CHAPTER 30
Immunology for clinical biochemists
Joanna Sheldon; Rachel D. Wheeler; Pamela G. Riches
CHAPTER OUTLINE
THE IMMUNE SYSTEM
Introduction
The human immune system monitors the internal and external environment at a molecular level and relays information within itself and to other systems regarding the molecular nature of the environment. Within this process, the system has evolved so that certain stimuli elicit a reaction (an immune response) while others are tolerated, eliciting no response. Microbes that invade our bodies can replicate every 20 min, giving them enormous potential to evolve more virulence. Humans reproduce approximately every 20 years with considerably less potential for population adaptation. However, an immune system that uses overlapping and interwoven processes, and can evolve by somatic mutation, allows an individual to adapt to a changing microbial environment. The immune response, like any other biological process, attempts to restore homoeostasis but may itself, if inappropriate or persistent, result in tissue damage and disease.
Over the last 20 years, there has been a rapid development of our knowledge of immunology, particularly at the biochemical and molecular level, in terms of the sophisticated intercellular interactions that accompany any immune response. This chapter will make no attempt to summarize this vast body of knowledge but rather aims to convey broad concepts in describing immune responses, the immune system that makes the responses and the relevance of these to clinical laboratory medicine. The spectrum of immunological diseases includes genetic deficiencies, autoimmune diseases, allergy, malignancy, inflammation and infection. Manipulation of immune responses by inducing immune activation or suppression has great importance in many areas, including immunization and transplantation.
Immune responses
There are two major types of immune response: innate and adaptive. The innate immune response shares many components with the adaptive response but differs fundamentally in that no specific antigen recognition is required. Instead, it involves recognition of molecular sequences that are widely expressed in nature, for example on microbial cells (lipopolysaccharide) and on damaged tissue (heat shock proteins).
The adaptive response is antigen driven and requires prior antigen exposure and recognition of the antigen by T and B lymphocytes through cell surface receptors. On first contact with any antigen, the system reacts relatively slowly and transient infection may occur. Immunologically, this process is termed immunization. The initial response is known as the primary immune response: it also involves acquisition of memory of the exposure. As a result, on second contact with an antigen there is a more rapid response (the secondary immune response) and there is no infection: the host has acquired immunity. These responses involve a combination of cells, tissues and soluble mediators that are described more fully below. In order to understand immune responses, it is first necessary to consider two important terms: antigen and clonality.
Antigens
Most biological materials, including proteins, polysaccharides, glycoproteins and polypeptides can be antigens. How the antigen influences the immune system is related to its reaction with lymphocytes, which will determine whether it stimulates an immune response (i.e. it is an immunogen) or is tolerated (i.e. it is a tolerogen). For example, human albumin is tolerated by the human immune system but on injection into a rabbit, it will stimulate an immune response such that the rabbit produces antibodies to the protein. These antibodies, which react with human albumin, but not, for example, with horse albumin or with the rabbit’s own albumin, are described as being specific for human albumin.
The antibodies produced are not directed against the entire molecule but to short, overlapping amino acid sequences termed epitopes. A large protein molecule such as albumin will have many hundreds of epitopes and, generally, the larger the protein, the more immunogenic it is.
Clonality
Both T and B lymphocytes have the property of antigen recognition through molecules expressed on their cell surfaces (antigen receptors). This is immunoglobulin (Ig) on B cells and the T cell receptor (TCR) on T cells. On any one cell, all the antigen receptors will be identical. Generally, this unique receptor is able to recognize one or a few epitopes. It is estimated that approximately 1010 different antigenic specificities may potentially be encountered during the lifetime of an individual, requiring approximately this number of unique receptors. The development of this very large number of lymphocyte receptors occurs before antigen contact by random rearrangements of the immunoglobulin and TCR genes. The molecular process by which this is achieved will be described in more detail in a later section. Immune responses are driven by the presence of antigen. Engagement of a compatible receptor with antigen provides signals for further proliferation and differentiation. The affinity of the antigen-receptor binding is crucial in determining the nature of that immune response. This is the molecular basis for the establishment of immune responses and immunological memory.
Additionally, in the B cell, the DNA that codes for the part of the Ig receptor that binds antigen is hypermutable, so that during proliferation, somatic mutations within the receptor DNA sequences occur frequently. These mutations may result in a receptor that has a higher affinity for the antigen and, as a result, the clone bearing that receptor will compete more successfully for the antigen and receive a stronger signal, so that it is preferentially selected and responds more quickly. The secreted immunoglobulin from the B cell will similarly show this higher affinity. This process is termed affinity maturation. Immunity to many potentially lethal or harmful human diseases, for example measles, polio and smallpox, is mediated by such high-affinity immunoglobulin (antibody).
The innate immune system
One of the major roles of the immune system is to neutralize and destroy invading pathogens. These pathogens – bacteria, viruses, fungi, protozoans and worms – vary in their size, typical route of entry into the body and mechanism for evading the immune system. Generally, they enter the body across the mucosal surfaces of the respiratory system and gut and across the skin; it is in these areas of the body where the innate immune system forms the first-line of defence from invading pathogens. The major components of the innate immune system are:
• mechanical barriers, e.g. intact skin, mucous membranes
• sebaceous secretions; contents include fatty and other acids (hence skin acidity)
• lysozyme (in tears)
• gut acidity
• urine acidity
• intestinal motility
• IGA (in tears, at mucosal surfaces)
• mucus
• ciliated surfaces
• normal bacterial flora
• acute phase proteins
• interferons
• other molecules (including defensins, lactoferrin, peroxidases)
• neutrophils and complement, although these overlap with the acquired immune system.
