CHAPTER 39
Therapeutic drug monitoring
CHAPTER OUTLINE
Pharmacokinetics and pharmacodynamics
Which drugs should be measured?
USE OF THERAPEUTIC DRUG MONITORING
Relevant clinical interpretation
PROVISION OF A THERAPEUTIC DRUG MONITORING SERVICE
PHARMACODYNAMIC MONITORING, BIOMARKERS AND PHARMACOGENETICS
Analgesic/anti-inflammatory drugs
Antiarrhythmics and cardiac glycosides
Anticonvulsants (antiepileptics)
INTRODUCTION
The aim of therapeutic drug monitoring (TDM) is to aid the clinician in the choice of drug dosage in order to provide the optimum treatment for the patient and, in particular, to avoid iatrogenic toxicity. It can be based on pharmacogenetic, demographic and clinical information alone (a priori TDM), but is normally supplemented with measurement of drug or metabolite concentrations in blood or markers of clinical effect (a posteriori TDM). Measurements of drug or metabolite concentrations are only useful where there is a known relationship between the plasma concentration and the clinical effect, no immediate simple clinical or other indication of effectiveness or toxicity and a defined concentration limit above which toxicity is likely. Therapeutic drug monitoring has an established place in enabling optimization of therapy in such cases.
Pharmacokinetics and pharmacodynamics
Before discussing which drugs to analyse or how to carry out the analyses, it is necessary to review the basic elements of pharmacokinetics and pharmacodynamics. Essentially, pharmacokinetics may be defined as what the body does to drugs (the processes of absorption, distribution, metabolism and excretion), and pharmacodynamics as what drugs do to the body (the interaction of pharmacologically active substances with target sites (receptors) and the biochemical and physiological consequences of these interactions). The processes involved in drug handling are summarized in Figure 39.1, which also indicates the relationship between pharmacokinetics and pharmacodynamics, and will now be discussed briefly. For a more mathematical treatment, the reader is referred to the pharmacokinetic texts listed in Further reading.
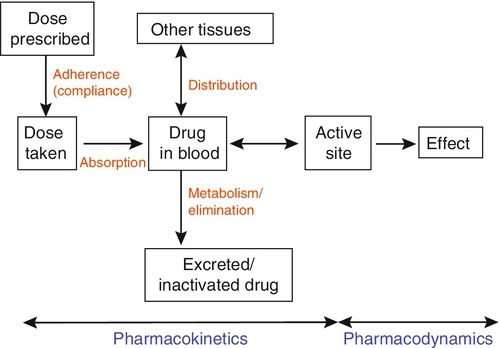
FIGURE 39.1 Processes involved in drug handling.
Adherence
The first requirement for a drug to exert a clinical effect is obviously for the patient to take it in accordance with the prescribed regimen. Patients are highly motivated to comply with medication in the acute stages of a painful or debilitating illness, but as they recover and the purpose of medication becomes prophylactic, it is easy for them to underestimate the importance of regular dosing. ‘Compliance’ is the traditional term used to describe whether the patient takes medication as prescribed, though ‘adherence’ has become the preferred term in recent years as being more consistent with the concept of a partnership between the patient and the clinician. ‘Compliance’, however, remains in widespread use. In chronic disease states such as asthma, epilepsy or bipolar disorder, variable adherence is widespread. The fact that a patient is not taking a drug at all is readily detectable by measurement of drug concentrations, although variable adherence may be more difficult to identify by monitoring plasma concentrations.
Absorption
Once a drug has been taken orally, it needs to be absorbed into the systemic circulation. This process is described by the pharmacokinetic parameter bioavailability, defined as the fraction of the absorbed dose that reaches the systemic circulation. Bioavailability varies between individuals, between drugs and between different dosage forms of the same drug. In the case of intravenous administration, all of the drug goes directly into the systemic circulation and bioavailability is 100% by definition, but different oral formulations of the same drug may have different bioavailability depending, for example on the particular salt or packing material that has been used. Changing the formulation used may require dosage adjustment guided by TDM to ensure that an individual’s exposure to drug remains constant. Other routes of absorption such as intramuscular, subcutaneous or sublingual may exhibit incomplete bioavailability. The total amount of drug absorbed can be determined from the area under the plasma concentration/time curve (see Fig. 39.2).
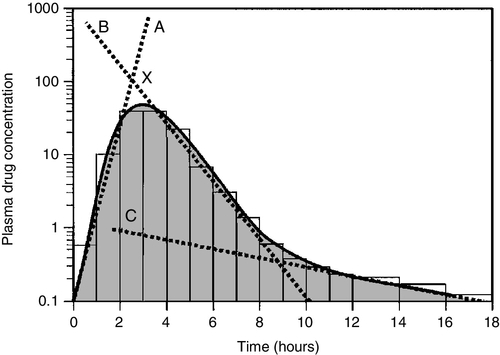
FIGURE 39.2 Concentration/time curve for a drug administered by any route other than intravenously. Line A represents the absorption phase; line B indicates the distribution half-life and line C the elimination half-life. At point X, absorption, distribution and metabolism are all in progress. The areas of the rectangles can be summed to estimate the total amount of drug absorbed, the ‘area under the curve’ (AUC) (hatched area).
Distribution
When a drug reaches the bloodstream, the process of distribution to other compartments of the body begins. The extent of distribution of a drug is governed by its relative solubility in fat and water, and by the binding capabilities of the drug to plasma proteins and tissues. Drugs that are strongly bound to plasma proteins and exhibit low lipid solubility and tissue binding will be retained in the plasma and show minimal distribution into tissue fluids. Conversely, high lipid solubility combined with low binding to plasma proteins will result in wide distribution throughout the body. The relevant pharmacokinetic parameter is the volume of distribution, which is defined as the theoretical volume of a compartment necessary to account for the total amount of drug in the body if it were present throughout the compartment at the same concentration found in the plasma. High volumes of distribution thus represent extensive tissue binding.
After reaching the systemic circulation, many drugs exhibit a distribution phase during which the drug concentration in each compartment reaches equilibrium, represented by Line B in Figure 39.2. This may be rapid (~ 15 min for gentamicin) or prolonged (at least six hours for digoxin). There is generally little point in measuring the drug concentration during the distribution phase, especially if the site of action of the drug for clinical or toxic effects lies outside the plasma compartment and the plasma concentration in the distribution phase is not representative of the concentration at the receptor.
Between-patient variations in volume of distribution caused by differences in physical size, the amount of adipose tissue and the presence of disease (e.g. ascites) can significantly weaken the relationship between the dose of drug taken and the plasma concentration in individual patients and hence increase the need for concentration monitoring.
Elimination (metabolism and excretion)
When a drug has been completely distributed throughout its volume of distribution, the pharmacokinetics enter the elimination phase (line C in Fig. 39.2). In this phase, the drug concentration falls owing to metabolism of the drug (usually in the liver) and/or excretion of the drug (usually via the kidneys into the urine or via the liver into the bile). These processes are described by the pharmacokinetic parameter clearance, which is a measure of the ability of the organs of elimination to remove active drug. The clearance of a drug is defined as the theoretical volume of blood that can be completely cleared of drug in unit time (cf. creatinine clearance, see Chapter 7). Factors affecting clearance and hence the rate of elimination include body weight, body surface area, renal function, hepatic function, cardiac output, plasma protein binding and the presence of other drugs which affect the enzymes of drug metabolism including alcohol and nicotine (tobacco use). Pharmacogenetic factors may also have a profound influence on clearance.
Rate of elimination is often expressed in terms of the elimination rate constant or the elimination half-life, which is the time taken for the amount of drug in the body to fall to half its original value and is generally easier to apply in clinical situations. Both the elimination rate constant and the elimination half life can be calculated from the clearance and the volume of distribution (see Pharmacokinetic texts in Further reading).
With some drugs (e.g. phenytoin), the capacity of the clearance mechanism is limited, and once this is saturated there may be large increments in plasma concentrations with relatively small increases in dose. This phenomenon makes these drugs very difficult to use safely without access to concentration monitoring.
Protein binding
Many endogenous constituents of plasma are carried on binding proteins in plasma (e.g. bilirubin, cortisol and other hormones), and drugs are also frequently bound to plasma proteins (usually albumin and α1-acid glycoprotein). Acidic drugs (e.g. phenytoin) are, in the main, bound to albumin, whereas basic drugs bind to α1-acid glycoprotein and other globulins.
The proportion of drug bound to protein can vary from zero (e.g. lithium salts) to almost 100% (e.g. mycophenolic acid). The clinical effect of a drug and the processes of metabolism and excretion are related to the free concentration of a drug, with drug bound to protein effectively acting as an inert reservoir. Variations in the amount of binding protein present in plasma can thus change the total measured concentration of the drug in the blood without necessarily changing the free (active) concentration, and this is a further reason why measured plasma concentrations may not relate closely to clinical effect. Other drugs or endogenous substances that compete for the same binding sites on protein will also affect the relationship between free and bound drug. For these reasons, it has been suggested that the free concentration of a drug rather than the total (free + protein-bound) concentration should be measured by a TDM service, at least for those drugs exhibiting significant protein binding, e.g. phenytoin. Despite the apparent logic of this idea, it has not been widely adopted because of methodological difficulties and continuing controversy about whether the theoretical benefits are realized in clinical practice. It is nonetheless important to be aware of the effects of changes in protein binding when interpreting total drug concentrations in plasma, especially:
• when there is a highly abnormal binding protein concentration in plasma; for example, in severe hypoalbuminaemia
• when a pathological state (e.g. uraemia) results in the displacement of drug from the binding sites.
The discussion above has outlined the pharmacokinetic factors that govern the relationship between dose and the drug concentration in the plasma or at the active site, and has indicated how these may vary between patients to produce poor correlation between the dose prescribed and the effective drug concentration at the site of action. In general, once steady-state has been reached, plasma concentrations in the individual should exhibit a constant relationship to the concentration at the site of action governed by the distribution factors discussed above, but this may not be the case if blood supply to target tissues is impaired (e.g. for poorly vascularized tissues or a tumour that has outgrown its blood supply (cytotoxic drugs) or a site of infection that is not well perfused (antibiotics)).
Pharmacodynamic factors
Pharmacodynamics is the study of the relationship between the concentration of drug at the site of action and the biochemical and physiological effect. The response of the receptor may be affected by the presence of drugs competing for the same receptor, the functional state of the receptor or pathophysiological factors such as hypokalaemia. Interindividual variability in pharmacodynamics may be genetic or reflect the development of tolerance to the drug with continued exposure. High pharmacodynamic variability severely limits the usefulness of monitoring drug concentrations as they are likely to give a poor indication of the effectiveness of therapy.
Which drugs should be measured?
