal convoluted tubules, the connecting tubules and the initial portion of the cortical collecting ducts. In these distal sites, calcium reabsorption is active and occurs against an electrochemical gradient.
Active calcium reabsorption is a multistep process with initial passage of Ca2 + across the luminal membrane through the epithelial channel TRPV5 (membrane calcium channel transient receptor potential cation channel, subfamily V, member 5), then cytosolic diffusion bound to vitamin D-sensitive calcium-binding proteins (calbindins), and finally active extrusion across the opposite basolateral membrane by a Na+, Ca2 +-exchanger (NCX1) and/or Ca2 +-ATPase (PMCA1b). This active calcium reabsorption is subject to hormonal regulation – principally by parathyroid hormone (PTH) and extracellular calcium itself, but possibly also by 1,25(OH)2D, calcitonin, oestrogens and androgens.
Urine calcium excretion is increased in subjects consuming a high protein diet, because of acid production in the metabolism of sulphur-containing amino acids and sequestration of calcium by sulphate in the urine, resulting in inhibition of calcium reabsorption. Sodium intake can also influence calcium excretion by affecting proximal tubular sodium and calcium reabsorption (increased sodium intake increasing calcium excretion). Other factors that increase renal tubular calcium reabsorption are hypovolaemia, alkalosis and thiazide diuretics, whereas volume expansion, acidosis and loop diuretics (e.g. furosemide) have the opposite effect.
Bone
In the mature adult, the movement of calcium into bone equals its rate of efflux and bone mass remains constant. The situation is different in both growth and senescence. In spite of this constancy of adult bone mass, there is an active exchange of calcium between bone and the ECF. This can take place as a result of bone remodelling (see Chapter 31), or it can be accomplished by a process of mineral exchange between bone and the ECF, without local changes in bone matrix. Bone remodelling accounts for the changes in bone density that take place with ageing or disease. In the healthy adult, about 5% of the entire skeleton is remodelled in one year. In contrast, radioisotope studies have indicated that 1–2% of total body calcium can be exchanged between the bone and ECF over a period of several days. The precise mechanisms of this exchange and the factors influencing it are not known, but the quantities of calcium involved suggest that it may be an important part of normal calcium homoeostasis.
Calcium losses in sweat are in the order of 0.3 mmol/24 h. Breast milk has a high calcium content (~ 7.5 mmol/L) and a breast-feeding woman can lose 4–8 mmol/24 h in her milk.
Regulation of calcium metabolism
Plasma calcium concentration is principally controlled by PTH and 1,25(OH)2D. Calcitonin may also be regarded as a calcitropic hormone, although it has no clearly defined physiological function in humans. Many other hormones, growth factors and cytokines can influence bone metabolism.
Parathyroid hormone
Parathyroid hormone is an 84-amino acid, single-chain polypeptide secreted by the parathyroid glands. The gene is on chromosome 11 and consists of three exons encoding a peptide of 115 amino acids (pre-pro-PTH), which is cleaved to produce pro-PTH (90 amino acids) and subsequently the mature peptide (84 amino acids), before secretion. Most of the biological activity of PTH appears to reside in the 34 amino acids at the N-terminal end of the peptide.
The principal regulator of PTH secretion is the ECF ionized calcium concentration: low concentrations stimulate secretion and high concentrations inhibit it. This regulation is mediated by the calcium sensing receptor (CaSR), a cell membrane-bound, G-protein-coupled receptor. The relationship between PTH secretion and ionized calcium concentrations is not linear. Marked hysteresis is evident from experiments where ionized calcium concentrations are raised or lowered. The PTH concentration at any given concentration of plasma ionized calcium is lower when the ionized calcium concentration is rising than it is when ionized calcium is falling (Fig. 6.2). Plasma PTH concentrations exhibit a diurnal variation. They are stable during the afternoon and evening, but then rise by about 50% to peak at around 02.00 h and subsequently fall to values approximately 50% below afternoon values around 09.00 h. The zenith to nadir difference (in healthy men) is ~ 2.3 pmol/L, using an intact PTH assay.
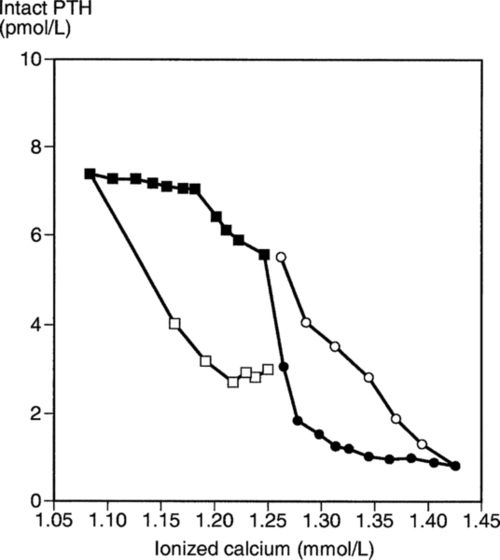
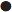 ), and during recovery from hypocalcaemia (
), and during recovery from hypocalcaemia ( ) and hypercalcaemia (
) and hypercalcaemia (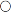 ). For any given ionized calcium concentration, the PTH concentration is lower when the ionized calcium is rising than when it is falling.
). For any given ionized calcium concentration, the PTH concentration is lower when the ionized calcium is rising than when it is falling. Many other factors have been shown to influence PTH secretion, including 1,25(OH)2D, adrenergic agonists, prostaglandins and magnesium, but whether any of these are physiologically important is debatable. Severe magnesium deficiency can produce a state of reversible hypoparathyroidism.
Parathyroid hormone binds to cell surface receptors in its target tissues. The PTH receptor has seven transmembrane domains, an extensive extracellular domain involved in hormone binding and an intracellular domain regulating intracellular signalling. In its two principal target tissues, bone and kidney, this results in activation of both adenylate cyclase (with production of cyclic adenosine monophosphate, cAMP) and phospholipase C (with production of inositol trisphosphate) and subsequent mobilization of intracellular calcium.
In bone, PTH receptors are present on cells of osteoblastic lineage. The osteoblasts in turn regulate osteoclast maturation through production of RANKL (receptor activator of nuclear factor κ-B ligand), which binds to the receptor, RANK, on pre-osteoclasts, leading to their maturation to osteoclasts. This process is modulated by osteoblast production of osteoprotegerin (OPG), which can also bind RANK-ligand, thus reducing its binding to RANK and so the stimulation of osteoclastogenesis.
Parathyroid hormone has three principal actions on the kidneys. It reduces proximal tubular reabsorption of phosphate, increases distal tubular reabsorption of calcium and increases the activity of the 25-hydroxyvitamin D 1α-hydroxylase enzyme in proximal tubular cells. It also decreases proximal reabsorption of bicarbonate, leading to a mild hyperchloraemic acidosis in states of PTH excess.
Measurement of circulating parathyroid hormone
The development of assays for PTH has greatly simplified the investigation of patients with abnormal plasma calcium concentrations, but these assays do have limitations. The parathyroid gland secretes both the intact 84-amino acid peptide and inactive PTH fragments. Hepatic and renal metabolism of intact PTH results in the appearance of mid-region and carboxy-terminal peptides in the circulation. In vivo, the principal biologically active peptide is intact parathyroid hormone, which is present at a low concentration (1–5 pmol/L) and has a short half-life (2–4 min). However, mid-molecule and carboxy-terminal fragments have significantly longer half-lives and accumulate in concentrations 5–20 times greater than that of the intact peptide. These inactive fragments are cleared by the kidneys and their concentrations increase substantially in the presence of renal insufficiency. Although these fragments are not biologically active, they may be immunoreactive, if they cross-react with the particular antibodies used in the immunoassay. Assays that detect inactive fragments, therefore, report higher PTH concentrations than those that detect the intact molecule.
In recent years, two-site and immunoassays of ‘intact’ PTH, which purportedly measure only the biologically active intact peptide, have been widely used. However, there is evidence that these first-generation assays for intact PTH also detect non-(1–84)-PTH peptides, in particular PTH 7–84, which may have different biological actions from the full-length peptide. Assays based on antibodies directed at the first four amino acids of PTH (second-generation intact PTH assays) reveal that non-(1–84)-PTH peptides account for about 15% of circulating immunoreactive PTH (as detected by first-generation assays) in healthy patients, and at least 30% in patients with either primary or secondary hyperparathyroidism. There is also evidence that some non-intact peptides might be biologically active, possibly antagonizing the actions of the intact hormone. Whether assays based on antibodies directed at the extreme N-terminus of PTH offer important clinical advantage in the management of either primary hyperparathyroidism or renal bone disease over the widely available first-generation intact PTH immunoassays remains uncertain.
Both mid-molecule and intact PTH assays show an increase in reference values with age, though this is more marked for the mid-molecule assays because of increased fragment accumulation secondary to the age-related decline in renal function. Because of the effect of renal function on measurements using mid-molecule and carboxy-terminal PTH assays, their interpretation must always take into account the patient’s estimated GFR. Owing to the heterogeneity of PTH immunoassays currently in use, it is important that laboratories use assay-specific data for reference ranges and the ranges of values to be expected in hyperparathyroidism, hypercalcaemia of malignancy, hypoparathyroidism and renal disease. Failure to do so may result in inappropriate management of patients with renal disease.
The action of PTH on the kidney causes cAMP to appear in the urine. ‘Nephrogenous’ cAMP has been used in the past as a surrogate measure of PTH. Its current use is limited to the evaluation of PTH resistance (see Appendix 6.3, below).
Classification of hyperparathyroidism
Increases in plasma concentrations of PTH are seen in a variety of circumstances. It is useful clinically to distinguish conditions in which the increased secretion of PTH is a normal physiological response to hypocalcaemia (secondary hyperparathyroidism) from those in which hypersecretion of PTH is the principal abnormality (primary hyperparathyroidism). If secondary hyperparathyroidism persists over long periods then the parathyroids can become hyperplastic. A gradual rise in plasma calcium accompanies this change and overt hypercalcaemia can develop. This is known as tertiary hyperparathyroidism, and arises chiefly in long-term dialysis patients, but it has also been described in patients with malabsorption syndromes. This classification is unsatisfactory in some respects. For example, the kidney transplant recipient with hypercalcaemia due to hyperparathyroidism that developed while on dialysis is difficult to categorize.
An alternative classification, which has the attraction of being more closely related to treatment stratagems, is to describe hyperparathyroidism in relation to the prevailing plasma calcium concentration. Thus, hyperparathyroidism can be described as hypocalcaemic, normocalcaemic or hypercalcaemic.
Vitamin D
Synthesis and metabolism
Vitamins are defined as essential organic compounds that the body cannot synthesize, and are therefore a vital component of the diet. The term ‘vitamin D’ is a misnomer as the majority of vitamin D is synthesized in the skin by the action of ultraviolet light on 7-dehydrocholesterol (Fig. 6.3). This produces cholecalciferol (vitamin D3). Vitamin D is present in a variety of foodstuffs. It can be added to food in the form of ergocalciferol (vitamin D2 – an additive to margarine and some breakfast cereals and, in the USA, to milk) or may occur naturally in animal products in the form of cholecalciferol. Fish oils are the richest dietary source of cholecalciferol.
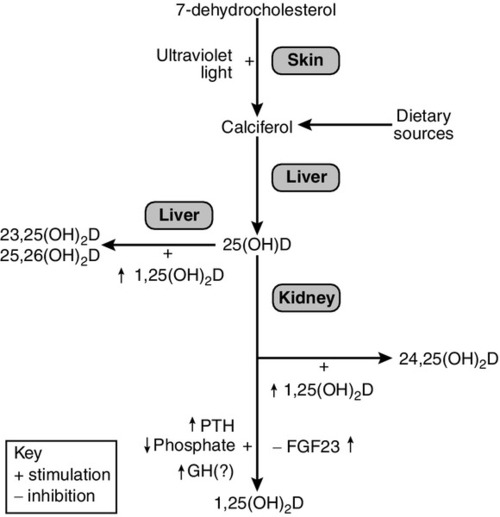
In most respects, these two forms of vitamin D have similar metabolism and action. The calciferols are fat soluble and large quantities can be stored in adipose tissue. They are virtually without biological activity unless they have been hydroxylated. Hydroxylation by the enzyme vitamin D 25-hydroxylase (CYP2R1), takes place in the liver producing 25-hydroxyvitamin D (25(OH)D, calcidiol), which is the principal circulating metabolite and the prohormone for 1,25(OH)2D (calcitriol). The dependence on dermal synthesis for provision of vitamin D is well illustrated by the marked variation in plasma 25(OH)D concentrations through the year in non-tropical areas of the world, where values are significantly higher in summer and autumn than in winter and spring. The vitamin D metabolites circulate in plasma bound to an α-globulin, vitamin D-binding protein and, to a lesser extent, to albumin (Table 6.2).
TABLE 6.2
Circulating concentrations of vitamin D metabolites
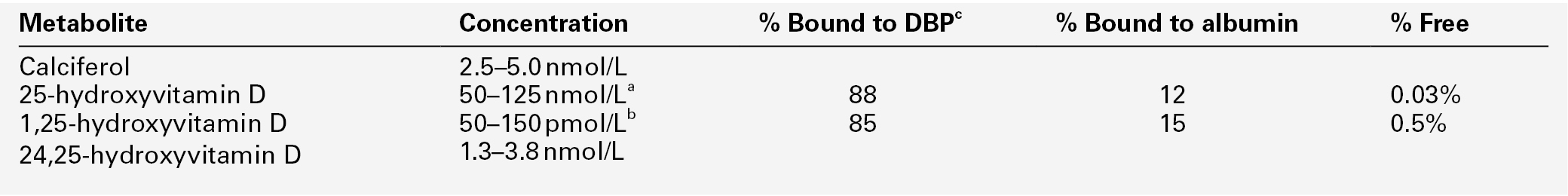
a Higher in summer/autumn than winter/spring;
b higher in pregnancy and during puberty.
c Vitamin D binding protein. Interconversions: 10 nmol ≡ 4 μg ≡ 160 international units.
Most of the circulating 25(OH)D is metabolized in the liver via intermediates such as 23,25(OH)2D and 25,26(OH)2D into inactive metabolites that are excreted into the bile. A small proportion of circulating 25(OH)D undergoes further hydroxylation in the cells of the proximal renal tubules to produce either 1,25(OH)2D or 24,25(OH)2D (secalciferol). The principal active metabolite of vitamin D is 1,25(OH)2D. Although circulating concentrations of 24,25(OH)2D are normally ten times greater than those of 1,25(OH)2D, it has no definite physiological role.
In contrast to hepatic 25-hydroxylation, renal 1α-hydroxylation is closely regulated, being enhanced by low circulating concentrations of phosphate and 1,25(OH)2D, and by high concentrations of PTH, and suppressed by high concentrations of the bone-derived hormone fibroblast growth factor 23 (FGF23). When the plasma concentration of phosphate is high and PTH low, 1α-hydroxylase activity (enzyme CYP27B1) decreases and that of 24-hydroxylase (enzyme CYP24B) becomes greater. Other factors may also influence renal hydroxylation of vitamin D. In vitro, low calcium can stimulate 1α-hydroxylation independently of PTH, although whether this is significant in vivo is not known. Direct or indirect effects of growth hormone, prolactin, oestrogens and calcitonin have also been described.
1,25-Dihydroxyvitamin D is inactivated by the formation of more polar metabolites, which are excreted primarily into bile. Over 96% of all vitamin D is ultimately eliminated through the bile into the faeces. Since this excretion is largely in the form of inactive metabolites, it is doubtful if there is any enterohepatic recycling of physiological importance.
Although the kidneys are undoubtedly the major source of production of 1,25(OH)2D, there is evidence from anephric haemodialysis patients that small quantities can be synthesized extrarenally. In pregnancy, 1,25(OH)2D is synthesized in the placenta, and in a variety of granulomatous diseases (sarcoidosis, tuberculosis, berylliosis) the macrophages of the granulomata can also 1α-hydroxylate vitamin D. In these circumstances, the synthesis is substrate dependent, meaning that the more 25(OH)D that is available, the more 1,25(OH)2D is synthesized.
Actions
Vitamin D metabolites act via a cytosolic receptor that is translocated to the nucleus where it regulates gene expression. The vitamin D receptor is similar in structure to the steroid and thyroid hormone receptors, having an amino-terminal domain, a central DNA-binding domain with ‘zinc fingers’ and a carboxy-terminal hormone-binding domain. 1,25-Dihydroxyvitamin D has an affinity for the vitamin D receptor that is approximately 1000 times greater than that of 25(OH)D, which in turn is an order of magnitude more potent than 24,25(OH)2D. However, the circulating concentration of 25(OH)D is substantially greater than that of 1,25(OH)2D, so it is possible that this metabolite has some biologically significant effect.
