CHAPTER 37
Lipids and disorders of lipoprotein metabolism
CHAPTER OUTLINE
Intermediate density lipoproteins
Assembly of apolipoprotein B-containing lipoproteins
High density lipoprotein metabolism
ENZYMES INVOLVED IN LIPOPROTEIN METABOLISM
Lecithin cholesterol acyltransferase
Acyl-CoA:cholesterol acyltransferase
TRANSFER PROTEINS INVOLVED IN LIPOPROTEIN METABOLISM
Cholesteryl ester transfer protein (CETP)
Phospholipid transfer protein (PTP)
RECEPTORS INVOLVED IN LIPOPROTEIN METABOLISM
Scavenger receptor class B type 1
Peroxisome proliferator-activated receptor family
OTHER PROTEINS INVOLVED IN LIPOPROTEIN SYNTHESIS, TRANSPORT AND METABOLISM
Microsomal triglyceride transfer protein
ATP binding cassette transporter family
Proprotein convertase subtilisin kexin 9
Sterol regulatory element binding proteins
Glycosylphosphatidylinositol-anchored HDL-binding protein 1
CLASSIFICATION OF LIPOPROTEIN DISORDERS
THE PRIMARY DYSLIPOPROTEINAEMIAS
Familial combined hyperlipidaemia
Familial hypertriglyceridaemia
Familial hypercholesterolaemia
Polygenic hypercholesterolaemia
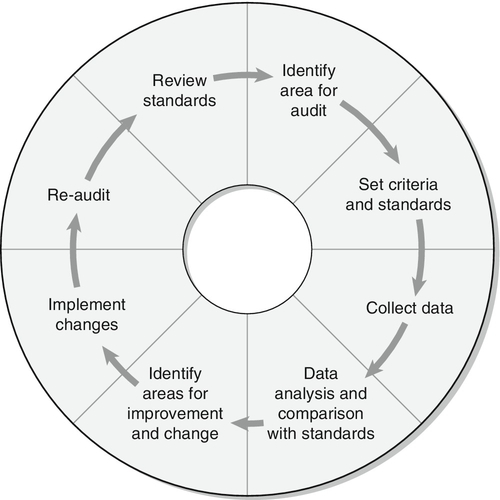
INVESTIGATION OF LIPID DISORDERS
High density lipoprotein cholesterol
Low density lipoprotein cholesterol
Post-heparin lipolytic activity
INTRODUCTION
‘Lipid’ is the term used to describe a number of substances of diverse chemical structure that bear little functional relationship to each other but which have in common the property of being soluble in organic solvents and virtually insoluble in water. Lipoproteins are macromolecular protein complexes that allow hydrophobic lipids to be transported within the hydrophilic environment of the circulation. Lipids are essential for health, but excessive concentrations of cholesterol and triglycerides in the circulation, whether due to lifestyle factors or to inherited disorders of lipoprotein metabolism, are major factors in the development of atherosclerosis and cardiovascular disease. These conditions are the focus of this chapter, although other lipids and their functions are discussed briefly.
LIPIDS
Lipids can be broadly divided into sterols, including cholesterol; fatty acids or substances containing fatty acids such as triglycerides and phospholipids; eicosanoids; the fat-soluble vitamins (A, D, E and K), and sphingolipids. The major classes of lipids and their principal functions are summarized in Table 37.1.
Sterols
Cholesterol
Cholesterol (Fig. 37.1) is the major sterol in humans, being present in all body cells and most body fluids. The majority of the cholesterol in the body is in the free, unesterified form; it is this form that is the structural component of cell membranes. Cholesteryl esters in normal cells represent a store for future use and appear microscopically as intracellular droplets. Cholesterol is present in the diet but most cholesterol in the body is made by de novo synthesis from acetate. The rate-limiting step in the synthetic pathway is the conversion of 3-hydroxy-3-methylglutaryl-coenzyme A (HMG-CoA) to mevalonate, catalysed by the enzyme HMG-CoA reductase. The liver is responsible for most cholesterol synthesis. Cholesterol is a precursor for the synthesis of gonadal and adrenal steroid hormones, vitamin D and bile acids.
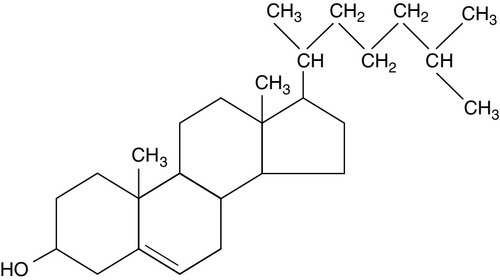
FIGURE 37.1 Structure of cholesterol.
Although only a small amount of the body’s cholesterol pool comes from dietary cholesterol, this has an important role in regulating the rate of cholesterol synthesis. The liver is the key organ in maintaining cholesterol balance; any excess cholesterol is excreted by the liver into the bile, either directly, or after conversion into bile acids.
Cholesterol and membranes
Cholesterol is a major component of cell membranes. Cholesterol and sphingomyelin form plasma membrane ‘lipid rafts’ or caveolae. Caveolae are cell surface invaginations found in differentiated cells and characterized by the presence of a protein, caveolin-1; they are sites where signalling molecules are concentrated. In order for these signalling molecules to function, the cholesterol concentration of the plasma membrane must remain constant. This is achieved by a regulatory system that senses the cholesterol content of the membrane and modulates the transcription of genes encoding proteins involved in cholesterol synthesis (e.g. HMG-CoA synthase and HMG-CoA reductase) and cholesterol uptake (e.g. the LDL receptor). The system that does this is a family of membrane-bound transcription factors called sterol regulatory element-binding proteins (see p. 721).
Phytosterols
Phytosterols are sterols derived by plants; they differ slightly from cholesterol. There are two general types: δ5-phytosterols (e.g. β-sitosterol) and 5α-reduced phytosterols, otherwise referred to as ‘stanols’. In natural foods, the sterols predominate. Both sterols and stanols are incorporated into commercially available foods promoted as cholesterol-lowering agents: they act by competing with cholesterol for absorption from the gut, and may lower plasma cholesterol concentration by 10–15%.
Fatty acids
Fatty acids (Fig. 37.2) have the general chemical formula RCOOH. Those relevant to human nutrition are the long chain (C12–C20) fatty acids containing even numbers of carbon atoms. They are further defined as saturated, for example stearic (C18:0); monounsaturated, for example oleic (C18:1), and polyunsaturated, for example linoleic (C18:2) and linolenic (C18:3), the second figures indicating the number of double bonds. In general, dietary saturated fatty acids originate from animals and the unsaturated fatty acids from plants. There are, however, exceptions, for example palmitic acid (C16:0) is saturated but derived from palm oil, and the ω-3 series, which are unsaturated but found in fish. The position of the carbon of the first double bond in the polyunsaturated fatty acids differentiates the ω-6 series (where the fist double bond starts beyond the 6th carbon atom from the methyl end of the molecule), from the ω-3 series, where it starts beyond the 3rd carbon atom.
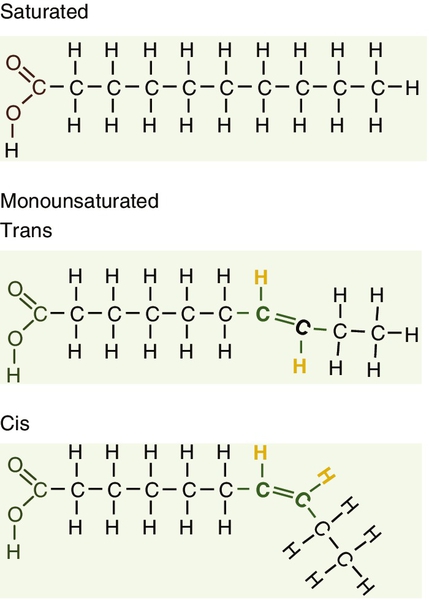
FIGURE 37.2 Structure of fatty acids.
The long chain fatty acids are oxidized for energy production by a process known as β-oxidation; this results in the sequential shortening of the chain by two carbon atoms and the production of acetyl-CoA. Small amounts of long chain fatty acids are elongated to very long chain fatty acids (VLCFAs), which have a structural function in certain specialized cells.
Triglycerides
Triglycerides (see Fig. 37.3) comprise three fatty acids esterified with a glycerol backbone. ‘Triacylglycerols’ is the correct chemical name but they are more commonly known as ‘triglycerides’ and this term will be used throughout this chapter. Triglycerides are the major dietary fat. They are hydrolysed in the gut by lipases to fatty acids and monoglycerides. The monoglycerides undergo re-esterification in enterocytes and subsequent incorporation into chylomicrons. The major sites of endogenous triglyceride synthesis are the liver and adipose tissue. In normal circumstances, hepatic triglyceride is secreted in very low density lipoproteins (VLDL). In certain pathological states, triglyceride accumulates in hepatocytes, leading to hepatic steatosis. Adipose tissue triglyceride represents the major energy store of the body. Fatty acids are mobilized from adipose tissue triglycerides by the action of hormone-sensitive lipase (HSL), which is activated by glucagon and adrenaline (epinephrine) and inhibited by insulin.
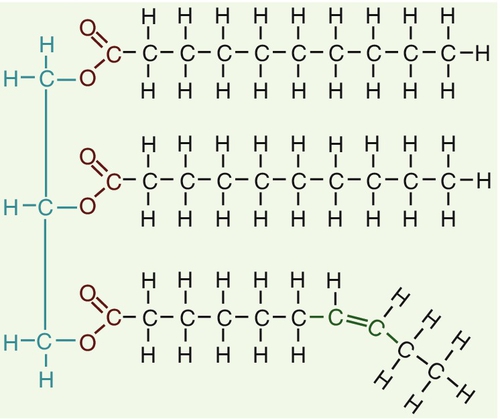
FIGURE 37.3 Structure of triglycerides.
Phospholipids
Phospholipids, like triglycerides, have a glycerol backbone; in phospholipids, this is esterified with two fatty acids and the third hydroxyl group is linked via a phosphodiester bond to an amino alcohol such as choline, serine or ethanolamine (Fig. 37.4). Phospholipids are therefore amphipathic molecules, with both hydrophilic (phosphate group) and hydrophobic (fatty acid) domains. This property is responsible for the capacity of phospholipids to solubilize other lipids and accounts for their location on the surfaces of lipoprotein molecules and in the cell membrane lipid bilayer.
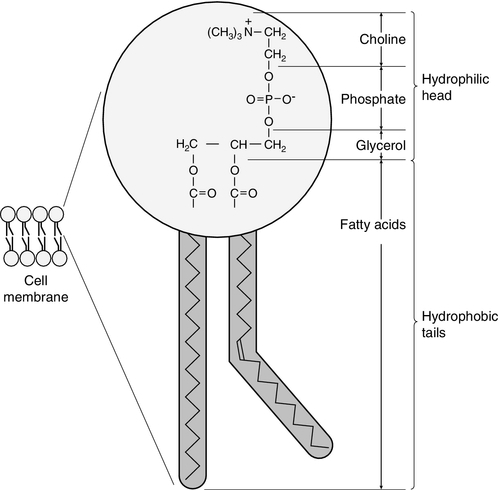
FIGURE 37.4 Structure of phospholipids.
Eicosanoids
This group of compounds takes its name from the systematic name of C20 (eicosa-) fatty acids from which they are derived. It includes prostaglandins, thromboxanes and leukotrienes. These were originally named because they were found in prostate, platelets (thrombocytes) and white cells (leukocytes), respectively. They have major effects on the immune response, reproductive function (including the induction of labour), cholesterol metabolism, smooth muscle function (causing vasoconstriction or dilatation), platelet aggregation and thrombosis.
Prostaglandins contain a substituted cyclopentane ring. Thromboxanes are oxygenated eicosanoids closely related to the prostaglandins. The rate-limiting step in the synthesis of both (see Fig. 37.5) is the phospholipase A2-mediated release of fatty acids from phospholipids. The principal precursor of prostaglandins is arachidonic acid (C20:4, eicosatetraenoic acid). The first two steps in their synthesis are catalysed by prostaglandin endoperoxide synthase, which has both cyclooxygenase and peroxidase activity. The major products of this enzyme’s activity are prostaglandin G2 (PGG2) and prostaglandin H2 (PGH2). These are both prostaglandins of the 2 series, having 2 carbon–carbon double bonds. Prostaglandin H2 is converted to thromboxane A2 (TXA2) in platelets and prostacyclin (PGI2) in the arterial wall. Arachidonic acid also serves as a substrate for various lipoxygenases, which generate the leukotrienes, inflammatory mediators and potent stimulators of muscle contraction. Eicosatrienoic acid (20:3) and eicosapentenoic acid (20:5), are alternative precursors for prostaglandin synthesis. The latter generates prostaglandins of the 3 series and leukotrienes of the 5 series.
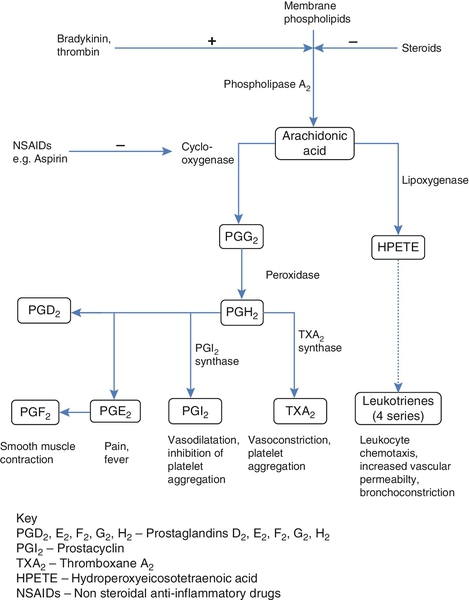
FIGURE 37.5 An outline of eicosanoid synthesis.
Eicosanoid imbalance is of interest in atherosclerosis because several recognized risk factors for atherosclerosis, such as smoking, hypertension and diabetes mellitus, are associated with changes in eicosanoid production.
Aspirin inhibits cyclooxygenase, thereby reducing prostaglandin, especially platelet TXA2, synthesis. This is the rationale for the use of aspirin in the prophylaxis of atherosclerotic vascular disease. Steroids directly inhibit the release of arachidonic acid from membrane phospholipids.
Sphingolipids
The backbone of sphingolipids is sphingosine or a similar long chain base. The sphingolipids vary in chain length from C14 to C20. Sphingosine itself has 18 carbon atoms and is formed from the condensation of palmitoyl CoA (C16:0) with serine. Another fatty acid chain is joined to sphingosine through an amide link to form a ceramide. Ceramides have a free hydroxyl group that allows reaction with another component. If this contains a phosphate group, then the resultant products are a type of phospholipid called sphingophospholids, also termed sphingomyelins (Fig. 37.6), which are essential components of nerve cells. If a carbohydrate group is attached to ceramide then glycosphingolipids, or simply glycolipids, are formed. These include cerebrosides, sulfatides, globosides and gangliosides. Glycosides of ceramide are referred to as cerebrosides: they are present in relatively high concentrations in brain. Many glycosphingolipids contain oligosaccharides; those that contain more than one molecule of sialic acid are referred to as gangliosides.
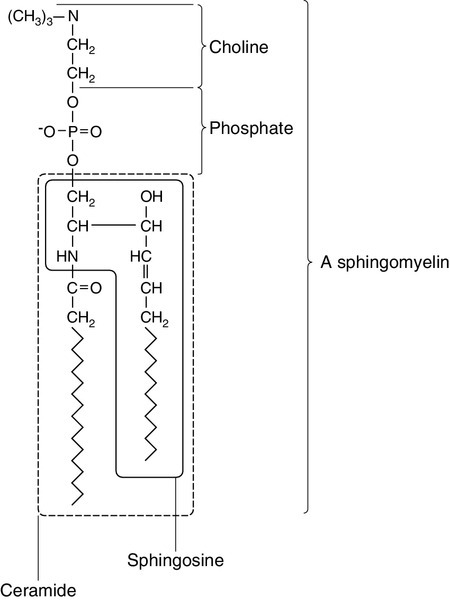
FIGURE 37.6 Structure of sphingolipids.
Nuclear lipids
The nucleus is a highly structured organelle. For a long time, it was thought that lipids, which quantitatively are a minor component of the nucleus, only served a structural role and originated in the cytoplasm. It is now recognized that lipids serve other functions including signalling and modulating. Lipid-metabolizing enzymes have been demonstrated to be present within the nucleus. Phospholipids are the predominant class of lipid in the nucleus, with lesser amounts of cholesterol, free fatty acids, diglycerides and sphingolipids. The nucleus is surrounded by the nuclear envelope, which comprises an outer nuclear membrane and an inner nuclear membrane. Cholesterol has a structural role in the outer nuclear membrane, which is continuous with the endoplasmic reticulum; the inner nuclear membrane is associated with the nuclear lamina and chromatin and is deficient in cholesterol. The inner and outer nuclear membranes are joined by the pore membranes at the nuclear pores, which are associated with the nuclear pore complexes that allow passive transfer of molecules < 50 kDa between the cytoplasm and nucleoplasm. The passage of larger molecules is energy dependent and requires a nuclear localization signal. Lipids with very long chain fatty acids are associated with the pore membrane–nuclear pore complexes and appear to be essential for maintaining their function.
LIPOPROTEINS
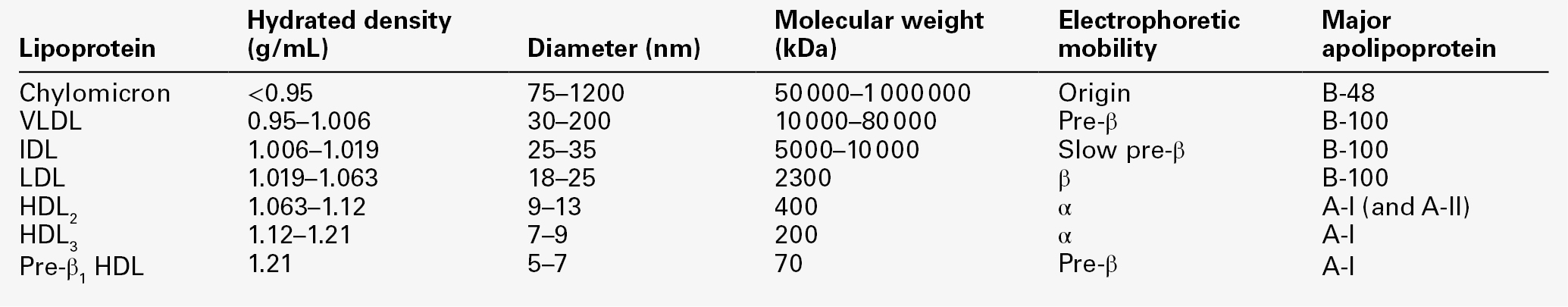
The lipoproteins are submicroscopic, macromolecular complexes of lipids (cholesterol, triglycerides, phospholipids) and proteins (apolipoproteins, enzymes), held by non-covalent forces. The basic structure of lipoproteins is a hydrophobic core of triglycerides and/or cholesteryl esters surrounded by a layer of amphipathic phospholipids, unesterified cholesterol and proteins (see Fig. 37.7). The hydrophilic surface protects the hydrophobic core from the aqueous environment. Lipoproteins differ in their relative concentrations of protein to lipid and in their constituent lipids and proteins (Table 37.2). The densities of lipoproteins are inversely related to their size. The lipoproteins can be classified on the basis of their size, density or protein composition. The nomenclature of the lipoproteins is based on their density: chylomicrons (< 0.95 g/mL); VLDL (0.95–1.006 g/mL); intermediate density lipoproteins (IDL) (1.006–1.019 g/mL); low density lipoproteins (LDL) (1.019–1.063 g/mL) and high density lipoproteins (HDL) (1.063–1.210 g/mL). The classes are not homogeneous; each represents a continuum of particles of differing size, density and fate and, in the case of VLDL, also of origin. The physicochemical characteristics of the principal lipoproteins are summarized in Table 37.3.