The above discussion provides a basis for determining which drugs are good candidates for therapeutic drug monitoring. As stated at the beginning of this chapter, the aim of TDM is the provision of useful information that may be used to modify treatment. For this reason, it is generally inappropriate to measure drug concentrations where there is a good clinical indicator of drug effect. Examples of this are the measurement of blood pressure during antihypertensive therapy; glucose in patients treated with hypoglycaemic agents; clotting studies in patients treated with heparin or warfarin, and cholesterol in patients treated with cholesterol-lowering drugs. While plasma concentration data for such drugs are valuable during their development to define pharmacokinetic parameters and dosing regimens, TDM is not generally helpful in the routine monitoring of patients. It may have a limited role in detecting poor adherence or poor drug absorption in some cases. However, where there are no such clinical markers of effect or where symptoms of toxicity may be confused with those of the disease being treated, concentration monitoring may have a vital role.
Therapeutic drug monitoring is useful only for drugs that have a poor correlation between dose and clinical effect (high pharmacokinetic variability). Clearly, if dose alone is a good predictor of pharmacological effect, then measuring the plasma concentration has little to contribute.
However, clinically useful TDM does require that there is a good relationship between plasma concentration and clinical effect. If drug concentration measurements are to be useful in modifying treatment, then they must relate closely to the effect of the drug or its toxicity (or both). This allows definition of an effective therapeutic ‘window’ – the concentration range between the minimal effective concentration and the concentration at which toxic effects begin to emerge – and allows titration of the dose to achieve concentrations within that window. Demonstration of a clear concentration–effect relationship requires low between-individual pharmacodynamic variability (see above), the absence of active metabolites that contribute to the biological effect but are not measured in the assay system and (usually) a reversible mode of action at the receptor site. Reversible interaction with the receptor is required for the intensity and duration of the response to the drug to be temporally correlated with the drug concentration at the receptor.
Many drugs have active metabolites, and some drugs are actually given as pro-drugs, in which the parent compound has zero or minimal activity and pharmacological activity resides in a product of metabolic transformation. For example, mycophenolate mofetil is metabolized to the active immunosuppressant mycophenolate. It will be clear that useful information cannot be obtained from drug concentration measurements if a substantial proportion of the drug’s effect is provided by a metabolite that is not measured and whose concentration relationship to the parent compound is undefined. In some cases (e.g. amitriptyline/nortriptyline), both drug and metabolite concentrations can be measured and the concentrations added to give a combined indication of effect, but this assumes that drug and metabolite are equally active. In other cases (e.g. carbamazepine and carbamazepine 10,11-epoxide), the active metabolite is analysed and reported separately.
Therapeutic drug monitoring is most valuable for drugs which have a narrow therapeutic window. The therapeutic index (therapeutic ratio, toxic-therapeutic ratio) for a drug indicates the margin between the therapeutic dose and the toxic dose – the larger, the better. For most patients (except those who are hypersensitive), penicillin has a very high therapeutic ratio and it is safe to use in much higher doses than those required to treat the patient, with no requirement to check the concentration attained. However, for other drugs (e.g. immunosuppressives, anticoagulants, aminoglycoside antibiotics and cardiac glycosides), the margin between desirable and toxic doses is very small, and some form of monitoring is essential to achieve maximal efficacy with minimal toxicity.
The criteria for TDM to be clinically useful are summarized in Box 39.1.
The list of drugs for which TDM is of proven value is relatively small (Box 39.2). Phenytoin and lithium are perhaps the best and earliest examples of drugs that meet all the above criteria and for which TDM is essential. The aminoglycoside antibiotics, chiefly gentamicin and tobramycin, also qualify on all counts. A number of other drugs that are frequently monitored fail to meet one or more of the criteria completely, and the effectiveness of TDM as an aid to management is therefore reduced. The evidence for the utility of monitoring many drugs is based more on practical experience than well-designed studies. However, for newer agents such as the immunosuppressants and antiretroviral drugs, there is good evidence supporting the benefits of TDM in improving clinical outcomes. (Further information is given in Individual Drugs, below.)
USE OF THERAPEUTIC DRUG MONITORING
Once the narrow range of drugs for which TDM can provide useful information has been defined, it should not be assumed that TDM is necessary for every patient on these drugs at every visit. For TDM to be used for maximum patient benefit and optimal cost-effectiveness, six important criteria must be satisfied each time a sample is taken. These are summarized in Box 39.3 and will now be discussed briefly.
Appropriate clinical question
The first essential for making effective use of any laboratory test is to be clear at the start what question is being asked. This is particularly true for TDM requests, and much time, money and effort is wasted on requests where the indication for analysis has not been clearly defined. If the question is uncertain, the answer is likely to be unhelpful.
The two main reasons for monitoring drugs in blood are to ensure effective therapy and to avoid toxicity. Effective therapy requires that sufficient drug reaches the drug receptor to produce the desired response (which may be delayed in onset). Where a drug is prescribed and the desired effect is not achieved, this may or may not be due to insufficient dosage, since there may be other reasons (individual idiosyncrasy, drug interactions etc.) for the lack of effect. Those at the extremes of age – neonates and the very elderly – have metabolic processes that render them differently susceptible even to weight-adjusted doses. For example, in neonates, the metabolism of theophylline is qualitatively as well as quantitatively different from that in older children. In the very elderly, there may be considerable alterations in absorption and also renal clearance. Drug interactions may also produce a reduced clinical effect for a given dose.
In patients on long-term therapy, once a steady-state concentration that produces a satisfactory clinical effect has been obtained, this concentration can be documented as an effective baseline for the individual patient. If circumstances subsequently change, then changes in response can be related back to both the dose and the plasma concentration of the drug. This is particularly important for psychotropic drugs. ‘Baseline’ concentrations may however change over time as disease processes develop, or gradually with increasing age or changes in drug metabolism.
The avoidance of iatrogenic toxicity is probably the most pressing case for the practice of TDM. The aim is to ensure that drug (or metabolite) concentrations are not so high as to produce symptoms/signs of toxicity. Since a narrow therapeutic index is a prerequisite for drugs suitable for TDM, it is inevitable that toxicity will occur in a small proportion of patients, even with all due care being exercised. Toxicity can never be diagnosed solely from the plasma drug concentration, and it must always be considered in conjunction with the clinical circumstances since some patients will show toxicity when their concentrations are within the generally accepted therapeutic range and others will tolerate concentrations outside the range with few or no ill-effects. The advantage of regular monitoring is that such circumstances may be recorded and the range for that individual adjusted accordingly.
There are two main factors that may lead to inappropriately high plasma drug concentrations. The first is an inappropriate dosing regimen, either due to a single gross error or (more often) a gradual build-up of plasma concentration either because the dose is slightly too high for the individual or because of the development of hepatic or renal insufficiency.
The second factor leading to toxicity is pharmacokinetic drug interactions. Patients are often treated with more than one drug, which can interfere with each other’s actions in a number of ways, for example:
• displacement from protein binding sites
• competition for hepatic metabolism
• induction of hepatic metabolizing systems
• competition for renal excretory mechanisms.
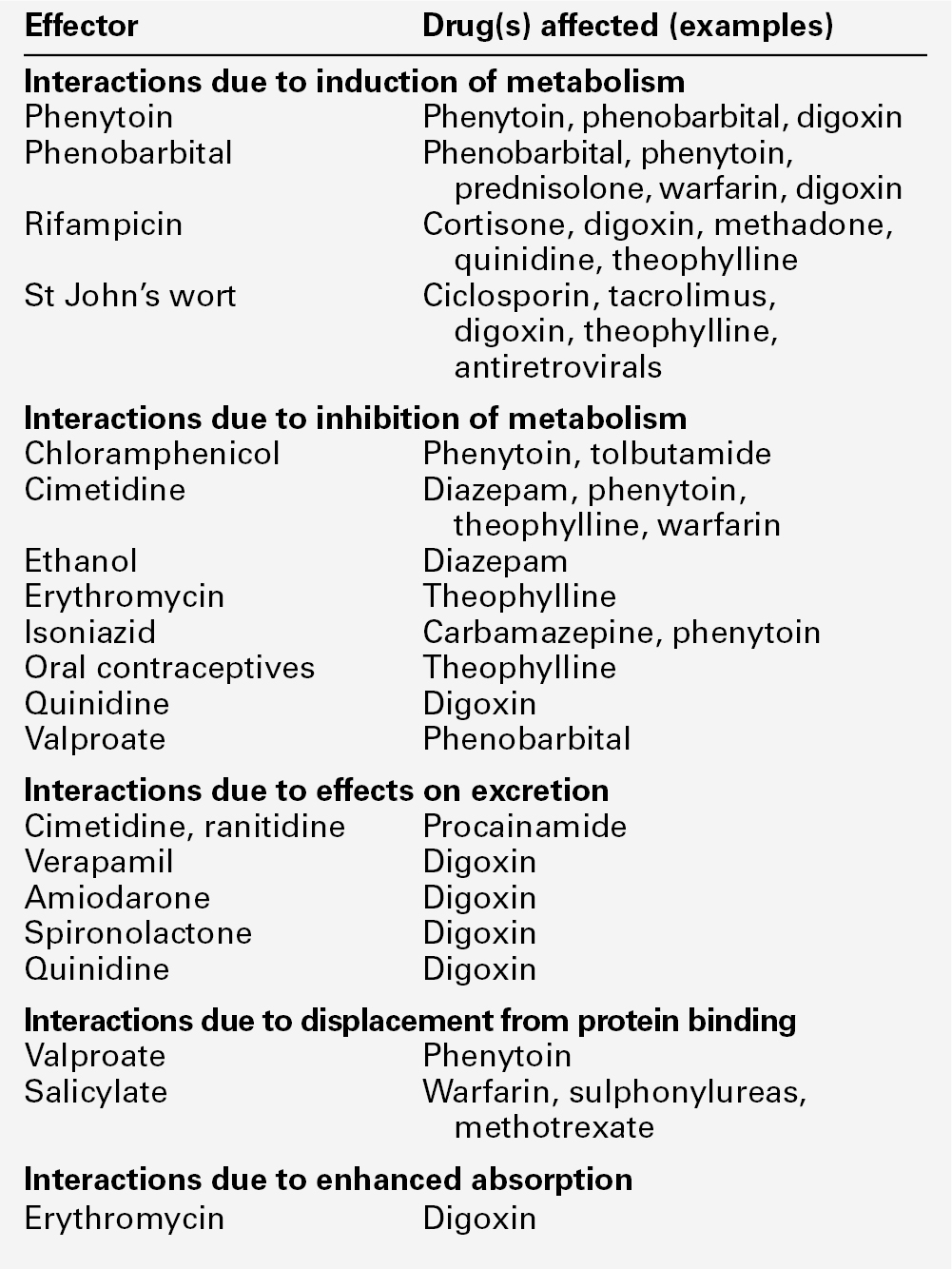
Examples of the more commonly encountered drug interactions involving drugs measured for TDM purposes are given in Table 39.1. There are, however, numerous other examples and the analyst faced with an unusual response or an inappropriate plasma concentration should seek a full drug history to assist in determining the explanation. This history should include specific enquiry about the use of alternative therapies, since many herbal and other non-pharmaceutical remedies (which may not be immediately mentioned by patients) may have significant effects on drug concentrations due to the induction of drug metabolizing enzymes. St John’s wort (Hypericum perforatum), a perennial herb with bright yellow flowers, is one example. It has been shown to induce the hepatic drug-metabolizing enzymes CYP3A4 and CYP2B6, and hence reduce the steady-state concentrations of many drugs.