The principal site of action of 1,25(OH)2D is the intestine, where it stimulates the production of the calcium channels TRPV6 and TRPV5, and the calcium-binding protein CaBP-9k. In bone, it is a potent osteolytic factor in vitro, acting through osteoblast RANK-ligand production to bring about fusion of osteoclast precursors to form mature osteoclasts. The physiological importance of its bone resorbing effect is uncertain. 1,25-Dihydroxyvitamin D also has direct effects on the liver and kidney. In the liver, it enhances the production of inactive 25(OH)D derived metabolites that are excreted in the bile, and in the kidney it stimulates the 24,25-hydroxylase – both these mechanisms protect against chronically high 1,25(OH)2D concentrations.
Several cells and tissues other than those involved in calcium and phosphorus metabolism, including macrophages, activated T-lymphocytes, the placenta and keratinocytes, express vitamin D receptors and/or the 1α-hydroxylase enzyme, suggesting that their functions might be regulated by components of the vitamin D system. Many malignant cells also express the vitamin D receptor. There is evidence that 1,25(OH)2D decreases the growth of several cancer cell lines, and slows or prevents the growth and/or development of cancers and autoimmune diseases in rodents. Genetic ablation of vitamin D signaling, by deleting the genes encoding either the vitamin D receptor or 1α-hydroxylase, leads to renin-dependent hypertension and an increased propensity to tumour development. The consequences of complete absence of vitamin D signaling in rodents, however, are likely to be very different from minor decreases in 25(OH)D in humans. At present, there is no robust clinical trial evidence that vitamin D supplements or bioactive vitamin D analogues favourably influence important outcomes such as cancer, hypertension or autoimmune diseases. Accordingly, vitamin D supplements are not indicated in the management of non-skeletal diseases.
Synthetic vitamin D analogues
A variety of 1,25(OH)2D analogues have been synthesized. 1α-Hydroxyvitamin D3 (alfacalcidol) is rapidly metabolized by the liver into 1,25(OH)2D and, although having roughly half the potency of 1,25(OH)2D on a weight basis, is similarly useful in conditions where the 1α-hydroxylation step is impaired. A similar preparation, 1α-hydroxyvitamin D2 (doxercalciferol) is also marketed. Other analogues such as 26,27-hexafluorocalcitriol are more potent calcaemic agents.
A number of analogues with attenuated calcaemic properties have also been developed. Examples include calcipotriol (used in the treatment of psoriasis), paricalcitol (used in some countries in the treatment of hyperparathyroidism in chronic renal failure) and eldecalcitol (which has been investigated in osteoporosis). The divergent effects of these analogues on cell differentiation, PTH secretion and calcium homoeostasis probably reflect pharmacokinetic differences rather than distinct 1,25(OH)2D-sensitive pathways, since there is no evidence that they act through different receptors.
Measurement of vitamin D metabolites
Measurements of 25(OH)D and 1,25(OH)2D can be of value in clinical practice, though the indications for 1,25(OH)2D measurement are relatively few (e.g. obscure causes of hypercalcaemia). 25-Hydroxyvitamin D is the best measure of vitamin D status. While its synthesis is dependent upon 25-hydroxylation in the liver, even advanced liver disease does not seem to be a limiting factor in its production. Free concentrations are normal in most patients with cirrhosis, though total concentrations may be low because of associated hypoproteinaemia. 1,25-Dihydroxyvitamin D is present in concentrations only 1000th of those of its precursor (see Table 6.2). Its production is closely regulated and so is not tied to concentrations of its precursor, except in patients with severe vitamin D deficiency or in granulomatous disease. Renal disease has a major impact on its circulating concentrations, as do states of parathyroid over- or underactivity.
A variety of assays have been used in the past, and a number have been withdrawn from the market because of unsatisfactory performance characteristics. Cross-calibration of available assays has not always been within acceptable limits. The reference method uses liquid chromatography with tandem mass spectrometry (MS/MS), but as this is an expensive assay to run, many laboratories use immunoassays based on automated platforms. The acceptability of this compromise depends on the performance characteristics and calibration of the particular assay platform. Some of the commercially available assays have limited ability to measure 25(OH)D2, which may be important when supplementation has been provided in the form of vitamin D2. It is evident that significant variation can occur in measurement of vitamin D metabolites, as a result both of differences between the available assays and in the implementation within individual laboratories.
Calcitonin
Calcitonin is a 32-amino acid peptide produced in humans in the parafollicular cells (C cells) of the thyroid gland. It contains a disulphide bridge and a proline-amide group at the C-terminus. Both of these and the full amino acid sequence are necessary for biological activity. It is metabolized in the kidneys and has a plasma half-life of approximately 5 min.
Secretion of calcitonin is stimulated by an increase in plasma calcium concentration and it is also released in response to a number of gut hormones (e.g. gastrin, glucagon, secretin, cholecystokinin-pancreozymin). It reduces plasma calcium concentration by a direct effect on osteoclasts, which have a cell surface calcitonin receptor linked to adenylate cyclase. It causes contraction of osteoclasts and an acute reduction in osteoclastic bone resorption. It also acts on the kidneys, where it decreases renal tubular reabsorption of calcium and phosphate.
Despite these clearly documented actions, its physiological significance is uncertain because its actions appear to be transient. Furthermore, chronic calcitonin deficiency (as in patients post-thyroidectomy) or excess (as in medullary carcinoma of the thyroid) do not result in significant changes in bone or mineral metabolism.
There are a number of different forms of calcitonin in the circulation, many of which are not biologically active. Thus, different immunoassays may detect different molecular species. The measurement of calcitonin does not form a significant part of the clinical evaluation of calcium metabolism, but it is an important tumour marker for medullary carcinoma of the thyroid.
Procalcitonin
Procalcitonin, the 116 amino acid peptide precursor of calcitonin, is produced not only by thyroid C-cells but also by the neuroendocrine cells of the lung and the intestine. The amount of procalcitonin produced by the cells of the lung and the intestine increases in response to proinflammatory stimuli, especially of bacterial origin. Measurement of procalcitonin can be used as a marker of severe sepsis and generally correlates well with the degree of sepsis.
Other hormones
A number of hormones influence calcium and bone metabolism in vitro, in normal physiology or in specific pathological circumstances. The oestrogen deficiency that develops at the time of the menopause results in increases in cytokine production which leads to increases in bone resorption. Direct effects of oestrogen on osteoclasts and on osteoblastic production of RANKL and osteoprotegerin are also important. Calcium absorption in the gut and reabsorption in the kidney are both reduced, and plasma calcium concentrations rise. These changes lead to a reduction in bone mass, but are reversible with oestrogen replacement therapy. Similarly, testosterone deficiency in men predisposes to osteoporosis, partly through loss of its direct anabolic effects on muscle and on osteoblasts, and partly through diminished conversion to oestrogen. Glucocorticoids can cause marked osteoporosis. These hormones act at a number of sites and reduce osteoblastic activity, induce apoptosis of osteoblasts and osteocytes, reduce intestinal calcium absorption, increase urinary calcium loss and probably increase bone resorption. In children, glucocorticoid excess causes marked growth retardation. Glucocorticoid deficiency may be associated with hypercalcaemia.
Growth hormone accelerates linear growth in children and increases plasma phosphate concentration by increasing renal tubular reabsorption of phosphate. These effects are probably mediated by growth hormone-induced changes in the production of insulin-like growth factor 1 (IGF-1, somatomedin C). There is controversy as to the effect of the growth hormone–somatomedin axis on bone density.
Subjects with high total body fat mass have been shown to have higher bone densities. This appears to be mediated by a number of hormonal mechanisms. Insulin is anabolic to bone in vitro, and obesity is usually associated with hyperinsulinaemia. At least two other anabolic peptides, amylin and preptin, are co-secreted with insulin and may contribute to the relationship. Leptin has been shown in vitro to stimulate osteoblast growth directly and to inhibit osteoclast development, so it is also likely to be an important factor. In contrast, when leptin is administered into the third ventricle of the brains of experimental animals, there is a profound reduction in appetite and fat mass, with resultant loss of bone mass. There is some evidence that central leptin administration activates the sympathetic nervous system and that this might also contribute to bone loss.
Thyroid hormones are potent stimulators of bone resorption, and thyrotoxicosis can be associated with decreased bone density and hypercalcaemia. Prostaglandins of the E series also stimulate bone resorption in vitro. Parathyroid hormone-related peptide (PTHrP) is a 141-amino acid peptide that is responsible for a large proportion of cases of humoral hypercalcaemia of malignancy. Eight of the 13 amino-terminal amino acids are identical to those found in PTH itself, and it appears to act on the PTH receptor. There is evidence for its production in both the fetal parathyroids and in the lactating breast, raising the possibility that it has a physiological role in these contexts. A carboxy-terminal fragment of PTHrP may act as an inhibitor of osteoclastic bone resorption. Direct measurement of PTHrP by immunoassay is possible, but is rarely necessary in clinical practice, since the malignant disease underlying humoral hypercalcaemia is frequently evident at the time that the elevated plasma calcium is recognized.
Biochemical assessment of calcium metabolism
In addition to the assays of calcitropic hormones discussed above, estimation of calcium concentrations in the circulation and of calcium fluxes in the gut and kidney are of value in assessing patients with disorders of calcium metabolism.
Plasma calcium
As indicated previously, total plasma calcium comprises protein-bound, complexed and ionized fractions. Thus changes in plasma protein concentrations or acid–base status will affect the relationship between ionized and total calcium. Total serum calcium remains the most commonly measured index. It should always be accompanied by a measurement of the serum albumin concentration from which a ‘corrected serum calcium’ can be calculated:

Laboratories should derive their own correction formulae based on their own particular calcium and albumin assays, as both the mean normal albumin concentration (assumed here to be 40 g/L) and the amount of calcium bound to each gram of albumin (assumed here to be 0.02 mmol/g) can vary. The corrected calcium is only an approximation, and when there are significant dysproteinaemias or acid–base disturbances, ionized calcium should be measured directly. This measurement is also useful in patients to whom large quantities of citrated blood products have been administered, since these individuals have an increased proportion of plasma calcium complexed to citrate. Ionized calcium is measured using an ion-selective electrode. Specimens must be obtained anaerobically to minimize changes in specimen pH. The pH is also usually measured and results adjusted to a pH of 7.4.
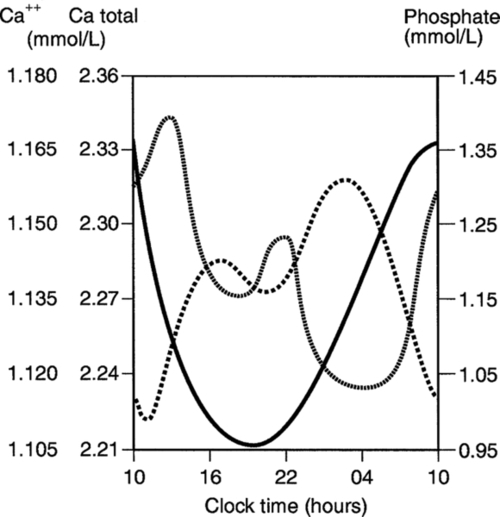
There are diurnal variations in both total and ionized plasma calcium concentrations (Fig. 6.4). Significant changes in plasma calcium take place following the ingestion of a calcium-rich meal or of calcium supplements. Posture and the application of tourniquets, both of which can influence plasma protein concentrations, may also influence total calcium estimations. Total plasma calcium rises significantly (by approximately 0.1 mmol/L) at the menopause.
Intestinal calcium absorption
This is not routinely measured in clinical practice, but knowledge of it is sometimes of value, e.g. in some patients with kidney stones and hypercalciuria. In the past, it was measured by metabolic balance, which required a period of 7–10 days as an inpatient, during which time subjects were given a diet with a constant calcium intake, and calcium absorption was estimated from precise measurements of faecal and urinary calcium output.
Calcium absorption can be measured with a variety of stable or radioisotope techniques in which an oral calcium load with a tracer is administered in the fasting state and absorption efficiency is estimated from the rate of appearance of the isotope in the blood. The size of the oral calcium load accompanying the isotope is important. Low doses reflect better the active duodenal transport, whereas high doses better reflect net intestinal absorption. The increment in urine calcium following the administration of a known calcium load has also been used as an index of calcium absorption (see Appendix 6.1, below). The clinical utility of these tests is limited and they are rarely used nowadays.
Urinary calcium
Calcium can be measured either in a 24 h urine or in urine collected after an overnight fast. In the latter instance, the subject empties the bladder on rising, has a glass of water and then collects the urine sample at any time between 30 min and 2 h later. In both instances, acid (usually 6 M hydrochloric acid) is added either before collection or subsequently, to prevent crystallization of calcium salts. Creatinine is usually measured on both 24 h and fasting urine samples – in the former case to allow assessment of completeness of collection and in the latter as a denominator in relation to which the concentration of calcium (and other urinary constituents) can be expressed.
The reference range for 24 h urine calcium is 1–7.5 mmol/24 h in men and 1–6.25 mmol/24 h in women. In the steady state, this reflects the absolute dietary intake of calcium and the net proportion absorbed from the intestine. Urine calcium excretion is usually increased with hypercalcaemia of any cause, and its measurement contributes little to the differential diagnosis of hypercalcaemia, except in the diagnosis of familial hypocalciuric hypercalcaemia.
Fasting urine calcium can be expressed as a simple molar ratio to creatinine (reference values 0.10–0.30 in adults). This is considered to be an index of bone resorption, but newer markers of bone resorption are preferred in clinical practice (see Chapter 31). If the calcium: creatinine ratio is multiplied by the plasma creatinine concentration, the resulting product is the calcium excretion per litre of glomerular filtrate (CaE). This is useful in assessing the contribution of abnormal renal tubular handling of calcium to disorders of plasma calcium homoeostasis (see Appendix 6.2, below).
Indices of bone turnover
Disorders of plasma calcium are often accompanied by abnormalities of bone turnover, so assessment of bone turnover can be useful. These measures are discussed in more detail in Chapter 31.
Hypercalcaemia
Hypercalcaemia develops when the rate of entry of calcium into the ECF from bone and gut exceeds the capacity of the kidney to excrete it. Diminished excretory capacity can be the result of either increased renal tubular reabsorption (an important component, for example, in the hypercalcaemia of primary hyperparathyroidism and familial hypocalciuric hypercalcaemia) or of a reduction in glomerular filtration rate (in sarcoidosis, for example, hypercalcaemia is more frequently encountered in subjects with impaired renal function). However, hypercalcaemia itself can affect these processes, thus initiating a vicious cycle with worsening hypercalcaemia. By its actions on the kidney, hypercalcaemia leads to polyuria, volume depletion and thus to a reduction in glomerular filtration rate and enhanced proximal tubular reabsorption of sodium, which carries calcium with it. This situation, termed ‘disequilibrium hypercalcaemia’, is a medical emergency. It is important both to recognize this and to differentiate it from ‘equilibrium hypercalcaemia’ (e.g. due to mild primary hyperparathyroidism or familial benign hypercalcaemia), where the plasma calcium remains stable.
Whether hypercalcaemia causes symptoms depends both upon the degree of elevation of plasma calcium and the rate at which it has risen. When plasma calcium is < 3.0 mmol/L, a large proportion of patients are asymptomatic. The clinical features of severe hypercalcaemia are summarized in Box 6.1.
Causes of hypercalcaemia
Box 6.2 lists the most frequently encountered forms of hypercalcaemia.
Primary hyperparathyroidism
Primary hyperparathyroidism is the most common cause of hypercalcaemia presenting outside hospital. Prior to the advent of large automated analysers, it was regarded as a rare disease, but is now a common incidental finding. Its prevalence is approximately 1 per 1000 and it is most frequently diagnosed in the 6th decade of life, when it is two or three times as common in women as in men. To some extent, this can be explained by the postmenopausal rise in plasma calcium concentration. The normal distribution curve is shifted to the right and, therefore, a higher proportion of postmenopausal women must necessarily have plasma calcium concentrations above the upper limit of normal for younger people. In younger patients, the sex incidences are equal.
In 90% of patients, primary hyperparathyroidism is attributable to a single parathyroid adenoma, and most of the remaining cases are attributable to four gland hyperplasia, sometimes as a part of the multiple endocrine neoplasia (MEN) syndrome type 1 (see Chapter 41). This syndrome accounts for the majority of patients with familial hyperparathyroidism. Parathyroid carcinoma, which usually presents with severe hypercalcaemia and very high PTH concentrations, accounts for < 1% of all cases.
Adenomatous primary hyperparathyroidism appears to have a clonal origin. Two genetic mechanisms have been elucidated (Table 6.3). The PTH gene is located on the opposite side of the centromere of chromosome 11 from the gene (PRAD1) that encodes cyclin D1, a proto- oncogene that regulates cell growth. In about 20% of sporadic parathyroid tumours, it has been shown that while one chromosome is intact, the other copy has undergone centromeric inversion, a rearrangement that places the PTH regulatory elements under the influence of cyclin D1, allowing deregulated parathyroid gland growth.