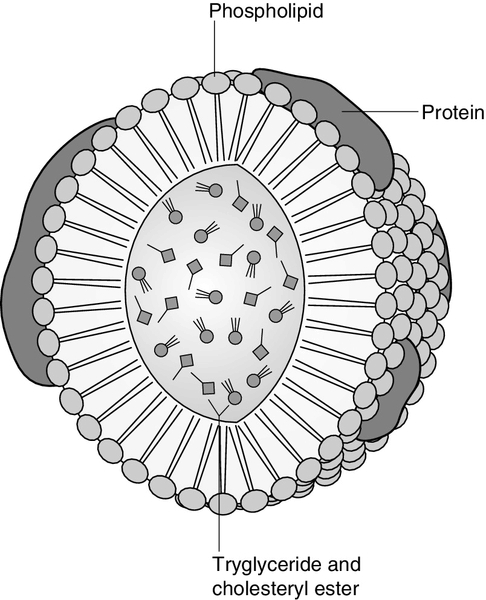
FIGURE 37.7 Generic lipoprotein structure.
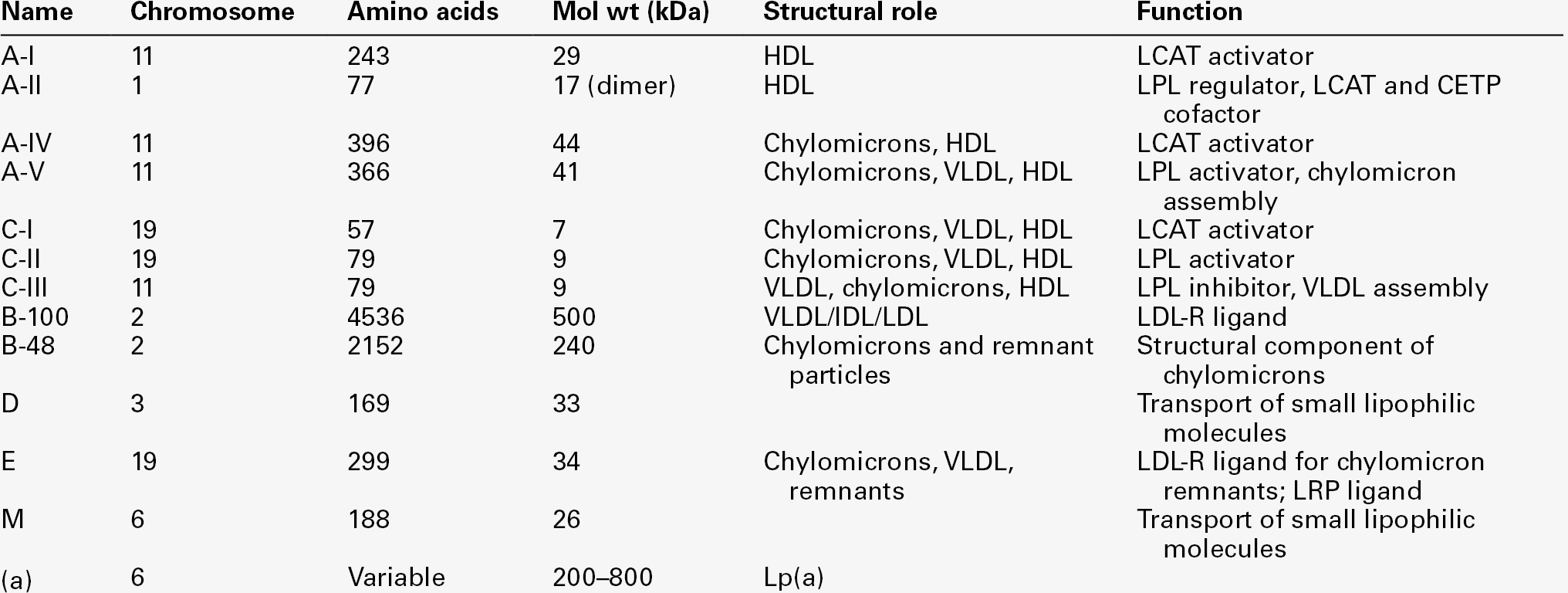
The apolipoprotein (apo) B-containing lipoproteins contain only one molecule of apo B per lipoprotein particle whereas multiple molecules of the other apolipoproteins are present in other lipoprotein particles.
Chylomicrons
Chylomicrons are the largest and most buoyant class of lipoprotein. The major protein component is apo B-48 but they also contain apo A-I, apo A-II and apo A-IV. After secretion, they acquire apo E and apo C from HDL. Chylomicrons are formed in the intestine and are the transport vehicle for dietary fat. The largest chylomicron particles have a diameter of over 1000 nm, whereas the smallest (75–200 nm) overlap with the apo B-100-containing lipoproteins. Some of the smaller particles within the chylomicron range are this size when they are secreted by enterocytes, while others represent partially delipidated ‘remnant’ particles. The core of chylomicrons is composed predominantly of triglycerides derived from the diet.
Very low density lipoproteins
These are the largest of the lipoproteins containing endogenously produced lipids. The major protein component of VLDL is apo B-100 but they also contain apo C-I, apo C-II, apo C-III, apo E and small amounts of apo A. Like chylomicrons, VLDLs acquire the majority of their component apo E and apo C from HDL in the circulation; the core of VLDLs is composed predominantly of triglycerides. In contrast to chylomicrons, the triglycerides in VLDL are endogenous in origin.
Intermediate density lipoproteins
These particles are produced during the conversion of VLDL to LDL; their densities lie between those of these lipoproteins. The core of IDLs contains cholesteryl esters and triglycerides.
Low density lipoproteins
These are the major cholesterol-containing lipoproteins and represent the end-product of VLDL catabolism. The core of LDLs comprises mainly cholesteryl esters; the protein component is apo B-100.
High density lipoproteins
These are the smallest and densest of the lipoproteins. They may be sub-classified on the basis of size, density, shape, surface charge and electrophoretic mobility, as well as apolipoprotein composition (Table 37.3). High density lipoprotein is usually divided into three major subclasses. Nascent, discoidal or pre-β1HDL comprises predominantly apo A-I and phospholipid. It is the preferred substrate for the ATP binding cassette transporter A1 (ABCA1), which actively exports free cholesterol from peripheral cells and macrophages. HDL3 is formed from pre-β1HDL by the acquisition of free cholesterol. It is the preferred substrate for lecithin cholesterol acyl transferase (LCAT), which esterifies free cholesterol, increasing the size of the particle and allowing the uptake of more free cholesterol, producing the larger and more cholesterol-rich HDL2.
High density lipoprotein particles may contain apo A-I alone (LpA-I), both apo A-I and apo A-II (LpA-I/A-II) or apo A-II alone (LpA-II). LpA-I predominates in HDL2, whereas LpA-I/A-II predominate in HDL3. LpA-II represents a very small proportion of both HDL2 and HDL3.
Lipoprotein(a)
Lipoprotein(a) (Lp(a)) consists of LDL with its apo B-100 bound by a disulfide bond to apolipoprotein(a) (apo(a)). It is thought to be assembled extracellularly, either in the circulation or on the surfaces of hepatocytes.
The plasma concentration of Lp(a) is genetically determined and is inversely related to the length of the apo(a), so that the greater the chain length, the lower the concentration. Epidemiological studies suggest that a high Lp(a) concentration is an independent risk factor for cardiovascular disease (CVD), particularly in subjects with familial hypercholesterolaemia.
Lipoprotein X
Lipoprotein X is a lipoprotein that is found only in the plasma of subjects with cholestasis or who have familial lecithin cholesterol acyltransferase deficiency. It is composed of phospholipids, free cholesterol and proteins; the major protein is albumin but small amounts of apo C and apo D are also present. It contains no apo B. Unlike all other lipoproteins, it migrates towards the cathode on agarose gel electrophoresis.
APOLIPOPROTEINS
The apolipoproteins are amphipathic; their hydrophobic regions interact with the lipids in the lipoprotein particle while their hydrophilic regions allow interaction with the aqueous environment. They have three functions: they provide the structural element to the lipoprotein particles, they act as ligands for specific receptors and they also act as activators or inhibitors of specific enzymes involved in lipoprotein metabolism.
On the basis of electrophoretic mobility, HDL and LDL were originally referred to as α- and β-lipoproteins. The nomenclature of the corresponding apolipoproteins has arisen from this, apo A being the apolipoprotein derived from HDL (α-lipoprotein) and apo B being derived from LDL (β-lipoprotein).
Apolipoprotein A
Apolipoprotein A-I
Apolipoprotein A-I (apo A-I) (molecular weight 29 kDa) is the major protein of HDL, constituting 70–80% of HDL protein. It is synthesized primarily in the liver and small intestine. In addition to its structural role in HDL, it is also an activator of lecithin cholesterol acyltransferase (LCAT). Reverse cholesterol transport is dependent on the ability of apo A-I to promote cellular cholesterol efflux, to bind to lipids, to activate LCAT and, within mature HDL, to interact with lipid transfer proteins and specific receptors. The gene for apo A-I (APOA1) is part of a gene cluster on the long arm of chromosome 11 that includes APOC3, APOA4 and APOA5.
Epidemiological studies have shown that plasma apo A-I concentrations, like those of HDL-cholesterol (HDL-C), are inversely related to cardiovascular risk.
Apolipoprotein A-II
Apolipoprotein A-II (apo A-II) (molecular weight 17 kDa, as homodimer) is also synthesized in the liver and, to a lesser extent, the small intestine. It accounts for about 20% of HDL protein. Some HDL contains apo A-I and apo A-II while some HDL contains apo A-I alone. A small amount of plasma apo A-II is associated with chylomicrons and VLDL. Apo A-II regulates lipoprotein lipase (LPL) activity, and is a cofactor for LCAT and for cholesteryl ester transfer protein (CETP). Like apo A-I, it appears to be inversely related to the risk of coronary disease. It may play a role in the remodelling of HDL, possibly by an effect on the reactivity of HDL towards lipid transfer proteins, enzymes and receptors, including the scavenger receptor B1 (SRB1) (see p. 719).
Apolipoprotein A-IV
Apolipoprotein A-IV (apo A-IV) (molecular weight 44 kDa) is synthesized only in the small intestine. It has been suggested that it may have a role in intestinal lipid transport, increasing the residence time of nascent chylomicron particles, allowing for greater expansion of their cores and thus capacity to transport triglycerides. The majority of apo A-IV in plasma exists in the free form. A small amount is associated with HDL and chylomicrons. In vitro, apo A-IV activates LCAT, although not as effectively as does apo A-I. It may also be necessary for the maximal activation of LPL by apo C-II. Over-expression of APOA4 in mice results in increased plasma concentrations of total- and HDL-cholesterol, and triglycerides; despite this, it protects against diet-induced atherosclerosis. In humans, apo A-IV deficiency has been reported in patients with apo A-I and apo C-III deficiency, and this may account for the fat malabsorption seen in affected individuals.
Apolipoprotein A-V
The APOA5 gene is expressed in liver, and apolipoprotein A-V (apo A-V), in contrast to other apolipoproteins, is present at very low concentrations in the plasma (approximately 5 nmol/L). It is found primarily in HDL. Apo A-V affects plasma triglycerides through an effect on the lipolysis of the triglyceride-rich lipoproteins, possibly by binding to the lipoprotein, endothelial proteoglycans and LPL and thus stabilizing the lipolytic machinery. Although plasma concentrations of apo A-V show little correlation with plasma triglyceride concentration or with prevalence of cardiovascular disease, genetic studies have shown polymorphisms in APOA5 to be strong determinants of both. Genetic variants in humans have been identified, in association with both high and low triglyceride concentrations. Deficiency leads to reduced LPL activity and a type V dyslipidaemia (p. 726).
Apolipoprotein B
This lipoprotein exists in two forms; apolipoprotein B-100 (apo B-100), which is made in the liver and is the structural protein of VLDL, IDL and LDL, and apo B-48, which is synthesized in the intestine and is incorporated into chylomicrons. Both apo B molecules remain with the lipoprotein particle in which they are secreted throughout the lifespan of that particle, unlike the other apolipoproteins, which readily transfer between different classes of lipoproteins. Increased plasma concentrations of apo B-containing lipoproteins confer an increased risk for the development of atheroma. Both forms of apo B are produced from the APOB gene; post-transcriptional editing of the mRNA in the intestine leads to the production of apo B-48.
Apolipoprotein B-100
Apolipoprotein B-100 (apo B-100) (molecular weight 500 kDa) is necessary for the assembly and secretion of VLDL. It contains several very hydrophobic areas that serve as strong lipid-binding domains. It also has several domains that could serve as binding sites for heparin-like molecules and form the basis for some of the cell surface interactions of the apo B-containing lipoproteins. In addition, apo B-100 contains an LDL receptor binding domain (amino acids 3100–3400), which allows the specific uptake of LDL by the LDL receptor.
Apolipoprotein B-48
The amino terminal 48% of apo B-100 forms apolipoprotein B-48 (apo B-48) (molecular weight 240 kDa). This apolipoprotein is produced from the APOB gene in the intestine by an mRNA editing process; a cytidine deaminase, APOB mRNA editing enzyme complex 1 (apobec-1), binds to and acts on the cytosine molecule at base 6666 of the mRNA to form a uracil. The editing enzyme complex is only found in intestinal epithelial cells. Its action results in the glutamine 2153 triplet of CAA being converted into the stop codon, UAA. Thus protein synthesis is prematurely terminated at amino acid 2152 and, as a result, apo B-48 does not contain the LDL receptor binding domain present in apo B-100.
Apolipoprotein C
There are three apolipoprotein Cs, all of which are synthesized in the liver. In plasma, they transfer between the triglyceride-rich lipoproteins (chylomicrons, VLDL and their remnants) and HDL.
Apolipoprotein C-I
Apolipoprotein C-I (apo C-I) (molecular weight 7 kDa) forms a minor component of VLDL, IDL and HDL; it acts as an activator of LCAT.
Apolipoprotein C-II
Apolipoprotein C-II (apo C-II) (molecular weight 9 kDa) is a component of chylomicrons and VLDL, in which it functions as an activator of LPL. Apo C-II is also found in IDL and HDL.
Apolipoprotein C-III
Apolipoprotein C-III (apo C-III) (molecular weight 9 kDa) is synthesized mainly in the liver and, to a lesser extent, the intestine. It forms a major structural component of VLDL but is also present in chylomicrons and HDL. It acts as an inhibitor of LPL, and has more recently been shown to promote hepatic assembly and secretion of VLDL. Apo C-III also inhibits hepatic uptake of chylomicron and VLDL remnant particles, possibly by preventing interaction of apo E on these remnant particles with the hepatic receptor. High plasma apo C-III concentrations are associated with high plasma triglyceride concentrations.
Null mutations have been reported which are associated with low plasma triglyceride and LDL, and high HDL concentrations. However, the APOC3 gene is close to the APOA1 gene, and both are deficient in some forms of apo A1 deficiency, which cause low plasma HDL and triglyceride concentrations.
Apolipoprotein D
Apolipoprotein D (apo D) (molecular weight 33 kDa) is a lipoprotein-associated glycoprotein, forming a minor component of HDL, VLDL, IDL and LDL. It transports small hydrophobic ligands, including sterols and cholesterol. Apo D is associated with increased activity of lipoprotein lipase, and missense mutations cause elevated triglycerides. The apo D concentrations in the hippocampus and cerebrospinal fluid (CSF) of patients with Alzheimer disease are increased.
Apolipoprotein E
Apolipoprotein E (apo E) is a 299 amino acid glycoprotein (molecular weight 34 kDa) synthesized by the liver and found in all classes of lipoproteins except LDL. It is involved in the control of chylomicron and VLDL remnant removal from the circulation. It also has antioxidant properties, and controls the efflux of cholesterol from cells, together with apo A-I. Apo E is a polymorphic protein: three common isoforms occur, which can be separated by isoelectric focusing and are designated apo E2, apo E3 and apo E4. Apo E2 differs from apo E3 by only a single amino acid, Cys being substituted for Arg at residue 158. Apo E4 also differs from apo E3 by only a single amino acid, Arg being substituted for Cys at residue 112. The apo E3/E3 phenotype is the most common, comprising 50–70% of the population, whereas the apo E2/E2 phenotype is the least common, occurring in about 1% of the population.
The apo E3 isoform is associated with normal chylomicron and VLDL metabolism. The E2 isoform does not function as an effective ligand for the receptor-mediated uptake of remnant particles, having less than 2% of normal apo E3 binding to the LDL receptor. As a result, remnant lipoproteins tend to accumulate in the plasma of individuals homozygous for apo E2/E2. The E4 isoform is associated with higher concentrations of LDL-cholesterol than E3. Apo E4 homozygotes are also at increased risk of Alzheimer disease. Apo E synthesis is upregulated to aid cell repair in response to cellular stress or injury. Apo E4 is more susceptible to proteolytic cleavage than E2 or E3. This results in accumulation of intracellular fragments that cause changes in the cytoskeleton and the formation of neurofibrillary tangles.
Apolipoprotein M
Apolipoprotein M (apo M) (molecular weight 26 kDa) fulfills the criteria for being an apolipoprotein as it is not found free in plasma but is predominantly associated with HDL. Like apo D, it is a member of the lipocalin family of proteins, which contain a binding domain for small lipophilic ligands, and may therefore have a role in transport of small lipid molecules. Although apo M is found in association with only 5% of HDL particles, where it may potentiate the antioxidant effect of HDL, its concentration is positively correlated with cholesterol concentration, suggesting that it may also have a role in cholesterol metabolism.
Apolipoprotein(a)
This apolipoprotein (apo(a)) is a large glycated protein of variable size (molecular weight 200–800 kDa). It contains multiple kinks in the polypeptide chain that are termed ‘kringles’. Apo(a) is a homologue of plasminogen; it contains a single copy of plasminogen kringle 5, multiple copies of plasminogen kringle 4 and an inactive protease domain. Kringle 4 shows wide variation in the number of repeats within the apo(a) molecule. In a subpopulation of LDL particles, apo(a) forms disulphide bridges with apo B-100 to form a distinct lipoprotein class termed lipoprotein(a) (Lp(a)) (see p. 708). The function of apo(a) is unknown. It has a strong homology with plasminogen and may interfere with fibrinolysis.
CHOLESTEROL ABSORPTION
Cholesterol of both dietary and biliary origin is absorbed in the upper jejunum facilitated by a specific transporter protein, the Niemann–Pick C1-like 1 (NPC1L1) protein. This has 50% homology with the product of the NPC1 gene, the Niemann–Pick C1 disease protein, which is involved in intracellular cholesterol trafficking. Niemann–Pick C1-like 1 protein transports plant sterols as well as cholesterol.
In small intestinal enterocytes, dietary cholesterol is released by the action of lysosomes and selectively esterified by acyl-CoA cholesteryl acyltransferase 2 (ACAT2) prior to incorporation into chylomicrons. Any excess free cholesterol in enterocytes, together with any absorbed plant sterol, is excreted back into the intestinal lumen. The apical excretion of cholesterol and other sterols back into the gut lumen is a function of two hemitransporters of the ATP binding cassette transporter family, ABCG5 and ABCG8. These are also responsible for the excretion of cholesterol and other sterols into the bile by hepatocytes. Expression of ABCG5 and ABCG8 in hepatocytes and enterocytes is under the control of the liver X receptor (LXR) and is induced by cholesterol feeding.