Therapeutic drug monitoring is particularly useful in confirming toxicity when both under- and overdosage with the drug in question give rise to similar clinical features, for example arrhythmias with digoxin, or fits with phenytoin, and where high drug concentrations give rise to delayed toxicity, for instance high aminoglycoside concentrations, which may give rise to irreversible ototoxicity if prompt action is not taken.
Where a patient is known to be suffering from toxicity and the plasma drug concentration is high, monitoring is often required to follow the fall in concentration following cessation of treatment. For example, in anticonvulsant overdose, it is important for the clinician to know when drug concentration(s) are likely to reach the therapeutic range, since reinstatement of therapy will then be required to prevent seizures.
Other important reasons for therapeutic drug monitoring include guiding dosage adjustment in clinical situations in which the pharmacokinetics are changing rapidly (e.g. neonates, children or patients in whom hepatic or renal function is changing) and defining pharmacokinetic parameters and concentration–effect relationships for new drugs.
The use of TDM to assess adherence (compliance) is to some extent controversial. Clearly, assessment of adherence could provide justification for monitoring every drug in the pharmacopoeia, at enormous cost and for little clinical benefit. Further, application of TDM in this situation is not simple. A patient with a very low or undetectable drug concentration is usually assumed to be non-compliant, but the situation is much less clear when a concentration that is only slightly low (based on population data) is found. Is the patient non-compliant or are other factors (such as poor absorption, induced metabolism or altered protein binding) leading to the low concentration? Much harm may be done in these situations by assumptions of poor adherence to therapy. Adherence can be assessed in other ways, by tablet-counting, supervised medication in hospital or the use of carefully posed questions that are non-judgemental, for example ‘How often do you forget to take your tablets?’ Such approaches are likely to be more effective than TDM in detecting and avoiding poor adherence to therapy, though TDM may have a role in patients with poor symptom control, who deny poor adherence despite careful questioning and interventional reinforcement.
Accurate patient information
Proper interpretation of TDM data depends on having some basic information about the patients and their recent drug history. The importance of this requirement has led many laboratories to design specific request forms for TDM. These are now being superseded by computerized request ordering systems that allow collection of essential items of information at the time of making the request, although it remains important to keep the requesting interface user-friendly and not too complex. Intelligent system design is necessary. Basic information requirements for TDM requests are summarized in Table 39.2.
TABLE 39.2
Information requirements for TDM requests
| Essential | Desirable | |
| Patient | Name | Weight |
| Age | Renal/hepatic function | |
| Gender | ||
| Hospital/health system ID number | ||
| Pathology/clinical details | ||
| Problem | Reason for request | |
| (e.g. poor response, ?toxic) | ||
| Therapy | Drug of interest dose formulation and route of administration duration of therapy date/time last given | Other drugs – list all |
Appropriate sample
The vast majority of TDM applications require a blood sample. In general, serum or plasma can be used, although for a few drugs that are concentrated within red cells, for example, ciclosporin, whole blood is preferable as concentrations are higher and partition effects are avoided. The literature is poor on the differentiation between serum and plasma samples and careful selection of sample tubes is necessary to avoid interferences by anticoagulants, plasticizers, separation gels etc, either in the assay system or by absorption of sample drug onto the gel or the tube, reducing the amount available for analysis. If in doubt, serum collected into a plain glass tube with no additives is usually safest. Haemolysis should be avoided, as it may cause in vivo interference if the drug is concentrated in erythrocytes, or in vitro interference in immunoassay systems. For drugs that are minimally protein bound, there need be no restrictions on the use of tourniquets; however, stasis should ideally be avoided for drugs which are highly bound to albumin, e.g. phenytoin. Care should also be taken to avoid contamination with local anaesthetics, for example lidocaine, which are sometimes used before venepuncture. Where intravenous therapy is being given, care must be taken to avoid sampling from the limb into which the drug is being infused.
Urine is of no value for quantitative TDM. Saliva may provide a useful alternative to avoid venepuncture (especially in children) or when an estimate of the concentration of free (non-protein-bound) drug is required. Saliva is effectively an in vivo ultrafiltrate and concentrations reflect plasma free drug concentrations quite well for drugs that are essentially unionized at physiological pH. Salivary monitoring is unsuitable for drugs that are actively secreted into saliva (e.g. lithium) and drugs that are strongly ionized at physiological pH (e.g. valproic acid, quinidine) as the relationship between salivary and plasma concentrations becomes unpredictable. Careful collection of the sample is required, and the mouth should be thoroughly rinsed with water prior to sampling. Sapid (taste related) or masticatory (chewing on an elastic band) stimulation of saliva flow is used to increase volume. The mucoproteins in saliva make the sample difficult to handle, and centrifugation is usually necessary to remove cellular debris. These and other problems have meant that salivary analysis has not been widely adopted for routine TDM.
It is normally necessary for the patient to be at steady-state on the current dose of drug, i.e. when absorption and elimination are in balance and the plasma concentration is stable. This is true except when suspected toxicity is being investigated, when it is clearly inappropriate to delay sampling until steady-state has been reached. The time taken to attain the steady-state concentration is determined by the plasma half-life of the drug, and the relationship between the number of half-lives which have elapsed since the start of treatment and the progress towards steady-state concentrations is shown in Table 39.3.
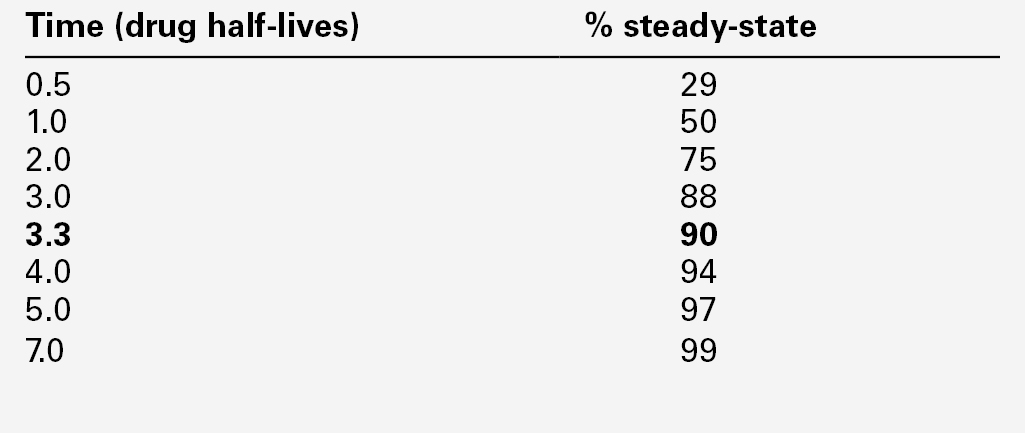
It is frequently stated that five half-lives must elapse before plateau concentrations are achieved, unless loading doses are employed when they are achieved much more rapidly. As shown in Table 39.3, the plasma concentration after 3.3 half-lives is 90% of the predicted steady-state concentration, and this may be taken as the minimum time for sampling for routine purposes after starting a drug or changing the dose. For drugs with a long half-life (e.g. digoxin or phenobarbitone), two weeks or more may be required before steady-state is achieved, especially if the drug is renally excreted and renal function is poor.
In neonates, the rapidly changing clinical state, degree of hydration and dosage requirements make the concept of steady-state a theoretical ideal rather than an attainable goal and there is little value in delaying measurements in the hope of attaining steady-state.
A further requirement for many drugs is for samples to be taken at the appropriate time following the last dose. The size of the fluctuations in plasma concentration between doses depends on the dosage interval and the half-life of the drug. Frequent dosing avoids large peaks and transient toxic effects but is unpopular with patients, difficult to comply with and more likely to lead to medication errors. Less frequent dosing can give rise to large fluctuations in plasma concentration. To some extent, these opposing considerations can be reconciled with the use of sustained-release preparations.
There is no single optimum time for taking samples in relation to dose. The most reproducible for sampling is immediately (< 30 min) before a dose (trough concentration), when the lowest concentrations in the cycle will be obtained. This is the optimum time if an indication of drug efficacy is required, and will show least between-sample variation in patients on chronic therapy. The use of peak and trough concentrations for detecting toxicity of aminoglycoside antibiotics has become less relevant with the advent of once-daily dosing regimens, but sampling at two hours post-dose for ciclosporin can give an excellent estimate of the probable area under the curve and has become a common and effective TDM technique, though sampling in the middle of the absorption/distribution phase in this way does require very accurate standardization of sampling time in relation to the last dose. In the case of digoxin, a specimen should not be taken within 6 h of the dose, since the digoxin absorption and distribution peak may be extremely sharp and high (Fig. 39.3) and serum digoxin concentrations in this period do not reflect tissue concentrations.
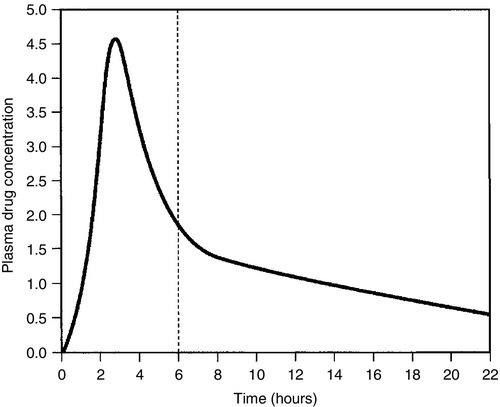
FIGURE 39.3 The concentration/time curve for a patient given digoxin orally. Note the sharp peak and relatively stable plateau concentration after 6 h.
Further details on optimal sampling times for individual drugs are given in the relevant sections of this chapter. When computer programmes are used for pharmacokinetic parameter optimization and dosage prediction, it becomes extremely important that dosage and sampling times are accurately known, as large prediction errors can result from inaccurate data.
Accurate analysis
Establishing a relationship between plasma drug concentrations and effect requires accurate, precise and reliable analytical methods. The choice of methods is vast, encompassing immunoassays (isotopic and optical), chromatography (e.g. gas liquid chromatography (GLC) and high performance liquid chromatography (HPLC), increasingly with mass spectrophotometric detectors) and a variety of novel techniques involving dry chemistry.
Selecting the most appropriate analytical methods is often challenging, and the choice depends on the availability of staff, expertise and equipment, the anticipated batch sizes, the range of drugs to be measured and the turnaround time required, There is no ‘best fit’ for every situation. Some guidelines are given in Table 39.4. Assays used in routine TDM must be specific, showing no interferences from endogenous compounds, drug metabolites or exogenous sources, and must also work with small sample sizes (certainly < 1 mL plasma/serum and ideally 10–100 μL).