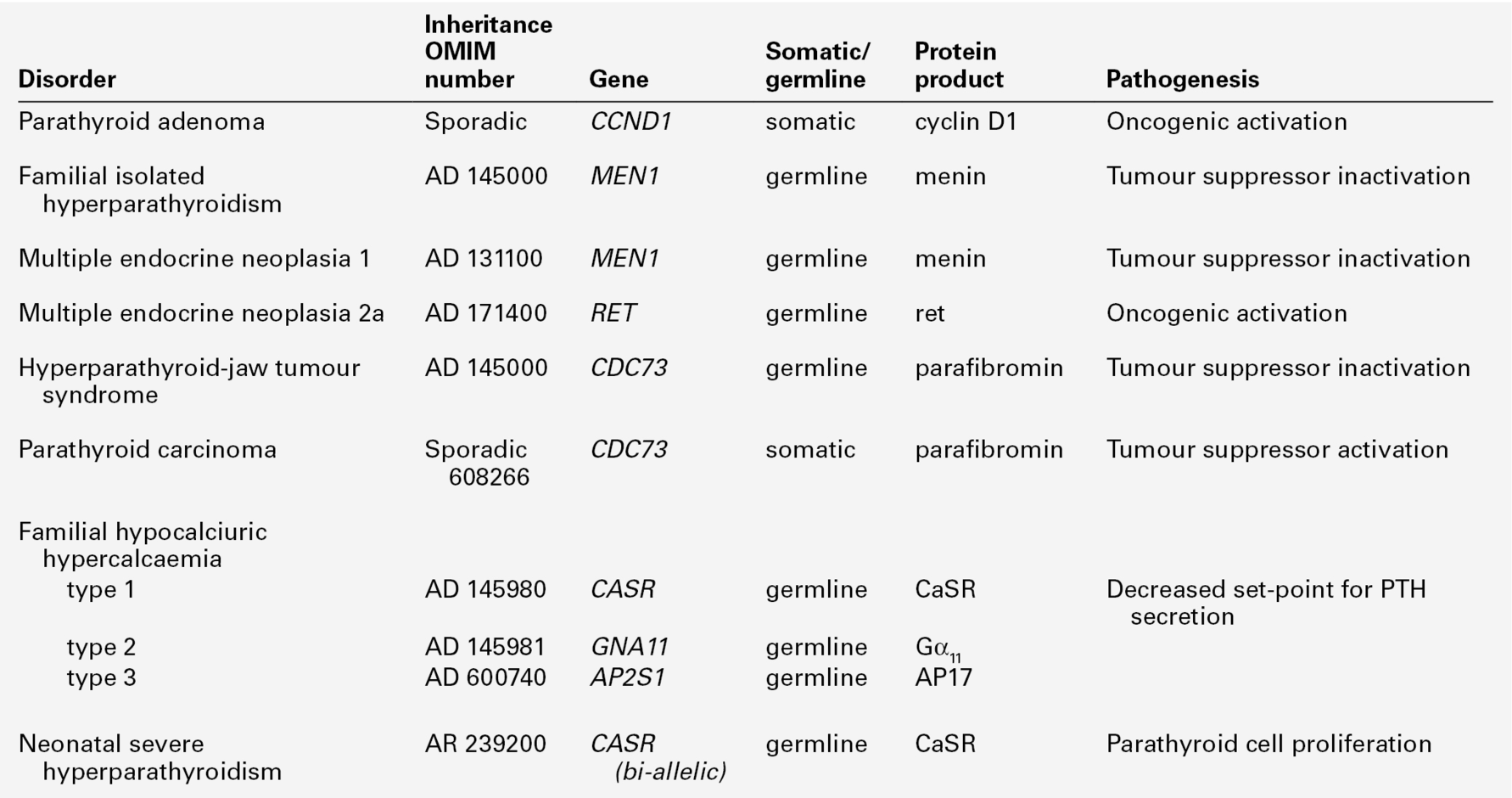
Germline mutations underlie several of the inherited syndromes that include primary hyperparathyroidism. The mutated gene in multiple endocrine neoplasia 1 (MEN1) is also located on chromosome 11, and encodes a protein, menin, that acts as a tumour suppressor. Rare instances have been reported of kindreds with germline MEN1 mutations in which primary hyperparathyroidism is the only clinical feature. Primary hyperparathyroidism is less frequently (~ 20%) a feature of the MEN 2 syndromes, which result from activating mutations of the gene encoding the RET tyrosine kinase receptor. Germline mutations in the tumour suppressor gene CDC73, which encodes the protein parafibromin, causes the inherited disorder, hyperparathyroidism-jaw tumour syndrome. Somatic mutations in this gene are also present in a high proportion of sporadic parathyroid carcinomas.
Primary hyperparathyroidism may present with the symptoms of hypercalcaemia outlined in Box 6.1, but frequently patients are symptom-free. The prevalence of osteoporosis is probably increased in patients with primary hyperparathyroidism, especially at skeletal sites enriched for cortical bone. The classic skeletal manifestations of primary hyperparathyroidism, which include subperiosteal phalangeal resorption, a ‘salt and pepper’ appearance in the skull and bone cysts (‘brown tumours’) of the long bones or jaw (collectively termed ‘osteitis fibrosa cystica’), are features of severe, longstanding disease, and are now uncommon in the developed world. Approximately 15% of patients have renal complications in the form of nephrolithiasis or nephrocalcinosis.
Familial hypocalciuric hypercalcaemia
Familial hypocalciuric hypercalcaemia (FHH) (also known as benign familial hypercalcaemia) is an autosomal dominant condition in which mild hypercalcaemia with relative hypocalciuria is present throughout life. Patients with this disorder are most frequently heterozygous for inactivating mutations in the calcium sensing receptor (CaSR). The ‘set-point’ for PTH release is raised, and mild PTH-dependent hypercalcaemia results. The CaSR is also expressed in the renal tubules, where it regulates renal tubular calcium reabsorption in a PTH-independent fashion. The inactivating CASR gene mutation promotes inappropriately avid tubular calcium reabsorption in the face of hypercalcaemia, explaining the relative hypocalciuria. Patients with FHH are usually asymptomatic, although non-specific symptoms of lethargy and polydipsia have been observed in some instances. There may be an increased incidence of pancreatitis, but the frequency of nephrolithiasis and peptic ulcer disease is the same as in the general population. In most cases, the condition follows a benign course and the greatest danger to patients is being misdiagnosed as having adenomatous primary hyperparathyroidism and thus having inappropriate parathyroidectomy. Familial hypocalciuric hypercalcaemia accounts for about 2% of asymptomatic hypercalcaemia.
Neonatal severe primary hyperparathyroidism occurs in newborns who have inherited two inactivating CASR mutations – one from each parent. It presents as severe, life-threatening hypercalcaemia. Urgent total parathyroidectomy is indicated.
Hypercalcaemia of malignancy
Malignancy can result in hypercalcaemia by four general mechanisms:
• as a result of osteolytic metastases that lead to local bone destruction
• osteolysis from bone marrow infiltration in haematological disorders
• more rarely, accelerated conversion of plasma 25(OH)D to 1,25 (OH)D2 by some haematological malignancies, leading to vitamin D-dependent hypercalcaemia.
Parathyroid hormone-related peptide is the main mediator of humoral hypercalcaemia of malignancy. It is synthesized and secreted by a number of tumours, particularly those of epithelial cell origin (e.g. squamous cell carcinoma of the bronchus, renal cell carcinoma, breast carcinoma) and produces biochemical changes similar to those of primary hyperparathyroidism, although the degree of hypercalcaemia is often more severe in HHM, and plasma 1,25(OH)2D concentrations tend to be normal or low rather than increased.
Hypercalcaemia associated with osteolytic metastases is most commonly seen in advanced breast cancer, but can occur with other solid tumours that spread to bone. Hypercalcaemia is very common in some haematological malignancies, particularly multiple myeloma and acute human T cell leukaemia–lymphoma virus (HTLV)-1-associated disease. The bone destruction is mediated by osteoclasts, activated by factors released by the tumour. A number of different cytokines have been implicated, including RANKL and TNF-α. In addition, bone formation may be impaired at sites of myelomatous skeletal disease, as a result of inhibition of Wnt signalling through the lipoprotein receptor-related protein 5 (LRP5). In contrast to the hypercalcaemia associated with metastases from solid tumours, hypercalcaemia associated with haematological malignancy typically responds rapidly to glucocorticoid treatment. In the rare cases of patients with hypercalcaemia associated with haematological malignancies that elaborate 1α-hydroxylase, the hypercalcaemia also responds rapidly to glucocorticoid treatment.
Granulomatous disease
The macrophages in granulomata occurring in sarcoid tissue, pulmonary tuberculosis and berylliosis are capable of 1α-hydroxylating 25(OH)D, independently of normal homeostatic regulation. The production of 1,25(OH)2D is dependent on disease activity and circulating 25(OH)D concentrations. Thus, during the summer months, patients with active disease have increased intestinal calcium absorption. This is usually evident as hypercalciuria, but some patients, usually those with some pre-existing renal impairment, become hypercalcaemic. 1,25-Dihydroxyvitamin D production can be rapidly suppressed (and hypercalcaemia corrected) by glucocorticoids, chloroquine or ketoconazole.
Vitamin D toxicity
Excessive intake of vitamin D or its analogues can produce hypercalcaemia. This may be iatrogenic or the result of self-medication. On withdrawal of vitamin D, hypercalcaemia resolves. The rate of reversal is much more rapid in patients intoxicated with alfacalcidol or calcitriol than it is in patients intoxicated by calciferol. In the former instances, plasma calcium reverses with a half-time of 1–5 days, but in the latter, the half-time is of the order of 10–30 days. Women being treated with vitamin D or its analogues for hypoparathyroidism may become hypercalcaemic during lactation or after stopping exogenous oestrogen therapy.
The vast majority of adult hypercalcaemic patients (> 98%) will have one of the above diagnoses, but there are rarer causes of hypercalcaemia (summarized in Table 6.4). Hypercalcaemia in dialysis and kidney transplant recipients is discussed in Chapter 31. Hypercalcaemia in infants and children is much less common than in adults, and a different range of diagnoses needs to be considered (Box 6.3).
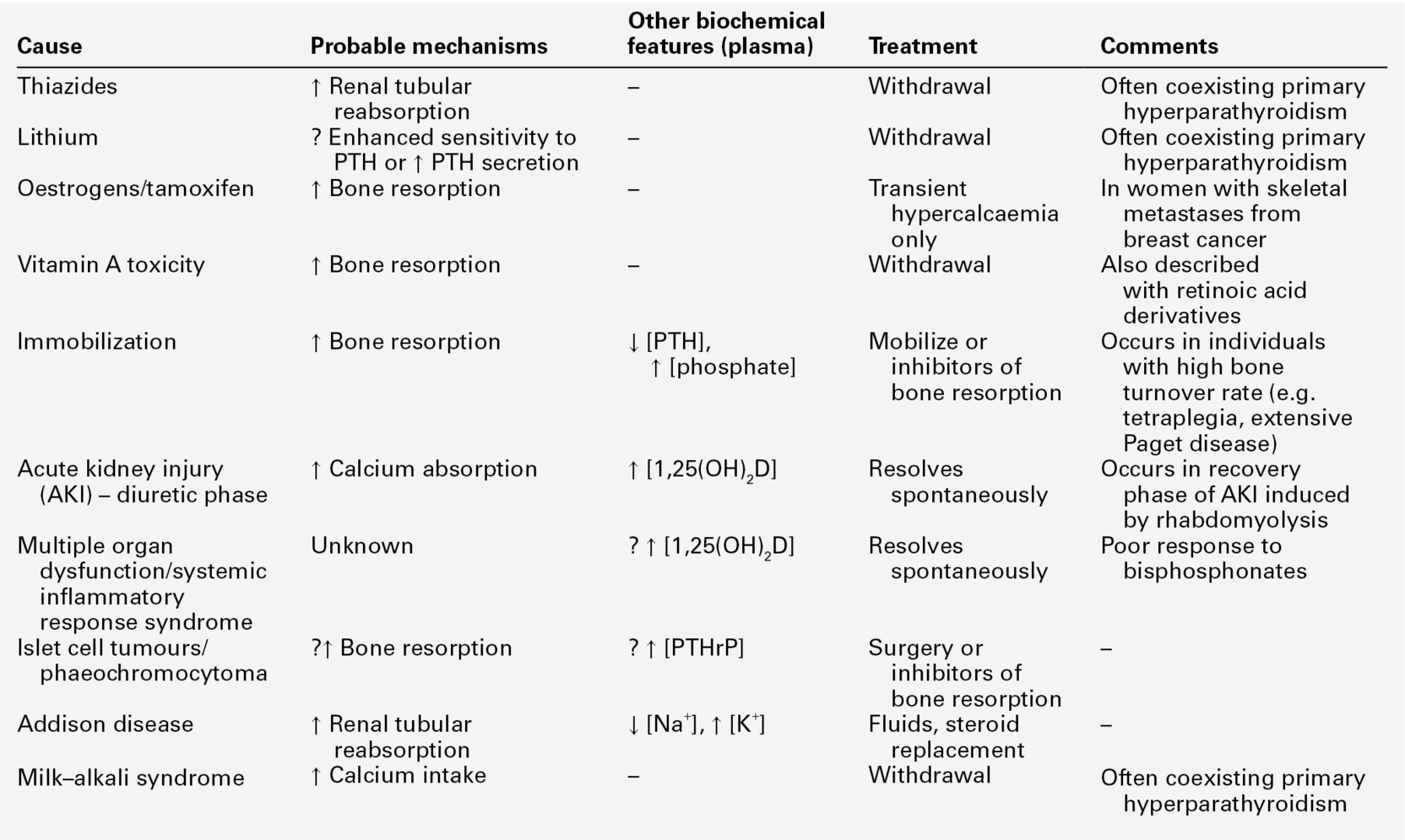
Investigation of hypercalcaemia
Figure 6.5 sets out an approach to the investigation of hypercalcaemia. Having confirmed that true hypercalcaemia exists, a clinical assessment will in many cases, point to the appropriate diagnosis by revealing evidence of underlying diseases or medications that may be contributing. The next stage is biochemical assessment, in which PTH measurement is pivotal. The currently available assays of intact PTH will usually produce either an elevated or suppressed value in hypercalcaemic subjects, thus establishing or excluding the parathyroids as the cause of the hypercalcaemia.
A normal intact PTH concentration in the face of an elevated plasma calcium may be consistent with either primary hyperparathyroidism or with FHH. The ratio of calcium clearance to creatinine clearance in a 2 h fasting urine collection is useful in distinguishing between these two possibilities – in FHH the ratio is usually < 0.01. However, clear distinction between adenomatous primary hyperparathyroidism and FHH is not always possible on biochemical grounds. Studying family members and/or sequencing the CASR gene may be necessary.
If plasma PTH concentration is low, then a variety of other investigations can be pursued. The order in which these are undertaken will be determined by the clinical clues. Cancer-associated hypercalcaemia is almost always a late complication of the disease, which is therefore either known or readily diagnosed when the hypercalcaemia is recognized. A search for malignancy may include radioisotope bone scanning, chest and abdominal imaging and serum and urine protein electrophoresis. Assays for PTHrP are now available, but are rarely needed in the evaluation and treatment of hypercalcaemia. Plasma PTHrP concentrations are elevated in most patients with humoral hypercalcaemia of malignancy, and are low in healthy subjects (< 2 pmol/L). Parathyroid hormone-related peptide concentrations are also increased in about half of women with lytic bone metastases from breast cancer, irrespective of whether they are hypercalcaemic. In difficult cases, measurement of 1,25(OH)2D may be of value. Elevated concentrations are seen in granulomatous disease, some lymphomas and sometimes in primary hyperparathyroidism, and subnormal concentrations are usually found in HHM. Measurement of 25(OH)D will allow confirmation of vitamin D intoxication, but not if it is caused by 1α-hydroxylated compounds such as alfacalcidol, calcitriol, paricalcitol or calcipotriol.
A number of investigations previously used in the differential diagnosis of hypercalcaemia have limited utility. Hypophosphataemia is a feature of both primary hyperparathyroidism and HHM, and phosphate concentrations tend to be increased in sarcoidosis and vitamin D toxicity or when there is renal impairment. Measurement of plasma chloride has been used because of the mild metabolic acidosis associated with primary hyperparathyroidism. While chloride concentrations do tend to be higher in primary hyperparathyroidism than in HHM, this measurement has very little clinical utility in individual patients. The same considerations apply to plasma magnesium concentrations. Although plasma magnesium is higher in FHH than in primary hyperparathyroidism, it is of limited diagnostic value. Since both PTH and PTHrP stimulate renal adenylate cyclase, measurement of cyclic adenosine monophosphate excretion has no discriminant value.
Skeletal radiology may be of help, showing myeloma deposits (that may be missed by bone scintigraphy) or showing evidence of other malignancy or revealing the changes of hyperparathyroidism. Occasionally, use is still made of the glucocorticoid suppression test (prednisolone 30 mg/day for a period of ten days). In primary hyperparathyroidism, no change in plasma calcium occurs, but in granulomatous disease, vitamin D intoxication and in haematological malignancies, a significant fall in plasma calcium usually occurs. The analysis of renal tubular calcium handling (see Appendix 6.2, below) can provide information on the mechanisms underlying hypercalcaemia, but does not help to differentiate primary hyperparathyroidism from HHM.