Cholesterol absorption reflects the imbalance between the movement of cholesterol across the brush border into enterocytes and its excretion by enterocytes back into the intestinal lumen. In normal individuals, the rate of cholesterol absorption correlates with the plasma sterol concentrations. The rate of cholesterol absorption is a predictor of benefit of statin treatment and response to ezetimibe (a selective inhibitor of NPC1L1). Those with higher rates of cholesterol absorption show reduced CVD benefit from statins, and increased cholesterol lowering with ezetimibe.
β-Sitosterolaemia
also referred to as phytosterolaemia, is an autosomal recessive disorder caused by mutations in ABCG5 or ABCG8, in which there is increased absorption of dietary non-cholesterol sterols, and consequently an increased concentration (30–100-fold) of these sterols in blood. A ‘normal’ Western diet contains similar amounts (200–500 mg/day) of cholesterol and of non-cholesterol sterols (mainly plant but also some fish sterols). Approximately 55% of the cholesterol is usually absorbed, but normally less than 1% of the non-cholesterol sterols. In the absence of a functional ABCG5/8 heterodimer, subjects overabsorb cholesterol and non-cholesterol sterols from enterocytes, because these transporters usually excrete some cholesterol and almost all non-cholesterol sterols back into the gut lumen. They also fail to excrete cholesterol and non-cholesterol sterols into the bile. These sterols become deposited at various sites in the body causing tendon xanthomas and premature atherosclerosis. Clinical features also include arthritis and haemolytic episodes. Treatment comprises a diet low in dietary sterols. Ezetimibe is also effective by inhibiting sterol absorption via NPC1L1.
TRIGLYCERIDE DIGESTION
Fat (triglyceride) digestion starts in the stomach, where lingual and gastric lipases hydrolyse 25–30% of ingested triglycerides into diglycerides and free fatty acids. In the duodenum, partially digested lipids mix with bile and pancreatic secretions. The latter contains a mixture of enzymes, including carboxyl ester lipase and pancreatic triglyceride lipase, which are capable of further hydrolysing dietary lipids to monoglycerides, free fatty acids, glycerol and cholesterol. The bile acids serve to solubilize the lipids and result in the formation of micelles, the contents of which are absorbed by enterocytes. The absorption of monoglycerides is by passive diffusion. Within the enterocytes, triglycerides are resynthesized from free fatty acids and either monoglycerides or glycerol.
BILE ACID METABOLISM
Approximately 500 mg of cholesterol is converted, in the liver, to bile acids each day. This replaces the bile acids lost in the stools and represents approximately 5% of the bile acid pool, as the enterohepatic circulation is 95% efficient. The synthesis and secretion of bile acids, together with hepatic cholesterol secretion into bile, represents the major pathway for the elimination of cholesterol from the body. The synthesis of the full complement of bile acids from cholesterol requires 17 enzymatic steps. It is under negative feedback control: accumulation of bile acids leads to reduced activity of the key enzymes 7α-hydroxylase and sterol 12α-hydroxylase. The products of the bile acid synthetic pathway are the primary bile acids, cholic and chenodeoxycholic acids. In the gut, these primary bile acids are converted into secondary and tertiary bile acids by the action of anaerobic bacteria. The bile acids present in bile are a mixture of primary, secondary and tertiary, the latter being a consequence of enterohepatic circulation.
A number of enzymatic defects in the synthetic pathway of bile acids have been described; in general, the earlier in the pathway, the earlier in life clinical problems become manifest and the greater their severity. One of these disorders, due to defective sterol 27-hydroxylase activity, is cerebrotendinous xanthomatosis (CTX). In this condition, cholesterol and its 5α-reduced derivative, cholestanol, accumulate in the blood and tissues. As a result of this accumulation, affected individuals develop tendon xanthomas like those found in familial hypercholesterolaemia. In CTX, however, these sterols also accumulate in myelin sheaths, leading to progressive neurological dysfunction.
The bile acids act to solubilize fats during absorption from the gut, but they also have a solubilizing action in the bile. If the proportions of bile acids, cholesterol and phospholipid in the bile are disturbed, there is an increased risk of gallstones being formed.
LIPOPROTEIN METABOLISM
Lipoprotein metabolism is summarized in Figure 37.8.
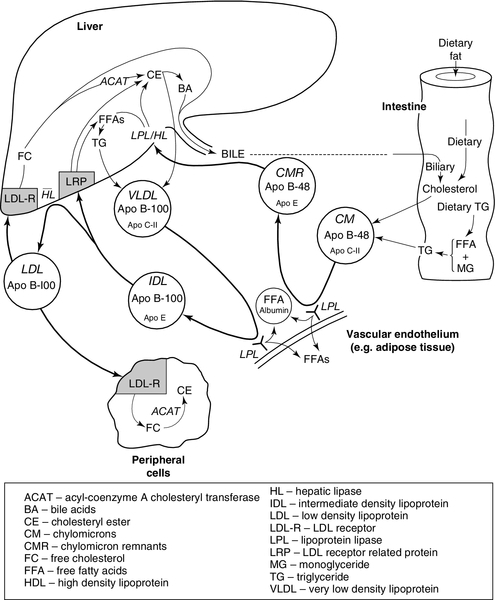
FIGURE 37.8 Outline of the metabolism of the apo B-containing lipoproteins.
Assembly of apolipoprotein B-containing lipoproteins
The assembly of the apo B-containing lipoproteins requires the coordinated synthesis of apo B and lipids. First, apo B has lipid added to it by microsomal triglyceride transfer protein (MTP) as it is being synthesized in the endoplasmic reticulum, to form a nascent lipoprotein particle. Further triglyceride may be added, forming VLDL2. In the next step, VLDL2 is exported from the endoplasmic reticulum by a membrane associated protein complex comprising a coatomere protein (COPII) and a GTPase (sar1b). The resulting sar1/COPII vesicles then fuse with the Golgi apparatus. Apo A-IV, apo C-III and apo A-I are added to the surface of the lipoprotein. In the presence of large amounts of free fatty acids and triglycerides, phospholipase D1 and extracellular signal-regulated kinase 2 (ERK2) increase the formation of lipid droplets that deliver lipids to the Golgi apparatus and promote the further addition of lipid to VLDL2, which is thereby converted to VLDL1.
In healthy individuals, most VLDL particles are small and relatively triglyceride-poor VLDL2. In conditions such as insulin resistance and type 2 diabetes there is increased production of larger, triglyceride-rich VLDL1, which in turn generate atherogenic remnants, small dense LDL particles and triglyceride rich HDL particles that are more readily catabolized, leading to low concentrations of circulating HDL.
Apolipoprotein B is constitutively synthesized, but the rate of production of apo B-containing lipoproteins is related to the availability of fatty acids for triglyceride synthesis. When there is a plentiful supply of fatty acids, the majority of the apo B that is synthesized is incorporated into lipoproteins; however, when fatty acids are in short supply, the apo B is degraded intracellularly.
Once lipoprotein particles are fully formed, they are transferred to the cell surface for secretion. Very low density lipoprotein particles (200 nm) and chylomicrons (1000 nm) are large in the context of classic transport vesicles (range 50–80 nm). The intracellular transport of chylomicrons and some VLDL particles relies on sar1/COPII vesicles. Defects in the SARA2 gene that codes for sar1b result in chylomicron retention disease, in which there is an inability to secrete chylomicron particles. Apo B-48 is absent from the circulation, but apo B-100 containing lipoproteins are still present, although in reduced amounts, as sar1b is also involved in later stages of the VLDL pathway. Newly synthesized VLDL is not necessarily secreted from the cell, and may undergo immediate degradation. Sortilins (see p. 721) are now recognized to be involved in routing newly synthesized lipoproteins either to intracellular degradation or for export into the circulation.
Exogenous pathway
Enterocytes absorb dietary cholesterol and triglyceride from the gut in the form of free cholesterol, fatty acids and monoglycerides. After re-esterification, cholesteryl esters and triglycerides (containing fatty acids of chain length C14 and greater) are incorporated into the cores of chylomicron particles. Enterocytes synthesize apo B-48 (the major protein of chylomicrons), apo A-I, apo A-II and apo A-IV that, together with phospholipids, form the surface layer of chylomicrons. Apolipoprotein B-48 is essential for chylomicron secretion. Secretory vesicles bud off from the Golgi apparatus and migrate to the basolateral regions of enterocytes. Here, they fuse with the plasma membrane and release chylomicrons into the intestinal lymphatics. The chylomicrons pass in the lymphatics to the thoracic duct and enter the circulation via the left subclavian vein.
There is only one apo B-48 molecule per chylomicron particle and it remains with the particle throughout its life until, as a chylomicron remnant, it is taken up by the liver. In contrast, multiple copies of the other apolipoproteins are present in a single chylomicron particle. These other apolipoproteins do not remain with a given chylomicron particle throughout its life but are exchanged with other lipoproteins. From the time of secretion, chylomicrons undergo constant modification, gaining apo C-II, apo C-III, apo E, phospholipids and cholesterol from HDL. The acquisition of apo C-II allows chylomicrons to interact with LPL, which is sited on the vascular endothelium, especially in adipose tissue and muscle. Lipoprotein lipase acts extracellularly to hydrolyse triglycerides within the chylomicron cores; the fatty acids thus released can be either utilized as an energy source or re-esterified and stored in adipose tissue as triglycerides. As hydrolysis proceeds, the cores of the chylomicrons reduce in size and excess surface components – phospholipids, free cholesterol, apo C-II and apo C-III – are transferred back to HDL. Apolipoprotein C-III may exert a modulating effect on LPL-catalysed chylomicron hydrolysis. Both apo C-II and LPL are necessary for normal chylomicron catabolism. Continuing loss of apo C-II as the mass of individual chylomicron particles is reduced eventually prevents further interaction with LPL, and chylomicron remnant particles are generated. These remnants have a relatively high content of cholesteryl esters and apo E.
Chylomicron remnants are normally cleared quickly from the plasma by the liver. They enter the space of Disse through the fenestrated sinusoidal endothelium, together with lipoprotein lipase. The space of Disse is rich in heparan sulfate proteoglycans (HSPRG) as well as hepatic lipase (HL) and apo E, which are secreted by hepatocytes. The proteoglycans and apo E bind the remnants that are further catabolized by HL and LPL, becoming enriched in apo E and further depleted in triglycerides. The remnants may then be taken up in a process directly mediated by HSPRG or via apo E binding to the LDL receptor related protein (LRP). This LRP mediated uptake may be of remnant particles alone, or of remnants bound to HSPRG. The LDL receptor is also able to uptake chylomicron remnants via apo E binding in a process independent of HSPRG.
The cholesteryl esters delivered to the liver by chylomicron remnant particles may be utilized for the synthesis of bile acids or membranes or be secreted in VLDL, while the apo B-48 undergoes degradation.
In summary, chylomicron metabolism essentially comprises two steps. In the first step, most of the fatty acids derived by peripheral lipolysis of chylomicrons enter adipocytes for storage as triglycerides or other cells for oxidation for energy production. A small fraction of the released fatty acids is bound to plasma albumin and transported in the blood to the liver and other tissues. The second step involves remnant particles delivering the remaining triglycerides and almost all the cholesterol to the liver.
Lipolysis in adipose tissue
Triglycerides in white adipose tissue, far from being an inert store, are continuously undergoing lipolysis and re-esterification. In the fasting state, and at times of increased energy demand, free fatty acids are released into the circulation and transported to other tissues. The lipolysis of triglycerides in white adipose tissue is initiated by adipose triglyceride lipase (ATGL). Its action results in the production of diglycerides and free fatty acids, but it has very little hydrolytic activity towards diglycerides. Hormone sensitive lipase, which until recently was thought to be the rate-limiting enzyme involved in the release of free fatty acids from adipose tissue, is actually rate limiting for the hydrolysis of diglycerides rather than triglycerides. The monoglycerides resulting from the action of HSL are acted on by a third enzyme, monoglyceride lipase (MGL).
Adipocyte lipolysis is controlled by a number of lipolytic and antilipolytic hormones, including catecholamines and insulin.
Endogenous pathway
Hepatocytes are the originators and often also the acceptors of particles involved in the endogenous pathway, which shows many points of similarity with the exogenous pathway.
The liver secretes VLDL, a triglyceride-rich lipoprotein. The triglycerides are produced either de novo by hepatocytes or taken up from the plasma. These triglycerides, together with cholesterol derived from chylomicron remnants or from de novo synthesis, are secreted with phospholipids and apo B-100 as nascent VLDL. Some apo C-I, apo C-II, apo C-III and apo E are also present in the nascent VLDL particle, but the majority of these apolipoproteins are probably acquired from HDL within the circulation in the same way as in the exogenous pathway. In situations where there is an excess of hepatic triglyceride, large VLDL particles are secreted. Large VLDL particles are also secreted in familial hypertriglyceridaemia, whereas in familial combined hyperlipidaemia (see p. 724), the rate of VLDL secretion is increased but a relative scarcity of triglycerides ensures that the individual VLDL particles are smaller and relatively poor in triglycerides.
The initial metabolic transformation of VLDL is a progressive LPL-mediated lipolysis analogous to the process involving chylomicrons. This requires apo C-II and produces cholesteryl esters and apo E-rich remnant particles. As in chylomicron metabolism, the surface components of VLDL are transferred back to HDL as the cores shrink. VLDL remnants comprise small VLDL particles and IDL. About half are cleared by the liver in a process that involves uptake by LRP, which recognizes apo E. The remainder undergoes further hydrolysis by HL to form LDL.
Low density lipoprotein is the major cholesterol-carrying lipoprotein in the plasma and usually accounts for 70% or more of the total plasma cholesterol. Virtually the only protein contained in the LDL particle is a single molecule of apo B-100, which acts as the ligand for the LDL receptor. LDL receptors are present on hepatocytes as well as the cells of peripheral tissues. Approximately 50% of plasma LDL uptake by the LDL receptor-mediated mechanism is hepatic. The major determinant of plasma LDL-C concentration is the number of functional LDL receptors.
Low density lipoprotein receptors recognize both apo B-100 on LDL and apo E on remnant particles and HDL. Once a lipoprotein has been bound to the receptor, the receptor-lipoprotein complex localizes in the coated pit region from where it is internalized by endocytosis. The LDL receptor is recycled while the lipoprotein undergoes lysosomal degradation to unesterified cholesterol and amino acids. The cholesterol thereby released is available for further metabolic transformations as well as to regulate the transcription and/or translation of the HMG-CoA reductase and the LDL receptor genes. The cholesterol may be re-esterified by the action of ACAT and stored, or may be utilized for bile acid, steroid or membrane synthesis.
Hepatic cholesterol trafficking
The liver is the key organ for the regulation of cholesterol; not only is it responsible for the majority of cholesterol synthesis but it also acquires cholesterol from all lipoprotein classes. The cholesterol secreted into bile is mostly derived from lipoproteins with only a small contribution coming from de novo hepatic synthesis or hepatic cholesteryl ester stores. This involves preferential trafficking of lipoprotein-derived cholesterol, which involves multiple cholesterol transport-related gene products, the expression of which is regulated by the concerted activity of sterol-activated transcription factors.
High density lipoprotein metabolism (see Fig. 37.9)
Assembly of lipoproteins
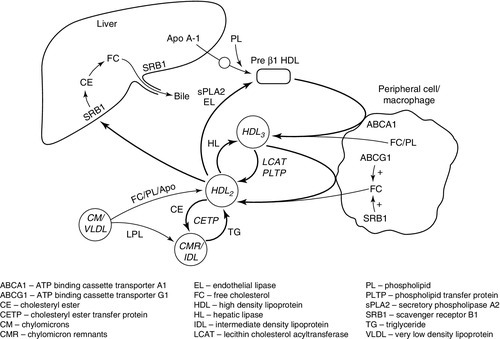
FIGURE 37.9 Outline of HDL metabolism and its role in reverse cholesterol transport.
Apo A-I is exported into the circulation by liver and intestinal cells. Here it may combine with phospholipids to form nascent (discoidal or pre-β1) HDL.
Pre-β1HDL may also be formed from spare surface components of triglyceride rich lipoproteins (VLDL, chylomicrons, and their remnants) following the action of lipases.
Cholesterol efflux
Except in steroidogenic tissues (adrenals and gonads), the catabolism of cholesterol in peripheral cells is limited to its partial degradation by 27α-hydroxylase. Excess free cholesterol has to be removed. This occurs by an active process, or by diffusion. Free cholesterol is actively exported by ATP binding cassette transporter A1 (ABCA1), which preferentially binds pre-β1HDL. Expression of ABCA1 is regulated by the liver X receptor/retinoid X receptor (LXR/RXR) system; the physiological ligand of LXR is oxysterol, the cellular concentration of which increases in parallel with that of cholesterol.
Diffusion is non-specific efflux of cholesterol from cell surface membranes to an extracellular ‘acceptor’. All HDL particles can act as acceptors of free cholesterol. This is a bidirectional process. Net efflux is increased by the activity of LCAT, which esterifies free cholesterol in lipoproteins, thereby increasing the concentration gradient between HDL and the cell membrane. Cholesterol efflux by diffusion is not as dependent on the activity of LCAT as previously thought, as it may also be facilitated by the SRB1 and the ATP binding cassette transporter G1 (ABCG1). The SRB1 binds preferentially to larger (HDL2) particles. In peripheral cells, SRB1 forms a hydrophobic channel through which free cholesterol can readily diffuse, enhancing the rate of efflux. The ABCG1 increases the concentration of free cholesterol within the plasma membrane and makes it more readily desorbed. This promotes the step which is normally rate limiting in cholesterol efflux by diffusion. Cholesterol efflux capacity correlates inversely with atherosclerosis, after adjustment for plasma HDL concentration. However, although both plasma apo A-I and HDL predict efflux capacity, they account for < 50% of the variation seen. This emphasizes the importance of overall flux through the pathway of reverse cholesterol transport rather than simply plasma HDL concentration in the development of atheroma.
Reverse cholesterol transport
Free cholesterol exported from peripheral cells or macrophages combines with apo A-I to form ‘nascent’ pre-β1HDL. Esterification of free cholesterol on the surface of the HDL particle by LCAT produces hydrophobic cholesteryl esters that migrate to the centre of the particle, releasing capacity to accept more free cholesterol, and transforming it into a larger, more spherical particle (HDL3). HDL3 accepts more cholesterol by active export or facilitated diffusion, and LCAT further increases particle size. More free cholesterol, phospholipids and apolipoproteins (C-II, C-III, E) are also transferred from triglyceride-rich lipoproteins (TGRLs) and their remnants as triglycerides are removed from these particles by the action of LPL. The HDL particle increases in size to become HDL2, which contains a greater proportion of apo A-II and is the preferred ligand for the hepatic SRB1 receptor. When HDL2 is bound, cholesteryl esters are taken up into the cell, converted to free cholesterol and may then be excreted in bile. Scavenger receptor B1 is present on the basolateral membrane of hepatocytes where it promotes influx of cholesterol, and on the canalicular membrane where it promotes efflux to bile. Cholesterol taken up into the hepatocyte by SRB1 may also be trafficked across the cell and pumped into bile by ABCG5/G8 at the canalicular membrane.
High density lipoprotein 2 may also transfer cholesterol to TGRLs in exchange for triglycerides, catalysed by CETP. This potentially ‘diverts’ cholesterol from the ‘normal’ reverse cholesterol transport pathway into potentially atherogenic lipoproteins, and may therefore be proatherogenic. However, in transferring cholesterol to the VLDL/LDL pathway, it allows for cholesterol clearance via hepatic LDL receptors, and may theoretically be antiatherogenic in situations where clearance of cholesterol from HDL via SRB1 is saturated. The triglycerides that enrich HDL as a result of this exchange are hydrolysed by lipases and this results in small dense HDL, which is more readily catabolized by the kidney, depleting plasma HDL. This CETP-mediated exchange is increased in the presence of increased circulating amounts of TGRLs and this accounts for the inverse relationship between plasma triglycerides and HDL. HDL2 is a substrate for hepatic and endothelial lipases (ELs) and secretory phospholipase A2 (sPLA2). These may regenerate pre-β1HDL or HDL3, but if HDL is triglyceride-enriched by excessive CETP activity then the HDL particle may disintegrate, promoting catabolism in the kidneys.