TABLE 39.4
Guidelines for choice of methods for therapeutic drug monitoring analyses
| Purpose for which required | Most appropriate method(s) |
| On-site analyses in clinics | Point of care immunoassay devices |
| Urgent analyses | Optical immunoassays |
| Batch analyses, single drug | Optical immunoassays, HPLC, GC, HPLC/mass spectrometry (MS) |
| Batch analyses, multiple drugs | HPLC, GC (optical immunoassays become uneconomic) |
| Single analyses with metabolite patterns | HPLC/MS or LC/MS |
For some drugs, no choice of method exists.
Most analytical methods determine the total concentration of drug in plasma or serum. As discussed above, it is sometimes useful to obtain an estimate of the free (non-protein-bound) drug concentration. The main methods for separation of bound and free drug prior to quantitation by conventional assays are equilibrium dialysis, ultrafiltration and centrifugation through a fine-mesh membrane. The latter two are non-equilibrium techniques and are therefore much quicker than equilibrium dialysis, which generally requires an overnight incubation. It is often difficult to assess the reliability of different procedures as no single method gives consistently accurate estimates of free drug concentration.
Relevant clinical interpretation
In the 40 years or so that TDM has been practised routinely, it has been shown that having drug concentration measurements available to clinicians does not in itself result in improved clinical care. Improved outcome depends on application of the analytical result to a specific clinical situation with appropriate expertise.
In particular, it is important to understand that the widely quoted (and just as widely misused) ‘therapeutic ranges’ for drugs represent a guide to the approximate concentrations that produce a therapeutic response in the majority of patients, rather than a set of inflexible concentration limits to which dosage regimens must be directed. ‘Target ranges’ has been suggested as a better term, which at least implies that these are aims, rather than that all concentrations within the specified range are therapeutic (and all outside are not). Many patients need plasma drug concentrations above the upper limit of the target range for effective therapy, and such concentrations must not provoke knee-jerk dosage reduction. Specialist clinicians usually appreciate this fact, but non-specialists frequently do not, and laboratory staff or pharmacists have an essential educational role here. Conversely, plasma drug concentrations below the lower limit of the target range may produce a satisfactory response in some patients, and arbitrary dose increases will merely increase the likelihood of toxicity without added benefit. In one of the earliest papers on TDM, Koch–Weser wrote: ‘Therapeutic decisions should never be based solely on the drug concentration in the serum’. The cardinal principle, often repeated but still forgotten, is to treat the patient, not the drug concentration.
Effective action taken
As implied above, drug concentrations above the target range do not invariably require a reduction in dosage. If the patient is symptom-free, a careful search for signs of toxicity should be made. If there is no evidence of toxicity, the patient may be best served by doing nothing, although for some drugs (e.g. phenytoin) continued monitoring for the development of long-term undesirable effects is advisable. Similarly, drug concentrations below the target range in a patient who is well and free from symptoms do not necessarily require an increased dose, although in some cases (e.g. digoxin), they may provide evidence that the drug is no longer effective, and that stopping it under medical supervision may be worth trying.
For most drugs, there is a relatively linear relationship between dose and plasma concentration at steady-state, the major exception to this being phenytoin (see later); thus doubling the dose should double the plasma concentration. The difficulty, therefore, is in determining the initial dose to use, especially where a rapid effect, for example the prevention of cardiac arrhythmias, is required. The aim then is to achieve a steady-state concentration rapidly, without waiting for five half-lives to elapse, particularly for drugs with long half-lives. For this reason a ‘loading’ dose of several times the daily maintenance dose is given, effectively reaching steady-state over two half-lives (Fig. 39.4). The calculation of the doses required for loading and maintenance of a steady-state concentration is relatively straightforward and the relevant equations are given in Appendix 39.1.
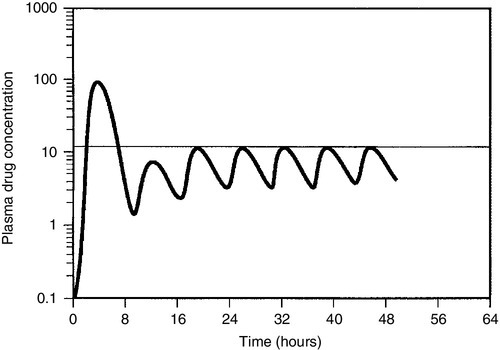
FIGURE 39.4 The effect of a loading dose on the rate of achieving a steady-state concentration
In practice, dosage prediction can be approached in two main ways. It can be estimated from average population parameter values using the equations in Appendix 39.1, or the measurement of one or more drug concentrations at appropriate times in the specific individual allows calculation of patient-specific parameters, which then allows further dosage optimization – TDM-assisted dosage optimization. The details are beyond the scope of this chapter and specialist pharmacokinetic texts (see Further reading) should be consulted. Numerous nomograms and software packages are available for dosage optimization based on TDM data and Bayesian principles. It is vital to ensure that the baseline data are accurate, for example ensuring that the time the drug was actually taken in relation to the sampling time is known (rather than when it was supposed to be taken).
PROVISION OF A THERAPEUTIC DRUG MONITORING SERVICE
The basic essentials for an effective TDM service are the availability of appropriate analytical methods and quality assurance procedures, specialist expertise and the ability to produce results within a clinically relevant time.
Staff
A TDM service requires experienced analysts who understand the basis of the procedures involved and are competent to advise on the analytical sensitivity and specificity of the methods being used and to maintain exacting standards of accuracy, precision and document control. In addition, it is essential that the service has access to specialist clinical advice from someone who understands the principles of pharmacokinetics and therapeutics – an appropriately trained laboratory scientist, a pharmacist or a specialist clinician. The best service is generally obtained with a team approach in which pharmacists, clinical scientists, technical and medical staff all play a role. Difficult cases can then be discussed before any samples are taken and advice obtained on whether concentration monitoring is indicated and on the appropriate timing of any samples required. Following analysis, results are scrutinized by members of the team and appropriate recommendations for action conveyed to the clinician.
However, this ideal degree of involvement is impractical in many hospital situations, and even more difficult for outpatient or primary care work. Provided the request form contains the necessary clinical information (see above), laboratory staff can check these details on receipt of the specimen and advise if the specimen or the reason for analysis is appropriate. Once the analysis is complete, all results (or just those which fall outside defined limits) can be assessed by specialist staff and communicated to the requesting clinician with the appropriate degree of urgency and with recommendations for action where appropriate.
Turnaround time
The ideal turnaround for TDM analyses requires that results are returned to the clinician before the next dose of the drug is due, in order that adjustments can be made immediately. This is not feasible in cases of three or four times daily dosage regimens but, provided that a responsive service is available, patients should not be exposed to inappropriate dosage regimens for more than 24 h after the specimen was taken.
Point-of-care testing
Point-of-care testing is appropriate in some clinical situations since the availability of a plasma drug concentration at the time of consultation allows the clinician to make immediate decisions on dosage adjustments based on objective data. The savings in patient time and improvements in clinic throughput have been shown to outweigh the extra costs of on-site testing in a number of settings. Several methods are available for on-site TDM testing, but need to be carefully evaluated to ensure that their analytical performance is adequate in the setting in which they are used.
Reporting
The reporting of a plasma concentration alone is of limited value to the clinician who is not fully familiar with TDM. Reports that contain information on the reason for the request, the dosage and timing along with the result and appropriate interpretative comments are much more useful. All urgent results should be telephoned and the fact noted. The provision of cumulative reports improves liaison between the laboratory and the clinician, and has been shown to improve patient care.
Units
There has been considerable debate about the appropriate units (mass or molar) for reporting measurements of drug concentration. Most of the toxicological literature has been in mass units, but molar units have theoretical advantages in making it easier to equate drug and metabolite concentrations. Uniformity of reporting is important for patient safety reasons, as confusion over units can result in dangerous misinterpretations. Within the UK, at least, there is now agreed consensus that SI mass units with the litre as the unit of volume should be used for all therapeutic drug assays, except for a specified few which have always been reported in molar units (methotrexate, lithium, thyroxine and iron).
Quality assurance
Measurements of plasma drug concentrations can only be used effectively to diagnose toxicity or under-dosage and to monitor treatment if the measurements are accurate and reproducible. The need for effective internal quality control and external quality assessment procedures for all methods used to produce results is therefore as great as in other areas of clinical biochemistry. Analytical laboratories should be accredited to ISO 15189.
Continuing education
A good TDM service provides an environment for education of trainee staff, not only from biochemistry but from clinical pharmacy and clinical pharmacology. The involvement of staff from other disciplines and their exposure to the analytical and interpretational aspects should be encouraged. Insistence on the provision of full clinical information and the feedback of interpretation on correctly documented cases can lead to considerable improvements in the effectiveness of the service.
PHARMACODYNAMIC MONITORING, BIOMARKERS AND PHARMACOGENETICS
Classic TDM uses drug concentration measurements in body fluids to guide optimization of therapy and minimize adverse consequences. In recent years, other methods of guiding drug therapy have been introduced, and though they do not fit the strict definition of TDM they merit discussion as they are becoming increasingly relevant to the provision of effective drug therapy.
Pharmacodynamic monitoring is the study of the biological effect of a drug at its target site, and has been applied to areas of immunosuppressive therapy and cancer chemotherapy. For example, the biological effect of the immunosuppressive calcineurin inhibitors ciclosporin A and tacrolimus can be assessed by direct measurement of calcineurin phosphatase activity. The main disadvantage of pharmacodynamic monitoring that has emerged to date is that the assays involved are often significantly more complex and time-consuming than the measurement of a drug by chromatography or immunoassay.
Any biochemical measurement that can be used to determine efficacy, extent of toxicity or individual pharmacodynamics for a therapeutic agent is called a therapeutic biomarker. Hitherto, most such markers have been markers of toxicity rather than of therapeutic efficacy (e.g. urinary N-acetylglucosaminidase as an index of tubular damage caused by nephrotoxic drugs), but there is increasing interest in biomarkers that give direct information about drug efficacy, for example red cell 6-thioguanine nucleotide concentrations in the assessment of thiopurine drug efficacy or measurement of inosine 5′-monophosphate dehydrogenase (IMPDH) activity as a marker of the IMPDH inhibitor mycophenolic acid. Biomarker monitoring can provide an integrated measure of all biologically active species (parent drug and metabolites) present, so that therapeutic ranges can be defined more closely. It is also often free from the matrix and drug disposition effects that frequently complicate drug concentration measurements.
Pharmacogenetic studies (studies of genetic influences on pharmacological responses) have wide-ranging clinical relevance. The enzymes responsible for metabolizing drugs exhibit wide interindividual variation in their protein expression or catalytic activity, resulting in quantitative and qualitative differences in drug metabolism between individuals. This variation may arise from transient effects on the enzyme, such as inhibition or induction by other drugs or metabolites, or may result from specific mutations or deletions at the gene level. Pharmacogenetic polymorphism is defined as the existence in a population of two or more alleles at the same locus that result in more than one phenotype with respect to the effect of a drug, the rarest of which occurs with a significant frequency (usually taken as 1%). The term pharmacogenomics is used to describe the range of genetic influences on drug metabolism, and the application of this information to the practice of tailoring drugs and dosages to specific individuals to enhance safety and/or efficacy. This practice, often referred to as ‘Personalized Medicine’, is a major growth area for 21st century medicine.