In summary, measurement of intact PTH, accompanied by clinical history and examination and targeted radiological investigations will allow the cause of hypercalcaemia to be ascertained quite quickly in the vast majority of patients.
Treatment of hypercalcaemia
Specific details of the treatment of hypercalcaemia are beyond the scope of this text, but its principles will be reviewed. In patients with mild asymptomatic primary hyperparathyroidism, long-term follow-up has revealed stability of plasma calcium and renal function, and low rates of disease complications. Consequently, some authorities feel that no intervention is necessary in patients who have not had renal stones and are at low fracture risk, although this remains an area of controversy. Either medical treatment with bisphosphonates or surgical correction are effective treatments for improving bone mineral density in patients with primary hyperparathyroidism who are at increased risk of fracture. When definitive treatment is necessary, as in patients who develop urolithiasis or severe hypercalcaemia (> 3 mmol/L), removal of the causative adenoma by an experienced surgeon is usually curative. Recurrence is rare, except patients in whom hyperparathyroidism is caused by multiglandular hyperplasia, as in the type 1 multiple endocrine neoplasia syndrome. Calcimimetic drugs, such as cinacalcet, which act as allosteric agonists of the CaSR present on the parathyroid chief cells, suppress PTH secretion and lower plasma calcium in PTH-dependent hypercalcaemia. Studies indicate that treatment with cinacalcet can maintain normocalcaemia for at least five years in the vast majority of patients with mild primary hyperparathyroidism, and that it is moderately effective in lowering plasma calcium in patients with severe, symptomatic PTH-dependent hypercalcaemia. Thus, the calcimimetics may acquire a role in the future in treating patients with severe, symptomatic primary hyperparathyroidism or parathyroid cancer in whom surgery has either failed or is not possible. In FHH, no therapy is required.
Individuals with disequilibrium hypercalcaemia are almost invariably volume depleted, and the first step in their management is rehydration with intravenous normal saline. This will be associated with increased renal blood flow and a resultant increased clearance of calcium through the kidneys. In those in whom hypercalcaemia is secondary to increased bone resorption, therapy with an inhibitor of bone resorption is appropriate. This is now satisfactorily achieved with intravenous infusions of bisphosphonates (e.g. pamidronate or zoledronate), with the majority of patients gradually returning to normocalcaemia within about a week.
In the past, forced saline diuresis has been used to increase renal clearance of calcium. This is potentially hazardous, particularly in the elderly, in whom congestive cardiac failure and electrolyte imbalances may cause significant morbidity. The infusion of phosphate, which can cause widespread metastatic calcification, is another dangerous and redundant therapy for hypercalcaemia. Steroids are helpful in haematological malignancies and in sarcoidosis.
Hypocalcaemia
Hypocalcaemia is a relatively uncommon finding, but true hypocalcaemia usually indicates a significant underlying abnormality requiring diagnosis and treatment.
Clinical features
Hypocalcaemia results in increased excitability of neuromuscular tissue. This may become clinically manifest as tetany, usually presenting as carpopedal spasm or, in milder cases, as paraesthesiae in the perioral region or fingers. Laryngeal stridor and seizures may also occur. The electrocardiogram shows prolongation of the QT and ST intervals, and patients may develop arrhythmias, heart block and congestive heart failure.
In longstanding hypocalcaemia, calcification of the basal ganglia, psychiatric disturbances and cataracts may occur. Hypocalcaemic children may show abnormalities in dental development.
Causes of hypocalcaemia
Causes of hypocalcaemia are set out in Box 6.4. Because hypoalbuminaemia is observed in patients with a wide variety of other diagnoses, it is mandatory to make allowance for the plasma albumin concentration or to measure ionized calcium when entertaining the diagnosis of hypocalcaemia. The common causes of chronic hypocalcaemia relate to abnormalities in the synthesis, secretion or action of PTH or 1,25(OH)2D, or both these hormones.
Hypoparathyroidism
The most common type of hypoparathyroidism is that occurring after surgery to the thyroid or, occasionally, to other structures in the neck. It varies widely in severity and, in some patients, is transient, with plasma calcium returning to normal in the weeks after surgery. Overt hypocalcaemia can be precipitated in previously asymptomatic patients with partial hypoparathyroidism by therapy with exogenous oestrogens or proton pump inhibitors.
Spontaneous hypoparathyroidism is rare. It may occur as an isolated disorder or be part of a congenital disease complex. The most frequently encountered forms are listed in Table 6.5.
TABLE 6.5
Genetic causes of hypoparathyroidism
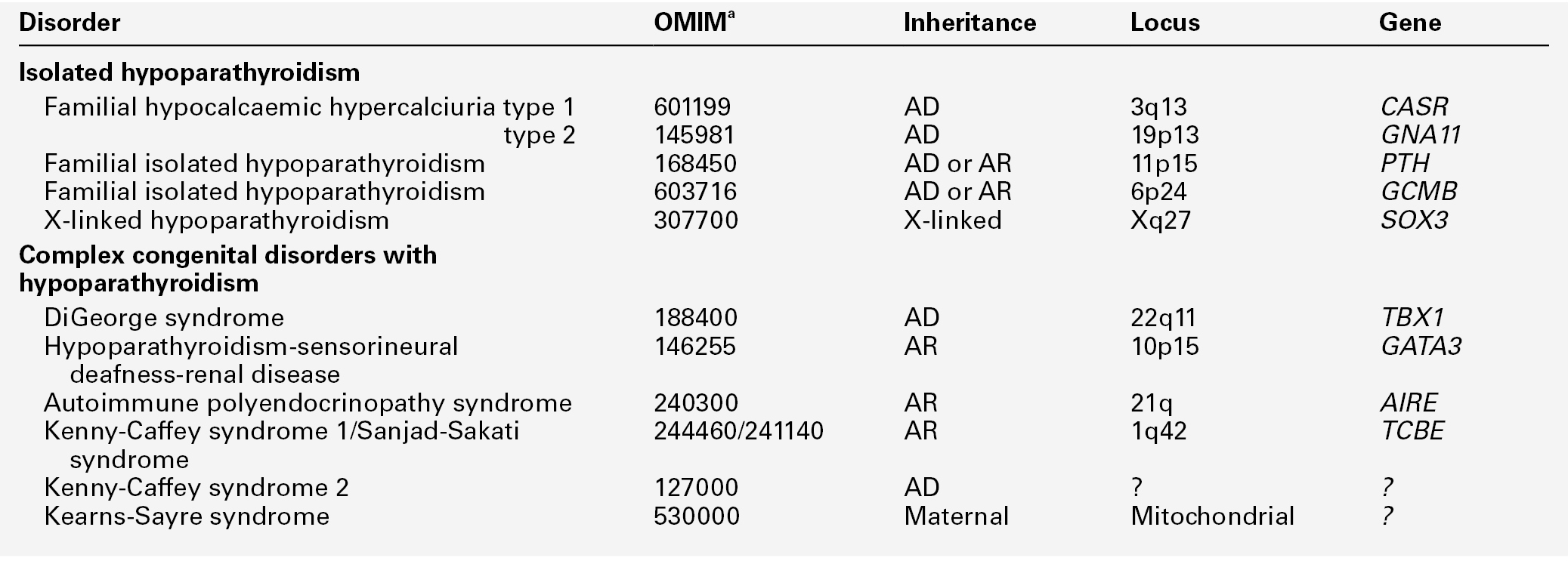
a Online Mendelian Inheritance in Man catalogue number. Note that the biochemical changes of hypoparathyroidism can be a mainfestation of the genetic pseudohypoparathyroid syndromes (Table 6.6) and genetic causes of hypomagnesaemia (Table 6.12). AD, autosomal dominant; AR, autosomal recessive.
The complex forms are usually the result of mutation in or deletions of genes encoding transcription factors regulating pharyngeal pouch development.
Idiopathic isolated hypoparathyroidism is also likely to have a genetic basis. Mutations in or near the genes PTH and GCMB (encoding ‘glial cell missing’; a master regulator of parathyroid development) have been described in kindreds with both autosomal recessive and autosomal dominant inheritance. A rare X-linked recessive form has also been described.
It is important to distinguish autosomal dominant hypocalcaemia with hypercalciuria (ADHH) from other forms of hypoparathyroidism. This is usually caused by activating mutations in the gene encoding the CaSR and is the genetic and phenotypic converse of familial hypocalciuric hypercalcaemia: PTH concentrations are low in the face of hypocalcaemia, and there is relative hypercalciuria. Most patients with this syndrome are asymptomatic or minimally symptomatic, and do not require treatment. Treatment to raise plasma calcium should not be given to individuals known to have ADHH unless they are symptomatic, because it causes hypercalciuria, which can lead to nephrolithiasis and renal impairment.
Autoimmune hypoparathyroidism may occur alone or in association with additional features, including mucocutaneous candidiasis and adrenal insufficiency, as a component of the autoimmune polyglandular syndrome type 1 (APS-1) or autoimmune polyendocrinopathy-candidiasis–ectodermal dystrophy (APECED) syndrome. This may be sporadic or familial with an autosomal recessive inheritance pattern. Less commonly, it may also be seen as part of the autoimmune polyglandular syndrome type 2 (APS-2), which is characterized by adult-onset adrenal insufficiency associated with type 1 diabetes and thyroid disease and is believed to be a polygenic disorder with apparent dominant inheritance. The parathyroid glands are an infrequent target for autoimmunity; antibodies directed against the parathyroid CaSR may have a direct pathogenetic role.
Hypoparathyroidism is characterized by hypocalcaemia, with plasma total calcium concentrations as low as 1.25 mmol/L being found. Urine calcium excretion is subnormal and the TmP/GFR (see p. 111) is increased, leading to hyperphosphataemia. Bone turnover is reduced, as are intestinal calcium absorption and circulating concentrations of both 1,25(OH)2D and intact PTH. Some PTH immunoassays are not able to distinguish low concentrations from those within the reference range.
Pseudohypoparathyroidism
This term refers to a heterogeneous group of rare conditions characterized by the combination of resistance to the actions of PTH and, variably, other glycoprotein hormones, and Albright hereditary osteodystrophy (short stature, obesity, short metacarpals and metatarsals, heterotopic ossification). These patients present with much the same clinical and biochemical findings as in hypoparathyroidism, but their concentrations of immunoreactive PTH are high rather than low. The resistance to PTH seems primarily to be in the kidneys, so some patients may have evidence of hyperparathyroid bone disease. The hypocalcaemia may be less severe than in hypoparathyroidism and may fluctuate. The current classification of the disorder is shown in Table 6.6.
TABLE 6.6
Classification and features of pseudohypoparathyroidism

a See Appendix 6.3 for testing protocol.
b Type Ia and pseudopseudohypoparathyroidism (PPHP) commonly occur within the same kindred.
The most common forms of the disorder, type Ia, Ib and pseudopseudohypoparathyroidism (PPHP), are caused by mutations in the GNAS gene. This encodes the signalling protein Gsα, that is part of the guanine nucleotide-binding protein complex linking the PTH receptor (and the receptors for other glycoprotein hormones) to the adenylyl cyclase catalytic unit. The phenotype that ensues is dependent upon the gender of the parent transmitting the mutant allele (genetic imprinting). Added complexity is provided by the fact that tissue- or cell-specific imprinting of Gsα occurs, thereby explaining the phenotypic differences in hormone resistance that are observed.
The diagnosis of pseudohypoparathyroidism and the classification of patients as type I or type II are dependent upon the modified Ellsworth–Howard test (see Appendix 6.3, below).
Vitamin D disorders
Hypocalcaemia is frequently encountered where abnormalities of vitamin D metabolism lead to failure to synthesize adequate quantities of 1,25(OH)2D. This can result from insufficient supplies of the precursors (privational vitamin D deficiency) or defects in hepatic 25-hydroxylation or renal 1α-hydroxylation (either inherited or acquired). Finally, there can be end-organ resistance to the actions of 1,25(OH)2D. In these disorders, in addition to hypocalcaemia, there is usually secondary hyperparathyroidism with hypophosphataemia and, in bone, osteomalacia or rickets. These disorders are discussed more fully in Chapter 31.
Other causes of hypocalcaemia
Acute hypocalcaemia can result from the release into the circulation of any factor capable of binding substantial amounts of calcium. In acute pancreatitis, free fatty acids can do this, whereas in acute rhabdomyolysis and in the tumour lysis syndrome, large quantities of intracellular phosphate are released. Incautious infusion or ingestion of phosphate can also result in hypocalcaemia by the same mechanism, and the infusion of large quantities of citrate as a result of blood transfusions has a similar effect. Some radiographic contrast dyes contain either citrate or the calcium chelator EDTA, and can also produce hypocalcaemia. These mechanisms, together with magnesium deficiency, which impairs both PTH secretion and action, contribute to the hypocalcaemia observed in 25–50% of patients admitted to intensive care units.
In patients with widespread osteoblastic metastases, modest hypocalcaemia may occur secondarily to increased incorporation of calcium into bone around the secondary deposits. Severe, prolonged hypocalcaemia may occur in patients following parathyroidectomy for longstanding hyperparathyroidism (hungry bone syndrome). Hypocalcaemia is sometimes seen in the neonatal period, being more common in premature infants and in the offspring of diabetic and hyperparathyroid mothers. Its aetiology is varied (see Chapter 25).
Investigation of hypocalcaemia
Figure 6.6 sets out an approach to the differential diagnosis of hypocalcaemia. Clearly, the first step is to confirm that hypocalcaemia is not just a reflection of low plasma albumin concentration. An ionized calcium measurement may be necessary, particularly in acutely ill patients in whom calcium binding to albumin or other components of plasma may be abnormal. A number of the causes of hypocalcaemia shown in Box 6.4 are easily distinguished clinically. The acutely ill patient is clearly distinguishable from someone with chronic hypocalcaemia. The family history may be contributory, there may be an abnormal phenotype, evidence of other hormonal abnormalities associated with autoimmune hypoparathyroidism, a past history of thyroid surgery, clinical evidence of osteomalacia etc. Data gained at this point may allow some of the investigations outlined below to be bypassed, or may direct the clinician towards specific diagnoses.
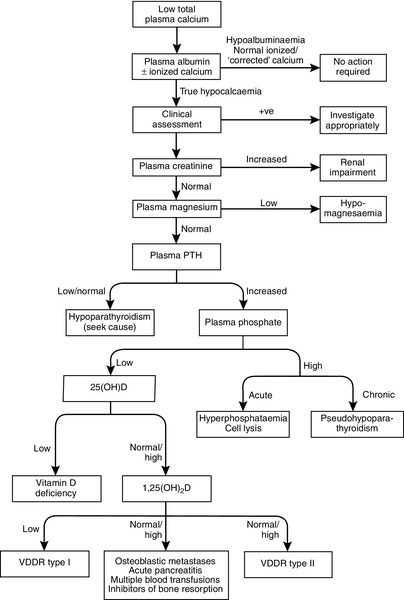
In the absence of such clues, serum creatinine and magnesium should be measured at an early stage, since renal failure and magnesium deficiency are relatively frequent causes of hypocalcaemia. Parathyroid hormone should also be assayed to determine whether parathyroid gland hypofunction is the cause. Serum phosphate measurement is useful, the phosphate concentration being high in hypoparathyroidism, syndromes of PTH resistance and when hyperphosphataemia is the primary disturbance, and (provided renal function is adequate) low in states of abnormal vitamin D activity and/or secondary hyperparathyroidism.
The plan of investigations outlined in Figure 6.6 is only a guide, and some patients will have multiple pathologies, which complicate the interpretation of their biochemistry. This is particularly true in those with acute illness or malignancy.
Some investigations not shown in this scheme may also be of value. A measurement of TmP/GFR is useful in confirming the diagnosis of hypoparathyroidism, and the modified Ellsworth–Howard test in the diagnosis of pseudohypoparathyroidism. Plasma alkaline phosphatase activity is normal or low in hypoparathyroidism, but elevated in most patients with secondary hyperparathyroidism.
Treatment of hypocalcaemia
A detailed discussion of treatment is beyond the scope of this text, but the principles involved will be outlined. Clearly, the underlying condition needs to be sought and, in some cases, appropriate treatment of that will correct the hypocalcaemia. In acute symptomatic hypocalcaemia, intravenous calcium treatment may be necessary. A typical regimen in an adult would be to give 10–20 mL 10% calcium gluconate over 5 min (2.26–4.52 mmol calcium), followed by a continuous intravenous infusion supplying 9–18 mmol calcium in 2 L of fluid over 24 h.
In vitamin D deficiency, treatment should be with vitamin D itself, for reasons of both cost and safety. Repletion of vitamin D does not bypass the body’s homoeostatic control of plasma calcium concentration, as will occur if 1α-hydroxylated vitamin D metabolites are used. In an adult, adequate vitamin D stores can be achieved by administering 50 000 IU cholecalciferol monthly.