High density lipoprotein therefore exists as a complex interchange of lipoproteins. ABCA1, ABCG1, LCAT and LPL all increase particle size, while CETP, SRB1, HL, EL and sPLA2 tend to reduce size, with net transport of free cholesterol from peripheral cells to the liver. These interactions also have the potential to ‘divert’ cholesterol to atherogenic TGRLs, with loss of apo A-I, especially in conditions where TGRLs are increased.
High density lipoprotein cholesterol (HDL-C) shows an inverse relationship with atherosclerosis in epidemiological studies, although this is not a straightforward as previously thought. Very high concentrations of HDL-C are associated with increased risk of atherosclerosis, which could be due to HDL being ‘trapped’ as a ‘dysfunctional’ pool in plasma rather than reflecting an increased flux through the reverse cholesterol transport pathway. This highlights the importance of qualitative changes in HDL that may affect its function, and the overall net flux through the reverse cholesterol transport pathway (see p. 714) as well as total plasma concentration.
High density lipoprotein has direct anti-atherogenic properties including:
• downregulation of the expression of adhesion molecules on the surface of the vascular endothelium
• inhibition of platelet aggregation
• prevention of the inhibition of nitric oxide synthase by oxidized LDL
• upregulation of ATP binding cassette transporter A1 (ABCA1) and activation of intracellular signalling leading to stimulation of cholesterol efflux.
In addition, paraoxonase, an ester hydrolase that is transported bound to HDL, has antioxidant properties.
ENZYMES INVOLVED IN LIPOPROTEIN METABOLISM
Lecithin cholesterol acyltransferase
Lecithin cholesterol acyltransferase (LCAT) is a glycoprotein that has both a phospholipase A2 and an acyltransferase action. It is essential for the normal maturation, interconversion and rearrangements of all lipoprotein classes and is involved in reverse cholesterol transport. It is synthesized by the liver and circulates in the plasma reversibly bound to HDL and LDL. Lecithin cholesterol acyltransferase catalyses the esterification of free cholesterol on the surfaces of lipoproteins. The preferred substrate for LCAT is HDL, which contains apo A-I, its most potent activator. The ability of apo A-I to activate LCAT depends on the lipid composition of HDL, being most effective in small HDL particles, which have a low ratio of phospholipid to apo A-I. The cholesteryl esters formed by the action of LCAT expand the cores of the HDL particles. Lecithin cholesterol acyltransferase activity may be a rate limiting step in reverse cholesterol transport, making it a potential therapeutic target in the prevention of atherosclerosis, although evidence as to the effect of LCAT on the development of atherosclerosis is conflicting. Both recombinant LCAT and small molecule activators of LCAT activity are being explored as possible therapies.
Mutations in the LCAT gene cause either familial LCAT deficiency, when there is no LCAT activity or fish eye disease, with partial LCAT deficiency (see p. 729).
Lipases
Lipoprotein lipase
Lipoprotein lipase (LPL) is an extracellular enzyme that is bound by the glycosaminoglycan heparan sulphate to capillary endothelial cells. It is present in large amounts in the capillaries of adipose tissue and muscle, both skeletal and cardiac. Lipoprotein lipase belongs to the triglyceride lipase gene family, which also includes HL, EL, and pancreatic triglyceride lipase (PTL). It is the major lipolytic enzyme involved in the intravascular metabolism of the triglyceride-rich lipoproteins. Myocytes and adipocytes secrete LPL in a catalytically inactive form, which is then transported to the capillary endothelial surface.
Along with HL and EL, LPL is a homodimer. Interaction of the dimer with heparan sulphate on the endothelial surface serves to anchor and stabilize the LPL. LPL monomers that are catalytically inactive are found in the circulation in association with remnant particles and may play a role in enhancing their clearance. Presence of apo C-II is required for full activity.
Lipoprotein lipase catalyses the partial hydrolysis of the core triglycerides of chylomicrons and VLDL to monoglycerides and fatty acids. The fatty acids are taken up by the tissue and either re-esterified and stored (in adipose tissue), utilized as an energy source (in muscle) or secreted (in lactating breast tissue). The monoglycerides are further hydrolysed to glycerol and fatty acids.
Lipoprotein lipase binds to heparin, which results in its release into the circulation. This is used in the assay of LPL activity (post-heparin lipolytic activity, PHLA). Lipoprotein lipase regulates the plasma concentrations of triglycerides and HDL. Individuals with low PHLA, such as those who are heterozygous for LPL deficiency, have high triglyceride and low HDL concentrations in plasma, and an increased risk of atherosclerosis.
A large number of mutations in the LPL gene have been described. Some are a cause of the familial chylomicronaemia syndrome (see p. 725), while others have less severe effects. It is estimated that 20% of patients with hypertriglyceridaemia are carriers of LPL gene mutations. The Asn291Ser mutation is present in 2–5% of Caucasians and is associated with a 31% increase in plasma triglyceride concentrations and an increased risk of coronary heart disease and type 2 diabetes. Asn291 is located in part of the molecule involved in homodimer formation, so it is likely that this mutation causes an increase in the relative amount of LPL present as inactive monomer. The risk of Alzheimer disease is also increased in Asn291Ser carriers.
Small amounts of LPL have been demonstrated to be present on arterial endothelium and also within the intima of arteries. LDL binds to LPL with an affinity similar to chylomicrons and VLDL. Although LDL is not the physiological substrate for LPL, the LPL present in the arterial wall may, by binding to LDL, increase the residence time of LDL in the arterial wall, thus promoting atherogenesis.
Hepatic lipase
This enzyme is a product of the same gene family as LPL. It is synthesized in the liver, where 95% remains, bound to the exterior of the hepatocyte membrane in the spaces of Disse; the remainder is found in the adrenals, ovaries and macrophages. HL has a role in the remodelling of chylomicron remnants, IDL, LDL and HDL and is involved in reverse cholesterol transport. Its concentration is inversely related to that of HDL. Its predominant activity on HDL is as a phospholipase, whereas in other lipoproteins it acts as a triglyceride lipase. In addition, HL may influence lipoprotein metabolism by means of its capacity to form a ‘bridge’ between lipoproteins and lipoprotein receptors.
Enzyme activity is lower in females than males and is modulated by insulin resistance, dietary fat intake, visceral obesity, physical activity, smoking and certain drugs. The size and buoyancy of LDL are inversely related to HL activity; when its activity is high, small, dense LDL particles are produced.
Like LPL, HL can bind to heparin, which results in its release into the circulation. This activity is utilized in HL assays.
Endothelial lipase
Endothelial lipase is the third member of the triglyceride lipase gene family and like the others it exists as a homodimer. It is produced by hepatic endothelial cells, and unlike HL and LPL it functions at the site of synthesis. It is also found in macrophages. Its predominant activity is as a phospholipase A1. Transcription is induced by cytokines and physical forces, which has led to the suggestion that it may play a role in the development of atheroma. Overexpression is associated with reduced plasma concentrations of HDL.
Lipase maturation factor 1
This membrane bound protein located in the endoplasmic reticulum is essential to the folding and assembly of the dimeric lipases (LPL, HL, EL). Loss of function mutations in LMF1 cause failure in the assembly of homodimers, thus preventing these lipases from attaining normal catalytic activity. This produces combined lipase deficiency with marked hypertriglyceridaemia.
Pancreatic triglyceride lipase
This enzyme, synthesized by pancreatic acinar cells, is essential for the digestion of dietary triglycerides. Enzyme activity requires formation of a complex with its protein cofactor, colipase. Following emulsification, the second step in the digestion of dietary triglycerides involves their hydrolysis to fatty acids and monoglycerides, which are incorporated into micelles with bile salts. Pancreatic triglyceride lipase (PTL) accounts for the majority of triglyceride lipase activity in the upper small intestine of adults.
The pancreatic acinar cells also synthesize two pancreatic lipase-related proteins (PLRP1, PLRP2) that have a high degree of structural and sequence homology to PTL. PLRP2 may play a role in dietary fat digestion in the newborn but no catalytic activity has, as yet, been demonstrated for PRLP1.
Hormone sensitive lipase
This enzyme is not related to any of the above lipases. It is a neutral intracellular lipase that has catalytic activity against diglycerides, monoglycerides and cholesteryl esters. It is predominantly located in adipose tissue. It is also expressed in the adrenals, testes and ovaries and, to a lesser extent, in skeletal muscle, cardiac muscle, macrophages and pancreatic islets. The activity of HSL is rate limiting in the release of fatty acids from adipose tissue. It is subject to a complex control mechanism, which involves both prolipolytic (ACTH and catecholamines) and antilipolytic (insulin) hormones, via cyclic AMP-dependent protein kinase phosphorylation. HSL interacts with adipocyte lipid binding protein (ALBP), which is a member of a family of lipid binding proteins that bind fatty acids and other hydrophobic ligands. In vitro, monoglycerides and fatty acids exert feedback inhibition on HSL; the interaction with ALBP, which sequesters these products, may prevent this happening in vivo. HSL is also responsible for most cholesteryl ester hydrolase activity in the adrenals.
Carboxyl ester lipase
This enzyme is a non-specific lipase capable of hydrolysing cholesteryl esters, triglycerides, diglycerides, monoglycerides, phospholipids, lysophospholipids and ceramide. It is the only enzyme in the gut with cholesteryl esterase action; all its other actions can also be effected by other enzymes, for example the triglyceride lipase action of pancreatic triglyceride lipase. Most carboxyl ester lipase (CEL) is synthesized by pancreatic acinar cells but it is also synthesized by the liver, macrophages and lactating breast tissue. In the intestine, CEL is attached to the brush borders of enterocytes by means of its heparin binding domain. It is taken up by enterocytes, possibly by endocytosis and may be involved in the intracellular processing of lipids during the assembly of chylomicrons. The fact that CEL acts on ceramide is important because ceramide can disrupt intracellular lipid and protein trafficking, resulting in a block in the assembly of large lipoproteins and their secretion by enterocytes; CEL prevents ceramide disrupting this process.
Vitamin A is ingested in the form of retinyl palmitate or retinyl acetate, which need to be hydrolysed prior to absorption. It is likely that this hydrolysis is a function of CEL in the neonatal period, although in the adult, other enzymes may be involved.
Lysosomal acid lipase
Lysosomal acid lipase (LAL) is secreted as a 399-amino acid precursor, which includes a 27-amino acid signal peptide for transport across the membrane of the endoplasmic reticulum. Further processing, including N-glycosylation in the endoplasmic reticulum and the attachment of mannose 6-phosphate residues in the Golgi, leads to lysosomal targeting. LAL is responsible for the breakdown of cholesteryl esters and triglycerides that are delivered to lysosomes as a result of receptor-mediated uptake of lipoproteins.
Wolman disease
is an autosomal recessive disease that presents in the neonate and is characterized by hepatosplenomegaly, steatorrhoea and abdominal distention, results from complete lack of LAL activity, causing massive accumulation of both cholesteryl esters and triglycerides in macrophages throughout the body. Affected children usually die before their first birthday. Enzyme replacement therapy has now been developed but its efficacy remains to be fully established.
Cholesteryl ester storage disease
is caused by partial deficiency of LAL. It is a rare autosomal recessive disorder, characterized by hepatomegaly, abnormal liver function tests, hypercholesterolaemia and premature atherosclerosis. The hepatomegaly is a consequence of accumulation of cholesteryl esters and triglycerides in both hepatocytes and Kupffer cells.
Phospholipase A2
Phospholipase A2 (PLA2) is one of a family of phospholipases, enzymes that hydrolyse phospholipids. It exists in five forms. Three are found within cells. Two are secreted, one of which, lipoprotein-associated PLA2 (LpPLA2) is also known as platelet activating factor acetyl hydrolase (PAF-AH). Lipoprotein-associated PLA2 promotes the hydrolysis of oxidized phospholipids in lipoproteins, generating lysophospholipids and proinflammatory oxidized fatty acids.
There is a correlation between plasma LpPLA2 concentration and atherosclerosis. It is unclear whether this association is causal, but inhibition of LpPLA2 is of potential therapeutic value, and clinical trials are underway.
Acyl-CoA:cholesterol acyltransferase
Acyl-CoA:cholesterol acyltransferase (ACAT) catalyses the esterification of cholesterol. Two forms have been identified: ACAT1 is found ubiquitously in the endoplasmic reticulum of cells; ACAT2 is predominantly found in the endoplasmic reticulum of the liver and intestine. ACAT1 is sensitive to the degree of membrane cholesterol enrichment, and it is thought that its function is to maintain the cholesterol content of cell membranes at an optimal level by catalysing the esterification of excess free cholesterol. ACAT2 is responsible for the secretion of cholesteryl esters into the apo B-containing lipoproteins.
TRANSFER PROTEINS INVOLVED IN LIPOPROTEIN METABOLISM
Cholesteryl ester transfer protein (CETP)
This is a hydrophobic glycoprotein that is secreted mostly by the liver. In the blood, it is mainly bound to HDL. It facilitates the transfer of cholesteryl esters from HDL to the triglyceride-rich lipoproteins and LDL, and of triglyceride from triglyceride-rich lipoproteins to HDL. The extent of this exchange depends on the plasma concentration of triglycerides; the more triglyceride is transferred into HDL, the more is available as a substrate for HL, and the greater the capacity for HDL to become more delipidated (and so more rapidly degraded). This is the explanation for the well-recognized inverse relationship between plasma triglyceride and HDL-cholesterol concentrations.
Cholesteryl ester transfer protein has been viewed as both anti-and pro-atherogenic. Its anti-atherogenic action is related to its role in reverse cholesterol transport: its pro-atherogenic action is related to its capacity to transfer cholesteryl esters from HDL to the atherogenic lipoproteins (VLDL, IDL and LDL). Under normal conditions, CETP-mediated cholesteryl ester transfer from HDL is predominantly to LDL. When the plasma concentration of VLDL is increased, as in type 2 diabetes, CETP-mediated cholesteryl ester transfer is preferentially directed towards the large VLDL particles, which consequently become cholesterol enriched and more atherogenic.
Although there is uncertainty about the potential effects of CETP activity on atheroma formation, it is a target for lipid-modifying drug therapy. Drugs that inhibit CETP may raise plasma HDL concentrations by over 100%. The results of clinical trials to see if this translates into effective reduction of clinical cardiovascular endpoints are awaited.
Phospholipid transfer protein (PTP)
This glycoprotein is a member of the same gene family as CETP. Along with other genes involved in lipid metabolism the PLTP gene is under control of the liver X receptor (LXR). Phospholipid transfer protein is important in the remodelling of HDL, the generation of pre-β1HDL, the transfer of surface lipids from the triglyceride-rich lipoproteins to HDL during intravascular lipolysis and the facilitation of HDL-mediated efflux of phospholipids and cholesterol from cells. PTP activity is higher in hypertriglyceridaemic than normotriglyceridaemic individuals, and is correlated with the degree of insulin resistance. Its activity has also been found to be increased in subjects with familial combined hyperlipidaemia.
Fatty acid transport proteins
Fatty acid transfer proteins (FATPs) are membrane proteins that facilitate the uptake of fatty acids by cells. They also have acyl-CoA synthase activity. They form a family of six proteins that differ in tissue expression, intracellular localization and responsiveness to insulin. FATP1 and FATP4 have been implicated in insulin resistance. The former is highly expressed in adipose tissue and skeletal muscle whereas the latter is principally expressed in the small intestine. FATP2 is located in peroxisomes of the kidney and liver. It is important for the formation and oxidation of very long chain fatty acyl-CoA and is involved in the activation of bile acids in the liver; it is therefore sometimes referred to as very long chain acyl-CoA synthase. FATP3 is located in mitochondrial membranes. FATP5 is located in the membrane of the endoplasmic reticulum and is also involved in the activation of bile acids. FATP6 is located in the plasma membrane of cardiac myocytes, where it probably plays an important role in the supply of fatty acids as an energy source.
RECEPTORS INVOLVED IN LIPOPROTEIN METABOLISM
The LDL receptor
This receptor is a transmembrane protein that can be expressed by most cell types under certain conditions. The LDL receptor (LDLR) binds two apolipoprotein ligands, apo B-100 and apo E and, for this reason, is sometimes referred to as the B-100/E receptor. Uptake of LDL via the LDLR is mediated through its interaction with apo B-100. Although VLDL and IDL also possess apo B-100, it is not accessible for binding: IDL binds to LDLR via apo E. Subclasses of HDL containing apo E can also bind to the LDLR. Indeed, lipoproteins that contain multiple copies of apo E bind to the LDLR with much greater affinity than does LDL.
The LDLR gene is located on chromosome 19. The LDLR protein contains 843 amino acids and comprises five domains (see Fig. 37.10). At the amino terminal end is the ligand binding domain (exons 2–6), which mediates binding to apo B-100 or apo E. Next, there is a domain (exons 7–14) homologous to the epidermal growth factor (EGF) precursor. This contains three EGF repeats separated by a region of beta sheet folded in a specific orientation called a ‘beta propeller’. At the acid pH found in endosomes, this domain undergoes a conformational change bringing the beta propeller adjacent to repeats 4 and 5 in the ligand binding domain and releasing bound LDL. It also serves to maintain the correct orientation of the ligand binding domain on cell surfaces. The specific orientation determined by this EGF domain is required for the binding of LDL to the receptor but not for the binding of apo E-containing lipoproteins. The third domain (exon 15) consists of a peptide chain enriched in serine and threonine to which sugar chains are attached. The fourth domain (exon 16) comprises hydrophobic amino acids that span the membrane and thus function as an anchor. The carboxy terminal portion constitutes the intracellular cytoplasmic domain (exons 17–18) and appears to be important in targeting the receptor to the coated pit region.
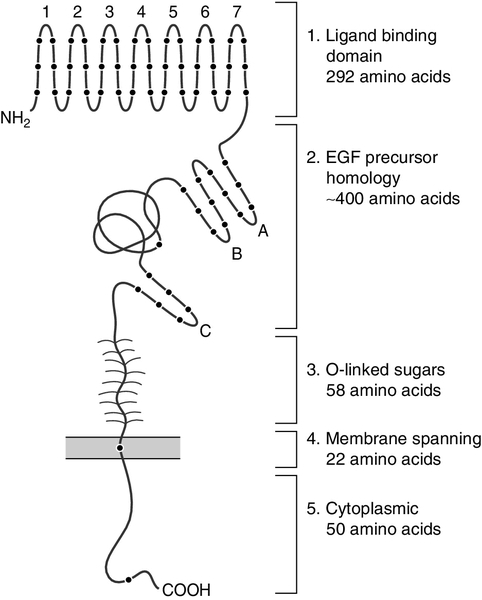
FIGURE 37.10 The LDL receptor. EGF, epidermal growth factor.
Expression of LDLR is under feedback control: in the steady state, the number of LDL receptors is controlled to allow uptake of sufficient cholesterol for cell growth and to balance losses. This feedback is by membrane bound transcription factors known as sterol regulatory element binding proteins (SREBPs) (see p. 721) and their associated insulin signalling (Insig) and SREBP cleavage activating proteins (SCAPs). The function of LDLRs is dependent not only on their binding to lipoprotein particles but also on their clustering in clathrin-coated pits for the subsequent internalization of the bound lipoproteins by endocytosis.