Determination of an individual’s ability to metabolize a specific drug may be performed either by administering a test dose of the drug or a related compound and measuring the metabolites formed (phenotyping), or by specific genetic analysis (genotyping). The information obtained can dramatically improve the clinician’s ability to select a drug dose appropriate to the specific requirements of the individual. For example, many of the isoenzymes of the cytochrome P450 superfamily responsible for drug oxidation show genetic polymorphisms that affect the extent of drug metabolism and produce differences in clinical response. The CYP2D6 isoform has more than 100 allelic variants, and metabolizes about a quarter of all drugs used in medicine, including many antiarrhythmics and psychoactive drugs. The rate of metabolism of a test dose of debrisoquine or dextromethorphan has been widely used for the determination of CYP2D6 phenotype and the differentiation of poor metabolizer (PM), extensive metabolizer (EM) and ultra extensive metabolizer (UEM) phenotypes. Alternatively, genetic analysis can be used to define the CYP2D6 genotype and identify the alleles associated with the PM phenotype (of which the most common are CYP2D6*3, *4, *5 and*6). Once determined, the phenotype can be used to guide dosing for any of the wide range of drugs metabolized by the CYP2D6 isoform, and ensure that lower doses are used for individuals with the PM phenotype, avoiding toxicity. The PM phenotype is found in 7-10% of Caucasians but less frequently in people of Asian origin.
Pharmacogenetic data have many other clinical applications, including anticoagulation (polymorphisms of the CYP2C9 isoform and the VKORC1 genes in assessing susceptibility to warfarin), oncology/immunosuppression (thiopurine methyltransferase polymorphisms and azathioprine therapy), psychiatry (CYP2D6 isoforms and rate of metabolism of some antidepressants), epilepsy, pain control and other areas. (See Chapter 43 for a further discussion of this topic.)
Integrating information
Pharmacogenetics and new biomarkers provide invaluable adjuncts to conventional drug concentration monitoring, and promise to deliver the ability to create individualized therapeutic regimens. However, integrating the information from all three strands is complex, and will require developments in decision support software and effective strategies for presenting the information in an accessible and comprehensible format to those involved in the provision of care. Pre-treatment pharmacogenetic profiling will allow identification of individuals who are likely to be particularly susceptible or resistant to a proposed regimen, allowing better selection of drug and initial dose. However, the effect of factors such as disease, age or drug interactions means that pharmacogenetics can never tell the whole story, and measuring the concentrations of drugs, metabolites or other biomarkers will still be necessary to complete the picture and deliver truly personalized medicine.
INDIVIDUAL DRUGS
Analgesic/anti-inflammatory drugs
Aspirin (acetylsalicylic acid)
Aspirin (acetylsalicylic acid) is a widely available drug used as a self-prescribed analgesic and as a prophylactic (at low dose) for thromboembolic disease. It is prescribed in relatively high doses for inflammatory arthropathies, since it has both analgesic and anti-inflammatory properties. Aspirin causes gastric erosions and, for this reason, is available in a variety of formulations designed to reduce this side-effect. Once absorbed, aspirin is rapidly converted to salicylate. Salicylate metabolism is complex and variable and its excretion is highly dependent upon urinary acidity, hence the concurrent ingestion of antacids can lead to lower plasma concentrations. The commonest symptom of toxicity is tinnitus, which occurs in the majority of patients once the plasma concentration exceeds 400 mg/L (2.9 mmol/L). Plasma salicylate concentrations may need to be monitored in a minority of patients to avoid toxicity. When used as an anti-inflammatory agent, plasma concentrations should lie between 150 and 300 mg/L (1.1–2.2 mmol/L), measured just before a dose. The half-life of salicylate varies widely between 3 and 20 h, depending on the dose and duration of therapy.
Value of monitoring
low.
Antiarrhythmics and cardiac glycosides
Amiodarone
Amiodarone is used in the treatment of intractable arrhythmias. It can be given intravenously or orally. It is strongly tissue bound and has a very high volume of distribution (about 5000 L), leading to a plasma half-life of approximately 50 days. There is considerable interpatient variability. Effective combined plasma concentrations of amiodarone and its active metabolite, desethylamiodarone, are in the range 0.5–2.5 mg/L (0.7–3.7 μmol/L). Most patients do not require concentration monitoring, but in some patients it may be useful to differentiate treatment failure from poor adherence or suboptimal dosing, while in others it may assist in the confirmation of concentration-related side-effects.
Value of monitoring
moderate.
Digoxin and digitoxin
The cardiac glycosides digoxin and digitoxin increase the force of cardiac contraction and increase cardiac output, and are used in the treatment of cardiac failure. They are also useful in the management of certain supraventricular arrhythmias. Digitoxin is structurally and functionally similar to digoxin, but is less frequently used in the UK.
Digoxin has a long half-life (20–60 h) and is usually given once daily. Clearance is primarily by the kidneys and a reduced glomerular filtration rate (GFR) can lead to toxicity. Effective plasma concentrations are 0.5–2.0 μg/L (0.6–2.6 nmol/L), with concentrations at the lower end of this range (0.5–1.0 μg/L; 0.6–1.3 nmol/L) recommended for the treatment of heart failure. Blood samples should be taken at least 6 h post-dose to allow time for distribution into tissues. Digoxin acts by inhibition of the Na+,K+-ATPase pump in the membranes of cardiac myocytes. Studies on red blood cells have shown that patients on long-term therapy adapt by production of additional pump activity. As a result, the interpretation of plasma concentrations is different depending upon whether therapy is newly instituted or long term. There is no clear relationship between digoxin concentration and therapeutic effect, but toxicity is a major problem and is more likely at plasma concentrations > 2.3 μg/L (3.0 nmol/L). Toxicity at any digoxin concentration is inversely proportional to plasma potassium concentration and, in the presence of hypokalaemia, toxicity may be observed at plasma digoxin concentration as low as 1.2 μg/L (1.5 nmol/L). The patient’s age and the severity of heart disease are independent risk factors for development of toxicity. Although the most common presentation of digoxin toxicity is bradycardia, there may also be other cardiac arrhythmias which, coupled with underlying cardiac pathology, may give rise to a confused clinical picture that may be clarified by the measurement of digoxin concentrations. Significant numbers of patients die with digoxin concentrations > 2.3 μg/L. The possible contribution of digoxin to the cause of death is not always suspected. It should be noted that, if a patient is treated with anti-digoxin antibodies to reverse toxicity (see Chapter 40), further measurements of digoxin concentration will not be possible using the commonly available immunoassays until the antibodies have been cleared, which may take up to a week.
It should be noted that in some patients, notably neonates, there exist in the plasma digoxin-like interfering substances (DLIS) that can lead to falsely elevated ‘digoxin’ concentrations when measured by immunoassay. Elevated DLIS concentrations are encountered in patients with volume-expanded conditions such as uraemia, essential hypertension, liver disease and pre-eclampsia, suggesting that DLIS may be a hormone involved in natriuresis. Falsely low digoxin values due to the presence of DLIS have also been reported. Commercially available immunoassays for digoxin vary in their specificity for DLIS and their ability to differentiate DLIS from exogenous digoxin. The presence of DLIS should be considered if digoxin concentrations are unexpectedly high.
Value of monitoring
moderate to high.
Disopyramide
Disopyramide is effective in controlling supraventricular and ventricular arrhythmias after myocardial infarction. Its use is limited by anticholinergic side-effects including dry mouth, blurred vision and difficulty in urination, and it is contra-indicated in prostatic enlargement. Monitoring can be helpful in ensuring efficacy and avoiding toxicity, though variable protein binding complicates the picture. Monitoring of free disopyramide concentration is recommended, although the assay is not always easily available. Target ranges for trough values are 1.5–5.0 mg/L (4.4–14.7 μmol/L) for total disopyramide and 0.5–2.0 mg/L (1.5–5.9 μmol/L) for free disopyramide.
Value of monitoring
moderate.
Flecainide
Flecainide is used to control serious symptomatic arrhythmias, junctional re-entry tachycardia and paroxysmal atrial fibrillation. It is eliminated via the kidneys (40–45%) and metabolized in the liver, where it is a substrate for the CYP2D6 isoform. The half-life is shortened and plasma concentrations are lower in extensive metabolizers. The usual target range is 0.2–1.0 mg/L (0.5–2.4 μmol/L), with most patients responding in the lower half of the range.
Value of monitoring
moderate.
Procainamide
Procainamide is used to control ventricular arrhythmias after myocardial infarction and atrial tachycardia. It is metabolized in the liver to N-acetylprocainamide (NAPA), which is also active, though slightly less potent. The rate of acetylation of procainamide to NAPA is genetically determined with a bimodal distribution, patients being classified as either slow or fast acetylators. Procainamide has a relatively short half-life (2–5.5 h) and is rapidly cleared. N-acetylprocainamide has a longer half-life (6–12 h) than the parent drug and renal impairment can result in the accumulation of toxic concentrations of the metabolite. Monitoring of procainamide should therefore include quantitation of both procainamide and NAPA. Trough samples are preferable. Procainamide concentrations should fall between 4 and 10 mg/L (17–42 μmol/L). Procainamide and NAPA concentrations combined should not exceed 30 mg/L (100 μmol/L).
Value of monitoring
moderate.
Anticonvulsants (antiepileptics)
Carbamazepine/oxcarbazepine
Carbamazepine is a drug of choice for the treatment of simple and complex focal seizures and generalized tonic–clonic seizures. It is also used for the prophylaxis of bipolar affective disorder and mania, and for pain relief in trigeminal neuralgia. Metabolism is primarily by hepatic oxidation. The major metabolite, the 10,11- epoxide, is active but is seldom measured routinely, which complicates the interpretation of plasma carbamazepine concentrations. The best available information suggests that carbamazepine, when prescribed alone, is effective at plasma concentrations of 4–12 mg/L (17–50 μmol/L). Nystagmus, ataxia and drowsiness may occur at concentrations > 12 mg/L and more serious toxicity > 15 mg/L (60 μmol/L). Protein binding is variable and drug concentrations in saliva have been studied but have not proved to be widely useful. A troublesome erythematous rash occurs in about 4% of patients, and other chronic side-effects including hepatic and haematological problems may occur, although these are not dose related. Carbamazepine induces its own metabolism, and therapy needs to be initiated at a low dose and built up slowly over weeks. The unpredictable relationship between dose and concentration, the narrow therapeutic index and the presence of numerous drug interactions that alter the pharmacokinetics support the requirement for therapeutic drug monitoring until stable therapy is established.
Oxcarbazepine is also licensed for the same indications as carbamazepine. It is effectively a pro-drug for 10- hydroxycarbazepine, which is active. The role of therapeutic monitoring has not been established.
Value of monitoring
high (carbamazepine); low (oxcarbazepine).