1α-Hydroxylated vitamin D derivatives are preferred in those conditions in which 1α-hydroxylation is impaired and pharmacological doses of vitamin D would otherwise be needed, such as hypoparathyroidism, pseudohypoparathyroidism and vitamin D-dependent rickets (VDDR) type I. Typical doses for an adult would be 1–4 μg/day of alfacalcidol or 0.75–2.25 μg/day of calcitriol. Calcium supplements are not usually required. The aim is to get the plasma calcium concentration just high enough to relieve symptoms, and this is usually achieved around the lower limit of the reference range. Running the plasma calcium concentration higher than this increases the risk of vitamin D toxicity.
PHOSPHORUS METABOLISM
Distribution of body phosphorus
Phosphorus accounts for about 1% of the weight of the elementary composition of the human body; about 23 mol in a 70 kg adult human. Of this, the bulk (85%) is in the skeleton and teeth (largely in the form of hydroxyapatite) and some 14% is located within the cells of the soft tissues. Within different cell types, the phosphorus content may vary from 300 to 1300 mmol/kg. Only 1% of total body phosphorus is present in extracellular fluids (Box 6.5).
In the blood, phosphorus is present in both organic forms (phosphoproteins, phospholipids etc.) and the inorganic form (as phosphate). The inorganic moiety, which is what is measured routinely in laboratories, exists predominantly in the form of the ions 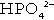 and
and 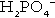 in a ratio of about 4:1. Some 15–20% of inorganic phosphate is non-covalently bound to plasma proteins and is not ultrafilterable.
in a ratio of about 4:1. Some 15–20% of inorganic phosphate is non-covalently bound to plasma proteins and is not ultrafilterable.
Intracellular phosphorus
Phosphorus is important in the metabolism of all cells. It is a key component of a wide array of biological molecules, including the phospholipids of cell membranes and intracellular organelles, nucleic acids, enzyme cofactors and glycolytic intermediaries. Phosphorylation or dephosphorylation induces dramatic shifts in the activity of many enzymes and cellular signalling proteins. Organophosphate compounds such as adenosine trisphosphate (ATP), creatine phosphate and diphosphoglycerate store chemical energy in their high energy phosphate bonds. Hydrolysis of ATP provides the main energy source for many metabolic processes and for muscle contraction. In muscle, ATP is replenished by the donation of phosphate groups from creatine phosphate. In the mitochondria, phosphate-containing proteins play essential roles in the electron transport system. Other phosphate-containing intracellular molecules include cyclic adenosine monophosphate and inositol trisphosphate, which act as second messengers.
Although organically-bound phosphorous concentrations within cells are much greater, non-organically bound intracellular phosphate also has important direct effects on cellular energy metabolism. For example, glucose uptake, lactate production and quantity of ATP all vary directly with the intracellular phosphate concentration. The amount of intracellular phosphate is also an important regulator of enzyme activity in the glycolytic pathway. Within red cells, the concentration of 2,3-diphosphoglycerate (2,3-DPG) plays a crucial role in oxygen availability to the tissues. In severe phosphate deficiency, 2,3-DPG synthesis is decreased, which increases the affinity of oxygen to haemoglobin and thus decreases the release of oxygen to the tissues.
Cytoplasmic inorganic phosphate concentrations are similar to those in the extracellular fluid (ECF) (1–1.3 mmol/L), but are not in simple equilibrium. The electrical potential of cells in relation to the ECF means there is a negative potential across the membrane, tending to repel phosphate ions from the cytoplasm. An active process pumps phosphate ions into cells against this membrane potential in order to maintain cytoplasmic phosphate concentrations.
Cytoplasmic inorganic phosphate concentrations change in response to shifts in the balance between the organic phosphorous pool and cytosolic phosphate. Examples of this are in skeletal muscle, when an increase in workload leads to the net degradation of creatine phosphate and generation of cytoplasmic phosphate. In cells in which glycolysis is taking place, such as erythrocytes, a rise in cytoplasmic hydrogen ion concentration stimulates glycolysis. Glycolytic intermediaries, which are organic phosphates, accumulate in the cytoplasm and cause a marked depletion of cytoplasmic phosphate.
Although cells can regulate their steady-state concentrations of phosphate metabolites in the face of changes in extracellular phosphate, severe hypophosphataemia can deplete intracellular phosphate. Large fluxes of phosphate across the cell membrane can, in turn, affect extracellular phosphate concentrations. Various intracellular metabolic disturbances, hormones and hydrogen ion shifts can all cause clinically significant redistribution of phosphate. Examples of mechanisms that permit shifts of phosphate into cells include systemic alkalosis, the stimulation of adrenoreceptors by catecholamines or sympathomimetic agents, and the insulin-mediated entry of glucose into cells.
Phosphate homoeostasis
The main sources of phosphate transfer to and from the plasma pool are the intestine, bone, soft tissues and the kidneys. Under steady-state conditions, the net intestinal phosphorus absorption equals the net urine excretion (Fig. 6.7). Small amounts of phosphate (~ 1 mmol/24 h) may be lost in the sweat. The phosphate content of breast milk is ~ 1.8 mmol/L, and a fully breast-feeding woman will lose ~ 1.4 mmol/24 h in the milk.
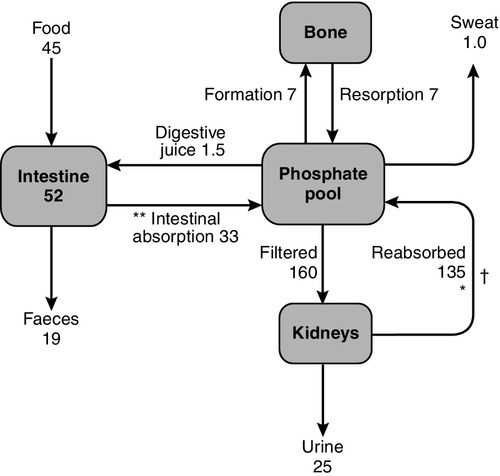
There are two interrelated mechanisms that react to variations in phosphate status in order to maintain phosphate homoeostasis, despite fluctuations either in supply (diet, intestinal absorption) or demand (growth, mineralization, cellular metabolism). These two mechanisms are mediated by:
• fibroblast growth factor 23 (FGF23), synthesis and release of which is stimulated by hyperphosphataemia.
1,25-Dihydroxyvitamin D regulates plasma phosphate primarily by its stimulatory action on intestinal phosphate absorption, whereas FGF23 primarily regulates plasma phosphate by decreasing renal tubular phosphate reabsorption. The two hormonal systems interact: FGF23 production is activated by 1,25(OH)2D, and in turn potently inhibits the activity of 1α-hydroxylase required for synthesis of 1,25(OH)2D. Fibroblast growth factor 23 is most highly expressed in bone, particularly by differentiated osteoblasts and osteocytes, but is also expressed in thymus, brain and heart. It signals through a co-receptor complex comprising the FGF receptor 1 and the β-glycosidase-like protein, klotho, and is catabolized intracellularly by subtilisin-like enzymes that cleave the native protein at a specific site to generate fragments that are thought to be biologically inert. Murine transgenic experiments demonstrate a critical role for FGF23 in phosphate homoeostasis: deletion of the FGF23 gene is accompanied by hyperphosphataemia, enhanced renal tubular reabsorption of phosphate, and elevated concentrations of 1,25(OH)2D; overexpression of the protein causes renal phosphate wasting, decreased concentrations of 1,25(OH)2D and osteomalacia. Fibroblast growth factor 23 induces phosphaturia by decreasing expression of the two active phosphate transporters in the luminal membrane of the proximal renal tubule, the types 2a and 2c sodium-phosphate co-transporters (NaPi-2a and NaPi-2c, respectively). It further lowers plasma phosphate by inhibiting the activity of renal 1α-hydroxylase and increasing the activity of the 25(OH)D-24-hydroxylase, thereby decreasing synthesis of 1,25(OH)2D, and intestinal phosphate absorption. The concentration of FGF23 rises in response to an increase in plasma phosphate as glomerular function declines, and, by directly promoting an increase in urinary phosphate excretion and indirectly reducing intestinal phosphate absorption, allows the maintenance of normal plasma phosphate concentration in early renal failure.
Fibroblast growth factor 23 is the best characterized of a group of circulating compounds that induce phosphaturia, collectively termed ‘phosphatonins’. Other putative phosphatonins, such as the skeletal product matrix extracellular phosphoglycoprotein (MEPE), and secreted frizzled-related protein 4, may be involved in the pathogenesis of pathological states of renal phosphate wasting, hypophosphataemia and osteomalacia (including X-linked hypophosphataemic rickets and oncogenic osteomalacia), but no evidence currently exists for a role of either in physiological phosphate homoeostasis.
Dietary phosphate and intestinal absorption
Phosphate is present in a wide range of foodstuffs, so that if the diet is adequate in other nutrients, it is usually also adequate in phosphate. A typical Western diet contains around 0.65 mmol/kg body weight/24 h. There is a considerable capacity to adapt to low phosphate intake, so it is difficult to establish a minimum dietary requirement. Hypophosphataemia resulting solely from inadequate dietary intake is extremely rare. A measurable increment in 1,25(OH)2D production, one of the adaptive responses, occurs when dietary phosphate is reduced to < 0.32 mmol/kg per 24 h.
Phosphate absorption takes place most efficiently in the duodenum and jejunum, though the ileum contributes most in absolute quantity because of its greater length. Approximately 67% of dietary phosphate is absorbed from the jejunum and ileum even under conditions of phosphate repletion, by a passive non-saturable mechanism. Thus, net absorption increases in parallel with dietary phosphate content. The active, 1,25(OH)2D-dependent, saturable component of phosphate absorption mainly increases jejunal uptake. The bioavailability of intestinal phosphate is reduced by a high calcium intake, as calcium forms insoluble complexes with phosphate in the intestinal lumen. Aluminium salts react similarly. Phosphate is secreted in the digestive juices at a rate of ~ 0.1 mmol/kg per 24 h, about 67% of which is reabsorbed from the intestine. Phosphate fluxes are summarized in Figure 6.7.
The renal tubular reabsorption of phosphate
A high proportion of the phosphate in plasma is ultrafilterable and about 75% of filtered phosphate is reabsorbed by the proximal tubules. A further 5–20% is reabsorbed in the distal tubules and/or cortical collecting ducts and the remainder (20–30 mmol/24 h in an adult) appears in the urine.
If phosphate is infused intravenously and the urinary excretion rate of phosphate measured, then above a certain plasma concentration of phosphate all additional increments in the filtered load will be paralleled by the same increment in urinary phosphate; that is, the relationship between the phosphate excretion rate and the plasma phosphate is linear, with a slope that equates to glomerular filtration rate at high (saturating) filtered loads. Extrapolating this line back to where the urine phosphate excretion rate is zero defines the threshold of glomerular filtrate (and hence plasma) phosphate concentration (TmP, tubular maximum for phosphate) that the renal tubular homoeostatic mechanism is seeking to maintain, in relation to glomerular filtration rate (GFR). In vivo, this can be determined by calculating the clearance of phosphate relative to that of creatinine on a fasting urine sample and using the nomogram of Walton and Bijvoet (1975), which relates this value to the prevailing plasma phosphate concentration (TmP/GFR). This is performed fasting to avoid changes in plasma phosphate due to food intake, and requires a urine sample and a plasma sample (see Appendix 6.4, for method).
Factors regulating TmP/GFR
A number of endocrine and non-endocrine factors are involved in the regulation of TmP/GFR and hence plasma phosphate (Table 6.7). The most important are FGF23 and PTH. Fibroblast growth factor 23 production is affected by perturbations in plasma phosphate, such that a rise in its concentration stimulates an increase in FGF23 production, which in turn increases urinary phosphate loss. It is not clear whether plasma phosphate concentrations directly affect FGF23 production, or do so by an as yet unknown intermediary pathway. Changes in plasma phosphate do not directly affect PTH secretion, but PTH acts on the renal tubules, to reduce TmP/GFR. Plasma phosphate concentrations are thus usually low when there is an elevation in PTH (e.g. in primary hyperparathyroidism or secondary hyperparathyroidism due to vitamin D deficiency) and renal function is normal. TmP/GFR is increased when PTH concentrations are low, or there is insensitivity to the hormone (e.g. in hypoparathyroidism or pseudohypoparathyroidism). Parathyroid hormone-related peptide has similar effects to PTH on renal phosphate handling. The phosphaturic effects of PTH and PTHrP result from action at multiple sites along the nephrons and are mediated through cAMP production.
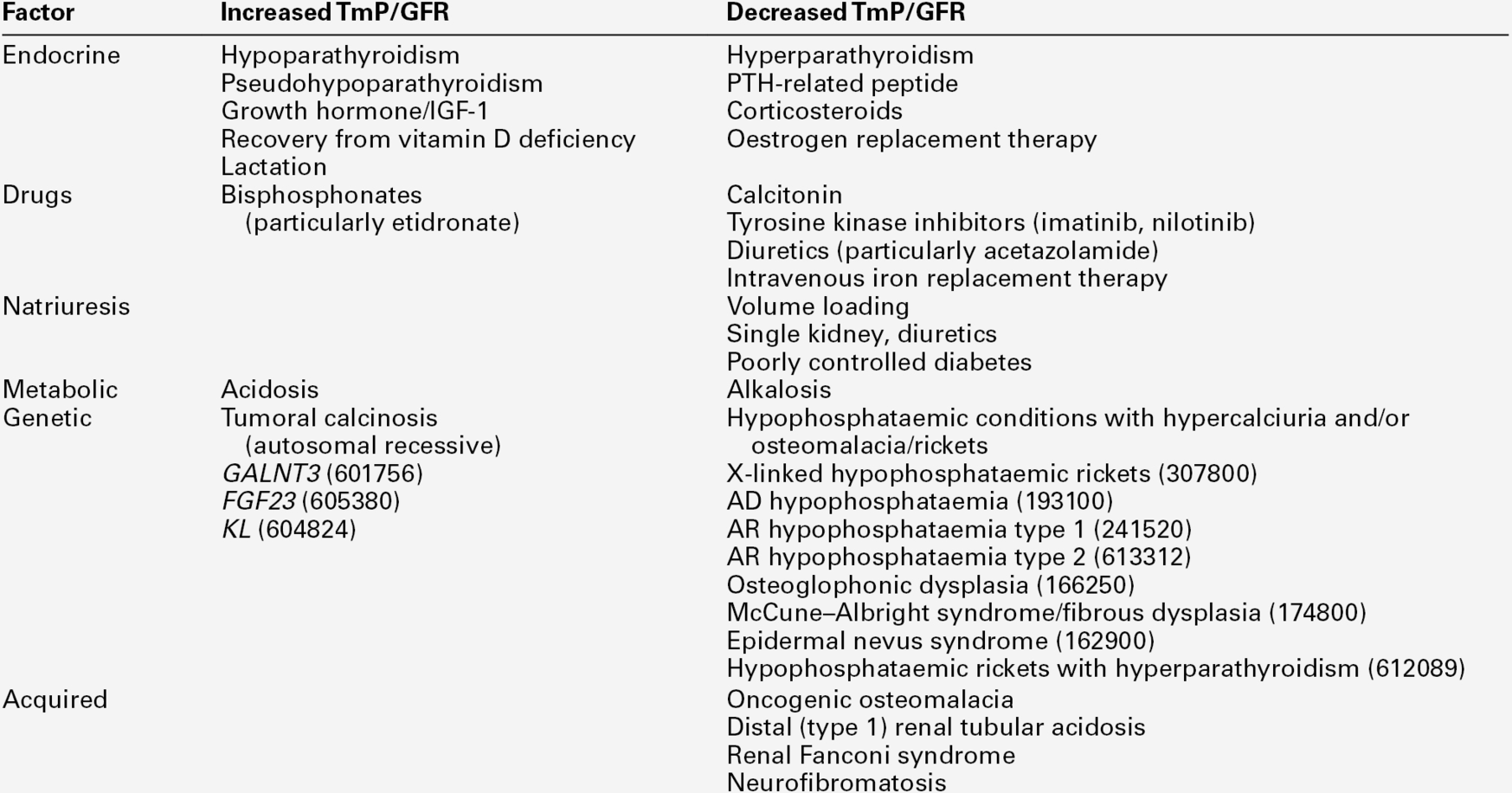
The high TmP/GFR seen in childhood and in acromegaly is mediated through insulin-like growth factor 1 (IGF-1). TmP/GFR is increased in thyrotoxicosis and decreased by corticosteroids.
The increase in plasma phosphate seen after the menopause is abolished by oestrogen replacement therapy. There is little evidence that 1,25(OH)2D greatly affects renal phosphate handling. Calcitonin, given in pharmacological doses, acutely decreases the TmP/GFR. In general, acidosis (whether respiratory or metabolic) reduces renal tubular phosphate reabsorption, whereas alkalosis has the opposite effect. Calcium itself can affect phosphate transport in the kidneys. In healthy subjects, this is difficult to demonstrate because of concurrent changes in PTH, but calcium infusion in hypoparathyroid subjects ultimately increases urinary phosphate losses and reduces plasma phosphate by reducing TmP/GFR.