Low density lipoprotein receptor-associated protein 1 (LDLRAP1) binds the LDLR, clathrin and the clathrin-associated adaptin protein complex 2 (AP-2), and is essential for the endocytosis of LDLRs into hepatocytes. Mutations in the LDLRAP1 gene cause the rare clinical phenotype of autosomal recessive familial hypercholesterolaemia.
LDL receptor-related protein
LDL receptor-related protein (LRP) is expressed by hepatocytes and has multiple ligands including apo E, HL and lipoprotein lipase. Following synthesis, LRP becomes associated with another protein, LDL receptor-related protein associated protein (LRPAP1), which acts as a chaperone to facilitate the passage of LRP to the cell surface. LRP is involved in the apo E-mediated uptake of remnant particles by the liver, and is sometimes referred to as the ‘apo E receptor’.
The apo E4 polymorphism has an impact on the uptake of cholesterol to neuronal tissue by LRP. This may partially explain the increased risk of Alzheimer disease in those with the apo ε4/ε4 genotype.
Scavenger receptor class B type 1
This is a 509-amino acid protein comprising short N- and C-terminal cytoplasmic and transmembrane domains and a large extracellular loop. Its amino acid sequence is 30% homologous with that of CD36 (see below). Like CD36, scavenger receptor class B type 1 (SRB1) can bind to a number of lipoproteins and modified lipoproteins (HDL, LDL, VLDL, oxidized LDL). It is present in a number of tissues but is most highly expressed in tissues that process HDL cholesterol and cholesteryl esters for excretion or as metabolic substrates, i.e. the liver and steroidogenic tissues. SRB1 binds HDL and mediates the selective uptake of cholesteryl esters from the cores of HDL particles to the plasma membrane without entailing the uptake and degradation of the whole HDL particle. This process is dependent on apo A-I. These cholesteryl esters are then hydrolysed extra- lysosomally by tissue-specific neutral cholesteryl hydrolases; in the adrenals this may be by HSL.
Scavenger receptor class B type 1 is also able to mediate the efflux of unesterified cholesterol from cells to lipoproteins. Thus, SRB1 is involved at both ends of reverse cholesterol transport; initially with the efflux of free cholesterol from peripheral cells to HDL and at the end in the delivery of cholesteryl esters and unesterified cholesterol to the liver for secretion into bile. The membrane domain-containing protein PDZK1 (the PDZ group of proteins are named from the initial letters of the first three proteins found to share this structural domain), controls the basal hepatocyte and enterocyte SRB1 content, whilst in the adrenals, ACTH binding to the melanocortin 2 receptor stimulates the expression of SRB1. This in turn promotes the uptake of cholesteryl esters from HDL, which provides a pool of free cholesterol within the adrenals for synthesis of glucocorticoids. In the liver, SRB1 is downregulated by fibric acid derivatives. Scavenger receptor class B type 1 has been shown to mediate the binding and endocytosis of lipopolysaccharides, suggesting that it may have a role in their cellular uptake and clearance in sepsis.
There is evidence that loss of function mutations in the SCARB1 gene that codes for SRB1 are associated with increased concentrations of HDL but a paradoxical increase in cardiovascular disease.
Other scavenger receptors
CD36 (which stands for Cluster of Differentiation 36) is a receptor found in caveolae in the cell surface plasma membrane. Like SRB1 it is a Class B scavenger receptor. CD36 can bind native and oxidized LDL. Together with the scavenger receptors A1 and A2 (SR-AI, SR-AII) it mediates the avid uptake of oxidized LDL by endothelial macrophages.
Peroxisome proliferator-activated receptor family
Nuclear receptors play a key role in glucose and lipid homoeostasis and in the inflammatory response. They are activated by a variety of ligands, including fatty acids. Among these receptors is the peroxisome proliferator activated receptor (PPAR) family. When they bind to a ligand, PPARs form heterodimers with the nuclear retinoid X receptor (RXR), which bind to response elements in the promoter regions of target genes, thus promoting their transcription. By this means, PPARs control the expression of genes that maintain lipid and glucose homoeostasis. This makes them a potential therapeutic target.
The PPAR family includes PPARα, PPARγ and PPARβ/δ. The fibrate family of lipid-lowering drugs are activators of PPARα and the thiazolidinediones, which are insulin sensitizing agents, are ligands for PPARγ. The intravascular lipolysis of lipoproteins is promoted by fibrates through increased LPL activity and reduced expression of the LPL inhibitor, apo C-III. Fibrates also induce the expression of apo A-V, which impacts on plasma triglyceride concentrations and of apo A-I and apo A-II, leading to increased production of HDL. Activation of PPARγ induces the expression of genes for proteins that control adipocyte fatty acid metabolism, including LPL and fatty acid transport proteins, thus facilitating the hydrolysis of lipoprotein triglycerides and promoting the storage of the released fatty acids as adipocyte triglycerides.
Other nuclear receptors
As well as PPARs, other nuclear receptors such as the liver X receptors (LXRs) and farnesoid X-activated receptor (FXR) have been demonstrated to exert control over lipid metabolism. The LXR is a sterol responsive nuclear receptor that is activated in response to excess of cholesterol within the cell. When activated, it triggers transcription of the inducible degrader of LDLR (IDOL); this causes ubiquitination of the cytoplasmic domain of LDLR, which promotes its destruction within the cell. Activated LXR also activates ABCA1 and ABCG1, which promotes cholesterol efflux from the cell. The net effect is to reduce cellular cholesterol uptake and increase efflux from the cell, this restoring sterol homoeostasis.
OTHER PROTEINS INVOLVED IN LIPOPROTEIN SYNTHESIS, TRANSPORT AND METABOLISM
Microsomal triglyceride transfer protein
Microsomal triglyceride transfer protein (MTP) is a member of a group of proteins that are able to transfer of lipids between membranes. MTP is an 894 amino acid (97 kDa) protein that forms a heterodimer with protein disulfide isomerase (PDI); the two subunits are held together by non-covalent interactions. This protein is present in high concentrations in the lumen of the endoplasmic reticulum and plays an important role in the maturation of the endoplasmic reticulum and its secreted proteins. Protein disulfide isomerase does not demonstrate isomerase activity when combined with MTP, but is required for retention of MTP in the endoplasmic reticulum at the site of apo B translocation. The promoter region of MTP is upregulated by cholesterol and downregulated by insulin. Microsomal triglyceride transfer protein binds and shuttles individual lipids between membranes.
Microsomal triglyceride transfer protein activity is essential for the synthesis of chylomicrons and VLDL. After the assembly of chylomicrons and VLDL within the lumen of the endoplasmic reticulum of enterocytes and hepatocytes respectively, they are transported to the Golgi apparatus and then secreted. Loss of function mutations in MTP cause abetalipoproteinaemia (see p. 724)
Microsomal transfer protein also transfers lipid to antigen presenting molecules on the surface of natural killer T cells. These mediate a number of autoimmune disorders, making MTP a pharmacological target, with inhibition having the potential to achieve both lipid lowering and anti-inflammatory effects. Inhibitors of MTP, which effectively lower plasma LDL-cholesterol concentrations, are in clinical trials, although it remains to be seen whether their use will be limited because of their tendency to cause an increase in hepatic steatosis.
ATP binding cassette transporter family
The ATP binding cassette (ABC) transporter family comprises more than 50 different proteins. Within this family, ABCG1, ABCG5, ABCG8 and ABCA1 are all sterol induced. ABCG5 and ABCG8 form a heterodimer, which mediates the excretion of absorbed plant sterols and cholesterol from enterocytes into the lumen of the gut and from hepatocytes into the bile. ATP binding cassette transporters A1 and G1 facilitate the export of cholesterol from peripheral cells, where its combination with apo A-I and phospholipids to produce HDL is the first step in reverse cholesterol transport.
Proprotein convertase subtilisin kexin 9
Proprotein convertase subtilisin kexin 9 (PCSK9) is one of nine members of the subtilisin protease family. The PCSK 9 gene is mainly expressed in liver and is regulated by sterol regulatory element binding proteins (see p. 721). The PCSK9 protein is synthesized as a 72 kDa protein that undergoes self-cleavage to a 63 kDa protein, which is found in the Golgi. Proprotein convertase subtilisin kexin 9 regulates LDLR activity by accelerating the degradation of mature receptors. It is expressed on the cell surface but also circulates free in plasma.
Gain of function mutations in the PCSK9 gene are a rare cause of autosomal dominant hypercholesterolaemia (familial hypercholesterolaemia). Loss of function mutations are associated with reduced plasma LDL-cholesterol concentration and lower than average risk of CVD. This makes both gene inactivation and immunological blockade of PCSK9 action attractive targets for lipid lowering therapy. Therapeutic agents are in clinical trials, and have been shown to reduce plasma LDL-cholesterol concentrations by > 50%.
Sterol regulatory element binding proteins
Sterol regulatory element binding proteins (SREBPs) are membrane-bound transcription factors that activate genes involved in cholesterol synthesis. They provide the means by which cellular cholesterol exerts negative feedback on cholesterol synthesis. There are three SREBPs: SREBP-2 activates genes involved in cholesterol synthesis, the other two (SREBP-1a and SREBP-1c) have more effect on the genes involved in fatty acid synthesis. When cholesterol is abundant in a cell, the SREBPs remain in the endoplasmic reticulum associated with an escort protein, SREBP cleavage activating protein (SCAP), and the endoplasmic reticulum retention protein, insulin signalling protein (Insig). When a cell is relatively cholesterol-depleted, SCAP dissociates from Insig and results in the release of SREBP-SCAP from the endoplasmic reticulum. To exert their action, SREBPs must undergo two-step proteolytic cleavage in the Golgi to release their N-terminal fragment. These fragments travel to the nucleus where they bind to the steroid response elements (SREs) in the promoter regions of a number of the genes encoding enzymes involved in cholesterol synthesis, including squalene synthase and HMG-CoA reductase, and the LDLR gene.
When intracellular cholesterol content is again adequate, the proteolytic release of the N-terminal fragment is blocked and the N-terminal fragments in the nucleus are degraded.
Both SREBP genes contain intronic sequences that code for a micro-RNA (miR-33). Micro-RNAs (miRNAs) are small non-coding RNAs approximately 22 nucleotides in length that bind to the three prime untranslated region (3′UTR) of mRNAs, causing their reduced translation or destruction. Micro-RNA-33 reduces translation of mRNA for ABCA1, ABCG1 and genes involved in fatty acid oxidation. This effect is complementary to the primary effect of SREBP on cholesterol and fatty acid synthesis and serves to further increase the accumulation of cholesterol and triglycerides within cells.
In the liver, insulin stimulates SREBP-1c production, and in states of hyperinsulinaemia with insulin resistance this may contribute to the accumulation of fat in hepatocytes, and drive increased VLDL synthesis. It is also possible that reduced ABCA1 expression caused by miR-33 may account in part for the low HDL seen in insulin resistant states.
These recent insights into control of lipid metabolism by miRNAs makes them potential therapeutic targets, and initial studies have shown that HDL can be increased by antisense oligonucleotides designed to block miR-33.
Sortilins
Sortilins are receptors that regulate intracellular transport by shunting proteins through secretory or endocytic pathways. They share in common a 700 amino acid extracellular domain which forms a ten bladed beta propeller and acts as a ligand binding site. This is designated the vacuolar protein sorting ten protein domain (VPS10P).
Genome wide association studies suggest that two of these receptors, sortilin and sorting protein-related receptor with type A repeats (SORLA), have a role in lipid metabolism and atherosclerosis, and may account for variability in cardiovascular risk. Reduced quantities of SORLA are associated with increased smooth muscle cell migration and lipid loading of macrophages in atheromatous plaques. This effect may be mediated by SORLA binding of LPL and apo A-V on the cell surface. Overexpression of the sortilin gene, SORT1 is associated with lower plasma LDL-cholesterol concentrations, either indirectly by reducing VLDL output from the liver, or directly by acting as an alternative hepatic receptor for LDL.
Glycosylphosphatidylinositol-anchored HDL-binding protein 1
Glycosylphosphatidylinositol-anchored HDL-binding protein1 (GPIHBP1) is a 184 amino acid protein that is a member of the GPI anchored Ly6 (lymphocyte antigen 6) group of proteins. It was originally identified as a protein capable of binding HDL. It is now recognized to be essential to the function of lipoprotein lipase. This protein binds LPL in the subendothelial space and transports it to the luminal surface of endothelial cells, where it anchors the LPL protein by means of the GPI domain. Genetic studies have shown that mutations in GPIHBP1, and mutations in LPL that make it unable to bind GPIHBP1, are both associated with increased plasma triglyceride concentrations.
Angiopoietin-like protein 3
Angiopoietin-like protein 3 (ANGPTL3) acts in the liver to promote the production and secretion of apo B containing lipoproteins. In the circulation, it inhibits the activities of LPL. It may also affect the activity of EL and thereby modulate HDL cholesterol concentrations. Loss of function mutations in the ANGPTL3 gene have been associated with a combined hypolipidaemia phenotype, with low plasma concentrations of cholesterol, HDL-cholesterol and triglycerides.
CLASSIFICATION OF LIPOPROTEIN DISORDERS
Fredrickson originally suggested dividing the hyperlipidaemias into five types (I–V) based on which of the apo B-containing lipoprotein classes were shown to be increased on paper electrophoresis of a fasting sample. This was subsequently modified by the World Health Organization, which introduced a subdivision of type II into two (types IIa and IIb) (Table 37.4).

The Fredrickson classification is limited in being only a description of the lipoprotein phenotype manifested in an individual, and only covers abnormal elevations of the apo B containing lipoproteins. It does not differentiate primary from secondary causes of any particular phenotype, and does not include deficiencies of the apo B containing lipoproteins or disorders of HDL metabolism. It soon became clear that the type I phenotype can be caused by any one of three inherited abnormalities, all of which result in a functional deficiency of lipoprotein lipase. Type III is caused by an inherited defect in apo E causing reduced binding to the LRP (apo E) receptor in the liver. The other types (IIa, IIb, IV and V) can each be a consequence of one or more inherited defects or may reflect secondary causes. Thus classifying an individual into one of these Fredrickson classes gives no indication as to the genetic basis of the manifested lipoprotein disorder, its mechanism, the need for family screening or the management required. It is now recognized that even the type I and type III phenotypes can have multiple other genetic causes due either to single gene disorders or the cumulative effect of multiple small genetic variants together with secondary environmental influences. For all these reasons, this classification has largely been superseded by a clinical and genetic classification, which relates to the molecular mechanism underlying the clinical phenotype. The genetic basis of the primary dyslipidaemias and the relationship, where known, to the corresponding clinical and Fredrickson phenotypes is given in Table 37.5.
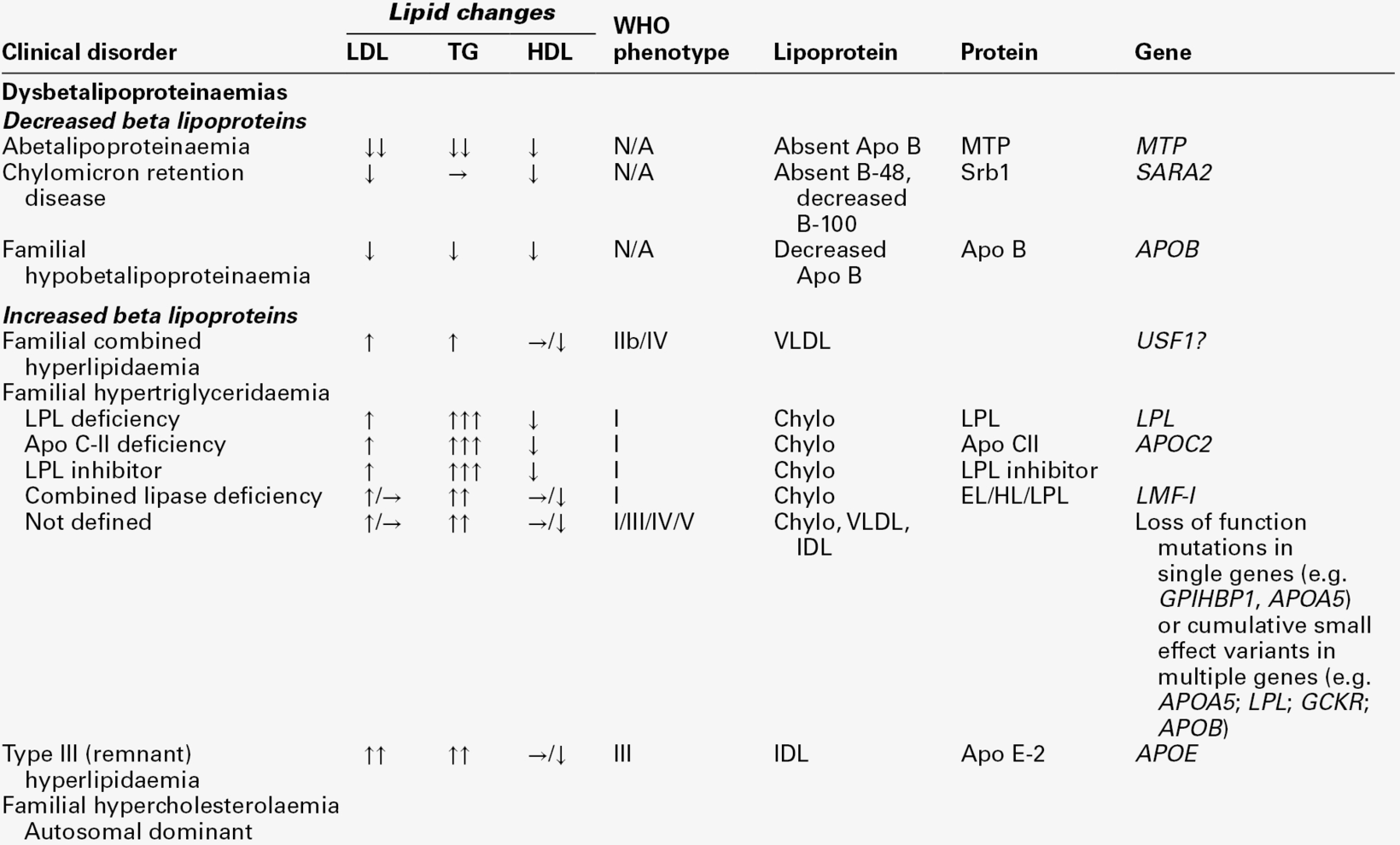
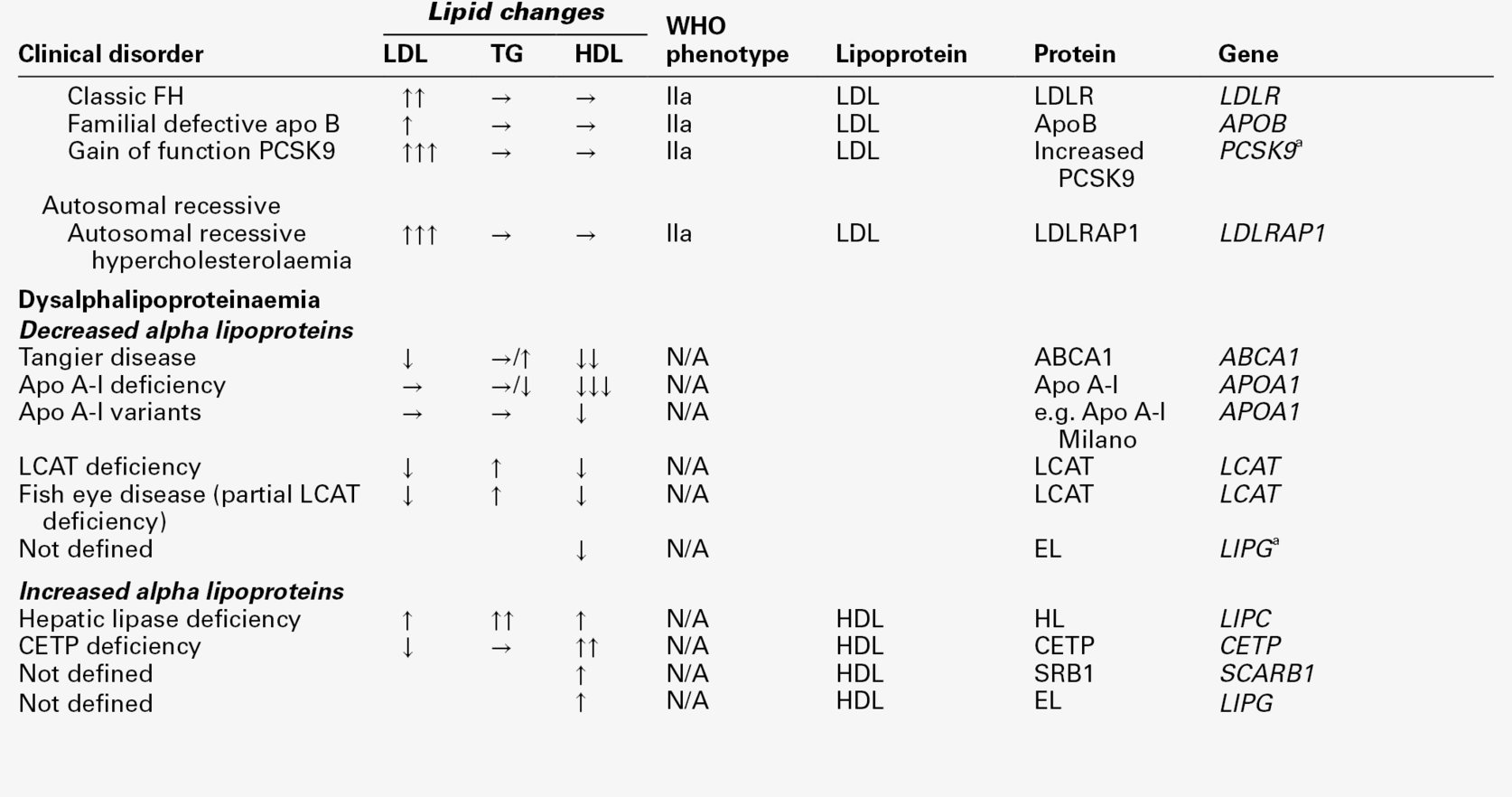
Lipids may be deposited in certain extravascular sites in the presence of significant hyperlipidaemia. In certain instances, the site and form of this lipid deposition may be characteristic of the underlying lipid abnormality (Table 37.6).