Intact mucosal surfaces give a high degree of protection against pathogens, and if breached, for example by surgery, burns or mechanical damage, significant infection can occur. The skin is relatively dry with a high salt content; sweat and sebaceous secretions contain fatty acids, triglycerides, lactic acid, amino acids and ammonia that have antimicrobial activity. Mucus and the cilia of the respiratory tract act together to trap and sweep inhaled particles towards the mouth and nose, where they can leave the body or where they can be swallowed to be inactivated by the gastric acid. The gastrointestinal tract has a combination of innate defence mechanisms such as the alkalinity of saliva and the strongly acidic conditions and enzymic activity of the stomach. Mucopolysaccharides in the secretions of the genital tract help make it inhospitable to pathogens. The eyes are bathed in tears containing the enzyme lysozyme, and the rate of tear production can increase to flush away any potential invading pathogens. Similarly, the urinary tract has a high flow rate that helps to keep it free from pathogens.
Many areas of the body are colonized with commensal organisms. It is estimated that a normal gut is colonized with 1014 bacteria, which form the normal (commensal) bacterial flora and produce chemicals that are antimicrobial and also compete with potentially pathogenetic organisms for essential nutrients. Commensal organisms may cause infection – usually called opportunistic infections – particularly if they get into areas where they do not normally live or if the immune system is compromised.
The innate immune system overlaps with the adaptive immune system (Fig. 30.1), with both systems using the complement cascade, cytokines, phagocytic cells, natural killer cells and acute phase proteins. These components can act alone in an innate response to destroy invading pathogens, but the mechanisms are non-specific and slow and are significantly enhanced by involvement of the adaptive immune system.
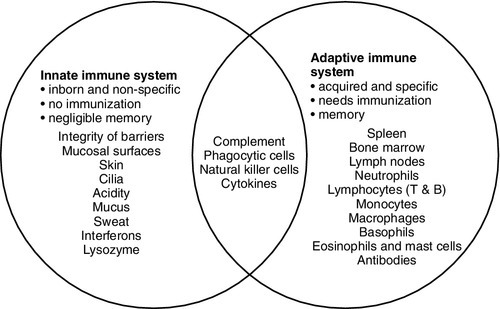
FIGURE 30.1 The overlap between the innate and adaptive immune systems.
The adaptive immune system
An adaptive immune response involves an integrated system, in which there are three elements.
2. Cells with specific functions, including specific antigen recognition and presentation.
3. Soluble mediators that participate in immune reactions and can interact with other systems of the body.
The components of the adaptive immune system are summarized in Table 30.1.
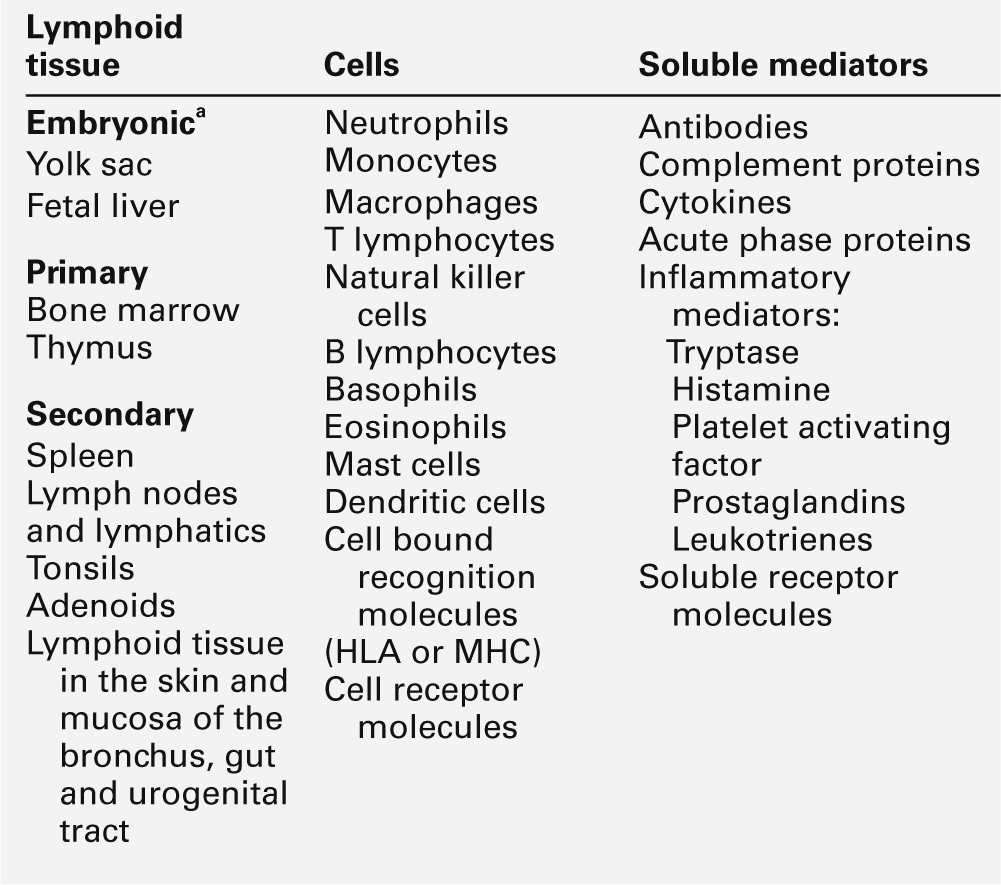
Lymphoid tissue
Lymphoid tissue is distributed around the body (Fig. 30.2), with the blood and the lymphatics forming the lymphoid circulatory system. The bone marrow and the thymus are primary lymphoid organs, the bone marrow being the major site of production of all the cells of the immune system. The bone marrow is also the site of maturation of B lymphocytes. Immature T lymphocytes leave the bone marrow to develop further in the thymus (a bilobed organ in the upper anterior mediastinum that diminishes in size with age). The secondary lymphoid organs are found throughout the body, but particularly at junctions of the lymphatics, for example in the groin and axillae. This arrangement enables specialized antigen-presenting cells (APCs) to show antigens to an enormous number of lymphoid cells as they ‘traffic’ around the immune system. Secondary lymphoid tissue can be diffuse aggregates of cells or can be more organized into identifiable areas, for example the tonsils and Peyer patches in the gut. Lymph nodes also have an identifiable structure, with B cells residing mainly in the outer cortex and T cells mainly in the paracortex. Upon antigen stimulation, primary follicles within lymph nodes develop into secondary follicles with germinal centres. Activation of the immune system causes the cells within the lymph nodes to differentiate and proliferate, resulting in enlargement of the lymph nodes (lymphadenopathy). This is sought on physical examination of patients as a sign of increased activity of the immune system, for example in infection and malignancy. A large proportion of lymphoid cells are found in the gut and respiratory systems (gut and mucosal associated lymphoid tissue: GALT and MALT), where the antigenic burden is high.
