CHAPTER 31
Metabolic bone disease
Timothy Cundy; Ian R. Reid; Andrew Grey
CHAPTER OUTLINE
Biochemical markers of bone turnover
Defective osteoblast function and osteomalacia
CHRONIC KIDNEY DISEASE – MINERAL AND BONE DISORDER
Bone disease after renal transplantation
BONE DISEASE IN PRIMARY HYPERPARATHYROIDISM
Clinical, biochemical and histological features
BONE BIOLOGY
The principal role of the skeleton is a structural one, maintaining body shape, providing protection for internal organs and, together with the neuromuscular system, making locomotion possible. It also has an important secondary role in mineral homoeostasis, functioning as a reservoir for calcium ions in particular. Metabolic bone diseases can affect both these functions.
Anatomy of bone
Macroscopic
The anatomist classifies bones as being either flat (e.g. skull, scapula, mandible, ilium) or long (e.g. the limb bones). Flat bones result from intramembranous ossification; long bones predominantly from endochondral ossification. A long bone consists of a shaft (diaphysis) broadening at either end into an epiphysis. The transitional zone between the diaphysis and the epiphysis is termed the metaphysis. On sectioning a long bone, two patterns of organization of bone tissue are found. The elements of bone can be packed together without intervening marrow spaces to form cortical or compact bone, or they can form an interlacing meshwork of trabeculae referred to as cancellous or trabecular bone. The diaphysis of the long bone consists mainly of cortical bone, whereas the metaphysis and epiphysis have a greater quantity of trabecular bone, enclosed within a thin cortical envelope. Some 80% of the weight of an adult human skeleton consists of cortical bone. However, the surface-to-volume ratio of trabecular bone is very much higher than that of cortical bone and it is metabolically much more active.
Microscopic
At a microscopic level, bone consists of matrix (~35% by volume), mineral (~60%) and cells (< 5%). The matrix is predominantly type I collagen fibres, usually organized in layers within which the fibres are parallel to one another. In adult bone, the fibre orientation varies from one layer to the next and this is referred to as lamellar bone. If deposited along a flat surface, the lamellae will be parallel to that surface, but in cortical bone they are concentrically oriented around a central blood vessel to form the Haversian canal system. When bone formation is rapid (e.g. during growth or fracture healing) collagen fibres may be laid down with more random orientation, producing woven bone.
The mineral phase of bone is hydroxyapatite (Ca10(PO4)6 (OH)2). This forms spindle-shaped crystals, which are found in association with the collagen and ground substance. Their orientation is usually parallel to that of the collagen fibres.
There are two general cell types in bone: osteoblasts and osteoclasts. Both are found on the bone surface at sites of active remodelling. Osteoblasts are also thought to be the precursor of two cells types that are more widespread: bone lining cells, which are found over inactive bone surfaces, and osteocytes, which are found in lacunae scattered throughout bone. They are thought to be osteoblasts that have been engulfed by the bone that they have formed. They have long cell processes, which are in contact with similar processes arising from other osteocytes or with those of the bone lining cells. The cell processes lie in an interconnecting network of canaliculi that extends throughout bone tissue. The bone lining cells and osteocytes thus delineate an extracellular fluid (ECF) space that is in contact with the bone surface. This space has a volume of 1–1.5 L and a surface area of several thousand square metres. This is the site of mineral exchange between ECF and bone. The size and nature of this exchange is unknown but the fluid within this space, the bone ECF, has an ionized calcium concentration of only 0.5 mmol/L – less than half of that elsewhere in the ECF.
Bone matrix proteins
Collagen
Almost 90% of the protein in bone matrix is type I collagen, which is synthesized by osteoblasts. It is a large molecule (MW > 300 000 Da) with a trimeric helical structure. Type I collagen is initially synthesized in the rough endoplasmic reticulum (RER) as a precursor molecule (type I procollagen) that combines two proα1(I) and one proα2(I) peptide chains (coded by COL1A1 and COL1A2, respectively) in a triple helix. Proα1(I) and proα2(I) have similar structures with a core triple helical domain of 1014 amino acids composed of uninterrupted Gly-X-Y tripeptide repeats, where Gly is glycine and X and Y are often proline or lysine, flanked by propeptides at both N- and C-terminal ends. During and after translation, the three chains undergo extensive modification. Prolyl-4-hydroxylase converts virtually all Y-position proline residues to 4-hydroxyproline, an alteration that is essential for thermal stability of the assembled trimer. In the absence of this modification the trimer melts (that is the individual chains unfold from the stable triple helix, at about 27°C, whereas with full hydroxylation, the melting temperature is about 42°C). Some Y-position lysine residues within the triple helical domain are hydroxylated by the enzyme lysyl hydroxylase-1, and glucose and galactose groups added by glycosyltransferases (Fig. 31.1). Hydroxylation of these triple helical residues is part of the pathway to form stable complex intermolecular crosslinks that provide the tensile strength in tissues. Most of these modifications are completed during translation and occur on the individual chains. If there is a delay in triple helix folding, the process can continue and the physical properties of the chains and molecules are altered and contribute to an osteogenesis imperfecta phenotype.
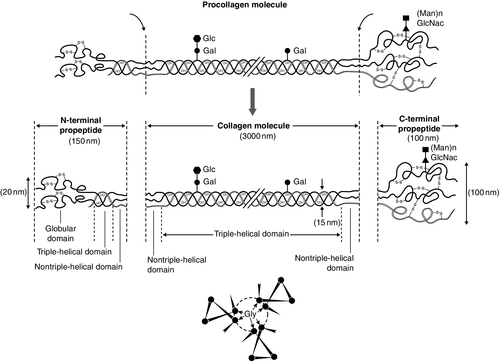
FIGURE 31.1 Type I collagen synthesis. Upper panel: schematic representation of the structure of the procollagen molecule, showing cleavage of the amino and carboxy terminal peptides. Glc, glucose; Gal, galactose; Man, mannose; GlcNac, N-acetylglucosamine. Lower panel: view along the axis of the triple helical structure of the collagen backbone, with every third amino acid a glycine residue forming the inner circle.
The three chains that form a [proα1(I)2 proα2(I)] trimer interact through regions in the carboxyl-terminal propeptide of each chain. The full length chain must be maintained in an unfolded state while the carboxyl-terminal propeptides fold, associate, and then begin the process of triple helix formation. Propagation of the collagen triple helix requires a number of enzymes and molecular chaperones to ensure correct folding and trimerization. These include peptidyl disulfide isomerase (PDI), which also forms part of the prolyl 4-hydroxylase complex, and is likely to involve prolyl peptidyl cis-trans isomerase B (also known as cyclophilin B). This protein can act on its own, to assist in the folding around prolyl residues, such as those in the carboxyl-terminal propeptide adjacent to cysteine residues, and as part of a complex that includes two additional proteins, cartilage-related protein and prolyl 3-hydroxylase, to modify certain triple helical prolines. The function of this last process is not yet entirely clear but when the complex is missing, the propagation of the triple helix is altered and modification of the chains increased.
Disulphide bonds between the carboxyl-terminal region of the chains act to secure the three chains in a trimer, a process requiring protein-disulphide isomerase. Lysine residues outside the major triple helical domain of type I collagen, needed for the formation of mature intermolecular crosslinks, are hydroxylated by lysyl hydroxylase-2. These complex modifications, which are necessary for correct folding, assuring thermal stability of the triple helix and crosslink formation between collagen molecules once they are secreted into the matrix, need to take place in an orderly and timely sequence, and various chaperone proteins, including HSP47 and FKBP65, help regulate this process. Procollagen trimers are then transported via the Golgi network and packaged into membrane-bound organelles where lateral aggregation, the initial phase of fibril formation, occurs. As secretion occurs, the procollagen molecules are further processed into mature type I collagen molecules by proteolytic cleavage of the N- and C-terminal propeptides (by the enzymes ADAMTS-2 (a disintegrin and metalloproteinase with thrombospondin motifs 2) and BMP1 (bone morphogenetic protein 1), respectively). Finally, the trimers are assembled into collagen fibrils and fibres and anchored in those positions by intermolecular lysine-derived crosslinks in a process that is begun by modification of specific residues by lysyl oxidase. These cross-links are initially reducible, but as tissue maturation proceeds, they are converted into non-reducible compounds including hydroxylysylpyridinoline (derived from three hydroxylysine residues) and the less abundant lysylpyridinoline (derived from two hydroxylysine residues and one lysine residue). The latter (also known as deoxypyridinoline) is present in the body almost exclusively in bone tissue and dentine, where it accounts for 21% of the total mature cross-links.
Non-collagenous proteins
The non-collagenous proteins of bone (proteoglycans, glycosylated proteins, RGD (arg-gly-asp)-containing glycoproteins and γ-carboxylated proteins) account for only 10–15% of bone protein mass but, because of their very much smaller size, are as common as collagen in bone on a molar basis. Most are synthesized by osteoblasts, but a number are produced elsewhere in the body and arrive in bone via the circulation. The latter proteins, which include albumin and platelet-derived growth factor, are usually negatively charged and become bound to hydroxyapatite. The most abundant of the bone-derived non-collagenous proteins are osteocalcin, also known as bone Gla-protein, and osteonectin, both of which have a high affinity for bone mineral.
Osteocalcin
is synthesized by osteoblasts as a 75-amino acid precursor known as pro-osteocalcin. The amino-terminal propeptide includes the substrate recognition site for the vitamin K-dependent γ-carboxylase enzyme which γ-carboxylates three glutamic acid residues (17, 21 and 24) in the central region of the osteocalcin molecule, following which the amino-terminal propeptide is cleaved, leaving the 49-residue osteocalcin molecule. The γ-carboxyglutamic acid residues are strongly calcium binding and allow the molecule to bind to hydroxyapatite. The process of γ-carboxylation seems to be impaired in the elderly.
The physiological role of osteocalcin is uncertain, but it probably contributes to the regulation of bone remodelling, since deletion of the osteocalcin gene in mice leads to increased bone density. Its synthesis is regulated by 1,25-dihydroxyvitamin D (1,25(OH)2D, calcitriol) and cortisol, and it is incorporated into bone matrix, being especially abundant in cortical bone.
Recently, its uncarboxylated form has been implicated in the regulation of intermediary metabolism, including fat mass, glucose tolerance and insulin secretion. The relevance to human physiology of these findings in genetically modified mice remains to be determined.
Other bone proteins
Several proteoglycans, macromolecules that contain acidic polysaccharide side chains, are present in bone. In the early stages of bone formation, versican, a dermatan sulphate proteoglycan, and the glycosaminoglycan, hyaluronan, are present and may act to delineate areas that are destined to become bone. Subsequently, two chondroitin sulfate proteoglycans, decorin and biglycan, are expressed and may influence cellular proliferation and differentiation in the newly forming bone. Fibromodulin, osteoglycin and osteoadherin are other proteoglycans that are expressed in bone matrix, but their functions are less well characterized.
The major glycoproteins present in bone are alkaline phosphatase (ALP), which is highly expressed on the osteoblast cell surface, and osteonectin, a phosphorylated glycoprotein that is also present in several other tissues that undergo rapid remodelling. In bone, osteonectin may regulate both cellular activity and matrix mineralization.
At least eight non-collagenous glycoproteins expressed in bone contain the arginyl-glycyl-aspartate (RGD) tripeptide as the consensus cell-attachment sequence (Table 31.1). Some are specific to bone (e.g. bone sialoprotein), but most are more ubiquitously expressed (e.g. fibronectin, osteopontin, thrombospondins, vitronectin). Although the role(s) of each individual protein in skeletal physiology is(are) uncertain, it seems very likely that they collectively serve to promote attachment of bone cells to bone matrix (and perhaps thereby regulate their function). The components of the matrix–cell interactions are clearly dependent upon the maturational stage of the remodelled bone. Three matrix non-collagenous proteins, matrix Gla-protein, protein S and osteocalcin (bone Gla-protein), undergo post-translational γ-carboxylation, a process that enhances calcium binding. These proteins may function physiologically as inhibitors of mineral deposition.
TABLE 31.1
Major glycoproteins containing the arginyl-glycyl-aspartate (RGD) cell-attachment motif in skeletal tissue
| Protein | Proposed function |
| Thrombospondins | Cell attachment, bind heparin, collagens, thrombin, fibrinogen, plasminogen |
| Fibronectin | Binds to cells, collagen, fibrin, heparin, gelatin |
| Vitronectin | Cell attachment, binds collagen and plasminogen |
| Fibrillin | Regulation of elastic fibre formation |
| Osteopontin | Binds to cells, inhibits mineralization, regulates cell proliferation and tissue repair |
| Bone sialoprotein | Binds cells, may inhibit mineralization |
| BAG-75 | Binds calcium,?cell attachment |
| Dentin matrix phosphoprotein 1 | Osteocyte-derived, regulates osteocyte maturation and FGF23 production |
Cellular elements of bone
Osteoblasts
Osteoblasts are the cells responsible for the synthesis and maintenance of bone matrix. They are derived from pluripotent mesenchymal progenitor cells that also give rise to adipocytes, chondrocytes and myocytes. Expression of the transcription factors cbfa1 and osterix is critical for commitment to the osteoblast lineage, while expression of peroxisome proliferation activating receptor-γ (PPAR-γ) favours adipocytic over osteoblastic differentiation.
A key molecular event in osteoblast proliferation, and perhaps survival, is signalling through the wnt-low density lipoprotein-related receptor 5 (LRP5)-β-catenin pathway. In this system, secreted wnt proteins bind to and activate LRP5 on the cell surface, which in turn activates a series of intracellular events that result in translocation of cytoplasmic β-catenin to the nucleus, where it activates transcription of genes critical for osteoblast growth and function. The pathway is further regulated by secreted proteins that inhibit wnt-induced activation of LRP5, such as sclerostin, Dickkopf (DKK) and frizzled. Thus, humans carrying mutations of components of this signalling pathway that decrease nuclear translocation of β-catenin have decreased bone mass and fractures as a result of impaired bone formation (e.g. WNT1 and most LRP5 mutations), while constitutive activation of the pathway (e.g. LRP5 mutations affecting one particular extracellular domain or SOST mutations) increases bone mass. Other LRP family members are also important in bone physiology. Mutations affecting one particular region of the extracellular domain of LRP4 result in impairment of the ability of sclerostin to inhibit bone formation, and thereby cause increased bone mass (sclerosteosis type 2).
Mature osteoblasts are metabolically very active and intimately involved in bone formation. They synthesize many of the matrix proteins referred to above. They have very high levels of alkaline phosphatase (ALP) activity associated with their cell membranes. This enzyme is necessary for normal bone mineralization to take place. Osteoblasts express receptors for parathyroid hormone (PTH) and 1,25(OH)2D, as well as a number of cytokines, growth factors and sex hormones. Osteoblasts also play a key role in regulating osteoclastic bone resorption, ensuring that the components of bone remodelling, formation and resorption are coupled. A critical mechanism by which osteoblasts regulate bone resorption is via the RANK–RANKL–OPG system (see below).
Osteocytes
Osteocytes and bone lining cells are terminally differentiated osteoblasts derived from mature osteoblasts that are no longer involved in active bone formation, and have become entrapped within the canaliculi of the bone matrix that they have produced. This process of osteocytogenesis is probably an active one, requiring cleavage of matrix proteins: mutations of the metalloproteinase MT1-MMP lead to osteocytes with fewer and shorter dendritic processes. As osteocytogenesis occurs, the osteoblast changes from a cuboidal cell to a typical osteocytic morphology, characterized by a stellate appearance and numerous cytoplasmic projections that permit the cell to communicate with cells on the bone surface and other osteocytes living within adjacent bone. Concurrently, the genetic signature of the cell changes from a typically osteoblastic one to that which reflects a mature osteocyte, in particular the expression of DMP1, SOST and FGF23. Osteocytes play a key role in the regulation of bone remodelling. At sites of skeletal microdamage, osteocytes undergo apoptosis and release apoptotic bodies expressing RANKL (see below), promoting the recruitment of osteoclasts to the area to initiate remodelling to allow repair of the damaged bone. Osteocytes also produce and release sclerostin, an endogenous inhibitor of osteoblast differentiation and function that acts to inhibit wnt-induced activation of LRP5. They also act as mechanosensory cells in bone, perhaps in tandem with osteoblasts lining the bone surface, with which they communicate. Finally, osteocytes are critically important for bone mineralization, by regulating phosphate metabolism at a whole organism level, principally by production of the phosphaturic hormone fibroblast growth factor 23 (FGF23), a process that involves the osteocyte products phosphate-regulating endopeptidase (PHEX) and dentine matrix phosphoprotein 1 (DMP1).
Osteoclasts
Osteoclasts are the cells that resorb bone. They are derived from cells of the monocyte/macrophage series. They are multinucleated and are usually found singly or in small numbers within resorption lacunae of their own making. They have large numbers of mitochondria and lysosomes. Activated osteoclasts possess a ‘ruffled’ border, adjacent to the bone being resorbed. The ruffled border is delimited by a ‘sealing zone’ at which the osteoclast is attached to the underlying bone. Bone resorption occurs as a result of the secretion of protons and proteolytic enzymes into the space between the ruffled border and the bone surface. Among these enzymes are tartrate-resistant acid phosphatase (a useful histological marker of osteoclasts) and cathepsin K.
Osteoclast development and function are regulated by osteoblast and osteocyte-derived cytokines, in particular osteoprotegerin (OPG) and receptor activator of nuclear factor-κB ligand (RANKL), which are members of the tumour necrosis factor (TNF) receptor superfamily. RANKL expressed on the osteoblast and osteocyte surface binds to, and activates its receptor – receptor activator of nuclear factor-κB (RANK) – which is expressed on the surface of osteoclast precursors. RANKL is also shed from the cell surface, and this soluble form of the protein may confer osteoclastogenic activity distant from the cell of origin, in particular the osteocyte. Interaction between RANKL and RANK promotes osteoclastogenesis. Osteoprotegerin is a secreted osteoblast product that acts as a decoy receptor for RANKL, and thereby functions as an endogenous inhibitor of osteoclastogenesis. Thus, the relative levels of expression by the osteoblast of RANKL and OPG determine the rate at which osteoclastogenesis occurs. Many of the systemic factors that alter bone resorption, such as PTH and 1,25(OH)2D (which increase osteoclast function) and sex hormones (which decrease it), do so by changing osteoblastic production of RANKL and/or OPG. An exception is calcitonin, whose receptor is expressed on osteoclasts, and which therefore inhibits bone resorption directly.