TABLE 37.6
Extravascular manifestations of hyperlipidaemia
| Extravascular site of lipid deposition | Type of hyperlipidaemia |
| Xanthelasma (periorbital) | Any form of hypercholesterolaemia; may occur with normal lipid concentrations; common in hyperlipidaemias due to cholestasis |
| Corneal arcus (around the iris of the eye) | Arcus senilis can occur with increasing age in subjects with normal lipid concentrations; if present in a subject < 45 years, probably denotes presence of significant underlying hyperlipidaemia (usually ↑ LDL) |
| Tendon xanthomas (occur in extensor tendons of hands, feet, Achilles tendon and patellar tendon) | Virtually pathognomonic of familial hypercholesterolaemia; also found in the very rare conditions, β-sitosterolaemia and cerebrotendinous xanthomatosis |
| Tuberous and tuberoeruptive xanthomas (occur over extensor surfaces of limbs, i.e. elbows/knees) | Remnant hyperlipoproteinaemia and familial hypercholesterolaemia |
| Palmar crease xanthomas | Remnant hyperlipidaemia |
| Eruptive xanthomas (occur in crops, particularly over the extensor surface of the elbows and over the buttocks) | Chylomicronaemia |
THE PRIMARY DYSLIPOPROTEINAEMIAS
A primary dyslipoproteinaemia is an inherited disorder of lipoprotein metabolism that may manifest as hyperlipidaemia, hypolipidaemia or normolipidaemia associated with lipoproteins of abnormal composition or an abnormal distribution of the normal lipoprotein classes.
Hypobetalipoproteinaemia
The term ‘hypobetalipoproteinaemia’ describes the situation when the total plasma cholesterol, LDL-cholesterol or apo B concentrations are less than the 5th centile. This may be secondary to an underlying disorder, for example fat malabsorption, or a consequence of an inherited defect in lipoprotein metabolism (primary hypobetalipoproteinaemia). Primary hypobetalipoproteinaemia encompasses three disorders: abetalipoproteinaemia, chylomicron retention disease and familial hypobetalipoproteinaemia.
Abetalipoproteinaemia
This is a very rare autosomal recessive condition, first described in 1950 by Bassen and Kornzweig. It typically presents in infancy with failure to thrive and chronic diarrhoea owing to fat malabsorption. Other features include acanthocytosis (caused by abnormal erythrocyte membrane lipid composition) and an atypical retinitis pigmentosa (as a result of vitamin A deficiency). Later manifestations are night blindness and neurological disability, particularly ataxia, caused by vitamin A and vitamin E deficiencies, respectively. The primary aim of treatment in these subjects is to ensure that fat-soluble vitamin intake is sufficient, which may require their parenteral administration.
There is a complete absence of all apo B-containing lipoproteins in the plasma, and no circulating apo B, whereas HDL and apo A-I concentrations are approximately 50% of normal. Plasma cholesterol concentration is low (typically 0.5–1.5 mmol/L) and plasma triglyceride concentration is very low (< 0.2 mmol/L). On microscopy, lipid droplets can be seen to accumulate intracellularly in hepatocytes and enterocytes, suggesting a defect in the assembly of the apo B containing lipoproteins. The cause is now known to be loss of function mutations in the MTP gene (see p. 720). In the presence of defective MTP, no apo B-containing lipoproteins are formed. The malabsorption of fat and the fat-soluble vitamins is a result of the failure of chylomicron formation.
Chylomicron retention disease
The inheritance of chylomicron retention disease is probably autosomal recessive. It is characterized by an absence of apo B-48 in plasma and lack of a postprandial lipaemic response. Affected individuals also have low LDL, HDL and fat-soluble vitamin concentrations and their LDLs are enriched in triglycerides. They have fat malabsorption and steatorrhoea and, without supplementation of fat-soluble vitamins, develop neurological dysfunction. Enterocytes of affected individuals contain fat droplets, and apo B-48 is demonstrable immunochemically within the enterocytes. Chylomicron retention disease is caused by mutations in the SARA2 gene. The protein product of the SARA2 gene is sar1b, which is involved in the transport of chylomicrons through the enterocyte secretory pathway (see p. 712).
Familial hypobetalipoproteinaemia
Familial hypobetalipoproteinaemia (FHBL) is an autosomal co-dominant disorder resulting from mutations in APOB generating premature stop codons and therefore truncation of apo B. These truncated forms have a reduced capacity for lipid binding and thus result in the secretion of smaller, denser, relatively lipid-poor lipoprotein particles. Lipoprotein particles containing the larger truncated forms (e.g. apo B-89 and apo B-75) fall into the VLDL fraction when secreted, whereas those containing the smaller forms (e.g. apo B-29) fall into the HDL fraction. There is a threshold in apo B size under which the apo B cannot form a lipoprotein; this threshold is between apo B-28 and apo B-29. Mutations producing truncated forms of apo B greater than apo B-48 are associated with the production of normal chylomicrons. Apolipoprotein B forms smaller than B-67 are incorporated into VLDL in the normal way but are not able to interact with LDL receptors.
Individuals heterozygous for FHBL associated with truncated apo Bs are often asymptomatic, but a small proportion have loose stools. The plasma LDL-cholesterol concentration in heterozygotes would be expected to be about 50% of that in unaffected family members, but concentrations of about a third of normal are actually observed, possibly because of reduced hepatic secretion or upregulation of LDL receptors that results in increased clearance. A high incidence of non-alcoholic fatty liver disease is reported in FHBL heterozygotes. Homozygous FHBL clinically resembles abetalipoproteinaemia.
Familial combined hyperlipidaemia
This condition is the most common lipid disorder in subjects presenting with ischaemic heart disease. It is inherited as an autosomal dominant trait, but usually does not become manifest until adulthood. The typical findings are a raised plasma apo B-100 concentration and an increase in plasma concentrations of LDL (high total cholesterol), VLDL (high triglycerides) or both. Different individuals within a kindred may have any of these phenotypes, and in a single individual, the phenotypic pattern may change over time. Plasma HDL-cholesterol concentration is usually low; this probably results from increased transfer of cholesteryl esters from HDL to the triglyceride-rich lipoproteins. The LDL particles in familial combined hyperlipidaemia (FCH) tend to be smaller and denser than usual; thus individuals with FCH display the ‘atherogenic lipid profile’ comprising high plasma triglyceride and low HDL-cholesterol concentrations and small, dense LDL particles.
Overproduction of apo B-100 occurs in FCH, so that affected individuals have raised plasma apo B concentrations even though their lipid concentrations may be normal. The development of hyperlipidaemia is dependent on an increase in the availability of hepatic triglycerides; this explains the observation that affected subjects are frequently obese, and may have other features of the metabolic syndrome. The phenotype expressed is thought to reflect the efficiency with which the VLDL is processed in affected individuals. Thus individuals who have one abnormal LPL gene (parents of children with LPL deficiency) may appear to have FCH because of the resultant slowed clearance of VLDL.
Genetic studies have suggested a link between FCH and the upstream transcription factor 1 (USF1) gene. This is a transcription factor responsible for upregulating the transcription of a number of genes involved in glucose and lipid metabolism, including those coding for apo A-V.
Familial hypertriglyceridaemia
Some families show Mendelian inheritance of hypertriglyceridaemia. In most cases this reflects an increase in VLDL, with moderately elevated triglyceride concentrations (4–10 mmol/L). In some cases, there is a more severe hyperlipidaemia (> 10 mmol/L) and fasting chylomicronaemia. The latter causes a Fredrickson type I phenotype, while less severe hypertriglyceridaemia generally causes a type IIb, IV or V pattern. Severe hypertriglyceridaemia is typically autosomal recessive and in addition to defects in LPL and apo C-II, which have long been recognized as causes of type I hyperlipidaemia (chylomicronaemia syndrome), it is now recognized that loss of function mutations in genes including APOA5, LMF1 and GPIHBP1 may produce a similar phenotype, often inherited in an autosomal recessive manner. By contrast, the cumulative effect of multiple small effect polymorphisms in genes including LPL, APOA5, GCKR and APOB, interact with environmental factors to produce more moderate hypertriglyceridaemia, and a variety of lipoprotein phenotypes. These include IIb, IV, V and, sometimes, type III. The environmental factors interacting with these genetic variants to produce moderate or severe fasting hypertriglyceridaemia are the same as those causing secondary hyperlipidaemia, e.g. diabetes, insulin resistance or excessive alcohol consumption. VLDL particles are larger than usual, and relatively deficient in apo B in familial hypertriglyceridaemia, which suggests that an important factor is overproduction of triglycerides by the liver. Chylomicronaemia syndrome, caused by autosomal recessive major loss of function mutations in single genes, often manifests in childhood, whilst other causes of familial hypertriglyceridaemia rarely manifest before adulthood.
In patients with a fasting plasma triglyceride concentration > 10 mmol/L, there is a risk of acute pancreatitis, and reducing this risk is usually the first priority of treatment, before addressing residual cardiovascular risk. Fibrates or ω3-fatty acids, in combination with a low fat diet and avoidance of excessive alcohol consumption, are usually effective. Plasma exchange, and intravenous insulin infusion (to increase lipoprotein lipase activity), have both been used in severe cases although the evidence for their long-term effectiveness is limited.
Chylomicronaemia syndrome
Chylomicronaemia syndrome manifests as eruptive xanthomata (see Fig. 37.11), lipaemia retinalis, hepatosplenomegaly and recurrent bouts of abdominal pain that may be symptoms of acute pancreatitis. Plasma triglyceride concentrations are markedly increased (> 10 mmol/L), HDL-cholesterol concentrations are very low and chylomicrons are present in the fasting state. Three inherited causes of this condition have been described: lipoprotein lipase deficiency, apo C-II deficiency and familial lipoprotein lipase inhibitor. Secondary causes, including excessive alcohol consumption and newly diagnosed type 1 diabetes, may produce a very similar phenotype in genetically predisposed individuals.
FIGURE 37.11 Eruptive xanthomata over the elbow.
Lipoprotein lipase deficiency
This is a rare autosomal recessive condition with an incidence of approximately 1 in 1 000 000. Presentation is usually in childhood with recurrent abdominal pain. Affected individuals may have low, normal or increased concentrations of immunoreactive LPL in post-heparin plasma but catalytic activity is undetectable. Gene replacement therapies for the most severe forms of this condition are undergoing clinical trials.
Obligate heterozygotes for LPL deficiency have been shown to have low plasma LPL activity: some may develop severe hypertriglyceridaemia, and a small number, pancreatitis.
Apo C-II deficiency
Homozygous apo C-II deficiency tends to present later and be somewhat milder than LPL deficiency. Apo C-II is detectable in about 50% of individuals with apo C-II deficiency, but the protein produced is unable to activate LPL. It is possible that the concentrations of the triglyceride-rich lipoproteins may reach a level at which LPL can hydrolyse them even in the absence of apo C-II: this could account for the milder form of the disease as compared with that seen with primary deficiency of the enzyme.
Relatives who are obligate heterozygotes for the abnormal gene usually have normal lipid and lipoprotein concentrations. This implies that up to a 50% reduction in the apo C-II concentration does not compromise the rate of chylomicron and VLDL clearance.
Familial lipoprotein lipase inhibitor
Families have been described whose affected members have chylomicronaemia apparently caused by an inhibitor of LPL. The defect appears to be inherited in an autosomal dominant fashion. The inhibitor has not been identified.
Remnant hyperlipoproteinaemia
The characteristic feature of remnant hyperlipoproteinaemia (RH) is cholesterol enrichment of the VLDL fraction, reflecting an accumulation of remnant particles, both chylomicron remnants and intermediate density lipoprotein. These remnants are responsible for the abnormal electrophoretic pattern, which gave the condition its former name of broad β disease (type III in the Fredrickson classification), since a broad β band is seen on electrophoresis instead of the usual two distinct pre-β (VLDL) and β (LDL) bands. Remnant hyperlipidaemia is also sometimes referred to as dysbetalipoproteinaemia. Subjects with this disorder show a mixed hyperlipidaemia, often with approximately equal plasma concentrations of cholesterol and triglycerides.
The predominant defect found in most subjects with the condition is the presence of apo E2, which displays a recessive mode of inheritance. While apo E2 homozygosity is essential for the accumulation of remnant particles, it is not sufficient for the manifestation of the condition. A super-imposed genetic or environmental factor is necessary before remnant hyperlipidaemia becomes manifest, such as the concomitant inheritance of another primary hyperlipidaemia or, more frequently, a secondary cause such as obesity, excessive alcohol consumption, diabetes or hypothyroidism. The 10% of individuals with RH who do not show homozygosity for apo E2/2 have a variety of molecular defects, including point mutations resulting in amino acid substitutions at residues 142, 145 or 146, or the insertional variant, apo E-Leiden; a few lack apo E entirely.
Remnant hyperlipoproteinaemia rarely presents before adulthood. It is associated with characteristic xanthomata occurring in the palmar creases (Fig. 37.12) which are virtually pathognomonic, and tuberous or tuberoeruptive xanthomata over the extensor surfaces of the elbows and knees (Fig. 37.13). There is an increased risk of premature atherosclerosis involving the peripheral vascular system as well as the coronary arteries.
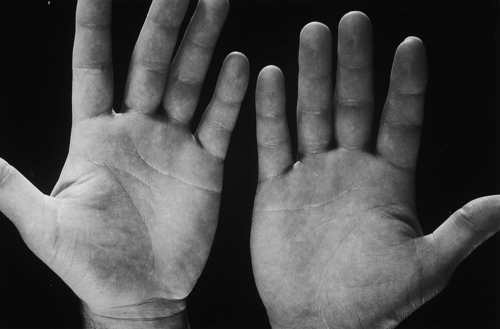
FIGURE 37.12 Xanthomata in the palmar creases.
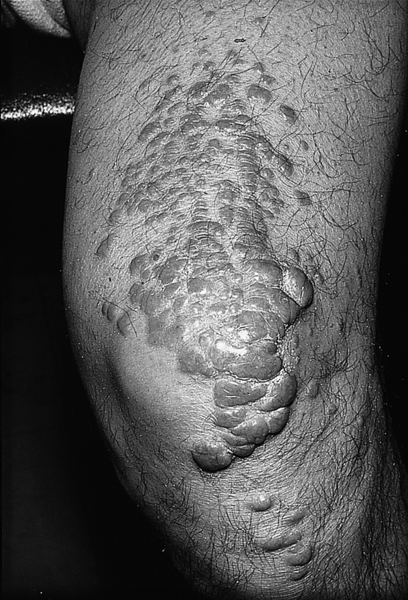
FIGURE 37.13 Tuberous xanthomata.
Familial hypercholesterolaemia
This term encompasses a group of disorders due to mutations causing reduced clearance of LDL by the LDL receptor resulting in marked hypercholesterolaemia and premature atherosclerosis. ‘Classic’ familial hypercholesterolaemia (FH) is due to mutations in the LDLR gene. Autosomal dominant mutations are now recognized in three genes causing conditions collectively referred to as autosomal dominant hypercholesterolaemia (ADH), which includes classic FH. A further gene locus has recently been associated with this clinical phenotype, but the causative gene has not yet been identified. A rare autosomal recessive condition has also been described with a similar clinical and biochemical phenotype. The four clearly defined disorders that result in FH are as follows:
• familial defective apolipoprotein B-100 – ADH 2
• gain of function mutation in PCSK9 – ADH 3
• loss of function mutation in LDLRAP1 – autosomal recessive hypercholesterolaemia.
Classic familial hypercholesterolaemia (FH)
This is the most severe of the hyperlipidaemias with respect to the propensity of affected, untreated individuals to develop atherosclerosis. It manifests in the heterozygous state as marked hypercholesterolaemia, owing to high LDL-cholesterol concentration, and premature cardiovascular disease. Heterozygous FH is one of the commonest inherited metabolic diseases, with a frequency of about 1 in 500 in most populations. Tendon xanthomata in the extensor tendons of the digits (Fig. 37.14) and in the Achilles tendons (Fig. 37.15) occur in approximately 70% of untreated heterozygotes and their presence in affected individuals or first-degree relatives aids the clinical diagnosis, which is based on criteria defined by the Simon Broome Trust (Box 37.1).
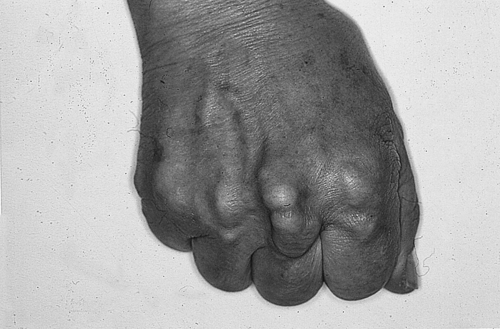
FIGURE 37.14 Xanthomata in the extensor tendons of the fingers.