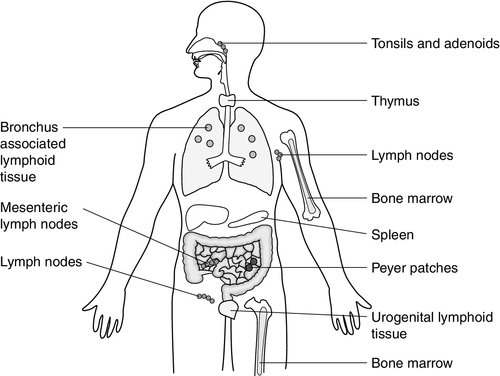
FIGURE 30.2 The lymphoid organs.
Although specific immune responses are made within lymphoid tissues, other non-lymphoid organs have a role to play; for example, the liver is the major site of production of the complement proteins and acute phase proteins.
Cells
All cells of the body interact with the immune system. For example, any damaged nucleated cell has the capacity to make cytokines, and red blood cells have complement and antibody receptors on their surface that, owing to their enormous numbers, represent an important removal mechanism for immune complexes. Immune cells circulating in the blood are represented by the white cell population. Normally, the total white cell count of an adult is 4–11 × 109/L. This total is made up of a number of different cell types as shown in Table 30.2; the differential white cell count represents the number and proportion of each of these types. Each cell type has particular functions within the immune system: the major functions are listed in Table 30.2.
TABLE 30.2
The major cells types, their approximate percentage of the differential white cell count and their major functions
| Cell type | Reference range (× 109/L) | Function |
| Total white cell count | 4–11 | |
| Neutrophil | 1.6–7.5 | Attracted to site of inflammation by chemotactic factors Most important cell type for phagocytosis and killing of bacteria Opsonization with antibody and complement enhances phagocytosis |
| Lymphocyte | 1.5–4.0 | Key role in orchestrating the immune response and producing soluble mediators of immunity |
| T cell (CD3 +) | 0.8–2.66 | T helper cells (CD4 +) control the development of an immune response by secreting cytokines Cytotoxic T cells (CD8 +) can directly kill cells, e.g. that are infected with virus Regulatory T cells modify immune responses |
| B cell (CD19 +) | 0.1–0.6 | Antibody production |
| Natural killer cell (CD56 +, CD16 +) | 0.05–0.60 | Can directly kill cells, e.g. infected with virus, tumour cells |
| Monocyte | 0.1–0.8 | Important phagocytic cells and can act as antigen presenting cells Cytokine production – particularly in inflammation |
| Basophil | < 0.1 | Major role is in allergic responses via release of mediators, e.g. histamine May be important in defending against parasites |
| Eosinophil | 0.04–0.45 | Some phagocytic function Major role in hypersensitivity reactions via release of variety of mediators Activation of mast cells |
Each cell type has a characteristic morphology that enables it to be identified in a stained blood film examined under a microscope. Figure 30.3 shows diagrams of the various types of white blood cell with their major progenitor cell types. The cell membranes are covered with molecules that enable the cells to interact with their environment. They may be constitutively expressed (i.e. expressed in the unstimulated state) and this expression be increased on activation, or they may be induced when a cell is activated. Cells have many thousands of molecules of a whole variety of cell receptors on their surfaces. The cell surface molecules are classified by their cluster of differentiation (CD) numbers, with a different CD number for individual types of cell membrane protein. (The designation + after a CD number indicates that a specific antigen is expressed by that cell. For example, all T cells are CD3 +; T helper cells are CD4 + .) The cellular expression of these markers is used to provide a more detailed picture of cell populations, for example whether they are immature or mature, whether activated, how they are functioning and whether they are normal or malignant. Aberrant expression of the CD markers is used to classify lymphoid malignancies while reduced expression of CD antigens can be used in the investigation of immunodeficiency, for example, CD4 counts to monitor HIV infection. The technique (known as immunophenotyping), uses specific antibodies to the CD markers and flow cytometry to count and quantify cell populations. There are approximately 400 different human proteins that have been assigned a CD number. The most important immunological markers, their major functions and the cells types on which they are predominantly expressed are shown in Table 30.3.