A number of other critical cellular and molecular events are now recognized to underpin osteoclastic bone resorption. The intracellular tyrosine kinase c-src regulates osteoclast motility and aspects of cytoskeletal function that are critical to formation of a functional resorption space; the αvβ3 integrin (vitronectin receptor) plays a key role in osteoclast attachment to bone; the cysteine protease cathepsin K and the type 7 transmembrane chloride channel (ClCN7) are each required for proteolytic degradation of bone matrix.
Bone remodelling and its regulation
In mature bone, there is a continuous process of removal and replacement of pockets of old bone. This is termed bone remodelling and is shown in schematic form in Figure 31.2. The process occurs at discrete sites on the bone surface and, in the mature skeleton, is probably an adaptive response to skeletal microdamage. Bone remodelling is achieved by teams of osteoclasts and osteoblasts acting within the anatomical structure known as the basic multicellular unit (BMU). Osteocytes play a critical role in initiating bone remodelling, and probably also in regulating the coordination of formation and resorption within the BMU. Thus, apoptotic osteocytes in an area of skeletal microdamage produce RANKL that directs osteoclastogenesis and initiation of bone remodelling to the damaged bone. At that site, the osteoclasts excavate a resorption pit, the depth of which is determined by the number and activity of the osteoclasts present. The motile osteoclasts are then replaced by osteoblasts, which proceed to fill the resorption pit with bone matrix. Mineralization of this new bone then occurs, lagging behind matrix synthesis by a period of about three weeks. Once the resorption pit has been filled, the osteoblasts either undergo apoptosis, return to their quiescent state as bone lining cells or survive as osteocytes within the bone lacunae.
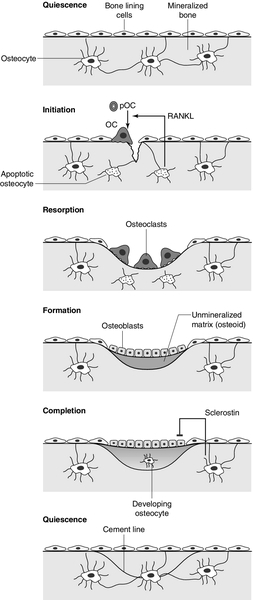
FIGURE 31.2 The bone remodelling cycle. Bone remodelling is initiated in response to skeletal microdamage, sensed by adjacent osteocytes which undergo apoptosis and produce the osteoclastogenic cytokine, receptor activator of nuclear factor-κB ligand (RANKL). Osteoclastic resorption of the damaged bone ensues, after which mature osteoblasts progressively fill the resorption cavity with osteoid, and coordinate its subsequent mineralization. Some of the osteoblasts develop into osteocytes within the lacunae of the newly formed bone. Sclerostin is produced by osteocytes adjacent to the remodelling space: it signals inhibition of bone formation and a return to a quiescent state. In skeletal homeostasis, the amount of new bone formed equals that resorbed during each remodelling cycle. OC, osteoclast; pOC, pre-osteoclast. (Courtesy of Dr Andrew Grey.)
An increase in bone turnover is characterized by a faster bone remodelling cycle initiation rate, rather than an increase in the duration of the cycle. The histological consequence of such an increase is that a greater proportion of bone surface is involved in remodelling at any one time. The time required to complete a full cycle of resorption and formation (σ) is in the order of 6–12 months. Inhibiting bone resorption, for example with pharmaceutical agents, rapidly reduces the initiation rate of new remodelling cycles, but those cycles already initiated go to completion. Thus there is a period (‘a transient’) of duration σ, when the bone formation rate exceeds the resorption rate. This ends when all those cycles initiated before the inhibition of bone resorption took place have completed their formation period. The bone formation rate at this stage will have fallen to match the new, lower, resorption rate.
Bone resorption and formation are thus very closely linked in normal bone and also in many metabolic bone disorders. As outlined earlier, the osteoclasts and osteoblasts clearly ‘communicate’ with each other. Osteoblastic regulation of osteoclast function is mediated in large part by the RANK–RANKL–OPG system, but the nature of the osteoclast-derived signal(s) that recruits and activates osteoblasts is not yet fully characterized. Osteocyte-derived sclerostin, an inhibitor of osteoblast differentiation, probably contributes to the regulation of the bone formation component of the remodelling cycle. Matrix-associated growth factors that are released from resorbed bone, such as transforming growth factor β (TGFβ), bone morphogenetic proteins (BMPs) and platelet-derived growth factor, may also constitute part of this signal. Finally, genetic evidence suggests that communication from osteoclasts to osteoblasts occurs via members of the ephrin (Eph) family of signalling proteins, specifically osteoclast-expressed ligand ephrin-B2 signalling through the osteoblast-expressed receptor EphB4 to activate osteoblast differentiation and bone formation.
During growth, the amount of bone deposited exceeds that which is removed and there are also changes in the size and configuration of bones (modelling). In senescence, because of factors including sex hormone deficiency, reduced physical activity and various endocrine and inflammatory diseases, the balance of formation and resorption is reversed and bone is lost. Age-related bone loss affects both cortical and trabecular bone, but early after the menopause, bone loss is particularly rapid from the axial skeleton, which has a greater proportion of trabecular bone.
A large number of factors have been found to influence bone remodelling (discussed in Chapter 6).
Biochemical markers of bone turnover
Bone turnover is a term referring to both osteoblastic bone formation and osteoclastic bone resorption. These processes can be assessed by bone histomorphometry, skeletal scintigraphy and by analysing the kinetics of intravenously administered isotopes of calcium. There are numerous biochemical markers of bone turnover, representing either products of bone cells or degradation products from the breakdown of bone collagen (Box 31.1). The number of potential markers available has increased considerably in recent years. In a small number of rare conditions (e.g. hypophosphatasia, osteopetrosis, Bruck syndrome and mild osteogenesis imperfecta), a specific pattern of bone turnover markers can be helpful in diagnosis. However, the main use of markers is to establish whether a high bone turnover state exists. High bone turnover, with its various adverse consequences, is a feature of many of the more common metabolic bone disorders, and a key target of currently available therapies.
Markers of bone formation
Alkaline phosphatase
Alkaline phosphatase is widespread in human tissues. Enzymes with this activity are coded for by genes at three separate loci, their respective products being referred to as intestinal, placental and bone/liver/kidney (or tissue non-specific) ALP. The latter gene is located on chromosome 1p36. The gene transcript has a number of glycosylation sites, and the bone, liver and kidney isoenzymes differ in their carbohydrate side chains.
The major limitation of total ALP as an index of bone formation is its multiple tissues of origin and hence its lack of sensitivity at low values. In health, plasma ALP activity is principally derived from the liver and osteoblasts in approximately equal proportions. If the activity of other hepatic canalicular cell enzymes is normal, then elevated total ALP activity usually reflects osteoblastic activity (or, more precisely, the number of active osteoblasts). If uncertainty persists in determining the origin of increased plasma ALP, then isoenzyme studies may be undertaken. The bone and liver isoenzymes have differing stabilities to heating (the bone fraction being more sensitive to heat inactivation) and different mobilities on electrophoresis. Immunoassays for bone-specific ALP use isoform-specific monoclonal antibodies; even so, there is up to 20% cross-reactivity with the liver isoform in these assays. Thus very high plasma activities of liver ALP, as occur in hepatobiliary disease, can still interfere with the measurement of bone-specific ALP. The activity of the bone isoenzyme has a diurnal variation in plasma, with peak values occurring around midnight and being some 25% higher than early morning values. Although the half-life in plasma of the bone isoenzyme is very short (~2 days), in metabolic bone disorders changes in ALP activity reflect slower processes – the acceleration or deceleration of the remodelling cycle – so the rate of change of plasma ALP is also much slower.
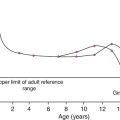
The marked elevation in plasma ALP activity seen in childhood and adolescence is due to an increase in the bone fraction. The plasma ALP activity closely parallels the growth velocity curve (Fig. 31.3). A significant (40–50%) rise in plasma ALP activity occurs at the menopause because of accelerated bone turnover. Plasma activity of the enzyme can also be increased after major fractures and reflects the increased cellular activity associated with healing. After hip fracture, for example, ALP activity reaches a peak at around four weeks, with values approximately double those on admission to hospital. Total plasma ALP activity is increased during pregnancy because of the appearance of the placental isoenzyme.
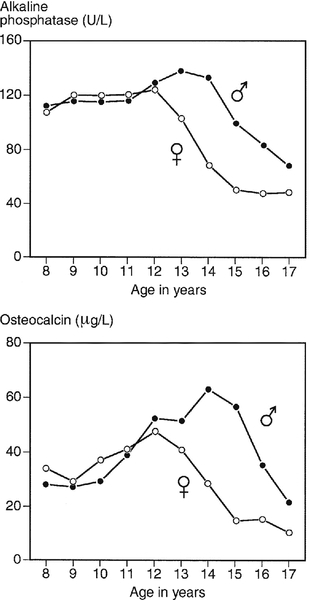
FIGURE 31.3 Bone turnover markers during growth. Mean values of plasma alkaline phosphatase (ALP) and osteocalcin in males ([closed circle]) and females ([open circle]) through childhood and adolescence. Peak values are reached at an average age of 12 years in girls and 14 in boys, coinciding with peak growth velocity. As growth velocity falls, both these indices decline toward adult values. (Data from Round JM 1973 Plasma calcium, magnesium, phosphorus and alkaline phosphatase concentrations in normal British school children. British Medical Journal 3: 173–140, and Johansen JS, Riis BJ, Podenphant J, Skakkeboek NE, Christiansen C 1987 Plasma bone Gla protein: a specific marker of bone formation. In: Christiansen C, Johansen JS, Riis BJ (eds). Osteoporosis, vol 2. Copenhagen: Osteopress ApS: 677–681.)
The entity of familial benign hyperphosphatasia is characterized by asymptomatic high ALP activities (predominantly of intestinal isoform) and dominant inheritance, but with no clinical evidence of bone disease. Benign transient hyperphosphatasaemia is a condition characterized by high concentrations of both bone and liver isoenzymes, first identified in children but now recognized in adults (see Chapter 13).
There are a number of causes of low ALP activities (hyophosphatasaemia), including vitamin C deficiency, hypothyroidism, starvation, zinc or magnesium deficiency, Cushing syndrome, Wilson disease and the genetic disorder, hypophosphatasia (discussed below).
Osteocalcin
Osteocalcin is produced by osteoblasts, and is generally regarded as a marker of bone formation, but it seems to be involved in the process of mineralization rather than matrix production. Plasma concentrations of intact osteocalcin are markers of bone formation, and circulating concentrations generally correlate well with histological measures of bone formation rate. Around 25% of circulating osteocalcin consists of intact osteocalcin, the remaining immunoreactivity comprising N-terminal, mid-region, mid-region-C-terminal and C-terminal fragments. When bone is resorbed, osteocalcin fragments are released, and the plasma concentrations of these fragments may thus reflect bone resorption. These fragments are cleared predominantly by the kidneys, so there is considerable immunological heterogeneity in plasma osteocalcin in patients with renal impairment. These factors may limit the usefulness of osteocalcin as a marker of bone formation.
Osteocalcin has a particular advantage in having greater sensitivity than ALP in the detection of low rates of bone formation. As with plasma ALP activity, plasma osteocalcin and growth rate are closely correlated (see Fig. 31.3), and there is a two-fold rise in plasma osteocalcin concentration at the menopause. Plasma osteocalcin concentrations fall markedly during pregnancy. Cord blood concentrations are two to three times higher than adult values. The half-life of intact osteocalcin in the circulation is about 5 min.
Plasma osteocalcin concentrations exhibit a diurnal variation with the peak in the early morning being about 15% higher than the nadir value, which occurs in the afternoon. Concentrations may also be depressed (by about 10%) by alcohol intake and are markedly suppressed (by about 50%) by glucocorticoids. Plasma concentrations rise when osteoblasts are stimulated by the administration of 1,25(OH)2D.
Procollagen 1 extension peptides
Antibodies have been raised to both the soluble C-terminal and N-terminal propeptides of procollagen, which appear in the circulation after cleavage from the newly formed collagen molecule. Their concentrations in the plasma thus reflect osteoblastic collagen synthesis. As they are not filtered by the kidney, variations in renal function should not affect their plasma concentrations. Degradation of the proteins takes place in hepatic endothelial cells (through the mannose 6-phosphate receptor in the case of procollagen 1 carboxy-terminal extension peptide (P1CP), and by scavenger receptors in the case of procollagen 1 amino-terminal extension peptide (P1NP)).
Procollagen 1 carboxy-terminal extension peptide is usually measured by enzyme linked immunosorbent assay (ELISA). It has a diurnal variation, with peak concentrations occurring at 04.00–08.00 h, and the nadir in the afternoon. There may be some contribution to circulating P1CP from non-osseous sources.
Procollagen 1 amino-terminal extension peptide has been more widely studied. It appears to be a more dynamic marker than P1CP or other formation markers. The peptide is stable, and the available assays are precise and responsive to common interventions in metabolic bone diseases. It is now regarded as the marker of choice to assess osteoblast activity. Of the various assays available for P1NP, some measure the intact propeptide (intact P1NP) while others (total P1NP) also detect a smaller antigen in serum. In many clinical situations, these assays give similar information, but renal insufficiency increases the concentrations of the smaller antigen, influencing both the apparent concentration of P1NP and assay calibration.
Markers of bone resorption
Hydroxyproline
Hydroxyproline is a major component of fibrillar collagen of all types, comprising ~14% of the total amino and imino acid content. It is produced by the post-translational modification of proline by the enzyme 4-prolyl hydroxylase. In plasma, hydroxyproline exists in protein-bound, peptide-bound and free forms. Most hydroxyproline is oxidized in the liver but a small proportion (~10%) is excreted in the urine. Approximately 90% of collagen-derived hydroxyproline excretion is in the form of peptides resulting from collagen degradation, with the remainder in the form of fragments of the N- and C-terminal procollagen peptides cleaved during collagen synthesis. When bone resorption is increased, urinary hydroxyproline excretion rises, because of the increased rate of catabolism of type 1 bone collagen. When bone turnover is low, urinary hydroxyproline is not a reliable index of bone resorption, since the other sources (for example C1q complement component) can contribute up to 50% of the total urinary hydroxyproline.
Like all urinary collagen breakdown products, hydroxyproline excretion can be measured in a 24 h urine collection or, more conveniently, in fasting, second voided morning samples, expressed as a ratio to the creatinine concentration. Dietary sources of hydroxyproline (e.g. gelatin) need to be eliminated immediately before and during the collection. The major limitations of hydroxyproline estimation as an index of bone resorption are interference from sources other than type 1 collagen, its lack of sensitivity at low concentrations, its day-to-day variability and its partial origin from bone formation.
Glycosylated hydroxylysine
Hydroxylysine – another amino acid unique to collagen-like peptides – is produced by the post-translational modification of lysine residues. These amino acids can then undergo further modification by glycosylation – giving rise to galactolysyl-hydroxylysine and glucosylgalactolysyl-hydroxylysine. The former predominates in the type 1 collagen of bone. Galactolysyl-hydroxylysine is not significantly metabolized before excretion in the urine (10% in free form, 90% peptide bound), and the urinary concentrations are not affected by diet, so it is a good marker of bone resorption. It can be measured by HPLC, but the assay is technically demanding.
Collagen cross-links
The amino acids hydroxylysylpyridinoline and lysylpyridinoline (also known as pyridinoline and deoxypyridinoline, respectively) are created by the covalent cross-linking that takes place when mature collagen molecules stabilize after forming the triple helical structure (Fig. 31.4). Since the cross-links are formed only in extracellular collagen fibrils, they appear in the circulation and the urine only as degradation products of mature matrix and do not reflect new collagen synthesis. In contrast to hydroxyproline, neither pyridinoline nor deoxypyridinoline occur in the collagen of normal skin. Pyridinoline is present in other connective tissues, notably cartilage and tendon, whereas the less abundant deoxypyridinoline is more specific to bone and dentine.
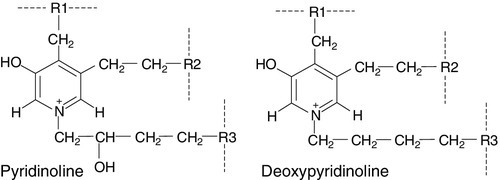
FIGURE 31.4 Molecular structure of pyridinoline cross-links. R1 and R2 are telopeptide sequences and R3 a helical fragment sequence; for free crosslinks, R1, R2 and R3 are: − CH(COOH)NH2.
In urine, both pyridinoline and deoxypyridinoline are present as both free amino acid derivatives (~40%) and as oligopeptide-bound fractions (~60%). The free forms can be measured directly, but the conjugated form has to be hydrolysed before assay. After acid hydrolysis, total urinary pyridinoline and deoxypyridinoline can be measured by HPLC, but ELISA immunoassays for the free amino acids or peptides containing the cross-links are now available.
The pyridinoline:deoxypyridinoline molar ratio in urine is similar to that found in bone (7:2), and the urinary excretion rate of the cross-links agrees well with radioisotopic and histomorphometric measures of bone resorption. The cross-links do not undergo further metabolism once released, and their rate of excretion is not influenced by dietary intake or moderate renal impairment. Random urine measurements (expressed relative to creatinine concentration) correlate closely with 24 h estimates. The elderly tend to have relatively greater excretion of the larger peptide-bound forms, and a correspondingly smaller proportion of the free pyridinoline and low molecular weight peptide forms.
Urinary pyridinoline and deoxypyridinoline concentrations appear to be good markers of bone resorption, and more sensitive than hydroxyproline in distinguishing small increases in bone resorption from normal. For example, when normal premenopausal and postmenopausal women are compared, hydroxyproline excretion in the latter group is about 50% above that of premenopausal women, whereas for urinary pyridinoline and deoxypyridinoline, excretion is increased by 100%.
Collagen telopeptides
Collagen cross-links form mainly between the short non-helical peptides at both ends of the collagen molecule (the N- and C-terminal telopeptides), which link via pyridinium or pyrrole compounds to the helical portion of an adjacent collagen molecule (Fig. 31.5). During collagen breakdown, N- and C-terminal telopeptide fragments, still attached by the pyridinium cross-link to the helical fragments of the nearby molecule, are released into the circulation and are cleared by the kidneys. Within the C-telopeptides of type I collagen is an aspartyl-glycine motif that can undergo β-isomerization to isoaspartyl-glycine. This process is believed to be linked to protein ageing, with younger bone having a greater proportion of the non-isomerized α-C-telopeptides, and older bone having a greater proportion in the β-isomerized C-telopeptide form. Various immunoassays have been developed to detect epitopes specific to particular fragments.