Ethosuximide
Ethosuximide is used in the treatment of absence seizures and is administered once daily as it has a long half-life. In most patients, therapy can be optimized on the basis of clinical response and EEG checks. Toxicity is readily recognizable clinically as anorexia, nausea, vomiting and dizziness. There is therefore no real need to measure plasma concentrations except in patients receiving multiple therapy to decide which drug is responsible for symptoms of toxicity. The therapeutic range is usually taken as 40–100 mg/L (280–710 μmol/L) with toxicity likely at concentrations > 150 mg/L (1060 μmol/L).
Value of monitoring
low to moderate.
Phenobarbital/primidone
Phenobarbital (phenobarbitone) is an anticonvulsant drug in its own right and also the active metabolite of primidone. It is effective but may be sedative in adults and cause behavioural disturbances in children, and is therefore not a recommended first- or second-line drug. Phenobarbital has a relatively long half-life and is cleared by both the renal and hepatic routes. There is a wide interindividual variation in handling and, for this reason, there are no well-defined therapeutic limits for plasma phenobarbital concentrations. The majority of well-controlled patients have plasma concentrations of 10–40 mg/L (40–160 μmol/L), although some individuals may require much higher concentrations owing to the development of tolerance. The toxic symptoms associated with inappropriately high dosage progress from increasing drowsiness to coma. Primidone therapy can be monitored by the phenobarbital concentration if required.
Value of monitoring
moderate (phenobarbital); low (primidone).
Phenytoin
Phenytoin is still widely used and is effective for the treatment of tonic-clonic and focal seizures, but is no longer a first- or second-line drug for the treatment of epilepsy. It is well absorbed, has a long elimination half-life and is cleared mainly by oxidation in the liver. Its enzymatic removal is, however, saturable and the concentration at which this occurs varies between individuals. Once saturation has occurred, there is an exponential (zero order) increase in plasma concentration (Fig. 39.5); saturation can occur within the therapeutic range, so small increments in dose can lead to large increments in plasma concentration and severe toxicity. In addition, phenytoin induces its own metabolism, leading to increased clearance and lower plasma concentrations after some weeks. Interaction with other drugs, particularly valproate, can also cause changes in clearance. The drug is highly protein-bound, and, at a given plasma total drug concentration, the lower the plasma protein concentration, the higher will be the free (effective) drug concentration. The unpredictable relationship between dose and plasma concentration, the narrow therapeutic index and the many clinically significant drug interactions mean that monitoring is essential for safe and effective use of the drug. The usual target range is 5–20 mg/L (20–80 μmol/L) but higher concentrations may be required in severe epilepsy. Neurotoxicity (nystagmus, dysarthria, ataxia) is usually concentration-dependent, though signs of chronic toxicity, for example gum hyperplasia, are not so clearly linked to concentration.
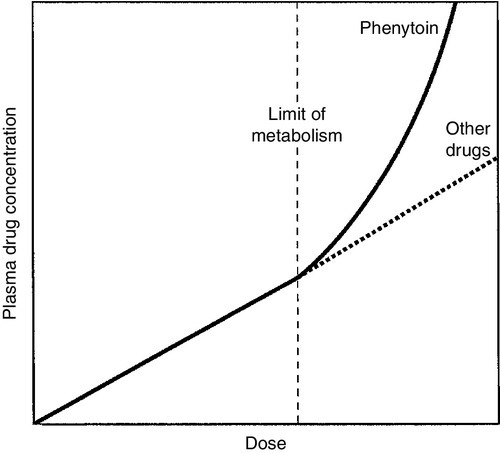
FIGURE 39.5 The relationship between dose and plasma concentration for phenytoin as compared with most other drugs (dotted line). The point where hepatic metabolism is saturated (at which divergence occurs) is specific to the individual.
Value of monitoring
high.
Valproate
Sodium valproate is a popular drug because it does not give rise to drowsiness or the CNS side-effects associated with other anticonvulsants, although it carries a higher risk of congenital malformation and should be used with caution in women of childbearing age. It is recommended as a first-line drug in most types of epilepsy and is used widely in children. It is also used in the treatment of bipolar disorder. However, there is no well-documented relationship between plasma valproate concentration and relief from seizures; indeed, seizures appear to be absent for some time following a dose of valproate when plasma concentrations are too low to measure. A number of cases of severe hepatocellular damage, some fatal, have been reported in patients receiving valproate alone. These appear to be idiosyncratic and are not related to plasma concentration. There is, therefore, no reason to measure plasma valproate in order to avoid hepatotoxicity; however, where the risk of liver damage is high, for example in young children or those with seizure disorders associated with metabolic or degenerative disorders, learning disorders or organic brain disease, baseline and follow-up liver enzyme measurements are indicated.
Valproate is highly protein bound (90–95%) and, when given together with other anticonvulsant drugs, for example phenytoin, can give rise to a (transient) increase in the concentration of free phenytoin. There may, on occasion, be some value in analysing valproate in order to adjust phenytoin or carbamazepine therapy or in the management of complex regimens, but the vast majority of valproate measurements are an unnecessary drain on resources.
Value of monitoring
low.
Newer anticonvulsant drugs
There is a now a plethora of newer antiepileptic drugs such as clobazam, clonazepam, felbamate, gabapentin, lamotrigine, levetiracetam, piracetam, retigabine, rufinamide, topiramate, vigabatrin and zonisamide. Data on concentration–effect relationships are limited, and TDM is not widely used outside specialist centres, since there are no accepted target ranges and most have a wide therapeutic range with overlap of toxic and non-effective concentrations. Tentative target ranges are available in the literature (see Further reading). Routine monitoring cannot be recommended at present, except for monitoring of lamotrigine concentrations on combination therapy because of the marked effect of drug interactions.
Value of monitoring
low to moderate.
Antidepressants and antipsychotic drugs
Tricyclic antidepressants (amitriptyline, clomipramine, dosulepin, doxepin, imipramine, lofepramine, nortriptyline, trimipramine)
The tricyclic antidepressants show wide interindividual genetic differences in metabolism, and for many of them a good relationship between plasma concentration and clinical effectiveness has been shown. Their use is diminishing in favour of selective serotonin release inhibitors (SSRIs), which are better tolerated and less toxic in overdose. Monitoring is potentially valuable to reduce the incidence of toxicity in susceptible patients.
Value of monitoring
moderate.
Selective serotonin release inhibitors (SSRIs) (citalopram, escitalopram, fluoxetine, fluvoxamine, paroxetine and sertraline)
Therapeutic drug monitoring for SSRIs is of little clinical importance in routine practice, primarily because of their much lower toxicity.
Value of monitoring
low.
Lithium
The lithium cation is of great clinical use in the management of bipolar disorder. Lithium is, however, extremely toxic, with a low therapeutic index. It is prescribed orally as uncoated lithium carbonate or as a slow-release preparation. The usual therapeutic range is 0.4–1.0 mmol/L in samples taken 12 h post-dose, with an upper limit of 0.8 mmol/L in the elderly. Concentrations up to 1.2 mmol/L may be required for the acute treatment of mania in younger patients. Since lithium is nephrotoxic and is excreted renally, excessive dosing can produce a vicious circle in which plasma concentrations increase, causing renal damage, excretion is reduced and concentrations increase further. Plasma concentrations > 1.5 mmol/L require intervention, and concentrations > 3.0 mmol/L on chronic therapy are potentially fatal. High concentrations in acute overdosage are less serious than on chronic therapy, since the tissues are not saturated and distribution will lead to a rapid fall in plasma concentration.
Patients newly started on lithium should be monitored regularly until the relationship between dose and plasma concentration is established. Thereafter, monitoring can be less frequent. Lithium competes with sodium for reabsorption in renal tubules, and alterations in sodium balance or fluid intake may precipitate toxicity. Patients on lithium who start diuretics, or develop diarrhoea and vomiting or renal problems should be assessed and their serum lithium measured. Lithium should not be stopped completely since acute withdrawal can give rise to severe psychiatric symptoms. Thyroid dysfunction (most often hypothyroidism), hyperparathyroidism and nephrogenic diabetes insipidus are recognized side-effects of lithium therapy. Lithium has a half-life of 20–40 h depending on the duration of treatment; steady-state concentrations are therefore obtained approximately seven days after the start of therapy.
Value of monitoring
high.
Other antidepressants
There is no good evidence for a significant relationship between drug concentration and therapeutic outcome for the tetracyclic antidepressants maprotiline, mianserin and mirtazapine, the monoamine oxidase inhibitors moclobemide and tranylcypromine or for trazodone and reboxetine.
Value of monitoring
low.
Antipsychotic drugs
The first-generation antipsychotic drugs such as haloperidol and the phenothiazines chlorpromazine, fluphenazine and perphenazine show marked variation in metabolism between individuals according to their CYP2D6 genotype. Drug and metabolites accumulate in poor metabolizers and overdosing leading to extra-pyramidal side effects and irreversible tardive dyskinesia may occur. If CYP2D6 genotyping is not available, TDM may be useful in assessing phenotype and guiding therapy.
The development of second generation antipsychotic agents, for example clozapine, olanzapine, quetiapine, amisulpride, aripiprazole and risperidone, has proved to be a significant advance in the treatment of schizophrenia. The atypical (or second-generation) antipsychotics have several therapeutic properties in common; however, they can differ significantly with regard to clinical potency and side-effects.
Therapeutic drug monitoring may assist in avoidance of extrapyramidal side-effects by maintaining minimum effective concentrations during chronic treatment, although for the majority of patients this is a matter of quality of life rather than safety. Monitoring is available from specialist centres in difficult cases. In the case of clozapine, there is a strong correlation between clozapine plasma concentrations and the incidence of seizures. Clozapine also carries a significant risk (∼3%) of agranulocytosis and regular (weekly at the start of therapy) differential white blood cell monitoring is required.
Value of monitoring
moderate (haloperidol, clozapine) to low.
Antimicrobial drugs
Most antimicrobial drugs are well tolerated and do not require therapeutic drug monitoring. The exceptions to this general principle include the aminoglycoside antibiotics, the glycopeptides vancomycin and teicoplanin, and chloramphenicol.
Aminoglycosides (amikacin, gentamicin, tobramycin)
The aminoglycosides are an important group of antimicrobial drugs used in the treatment of severe systemic infection by some Gram-positive and many Gram-negative organisms. Amikacin, gentamicin and tobramycin are also active against Pseudomonas aeruginosa. Streptomycin is active against Mycobacterium tuberculosis and is now generally reserved for tuberculosis (see below). Neomycin is too toxic for systemic use and can only be used for topical application (skin, mucous membranes). The parent compounds are produced by moulds of the Streptomyces family (streptomycin, tobramycin, neomycin) or the Micromonospora family (gentamicin). The different host organisms account for the variation in spelling of the suffix.
The aminoglycoside antibiotics exhibit relatively simple pharmacokinetics. They are large, highly polar molecules with very poor oral bioavailability, and must be given parenterally. They are not protein bound and not metabolized, and are excreted through the kidneys. The plasma half-life is 2–3 h, except when renal function is impaired, but the drug may accumulate in tissues. If therapy is continued for longer than a week, tissue sites become saturated and plasma concentrations may rise.