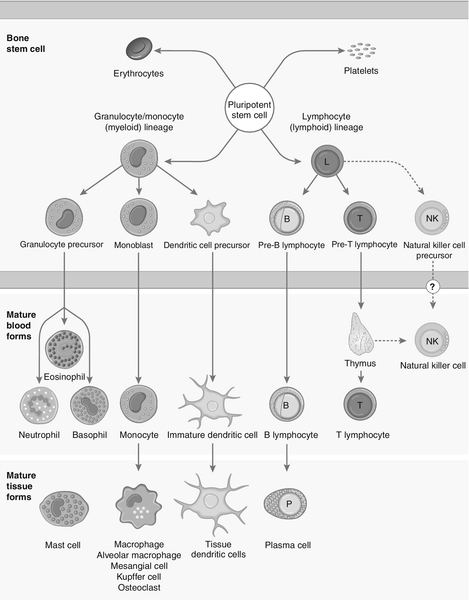
FIGURE 30.3 Major types of white blood cell and their generation.
TABLE 30.3
The major CD phenotypes, their normal functions and cell types where they are normally expressed
| Phenotype | Function of protein | Cells where expressed |
| CD3 | Part of the T cell receptor | All T cells (pan T cell marker) |
| CD4 | Helps T cells to hold on to antigen-presenting cells (HLA class II) | T helper cells |
| CD8 | Helps T cells to hold on to antigen-presenting cells (HLA class I) | Cytotoxic T cells |
| CD19 | B cell co-receptor | B cells |
| CD20 | B cell activation and differentiation | B cells |
| CD45 | Cell differentiation, signal transduction and lymphocyte activation | All white blood cells (leukocyte common antigen) |
| CD16 | Binds Fc γ RIII (binds IgG Fc) | Natural killer cells, neutrophils, monocytes |
| CD32 | Binds Fc γ RII (binds IgG Fc) | Neutrophils, macrophages, B lymphocytes, eosinophils |
| CD64 | Binds Fc γ RI (binds IgG Fc) | Monocytes, macrophages, activated neutrophils |
| CD89 | Binds Fc α R (binds IgA Fc) | Monocytes, macrophages, neutrophils |
| CD21 | CR2 (binds C3d) | B cells (some) |
| CD35 | CR1 (binds C3b, C4b) | Neutrophils, monocytes, B cells, erythrocytes |
| CD25 | IL-2R α chain | Activated T and B cells |
| CD56 | Natural killer cells | |
| CD40 | Binds to CD40 ligand (CD154); B cell proliferation and class switching | B cells, antigen-presenting cells |
| CD11a (+ CD418) | Cell adhesion (to other cells) | Leukocytes |
| CD11b (+ CD18) | Cell adhesion (to other cells) CR3 | Monocytes, neutrophils, natural killer cells |
| CD11c (+ CD18) | Cell adhesion (to other cells) CR4 | Monocytes, neutrophils, natural killer cells, some T and B cells |
| CD18 (+ CD 11 a, b and c) | Cell adhesion (to other cells) | Leukocytes, monocytes, neutrophils, natural killer cells, some T and B cells |
| CD62E | E selectin cell adhesion (to other cells) | Endothelium, platelets |
| CD62L | L selectin cell adhesion (to other cells) | Neutrophils |
| CD62P | P selectin cell adhesion (to other cells) | Endothelium |
| B cell proliferation and class switching | Activated T cells |
IL, interleukin; HLA, human lymphocyte antigens.
The cells of the immune system arise from haematopoietic stem cells in the bone marrow. Individual stem cells respond to a number of positive or negative feedback mechanisms, for example colony stimulating factors, and are driven to proliferate and differentiate along committed pathways to give rise to populations of lymphocytes, granulocytes, APCs etc. The major processes are outlined in Figure 30.3.
Neutrophils
Neutrophils, in common with basophils and eosinophils, can be shown to contain a large number of cytoplasmic granules when stained and examined by light microscopy and are thus termed granulocytes. They have a multilobed nucleus (polymorphonuclear or PMN, hence their alternative name, polymorphs) and are the most abundant white cells in the circulation. They show high expression of adhesion molecules that enable them to roll along blood vessel walls. If expression of adhesion molecules on the vascular surface is upregulated, for example because of tissue damage or infection, neutrophils are attracted to the site of inflammation (by chemotactic factors) and leave the blood stream and enter the tissues to engulf the invading pathogens or damaged tissue components, and destroy them, in a process termed phagocytosis.