Various drugs may induce changes in TmP/GFR. In general, these changes are small, the exceptions being glucocorticoids and acetazolamide, which reduce TmP/GFR, and the bisphosphonate, etidronate which, when given at doses of 10 mg/kg, causes a dose-dependent increase in TmP/GFR and plasma phosphate.
Renal phosphate handling may also be affected by haemodynamic changes. Patients with a single kidney (e.g. transplant recipients or live kidney donors) have a high filtered load per nephron and lower TmP/GFR as an adaptation. Phosphaturia also accompanies the natriuresis that follows volume loading, poorly controlled diabetes or diuretic use, because renal phosphate transport is related to sodium transport. Parathyroid hormone is needed for the full expression of this phenomenon.
Disorders of renal phosphate metabolism
There are a number of conditions that share as a central feature a combination of low TmP/GFR, hyperphosphaturia, hypophosphataemia and osteomalacia: the defect may be inherited or acquired. The clinical manifestations depend on the severity of the renal phosphate leak, the degree of response of the 1α-hydroxylase enzyme to the ensuing hypophosphataemia, the age at onset and associated defects. In some instances (e.g. in some patients with idiopathic hypercalciuria), the primary defect appears to be a low TmP/GFR with modest hypophosphataemia and, in response to this, there is an appropriate increase in 1,25(OH)2D production. Intestinal absorption of calcium and phosphate is enhanced and the extra calcium that is absorbed appears in the urine, predisposing to renal stones.
There are several syndromes of hyperphosphaturia and hypophosphataemia, accompanied by inappropriately low concentrations of 1,25(OH)2D and osteomalacia, including autosomal dominant hypophosphataemic rickets (ADHR), X-linked hypophosphataemic rickets (XLH) and oncogenic osteomalacia (OOM) (Table 6.7). In most instances, an abnormality in FGF23 metabolism underlies the phenotype.
Oncogenic osteomalacia is a paraneoplastic syndrome, in which tumours of mesenchymal origin elaborate and secrete excess amounts of FGF23, and possibly other phosphatonins, such as MEPE or secreted frizzled-related protein 4. Detection and removal of the tumour reduces FGF23 concentrations and corrects the biochemical and skeletal phenotype. In autosomal dominant hypophosphataemic rickets, affected individuals harbour a mutation in the gene encoding FGF23, which renders the full-length protein resistant to normal (inactivating) proteolysis. Patients with X-linked hypophosphataemia carry a mutation in the PHEX (phosphate-regulating gene with homologies to endopeptidases on the X chromosome) gene, which encodes an endopeptidase that is believed to sequester and stabilize a secreted FGF23-stimulating bone matrix protein. Inactivation of PHEX allows accumulation of the matrix-bound FGF23-stimulating protein and an increase in local, and then systemic, FGF23 production. Whether the PHEX endopeptidase also inactivates FGF23 in vivo is currently uncertain, but thought to be unlikely.
In all three of these phosphaturic syndromes, circulating concentrations of FGF23 are typically elevated. Circulating concentrations of FGF23 can also be elevated in patients with polyostotic fibrous dysplasia/McCune–Albright syndrome (caused by somatic activating mutations in GNAS1) in 50% of whom renal phosphate wasting occurs. Two autosomal recessive conditions are associated with FGF23-dependent hypophosphataemia. In type 1, autosomal recessive hypophosphataemic rickets (ARHR), inactivating mutations of DMP1, which encodes an osteocyte protein that restrains FGF23 production, produce a phenotype similar to that of ADHR. Inactivating mutations of ENPP1, which encodes an enzyme important in regulating hydroxyapatite deposition, cause type 2 autosomal recessive hypophosphataemic rickets. This is a rare syndrome of generalized arterial calcification of infancy and, in those who survive to adulthood, FGF23-dependent hypophosphataemia.
The recessively inherited condition, hereditary hypophosphataemic rickets with hypercalciuria, is caused by inactivating mutations in the gene SLC34A3, which encodes the NaPi-2c transporter. In this hypophosphataemic disorder, which is not mediated by FGF23, chronic hypophosphataemia is accompanied by an appropriate increment in 1α-hydroxylase activity and plasma 1,25(OH)2D. In a few patients, a milder phenotype of hypophosphataemia with nephrolithiasis and osteopenia has been associated with mutations in NaPi-2a. The bone diseases associated with hypophosphataemia are discussed in Chapter 31.
Plasma phosphate concentrations
There are marked age-related changes in plasma phosphate, with the highest concentrations seen in early infancy when growth velocity is highest. Throughout childhood and adolescence, plasma phosphate concentrations remain higher than in adults. In adulthood, a decline in fasting plasma phosphate concentrations is seen in men after the age of 40. In women, there is little change, but a small increment (~0.07 mmol/L) occurs after the menopause. Plasma phosphate does not change during pregnancy, but is increased during lactation. The age- and lactation-related changes in plasma phosphate are mediated through changes in TmP/GFR (Table 6.8).
TABLE 6.8
Plasma phosphate concentrations according to age and sex
| Age | Sex | Reference range for plasma phosphate (mmol/L) |
| Last trimester in utero | M, F | 0.8–1.4 |
| 1 day | M, F | 1.0–2.6 |
| 1 day–1 month | M, F | 1.8–3.2 |
| 1–12 months | M, F | 1.4–2.1 |
| 1–14 years | M, F | 1.2–1.7 |
| 14–18 years | M, Fa | 1.0–1.6 |
| 18–45 years | M, F | 0.8–1.4 |
| > 45 years | F | 0.8–1.4 |
| > 45 years | M | 0.7–1.2 |
| Lactation | F | 1.1–1.5 |
a The decline in plasma phosphate concentrations from adolescent to adult values occurs, on average, two years earlier in girls than boys. Adult concentrations are reached around age 15 in girls and age 17 in boys. This parallels the earlier start and finish to the adolescent growth spurt in girls.
There is marked diurnal variation in plasma phosphate. During the day, concentrations are substantially higher in the mid-afternoon than in the early morning and there is a second peak in the early hours of the morning (see Fig. 6.4). This early morning peak is attenuated with age. Plasma phosphate rises after food and subsequently falls as phosphate either enters cells or is excreted. The fall in plasma phosphate after a meal is partly attributable to insulin-dependent stimulation of glycolysis, which increases intracellular phosphate utilization.
Hyperphosphataemia
The major causes of hyperphosphataemia are listed in Box 6.6. Hyperphosphataemia may arise through an increase of phosphate input into the blood or decreased excretion. The latter may result from a reduction in GFR or an increase in TmP/GFR. Conditions in which the TmP/GFR is raised, generally cause modest hyperphosphataemia (plasma phosphate 1.4–2.4 mmol/L). Tumoral calcinosis in non-uraemic subjects is a rare recessively inherited disorder associated with a high TmP/GFR, elevated 1,25(OH)D2 concentrations and hyperphosphataemia; a biochemical phenotype that is the mirror image of autosomal dominant hypophosphataemic rickets. It is most commonly caused by inactivating mutations in the GALNT3 gene. GALNT3 encodes a glycosyltransferase known as ppGalNac-T3. This enzyme O-glycosylates the subtilisin-like proprotein convertase recognition site in the FGF23 protein, thereby protecting FGF23 from proteolytic cleavage and permitting intact FGF23 to be secreted. In the absence of this enzyme, most FGF23 is processed into inactive fragments before secretion, leading to low or undetectable circulating concentrations of biologically active FGF23. Tumoral calcinosis can also result from mutations in the genes encoding FGF23 or klotho, leading to impaired secretion of FGF23, and resistance to the actions of FGF23, respectively. A feature of tumoral calcinosis is the presence of large periarticular calcific masses, which are subject to breakdown and chronic inflammation.
Renal impairment, whether acute or chronic, can cause hyperphosphataemia, but this does not usually occur until the GFR falls below 30 mL/min. As renal function deteriorates further, plasma phosphate rises sharply and may reach 6 mmol/L in established renal failure. Other abnormalities of phosphate metabolism that present early in the course of chronic kidney disease are important in the aetiology of hyperparathyroidism and are discussed further in Chapter 31.
The diet rarely contains sufficient phosphate to cause significant postprandial hyperphosphataemia, but excessive input of phosphate can occur with the use of intravenous phosphate salts or phosphate enemas. The latter is particularly liable to occur with children unless the dose of phosphate in the enema is reduced. Massive cell death with release of intracellular phosphate can cause hyperphosphataemia. Examples of the latter include rhabdomyolysis and the tumour lysis syndrome, seen, for example, after the initiation of chemotherapy in childhood leukaemia. It should be emphasized that, as with hypercalcaemia, an increased input of phosphate into the circulation is much more likely to cause severe hyperphosphataemia if there is coexistent renal impairment and thus a reduced capacity to excrete phosphate. This is often the case in critically ill patients.
Plasma phosphate may be artefactually raised (by ~ 30%) in very haemolysed samples or if separation from red cells is delayed. Pseudohyperphosphataemia has also been described with certain chromogenic assay methods of automated analysis in patients with paraproteinaemia or hypertriglyceridaemia.
Consequences of hyperphosphataemia
No specific symptoms are directly attributable to hyperphosphataemia. However, when the product of the calcium and phosphate concentrations in blood exceeds the solubility product (approximately 4.85 × 10− 6 molar units), soft tissue (metastatic) calcification occurs. This is seen most frequently in chronic kidney disease, where calcification occurs in blood vessels, the skin, heart, lungs, kidneys, conjunctivae and around joints. Interstitial calcification of the kidneys is of particular importance, since it impairs renal function further and reduces the ability to excrete phosphate loads. The deposition of calcium phosphate salts in the skin is thought to contribute to the pruritus of chronic uraemia, a symptom that may improve with better control of hyperphosphataemia. The syndrome of progressive ischaemic skin ulceration (calciphylaxis) in patients with established renal failure has also been attributed to calcium phosphate deposition. In this syndrome, ulcers spread rapidly with secondary infection of the necrotic tissues. Histologically, there is extensive arterial and arteriolar calcification and thrombosis. These lesions do not respond to local therapy, but do heal after parathyroidectomy (which reduces the [calcium] × [phosphate] product acutely). Urgent parathyroidectomy is indicated in this condition.
If plasma phosphate rises acutely, then plasma calcium concentrations fall by a mass action effect; this is seen, for example, with the tumour lysis syndrome and rectal or intravenous phosphate administration. This was the basis of the former use of intravenous phosphate in the management of malignant hypercalcaemia. However, the perils of soft tissue calcification induced by this treatment outweigh any benefit.
Diagnostic approach to hyperphosphataemia
The clinical history should reveal excessive exogenous phosphate supply (intravenous or rectal). Massive cell death in, for example rhabdomyolysis or the tumour lysis syndrome, should also be evident from the clinical setting. Rhabdomyolysis can be confirmed by measurement of serum creatine kinase activity.
Diminished excretion of phosphate due to severe renal impairment is evident from measurement of the plasma creatinine. The most convenient method of detecting changes in TmP/GFR is to use the nomogram of Walton and Bijvoet (see Appendix 6.4, below). Other measures of urinary phosphate clearance have theoretical failings compared with the TmP/GFR. In interpreting the result, it is important to bear in mind the normal age-related changes in this index. There is no diagnostic value in measuring the 24 h urine phosphate excretion which, in steady-state conditions, reflects the product of dietary phosphate content and fractional phosphate absorption.
Therapeutic approach to hyperphosphataemia
Hyperphosphataemia due to a raised TmP/GFR is rarely high enough to require specific action. Phosphate control is, however, important in patients with established renal failure, both to limit damaging metastatic and vascular calcification and to control the development of hyperparathyroidism. There are various salts of aluminium, magnesium and calcium which, when administered orally, bind phosphorus in the intestine and limit its absorption. Aluminium hydroxide is the most effective of these agents, but significant quantities of aluminium, which has toxic effects on bone, may be absorbed. Calcium-based phosphate binders (such as calcium acetate or calcium carbonate) are generally used in preference to aluminium salts. However, calcium-based phosphate binders tend to worsen hypercalcaemia in dialysis patients, and have been linked to adverse vascular outcomes, so calcium-free phosphate-binding agents, such as lanthanum carbonate and sevelamer hydrochloride are increasingly used. In dialysis patients, inadequate phosphate control may be due to a number of factors (see Box 31.6).
Acute kidney injury can complicate the tumour lysis syndrome and is attributable to phosphate and urate released from cells. Maintaining a brisk saline diuresis at the time chemotherapy is begun reduces the risk of developing acute kidney injury.
Hypophosphataemia
Mechanisms
Hypophosphataemia can arise by any one or combination of three mechanisms:
• inadequate phosphate absorption from the intestine
• shifts of phosphate from extracellular fluid into cells
• abnormal urinary phosphate losses (Table 6.9).
TABLE 6.9
Mechanisms underlying common causes of hypophosphataemia

a Contribution of phosphate malabsorption depends on 1,25(OH)2D response to hypophosphataemia.
As indicated earlier, it is very rare that the diet is so inadequate in phosphate that, on its own, it can cause hypophosphataemia. However, chronic malnutrition can result in a deficit of whole body phosphate such that, when conditions occur in which there is a shift of phosphate from the extracellular to the intracellular compartment, such as during refeeding, profound hypophosphataemia may develop. Inadequate dietary phosphate may thus be a predisposing factor to, rather than a direct cause of, hypophosphataemia. Clinically significant hypophosphataemia due to the use of phosphate-binding antacids has been described, but this is now rare because of the availability of alternative therapies for peptic ulcer disease.
Hypophosphataemia can result from the acute shift of phosphate into cells. Less than 1% of total body phosphate is in the extracellular space, so small shifts of no more than 6 or 7 mmol can induce significant changes in plasma phosphate concentrations. If this occurs without prior intracellular phosphate depletion, then the condition is usually benign and self-limiting. The most frequently encountered conditions in which this phenomenon occurs are the following.
• Respiratory alkalosis. Hyperventilation due to any cause will reduce the carbon dioxide content of the blood and cells. Intracellular hydrogen ion concentration falls, which activates the enzyme phosphofructokinase, which in turn accelerates phosphorylation of glucose. Phosphate is taken up rapidly from the plasma and hypophosphataemia results. Examples of clinical situations in which this mechanism may occur include liver failure, severe burns, salicylate poisoning, alcoholic ketoacidosis, alcohol withdrawal and sepsis. Hyperventilation in these situations may be the result of either direct metabolic effects or simply fear and pain. It is commonly observed in hospitalized patients, but is typically mild and transient and does not require specific treatment; correction of the underlying cause of hyperventilation also corrects the hypophosphataemia.
• Increased muscle uptake. Prolonged intense exercise can increase uptake of phosphate into muscle in an attempt to replenish creatine phosphate stores. This has caused profound hypophosphataemia in marathon runners.
Increased urinary phosphate loss is usually a chronic phenomenon resulting from a low TmP/GFR and gives rise to moderate hypophosphataemia and osteomalacia (see above, and Chapter 31). In uncontrolled diabetes, osmotic diuresis causes loss of phosphate in the urine, and this contributes to the occurrence of hypophosphataemia once insulin therapy is begun. A reduction in TmP/GFR also contributes to the hypophosphataemia that can occur in paracetamol overdose, even in the absence of significant hepatic damage. A reduction in TmP/GFR, along with other renal tubular function abnormalities, may be seen in patients with severe alcohol dependency. It reverses on abstinence.
Consequences of hypophosphataemia
Mild to moderate hypophosphataemia (0.35–0.80 mmol/L in an adult) is not harmful in the short term, but if chronic it may induce osteomalacia. When plasma phosphate concentrations fall below 0.35 mmol/L, the syndrome of acute phosphate deficiency may develop. The major manifestations are listed in Table 6.10.
TABLE 6.10
The acute phosphate deficiency syndrome
| System affected | Mechanism/site | Clinical consequence |
| Haemopoietic system | ↓ 2,3-DPG in red cells, reduced O2 release to peripheral tissues | Tissue hypoxia |
| ↑ Red cell fragility | Haemolysis (plasma phosphate < 0.1 mmol/L) | |
| Abnormal leukocyte function | ↓ Resistance to infection | |
| Abnormal platelet function | ↓ Platelet survival, abnormal clotting | |
| Striated muscle | ↓ Contractility of diaphragm, heart, skeletal muscles | Respiratory failure ↓ Stroke work Stiffness, weakness, debility |
| Rhabdomyolysis | Muscle pain, weakness | |
| ↑ Plasma creatine kinase | ||
| Nervous system | Brain | Lethargy, confusion, irritability, dysarthria, tremor, seizure, coma |
| Peripheral nerves | Paraesthesiae | |
| ↓ conduction velocity | ||
| Gastrointestinal | Smooth muscle | Gastric atony, ileus |
Diagnostic approach to hypophosphataemia
The acute phosphate deficiency syndrome often arises in patients who are critically ill from other causes, and the symptoms of phosphate deficiency may mimic features of these illnesses. Examples of this include deteriorating mental function simulating encephalopathy in the hepatic failure patient, and tremor and irritability simulating alcohol withdrawal symptoms. The detection of severe phosphate deficiency thus depends on anticipating the circumstances in which it might occur and requesting laboratory confirmation. In these circumstances, measurement of plasma phosphate is usually sufficient, but assessments of recent dietary and intravenous phosphate intake may also be helpful. In all patients with severe acute hypophosphataemia, phosphate disappears from the urine so little information can be gained from its measurement.