These drugs exhibit significant systemic toxicity at plasma concentrations just above those necessary for bactericidal activity. The main toxic effects are nephrotoxicity and ototoxicity. Nephrotoxicity further reduces the ability to excrete aminoglycosides, and a vicious cycle may be precipitated. Nephrotoxicity is often reversible, as is mild ototoxicity, but severely damaged cochlear hair cells cannot be replaced and patients may be left with irreversible hearing loss and disturbed balance.
The drug concentration at which bactericidal effects are achieved (the minimum inhibitory concentration, MIC) is relatively easy to determine in vitro but often has little relevance in vivo, owing to variable penetrance of the drug to the site of infection and differing conditions at the infection site. Aminoglycosides also show a marked post-antibiotic effect – suppression of bacterial growth persists for some time after the drug is no longer present in the plasma. This makes definition of target plasma concentrations difficult, and this difficulty has been compounded in recent years by changes in the ways in which aminoglycosides are administered.
Until the mid-1990s, aminoglycosides were administered every 8 or 12 h, to give relatively stable plasma concentrations in view of their short half-life. It has become clearer that less frequent dosing (every 24 h or more) produces higher peak concentrations, which enhance bacterial kill, and lower trough concentrations, which reduce the systemic toxicity. Such regimens are more convenient, less toxic, reduce adaptive resistance and are generally more suitable in patients with normal renal function. The approach was originally devised as once daily dosing, but a more accurate term is probably ‘extended dosing interval’, in which a plasma concentration measurement is used to design an individual dosing interval which reflects the patient’s needs and renal function. The Hartford nomogram (Nicolau et al. 1995, see Further reading) is an example of this approach, but local guidelines should be consulted on dosage and serum concentrations. Monitoring is essential to achieve effective therapy while avoiding toxicity, particularly in infants, the elderly, the obese and patients with cystic fibrosis, if high doses are being used or if renal function is impaired.
Value of monitoring
high.
Glycopeptides (vancomycin and teicoplanin)
The glycopeptide antibiotic vancomycin is used intravenously in endocarditis and other serious infections caused by Gram-positive cocci including multi-resistant Staphylococci (MRSA). It is also used orally in the treatment of antibiotic-associated (pseudomembranous) colitis. Like the aminoglycosides, the glycopeptides are poorly absorbed, not metabolized, excreted renally and are potentially nephrotoxic and ototoxic. Indications for monitoring have been controversial, but there is definitely a role for monitoring in patients with poor renal function to achieve maximum effect with minimal toxicity. Teicoplanin is similar to vancomycin, but has a longer duration of action such that once-daily dosing is sufficient. No relationship between plasma concentration and toxicity has been established, and teicoplanin is not normally monitored.
Value of monitoring
moderate (vancomycin), low (teicoplanin).
Chloramphenicol
Chloramphenicol is a powerful broad-spectrum antibiotic that carries a risk of serious haematological side-effects when given systemically. It is used to treat life-threatening infections such as cholera, typhoid fever, resistant Haemophilus influenzae, septicaemia and meningitis. Chloramphenicol is metabolized in the liver to the active glucuronide, and peak plasma concentrations (2 h post-dose) in the range 10–25 mg/L (30–77 μmol/L) are desirable. Concentration monitoring is essential in neonates and recommended in children under four years, the elderly and patients with hepatic impairment.
Value of monitoring
moderate.
Antifungal drugs
The successful management of invasive fungal infections remains challenging to clinicians, and the incidence of invasive mycosis has risen in parallel with the rise in the population of immunocompromised patients. The morbidity and mortality caused by these infections remains high, and TDM has a role in ensuring that adequate drug concentrations are attained at the site of infection without systemic toxicity. Four triazole antifungal drugs (fluconazole, itraconazole, posaconazole and voriconazole) have been approved for use. They show marked inter- and intraindividual variations in blood drug concentrations after dosing, owing to variable absorption from the gut (itraconazole, posaconazole) and polymorphism in the CYP2C19 enzyme (voriconazole). The pyrimidine analogue flucytosine also exhibits wide interpatient variation in blood concentration owing to variation in renal elimination. The high pharmacokinetic variability and need for optimal adjustment of drug exposure to ensure that infections are treated adequately are arguments in favour of TDM, but although concentration–effect relationships have been demonstrated for the triazoles and flucytosine, optimal target blood concentrations have not been definitively established and concentration measurements in clinically relevant timescales are not yet readily available. Itraconazole and flucytosine concentrations should be measured routinely in all patients during the first week of therapy and in patients with poor responses. Monitoring of posaconazole and voriconazole should be considered in patients who are not responding to therapy, those with gastrointestinal dysfunction, children and those taking drugs which interact with triazole antifungal agents.
Value of monitoring
moderate.
Antitubercular drugs
The treatment of active tuberculosis (TB) always requires the use of multiple antibacterial agents. It is usually treated in two phases – an initial phase using four drugs and a continuation phase using two drugs. The most frequently used regimen is isoniazid, rifampicin, pyrazinamide and ethambutol (or, rarely, streptomycin), followed by isoniazid and rifampicin. Isoniazid and rifampicin are the key components of this regimen, and both drugs show significant pharmacokinetic variability. The majority of patients are completely cured by standard regimens, and TDM has no role in dosage optimization in these patients. However, patients who are slow to respond to treatment, have drug-resistant TB, are at risk of drug–drug interactions or have concurrent disease states (e.g. acquired immunodeficiency syndrome) that significantly complicate the clinical picture may require individualization of drug therapy, and may benefit from TDM. Early intervention guided by concentration monitoring may prevent the development of further drug resistance. The pharmacokinetic interactions between antitubercular drugs and other medications can often be of considerable clinical concern. Rifampicin is a potent inducer of cytochrome P450 CYP3A and decreases plasma concentrations of the HIV-protease inhibitors significantly; isoniazid is a P450 enzyme inhibitor. Patients with HIV are at particular risk for drug–drug interactions.
When TDM is justified, the 2 h post-dose concentrations of isoniazid, rifampicin, pyrazinamide and ethambutol are usually most informative. Unfortunately, low 2 h values do not distinguish between delayed absorption or malabsorption and non-adherence. In such cases, a second sample, often collected at 6 h post-dose, provides additional information. Trough concentrations of many anti-TB drugs are below the limit of detection of current assays and have limited relevance. Plasma concentrations required for effective therapy, especially on multi-drug regimens, are only partially known, as detailed pharmacokinetic and pharmacodynamic information in humans is lacking. There is a clear dose–response relationship for rifampicin and pyrazinamide and low isoniazid concentrations are also associated with poorer outcomes. Second-line TB drugs such as p-aminosalicylic acid, cycloserine and ethionamide are not usually monitored. Streptomycin is used against resistant organisms. It is nephrotoxic and should be used with great care in patients with renal impairment; concentration monitoring is essential in such patients.
Value of monitoring
moderate.
Antiretroviral drugs
Several classes of antiretroviral drugs are used in the management of infection by HIV:
• non-nucleoside reverse transcriptase inhibitors, e.g. efavirenz, etravirine and nevirapine
• protease inhibitors, e.g. atazanavir, darunavir, fosamprenavir, indinavir, lopinavir, nelfinavir, ritonavir, saquinavir and tipranavir
• fusion inhibitors, e.g. enfuvirtide
• entry inhibitors, e.g. maraviroc
• integrase inhibitors, e.g. raltegravir.
The nucleoside reverse transcriptase inhibitors (NRTIs) are pro-drugs that require activation by intracellular phosphorylation. Plasma NRTI concentrations therefore do not correlate with clinical effect and NRTIs are not suitable for TDM.
Non-nucleoside reverse transcriptase inhibitors (NNRTIs) display highly variable pharmacokinetics; the cytochrome P450 isoenzyme involved in their metabolism (CYP2B6) has a polymorphic variant resulting in slower metabolism and higher plasma concentrations. The protease inhibitors (PIs) also have highly variable pharmacokinetics, but their plasma concentrations have been shown to correlate with virological response. Therapeutic drug monitoring for NNRTIs and PIs may have an important adjunctive role in individualizing therapy in high-risk patients or those with PI resistance. Adherence remains a major problem, especially in developing countries where patient understanding is often poor. Concentrations are normally monitored 1h post-dose. Protease inhibitors and NNRTIs can be quantitated by liquid chromatography tandem mass spectrometry (LC-MS) in a single run.
Value of monitoring
low to moderate.
Antineoplastic drugs
Despite their considerable toxicity, there are no controlled trials that indicate clear advantages to routine concentration monitoring of anticancer drugs, with the exception of methotrexate (see below). Antineoplastic agents are often used in combinations of up to four drugs, which makes the establishment of therapeutic ranges difficult. Areas under the concentration–time curve give a better indication of total exposure to the drug than isolated plasma concentrations, but are more difficult to apply routinely. Pharmacokinetic optimization protocols for many classes of cytotoxic compounds have been used in some specialized centres and a role for monitoring has been proposed for some newer drugs such as the tyrosine kinase inhibitor imatinib in chronic myeloid leukaemia.
Value of monitoring
low (except methotrexate – see below).
Methotrexate
Methotrexate is widely used in a variety of chemotherapeutic regimens for malignancies, where it is given in high doses by the intravenous route over periods of 6–12 h. It is also used orally at low dose (7.5–25 mg weekly) in Crohn disease, rheumatoid arthritis and as an antipsoriatic agent.
Methotrexate is an inhibitor of folic acid metabolism. The rationale behind high-dose therapy is that a short period of exposure to high concentrations of drug will eliminate rapidly dividing cells, while sparing those growing more slowly. At the end of the infusion, methotrexate concentrations should fall rapidly. Where this is not the case, or where the rate of infusion produces too high a plasma concentration, folinic acid (leucovorin) is infused to provide a source of folate and reverse toxic effects. Measurement of methotrexate concentrations may therefore be required in two circumstances – to allow adjustment of infusion to maintain a steady plasma concentration at 10–4 M and to ensure that the concentrations at 24 and 48 h after the start of the infusion have fallen to < 5–10 μmol/L and < 0.5–1.0 μmol/L, respectively, depending on the protocol. A rapid turnaround time is required. Folinic acid may interfere in some assay systems, complicating measurements made after treatment has been given. Methotrexate is metabolized to 7-hydroxymethotrexate and both the parent drug and metabolite are excreted via the kidneys and are nephrotoxic due to their low solubility; prehydration, alkalinization and good renal function are essential for the safe use of this drug. Hepatotoxicity has been reported in a number of patients. Monitoring of liver function and the plasma concentration of the N-terminal propeptide of collagen type III to detect hepatic fibrosis is advised in patients on long-term oral treatment; renal function and white blood cell counts should also be monitored. Methotrexate concentration monitoring is not usually necessary in patients on low-dose regimens.
Value of monitoring
high (on high-dose regimens).
Bronchodilator drugs
Theophylline/caffeine
Theophylline is a bronchodilator used for the relief of asthma that has a number of other pharmacological effects, notably the stimulation of cardiac output and the dilatation of peripheral blood vessels. It is no longer a first- or second-line drug in asthma, but is still useful in patients who have difficulty with inhalers and those with predominantly nocturnal symptoms. Theophylline has a short half-life (3–13 h in adults), but the availability of sustained-release preparations compensates for this and allows dosing at 12 or 24 h intervals. It also has a narrow therapeutic window and wide pharmacokinetic variability, so it is important to have access to TDM if the drug is to be used effectively and safely.