The stages in phagocytosis are as follows.
2. At the site of injury, the particle or microorganism, coated with antibody and complement, adheres to the phagocyte via C3b and Fcγ receptors.
3. The adherence of the particle to the phagocyte results in the activation of the phagocyte’s membrane, initiating a change in its shape and leading to internalization of the foreign material (phagocytosis: literally, ‘eating’ the particle).
4. As the particle is taken into the cell, it becomes surrounded with a membrane, forming a phagocytic vesicle or phagosome.
5. The phagosome fuses with lysosomes to form a phagolysosome, in which the microorganism is killed, and it, or other ingested material, is digested.
6. Completely digested material is reused by the cell; incompletely digested material is released (sometimes accompanied by potentially harmful enzymes).
If phagocytic cells are incubated in vitro with, for example, bacteria, phagocytosis will occur, but the process is significantly enhanced if the bacteria are coated with complement or antibody. This coating is called opsonization and, in simple terms, it enables phagocytic cells to adhere to the marked particles (giving the process specificity) and enables them to change shape to engulf the particles. Many cell types are capable of phagocytosis, the ‘professional’ phagocytic cells being neutrophils and cells of the monocyte/macrophage lineage. Many macrophages home to particular tissues where they develop highly specialized functions, e.g. Kupffer cells (in the liver), mesangial cells (kidneys), microglia (brain) and Langerhans cells (epidermis).
Basophils and eosinophils
These two cell types are also granulocytes and are particularly important in defending against multicellular pathogens, for example parasitic worms. This defence mechanism relies on the mediators in the cytoplasmic granules rather than the phagocytic functions of the cells. A close relation of the basophil, the mast cell, is tissue-based and important in allergic responses.
Monocytes
Monocytes are immature macrophages that are travelling from their site of production in the bone marrow through the bloodstream to the tissues where they will become fixed macrophages. Potentially, they are phagocytic cells but they do not seem to act as phagocytes in the bloodstream. They have receptors that recognize many bacterial products so are very effective cells in innate immunity. Engagement of these receptors stimulates release of proinflammatory cytokines such as interleukin 1 (IL-1), IL-6 and tumour necrosis factors (TNFs). These cytokines are able to drive many aspects of inflammation and specific immunity. Monocytes therefore provide a valuable second-line defence mechanism once infectious agents have breached the mucosal defences and entered the bloodstream.
Lymphocytes
The major subpopulations of lymphocytes are B and T cells. A haemopoietic stem cell first gives rise to a lymphopoietic stem cell, which in turn becomes committed to the B or T cell lineage. Lymphocytes are further divided into subsets on the basis of the presence of particular cell surface molecules (see Table 30.3). B cells (CD19 +) can differentiate into antibody (immunoglobulin)-producing plasma cells in response to exposure to antigen. T cells leave the bone marrow to mature in the thymus. They undergo further differentiation to become either T helper cells (CD4 +) that ultimately control the immune response, through cytokine production or cytotoxic T cells (CD8 +) that have an important role in killing viruses, fungi and infected cells. Other T cell types are becoming increasingly well recognized as having important immunological roles; for example, that of regulatory T cells (Treg) in the modulation of autoreactive T cells, and of Th17 (T helper) cells in inflammatory responses.
There are two main types of T helper cells (Th1 and Th2), which are defined by the cytokines they produce. Th1 cells predominantly produce IL-2, TNFα/β and γ-interferon, and drive cell-mediated immunity by cytotoxic T cells, macrophage stimulation and inflammation, and production of immunoglobulins, such as IgG1 and IgG3 by B cells. Th2 cells predominantly produce IL-4, 5, 6, 10 and 13, and are important in driving IgE responses and the growth of mast cells and eosinophils.
Natural killer (NK) cells (CD16 + and CD56 +) are cytolytic cells of the lymphocyte lineage, acting in a slightly different way from the cytotoxic Tc cells.
B and T lymphocytes are at the heart of the adaptive immune response by virtue of their antigen receptors (Ig and T cell receptor, TCR). A comparison of the B and T cell receptors is shown in Table 30.4. The property of antigen binding is also shared by the human leukocyte antigens (HLA). These three sets of antigen-binding molecules are all products, or partial products, of the immunoglobulin gene superfamily; they are all composed of one or more homologous domains of approximately 110 amino acids, as illustrated in Figure 30.4.