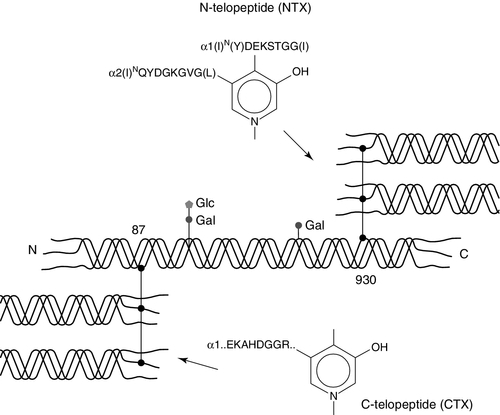
FIGURE 31.5 Intermolecular cross-linking in type I collagen and the location of the peptide sequences of the N-terminal (NTX) and C-terminal (CTX) immunoassays.
The urinary N-telopeptide (NTX) assay is an ELISA based on a monoclonal antibody that recognizes a conformational motif in the α2-chain that is linked to a pyridinium cross-link. Although it does not recognize the pyridinoline or deoxypyridinoline cross-link itself, it is specific for the cross-linking site of bone collagen. The assay requires no hydrolysis. The measurement is usually performed on a second void, morning urine sample, and the results are expressed as bone collagen equivalent, corrected for urinary creatinine concentration. A serum assay has also been developed, which correlates well with urinary concentrations.
Immunoassays for the C-terminal telopeptide cross-linked region of type I collagen (CTX) have also been developed. The monoclonal antibody recognizes an octapeptide sequence of the α1 chain containing the lysine in the C-terminal telopeptide that is involved in intermolecular cross-linking. Immunoassays that are specific for either α-CTX or β-CTX have been developed, and can measure these fragments in both serum and urine. Serum β-CTX is generally regarded as the marker of choice for assessing bone resorption. It should be measured fasting at 08.00–09.00 h, since it shows diurnal variation and is suppressed by food.
Tartrate-resistant acid phosphatase
The acid phosphatases are lysosomal enzymes that hydrolyse phosphomonoesters at low pH, releasing phosphoric acid. They are produced in a number of tissues, including bone, prostate, platelets and erythrocytes. Five isoenzymes are detectable in plasma. The different acid phosphatase isoenzymes differ in tissue of origin, chromosomal origin, molecular size and electrophoretic mobility. Based on the latter, the plasma isoenzymes have been classified as types 1–5. According to their sensitivity to inhibition by L(+)-tartrate, the isoenzymes are further classified as tartrate sensitive (TSAP) and tartrate resistant (TRAP).
Prostatic acid phosphatase (type 2b) activity is typically raised in plasma in carcinoma of the prostate. Type 5 acid phosphatase is produced by macrophages (TRAP5a) and osteoclasts (TRAP5b). TRAP5a activity in plasma is elevated in Gaucher disease and hairy cell leukaemia.
Tartrate resistant 5b differs from TRAP5a by not having sialic acid residues. It can be measured in serum by a variety of kinetic assays or by immunoassay. Measurement has some value in the diagnosis of osteoclast-rich forms of osteopetrosis. In most forms of this disorder, the number of osteoclasts in bone is actually increased – although they are ineffective at resorbing bone – and plasma activities of TRAP5b are increased. In most other bone disorders, TRAP5b plasma concentrations correlate well with other indices of bone resorption and with histological measurements of osteoclastic activity. Unlike other resorption indices, TRAP5b reflects osteoclast number and/or activity, rather than matrix breakdown. It may find a particular role in situations where these are dissociated, such as in examining the actions of drugs that inhibit osteoclast activity without reducing their number (e.g. cathepsin K inhibitors). Plasma acid phosphatase concentrations are also increased in bisphosphonate-induced osteosclerosis.
New markers
It is now possible to measure circulating cathepsin K concentrations. Preliminary reports indicate that these are higher in subjects with fractures, and are related to other turnover markers. Assays are also available for circulating osteoprotegerin, soluble RANKL, and sclerostin. These factors all act locally in bone, and the relevance of circulating concentrations to judging their bioactivity in the bone micro-environment remains to be determined. These new markers are used mainly for research purposes at present, and their clinical utility is uncertain.
Variation in bone turnover markers
Many of the markers described are now widely used in research, and are finding a place in the management of some metabolic bone diseases. In general, they correlate well with assessments of turnover based on histomorphometry or calcium kinetic studies. The newer assays are more specific to bone and, in some cases, are more sensitive to perturbations, but their usefulness is limited by the day-to-day biological variability (as well as analytical imprecision). Stability is also an important issue for some analytes. In general, plasma markers of bone resorption show less day-to-day variability than urinary markers. Urinary resorption markers are usually measured on fasting second void urine samples to minimize the effect of diet and results expressed as a ratio to creatinine concentration. Thus, artefact can arise if muscle mass is significantly altered from normal.
Most markers of bone turnover show significant diurnal variation with highest values in the early morning hours and lowest values during the afternoon or night. The amplitude of this variability is around 15–30%. Therefore, resorption markers are best assessed in the early morning, after overnight fasting.
Bone turnover may also vary with the menstrual cycle: formation markers are higher in the luteal phase, whereas resorption markers are higher during the follicular phase. The amplitude of these changes is ~10%. After the menopause, there is an increase in bone turnover – resorption markers increase by 30–50% within 12 months of the menopause, and there are similar, but later, increases in formation markers. A less marked increase in bone turnover markers is also seen in men after the age of 50 years. Compared with adult values, all turnover markers are much higher during childhood and puberty, because the skeleton is growing and modelling, as well as remodelling (see below). It is thus important that age-appropriate reference ranges are used for all these assays.
Bone markers increase by ~50% two to four weeks after a fracture, and can take six to nine months to return to normal.
OSTEOPOROSIS
Osteoporosis is a reduction in bone density to the point that fractures occur as a result of the minor trauma that is a normal part of everyday life. At the microscopic level, osteoporotic bone is normally mineralized but reduced in volume. There is a generalized thinning of trabecular elements with the total loss of some trabeculae, greatly reducing the strength of bone (Fig. 31.6) and a reduction in cortical width in long bones. This loss of the normal microarchitecture emphasizes the need for prevention of bone loss, since restoration of the lost structure is unlikely to be achievable with pharmacological agents.
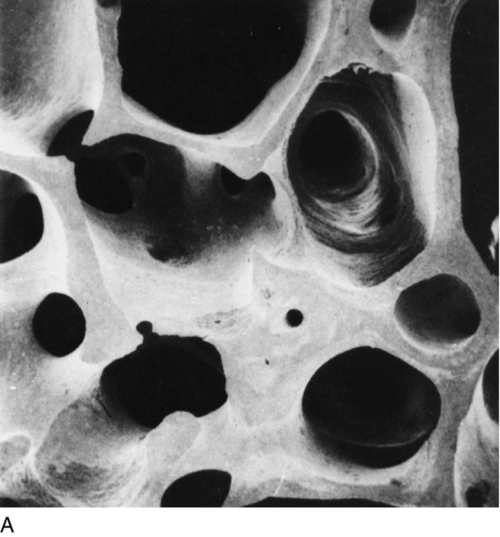
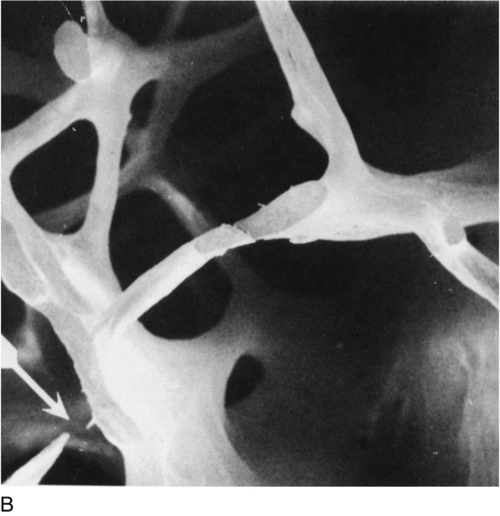
FIGURE 31.6 Low-power scanning electron micrographs of iliac crest biopsies at the same magnification. (A) Taken from a healthy 47-year-old woman; (B) from an osteoporotic woman of 75 years. In comparison with (A), there has been thinning of the trabeculae with loss of some trabecular elements and discontinuity (arrowed) of some of those which remain. (From Dempster D W et al. 1986 A simple method of correlative light and scanning electron microscopy of human iliac crest bone biopsies. Journal of Bone and Mineral Research 1:21–25, with permission.)
Osteoporosis can also be defined using bone mineral density (BMD) criteria. Thus, osteoporosis is defined as a BMD T-score less than − 2.5, a BMD that is more than 2.5 standard deviations below the mean value in the young healthy population. Using this diagnostic criterion, ~50% of women have osteoporosis by the age of 80 years. Osteopenia is defined as a BMD T-score that is 1–2.5 standard deviations below the mean value in the young healthy population. Low bone mineral density is an important predictor of future fractures. Loss of BMD is sometimes pathological, but more frequently occurs as a normal part of ageing. With age, the efficiency with which osteoblasts refill resorption cavities is reduced, and there is also an increase in bone resorption associated with hypogonadism.
The important clinical consequence of osteoporosis is fracture. Common osteoporotic fractures include those of the hip, forearm, vertebral body, humerus, ribs and pelvis, while digital, skull, facial and fibular fractures are generally not osteoporotic in origin. The epidemiology of osteoporotic fractures reflects the importance of age and sex in the pathogenesis of the disease. The incidence of vertebral and hip fractures increases exponentially in later life in both men and women, although fracture rates at these sites in men increase about 10 years later (at age 75) than those in women.
Causes of osteoporosis
Box 31.2 sets out a list of risk factors for, and causes of, osteoporosis. Some of these (for example age and family history) are not modifiable. Whether or not an individual will develop osteoporosis depends on two factors: the peak bone mass, typically attained by age ~20 years, and the rate of loss of bone in later life. Probably the strongest influence on peak bone density is genetic – there is a close relationship between an individual’s BMD and that of his parents, and bone density is reduced in the children of patients with osteoporosis.
Because of the evidence for a strong heritable component to peak BMD, there has been intense interest in identifying genes that regulate skeletal health. Thus, a large number of genome-wide association studies (GWAS) have been performed in recent years. These analyses have identified more than 20 genes that are associated with alterations in BMD. Several of these genes cluster into four biological pathways: the vitamin D endocrine pathway, the oestrogen endocrine pathway, the wnt-β-catenin signalling pathway and the RANKL–RANK–OPG pathway. However, collectively these genes account for only a few percent of the variability in BMD. So, at present, it seems unlikely that genetic testing will improve the predictive power of algorithms used to identify patients at high fracture risk. Genetic factors may also underlie the significant racial differences in bone density and fracture incidence. Individuals of African or Polynesian ancestry have higher bone mass than Europeans and Asians.
Body weight is an important influence on bone density, with heavier people having greater bone mass, and lower fracture risk than those who are thin and/or underweight. The relationship between body weight and bone density is probably mediated in several ways, including mechanical loading of bone, the skeletal actions of hormones affected by nutritional status, and the actions of adipokines produced by adipose tissue.
Despite continued argument as to what constitutes an adequate calcium intake at different stages of life, the impact of calcium intake on bone density and fracture risk is probably slight at intakes > 10 mmol (400 mg)/24 h (populations in Asia and Africa appear to maintain satisfactory bone health on intakes of < 10 mmol/24 h). Vitamin D insufficiency (plasma 25-hydroxyvitamin D < 50 nmol/L), or deficiency (< 25 nmol/L), which is common in the contexts of skin pigmentation and sunlight deprivation, may contribute to reduced bone mass.
There has been extensive study of the relationship of physical activity to bone density. Cross-sectional studies are subject to various biases, but most prospective intervention studies indicate that this variable contributes only moderately to the differences in bone density that exist in postmenopausal women. However, prospective studies do indicate that weight-bearing exercise increases bone mass in the pre-pubertal skeleton.
Age has an important effect on bone density in both sexes, and also contributes to skeletal fragility, independently of BMD. In the proximal femur, loss of BMD begins in both sexes in the third and fourth decades, whereas in women BMD in the lumbar spine is probably stable until the perimenopause.
The loss of sex hormones has a deleterious effect on bone mass at whatever time it occurs. In both men and women it is oestradiol deficiency that appears to be critical. Men with inactivating mutations in the genes encoding either the aromatase or the oestrogen receptor have low BMD, despite the presence of normal or high testosterone concentrations. In women, late menarche and episodes of amenorrhoea with oestrogen deficiency reduce bone mass in much the same way as early menopause. Amenorrhoea with oestrogen deficiency can arise from a number of causes (Box 31.3). In the underweight, the low body weight itself has an effect on bone density. Adipose tissue is an important site of oestrone production, and high fat mass is associated with higher circulating concentrations of a number of bone anabolic factors including insulin, amylin and leptin. Cigarette smoking accelerates the metabolism of oestrogens to biologically inactive forms, is associated with reduced body weight and early menopause and may directly inhibit osteoblastic function: consequently, BMD tends to be lower in smokers than in non-smokers.
Various pathological conditions can also have significant effects on BMD (see Box 31.2). Alcohol appears to act as an osteoblast toxin and high intakes are also associated with both liver disease and hypogonadism. Glucocorticoids cause bone loss through inhibition of osteoblastic and osteocytic activity, intestinal calcium absorption and renal tubular calcium reabsorption. Glucocorticoids may directly or indirectly stimulate both hyperparathyroidism and bone resorption and, in men, are associated with reduced plasma testosterone concentrations.
Investigation and diagnosis
In recent years, there has been a move to characterize osteoporosis as a disease, principally so that it will be taken seriously by doctors, patients and regulatory authorities. However, this can be counterproductive to an understanding of the nature of the condition, since the process of bone loss is universal. Therefore, the key issue is often not so much one of diagnosis (required in the context of a disease), but one of assessing fracture risk, and then determining which interventions are cost-effective for a given level of risk. Fracture risk can be assessed from clinical risk factors (discussed above) and measurement of BMD.
Clinical risk factors for fracture
The risk factors of greatest importance include age, weight, family history and smoking. However, the most powerful predictor of future fractures is a past history of fracture. The presence of a deformed vertebra on a lateral spine or chest X-ray increases future fracture risk as much as five-fold, and a similar effect is seen with a past history of fractures at other sites. Therefore, treatment is sometimes indicated following fracture, even if the BMD is not particularly low.
Bone densitometry
There is a continuous, inverse, relationship between fracture risk and BMD. The best validated methodology for assessing BMD is dual-energy X-ray absorptiometry (DEXA). Using this technology, a difference in bone density of one population standard deviation (about 10%) is associated with a two-fold difference in fracture risk. Most sites of bone density measurement give comparable prediction of global fracture risk. However, fracture at a given site is best predicted by bone density measurement at that site: for example, hip bone density predicts hip fracture risk better than measurements elsewhere. The sites most commonly assessed are the hip and spine. The fact that hip fractures are numerically the most important osteoporotic fractures argues in favour of using the hip as the preferred measurement site. However, the precision of measurement is greater in the spine, and the trabecular bone of the spine is more rapidly responsive to both disease and to therapeutic interventions. With advancing age, the value of vertebral scans diminishes because of artefacts associated with degenerative joint and disc disease. Because of these conflicting issues, it is common practice to measure bone density in both the lumbar spine and proximal femur. In the past, the forearm was a common site for measurement because that was all that was technically feasible. Modern densitometers can also measure the forearm, but there is no reason to prefer it to the spine and hip when these measurements are available.
Absolute values of bone density are not comparable between different anatomical sites, measurement techniques and make of densitometer. This problem has been addressed by reporting bone densities in terms of the young healthy population range, using standard deviation units (T-scores). The DEXA-based ‘definition’ of osteoporosis can be used as the threshold for intervention. However, it does not take into account the fact that clinical risk factors are multiplicative with the risk estimated from bone density measurement. For example, a T-score of − 2.5 in an 80-year-old woman with a history of vertebral fracture is associated with a considerably higher risk of fracture over the next year than is the same density in a 50-year-old woman with no fracture history.
It is now standard practice to combine clinical risk factors with BMD in algorithms that produce estimates of absolute fracture risk. Examples, freely available via the internet, include the WHO-sponsored FRAX and the Garvan Fracture Risk Predictor. A 10-year hip fracture risk of > 3% or an osteoporotic fracture risk of > 15–20% are typically taken as indications for pharmaceutical intervention, but these thresholds are obviously influenced by the cost and safety of the interventions, balanced against their effectiveness. Of the other modalities available for assessment of BMD, computerized tomography (CT) scanning is rarely used because of the higher radiation exposure involved and ultrasound-based methods (e.g. of the os calcis) have not been as extensively validated.
Biochemical investigation
The investigations necessary in a patient who has osteoporosis will depend upon the extent to which the low bone mass can be accounted for by information already available. Thus, in an elderly subject or someone who has received long-term high-dose glucocorticoid therapy, there may be very little need for further investigations. However, in a younger individual whose bone density is clearly subnormal without apparent cause, a more extensive investigation for an underlying abnormality is appropriate. The investigation thus depends on the clinical context (see Box 31.2). Measurements of sex hormone concentrations (particularly in men), thyroid, renal and liver function tests, plasma and urinary cortisol, plasma calcium, albumin and phosphate, 25-hydroxyvitamin D (25(OH)D), serum and urine protein electrophoresis, coeliac disease markers, serum tryptase (to diagnose mastocytosis) and full blood count may be appropriate.
There remains much debate about the utility of measuring bone turnover markers in the management of osteoporosis (Table 31.2). The potential uses include both prognosis (prediction of fracture, selection of patients for treatment, prediction of response to treatment) and therapeutics (selection of treatment, monitoring adherence to, and effectiveness of treatment). At present, there is little convincing evidence for use of biochemical markers in most clinical settings, but these are active areas of research and more data will emerge.