In the case of chronic hypophosphataemia causing osteomalacia, the critical investigation is determination of the TmP/GFR. An inappropriately low TmP/GFR should prompt evaluation for one of the causes of renal phosphate wasting, discussed under Disorders of renal phosphate metabolism (see p. 112). Measurement of serum concentrations of FGF23 can be helpful in identifying FGF23-dependent causes of hypophosphataemia. Development of FGF23 immunoassays is continuing; there is an assay that measures the full-length peptide only and a C-terminal assay that recognizes both full length and the cleaved C-terminal fragment.
Nuclear magnetic resonance has been used to detect changes in intracellular phosphate metabolism and has proved particularly valuable in research into muscle diseases. The technique is not, however, generally available for clinical practice. Isotopic tests of intestinal phosphate absorption have also been used in research, but have little practical clinical application.
Therapeutic approach to hypophosphataemia
Mild to moderate acute hypophosphataemia secondary to redistribution (plasma phosphate 0.35–0.80 mmol/L in adults) is usually transient and requires no specific treatment. Severe hypophosphataemia (< 0.35 mmol/L) occurs only when there has been a cumulative net loss of ~ 100 mmol phosphate. Symptoms of hypophosphataemia appear when net losses reach ~ 300 mmol. However, the deficit in total body phosphate cannot be predicted reliably from the plasma phosphate concentration.
Treatment may be given orally in the form of sodium or potassium hydrogen phosphate (100 mmol phosphate daily in divided doses for a week), but often with critically ill patients, intravenous replacement is necessary, particularly in those with symptomatic acute hypophosphataemia. Intravenous therapy can safely be undertaken using monobasic potassium phosphate (KH2PO4) in a dose of 50–100 μmol/kg body weight in half normal saline by continuous infusion over 12 h. This can be repeated every 12 h with monitoring of plasma calcium, phosphate and potassium. Intravenous administration can be stopped when plasma phosphate has risen above the symptomatic watershed of 0.35 mmol/L, assuming the rest of the deficit can be replaced orally.
Prevention is appropriate for at-risk patients. For example, many patients receiving parenteral nutrition require about 0.5 mmol phosphate/kg body weight/24 h, and occasionally more. Malnourished alcoholic patients receiving intravenous glucose should receive phosphate supplementation too.
The complications of intravenous phosphate administration include hyperphosphataemia, hypocalcaemia, hyperkalaemia and acidosis. Oral phosphate is a laxative that may induce diarrhoea.
MAGNESIUM METABOLISM
Most magnesium is in the mineral phase of bone or within the cells of the soft tissues. After potassium, it is the most abundant intracellular cation. Within cells, magnesium is a cofactor in over 300 enzymatic reactions, involving energy metabolism, control of various calcium and potassium channels, membrane stabilization and neuromuscular excitability, protein and nucleic acid synthesis and oxidative phosphorylation. It is of particular importance in those processes involving the formation and utilization of ATP. All enzymatic reactions involving ATP have an absolute requirement for magnesium. Less than 0.5% of total body magnesium is present in the plasma, although, for practical purposes, measurements of plasma ionized or total magnesium are the only widely used methods for determining magnesium status. However, such measurements may not accurately reflect whole body magnesium status. The body distribution of magnesium in an adult is summarized in Table 6.11. The magnesium content of infants is lower than adults (approximately 10 mmol/kg body weight, compared with 16 mmol/kg body weight in adults).
TABLE 6.11
Magnesium content and distribution in a typical 70 kg adult
| Total (mmol) | Concentration | |
| Bone | 600 | 100 mmol/kg dry weight |
| Intracellular | ||
| Soft tissues | 500 | 12–18 mmol/kg |
| Red cells | 5.5 | 2.7 mmol/L |
| ECF | ||
| Interstitial fluid | 7.0 | 0.7 mmol/L |
| Plasma | 2.5 | 0.8–1.0 mmol/L |
| Ionized | 1.5 | 0.5–0.6 mmol/L |
Plasma magnesium
In plasma, ~ 60% of the total magnesium is in the ionized form and ~ 15% complexed (with phosphate, citrate or bicarbonate), the remainder (~ 25%) being protein-bound, mainly to albumin. Plasma magnesium concentrations can be adjusted for albumin concentration by the formula:
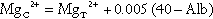 ; where
; where  is the corrected value and
is the corrected value and  is the measured total value. The ionized and complexed magnesium fractions are ultrafilterable. The precise distribution of magnesium in the various plasma fractions may, as in the case of calcium, be altered by changes in protein and hydrogen ion concentrations. Plasma magnesium concentrations do not show any major changes with age or between the sexes, apart from a small increment at the menopause of ~ 0.06 mmol/L. Plasma magnesium falls to premenopausal concentrations with oestrogen replacement, in the same way that calcium does.
is the measured total value. The ionized and complexed magnesium fractions are ultrafilterable. The precise distribution of magnesium in the various plasma fractions may, as in the case of calcium, be altered by changes in protein and hydrogen ion concentrations. Plasma magnesium concentrations do not show any major changes with age or between the sexes, apart from a small increment at the menopause of ~ 0.06 mmol/L. Plasma magnesium falls to premenopausal concentrations with oestrogen replacement, in the same way that calcium does.
Magnesium homoeostasis
Magnesium is widely distributed in foods. Being the mineral ion of chlorophyll, green vegetables are an important source. Daily intakes range from 6 to 20 mmol, with a median of ~ 12 mmol. Hard water contains an appreciable amount of soluble magnesium (up to 5 mmol/L), which may be more available than some of the intracellular magnesium in particular foods. Food processing can remove much of the magnesium from cereals and carbohydrate foodstuffs. The wide reference range for 24 h urinary magnesium excretion (2.0–7.5 mmol) largely reflects different magnesium intakes.
Of the typical dietary intake of 12 mmol, 6 mmol is absorbed, mainly in the small intestine. There are two separate transport systems. One, an active transport, is saturated at low intraluminal concentrations; the other is a passive diffusion mechanism that absorbs a constant fraction (~ 7%) of ingested magnesium. Some magnesium can be absorbed from the large bowel, as demonstrated by the occurrence of hypermagnesaemia after the use of magnesium-containing enemas. About 2 mmol magnesium is secreted into the intestine, thus the net daily intestinal absorption is ~ 4 mmol, which is balanced by excretion of this amount into the urine.
Approximately 75% of the total plasma magnesium is filtered through the glomeruli. Of that, ~ 20% is reabsorbed in the proximal tubules by paracellular absorption. The majority of the ultrafilterable magnesium (~ 65%) is reabsorbed in the cortical thick ascending limb of the loop of Henle. This is a passive paracellular process, driven by a positive transepithelial voltage gradient and mediated by a tight-junction Mg2 + pathway, which involves the molecules claudin-16 and claudin-19.
The distal convoluted tubules regulate the final urinary magnesium excretion by active transcellular reabsorption of Mg2 + (~ 5–10% of ultrafilterable magnesium). Active magnesium reabsorption is a multistep process. A negative membrane potential maintained by the voltage-gated K+ channel Kv1.1 provides the driving force for Mg2 + transport across the apical, epithelial channel, TRPM6 (transient receptor potential cation channel M6). Epidermal growth factor regulates active Mg2 + transport through TRPM6 in a paracrine/autocrine manner. The Na+,K+-ATPase situated in the basolateral membrane generates a local negative membrane potential helping to drive Mg2 + across the TRMP6 channel in the basolateral membrane. The transcription factor HNF1β (hepatocyte nuclear factor 1β) affects the expression of the regulatory protein γ-subunit that binds and modulates Na+K+-ATPase. There is a probable active extrusion mechanism, either via a Mg2 + pump or a Na+–Mg2 + exchanger, with the transmembrane protein CNNM2 (cyclin M2) a likely candidate.
There is no significant reabsorption in the collecting ducts. Between 3% and 5% of the filtered magnesium finally appears in the urine. The kidney has a maximal limit for tubular reabsorption (TmMg), above which all the ultrafilterable magnesium is excreted. Calculating either the TmMg or the fractional urinary magnesium excretion from magnesium infusion experiments can be helpful in characterizing genetic hypomagnesaemic disorders.
Renal magnesium clearance is increased by osmotic diuretics, loop diuretics and thiazides. The kidneys of the premature newborn differ from those of adults in that the proximal tubules can reabsorb up to 70% of the ultrafilterable magnesium.
There are clear homoeostatic mechanisms for regulating magnesium status, although the magnesium ‘sensor’ has not been characterized. The kidneys appear to be the prime organs in the fine regulation of magnesium homoeostasis. In times of magnesium deprivation, the kidneys can reduce magnesium excretion to < 1 mmol/24 h. There is intestinal adaptation too, with the proportion of dietary magnesium absorbed higher (up to 75%) when the diet is magnesium-depleted, but lower (down to 25%) when the load is greater. Intestinal magnesium absorption can be inhibited by binding agents such as cellulose phosphate. Faecal magnesium output is normally < 15 mmol/24 h.
Magnesium appears to regulate parathyroid hormone secretion in a manner similar to calcium, but is only one-half to one-third as potent. Parathyroid hormone secretion is thus stimulated by modest hypomagnesaemia and suppressed by hypermagnesaemia. Paradoxically, profound hypomagnesaemia actually inhibits parathyroid hormone secretion. Although PTH may enhance the renal tubular reabsorption of magnesium, it is probably of little importance in the overall regulation of magnesium metabolism.
Magnesium losses through sweat may be great. At high temperatures, 10–15% of total magnesium output can be found in the sweat. Typical magnesium fluxes in an adult are illustrated in Figure 6.8. The magnesium content of breast milk is ~ 1.6 mmol/L, and a breast-feeding woman may lose up to 1.2 mmol/24 h through this route.
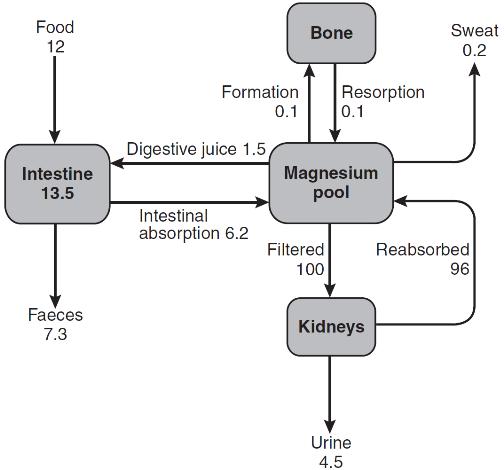
Hypomagnesaemia
Causes
Hypomagnesaemia may arise through inadequate absorption (or intake), by excessive urinary or gastrointestinal losses, or by redistribution of magnesium from extracellular to intracellular sites. Hypomagnesaemia in adults is usually an acquired condition, the major causes of which are listed in Box 6.7. It is important to recognize that several drugs can cause symptomatic hypomagnesaemia, and that withdrawal of the drug concerned will resolve the problem. Most drugs associated with hypomagnesaemia affect renal tubular magnesium reabsorption. The exceptions are proton pump inhibitors (PPI), which probably affect intestinal transport or increase intestinal loss. This is a class effect and hypomagnesaemia will recur if a second PPI is substituted for the first. In adults, severe symptomatic hypomagnesaemia (< 0.35 mmol/L) is seen in patients having platinum-based chemotherapy or taking proton pump inhibitors, or who are very unwell in intensive care settings.
There are a number of familial disorders (predominantly affecting renal tubular magnesium handling) that have helped understanding of magnesium homoeostasis (Table 6.12). The recessively-inherited forms tend to be more severe and present in infancy or childhood. The dominantly inherited forms tend to be milder (although hypomagnesaemia can be severe with KCNA1 mutations, plasma calcium is often unaffected).
TABLE 6.12
Genetic causes of hypomagnesaemia
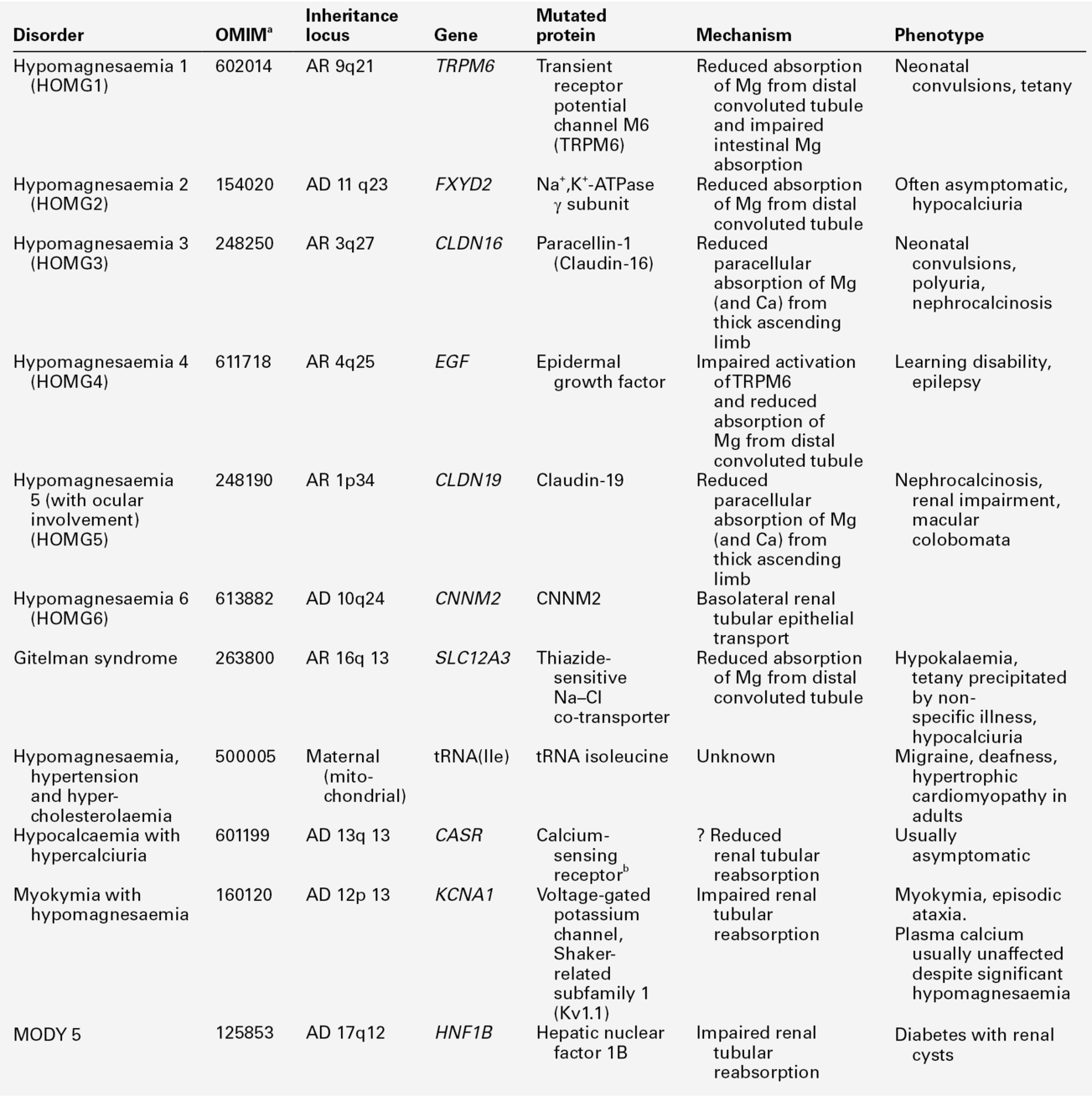
a Online Mendelian Inheritance in Man phenotype catalogue number.
b Indicates an activating mutation; all others are thought to be inactivating.
Consequences
Symptoms directly attributable to hypomagnesaemia occur at plasma concentrations < 0.5 mmol/L and include anorexia, nausea, tremor, apathy, depression, agitation and confusion. Severely hypomagnesaemic patients usually have coexistent hypokalaemia and a cellular potassium deficit; probably because of reduced activity of the magnesium-dependent Na+,K+-ATPase system of cell membranes. Potassium is lost from cells into the ECF and is then lost in the urine because magnesium deficiency also impairs the renal conservation of potassium. If severe hypomagnesaemia is prolonged then hypocalcaemia may also develop, principally because of impaired parathyroid hormone secretion. With the development of hypocalcaemia, neuromuscular signs such as muscle weakness, tremor and twitching, a positive Chvostek sign, delirium, convulsions and ultimately coma may occur.