TABLE 30.4
Comparison of B cell receptor and T cell receptor
| B cell receptor (membrane-bound IgM) | T cell receptor | |
| Similarities | Variation created by gene rearrangement | |
| Part of a protein complex including accessory chains and signalling adaptors | ||
| Variable and constant domains | ||
| Differences | Two heavy chains and two light chains | Two polypeptide chains (αβ or γδ) |
| Somatic hypermutation allows affinity maturation | No somatic hypermutation | |
| Class switching from IgM to other immunoglobulin classes | No class switching | |
| Binds directly to specific antigen | Binds to specific antigen only when bound by appropriate HLA molecule |
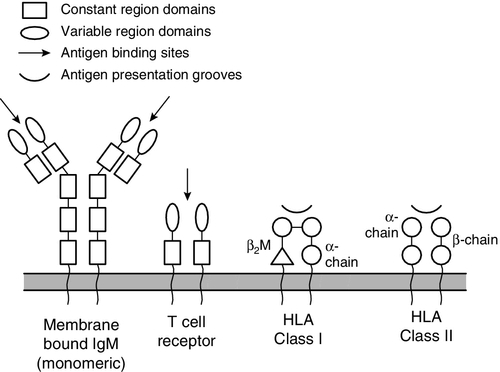
FIGURE 30.4 The antigen presenting and binding molecules. β2M, β2-microglobulin; HLA, human leukocyte antigen.
Antigen recognition
Immunoglobulins
Immunoglobulins are proteins produced by cells of the B-lymphocyte lineage, either in a membrane-bound form as an antigen receptor, or as a secreted product that shows antibody activity. The membrane-bound and secreted forms differ only in the terminal amino acid residues that serve to anchor the protein into the membrane or allow secretion. The basic monomeric immunoglobulin unit consists of two identical heavy chains and two identical light chains arranged in a Y-shape (see Fig. 30.5). In humans, there are five possible heavy chains (γ, α, μ, δ and ε) that give the five immunoglobulin classes (IgG, IgA, IgM, IgD and IgE). In addition, there are four subclasses of IgG and two subclasses of IgA. There are two light chain types (κ and λ).
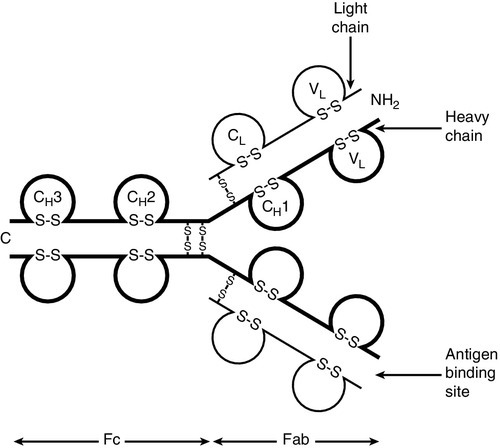
FIGURE 30.5 The basic structure of an immunoglobulin molecule.
The class (or subclass) of the heavy chains and type of light chains is determined by the common amino acid sequences in areas of the molecule called the constant domains. The light chains have one constant domain and the heavy chains have three or four constant domains, depending upon the class; it is these areas that confer functionality to secreted immunoglobulin molecules. Each chain also has a single domain of great variability in the amino acid structure, called the variable domain. The heavy and light chains are arranged such that their variable domains are adjacent, forming a groove in which antigen binding occurs.
If every antigen recognition receptor on B and T cells were encoded on a one gene/one enzyme principle, these receptors alone would require more than the total DNA present within individual cells. The enormous scope for receptors to recognize antigens is achieved by a mechanism of genetic recombination.
In humans, the gene for the production of the immunoglobulin heavy chain is located on chromosome 14. In the germ line, there are large numbers (estimated 250–1000) of variable (V) gene segments followed by small numbers of D (∼ 12) and J (∼ 4) gene segments. Finally, there is the constant (C) gene segment that can encode for any of the immunoglobulin heavy chains. A diagram of the immunoglobulin heavy chain gene is shown in Figure 30.6. Processing of this genetic material occurs by deletion of the introns (the non-coding DNA) and splicing together of the exons (the coding portions of the gene). One of the many V-region genes is selected and joined to one of the D-region genes, which is joined to one of the J-region genes. This VDJ sequence is then joined to the constant region genes, typically starting with the μ gene, which is the first downstream heavy chain gene in the sequence, resulting in the production of IgM. The genes for the κ and λ light chains are located on chromosomes 2 and 22, respectively, and are similar to the heavy chain gene except that they do not have a D region. Diversity of the variable region is generated by the large number of possible combinations between the V, D and J or V and J regions of the genes, variation in how these genes are spliced and by somatic mutation generated during cell development.
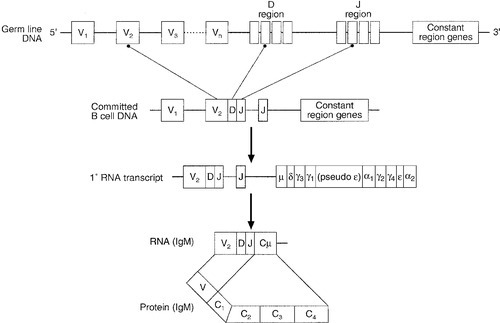
FIGURE 30.6 The immunoglobulin heavy chain gene.