TABLE 31.2
Potential uses of bone turnover markers in osteoporosis
| Clinical question | Utility of test |
| Diagnosis of osteoporosis or prediction of bone mass | None |
| Prediction of fracture and selection of patients for therapy | Possibly useful, but not yet validated |
| Selection of type of therapy | Theoretically appealing, not yet validated |
| Monitoring adherence to treatment | Useful |
| Monitoring effectiveness of therapy | Possibly useful, in conjunction with bone mass measurement |
| Predicting change in bone mass on treatment | Reasonable correlation in some studies |
| Determining duration of ‘drug holiday’ in patients on long-term antiresorptive therapy | Useful |
There has been concern that long-term suppression of bone turnover with potent bisphosphonates might increase the risk of osteonecrosis of the jaw and subtrochanteric femoral fractures. While the significance of these issues continues to be debated, ‘drug holidays’ after 5–10 years of bisphosphonate therapy are now commonly recommended; bone turnover markers have a role in deciding when treatment should be reintroduced.
There is limited value in measuring 24 h urine calcium in patients with osteoporosis. The urine calcium excretion reflects an interaction of dietary calcium intake, fractional intestinal absorption, bone resorption and renal tubular reabsorption, and it is not possible to tease these apart without other investigations. The calcium:creatinine ratio in a fasting, second-voided urine sample in the morning is a poor guide to the rate of bone resorption, but does permit calculation of indices of renal calcium handling – though these are seldom needed in managing osteoporosis.
In a few centres, the efficiency of intestinal calcium absorption is measured. However, there are practical difficulties in the performance of these tests (see Chapter 6) and there is no clear evidence that this approach is a useful guide to treatment selection or improves treatment outcomes.
Other investigations
The clinical history should include an assessment of recent and past dietary calcium intake. The presence of fractures should be confirmed radiologically and the radiographs scrutinized for evidence of other abnormalities, particularly the possibility of malignancy. Isotope scanning is also useful in detecting metastases. Bone biopsy is needed rarely, to diagnose pathologies such as osteomalacia or mastocytosis, and is reserved for patients with atypical presentations in whom there is a high index of suspicion of some other underlying abnormality (Appendix 31.1).
Treatment
Treatment of osteoporosis involves a number of strategies, all of which are aimed at reducing the individual’s future risk of fracture. They can be divided into lifestyle changes (many of which are applicable to the whole population), oral calcium and vitamin D supplementation (in subjects with low intakes) and pharmacological measures. The latter are more expensive and more likely to be associated with side-effects, so are reserved for those with a moderately high absolute risk of fracture. In addition, minimization of exposure to drugs (particularly glucocorticoids) that are toxic to the skeleton is important.
Lifestyle modifications
Lifestyle interventions that can be recommended to all patients include maintenance of ideal body weight throughout life, maintenance of normal sex hormone concentrations from puberty until the late forties (as evidenced in women by regular menstruation), regular weight-bearing exercise, avoidance of smoking and avoidance of high alcohol intake. Exercise may be particularly important during childhood and adolescence, when the skeleton is very responsive to the loads placed upon it. Vigorous exercise interventions in postmenopausal women produce changes in bone density of only a few percent, but regular exercise, by increasing fitness, may also decrease an individual’s risk of falling.
Calcium and vitamin D
While the fundamental building block of bone is collagen, calcium is also an important constituent, and its supply in the diet has been suggested to be a limiting factor for bone growth and maintenance. The average calcium intake in Western postmenopausal women is 17.5–22.5 mmol (700–900 mg)/24 h, and there is evidence that increasing this by a further 25 mmol (1 g)/24 h slows postmenopausal bone loss by ~50%, though calcium supplementation alone is not able completely to prevent postmenopausal bone loss. Fracture risk appears to be reduced by ~10% with calcium supplements. However, the trials which suggest this fracture prevention also demonstrate an increase in risk of myocardial infarction of about 25% and of stroke of about 15%, effectively negating any potential benefit. These risks have not been demonstrated with dietary calcium intake, so modest augmentation of the diet may be a safer course to follow than the use of mineral supplements.
Vitamin D deficiency is extremely common among the frail elderly as a result of low levels of exposure to sunlight. If severe, it sometimes accelerates bone loss and it may also contribute to muscle weakness, increasing the risk of falls. If suspected, it can be confirmed by measurement of serum 25(OH)D. The current consensus is that optimum plasma concentrations of this compound are at least > 50, and probably > 70 nmol/L, although some authorities advocate concentrations as high as 100 nmol/L. Vitamin D deficiency can be prevented or treated with oral calciferol (at least 500–1000 U/day or, more conveniently, 50 000 U/month). There is evidence from randomized controlled trials in markedly deficient frail elderly subjects, that the provision of calcium and calciferol supplementation reduces hip fracture risk by more than a quarter. The use of the potent metabolites of vitamin D, such as calcitriol, is more expensive, more likely to cause hypercalcaemia or hypercalciuria and is of uncertain clinical benefit.
Pharmacological management
Bisphosphonates
The pharmacological management of osteoporosis is currently dominated by the bisphosphonates. These are relatively simple phosphate salts that have a very high affinity for the surface of bone but are very poorly absorbed from the gastrointestinal tract. Thus, only 1–2% of an oral dose is absorbed, about half of which is rapidly deposited on the bone surface, the balance being excreted unchanged in the urine. When osteoclasts resorb bone, they ingest the bisphosphonate and are effectively poisoned by it. This reduces the amount of bone resorption (bone resorption markers fall by 50–80%) with a consequent redressing of the imbalance between bone formation and resorption. Bisphosphonate remains present on the bone surface for many years and is gradually incorporated into the structure of the bone, so that it can inhibit remodelling cycles that occur years after the time of dosing. Thus, bisphosphonates have a long duration of action and intermittent administration is effective. Zoledronate, for instance, can be given as infrequently as once every 2–3 years by intravenous infusion and produces changes in bone density comparable to or greater than those seen after weekly oral dosing with alendronate.
The low oral bioavailability of bisphosphonates is a critical issue in their use. They must be taken fasting, with water alone, if they are to be absorbed at all. Aminobisphosphonates, such as alendronate and risedronate, can cause oesophagitis and gastric ulceration, so patients should not lie down for 30–60 min after oral dosing, since this may permit reflux of the tablet into the oesophagus. This does not appear to be such a problem for the less potent, non-amino bisphosphonate, etidronate. A common phenomenon after first exposure to intravenous (and occasionally oral) nitrogen-containing bisphosphonates is an ‘acute phase response’ of fever, myalgia and headache that lasts 24–48 h. This is mediated by the release of TNFα and interleukin (IL)-6 from peripheral blood T cells. While sometimes unpleasant, the reaction is transient, and tends not to recur with subsequent doses.
Potent bisphosphonate use in postmenopausal women results in an increase in bone density comparable to that produced by hormone replacement therapy. Bone resorption is reduced by more than one half, and the risk of hip, spine and forearm fractures is lowered by ~50%. While there are a number of trials assessing the effects of bisphosphonates on fracture risk over periods of three to four years, there are only limited data available from longer-term studies. The available data suggest that anti-fracture efficacy is maintained with long-term use.
Of the currently available bisphosphonates, the evidence for anti-fracture efficacy is strongest for alendronate (most conveniently given as a weekly dose), risedronate (weekly) and intravenous zoledronate (annually), each of which has been demonstrated to prevent both vertebral and non-vertebral fractures in osteoporotic and glucocorticoid-treated patients and in men as well as postmenopausal women. Thus, these bisphosphonates are currently the first choice therapy in all forms of osteoporosis.
Following discontinuation of bisphosphonates, bone resorption rises to some extent, but even several years after discontinuation of long-term use of alendronate and zoledronate, bone turnover markers do not return to baseline. The same is true of bone density, which declines only slowly after withdrawal of these agents. This prolonged duration of action is a consequence of the long residence time of bisphosphonates in bone, and it raises important (as yet unanswered) questions about the optimal duration of continuous therapy. There is general agreement that these agents should be used continuously for periods of up to five years. Beyond that, some experts reduce the dose, whereas others opt either for no change or for a period off treatment. There is no clear evidence at present to determine what is optimal. Risedronate and ibandronate have a more rapid offset of effect, and will often require re-institution of therapy within 6–12 months of treatment cessation.
Hormone replacement therapy
Hormone replacement therapy (HRT) was the most widely used intervention for the prevention and treatment of osteoporosis for many years, but was displaced by the aminobisphosphonates, because of their demonstrated anti-fracture efficacy, and because the Women’s Health Initiative produced a marked change in attitudes to hormone replacement therapy. On the one hand, it clearly demonstrated that HRT reduces the incidence of fractures, including hip fractures. On the other, it raised concerns regarding safety, particularly with respect to cardiovascular and cerebrovascular disease, breast cancer and venous thromboembolism. Public and professional enthusiasm for the use of HRT in the management of osteoporosis waned, because the older age group in whom osteoporotic fracture risk is high is the same group in whom vascular disease is a major concern. If early postmenopausal women require interventions for osteoporosis, HRT may still be a reasonable option, although the balance of risks and benefits of HRT in younger (< 50 years) postmenopausal women is uncertain, and the bisphosphonates are also suitable for use in this context. The cardiovascular and skeletal effects of transdermal oestrogen preparations have not been assessed in large randomized studies. Unlike the bisphosphonates, oestrogen effects on bone disappear within months of treatment discontinuation, so long-term reductions in fracture risk are unlikely to occur.
Selective oestrogen receptor modulators
Selective oestrogen receptor modulators (SERMs) are a newer class of pharmaceuticals with mixed oestrogen agonist/antagonist activities that vary from tissue to tissue. Thus, the prototypic SERM, raloxifene, acts as an oestrogen agonist on bone, but as an antagonist in the breast and endometrium. Raloxifene reduces bone resorption and increases bone density, but is less potent than either oestrogen or bisphosphonates in this regard. It decreases the incidence of vertebral fractures but it has not been shown to have any effect on non-vertebral fractures. It substantially reduces the incidence of breast cancer, and does not significantly influence risk of vascular disease. Basodoxifene has similar effects and is being developed for co-administration with oestrogen, thus augmenting its bone effects, while the anti-oestrogen actions of the SERM block the proliferative effects of the oestrogen on breast and endometrial cells. Lasofoxifene is the only SERM to be shown to reduce risk of non-vertebral factors, but it is not clear that this drug will reach the market.
Parathyroid hormone
Parathyroid hormone (PTH) preparations, either PTH(1-34) (teriparatide) or PTH(1-84), were the first anabolic therapies to be approved for treatment of osteoporosis. When given as a daily injection, they produce substantial increases in BMD at skeletal sites containing predominantly trabecular bone, with less marked increases in cortical BMD, and reductions in fracture incidence that tend to be greater than are seen with other currently available agents. PTH use is restricted to a period of 1.5–2 years because of the high incidence of osteosarcoma in rat toxicology studies, although this problem has not been an issue in human use. Unlike the antiresorptive agents, it causes sustained increases in markers of both formation and resorption. Practices for combining PTH with antiresorptives vary; co-administration with alendronate may blunt its anabolic effects, though this appears not be the case for combination with zoledronate. The anabolic effects dissipate rapidly after treatment discontinuation, so locking in gains in BMD with subsequent antiresorptive therapy is important.
Other pharmacological treatments
The divalent cation strontium (in the form of strontium ranelate) has been shown to prevent both vertebral and non-vertebral fractures, though effects on hip fracture were only seen in post hoc analyses. The mechanism of action of strontium on bone is not understood, but some of the increase in BMD it produces is artefactual, resulting from replacement of calcium ions by the heavier strontium ions in the hydroxyapatite. BMD measurements in strontium-treated patients, should be adjusted for skeletal strontium content.
Denosumab is a monoclonal antibody against the osteoclastogenic factor, RANKL. It produces profound reductions in both bone resorption and bone formation, resulting in increases in BMD and decreases in fracture rates comparable to those resulting from the use of potent bisphosphonates. It is given by six-monthly subcutaneous injection but there is a rapid loss of effect 6–8 months after an injection, with an overshoot in bone turnover during this time. Therefore, maintaining continuity of treatment is very important to achieve anti-fracture efficacy.
Cathepsin K is the principal osteoclastic enzyme involved in the degradation of type I collagen. It is therefore an obvious target for an antiresorptive agent. Several early studies in this area were abandoned because of insufficient specificity of the investigation compounds. The cathepsin K inhibitor odanacatib, has demonstrated sustained year-on-year increases in bone density in phase 2 studies. A large phase 3 study to determine anti-fracture efficacy is nearing completion. Tartrate resistant 5b concentrations are not suppressed (as osteoclasts remain numerous on the bone surface, although their bone resorbing activity is markedly curtailed) and the suppression of bone turnover markers with odanacatib is less marked than seen with other antiresorptive agents; in particular the suppression of bone formation appears to diminish after 2–3 years. Discontinuation of treatment is accompanied by a rapid rise in bone turnover markers.
In recent years, the wnt-LRP5 pathway has emerged as a critical regulator of bone formation. Activity of this pathway is regulated by sclerostin, an inhibitor secreted by the osteocyte. A number of studies are exploring the efficacy of monoclonal antibodies directed against sclerostin as bone anabolics. Phase 1 and 2 studies suggest that these could be the most effective bone anabolic drugs yet developed; results of phase 3 trials are awaited.
In the past, sodium fluoride and calcitonin were widely used to treat osteoporosis but good evidence of anti-fracture efficacy is lacking for both and fluoride interferes with normal bone mineralization. As a result, both agents have dropped out of common usage.
Biochemical responses to treatments
Antiresorptive agents such as HRT, SERMs and the bisphosphonates produce an early (within weeks) reduction in indices of bone resorption, followed by a slower fall in indices of bone formation (Fig. 31.7). Similar effects are observed in hypogonadal men treated with testosterone replacement. These effects are sustained for the duration of therapy and, in the case of potent bisphosphonates, which have long skeletal retention times, may persist for considerable periods of time (months or years) after therapy is stopped. The most potent currently available bisphosphonates may reduce markers of bone resorption by up to 80% from baseline levels. Denosumab can produce even larger decreases in bone resorption, most patients having undetectable resorption markers in the first weeks after injection. In contrast, the cathepsin K inhibitor odanacatib only reduces resorption markers by about 50%, with smaller declines in formation markers. Plasma calcium and phosphate concentrations can also fall during the early phase of antiresorptive therapy but usually recover without specific intervention.
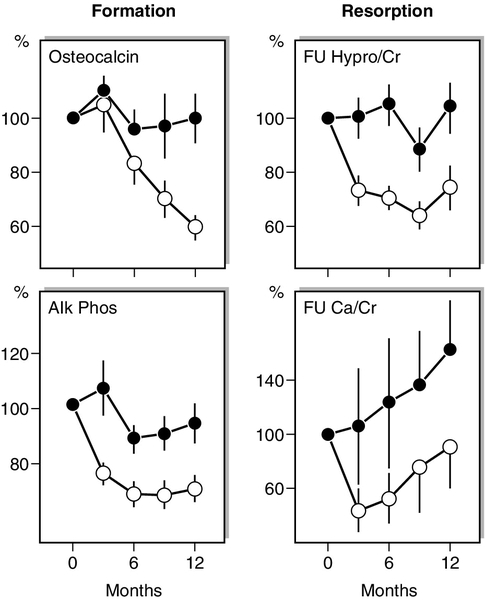
FIGURE 31.7 Changes in mean (± SD) biochemical indices of bone formation and resorption in postmenopausal women receiving either placebo ([closed circle]) or a regimen of continuous oestrogen and progesterone ([open circle]). The group receiving active treatment show significant suppression of both bone resorption and formation. There is a tendency to reach a plateau in these indices as a new steady state is reached. Note that the variance in fasting urine calcium/creatinine ratios (FU Ca/Cr) is considerably greater than that in fasting urine hydroxyproline/creatinine (FU Hypro/Cr). (Data from Christiansen C, Riis BJ 1990 17β estradiol and continual norethisterone: a unique treatment for established osteoporosis in elderly women. Journal of Clinical Endocrinology & Metabolism 71:836–841.)
Intermittent PTH treatment produces biochemical changes characterized by an initial increase in markers of bone formation (P1NP, e.g. typically doubling), reflecting primary activation of osteoblasts. A smaller increase in osteoclastic activity follows as a secondary, coupled phenomenon. Plasma 1,25(OH)2D concentration increases by ~25% and there may be small falls in 25(OH)D. Plasma calcium concentration may increase and plasma phosphate decrease during treatment because PTH also stimulates renal tubular calcium reabsorption and phosphaturia (directly) and intestinal calcium absorption (indirectly). Occasionally, a reduction in PTH dose may be required because of mild hypercalcaemia. Early data with anti-sclerostin antibodies demonstrate large early increases in formation markers and, surprisingly, some inhibition of bone resorption markers. Over a period of months, both changes tend to revert towards baseline values.
Treatment with strontium produces inconsistent changes in markers, smaller in magnitude than most agents discussed above. Notably, strontium interferes in many routinely available assays for calcium concentration.
Growth hormone replacement in adults who are deficient improves bone mass, primarily by stimulating cortical bone accretion. Growth hormone treatment induces an early increase in bone resorption markers, and later increases (~two-fold) in bone formation markers that reach a peak after a year of treatment, and then gradually decline. The peak in osteocalcin is delayed relative to the other formation markers.
OSTEOMALACIA
Osteomalacia is the term used to describe the disorder arising from defective mineralization of bone. It is a histological diagnosis, based on the volume of unmineralized osteoid, its extent over bone surfaces, its thickness and the rate of mineralization, as assessed by tetracycline labelling. If a mineralizing defect is a diagnostic possibility in an unusual case, then biopsy should be undertaken (Appendix 31.1). In adults, the radiographic appearances are generally non-specific, showing generalized osteopenia or (in severe cases) vertebral collapse. Looser zones (short radiolucent lines through the cortex, at right angles to the shaft) are very suggestive of osteomalacia, but occur in only a minority of patients.
When osteomalacia arises before growth is complete, the clinical and radiographic features differ from those seen in adulthood, and the condition is termed rickets. The histological processes occurring in osteomalacia and rickets are similar. In order for newly formed osteoid to mineralize, adequate supplies of mineral (calcium and phosphate) must be available and the function of the osteoblasts that regulate the mineralization process must be normal. The causes of osteomalacia are conveniently categorized into those where there is inadequate supply of mineral and those where osteoblast function is defective. The former group may be further categorized into those where inadequate calcium supply is the critical factor (calciopenic) and those where inadequate phosphate supply is critical (phosphopenic). Calciopenic forms are frequently associated with abnormalities of vitamin D metabolism. Biochemical investigations can suggest that osteomalacia is present and indicate possible aetiologies, but cannot prove its presence. However, the biochemical findings in calciopenic and phosphopenic forms of osteomalacia are often very characteristic, so that bone biopsy is not necessary in routine cases.