The effects of hypomagnesaemia on the excitability of nerve, muscle and cardiac cells appear not to be direct since, as far as is known, magnesium is not involved in the normal action potential, in conduction or contraction. The effects appear to be mediated by inhibition of the Na+,K+-ATPase enzyme and, in practice, it is difficult to distinguish direct neuromuscular and cardiac effects of severe hypomagnesaemia from those secondary to alterations in potassium and calcium metabolism.
Cardiac effects
Hypomagnesaemia of a more modest degree (0.5–0.8 mmol/L) is not uncommon and, while it is not associated with obvious symptoms, it is not necessarily benign. In patients admitted to coronary care units, there is a relationship between hypomagnesaemia and the incidence of serious dysrhythmias (including multifocal atrial tachycardia, ventricular tachycardia, ventricular fibrillation and torsades de pointes). However, hypomagnesaemia is strongly associated with hypokalaemia, and this is itself a potent cause of dysrhythmias. The association between low plasma concentrations of magnesium and potassium is again explicable by reduced membrane ATPase activity, a loss of intracellular potassium and subsequent urinary losses. The ratio of intra- to extracellular potassium, which largely determines the resting membrane electrical potential, is reduced with a consequent increase in electrical excitability. Hypokalaemia can be refractory to replacement therapy while magnesium deficiency persists. Some cardiac dysrhythmias are less responsive to antidysrhythmic therapy in the presence of hypomagnesaemia. In addition to the dysrhythmias, magnesium depletion is associated with other electrocardiographic abnormalities, including PR and QT interval prolongation and T-wave flattening. Following acute myocardial infarction, plasma magnesium concentrations typically fall and are at their lowest 12–24 h after admission to hospital. Drug therapy, particularly non-potassium-sparing diuretics, may exacerbate this hypomagnesaemia by increasing urinary losses.
Diagnostic approach to hypomagnesaemia
Plasma magnesium concentration remains the only widely available measure of magnesium status. Attempts to assess intracellular magnesium more reliably have included the measurement of erythrocyte, monocyte and muscle cell concentrations, but these experimental approaches are not widely available. Hypomagnesaemia is common in the critically ill, occurring in up to 50% of such patients. However, only about 25% have low ionized magnesium concentrations.
A magnesium retention test, which aims to detect a total body deficit, has been proposed. The urinary excretion of magnesium is measured over 24 h following the parenteral administration of 0.1 mmol/kg magnesium (in the form of magnesium sulphate). Normally 75–80% of the dose is excreted, but less if there is magnesium deficiency (see Appendix 6.5, for method).
To assess renal magnesium handling in patients with suspected renal tubular abnormalities, the fractional urinary magnesium (FEMg) can be estimated on fasting urine (U) and plasma (P) samples:
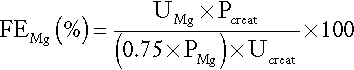
Values > 2%, while the patient is hypomagnesaemic, suggest renal magnesium wasting. Renal tubular magnesium handling can be assessed more rigorously by magnesium infusion testing. It is possible to combine an infusion test to determine TmMg with a magnesium retention test to determine if there is a magnesium deficit (see Appendix 6.6, for a suggested protocol).
Where hypomagnesaemia is due to inadequate intake or malabsorption, urinary magnesium falls rapidly to < 0.7 mmol/24 h from the usual 2–7 mmol/24 h.
The main diagnostic step must remain thinking of hypomagnesaemia as a potential problem and making a request for plasma measurement. Studies comparing physician-initiated testing and ‘routine’ measurement have shown that hypomagnesaemia is very common in hospitalized patients, that it is frequently associated with hypokalaemia and that only about 10% of hypomagnesaemic patients are identified by physician-initiated requests. This has been used as an argument for including magnesium among the routine hospital analyses of plasma ‘electrolytes’.
The ingestion of large quantities of magnesium (in the form of antacids or laxatives) can induce diarrhoea. In magnesium-related diarrhoea, the faecal magnesium content is high (> 15 mmol/24 h). This measure is of occasional value in the diagnosis of factitious diarrhoea.
Therapeutic approach to hypomagnesaemia
Conditions that may benefit from magnesium repletion are listed in Box 6.8. The amount and rate of magnesium administration depends on the cause and severity of the magnesium depletion and on renal and intestinal function.
Symptomatic deficiency is best treated by the intravenous route. Adults with good renal function should be given 12 mmol magnesium (in the form of magnesium sulphate in saline or 5% dextrose) over 2–3 h and a further 12 mmol infused slowly over the following 24 h. This regimen may need to be repeated over several days, as the tissue magnesium deficit takes longer to correct than the plasma concentration. If the plasma concentrations are increased too much, urinary magnesium losses increase. In infants, replacement is given in a similar manner, but the dosage should be reduced to 0.2–0.3 mmol/kg. Plasma magnesium concentrations need to be monitored during and after replacement therapy. The intramuscular route can also be used for replacement, but the injections are painful.
Where feasible, magnesium supplements may also be given orally (5–10 mmol/24 h, in divided doses). Suitable preparations include magnesium oxide and its sulphate and carbonate salts, which are available as antacids or purgatives. Higher doses may cause diarrhoea.
With drug-induced hypomagnesaemia, the offending agent should of course be withdrawn.
Hypermagnesaemia
The kidneys normally excrete excess magnesium very efficiently and can handle loads of up to 400 mmol/24 h, so hypermagnesaemia as a clinical problem is largely restricted to patients with acute or chronic renal failure. Hypermagnesaemia is, however, seen in familial hypocalciuric hypercalcaemia (FHH), and can also occur with magnesium-containing enemas and cathartics. In subjects with renal failure, the potential sources of excess magnesium loads are the dialysate, magnesium-containing antacids (as phosphate-binding agents) and, in acute kidney injury, release of magnesium from tissues.
Hypermagnesaemia in patients with renal impairment can also occur with the use of citrate-gluconic acid solutions. These solutions, which contain substantial quantities of magnesium salts, are used either for bladder irrigation or are infused into the renal pelvis through a nephrostomy tube for the dissolution of renal stones.
Hypermagnesaemia in the order of 1.5–2.5 mmol/L may be associated with hypotension but is commonly asymptomatic. In the range 2.5–5.0 mmol/L, areflexia may be present and electrocardiographic changes occur (prolonged PR and QRS interval, peaked T waves). At higher concentrations, respiratory paralysis and cardiac arrest ensue: concentrations > 8 mmol/L (almost always iatrogenic) are frequently fatal. At these very high plasma concentrations, magnesium has direct effects on neuromuscular cells; inhibiting acetylcholine release and causing peripheral blockade. Use is made of this in the pharmacological management of seizures associated with eclampsia of pregnancy, where an effective remedy is the parenteral administration of magnesium sulphate to maintain plasma concentrations at 3–4 mmol/L.
The treatment of hypermagnesaemia initially consists of cessation of magnesium administration. In an emergency, intravenous calcium gluconate (10 mL, 10% solution; 2.2 mmol calcium) will reverse the effects of hypermagnesaemia. Dialysis is also effective.
CONCLUSION
The physiology and pathology of calcium, phosphate and magnesium are closely related. The principal hormones involved in calcium and phosphate homoeostasis are 1,25-dihydroxyvitamin D, FGF23 and parathyroid hormone. The great majority of calcium and phosphate is present in the skeleton, as is over half the magnesium, but all three ions have many other vital functions.
Pathological increases and decreases in concentrations of calcium, phosphate and magnesium can occur, and all can have serious consequences. For each disorder, a relatively small number of conditions account for the majority of presentations.
Further reading
APPENDIX 6.1 CALCIUM ABSORPTION TEST
This test was originally described by Broadus et al. (1978) for use in the investigation of hypercalciuria, but it can also be used to estimate intestinal calcium absorption in other contexts. The patient takes a low calcium diet (10 mmol (400 mg)/24 h) for one week and is studied after an overnight fast (distilled water only). A water intake of 250 mL/h is maintained from 1 h before the test until its completion. The test consists of three consecutive 2 h urine collections for measurement of calcium and creatinine, with a plasma calcium and creatinine measurement at the midpoint of the first and third of these collections. At the beginning of the second urine collection, a 1 g (25 mmol) oral calcium load is taken.
Interpretation
Reference values:
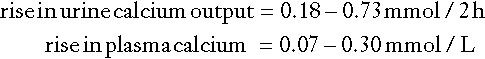
Reference
Broadus AE, Dominguez M, Bartter FC. Pathophysiological studies in idiopathic hypercalciuria: use of an oral calcium tolerance test to characterize distinctive hypercalciuric subgroups. J Clin Endocrinol Metab 1978;47:751–760.
APPENDIX 6.2 ANALYSIS OF TUBULAR HANDLING OF CALCIUM
The relationship between urine calcium excretion per litre of glomerular filtrate (CaE) and plasma calcium has been established from calcium infusion experiments in healthy subjects. The renal tubular handling of calcium can be assessed by reference to this normal relationship. CaE is estimated from:
obtained from the second void sample after rising in the morning, while still fasting. All these concentrations are expressed as mmol/L. The units of CaE are mmol/L glomerular filtrate.
Interpretation
Hypercalcaemic patients falling to the right of the reference range (Fig. 6.9) are excreting less calcium than expected for their prevailing plasma calcium concentration. Provided that this is documented at a time when the patient is not volume depleted, it can then be inferred that abnormally high renal tubular calcium reabsorption is contributing to the patient’s hypercalcaemia. Typically, this is seen in primary hyperparathyroidism, familial hypocalciuric hypercalcaemia and malignant hypercalcaemia due to PTHrP. Similar considerations apply in the investigation of hypocalcaemia. Patients in whom CaE falls to the left of the reference range are excreting more calcium than would be predicted from the low plasma calcium concentration, and it can be inferred that abnormally low renal tubular calcium reabsorption is contributing to the hypocalcaemia (typically seen in hypoparathyroidism).
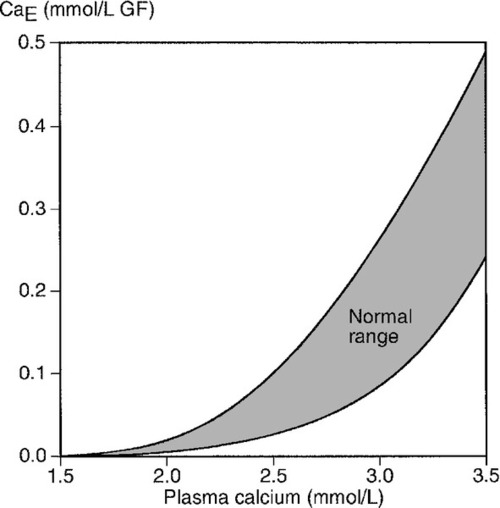
APPENDIX 6.3 CLASSIFICATION OF PSEUDOHYPOPARATHYROIDISM
This protocol is a modification of the Ellsworth–Howard and Chase–Auerbach tests. It measures the response of urine cAMP and TmP/GFR to an injection of synthetic human PTH (1–34).
The test is performed in a fasting patient, who ingests 250 mL/h of water between 06.00 and 12.00 noon. Two 30 min urine collections are collected before 09.00, at which time PTH (1–34), 0.625 μg/kg (maximum dose 25 μg) is infused intravenously over 15 min. Consecutive urine collections are made from 09.00–09.30; 09.30–10.00; 10.00–11.00 and 11.00–12.00, with blood samples taken at 09.00 and 11.00.
Analysis
Urine specimens: cAMP, creatinine, phosphate concentrations.
Blood specimens: creatinine, phosphate concentrations.
cAMP excretion is expressed as nmol/L glomerular filtrate (GF):
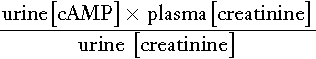
Urine and plasma creatinine concentrations are expressed in the same units.
Phosphate excretion expressed as TmP/GFR (mmol/L glomerular filtrate; see Appendix 6.4).
Interpretation

Reference
Mallette LE, Kirkland JL, Gagel RF et al. Synthetic human parathyroid hormone (1–34) for the study of pseudohypoparathyroidism. J Clin Endocrinol Metab 1988;67:964–72.
APPENDIX 6.4 ESTIMATION OF TMP/GFR
A fasting, second voided urine sample is obtained in the morning. The convention is that this should be a 2 h collection, but the timing is in fact immaterial since time does not enter into the calculation. A plasma sample is obtained at the same time.
Measurements: plasma phosphate and creatinine; urine phosphate and creatinine (all expressed in the same units).
The ratio of the clearance of phosphate (Cphosphate) to the clearance of creatinine (Ccreatinine) is calculated:
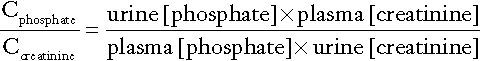
The tubular reabsorption of phosphate (TRP) = 1 − (Cphosphate/Ccreatinine).
TmP/GFR is read from the nomogram which relates the prevailing plasma phosphate concentration [PO4] to TRP (Fig. 6.10).
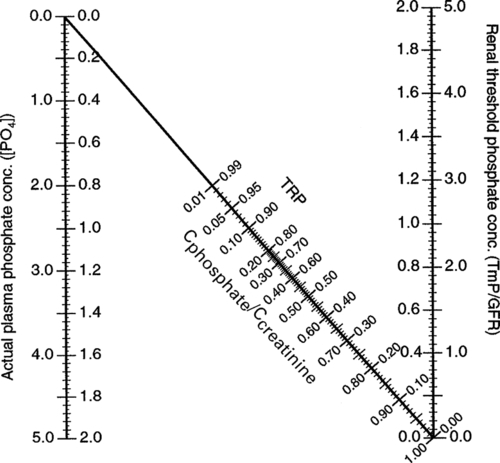
A straight line through the appropriate values of plasma phosphate and TRP (or Cphosphate/Ccreatinine) passes through the corresponding value of TmP/GFR. It should be noted that TmP/GFR and plasma phosphate concentrations are expressed in the same units. Two scales have been given: the 0.0–2.0 scale is suitable for estimating values of TmP/GFR close to the reference range expressed in SI units (0.80–1.35 mmol/L glomerular filtrate) and the 0.0–5.0 scale for values expressed in mass units (2.5–4.2 mg/100 mL glomerular filtrate).
APPENDIX 6.5 MAGNESIUM RETENTION TEST
Suggested schema for clinical use of magnesium tolerance test in normomagnesaemic patients:
1. Collect baseline urine (spot or timed) for magnesium/creatinine molar ratio
2. Infuse 0.1 mmol MgSO4/kg lean body weight in 50 mL dextrose saline over 4 h
3. Collect urine for 24 h (starting with infusion) for measurement of excretion of magnesium and creatinine (Cr) (both in mmol)
4. Calculate % Mg retained using the following formula:
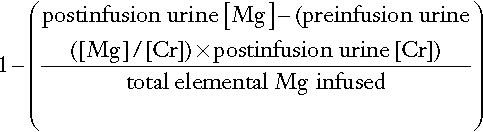
5. Criteria for Mg deficiency: > 50% retention at 24 h.
Reference
From Ryzen E, Elbaum N, Singer FR, Rude RK. Parenteral magnesium tolerance testing in the evaluation of magnesium deficiency. Magnesium 1985;4:137–47. With permission of the publishers Karger, Basel.
APPENDIX 6.6 RENAL TUBULAR REABSORPTION OF MAGNESIUM
1. A fasting urine and blood sample are obtained after an overnight fast.
2. A loading dose of 0.4–0.8 mmol magnesium is infused (1–2 mL 10% MgSO4 solution), and then a continuous infusion of 8 mmol over 2 h (in 50 mL 5% dextrose) is begun.
3. Over the third and fourth hours of the test, 16 mmol magnesium are infused.
4. For the fifth and sixth hours of the test, 32 mmol magnesium are infused. (Quantities shown are for an adult.)
5. Urine specimens are collected hourly for the measurement of magnesium and creatinine concentrations. Blood specimens are collected at the midpoint of each hour for the measurement of magnesium and creatinine.
6. A graph is plotted of magnesium excretion/litre of glomerular filtrate (Mgexcr: y-axis) vs ultrafilterable plasma magnesium (ufMg: x-axis).
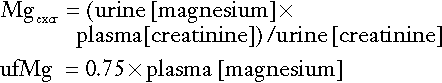
The graph should show a line roughly parallel to the line of identity. The TmMg is the vertical distance between these parallel lines. A typical normal value is ~ 0.6 (meaning that when ufMg is less than 0.6 mmol/L, virtually all filtered magnesium is reabsorbed). In patients with impaired renal tubular reabsorption, TmMg values may be close to zero.
It is possible to combine both retention and tubular reabsorption tests in hypomagnesaemic patients, if the volume of each of the six urine samples is measured.
In addition to calculating magnesium excretion/litre of glomerular filtrate (as above) for each collection, the total amount of magnesium excreted over the 6 h of the test can be calculated. A collection is then made of the next 18 h urine to calculate 24 h urine excretion for determination of magnesium retention (as in Appendix 6.5).