In any cell, there is allelic exclusion, so that only one of each pair of chromosomes coding for immunoglobulin is translated. Each cell, theoretically, has two chances for each gene to be successfully rearranged. The products of unsuccessful rearrangements are secreted by the cell and degraded or cleared by the kidneys. The immunoglobulin heavy chain gene(s) is rearranged first, followed by κ-gene rearrangement. In about 40% of cells, the κ rearrangements are abortive and the cell then proceeds to λ-gene rearrangement. A small excess of free light chains (polyclonal) is produced. The immunoglobulin molecule is then assembled and expressed on the surface of B cells. The cell is now committed to a light chain type and unique variable region specificity (also called the idiotype). The organization of the heavy chain gene enables the variable region to be spliced onto a different heavy chain by the process of ‘class switching’. This occurs after interaction with antigen, when cells that show appropriate recognition of the antigen are driven to proliferate. IgM is the antibody produced in primary immune responses; IgG (or IgA or IgE), produced after class switching, is produced in secondary immune responses, as shown in Figure 30.7. Some cells will mature into memory cells and others into end-differentiated plasma cells. Plasma cells are typically non-proliferative and have little membrane-bound immunoglobulin but secrete large amounts.
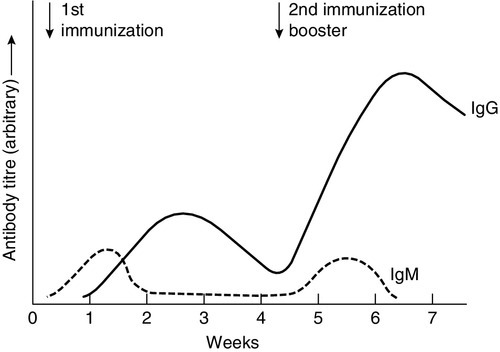
FIGURE 30.7 Antibody production in primary and secondary responses.
IgA and IgM both occur as oligomers of the basic unit. Combination is facilitated by the addition of a short polypeptide chain called the J-chain. This J-chain should not be confused with the J gene segments coding for part of the variable region of the immunoglobulin molecule – the two bear no relationship. In the plasma, IgM occurs as a pentamer. IgA can occur as a dimer and this is the predominant form in secretions. A glycoprotein called ‘secretory piece’ is synthesized by mucosal epithelial cells and integrated into secreted IgA molecules, making them more resistant to degradation by the mucosal environment.
The major properties of the immunoglobulins are shown in Table 30.5.

Under normal circumstances, there is enormous microheterogeneity within immunoglobulin molecules. This results in polyclonal immunoglobulins, representing all classes, light chain types and a large number of different idiotypes, each produced by a different B cell clone, even to a single antigen. Monoclonal immunoglobulins, which consist of a single heavy chain class, light chain type and idiotype, result from the proliferation of a single clone of B cells. In fact, polyclonal immunoglobulins comprise very small amounts of many thousands of monoclonal immunoglobulins.
Each class and subclass of immunoglobulin has its own function or group of functions. IgG is the most abundant antibody in the circulation and also diffuses into the extravascular compartment bathing all the tissues. It is produced in secondary immune responses and is vital for immunological memory. There are four subclasses, with IgG1, IgG2, IgG3 and IgG4 being approximately 65%, 25%, 7% and 3%, respectively, of the total IgG. IgG1 and IgG3 responses mature earliest, while IgG2 and IgG4 responses mature more slowly. IgG1, IgG2 and IgG3 are all potent activators of the classical complement cascade. IgG (bound to an antigen) binds to the Fcγ receptors I, II and III on neutrophils, macrophages etc. to facilitate and enhance phagocytosis. IgG has a half-life of approximately 21 days, but this depends upon the plasma concentration, catabolism being increased with raised IgG concentrations and decreased with low IgG concentrations.
IgA is the major immunoglobulin in mucosal secretions and is also produced in secondary immune responses. It is most important in binding and neutralizing organisms at mucosal surfaces, without interacting with other immunological systems.
IgM is the antibody of the primary immune response, culminating in the process of class switching, inducing the B cells to make the antibodies of the secondary responses, IgG or IgA. The large pentameric structure of IgM gives good potential for antigen binding and its high molecular weight keeps it restricted to the vascular compartment. The half-lives of IgA and IgM are independent of their plasma concentrations and are approximately five days for both.
IgD is detectable on B cells early in their development, but its concentration in plasma is normally very low. Its exact function in not known, but it is likely that it is important in processing antigens in immature B cells.
The plasma concentration of IgE is very low; it relies on its binding to the Fcε receptor on mast cells and basophils for its activity. The release of inflammatory mediators within these cells is precipitated by binding of the surface-bound IgE to a specific antigen or allergen.
T cell receptors
The T cell receptor is somewhat simpler. It is a heterodimer in which each polypeptide chain is composed of two domains, one variable and one constant. There are four possible polypeptide chains, α, β, γ and δ. A receptor is made up of a pairing of either α with β or γ with δ. In no cell are both types expressed, nor are other pairings possible (e.g. α with δ). The variable domains are similarly constructed from gene segments, as described for immunoglobulin receptors (V, D and J for β and δ; V and J for α and γ), and then spliced into the corresponding constant regions.
The T cell receptor differs from the immunoglobulin receptor in that it has no hypermutable sequences and therefore does not show affinity maturation. Also, it can only recognize antigen that is bound to an HLA (human leukocyte antigen) molecule expressed on the surface of an appropriate antigen presenting cell (APC).
Human leukocyte antigens (HLA)
The major histocompatibility complex (MHC) is a group of molecules that are integral to the development of adaptive immune responses. In humans, the genes for these molecules, which are designated human leukocyte antigen (HLA), are on chromosome 6. They are expressed on the cell surface and bind peptides in an antigen-binding groove, where they are recognized by T cells via the T cell receptor. There are two types of HLA, class I and class II, summarized in Table 30.6.