Calciopenic osteomalacia
The major causes of calciopenic osteomalacia are summarized in Box 31.4.
Vitamin D deficiency
Rickets and osteomalacia were endemic in the UK and many other European countries from the 17th century until the mid-20th century. Crucial scientific observations made in post-war Vienna in 1919–1922 established the role of vitamin D in the aetiology of privational osteomalacia. Although it was clearly demonstrated at that time that it was possible to cure privational rickets by exposing children to sunlight, as well as by the administration of a fat-soluble food factor, the dominance of nutritional theory led to the active factor being termed a vitamin, that is, an obligatory food component that the body cannot synthesize. It is now clear that under the influence of ultraviolet light, the skin can synthesize vitamin D3 (cholecalciferol), the precursor of the prohormone 25(OH)D, and that dietary sources of vitamin D (mainly vitamin D2, ergocalciferol) are critical only when exposure to sunlight is significantly limited.
Diminished exposure to ultraviolet light through industrial pollution and the practice of infant swaddling may have been important during the years that rickets was endemic, and would account for its greater prevalence in more northerly cities in the UK. In the present day, vitamin D-deficient osteomalacia is observed most commonly in individuals who are sunlight deprived because of skin pigmentation, whose cultural practices involve sunlight deprivation, the malnourished and the institutionalized elderly. Osteomalacia occurring in patients with malabsorption syndromes or after partial gastrectomy is not so easily explained, but does suggest that a nutritional factor is important. This factor is most probably calcium.
Reduced intake of calcium, or impaired calcium absorption, initially causes a small fall in plasma ionized calcium concentration and compensatory secondary hyperparathyroidism. As a consequence of this, a greater proportion of 25(OH)D is metabolized to 1,25(OH)2D – the active hormone – rather than to inactive metabolites such as 24,25(OH)2D3 and 25,26(OH)2D3. In the short term, the increased 1,25(OH)2D production, by increasing fractional absorption of calcium from the intestine, corrects the hypocalcaemia at the expense of modest hyperparathyroidism. However, 1,25(OH)2D has an additional effect upon the hepatic metabolism of 25(OH)D, accelerating the metabolic clearance of 25(OH)D to its inactive dihydroxylated forms (see Fig. 6.3, p. 97). In the chronic situation, the sustained increase in 1,25(OH)2D production eventually depletes the available 25(OH)pool. Once the concentrations of 25(OH)have fallen beyond a critical point, 1,25(OH)2D synthesis itself may become insufficient to sustain adequate calcium absorption, so that there is overt hypocalcaemia. Plasma phosphate is also reduced because of diminished intestinal absorption and the renal effects of hyperparathyroidism. With insufficient calcium and/or phosphate available for mineralization, osteomalacia follows.
Osteomalacia and low plasma 25(OH)D concentrations are also found in some patients on long-term anticonvulsant therapy. Hepatic enzyme induction by the anticonvulsant drugs and accelerated clearance of 25(OH)D are important, but the people most likely to develop this syndrome are institutionalized patients who also have limited exposure to sunlight. Intestinal calcium transport can be impaired directly by phenytoin. A patient from Nigeria with severe rickets/osteomalacia and a very low serum 25(OH)D concentration was found to be homozygous for an inactivating mutation in the gene CYP2R1, that encodes the enzyme mainly responsible for 25-hydroxylase activity.
Osteomalacia in people of South Asian origin is multifactorial. As well as diminished dermal synthesis of vitamin D, the traditional diet has a high content of calcium-binding factors such as phytate, which limit the bioavailability of dietary calcium. In groups with a high prevalence of osteomalacia, the disease may be exacerbated by lack of sunshine in winter and at times of increased vitamin D requirements: infancy, adolescence and pregnancy.
The clinical features of privational osteomalacia in adults include bone pain, proximal myopathy and fractures. Bone pain and myopathy are often overlooked and their severity only appreciated when a cure has been effected. The aetiology of the myopathy is unknown. It is not a feature of hypocalcaemia, and does not invariably occur in other types of osteomalacia. In children, rickets may additionally result in difficulty with walking, growth retardation, bowed legs and enlargement of the costochondral junctions.
Defects in 1,25-dihydroxyvitamin D synthesis or action
Osteomalacia and rickets can result from deficiency of the renal 1α-hydroxylase enzyme and thus failure to synthesize 1,25(OH)2D, or from defective action of 1,25(OH)2D, owing to abnormalities of the receptor. Deficiency of the renal 1α-hydroxylase enzyme can be acquired, as in chronic kidney disease (CKD, see below), or can occur as a rare autosomal recessive disorder known as vitamin D-dependent rickets (VDDR) type I. Clinically, these children develop florid rickets in the first two years of life. The syndrome of VDDR type II is also a rare autosomal recessive disorder, often associated with alopecia totalis, but otherwise with a clinical picture similar to VDDR type I. It is caused by an abnormality in the receptor for 1,25(OH)2D, arising from mutations either in the DNA-binding or the hormone-binding domain, and there is target organ resistance to the hormone. Acquired resistance to 1,25(OH)2D has been seen in untreated coeliac disease, where the intestinal mucosa is very abnormal and the absorption of calcium and phosphate impaired.
Laboratory investigation
The relevant laboratory investigations are summarized in Table 31.3. It is important to note that the plasma calcium concentration may be normal in the early phase of privational vitamin D deficiency. Hypophosphataemia due to hyperparathyroidism and reduced intestinal phosphate absorption can occur in any type of calciopenic osteomalacia. However, there are occasional patients with an apparent renal resistance to the effects of PTH who do not become hypophosphataemic when deficient in 25(OH)D. These patients have marked hypocalcaemia and hyperphosphataemia with increased plasma ALP activities.
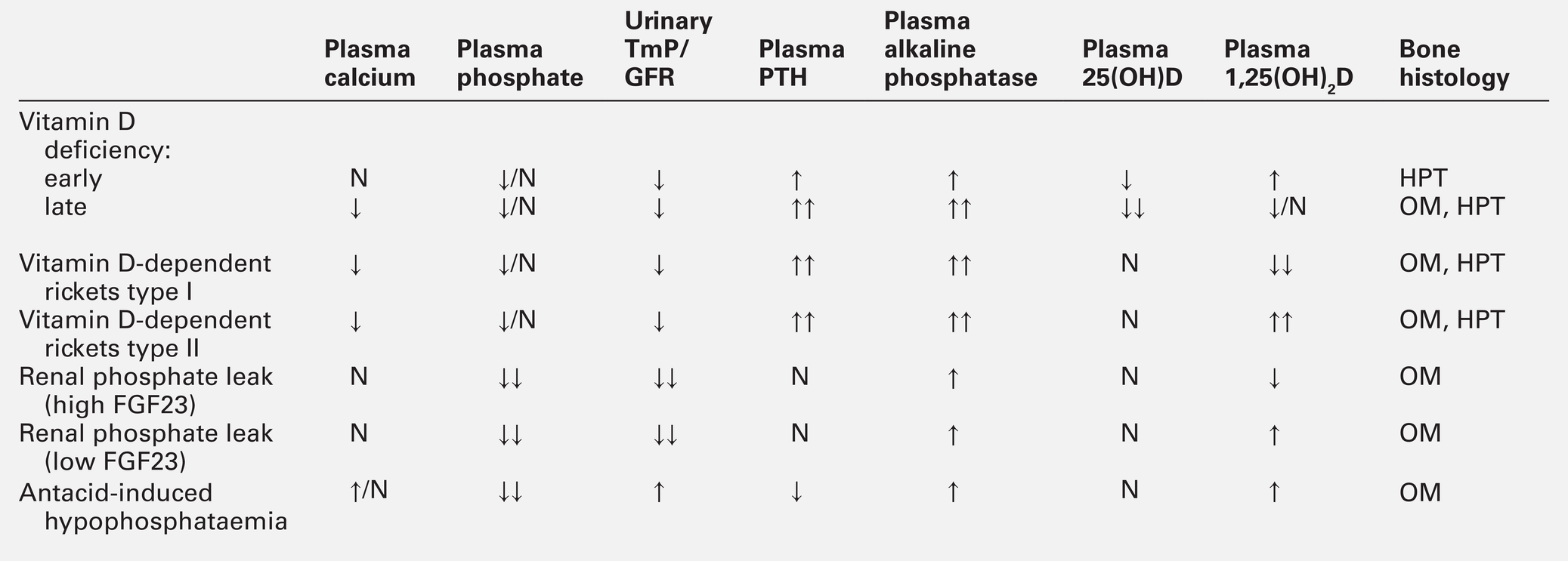
All bone turnover markers, apart from plasma osteocalcin, are increased in vitamin D deficiency osteomalacia. When assessing plasma phosphate and ALP concentrations, it is important to bear in mind the normal age-related ranges for these indices. The key diagnostic investigation in privational vitamin D deficiency is measurement of serum 25(OH)D concentration. Measurement of 1,25(OH)2D is indicated in infants with rickets who have no medical or social risk factors for privational vitamin D deficiency and who are hypocalcaemic despite normal plasma 25(OH)D concentrations.
Responses to therapy
Vitamin D deficiency should be treated with calciferol. Single high-dose treatment (300 000–600 000 units orally in an adult) may be more effective than low-dose long-term replacement and reduces problems of adherence to therapy. Patients with chronic conditions can be given weekly or monthly treatment with smaller doses, or annual prophylaxis in mid-autumn. There is a marked tendency for spontaneous improvement in the spring and summer. Attention needs also to be directed to underlying problems such as malabsorption. Vitamin D therapy is ineffective if the dietary calcium content is very low.
Effective therapy is followed by changes in the plasma biochemistry. Plasma calcium concentration rises and hyperparathyroidism is corrected. A marked rise in plasma phosphate concentration occurs after initiation of therapy and peaks at 3–5 weeks; thereafter plasma phosphate gradually declines, reaching the concentration which is normal for that individual after about 6–9 months. Plasma 1,25(OH)2D concentration is also supranormal during this period, but there is no clinical need to measure 1,25(OH)2D in this circumstance. Plasma ALP activity often flares during the first two to three weeks but then declines exponentially with a half-time of 50–100 days. A similar early increase in plasma concentrations of procollagen 1 extension peptides can also be seen. Osteocalcin concentrations also rise transiently, and peak about two weeks after treatment is started.
Vitamin D-dependent rickets type I is best treated with oral calcitriol (50 ng/kg per day) or alfacalcidol (100 ng/kg per day). Vitamin D-dependent rickets type II may respond to a combination of calcium supplementation and treatment with very high doses of calcitriol (60 μg/kg perday) or alfacalcidol (100 μg/kg per day).
Any patient being treated continuously with pharmacological doses of any form of vitamin D is at risk of vitamin D toxicity, and plasma calcium concentrations need to be monitored and dosages adjusted accordingly. Toxicity is particularly liable to occur as bone disease heals and therapeutic requirements fall.
Phosphopenic osteomalacia
In this group of conditions, the low plasma phosphate concentration is the main factor underlying the osteomalacia. Very rarely, this may result from the ingestion of phosphate-binding antacids, but most commonly it is due to renal tubular dysfunction and phosphaturia (manifest as a low TmP/GFR, see Table 31.3). The disorders can be classified into genetic and acquired forms and by whether or not there are other associated renal tubular defects (aminoaciduria, bicarbonate loss with systemic acidosis, glycosuria and hypokalaemia: the Fanconi syndrome). The most frequently encountered forms are listed in Box 31.5, and their aetiology discussed in Chapter 6.
The presentation of the inherited syndromes is usually in infancy with a clinical picture similar to that of rickets due to privational vitamin D deficiency. The most common form of inherited hypophosphataemic rickets, the X-linked type, is exceptional in that myopathy is absent and that marked ligamentous calcification can occur. Male children are more severely affected than females and short stature is a prominent feature.
Adult onset hypophosphataemic osteomalacia is usually associated with mesenchymal tumours (oncogenic osteomalacia) that are often difficult to identify because of their small size. These tumours secrete phosphatonins, such as FGF23, that act on the renal tubules to induce phosphaturia. If a tumour is solitary and can be removed surgically, then complete resolution of the osteomalacia occurs. Tumours can be identified by either magnetic resonance imaging (MRI) or scintiscanning using isotope-labelled somatostatin or sestamibi, but in a significant minority of cases even these sophisticated techniques may be inadequate. Plasma concentrations of FGF23 are usually increased, and fall after successful resection of the tumour.
Heavy metal nephropathy or light chain deposition in the renal tubules can cause an acquired form of the Fanconi syndrome with hypophosphataemic osteomalacia.
Laboratory investigation
The occurrence of rickets or osteomalacia in an individual without evidence of vitamin D deficiency should prompt consideration of phosphopenia as a cause. A key, and often overlooked, investigation is measurement of the plasma phosphate which, if low, should prompt evaluation of renal phosphate handling. The typical laboratory findings are listed in Table 31.3. The critical biochemical features that distinguish osteomalacia caused by hypophosphataemia due to a renal phosphate leak from that caused by vitamin D deficiency are:
• normal plasma 25(OH)D concentration
• low TmP/GFR, which persists after treatment of the bone disease
• absence of hyperparathyroidism.
Plasma FGF23 measurements are helpful in the diagnosis of oncogenic osteomalacia and some of the genetic disorders. Plasma calcitriol concentrations are usually low in hypophosphataemic osteomalacia that is the result of excess FGF23 activity, but high in conditions such as Dent disease or hereditary hypophosphataemic rickets with hypercalciuria, where the primary defect lies in renal tubular phosphate transporters.
Special tests are required for the diagnosis of the rare inherited and acquired metabolic disorders in which hypophosphataemic osteomalacia is part of a more generalized Fanconi syndrome.
Treatment
When hypophosphataemic osteomalacia occurs with inappropriately low concentrations of calcitriol (usually in the context of increased FGF23 activity) then it is best treated with phosphate supplements and 1α-hydroxylated vitamin D derivatives. Commercial preparations of sodium dihydrogen phosphate, are available but the precise form of phosphate is not important. Phosphate (total 30–120 mmol/day) has to be given frequently through the day to be effective, because of its rapid loss through the kidneys. Phosphate therapy alone induces hyperparathyroidism, so vitamin D therapy, which offsets this and enhances intestinal phosphate absorption, is also used. These patients are vitamin D resistant, so if calciferol (vitamin D2) is used, pharmacological doses are required and there is a danger of prolonged vitamin D toxicity. Alfacalcidol or calcitriol are therefore preferred to the parent compound. The histological responses to alfacalcidol or calcitriol also seem to be superior to those seen with calciferol. The therapy should aim to keep plasma phosphate around 1.0 mmol/L (measured 1–2 h after a dose), but to avoid hypercalciuria and hypercalcaemia. There is an increased risk of metastatic calcification, particularly in the kidney. The risk of nephrocalcinosis is related primarily to the amount of phosphate given, and its occurrence suggests that the dose of phosphate should be reduced (or stopped in the X-linked form when growth is complete). Healing of the bone disease is accompanied by a fall in plasma ALP activity.
Hereditary hypophosphataemic rickets with hypercalciuria is caused by mutations in the renal tubular phosphate transporter NaPi2c. FGF23 concentrations are not increased and plasma calcitriol is appropriately raised (given the hypophosphataemia). The bone disease can be cured with phosphate alone. In some conditions, the Fanconi syndrome responds to treatment of the underlying disorder (e.g. Wilson disease).
Antacid-induced hypophosphataemic osteomalacia is treated by withdrawal of antacids and the short-term prescription of phosphate supplementation.
Osteomalacia and acidosis
Longstanding metabolic acidosis is also associated with osteomalacia. The clinical situations in which this occurs most commonly are types 1 and 2 renal tubular acidosis and patients with bladder resection and ureterosigmoidostomy. The physiological basis of the osteomalacia is incompletely understood. At high [H+], the renal 1α-hydroxylase enzyme is inhibited, so the increase in plasma 1,25(OH)2D concentrations induced by phosphate depletion is less than expected. However, metabolic acidosis reduces TmP/GFR and, in the short term, stimulates 1,25(OH)2D synthesis. The mechanism of induction of osteomalacia may thus be similar to that seen in simple vitamin D deficiency. In keeping with this, plasma 25(OH)D concentrations have been reported to be low in many of the published cases of osteomalacia associated with ureterosigmoidostomy. In type 2 renal tubular acidosis, the hypocalcaemia and hypophosphataemia accompanying the Fanconi syndrome could also be important contributory factors. Treatment with alkali alone (sodium bicarbonate) can improve the osteomalacia, but in the limited studies published, has not produced complete healing.
Defective osteoblast function and osteomalacia
Osteomalacia can arise from either congenital or acquired abnormalities of osteoblast function. The acquired forms relate to aluminium or the use of high-dose etidronate in the treatment of Paget disease. In both these conditions, osteoblast numbers are reduced, plasma ALP activity is low and the osteomalacia is resistant to treatment with vitamin D.
Fluoride therapy, or endemic fluorosis, produces a mineralization defect despite abundant osteoblasts being present.
Hypophosphatasia
This is a rare familial disorder with autosomal recessive inheritance arising from mutations in the tissue non-specific ALP gene (ALPL). There are varying degrees of clinical severity, depending on the types of mutation and the residual activity of the enzyme. The clinical presentation can vary from a lethal perinatal form through to a relatively benign disorder presenting in adulthood (Table 31.4). Heterozygous carriers can be asymptomatic or have a mild form of the disease. In the childhood form, the diagnosis is made usually after the age of six months of age because of orthopaedic abnormalities that resemble rickets. Other features include premature loss of the deciduous teeth, premature closure of the cranial sutures and growth retardation.