In outpatients, adherence has been shown to be erratic, with as few as 30% of patients taking the drug as prescribed. In such cases, TDM can contribute little unless the patient is toxic or grossly non-compliant. However, in inpatients, and especially those receiving intravenous therapy, TDM can be of great assistance in dosage optimization and in confirming toxicity. Steady-state trough concentrations are the most reproducible for monitoring, except when toxicity is suspected.
Theophylline is metabolized in the liver by a variety of pathways, including oxidation via the cytochrome P450 system (CYP1A2/CYP2E1), which is subject to induction by drugs such as phenytoin. In addition to this, theophylline shows dose-dependent excretion. It is therefore preferable that those involved in its monitoring are familiar with the pharmacokinetic optimization programmes that are available.
The target range in adults is 10–20 mg/L (55–110 μmol/L). The range is lower in neonates (5–15 mg/L; 25–80 μmol/L). This is partly due to sensitivity and partly because babies under six months old metabolize theophylline in a different manner to adults, converting a proportion to caffeine, an active metabolite. After six months, caffeine production is replaced by the adult pattern of metabolism to a variety of methylated urates, including 3-methylxanthine, which is also active. Caffeine has now taken over as the drug of choice to treat neonatal apnoea, as dose regimens are simpler and the effects are more predictable. Caffeine concentrations in the range 5–20 mg/L (25–100 μmol/L) are generally associated with response in neonatal apnoea, but the lower toxicity and simpler pharmacokinetics mean that TDM is rarely required.
Side-effects of theophylline are relatively frequent – mild or moderate effects such as nausea, headache and jitteriness are common within the target range, and more serious effects (tremor, agitation, insomnia, diarrhoea, palpitations, seizures and cardiac arrhythmias) occur with increasing frequency at plasma concentrations > 20 mg/L (110 μmol/L). Caffeine produces much less tachycardia and fewer fits.
Oral therapy with theophylline may involve the free base or any of a number of salts. For intravenous therapy, a water-soluble salt such as aminophylline (theophylline ethylenediamine) is used (aminophylline is approximately 80% w/w theophylline).
Value of monitoring
high (theophylline); low to moderate (caffeine).
Immunosuppressants
Monitoring of immunosuppressants has become an essential adjunct to drug therapy for organ transplant patients because of the relatively narrow therapeutic index of these agents. Therapeutic drug monitoring is important for three of the major immunosuppressants in current use: ciclosporin, tacrolimus, and sirolimus. All three of these drugs are lipophilic and are concentrated in red cells, so whole blood samples are commonly used for analysis. The indications for and approach to monitoring of mycophenolic acid are more controversial.
Ciclosporin
Ciclosporin is a cyclic peptide, widely used for prevention of graft rejection following kidney, liver, heart, lung, heart–lung, bone marrow or pancreas transplants. It is also used in lower doses as an immunosuppressant in severe psoriasis, atopic dermatitis, nephrotic syndrome, ulcerative colitis and rheumatoid arthritis, and in the prophylaxis and treatment of graft-versus-host disease. Ciclosporin inhibits calcineurin phosphatase and limits T cell activation, producing immunosuppression without myelosuppression. However, it is nephrotoxic, and the major difficulty with its use lies in balancing the risks of undertreatment, leading to rejection of the organ, and overtreatment, which can cause kidney damage. This is particularly relevant in renal transplantation, where it is essential to determine whether a rising plasma creatinine concentration is due to nephrotoxicity or rejection.
The pharmacokinetics of ciclosporin vary widely between patients, and target plasma concentrations are dependent on the transplant type, time after transplantation, sample time post-dose, analytical method used and other drugs administered. It is not possible to give specific guidance here and local protocols should be consulted. The drug is highly lipophilic and accumulates in red cells, so EDTA whole blood is the preferred matrix for assay. Trough (pre-dose) concentrations have traditionally been used; although concentrations at 2 h post-dose (C2) give a better indication of ciclosporin exposure, they do require precise timing. Immunoassays and LC-MS assays have been used. Ciclosporin is metabolized in the liver by CYP3A, producing more than 30 metabolites that have variable cross-reactivity in immunoassays, explaining some of the differences in apparent ciclosporin concentrations determined by different assays.
Value of monitoring
high.
Sirolimus
Sirolimus is a macrolide antibiotic, also known as rapamycin. It is structurally similar to tacrolimus, but has a different mode of action. Both sirolimus and tacrolimus bind to a binding protein (FK binding protein, FKBP), but the sirolimus-FKBP complex then binds to a protein kinase, mammalian target of rapamycin (mTOR). This interaction blocks signal transduction in the second phase of T cell activation. Sirolimus reduces acute rejection when co-administered with a calcineurin phosphatase inhibitor and steroids, and allows earlier withdrawal of ciclosporin When sirolimus is given concurrently with the microemulsion formulation of ciclosporin, the absorption of sirolimus is markedly enhanced, almost doubling the effective dose, so it is recommended to give sirolimus 4 h after ciclosporin. The drug is metabolized in the liver by CYP3A, and is also a substrate for the P-glycoprotein transporter system. Sirolimus is not nephrotoxic so is particularly useful in kidney transplantation, but has a range of other adverse effects, including lipid disturbances, anaemia, leukopenia and thrombocytopenia and impaired wound healing. Over-suppression of the immune system is associated with infection and neoplasia. Blood concentrations are a better guide to effect than dose is, and concentration monitoring is advised, particularly in patients with hepatic impairment or on interacting drugs. Pre-dose (trough) concentrations measured on EDTA whole blood should be in the region of 4–12 μg/L when the drug is being given with ciclosporin and 12–20 μg/L when ciclosporin has been withdrawn. Liquid chromatography or LC-MS are the preferred analytical methods, but an immunoassay is also available.
Value of monitoring
high.
Tacrolimus
Like ciclosporin, tacrolimus is a calcineurin phosphatase inhibitor used to prevent or treat graft rejection in kidney, liver and heart transplant patients. It is also metabolized in the liver by CYP3A and is concentrated in red blood cells, so EDTA whole blood samples are used for monitoring. Unlike ciclosporin, it is not intrinsically nephrotoxic, but does cause renal vasoconstriction, which leads to chronic graft dysfunction. Adverse effects are minimized by using the lowest dose consistent with efficacy and by the use of combined therapy with other drugs such as mycophenolic acid, allowing dose minimization for all drugs. Monitoring is essential, particularly in hepatic impairment and during or following treatment with drugs that induce or inhibit metabolism. Target concentration ranges vary with transplant type, other drug therapy and analytical method, but are typically in the range 5–15 μg/L. Immunoassays or LC-MS are used.
Value of monitoring
moderate to high.
Mycophenolic acid
Mycophenolic acid (MPA) is increasingly used in solid organ transplantation as an antiproliferative immunosuppressant. It is used for the prophylaxis of acute rejection in kidney, liver and heart transplantation in combination with calcineurin phosphatase inhibitors (ciclosporin/tacrolimus) and corticosteroids. It is given as the pro-drug mycophenolate mofetil (MMF), or as the enteric-coated sodium salt of MPA. It is rapidly absorbed and mainly excreted renally, with < 10% hepatic metabolism to the inactive glucuronide. Concentrations in plasma are relatively high (1–3 mg/L; 3–9 μmol/L) and trough plasma (EDTA or heparin) samples are often used for monitoring, although the optimum measures of MPA exposure are still the subject of considerable debate as trough plasma concentrations vary widely. Limited-sampling strategies to give a better estimate of the area under the concentration–time curve have been advocated. The general routine use of MPA monitoring in solid-organ transplantation is not currently recommended, though monitoring has a role in specific patient populations, including those at increased immunological risk or those with changing renal or hepatic function.
Value of monitoring
low to moderate.
Opiate and opioid drugs
Methadone/buprenorphine
Methadone and buprenorphine are synthetic opioids used as adjunct therapy in the treatment of opiate dependence. The primary requirement in this setting is to confirm that patients have been taking their medication, rather than selling it, since these drugs have a street value. Qualitative testing of urine is used to achieve this. For patients on methadone therapy, urine is often tested for the methadone metabolite EDDP (2-ethylidine-1,5-dimethyl-3, 3-diphenyl pyrrolidine), as this guards against adulteration of urine specimens with the medication and confirms that drug has actually been ingested. Monitoring of plasma concentrations has not yet been shown to be routinely useful, despite significant pharmacokinetic variation and much recent interest in dosage optimization for opiate substitutes.
Value of monitoring
low.
Morphine
Morphine is used as a short-term analgesic, often as patient-controlled analgesia following surgical procedures, and for long-term analgesia in conditions with protracted pain, for example the later stages of malignant disease. Morphine kinetics are complex and absorption can be erratic. There is also an active metabolite, morphine 6-glucuronide (M6G), with analgesic properties superior to morphine. Heroin (diacetylmorphine) is converted rapidly to morphine via a short-lived monoacetylmorphine intermediate. Until recently, morphine was not part of the repertoire of drugs for which therapeutic monitoring was advocated, but there is now evidence that, in patients in whom analgesia is difficult to achieve, plasma morphine concentrations can be useful. The method of choice is high performance liquid chromatography – mass spectrometry, since the active metabolites M6G and codeine can also be determined; this is of particular use in patients with renal insufficiency in whom there may be accumulation of M6G.
Value of monitoring
low to moderate.
ACKNOWLEDGEMENT
I am grateful to Dr Michael J. Stewart who was the author of this chapter in previous editions of the book.
Further reading
General pharmacokinetics
Analytical methods
Antiarrhythmics/cardiac glycosides
Anticonvulsants
Antidepressants/antipsychotics
Antimicrobial drugs
Dasgupta A. Advances in antibiotic measurement. Adv Clin Chem. 2012;56:75–104.
Antifungal drugs
Antitubercular drugs
Antiretrovirals
Antineoplastic drugs
Immunosuppressants
Methadone/buprenorphine
APPENDIX 39.1 CALCULATIONS FOR THE DETERMINATION OF DOSE REQUIREMENTS TO ACHIEVE STEADY-STATE CONCENTRATIONS
The loading dose depends on the volume of distribution (Vd). Hence, to calculate a loading dose use:
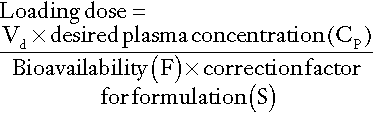
The maintenance dose required for steady-state depends on the clearance and is calculated by:
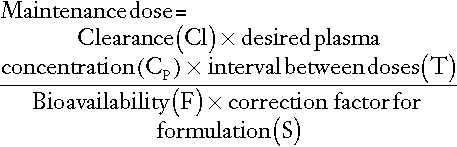
Average population values for clearance, volume of distribution, bioavailability and formulation correction factor (if relevant) may be found in reference texts.