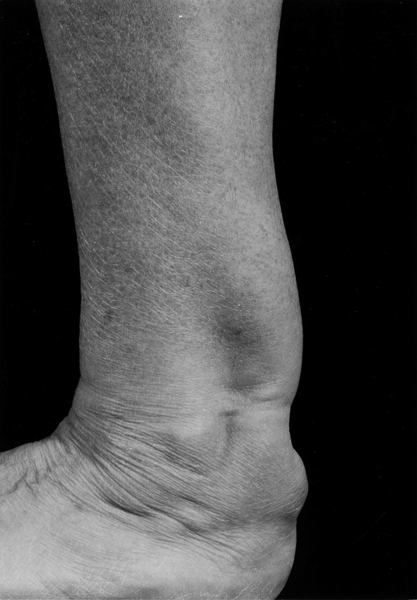
FIGURE 37.15 Xanthomata in the Achilles tendon.
Based on the frequency of the heterozygous state, homozygous FH would be expected to have an incidence of one in 1 000 000. Most apparently homozygous FH subjects are actually compound heterozygotes. The hypercholesterolaemia is far more severe in subjects with two mutant genes; cutaneous xanthomata often occur in childhood and coronary artery disease may present during the first decade of life. LDL apheresis has now become standard treatment in these patients in addition to lipid lowering drugs and diet. This can significantly increase life expectancy, particularly if the condition is diagnosed at a young age.
Classic FH, which accounts for the defect in 80–90% of patients with familial hypercholesterolaemia, is due to mutations of the LDL receptor gene. Around 20 common mutations account for 50% of cases, but over 1200 mutations have now been described, including a number of pathogenic intron mutations.
In vitro, it has been demonstrated that cells from FH heterozygotes have about half the number of functional LDL receptors than do cells from normal individuals. The effect of this deficiency of functional LDL receptors in vivo is that plasma LDL-cholesterol concentrations are increased to approximately twice the usual level, owing to a combination of reduced LDL uptake and LDL overproduction. The overproduction of LDL results from defective hepatic IDL uptake by the LDL receptor so that extracellular IDL to LDL conversion is increased. Hepatic cholesterol synthesis is also upregulated because of reduced LDLR-mediated cholesterol uptake.
Familial defective apolipoprotein B-100 (FDB)
This is an autosomal co-dominant disorder that results from an abnormality in the LDLR-binding domain of apo B-100. Most cases are due to mutations in the APOB gene at the codon for amino acid 3500 where Arg to Gln and Arg to Trp substitutions have been described. The arginine to glutamine substitution has been shown to change the conformation of the C-terminal tail, which results in reduced binding to the LDLR. Other mutations have been described that have a less severe effect on the ability of apo B to bind to LDL receptors. FDB is phenotypically very similar to classic FH; affected subjects have elevated plasma total and LDL cholesterol concentrations but the hypercholesterolaemia is usually less severe and tendon xanthomas are less common.
Gain of function mutation in PCSK9
This variant of FH was characterized by studying families that phenotypically were similar to classic FH and FDB. It was shown to be caused by gain of function mutations in the gene coding for proprotein convertase subtilisin kexin 9 (PCSK9). These mutations increase the rate at which PCSK9 degrades LDL receptors, resulting in reduced receptor expression on the cell surface. These mutations tend to be associated with a more severe clinical phenotype, with affected individuals showing higher plasma LDL-cholesterol concentrations and more aggressive vascular disease than seen in classic FH.
Autosomal recessive hypercholesterolaemia
Patients with autosomal recessive hypercholesterolaemia (ARH) phenotypically resemble classic FH homozygotes. They have large xanthomas (tendon, tuberous, planar) that present in childhood, and develop premature atherosclerosis, particularly of the coronary and carotid arteries. They also develop aortic stenosis. Plasma LDL-cholesterol concentrations in patients with ARH are generally higher than those found in classic FH but not as high as those seen in homozygous FH.
Autosomal recessive hypercholesterolaemia results from a mutation in the LDLRAP1 gene. LDLRAP1 mediates normal LDL receptor-mediated endocytosis after LDL is bound to the receptor, and this process is defective in ARH. Obligate heterozygotes for ARH who carry a single pathogenic LDLRAP1 mutation have normal plasma LDL-cholesterol concentrations.
Polygenic hypercholesterolaemia
Hypercholesterolaemia is more common in Western societies than would be expected, given the frequency of the individual monogenic disorders discussed above. The remainder of individuals with primary hypercholesterolaemia are therefore referred to as having polygenic hypercholesterolaemia. The term ‘polygenic’ is used because the individuals’ genetic background affects the extent to which lipoprotein metabolism will be influenced by environmental factors.
The monogenic disorders are associated with the same increased risk for atherosclerosis in different populations. However, the frequency with which polygenic hypercholesterolaemia occurs in different populations varies widely, as does the incidence of coronary artery disease. Populations with a high mean plasma cholesterol concentration have a high rate of coronary artery disease. It is believed that the difference between populations in the frequency of hypercholesterolaemia reflects one or more environmental factors, the most obvious being diet and, in particular, the saturated fat content of the diet.
The frequency with which polygenic hypercholesterolaemia is diagnosed in a population depends on the value taken as the upper cut-off for a ‘normal’ cholesterol. If 5 mmol/L is used, about 50% of most adult populations will be classified as hypercholesterolaemic, as the population mean cholesterol concentration is typically 5.5–6.0 mmol/L in many Western countries. At this level, CVD risk is already twice that of a population with a mean cholesterol < 4.0 mmol/L, so a statistical definition of ‘normal’ based on population values is unhelpful. Laboratories will generally refer to ‘ideal’ cholesterol targets. In the UK, these are currently total cholesterol < 4.0 mmol/L and LDL-cholesterol < 2.0 mmol/L.
Dysalphalipoproteinaemias
The dysalphalipoproteinaemias are disorders of the synthesis and secretion of apo A-containing lipoproteins. With the exception of apo A-I Milano (see below), all other genetic causes of familial hypoalphalipoproteinaemia are associated with some degree of increased cardiovascular risk.
Abnormal apolipoprotein A structure
Population screening for apo A-I structural variation has revealed at least 11 variants. The best described is apo A-I Milano, which is associated with a low HDL (HDL2) concentration but no increased incidence of atherosclerosis. Apolipoprotein A-I Milano results from substitution of cysteine for arginine at position 173, which has the effect of changing the physical property of one of the amphipathic helical regions involved in lipid binding and also allows disulphide bonding to other proteins. Dimers with apo A-II and apo E have been observed. In addition, LCAT activity is reduced.
Apo A-I deficiency
At least three types of apo A-I deficiency have been described, all associated with corneal clouding and premature coronary heart disease. Heterozygotes have 50% of normal HDL concentrations. HDL is virtually undetectable in homozygotes. In one type there is deficiency only of apo A-I, another also has deficiency of apo C-III and has low plasma triglyceride concentrations. The APOC3 gene is 3’ to the APOA1 gene but in the opposite orientation. These two genes are normally convergently transcribed from opposite DNA strands. In patients with combined deficiency of apo A-I and apo C-III, an inversion of approximately 5.5 kb containing portions of the APOA1 and APOC3 genes has been identified. In a third form, there is deficiency of apo A-I, apo C-II and also apoA-IV. These may show fat malabsorption due to the deficiency of apo A-IV.
Disorders of HDL metabolism (Box 37.2)
Tangier disease
Tangier disease is a rare autosomal recessive condition characterized by storage of cholesteryl esters in macrophages. This is responsible for the characteristic orange appearance of the tonsils. Splenomegaly also occurs, and is often accompanied by mild thrombocytopenia and reticulocytosis, but hepatomegaly and lymphadenopathy are less constant features. Corneal clouding may occur and most affected individuals have some neurological dysfunction.
Total plasma cholesterol concentrations are low (typically < 3 mmol/L) but, unlike the apo B deficiency states, plasma triglyceride concentrations are normal or increased. Virtually no HDL-cholesterol is present (< 0.1 mmol/L) and only pre-β1HDL particles are detectable in plasma. The concentrations of apo A-I and apo A-II are approximately 1–3% and 5%, respectively, of values found in normal individuals. An increase in pro-apo A-I can be demonstrated by isoelectric focusing but rapid catabolism of pro-apo A-I results in reduced conversion of pro-apo A-I to the mature form and consequently low plasma concentrations of apo A-I. Although it appears that Tangier patients are at increased cardiac risk, the level of risk varies between affected families and is not as high as predicted from epidemiological studies based on their plasma HDL-cholesterol concentrations. This may be related to their low LDL-cholesterol concentrations, but thrombocytopenia and relative platelet hyporeactivity may also be important factors.
Tangier disease is caused by the loss of function of the cholesterol-efflux regulatory protein, ABCA1, resulting from mutations in the ABCA1 gene. Individuals homozygous for ABCA1 mutations are at increased risk of coronary artery disease compared with unaffected individuals and heterozygous individuals are at intermediate risk.
Familial lecithin–cholesterol acyltransferase deficiency
Familial LCAT deficiency is an autosomal recessive condition. The gene for LCAT is located on chromosome 16: mutations in this gene that are associated with no LCAT activity result in familial LCAT deficiency. The clinical features of this condition include corneal opacities, haemolytic anaemia, proteinuria, high plasma triglyceride and low HDL-cholesterol concentrations. Despite the low HDL-cholesterol concentrations, premature atherosclerosis is rarely seen. Heterozygous carriers lack clinical features but have HDL-cholesterol concentrations about 50% of normal.
Lecithin-cholesterol acyltransferase deficiency results in an inability to esterify free cholesterol in plasma and hence the accumulation of free cholesterol in all lipoprotein fractions. Lipoprotein particles may also be of abnormal size or shape as well as composition; thus the HDL particles are either disc shaped or unusually small spherical particles similar to newly secreted HDL. Lipoprotein X is detectable on electrophoresis.
Fish eye disease
Fish eye disease is an autosomal recessive disorder resulting from mutations in the LCAT gene. In contrast to familial LCAT deficiency, there is detectable LCAT activity. Corneal opacities that give the eyes the appearance of those of dead fish have been described in association with low HDL-cholesterol concentrations (approximately 10% of normal). LDL is enriched in triglyceride; the concentration of VLDL is increased but the composition is normal.
Hepatic triglyceride lipase deficiency
Several familial cases of HL deficiency have been reported and inheritance appears to be autosomal recessive, with heterozygotes showing an intermediate phenotype. Homozygotes have severe hypertriglyceridaemia, hypercholesterolaemia, and increased IDL concentrations, producing a phenotype similar to type III or remnant hyperlipidaemia. HL deficiency is distinguished by an absence of post heparin HL activity. Apolipoprotein A-I and HDL-cholesterol concentrations are slightly raised, with HDL particles being abnormally triglyceride-rich HDL2. Premature vascular disease has been reported in HL deficiency, although the exact role of HL in the development of atherosclerosis remains uncertain, as increased HL activity (e.g. secondary to anabolic steroid use) may also result in increased vascular disease.
Cholesterol ester transfer protein deficiency
Several mutations of the CETP gene that cause CETP deficiency have been described. Homozygous CETP-deficient subjects have high plasma concentrations of HDL-cholesterol, apo A-I, apo A-II and apo E. Plasma HDL-cholesterol concentrations are usually > 3 mmol/L in homozygous subjects who have a complete absence of CETP activity, values of 2–2.5 mmol/L are typically seen in heterozygotes. These subjects also have an approximately 40% reduction in LDL-cholesterol and apo B. Cholesterol ester transfer protein deficiency is relatively common in Japan, where it accounts for about half of all cases of hyperalphalipoproteinaemia, but it is rare in Caucasians. There is evidence that, when associated with a high plasma HDL concentration, CETP deficiency protects against the development of atheroma. On this basis, drugs that inhibit CETP have been developed: these produce an increase in plasma HDL-cholesterol concentration of over 100% and fall in LDL-cholesterol concentration of around 40%. Because of the complex relationship between CETP activity, HDL concentrations and atherosclerosis, the effect of these agents on cardiovascular outcomes will need to be tested in clinical trials before it is clear whether or not CETP inhibition has therapeutic value.
ACQUIRED HYPERLIPIDAEMIAS
Acquired, or secondary, hyperlipidaemia is caused by alterations in lipoprotein metabolism resulting from another disease state or from drug therapy. Treatment of the underlying condition or discontinuation of the precipitating drug may correct the hyperlipidaemia. However, in some cases, such as chronic kidney disease or diabetes, where metabolism remains disturbed despite treatment, hyperlipidaemia may persist. In these conditions the increased vascular risk is a function of qualitative as well as quantitative changes in plasma lipoproteins. In particular, LDL exists in a small, dense form which is more readily oxidized and consequently more atherogenic. Cardiovascular risk therefore remains higher than is predicted from cholesterol and triglyceride measurements alone. The secondary hyperlipidaemias are summarized in Box 37.3.
Xanthomata may occur in acquired hyperlipidaemia just as in the primary lipid disorders with the same pattern of lipid abnormalities: thus eruptive xanthomata occur in the chylomicronaemia syndrome, whatever its aetiology. Florid cutaneous lipid deposition may occur in the presence of abnormal lipoproteins such as lipoprotein X in cholestasis (see below) or where monoclonal immunoglobulins bind lipoproteins or their receptors and interfere with lipid metabolism, as may occur in monoclonal gammopathy of uncertain significance (MGUS) and multiple myeloma.
Diabetes mellitus
Poorly controlled diabetes gives rise to hypertriglyceridaemia. In both types of diabetes, there is insulin deficiency, either absolute in type 1 diabetes or relative in type 2 diabetes. Insulin activates lipoprotein lipase and thereby enhances clearance of triglyceride-rich lipoproteins, but has the opposite effect on the HSL of adipose tissue. Thus, in insulin deficiency there is, in addition to reduced clearance, an increased influx of free fatty acids to the liver, leading to increased hepatic triglyceride synthesis.
In well-controlled diabetes, although plasma total cholesterol and triglyceride concentrations may be normal, there often remains a significant dyslipidaemia, sometimes referred to as an ‘atherogenic lipoprotein phenotype’. Plasma HDL-cholesterol concentration is usually low (< 1.1 mmol/L) and triglyceride concentration elevated (> 1.7 mmol/L). This pattern is indicative of the presence of small, dense LDL particles, which are more susceptible to oxidation and more atherogenic. It has been demonstrated that apolipoproteins A-I, A-II, B, C-I and E become glycated in patients with diabetes: it is possible that this glycation affects the normal uptake of remnant particles, resulting in their persistence in the circulation with atherogenic consequences.
The genetic background on which a secondary hyperlipidaemia is superimposed will affect its severity; for example, diabetic patients who manifest fasting chylomicronaemia are likely to have an underlying primary hypertriglyceridaemia.
Hypothyroidism
Various lipid abnormalities may occur in patients with untreated hypothyroidism, but the commonest is an increase in plasma LDL-cholesterol concentration. This is a consequence of a reduction in LDL receptors, resulting in reduced clearance of LDL. Lipoprotein lipase activity may also be impaired in hypothyroidism, which explains the hypertriglyceridaemia that sometimes occurs.
Since both overt and subclinical hypothyroidism are relatively common, it is imperative that all subjects found to have hyperlipidaemia are screened for hypothyroidism. If they are found to be hypothyroid, treatment with thyroxine should be instituted. If the subject does not have cardiovascular disease, the lipid profile should be rechecked once the euthyroid state is regained before a decision on lipid-lowering treatment is made. If the subject already has cardiovascular disease when hypothyroidism is diagnosed, it may be unwise to delay lipid lowering therapy, but it should be instituted with care since hypothyroidism increases the risk of statin-induced adverse muscle effects.
Nephrotic syndrome
Longstanding nephrotic syndrome is associated with accelerated atherosclerosis; thus lipid-lowering treatment will usually be required. There is evidence that, in the nephrotic syndrome, the hyperlipidaemia may worsen renal function: oxidized LDL has been shown in vitro to affect mesangial cells and accelerate glomerulosclerosis. The hyperlipidaemia occurring in the nephrotic syndrome is most commonly hypercholesterolaemia, but mixed hyperlipidaemia also occurs. The hypercholesterolaemia is a result of hepatic overproduction of apo B-100 as part of the generally increased hepatic protein synthesis typical of the condition. Hydroxymethylglutaryl-CoA (HMG-CoA) reductase activity is also increased. The increase in triglyceride results from reduced removal of chylomicrons and VLDL due to impaired lipoprotein lipase activity. HDL may be lost in the urine and, dependent on whether or not increased synthesis matches the rate of loss, the plasma HDL-cholesterol concentration may be low, normal or occasionally high.
Chronic kidney disease
Patients with chronic kidney disease (CKD) are at very high risk of cardiovascular disease. The lipid abnormality most commonly seen is hypertriglyceridaemia; the lipoprotein profile is characterized by a high concentration of intestinally derived apo B-48-containing lipoproteins and their remnants. The composition of the lipoproteins is also changed. The apo C-III concentration of the triglyceride-rich lipoproteins is increased and, since apo C-III inhibits both lipoprotein lipase and the hepatic uptake of chylomicron and VLDL remnants, this results in increased plasma triglyceride concentrations. Although total cholesterol may be relatively ‘normal’, much of the LDL is small and dense and therefore is particularly atherogenic. Although hypertriglyceridaemia is usually present, the most extensive evidence for reduction in cardiovascular disease risk by lipid modification is through use of HMGCoA reductase inhibitors (statins), which reduce LDL, even though they have comparatively little effect on plasma triglycerides.
Renal transplantation
After transplantation, the hyperlipidaemia that accompanies impaired renal function may be corrected, but this is not always the case. In addition, immunosuppressant therapy, including corticosteroids, may itself cause hyperlipidaemia. The degree of hypercholesterolaemia is often greater with sirolimus and ciclosporin than with tacrolimus. Thus, subjects post-transplant remain at high risk of cardiovascular disease, and since they will have been hyperlipidaemic prior to transplantation, therapy to improve their lipids is warranted. Caution needs to be taken, however, because of the well-documented interactions between ciclosporin and both statins and fibrates.
Liver disease
The liver plays a central role in lipoprotein metabolism. In addition, the only physiological means the body has of excreting cholesterol is by its hepatic secretion into the bile.
Cholestasis is frequently accompanied by a mixed hyperlipidaemia due to the accumulation of remnant lipoproteins. Lipoprotein X is found uniquely in cholestasis. This lipoprotein contains bile acids, apo C, apo D, albumin and cholesterol, and while it falls within the LDL density range, unlike all the other lipoproteins, it migrates towards the cathode on lipoprotein electrophoresis. The atherogenic potential of lipoprotein X is undefined; when present in massive amounts (cholesterol > 50 mmol/L), lipoprotein X has been associated with hyperviscosity. Plasma Lp(a) concentration is low and HDL-cholesterol is high in cholestasis; these abnormalities may, in part, explain the fact that cholestasis is not generally associated with a high cardiovascular risk.
Non-alcoholic fatty liver disease (NAFLD) is a manifestation of the metabolic syndrome and, as such, is associated with an increased cardiovascular risk. The dyslipidaemia associated with NAFLD is either hypertriglyceridaemia or mixed hyperlipidaemia; it is frequently associated with small, dense LDL particles. Where the condition runs in families, it is difficult to distinguish from familial combined hyperlipidaemia. Subjects with NAFLD may have abnormal plasma liver enzyme activities (particularly increased aminotransferases) and caution therefore needs to be exercised when starting lipid-lowering agents. However, owing to the increased cardiovascular risk, treatment is warranted. Statins are generally the drug of first choice unless severe hypertriglyceridaemia is present creating a risk of pancreatitis. Although liver enzymes should be monitored, treatment is usually safe, and in some individuals, normalization of the lipid profile results in a reduction in the aminotransferase activities.
Alcohol
Alcohol causes hypertriglyceridaemia in susceptible individuals. This results from a combination of increased production and impaired removal of VLDL. In severe cases, this may result in chylomicronaemia, which may result in acute pancreatitis.