TABLE 31.4
Clinical syndromes of hypophosphatasia
| Presentation | Clinical features |
| Perinatal | Stillbirth or neonatal death; absence of mineralized skeleton |
| Infantile | Diagnosed < 6 months; rickets, flail chest, hypercalcaemia/hypercalciuria, pyridoxine-responsive seizures |
| Childhood | Premature loss of primary teeth, rickets, hypercalcaemia/hypercalciuria |
| Adult | Middle age; recurrent stress fractures; pathological fractures |
| Odontohypophosphatasia | Premature loss of primary teeth only |
The characteristic biochemical findings are reduced plasma ALP activity (both total and bone ALP) with concomitant increases in endogenous substrates of the enzyme. These include plasma pyridoxal 5′-phosphate, inorganic pyrophosphate and phosphoethanolamine. An elevated plasma pyridoxal 5′-phosphate concentration is specific to hypophosphatasia (provided the patient is not taking vitamin B supplements), but urine phosphoethanolamine excretion can be modestly increased in a variety of other disorders. Severely affected children may have hypercalcaemia and hypercalciuria. Plasma phosphate concentrations also tend to be above average for age (due to an increase in the TmP/GFR), and PTH concentrations relatively low. Antenatal diagnosis of the severe form is possible by measurement of amniotic fluid ALP.
The bone shows osteomalacia with reduced osteoblast numbers. Treatment options are limited, but there are case reports of improvement of bone disease with parathyroid hormone treatment, and in severe infantile disease with bone marrow transplantation. The most promising treatment for severe hypophosphatasia is enzyme-replacement therapy, using a fusion protein (named asfotase alfa) that comprises the TNSALP ectodomain, the constant region of the human IgG1 Fc domain and a deca-aspartate motif.
CHRONIC KIDNEY DISEASE – MINERAL AND BONE DISORDER
Bone diseases complicating CKD emerged as major problems with the development of dialysis programmes, which permitted the prolonged survival of patients with established renal failure. The major aetiological factors are disturbances of the parathyroid hormone–calcitriol axis and skeletal retention of aluminium. Renal bone disease is not uniform in its features because the effects and interactions of these factors differ at various stages of its evolution. The disease can be modified substantially by a variety of treatments, from changes in dialysate composition, diet or drugs to renal transplantation. Chronic kidney disease – mineral and bone disorder (CKD-MBD), previously called renal osteodystrophy, can be regarded as a dynamic process that is affected by both patient characteristics and a number of pathophysiological and iatrogenic factors. These are illustrated in Figure 31.8.
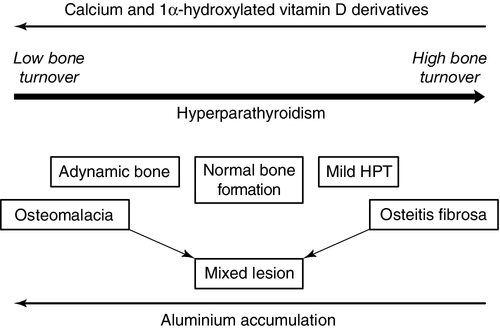
FIGURE 31.8 Chronic kidney disease mineral and bone disorder is a dynamic process. The main influences determining the nature of the bone histology are the degree of hyperparathyroidism (HPT) and the degree of aluminium accumulation in bone. Hyperparathyroidism can be suppressed to varying degrees by the manipulation of dietary and dialysate calcium and the use of 1α-hydroxylated vitamin D derivatives, or by parathyroidectomy. Aluminium accumulation also favours low bone turnover and, in significant quantities, can cause osteomalacia.
Aetiology
Parathyroid hormone–calcitriol–FGF23 axis
Mild hyperparathyroidism can be detectable when the glomerular filtration rate falls to < 50 mL/min. The hyperparathyroidism is secondary to a modest fall in plasma ionized calcium, resulting from a reduction in intestinal calcium absorption that, in turn, is due to a decline in the production of 1,25(OH)2D because of reduced renal 1α-hydroxylase activity. This arises initially not because of the loss of functioning nephrons, but because of phosphate retention. Plasma phosphate concentrations are not increased and may even be low because of secondary hyperparathyroidism, but there is saturation of the intracellular phosphate pool that ‘senses’ phosphate requirements. The high intracellular phosphate is responsible for the reduction in renal 1α-hydroxylase activity and therefore 1,25(OH)2D synthesis. Rising FGF23 concentrations in response to phosphate retention may mediate this early reduction in 1α-hydroxylase activity: increases in plasma FGF23 concentrations seem to precede the development of hyperparathyroidism. Reduction in the intake or the intestinal absorption of phosphate at this early stage reverses all these abnormalities: 1,25(OH)2D concentration in the blood rises, intestinal calcium absorption increases and hyperparathyroidism is suppressed.
As kidney disease progresses, the disturbances in the PTH-1,25(OH)2D axis become more marked. When the glomerular filtration rate falls to < 30 mL/min, hyperphosphataemia develops, as the number of functioning nephrons becomes too low to cope with the phosphate load, despite lowering of the TmP/GFR. Progressive nephron loss and metabolic acidosis further impair 1,25(OH)2D synthesis. Hypocalcaemia develops and hyperparathyroidism worsens, so by this stage, bone histology in most subjects will show signs of hyperparathyroidism that cannot be reversed by restricting the intake and absorption of phosphate.
Untreated, ~40% of patients reaching established renal failure will have significant parathyroid bone disease, as judged by the presence of marrow fibrosis on bone biopsy. If there is coincident vitamin D deficiency, then florid osteomalacia with hyperparathyroidism can arise.
The interactions between PTH and 1,25(OH)2D are complex. 1,25(OH)2D modifies PTH secretion indirectly through its actions on plasma ionized calcium, but it can also act directly on the parathyroids and inhibit the transcription of pre-pro-PTH mRNA. In established renal failure there are fewer receptors for 1,25(OH)2D in the nuclei of the parathyroid cells, as well as reduced circulating concentrations of 1,25(OH)2D. The loss of this direct effect upon the parathyroids is an important factor in the development of parathyroid hyperplasia. An additional effect of importance is the change in the ‘set point’ for PTH secretion. This is defined by the concentration of plasma ionized calcium that will suppress PTH secretion by 50% as calculated from calcium infusion experiments. Calcitriol deficiency shifts the set point to the right, that is, a higher ionized calcium has to be achieved before PTH secretion is suppressed.
Aluminium retention
Aluminium retention was recognized as a significant cause of skeletal disease in the late 1970s, when an unusual type of osteomalacia became endemic in haemodialysis units in certain geographical locations, such as Newcastle-upon-Tyne. Aluminium sulphate is added to water as a flocculent during the purification process in order to precipitate organic matter. The quantity of aluminium required for this process varies with environmental factors, so the concentration in tap water may range from 5–30 μmol/L. When the dialysate aluminium content is > 2 μmol/L, there is a substantial transfer from dialysate to plasma. Aluminium is normally excreted by the kidneys, so, in a patient with no renal function, it accumulates in various tissues, including the brain, bone and the parathyroids. The aluminium content of the dialysate can be reduced to < 2 μmol/L by reverse osmosis treatment and it is now standard to use dialysates prepared this way. The oral ingestion of aluminium hydroxide as a phosphate-binding agent can also contribute to aluminium loading.
In bone, aluminium is taken up at the bone–osteoid interface and has a marked toxic effect upon osteoblasts, inhibiting the rate of new matrix synthesis and, at higher doses, inhibiting mineralization. Aluminium can also accumulate in the parathyroids, and patients with severe aluminium-related disease often have low plasma PTH concentrations.
Clinical features
Hyperparathyroidism in the early stages of CKD-MBD is asymptomatic, but with advanced disease, bone pain and proximal myopathy are the predominant symptoms. Because of its insidious onset, it tends to be under-reported and patients may only notice that they have had it when adequate treatment is given and the pain relieved. Fractures are rare in parathyroid disease, but in adolescents and children, horrifying deformities can develop very rapidly as a result of the erosion and weakening of the metaphyses of the long bones. Hyperparathyroidism can impair the effectiveness of erythropoietin therapy.
The full-blown syndrome of aluminium-induced osteomalacia comprises generalized skeletal pain, frequent fractures and disabling proximal myopathy. With very high total body aluminium loads, hypochromic anaemia and dementia (‘dialysis dementia’) may occur. Because of increased awareness of aluminium, this presentation is uncommon nowadays. Milder degrees of aluminium-related bone disease and a mixed form of aluminium and parathyroid disease are recognized. These forms of disease give rise to fewer symptoms. Aluminium accumulates more readily in the bones of children and adolescents with CKD than in older patients, and also more readily in the bones of patients with CKD secondary to type 1 diabetes.
In recent years, changes in the therapy for CKD-MBD, and changes in dialysis techniques resulting in more active suppression of hyperparathyroidism, have meant that the more florid forms of CKD-MBD have become less common. A consequence of therapeutic measures to suppress hyperparathyroidism has been the emergence of a low turnover state called ‘adynamic bone disease’. The clinical sequelae are not clear, but there are suggestions that the risk of fracture is higher and that, in children, growth may be impaired. The strategy of maintaining higher plasma calcium concentrations to suppress hyperparathyroidism also increases the ECF [calcium] × [phosphate] product, and thus the risk of metastatic or vascular calcification. With an increasing number of older patients being taken onto dialysis programmes, osteoporosis with a high incidence of fractures has also emerged as a significant clinical issue.
Investigations
Biochemical measurements in serum are useful in assessing renal bone disease, but even with the help of radiographic assessments they cannot always provide an unequivocal account of events in bone. CKD-MBD is still, therefore, the commonest indication for bone biopsy, which remains the reference standard for other tests.
Calcium
Plasma calcium concentration (either total or ionized) is usually normal in early renal impairment but falls as renal function deteriorates (glomerular filtration rate < 30 mL/min). Dialysis partially corrects the hypocalcaemia. In long-term dialysis patients, hypercalcaemia may develop and can arise from one or more of a number of causes (Box 31.6). With the move away from using aluminium-based to calcium-based phosphate binders, the prevalence of hypercalcaemia in dialysis-treated patients has increased.
Phosphate
Plasma phosphate concentration is normal or reduced in early renal disease, but hyperphosphataemia occurs when renal impairment is advanced. In dialysis-treated patients, hyperphosphataemia is inevitable and it is almost impossible to maintain ‘normal’ plasma phosphate concentrations (0.8–1.4 mmol/L). Phosphate should be kept in the range 1.4–2.0 mmol/L, although even this can be difficult. Phosphate concentrations < 1.4 mmol/L can contribute to impaired bone mineralization, whereas at > 2.0 mmol/L there is a risk of metastatic calcification (see Chapter 6). Failure to maintain plasma phosphate below this concentration can be due to a variety of reasons (see Box 31.6).
Parathyroid hormone
After secretion of the intact molecule, PTH undergoes cleavage into amino-terminal and carboxy-terminal fragments. The latter are metabolized and excreted predominantly by the kidney, so that immunoassays that are not specific for the intact molecule or the amino terminal are liable to detect high concentrations of circulating PTH, whether or not glandular secretion is increased (see Chapter 6). It may be difficult to interpret a single measurement of plasma PTH in an individual. Nonetheless, in population studies the patients with the most severe parathyroid bone disease have the highest plasma PTH concentrations, irrespective of the assay used, and plasma PTH correlates well with histological indices of bone resorption (and formation). The newer assays for ‘intact’ PTH perform well in established renal failure but these assays also measure a large PTH fragment (probably PTH 7-84). New two-site chemiluminescence and immunoradiometric assays specific for PTH 1-84 are increasingly used in patients with renal impairment.
FGF23
By the time patients reach dialysis, plasma FGF23 concentrations are extremely elevated (commonly ~1000 times normal). This may reflect delayed clearance as well as increased production of FGF23, but almost all the circulating FGF23 seems to be biologically active. The prognostic value of FGF23 measurements in patients with CKD is beginning to be explored, but at present there is no indication for its routine measurement.
Alkaline phosphatase
The bone isoenzyme is neither dialysable nor filterable by the kidney, so its plasma concentration (and thus activity) is not modified by variations in renal function. Plasma alkaline phosphatase activity is increased when bone turnover is high and, in general, reflects the degree of hyperparathyroidism. If the plasma PTH concentration is clearly elevated, a high ALP is strongly predictive for high-turnover renal bone disease. Aluminium deposition affects osteoblast function so that plasma ALP activity is often normal, or only slightly above the reference range, in aluminium-related disease. The activity of ALP gives no indication of the severity of this type of bone disease.
Other markers of bone turnover
The markers of bone resorption based on urine measurements are obviously of limited use in CKD. Markers such as osteocalcin, hydroxyproline, pyridinoline, deoxypyridinoline and cross-linked C-telopeptide of type I collagen (ICTP) are wholly or partially cleared by the kidneys, so in CKD their plasma concentrations are greatly increased because of accumulation of inactive fragments. In population studies, correlations with histological indices of bone resorption or formation can be demonstrated, but in practice they are of little diagnostic value in individual patients. Concentrations of the formation markers P1NP and P1CP are not so affected by CKD, but correlate less well than ALP with histological indices. Tartrate-resistant acid phosphatase 5b is unaffected by CKD and correlates with histological measures of bone resorption. It is elevated in high-turnover bone disease.
Aluminium
Provided that care is taken in the collection and laboratory handling of specimens to avoid contamination, aluminium concentrations in plasma reflect the recent level of exposure to this element. Because of the rapid uptake of aluminium into tissues, plasma concentrations do not reliably indicate the total body burden. The desferrioxamine test was developed in an attempt to measure the total body aluminium load. Desferrioxamine mobilizes aluminium from bone and other tissues, so after its intravenous administration to dialysis patients, plasma aluminium concentrations increase (see Appendix 31.2 for protocol). The increment in plasma aluminium correlates reasonably (r = 0.6–0.7) with the degree of aluminium loading as assessed by bone biopsy, but the test has insufficient sensitivity and specificity to be used on its own. However, an increment of > 7.5 μmol/L in the presence of low plasma PTH is very suggestive of aluminium loading and an indication for bone biopsy to establish the extent of aluminium accumulation. A recommended protocol for surveillance for aluminium overload in dialysis patients is to measure serum aluminium every 4–6 months, seeking to maintain plasma concentrations at < 1.9 μmol/L.
Radiology, scintigraphy and densitometry
Radiology is useful in the diagnosis of parathyroid bone disease. Subperiosteal erosions of the phalanges and/or the distal ends of the clavicle are virtually pathognomonic. Other radiographic manifestations of hyperparathyroidism include osteosclerosis (commonly in the spine) and, more rarely, periosteal new bone formation. Aluminium-related disease has no specific X-ray findings, but pathological fractures and radiolucency of bones are typical if there is osteomalacia. Spinal bone density measurements are often increased in hyperparathyroid bone disease and reduced in aluminium-related disease, but these measurements are not of diagnostic value. The scintigraphic changes of hyperparathyroidism add little to the information from other diagnostic modalities.
Bone histology
Bone biopsy after double tetracycline-labelling, with the preparation of undecalcified sections for quantitative histomorphometry, remains an important investigation. Severe hyperparathyroidism can be demonstrated satisfactorily by radiographic and biochemical means, but mineralization defects, low bone turnover and aluminium loading are difficult to quantitate accurately without biopsy. Florid forms of aluminium-related disease are now uncommon, but a mixed form of bone disease with hyperparathyroidism and aluminium retention together, which can only be diagnosed by biopsy, is recognized. Bone histomorphometry permits classification of an individual into one of several diagnostic categories described according to the parameters of turnover (low, normal or high), mineralization (normal or abnormal) and volume (low, normal or high). CKD-MBD remains one of the commonest indications for bone biopsy (Appendix 31.1).
Treatment
Hyperparathyroidism
Vitamin D derivatives
Abnormalities of vitamin D metabolism are critical in the genesis of hyperparathyroidism, and vitamin D, its metabolites or derivatives are successful in reversing hyperparathyroidism at least in the short term. All forms of vitamin D are effective, but forms that require 1α-hydroxylation (such as calciferol or 25(OH)D) have to be given in pharmacological doses in order to achieve the desired effects. The 1α-hydroxylated forms, 1,25(OH)2D, alfacalcidol (1αOHD3) or doxercalciferol (1αOHD2) are effective at much lower doses and are preferred. Their main advantages are their very quick onset and cessation of action: if vitamin D toxicity occurs with these compounds, it reverses rapidly. Although this is a distinct advantage, toxicity can also arise abruptly. Intestinal calcium and phosphate absorption are increased, the [calcium] × [phosphate] product in plasma increases and, with it, the risk of metastatic and vascular calcification. This can accelerate the decline in renal function in patients with moderate CKD who have not yet reached dialysis and increases the risk of cardiovascular disease.
How well hyperparathyroidism is suppressed depends mainly on the increment in plasma calcium that can be safely attained. Patients who have high or high-normal plasma calcium concentrations at the outset of therapy are therefore likely to tolerate only low dosages. 1,25(OH)2D may be given intravenously (usually thrice weekly). The object is to obtain transiently high plasma 1,25(OH)2D concentrations and to maximize the directly suppressive effects of 1,25(OH)2D upon PTH synthesis. However, the degree of suppression of hyperparathyroidism obtained by this method remains closely correlated with the increment in plasma calcium. In patients with established hyperparathyroidism, successful treatment with vitamin D derivatives is accompanied by reductions in plasma PTH concentration and ALP activity. Alkaline phosphatase may rise in the first few weeks of therapy, but then declines exponentially with a half-time of around 50 days. As healing takes place, vitamin D toxicity is likely to occur, so dosage reductions are required. All patients taking vitamin D derivatives require monitoring of plasma calcium, phosphate and ALP.
There is increasing use of an emerging group of bioactive vitamin D analogues with supposedly less marked calcaemic activity than calcitriol (e.g. paricalcitol and maxacalcitol). It is hoped that these agents may allow more effective control of parathyroid gland hyperplasia with less hypercalcaemia, although as yet there is little evidence that they are superior to calcitriol, alfacalcidol or doxercalciferol.
Phosphate metabolism
Control of plasma phosphate is important in slowing the development of hyperparathyroidism and is approached by dietary restriction, the use of phosphate-binding agents and adequate dialysis. The concentration of calcium in the dialysate can also be manipulated to influence hyperparathyroidism.
Parathyroidectomy
Parathyroidectomy is very effective in patients with severe hyperparathyroidism and produces a quicker and more complete resolution of parathyroid bone disease than can be achieved with alfacalcidol or calcitriol. With the abrupt cessation of PTH-driven bone resorption after surgery, there follows a period when bone formation remains very rapid, before it, too, slows to match the new, lower resorption rate. During this period, calcium and phosphate are taken up very rapidly from the blood into bone and profound hypocalcaemia (and hypophosphataemia) can occur – the ‘hungry bone syndrome’. This can be so severe as to cause hypocalcaemic fits. Patients undergoing parathyroidectomy are therefore treated pre- and postoperatively with 1α-hydroxylated forms of vitamin D and may require intravenous or oral calcium. After parathyroidectomy, plasma ALP activity may show an early rise but then suppresses rapidly, with a half-time of around 23 days, as bone disease heals. The more rapid suppression of ALP after parathyroidectomy than after alfacalcidol or calcitriol reflects the more rapid reduction in PTH.