Epidemiological studies have suggested that moderate alcohol intake (no more than 1–2 units per day) is associated with a lower mortality than either higher alcohol intakes or abstinence from alcohol. Such moderate intake is associated with an increase in plasma HDL-cholesterol concentration, which may be responsible for the apparent cardiovascular protection.
Drug-related hyperlipidaemia
Various drugs, including antihypertensive agents (β-blockers, thiazides), corticosteroids, sex steroids, immunosuppressants, second generation antipsychotics and antiretroviral drugs, can affect lipoprotein concentrations.
The effect of antihypertensives on the lipid profile has been extensively studied since early hypertension trials demonstrated that effective lowering of blood pressure reduced the incidence of strokes but not that of coronary heart disease. The effect of β-blockers depends on their selectivity: non-selective (e.g. propranolol) and β1-selective (e.g. atenolol) β-blockers raise plasma triglycerides and lower HDL, whereas β-blockers with intrinsic sympathomimetic activity (e.g. pindolol) are lipid neutral. The hyperlipidaemia associated with diuretic therapy appears to be caused by the unopposed α-adrenergic activity elicited by these drugs; the low dosages (e.g. 2.5 mg of bendroflumethiazide) now used for treatment of hypertension have a negligible effect on the lipid profile.
Glucocorticoid treatment results in an increase in plasma LDL, HDL and triglyceride concentrations. Oestrogens increase hepatic VLDL secretion and increase HDL (HDL2). In postmenopausal women, they also tend to reduce LDL. Progestogens, on the other hand, tend to reduce HDL (especially HDL2) and raise LDL. With both oral contraceptives and hormone replacement therapy, the magnitude of the changes in the lipoproteins depends on the dose, the route of administration, the particular oestrogen and progestogen involved and whether or not there is an underlying primary hyperlipoproteinaemia. The use of oral oestrogen results in exposure of the liver to supra-physiological doses of oestrogen, with stimulation of hepatic lipoprotein synthesis. Severe hypertriglyceridaemia may be precipitated by the use of oral oestrogen in susceptible individuals, who may have a normal plasma triglyceride concentration or only mild hypertriglyceridaemia when not taking oestrogen or using topical (patch) oestrogen hormone replacement therapy. Of the progestogens, the 19-nortestosterone derivatives (e.g. norethisterone, levonorgestrel) are considerably more androgenic than the C21 progestogens (e.g. medroxyprogesterone acetate, dydrogesterone) and result in greater reductions in plasma HDL concentrations.
Tamoxifen, a selective oestrogen receptor modulator that has agonist and antagonist oestrogen effects, has a predominantly agonistic effect on the liver and thus can cause severe hypertriglyceridaemia.
Among immunosuppressant drugs, both the calcineurin inhibitors (ciclosporin, tacrolimus) and the non-calcineurin inhibitor, sirolimus, can cause hyperlipidaemia, which, in most individuals, is an isolated increase in plasma LDL concentration.
Hypertriglyceridaemia is a common side-effect of second generation antipsychotic drugs including clozapine, olanzapine and quetiapine. This may be severe, and cases of pancreatitis, thought to result from the hypertriglyceridaemia, have been reported.
Antiretroviral drug treatment is frequently associated with dyslipidaemia. The combination of fat redistribution, insulin resistance and dyslipidaemia seen in subjects on antiretroviral treatment is referred to as ‘the lipodystrophy syndrome’. The fat redistribution is characterized by loss of subcutaneous fat and accumulation of intra- abdominal fat. This is associated with regimens containing protease inhibitors and nucleoside reverse transcriptase inhibitors, whereas the dyslipidaemia and insulin resistance are associated particularly with protease inhibitors. Care is required in treating dyslipidaemia in these individuals as a number of antiretroviral therapies interact with lipid lowering treatments.
ACQUIRED HYPOLIPIDAEMIA
Acquired or secondary hypolipidaemia is much less common than its hyperlipidaemic equivalent. It occurs with cachexia, malabsorption, hyperthyroidism, malnutrition, liver failure and some forms of malignancy. It is of no direct clinical consequence, being only a manifestation of the underlying condition.
Drug therapy can also cause hypolipidaemia. Excessive thyroxine will cause a reduction in total cholesterol, mimicking that seen with thyrotoxicosis. Drugs including fibrates and thiazolidinediones have been reported occasionally to cause an unpredictable and profound paradoxical fall in plasma HDL-cholesterol concentration in susceptible individuals.
INVESTIGATION OF LIPID DISORDERS
The simple appearance of a serum or plasma sample may indicate a lipid disorder. Chylomicrons and VLDL are large enough to scatter light. Chylomicrons are less dense than plasma and will form a layer on the surface of a sample left standing at 4°C overnight. If present in very large amounts, VLDL will make the sample appear opalescent.
Total cholesterol
This is the most commonly used single measure of lipid status. It has been used extensively in epidemiological studies, and may have some use in monitoring treatment. However, it is inadequate on its own for the diagnosis of lipid disorders, or as the only measure prior to starting treatment. A full fasting lipid profile including triglyceride and HDL-cholesterol should be measured at least once to avoid missing significant dyslipidaemias with normal total cholesterol. LDL-cholesterol, non-HDL-cholesterol and apo B measurements may have benefits in assessing cardiovascular risk and the adequacy of treatment.
Cholesterol is generally measured by enzymatic methods using cholesterol oxidase. Although analytical CV is low (typically < 3%), intraindividual biological variation is around 5% so it is important to appreciate that the critical difference between consecutive measurements in monitoring therapy may be around 0.8 mmol/L. Fasting or non-fasting makes little difference (± 3%) to measurements.
Triglycerides
Triglyceride increases up to 2–3-fold after a meal, and so samples should be taken after an overnight (> 12 h) fast in order to obviate difficulties of interpretation resulting from the presence of chylomicrons or chylomicron remnants. Most triglyceride methods involve hydrolysis of triglycerides and measurement of the free glycerol released. Glycerol blanking is not routinely performed. In certain circumstances, such as patients with uncontrolled diabetes mellitus, on haemodialysis, or with the rare X-linked recessive disorder glycerol kinase deficiency, the glycerol content of the sample may be significantly raised. In these situations, high concentrations of glycerol in the plasma will produce falsely high triglyceride results, and glycerol blanking is essential to obtain a valid result.
Plasma triglyceride concentration has a much greater biological variation than does that of cholesterol, at ~ 20% even in fasting samples.
High density lipoprotein cholesterol
Plasma HDL concentration is generally measured by direct (homogeneous) methods that rely on antigen/antibody complex formation, or polyethylene glycol (PEG) modified enzymes that react selectively with cholesterol in HDL. These assays can provide reliable results in the presence of triglyceride concentrations up to 10 mmol/L.
Low density lipoprotein cholesterol
LDL-cholesterol (LDL-C) can be derived by substituting the results of the analysis of total cholesterol (TC), HDL-cholesterol (HDL-C) and triglyceride (TG) (fasting) into the Friedewald formula:
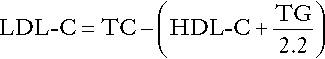
(all measurements being in mmol/L).
The Friedewald formula assumes that most of the triglyceride in the plasma is in VLDL and that there is a molar ratio of 5:1 of triglyceride to cholesterol in the VLDL fraction. For this reason, the Friedewald formula is not applicable in subjects with remnant hyperlipidaemia in whom the remnant lipoprotein fraction is cholesterol-enriched. In addition, it is not applicable to subjects with plasma triglyceride concentrations > 4.5 mmol/L since, at these levels, VLDL contains a greater proportion of triglyceride, and so the formula overestimates VLDL-cholesterol and underestimates LDL-cholesterol. Although most laboratories report derived plasma LDL-cholesterol concentrations on samples with triglycerides up to 4.5 mmol/L this tendency for VLDL to contain a greater proportion of triglyceride is sometimes present at lower concentrations of triglycerides, to a degree with may be clinically significant. The error in calculated LDL-cholesterol is > 10% in more than 30% of subjects with a plasma triglyceride concentration of 2.3–3.4 mmol/L and in > 40% of subjects with triglycerides of 3.4–4.5 mmol/L.
Directly LDL-cholesterol assays are being used increasingly. In general, they compare well with LDL-cholesterol as measured using ultracentrifugation. They have the advantage of being able to measure LDL-cholesterol in non-fasting samples, but because they are immunoseparation methods, have the disadvantage of higher cost compared with the simpler enzymatic measurement of total and HDL-cholesterol, and triglyceride.
Plasma LDL-cholesterol concentration is increasingly used in epidemiological and therapeutic studies and treatment goals based on LDL-cholesterol have been incorporated into most national guidelines.
Non-HDL-cholesterol
Non-HDL-cholesterol is derived simply using the formula (total cholesterol – HDL-cholesterol). The major difference from LDL-cholesterol is that it includes cholesterol in VLDL. In some epidemiological studies it has been shown to predict vascular risk almost as well as apo B and better than LDL-cholesterol. This may simply be because VLDL is atherogenic in its own right, but also because non-HDL-cholesterol may reflect LDL particle number better than measurement of LDL-cholesterol concentration alone.
Other advantages of non-HDL-cholesterol are that it can be measured in non-fasting samples, and that its measurement has better precision and accuracy than Friedewald derived LDL-cholesterol. It is also cheaper than measurement of apo B. It has been adopted in a number of current guidelines alongside LDL-cholesterol.
Apolipoproteins
Apolipoprotein A-I
Apolipoprotein A-I measurement has been advocated as an alternative to HDL-cholesterol, but it is generally more expensive, and clear advantage over measurement of HDL-cholesterol with modern homogeneous assays has not been demonstrated.
Apolipoprotein B
Apolipoprotein B measurement does serve a useful purpose in the investigation of lipid disorders. As there is only one apo B molecule in each apo B containing lipoprotein particle, measurement of apo B provides a measure of particle number. There is epidemiological evidence that this may provide a better predictor of cardiovascular risk and response to treatment than measurement of LDL-cholesterol (directly measured or derived). This is because small dense LDL particles will tend to result in lower concentrations of LDL-cholesterol, but are relatively more atherogenic than less dense, cholesterol-enriched LDL.
Apolipoprotein B measurement may be helpful in diagnosis, particularly in patients with mixed hyperlipidaemia. Apo B is often elevated in familial combined hyperlipidaemia, and low in proportion to total cholesterol in remnant hyperlipidaemia.
Apolipoprotein B and non-HDL-cholesterol show similar ability to predict cardiac risk in population studies. Apolipoprotein B may, however, be superior in patients with a large proportion of small dense LDL including those with type 2 diabetes and the metabolic syndrome.
Apolipoprotein E
Although it is not essential for managing patients, knowledge of the apolipoprotein E phenotype (based on electrophoresis) or confirmation of an ε2/ε2 genotype is sometimes helpful in confirming a diagnosis of remnant hyperlipoproteinaemia. It should also be remembered that the finding of an ε2/ε2 genotype on its own does not establish the diagnosis as other factors need to be present before the typical clinical phenotype becomes manifest.
Lipoprotein(a)
Standardization has only become available for Lp(a) over the last few years. Very wide variation between methods made the assay of limited utility in the past. It is important that assays are independent of the number of kringle 4 repeats in the highly variable region of the molecule. Plasma Lp(a) concentration is normally < 30 mg/dL but shows a highly positively skewed distribution. High concentrations are associated with an increased risk of cardiovascular disease, and the greatest utility of measurement is in assessing risk in those with high plasma LDL-cholesterol concentration.
In patients with familial hypercholesterolaemia who have very high Lp(a) concentrations (> 60 mg/dL), LDL apheresis, which selectively removes apo B containing lipoprotein particles, is occasionally used, as statins have very little impact on Lp(a).
Post-heparin lipolytic activity
The intravenous injection of heparin leads to the release into the circulation of both LPL and HL. Measurement of LPL activity is useful in the differential diagnosis of severe hypertriglyceridaemia where chylomicronaemia is present (type I hyperlipidaemia). This may be due to a deficiency of lipoprotein lipase.
Lipoprotein separation techniques
Ultracentrifugation
Preparative ultracentrifugation is the reference procedure for isolating lipoproteins; it involves sequential isolation of lipoprotein classes from plasma after adjustment of the density with sodium chloride, potassium bromide, sodium bromide or mixtures of these salts. This is a time-consuming procedure and may take up to five days for isolation of all subclasses to be completed. Losses may occur at all stages and artefacts may also be introduced. It is generally reserved for research studies.
Lipoprotein electrophoresis
Electrophoresis on cellulose acetate, paper, agarose and polyacrylamide has been used as a means of separating the lipoprotein classes and, indeed, the separation pattern on paper electrophoresis was the basis of the Fredrickson classification of lipid disorders. For therapeutic purposes, the Fredrickson classification has been replaced by defining the genetic basis of the hyperlipidaemia. It may still be of use in the diagnosis of remnant hyperlipidaemia with its broad β band, and may be helpful in deficiency states; for example, in demonstrating the lack of pre-β and β bands in abetalipoproteinaemia.
Genotyping
Genetic testing is particularly used for the diagnosis of remnant hyperlipidaemia, and familial hypercholesterolaemia (FH). Homozygosity for the ε2 allele of the apolipoprotein E gene is seen in remnant hyperlipidaemia. FH is due to mutations in LDLR, APOB, PCSK9 or LDLRAP1 genes.
Genotyping will also be used increasingly to confirm the diagnosis of rare monogenic lipid disorders. In addition, genome-wide association studies are starting to identify single nucleotide polymorphisms (SNPs) in multiple genes that may have minor effects individually but clinically significant effects cumulatively. This is starting to provide insight into the genetic basis of many common lipid and lipoprotein phenotypes, but has not yet found application in routine practice.
A further application of genetic testing is in tailoring therapy. A SNP in SLCO1B1 has been described which is associated with a four-fold increased risk increased risk of statin toxicity in heterozygotes and 16-fold increased risk in homozygotes. This gene codes for an organic anion transporter protein (OATP1B1) that is responsible for the uptake of statins to their site of action in the liver. Deficiency results in reduced efficacy and increased plasma levels with consequent increased risk of myositis. Fluvastatin is not taken up via this mechanism, and may be more effective and better tolerated in individuals with this polymorphism.
TREATMENT OF HYPERLIPIDAEMIA
Management of patients with hyperlipidaemia requires a full history, including a personal history of cardiovascular disease and conditions that can cause dyslipidaemias, family history of cardiovascular disease (with age of onset), lifestyle factors and drugs that may produce lipid abnormalities. Clinical examination and a detailed family history may suggest a specific genetic diagnosis, which may be confirmed by lipid, lipoprotein or genetic testing. Genetic lipid disorders may require more aggressive treatment, particularly if they suggest (e.g. in FH) that cholesterol levels have been elevated since soon after birth. They may also indicate the need for family screening.
All patients should receive lifestyle advice (including advice concerning smoking cessation where relevant). The aim of dietary advice is both to achieve weight loss (where needed) and an appropriate balance of nutrients including control of the amount of fat, and especially saturated fat, in the diet. Causes of secondary dyslipidaemia should be treated: correction of untreated hypothyroidism or newly diagnosed diabetes may produce a marked fall in plasma lipid concentrations.
In patients with fasting serum triglyceride concentrations of > 10 mmol/L, there is a risk of acute pancreatitis, and treatment to lower the triglycerides should normally be the initial priority. Fibric acid derivatives (fibrates) and high doses of fish oils (e.g. Omacor®) are generally the first-line treatments. Some patients with type 2 diabetes may benefit from the introduction of insulin.
In other cases, treatment is based on serum lipid concentrations and an assessment of a patient’s overall cardiovascular risk. Reduction in cardiovascular risk is the main aim, and statins are generally the first-line treatment. They have the most extensive evidence base and are generally used even if triglyceride concentrations are slightly or moderately raised, so long as this is not high enough to cause pancreatitis.
The highest priority is to treat those with clinical evidence of vascular disease (including cerebrovascular and peripheral vascular as well as coronary disease, i.e. secondary prevention). Primary prevention (i.e. in the absence of vascular disease) is targeted to those at the highest risk. This includes those with genetic hyperlipidaemias (e.g. FH), those with conditions known to cause qualitative lipoprotein changes that increase risk (e.g. diabetes, when treatment is generally recommended above the age of 40), and those estimated to be at high risk of cardiovascular disease based on lipid results and cardiovascular risk factors. Various risk calculation tools are available for calculation of ten-year or lifetime risk, based on epidemiological data (see Chapter 38 for a more detailed discussion).
The aim of treatment is ideally to reduce serum total cholesterol concentration to < 4 mmol/L and LDL-cholesterol concentration to < 2 mmol/L. Targets for non-HDL-cholesterol of < 2.5 mmol/L and apo B of < 0.8 g/L are also recommended. These targets recognize that residual risk of cardiovascular event after LDL-cholesterol reduction with statins depends on LDL particle number, which may better be reflected by measuring plasma apo B concentration.
If targets are not achieved, then fibrates, ezetimibe or bile acid sequestrants are all options, usually in addition to a statin, although none have such a robust evidence base as statins for reducing cardiovascular events. Use of lipid lowering drugs in combination is usually safe although care must be taken to identify patients who are at increased risk of skeletal myopathy.
In patients with FH, drug treatment is generally considered by the age of ten, with the aim of reducing serum LDL-cholesterol concentration by > 50%. In those with a more severe phenotype or with homozygous FH, LDL apheresis is a treatment option. New agents such as MTP and PCSK9 inhibitors, and apo B mRNA antisense oligonucleotides are in development or clinical trials.
A summary of drugs used to treat hyperlipidaemia, and of those currently in development or clinical trials, is given in Table 37.7.
TABLE 37.7
Drugs used in the treatment of dyslipoproteinaemias

a LRCCT, Lipid Research Clinics Coronary Primary Prevention Trial
b CTT, Cholesterol Treatment Trialists collaboration (a meta-analysis of the major statin trials)
Side-effects of statins and other lipid lowering treatments include elevation of transaminases and muscle effects ranging from an asymptomatic slight rise in creatine kinase (CK), through myalgia or myositis, with or without a rise in CK, to, rarely, rhabdomyolysis. Patients should be warned of possible effects and liver enzymes should be monitored. Drug interactions (e.g. with immunosuppressant drugs) increase the risk of myositis. Genetic variants may also increase the risk of muscle side-effects (see p. 734).
While targeting those at highest risk, and treating them aggressively with lipid lowering drug treatment, is highly effective in individuals, it is important to recognize that the greatest impact on population rates of CVD would still to be made by changing the diet and lifestyle habits of the population as a whole.
CONCLUSION
The principal lipids present in the blood are cholesterol, triglycerides and phospholipids. They are transported, in association with various apolipoproteins, in lipoprotein particles. Lipids have essential functions in cellular structure and metabolism but, at present, the major clinical interest in them derives from the relationship between the plasma cholesterol concentration (specifically LDL-cholesterol) and the risk of developing atherosclerosis.
Lipid metabolism is complex; there is a continuous flux of lipoprotein particles between the blood and tissues, and of the lipid and protein components between lipoprotein particles. Abnormalities of plasma lipids and of lipoprotein composition occur frequently. They can arise as a result of a disease that secondarily affects lipid metabolism (e.g. diabetes, hypothyroidism) or be primary, that is, genetically determined.
In the genetic hyperlipidaemias, appropriate investigation will define the nature and severity of the disorder and provide a rational basis for treatment. Greater understanding of the details of lipoprotein metabolism will continue to identify new therapeutic targets and bring the potential for effective new therapies. In the majority of cases, the aim is to reduce the risk of cardiovascular disease, and therefore specific therapy for the hyperlipidaemia needs to be accompanied by action against all the other pertinent risk factors.
ACKNOWLEDGEMENT
The author wishes to acknowledge Dr Christine Marenah, who was the author of this chapter in previous editions of the book.