Hyperparathyroidism can recur after parathyroidectomy and there is continuing debate over the choice of procedure – subtotal or total parathyroidectomy – and, in the subtotal procedure, about the size of the remnant and whether to leave it in situ or to transplant it to the sternomastoid or forearm muscles. One concern about the rapid suppression of parathyroid hormone, whether by parathyroidectomy or by the use of 1α-hydroxylated vitamin D compounds, is the ease with which aluminium is retained in bone when turnover is very slow. The aluminium–osteomalacia syndrome can certainly be precipitated by parathyroidectomy, hence the importance of making sure the bone is free of aluminium before undertaking this procedure.
Calcimimetic agents
An important development in the treatment of hyperparathyroidism secondary to CKD has been the use of cinacalcet, an allosteric agonist of the ionized calcium-sensing receptor that is expressed on parathyroid cells. With cinacalcet treatment, reductions of ~40% in PTH concentrations are reached after about three months of treatment, and are maintained for at least two years. Reductions in plasma calcium and phosphate of 6–8% are seen and the [calcium] × [phosphate] product decreases by ~15%.
Aluminium toxicity
Aluminium toxicity is treated by withdrawing all parenteral sources of aluminium and stopping aluminium-containing phosphate binders. The chelating agent desferrioxamine effectively removes aluminium from bone and other tissues. In order to minimize the risk of desferrioxamine-related cerebral, auditory and visual side-effects, and siderophore-mediated opportunistic infections, the chelator should be used at low doses (5 mg/kg) and administered only once weekly – typically for 4–6 months. In patients not requiring dialysis, most of the aluminium–desferrioxamine complex is excreted in the urine and can be monitored by urinary aluminium measurements. Prolonged desferrioxamine treatment can result in iron depletion.
Bone disease after renal transplantation
In the first two weeks following successful renal transplantation, plasma FGF23 concentrations fall very rapidly and 1,25(OH)2D concentrations rise. Hyperparathyroidism resolves slowly after transplantation and may be expressed as post-transplant hypercalcaemia. Hyperparathyroidism, persistent FGF23 elevation and steroid treatment all contribute to hypophosphataemia in transplant recipients (see Chapter 6). Bone resorption, stimulated by hyperparathyroidism, may be high after transplantation, but corticosteroids, given for immunosuppression, suppress bone formation. Transplant recipients may thus lose bone and become osteoporotic. The suppressive effect of steroids on osteoblasts means that the markers of osteoblast function, ALP activity and osteocalcin are not reliable guides to bone resorption in these circumstances.
BONE DISEASE IN PRIMARY HYPERPARATHYROIDISM
Bone disease was one of the complications of primary hyperparathyroidism that led to the recognition of this disorder in the 1920s, but in Western countries florid parathyroid bone disease is now uncommon. The availability of plasma calcium measurements on modern biochemical analysers has identified a large population with asymptomatic hyperparathyroidism, and early surgery in symptomatic patients probably accounts for the infrequency of severe skeletal manifestations.
Clinical, biochemical and histological features
Since bone is one of the target tissues for PTH, it is not surprising that the bones of almost all patients with hyperparathyroidism show some histological evidence of increased parathyroid activity. In asymptomatic or mildly affected individuals, this amounts only to increases in the extent of active formation and resorption surfaces. This is reflected in modest increases in biochemical markers of bone turnover. Mild primary hyperparathyroidism causes a preferential loss of cortical bone, and may be associated with an increased fracture risk. Both antiresorptive therapy and surgical correction of the disease prevent progressive bone loss.
Much less commonly, patients with primary hyperparathyroidism manifest overt parathyroid bone disease (osteitis fibrosa) characterized by marked increases in osteoclastic bone resorption and increases in bone formation rate, with a resulting increase in osteoid surface extent and marrow fibrosis. Patients experience generalized bone pain, often worse when standing, bone tenderness and proximal myopathy. Radiographic signs can be helpful, subperiosteal erosions being pathognomonic of parathyroid bone disease. Spinal osteosclerosis can occur in primary hyperparathyroidism, but is seen less frequently than in uraemic hyperparathyroidism. Cystic lesions filled with osteoclasts, and marrow fibrosis (‘brown tumours’), are a feature of bone disease in severe primary hyperparathyroidism and can be sites of pathological fracture.
The presence of bone abnormalities tends to be associated with more aggressive disease with a shorter duration of symptoms, higher plasma concentrations of calcium and PTH and larger, more rapidly growing brown tumours. Occasionally primary hyperparathyroidism can enter an accelerated phase with a marked increase in the rate of bone resorption and disequilibrium hypercalcaemia (acute parathyroid crisis).
Vitamin D insufficiency occurs commonly in patients with primary hyperparathyroidism, and is probably associated with larger parathyroid adenomas and higher rates of bone turnover. Accelerated hepatic metabolism of 25(OH)D may contribute to the lower vitamin D concentrations in this situation. Maintenance of vitamin D sufficiency may restrain the progression of the parathyroid disease. In patients with mild hyperparathyroidism and low plasma 25(OH)D, supplementation with vitamin D does not usually exacerbate the hypercalcaemia and may, indeed, lower PTH and bone turnover.
Treatment
Most patients with mild primary hyperparathyroidism do not require specific therapy. All patients with severe primary hyperparathyroidism or osteitis fibrosa require parathyroidectomy to correct the disease, but these patients are at risk of developing the ‘hungry bone syndrome’ with postoperative hypocalcaemia and hypophosphataemia. This can be ameliorated by treatment for 1–2 weeks prior to surgery with low doses of 1α-hydroxylated vitamin D derivatives. Following successful surgery, plasma calcium concentrations fall, but there can be secondary hyperparathyroidism as the remaining normal parathyroid glands respond to the fall in plasma calcium. The changes in plasma calcium following surgery are therefore a better immediate guide to the success of the procedure than changes in plasma PTH concentration. Nonetheless, intraoperative measurement of PTH can provide useful information as to whether the abnormal parathyroid tissue has been resected – a fall of > 50% in the PTH concentration within 20 min of parathyroid gland removal is highly predictive of long-term surgical cure. Plasma ALP may flare in the 1–2 weeks following surgery, and then falls in a similar manner to that seen after parathyroidectomy in uraemic hyperparathyroidism. The biochemical markers of bone resorption fall early after successful surgery.
PAGET DISEASE OF BONE
Epidemiology
Although Paget disease of bone is, next to osteoporosis, the metabolic bone disorder most frequently encountered in general medical practice, it is in many ways quite mysterious. The disease is characterized by extreme metabolic disturbance of one (monostotic) or more (polyostotic) bones in the skeleton, while the uninvolved bones are quite normal. This patchy, asymmetrical distribution of disease within the skeleton is unexplained, as is its striking geographical distribution. It is most common in people of European origin living in temperate zones and is common in the UK, the USA, Australia, New Zealand and western Europe. It has been described in African-Caribbean peoples living in the UK and in African-Americans, but it is rare in groups native to areas where it is common among whites, such as native North Americans, Australian aborigines and the Maori of New Zealand. Even in countries where the disease is widespread, there are well-documented variations in prevalence. For example, in the UK, the prevalence in parts of Lancashire is two to three times greater than elsewhere. Families with many members affected, and a dominant pattern of inheritance, are not uncommon.
Aetiology
The cause of Paget disease is unknown, but both genetic and environmental factors appear to be important. In about a third of patients with a positive family history (and a much lower proportion of patients without a positive family history), Paget disease is associated with mutations in the gene SQSTM1. This gene encodes the p62 protein, which forms part of the scaffold linking the RANK receptor on osteoclasts to the nuclear transcription factor NF-κB. The majority of Paget disease-associated mutations identified to date (currently numbering ~30) have been in the ubiquitin-binding domain of the molecule. Patients with SQSTM1 mutations tend to have disease that is more extensive, more symptomatic and is of earlier onset than in patients without such mutations. This is particularly the case with mutations that result in truncation of the p62 protein. The commonest SQSTM1 mutation is P392L which is found all around the world. Haplotype evidence suggests this is a founder mutation distributed by European colonists. The rare dominantly inherited syndrome of inclusion-body myopathy, Paget disease and frontotemporal dementia (OMIM 605382) is associated with mutations in the VCP gene, encoding the valosin-containing protein. This protein is also a component of the scaffold linking RANK to NF-κB.
Genome-wide association studies of patients negative for SQSTM1 mutations have indicated that polymorphisms at a number of genetic loci are statistically associated with Paget disease. Several of the candidate genes encode proteins known to be important in osteoclast physiology, such as M-CSF, a cytokine essential for osteoclast differentiation, DC-STAMP, a cell surface protein essential for the formation of multinucleated osteoclasts and RANK, a receptor essential for osteoclast differentiation and bone resorption.
Recent epidemiologic data indicate that in the past 40 years, there has been a significant reduction in the prevalence of Paget disease in high-prevalence regions such as the UK and New Zealand, and that extensive polyostotic involvement has become less common. These observations suggest that in addition to genetic influences an important environmental factor is involved in the causation of Paget disease. Research has concentrated on a potential viral cause as the environmental agent. Both measles and canine distemper virus have been suggested as culprits, but the laboratory evidence is inconclusive.
Natural history
Many people have the disease without symptoms, as shown by the frequent incidental finding of either an isolated elevation in plasma ALP activity in ‘routine’ blood tests or pagetic changes in bone when radiographs are taken for other indications. Radiographic surveys have shown that in endemic areas of the world, the prevalence increases with age, and suggest that disease before the age of 40 is rare. It appears to arise more or less simultaneously at several skeletal sites. In some patients, distinct phases can be discerned. Bone lysis and progression of lytic lesions may predominate, particularly in the skull and long bones: this is presumed to be early disease. In the later stages, in some patients, osteoclast activity dies out, the bones become intensely sclerotic and ultimately osteoblast activity declines too, leaving abnormal but metabolically quiescent bone. What proportion of patients go through this cycle and how long it takes are not known. The majority of patients have mixed disease with similar increases in both resorption and formation of bone. Observations of plasma ALP activity in untreated subjects show an unpredictable variation from year to year, with an overall tendency to rise with time until it becomes stabilized within a range characteristic for an individual.
Pathology
Paget disease is characterized by massively increased turnover in affected bones. It is thought to be primarily a disorder of osteoclasts. The number and size of the osteoclasts are greatly increased and they contain numerous large nuclei. Resorption surfaces are increased in extent. The coupling of bone resorption to formation is, however, more or less maintained, and many of the histological features of Paget disease relate to the massive increase in bone formation. Osteoblast numbers, the extent of bone-forming surfaces and the rate of new bone formation are all increased. At very high rates of bone formation, collagen fibres are not laid down in the usual orderly lamellar pattern, but more randomly, resulting in new bone of abnormal structure (visible as a mosaic pattern under polarized light), known as ‘woven bone’. This bone has a larger volume than lamellar bone, hence pagetic bone shows an increase in trabecular bone volume. Collagen fibres are also secreted by osteoblasts into the marrow space, giving rise to marrow fibrosis.
Clinical features
Bone pain is a common feature. Pagetic bone pain is characteristically chronic and persistent rather than episodic, worse at rest and relieved by movement. It responds rapidly to antipagetic therapy. Because pagetic bone is soft and readily deforms, secondary osteoarthritis is very common. This is particularly so in the hip joints (from pelvic or femoral involvement). Pathological fractures of long bones (particularly in areas where lysis dominates) are common. Pagetic bone is very vascular and the high blood flow causes an elevation of skin temperature over the affected bone. In severe polyostotic disease in the elderly, the quantity of blood shunted through bone may precipitate heart failure. Spinal disease can cause a reversible myelopathy through a vascular steal phenomenon. Involvement of the otic capsule causes deafness. The most feared complication is the development of osteosarcoma, but fortunately this is rare. Osteosarcoma appears to arise from somatic mutations.
Investigations
Radiology
The radiographic appearances of bone reflect the histological events. There are areas of focal bone resorption (lytic lesions). In long bones and in the skull, flare-shaped lytic fronts can progress through bone: typical rates of progression are 0.8–1.2 cm/year. The increased bone formation is reflected by loss of the normal trabecular pattern, sclerosis and an overall enlargement of bone dimensions. Deformity, arthritis and fracture are common secondary events visible on radiographs. Incomplete cortical ‘fissure’ fractures of the convex surface of deformed long bones often precede completed fractures. Isotope scanning is the best method of determining the extent of the disease.
Biochemical tests
Bone turnover markers
Biochemical tests are very useful in monitoring the progress of Paget disease and its response to therapy. The markers in use are not specific to the disease, but are simply markers of increased rates of bone formation and resorption. The concentrations of plasma ALP and other markers reflect both the extent and the activity of the disease. Hence the greatest values are seen in patients with widespread active polyostotic disease. In such patients, the turnover markers are elevated to a greater extent than in any other metabolic bone disorder. In patients with monostotic disease, bone turnover markers need to be interpreted with caution; although increased for that individual, they may not be outside the reference range.
All bone turnover markers are increased proportionately apart from osteocalcin, which is not increased to the extent expected from ALP measurements. Because of its rapid turnover, much of pagetic bone is ‘young’ and thus has a greater proportion of the linear, non-isomerized α-C-telopeptide collagen cross-links, and a smaller proportion of the β-isomerized C-telopeptides typical of older bone. This is reflected in the results of assays for α-CTX and β-CTX – patients with Paget disease having a greater than usual proportion of the former.
Alternative methods of monitoring disease activity can be employed. Over the lower limb and the forearm, skin temperature is a useful guide to disease activity, provided the disease is unilateral so that there is a normal limb for comparison. The skin temperature at the same point on opposite limbs in normal subjects does not differ by more than 0.5°C. The skin overlying a tibia affected by lytic Paget disease can be up to 4°C warmer than the unaffected limb opposite. Alternatively, the uptake of isotope on a bone scintiscan gives a measure of disease activity.
Since bone resorption and formation are closely linked, there is, not surprisingly, a strong correlation between plasma ALP activity and bone resorption markers in the untreated state. It is arguable as to whether, in the presence of symptoms, signs and radiographic evidence of Paget disease, it is necessary for diagnostic purposes to measure resorption markers if the plasma ALP is elevated and other liver-derived enzymes are normal. The smaller coefficient of variation in ALP measurements at times when disease activity is stable (~10%), compared with that of urine resorption markers, is an additional argument in favour of using the former as the main index of disease activity. A potential advantage of measuring resorption markers is the earlier detection of relapse after treatment, but the greater imprecision of the resorption markers means that in practice there is little advantage over measuring formation markers.
In response to treatment with potent bisphosphonates, the bone markers that appear to perform best (i.e. show the greatest changes) are total ALP, bone ALP and P1NP. The latter probably performs best in terms of pre-treatment values being clearly elevated in disease of limited extent, and showing the greatest changes with treatment and on relapse of disease. Using the same criteria, the best performing resorption marker is the urine N-telopeptide/creatinine ratio.
Plasma and urinary calcium
Plasma and urinary concentrations of calcium (and phosphate) are usually normal in Paget disease, but patients with very extensive and active disease are prone to develop hypercalciuria and even hypercalcaemia if immobilized. This results from increased bone resorption and a relative fall in bone formation, detectable by an increase in bone resorption markers and a fall in ALP activity.
Responses to treatment
There is no cure for Paget disease, but the metabolic activity of the affected bone can be reduced by the use of inhibitors of bone resorption – nowadays bisphosphonates are used almost exclusively. The main indication for treatment is bone pain. In practice, it can be difficult to distinguish pagetic from arthritic pain, so a trial of therapy may be given. Prompt (and prolonged) relief after treatment suggests that Paget disease was the main cause of pain. Myelopathy secondary to vascular steal is an indication for immediate treatment. Bisphosphonates are commonly give in preparation for joint replacement surgery, where one of the bones concerned is involved with Paget disease, in the hope that this will reduce the vascularity of the bone and make the surgery easier. There is, as yet, no clear evidence that aggressive treatment to reduce bone turnover to normal prevents fracture or the development of late complications such as deafness or arthritis, though it is probable (comparing radiographic series from the pre-bisphosphonate era and now) that deformity can be prevented.
In response to bisphosphonate treatment, there is an early fall in urine resorption markers reflecting the reduction in osteoclastic activity. This rapid fall is complete by four days after intravenous treatment. Bone formation falls secondarily to this, so that the decline in plasma ALP lags behind that of bone resorption markers by several months. Ultimately, a new steady state is reached where resorption and formation are matched (Fig. 31.9). When the treatment is with oral bisphosphonates, a similar pattern is seen. Both bone resorption and bone formation markers fall in a monoexponential manner with a half-time of around two weeks, but the response of the formation markers lags behind by two to three weeks.
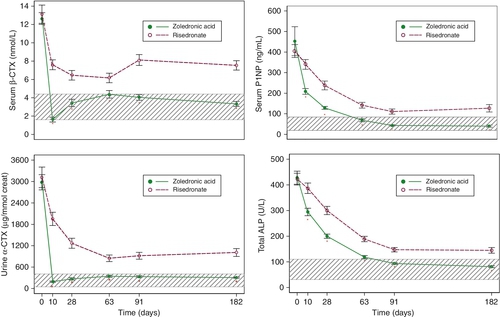
FIGURE 31.9 The treatment of Paget disease. Effects of a single infusion of zoledronate 5 mg or oral risedronate 30 mg/day for two months on bone turnover in patients with Paget disease. The very rapid drop in bone resorption markers (serum β-CTX and urine α-CTX) is followed by a slower fall in the formation markers (serum P1NP and ALP). Zoledronate produces a more profound and sustained suppression of bone turnover. Hatched areas indicate normal reference intervals. (Based on data from Reid et al. 2005 in N Engl J Med 353: 898–908.)
During the period early after initiation of therapy where resorption has fallen but formation remains high, there is a considerable uptake of calcium and phosphate into bone, thus some degree of secondary hyperparathyroidism is usual. Following intravenous bisphosphonates, this is evident some 2–4 weeks after the infusions but it is not of any great consequence in most patients. Plasma osteocalcin concentrations may actually increase by 30–40% within the first two weeks of treatment, probably because the secondary hyperparathyroidism stimulates an increase in 1,25(OH)2D synthesis and this, in turn, stimulates osteocalcin production. Osteocalcin concentrations do subsequently decline with the fall in bone formation. Because of these disparate responses and the poor correlation with ALP in the untreated state, plasma osteocalcin measurements are not used in the management of Paget disease.
Treatment with calcitonin produces reductions in both plasma ALP activity and urinary hydroxyproline excretion to an average of 50% of the pretreatment values, within six months of starting treatment. Continued use of calcitonin produces no further improvement – the ‘plateau’ phenomenon – and discontinuation of treatment is followed by prompt relapse.
The bisphosphonates have made the use of calcitonin and other treatments obsolete, since they are far more effective and have fewer side-effects. A large number of bisphosphonates have been used. The more recently developed, such as zoledronate, are substantially more potent and have a long duration of action. Patients whose turnover markers suppress to the lowest values are likely to remain in biochemical ‘remission’ the longest. For example, if plasma P1NP is suppressed to < 40 μg/L six months after treatment with intravenous zoledronate, then > 90% of patients will remain in biochemical remission 6 years later. If P1NP suppresses only to the 60–80 μg/L range, then only ~60% will remain in remission. If treatment is discontinued, all patients seem ultimately to relapse – although in patients with limited disease, treatment with potent bisphosphonates can produce remissions of ten years or more.
Relapse is first detectable biochemically by an increase in markers of bone resorption. Retreatment when a relapse has occurred produces a further suppression of bone turnover markers, but it is proportionately smaller, so the previous nadir is reached but concentrations are not usually reduced much further. The bisphosphonates, though effective orally, are poorly absorbed from the gut, so short intravenous regimens are frequently employed. The more potent amino-bisphosphonates (such as pamidronate, ibandronate and zoledronate), when given intravenously for the first time, often provoke an acute febrile reaction.
Etidronate, the first bisphosphonate to be developed, has a toxic effect upon osteoblasts and impairs mineralization when given in high doses (> 10 mg/kg per day) for > 3 months. Etidronate also has a much greater effect on the TmP/GFR than the other bisphosphonates, so that hyperphosphataemia is not uncommon during treatment. It is little used nowadays as the newer bisphosphonates are significantly more effective and longer lasting.
BONE TURNOVER AND BONE DISEASE IN CHILDREN
In infants, children and adolescents, bone turnover, as assessed by biochemical markers, is at much higher levels than in healthy adults. In general, the concentrations reflect growth velocity. Figure 31.3 illustrates age-related changes in ALP and osteocalcin: the mean values at peak growth velocity are 3–5-fold higher than mean adult values. For P1NP, mean values at peak growth velocity are 6–10-fold higher than mean adult values. Turnover markers cannot accurately predict the accrual of bone during growth. The interpretation of bone marker data in individual children is difficult as the growth rate, nutritional status, sex, age, pubertal stage and renal function of the child all need to be taken into account.
There is relatively little information about very early life, but in healthy infants the urine marker of bone resorption NTX is low at birth, increases dramatically in the first ten days of life, remains very high for the next three months of life and then decreases to values similar to those at birth and one year of age.
Because new bone formed during growth is necessarily immature, the α-CTX isomer of the C-telopeptide predominates when this index is measured. Abnormalities in bone markers can be seen in various acquired and genetic disorders that affect bone directly or indirectly, and these are occasionally of diagnostic value (Tables 31.5 and 31.6).
TABLE 31.5
Bone turnover abnormalities in paediatric bone diseases
| Disorder | Abnormalities |
| Prematurity | Bone ALP and osteocalcin relatively reduced, but increase after several weeks |
| Malabsorption/malnutrition | All turnover markers relatively reduced |
| Growth hormone deficiency | Relative reduction in formation markers – which increase with growth hormone treatment |
| Glucocorticoid use | Relative reduction in both formation and resorption markers |
| Vitamin D deficiency | Increased ALP and P1NP, but osteocalcin relatively low: resorption markers increased |
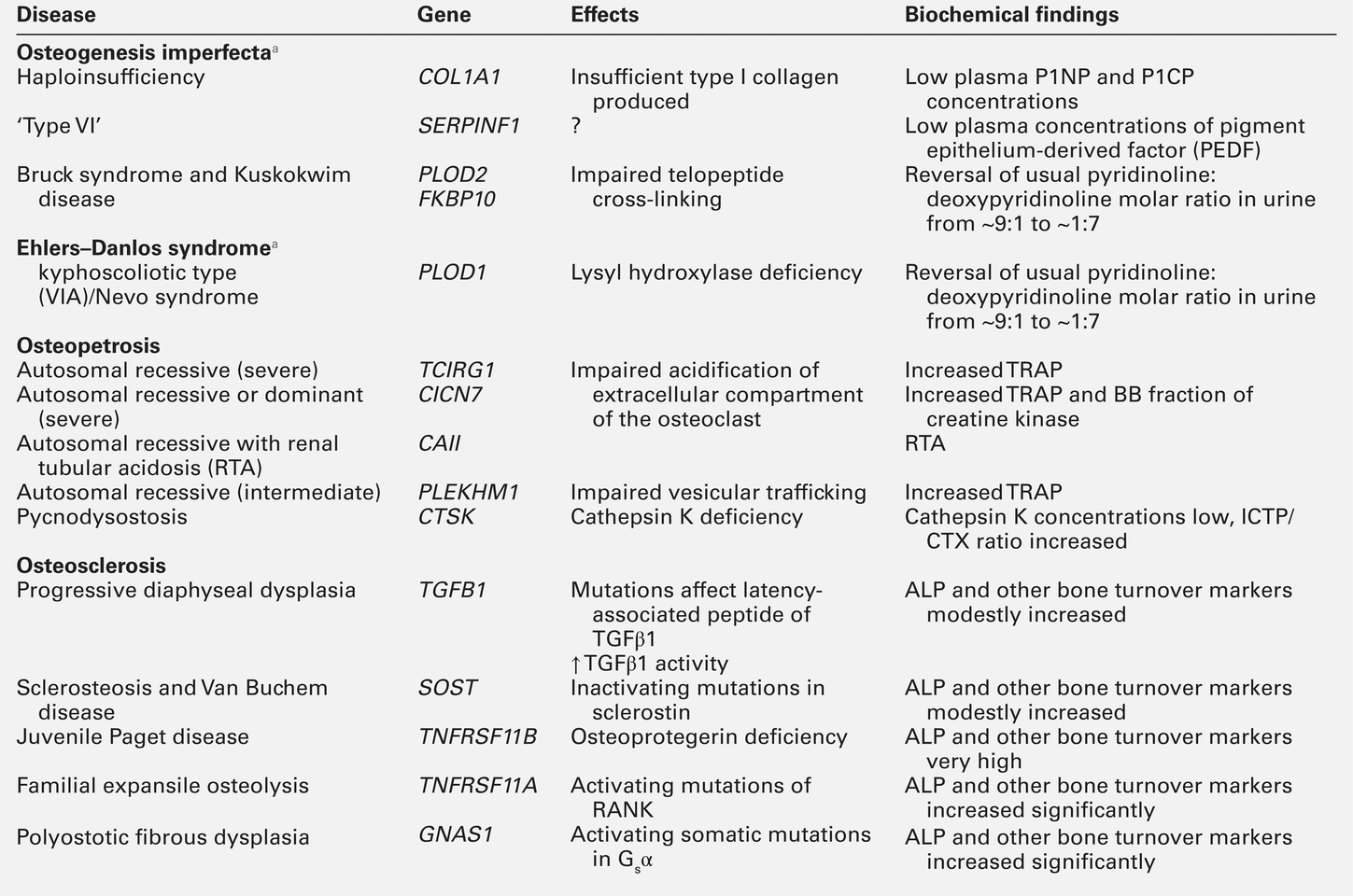
GENETIC BONE DISEASES
There are numerous genetic bone diseases. The discussion below refers only to those conditions not covered above, for which there is a clear role for the clinical biochemistry laboratory in diagnosis (see Table 31.6).
Osteogenesis imperfecta
The term osteogenesis imperfecta (OI) encompasses a group of genetic disorders usually defined by recurrent fractures, low bone mass and skeletal fragility. Most cases are dominantly inherited and result from mutations in the genes encoding type I collagen (COL1A1 and COL1A2). The condition is not synonymous with type I collagen abnormalities, as a number of genetic disorders affecting type I collagen or its post-translational modification that have skeletal phenotypes are not usually included in the OI rubric. These include Caffey disease, and the type VI and VII variants of the Ehlers–Danlos syndrome. In addition, some genetic disorders of skeletal fragility included in classifications of OI are the consequence of mutations in key osteoblast genes (LRP5, WNT1 and SP7) that code for proteins concerned with matrix homeostasis, and are not directly related to collagen metabolism and matrix structure.
More than 200 mutations in the collagen genes COL1A1 and COL1A2 that cause OI have been identified. All the mutations result in decreased synthesis and secretion of normal type I procollagen. When no abnormal procollagen is synthesized, the phenotype is generally mild: this is the commonest form of the disease, arising where a mutation in one of the COL1A1 genes produces a null allele (haploinsufficiency). When abnormal molecules are synthesized in addition to normal type I collagen, the phenotype can range from very mild to lethal, depending on the nature and location of the mutation.
The diagnosis of OI is predominantly based on clinical assessment (including family history) and imaging. Biochemical tests are helpful with excluding differential diagnoses, but with a few exceptions are not generally specific. Ultrasound and radiographic examinations are useful for intrauterine diagnosis of severely affected fetuses. The quantity and some structural characteristics of type I collagen produced by cultured skin fibroblasts can be assessed in vitro.
The usual biochemical indices of metabolic bone disease are normal in osteogenesis imperfecta, though resorption markers are often increased when the patient is immobilized. In patients with haploinsufficiency, the bone formation markers plasma procollagen C-terminal peptide and procollagen N-terminal peptide (which are derived from type 1 collagen) are often relatively low compared to the other formation markers such as bone ALP.
The genetic bases of a number of recessively-inherited OI variants have been identified. The majority of the genes involved code for proteins that regulate the post-translational modification of type I collagen, and there are some instances where specific biochemical tests can help. In patients with Bruck syndrome (OI with contractures), where there is defective cross-linking of collagen (owing to mutations in FKBP10 or PLOD2) the usual ratio of pyridinoline to deoxypyridinoline in urine is reversed. Another recessive OI variant results from mutations in SERPINF1 that encodes pigment epithelium-derived factor (PEDF), a secreted glycoprotein of as yet uncertain function in bone. Pigment epithelium-derived factor can be measured in normal serum and is undetectable in the serum of patients with this OI variant.
Bisphosphonate treatment is now widely used in children with more severe forms of osteogenesis imperfecta. During growth, osteoclasts remove bone from the endosteal surface of the diaphysis and the primary spongiosa in the metaphysis, as part of the modelling process. Bisphosphonates inhibit this osteoclastic resorption, with the end result that cortices are thicker and trabecular number is increased. Bone pain is reduced, the bones are stronger and fracture less frequently. The benefit of bisphosphonates in children and adults with milder forms of osteogenesis imperfecta is less certain.
High bone mass
High bone mass disorders are classified according to whether there is excessive bone formation (osteosclerosis) or a failure of bone resorption (osteopetrosis).
Osteopetrosis
Osteopetrosis is further categorized according to whether there are normal or increased numbers of osteoclasts: osteoclast-rich (the majority of cases) or osteoclast-poor. Osteoclast-rich osteopetrosis arises because of mutations in a number of genes encoding proteins important in generating and excreting hydrogen ions onto the bone surface sealed beneath the osteoclast. The genes usually involved are TCIRG1, CICN7, PLEKHM1 or CAII. Osteoclasts are typically abundant, but their capacity to resorb bone is impaired. Severe osteopetrosis typically becomes manifest during infancy. The bone marrow space is occluded so the infant develops leukoerythroblastic anaemia with bruising and bleeding and extramedullary haematopoiesis. Growth is poor and blindness and deafness occur because of cranial nerve entrapment. The radiographic features are diagnostic. The plasma calcium concentration may vary and seems to reflect the dietary calcium intake. Restriction of dietary calcium may precipitate tetany, presumably because of defective osteoclastic bone resorption. Urinary calcium excretion is often low. Plasma total and tartrate-resistant acid phosphatase activities are increased, although ALP is normal. This condition can be cured by successful bone marrow transplantation, which permits colonization of the host marrow by normal osteoclast precursors.
Carbonic anhydrase II deficiency, resulting from mutation in the CAII gene, is a congenital disorder comprising osteopetrosis, renal tubular acidosis and cerebral calcification. Although haematopoiesis is not affected, cranial nerve entrapment may be a feature. The laboratory findings include a metabolic acidosis and greatly reduced activity of the enzyme carbonic anhydrase II in erythrocytes. This enzyme is widely distributed and is found, among other places, in normal osteoclasts.
The osteoclast-poor forms arise from inactivating mutations in TNSF11 (encoding RANKL) or TNFRSF11A (encoding RANK). Children with the latter also have hypogammaglobulinaemia and commonly develop severe but transient hypercalcaemia after bone marrow transplantation.
Progressive diaphyseal dysplasia
This osteosclerotic condition results from mutations in the gene encoding TGFβ. It is characterized by new bone formation, affecting predominantly the diaphyses of the long bones and the skull. The clinical features may include difficulty in walking, muscle wasting and leg pain. Laboratory findings of note are increased plasma ALP activity and other bone turnover markers.
Familial or idiopathic hyperphosphatasia (juvenile Paget disease)
This is a very rare, recessively inherited disease that results from deletion of, or inactivating mutations in, the gene for osteoprotegerin (TNFRSF11B). The onset is in infancy with skeletal abnormalities including dwarfism, bone fragility, a large head and anterior bowing of the limb bones. The bone shows greatly increased cellular activity. The characteristic biochemical changes are very high levels of plasma ALP activity and all other bone turnover markers.
Other disorders
Familial expansile osteolysis and related disorders
These rare, dominantly inherited conditions result from activating mutations in the signal peptide domain of RANK. Painful, progressively worsening expansile lytic lesions develop in bone. The characteristic biochemical changes are a sustained rise in plasma ALP activity and all other bone turnover markers.
Fibrogenesis imperfecta ossium
Fibrogenesis imperfecta ossium is a disorder of bone matrix in which collagen is laid down in a haphazard fashion. The onset is after the age of 50 years and it presents with severe bone pain and tenderness. The plasma ALP activity is consistently elevated.
Polyostotic fibrous dysplasia
Polyostotic fibrous dysplasia is characterized by well-circumscribed areas of marrow fibrosis that replace bone and give the radiographic appearance of cysts, through which pathological fractures may occur. Plasma ALP and urinary hydroxyproline are variably increased, depending on the extent of the disease. The major biochemical interest in this disorder is its association with endocrine diseases such as thyrotoxicosis, acromegaly, Cushing syndrome, precocious puberty and hypophosphataemic osteomalacia (McCune–Albright syndrome). The endocrine disorders are the consequence of constitutive (non-ligand-dependent) activation of the adenylate cyclase second messenger system. They arise from somatic mutations in the gene for the α subunit of the stimulating guanine binding protein (Gsα), which forms a critical part of the adenylate cyclase activating system, linking the receptor to the second messenger.
CONCLUSION
Bone is a metabolically active tissue. In mature bone, there is a continuous cycle of bone resorption and replacement. Osteoblasts are responsible for the synthesis of bone matrix and osteoclasts for bone resorption. The important role of osteocytes is increasingly recognized. Normal bone formation, metabolism and repair depend on the coordinated activity of these cells and the integrity of the homoeostatic mechanisms for calcium and phosphate, which primarily involve parathyroid hormone, FGF23 and vitamin D.
Metabolic bone disease can be due to intrinsic abnormalities of the cellular elements of bone or to abnormalities of the homoeostatic mechanisms for calcium and phosphate.
The commonest metabolic bone diseases are osteoporosis – a generalized reduction in bone density due to an imbalance between the rates of bone formation and resorption, which can have several causes, of which oestrogen deficiency in postmenopausal women is particularly important; osteomalacia, a defect of mineralization related either to a lack of calcium or phosphate or to defective osteoblast function; CKD-MBD, a complex bone disease occurring in patients with chronic kidney disease, and Paget disease, a condition of unknown aetiology characterized by greatly increased osteoclastic activity and disordered new bone formation.
Biochemical measurements are of considerable importance in the diagnosis of metabolic bone diseases and in the assessment of the response of patients to treatment.
Further reading
APPENDIX 31.1 INDICATIONS FOR DIAGNOSTIC TRANSILIAC BONE BIOPSY
• Excessive skeletal fragility in unusual circumstances (e.g. in a young adults).
• When a mineralizing defect is a diagnostic possibility.
• Characterization of bone lesion in CKD-MBD.
• Diagnosis and assessment of response to treatment in vitamin D-resistant osteomalacia and related disorders.
• When a rare metabolic bone disease is suspected.
To get maximum information from a biopsy, fluorochrome labelling of bone is advised. Tetracycline (250 mg q.d.s.) or demeclocycline (150 mg q.d.s.) should be administered by mouth for two days, 18 and 17 days before the biopsy, and again five and four days before the biopsy. Tetracyclines are taken up into the mineralization front and ‘double labelling’ permits calculation of bone formation and mineralization rates. The biopsy should be collected into phosphate-buffered formalin or 70% alcohol and not decalcified. An experienced bone histomorphometrist is needed to interpret the biopsy.
APPENDIX 31.2 PROTOCOL FOR DESFERRIOXAMINE TEST IN DIALYSIS PATIENTS
Desferrioxamine (DFO) 5 mg/kg body weight is administered i.v. (in 100 mL 5% dextrose during the last 60 min of a dialysis session in haemodialysis patients). The plasma aluminium concentration is determined before this session and before the subsequent session (i.e. approximately 44 h after DFO administration).
An increment in plasma aluminium > 7.5 μmol/L suggests aluminium accumulation in bone. When coupled with a low concentration of PTH (< 15 pmol/L with an intact PTH assay), it is indicative of a high total aluminium burden, and has a sensitivity of 87% and specificity of 95% for the diagnosis of aluminium-related bone disease.
If the plasma PTH is > 65 pmol/L, patients are not prone to the effects of aluminium at the bone mineralization front.