TABLE 30.6
Comparison of HLA class I and class II
| Class I | Class II | |
| Structure | Variable heavy chain + invariant chain (β2-microglobulin) | Two variable chains, α and β |
| Expression | Most nucleated cells | Antigen presenting cells; may be induced on other cell types |
| Antigen presented | Intracellular antigen, i.e. viral, self 8–9 amino acids |
Extracellular antigen, e.g. microbes or their products 14–22 amino acids |
| Gene loci | HLA-A, B, C | HLA-DQ, DR, DP |
| Responding T cell | CD8 + T cells | CD4 + T cells |
HLA molecules bind antigen but they are different from the receptors on B and T cells in that there is no genetic re-arrangement. Instead, the HLA genes are highly polymorphic, i.e. their nucleotide sequences vary between individuals giving different alleles, summarized in Figure 30.8.
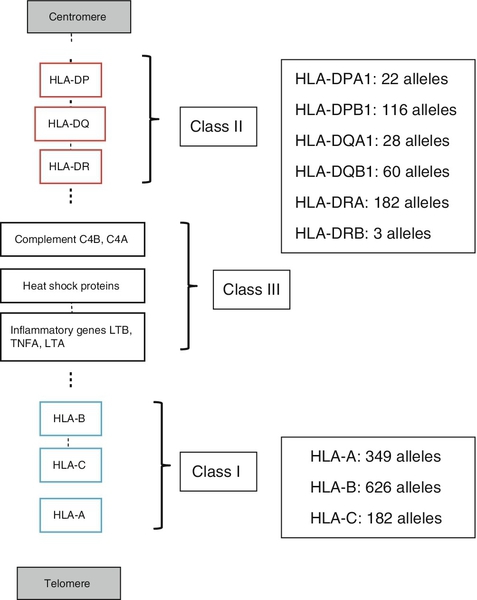
FIGURE 30.8 Simplified overview of the MHC locus on human chromosome 6. The MHC locus is 4 MB and encodes ∼ 300 genes. Dashed lines are used to indicate that other genes lie between the ones shown.
HLA class I molecules are widely expressed on most nucleated cells. They comprise a small non-membrane polypeptide, β2-microglobulin (the invariant chain of MHC class I) covalently linked to a heavy chain, an integral membrane protein with external domains. The variation within MHC class I arises from polymorphisms within genes for the heavy chains. There are three loci for class I (HLA-A, HLA-B and HLA-C). Each individual inherits A, B and C from each parent and, unlike the situation with immunoglobulins and the TCR, there is no allelic exclusion: both sets of genes are translated. All types occur on any one cell. Because of the polymorphisms, it is likely that any individual will be heterozygous at each locus; therefore six products will be formed.
HLA class II molecules are primarily expressed on immune cells, particularly those able to present antigen. They may be inducible on many other cell types. They comprise two transmembrane polypeptides (α and β chains) of similar size. As for class I, there are three loci (HLA-DR, HLA-DQ and HLA-DP). Again, the likelihood is that these properties will result in at least six distinct receptors. The class II system is, however, more complex. During evolution, there has been replication of some loci such that it is possible to express a number of different DR and DQ types, leading to increased diversity. Even with the variations described above, it is apparent that the total number of different HLA types expressed in any one individual is relatively small compared with immunoglobulins and TCRs.
Antigen presentation
Unlike immunoglobulin and the TCR, HLA molecules are loaded with peptide antigen intracellularly during molecule assembly, so that prior ‘loaded’ HLA is expressed on the cell surface. Indeed, it would appear that antigen-binding is essential for stability of the molecule and that HLA without antigen is intrinsically unstable. Antigen binds in grooves formed by the folding of class I molecules and the juncture of the α and β chains in class II. The source of the antigen is different for each class. Class I molecules bind antigens synthesized intracellularly, e.g. self-peptides or viral antigens; if these antigens are able to stimulate an immune response it is via CD8 + T cells, resulting in destruction of the target cell. Class II molecules bind antigen that was derived extracellularly, e.g. microbes or microbial products. These antigens are internalized within the APC, where they are subjected to partial proteolytic degradation and then bound to the class II molecules for antigen presentation. These antigens are recognized by CD4 + T cells and stimulate an immune response.
B cells can act as antigen presenting cells. Their surface immunoglobulins are able to react with free antigen molecules, forming complexes that are taken into the cells, where the antigen is degraded into peptides. These peptides can combine with HLA class II molecules to allow antigen presentation to T cells.
Cellular immune activation
Antigen binding alone will rarely stimulate an immune response. Some very repetitive antigen sequences, for example polysaccharides, may directly stimulate B cells. To respond to most antigens, however, both T and B cells require additional signaling from cytokines or contact with other cell types mediated through accessory or co-ligatory molecules expressed on these cells. Accessory cell interactions are many and complex and the reader is referred to dedicated immunological texts for an account of these.
Complement
The complement system
The complement system consists of approximately 30 plasma proteins, normally present in inactive forms (zymogens). Activation results in the generation of a cascade of enzymes that leads to activation of the terminal cytolytic pathway (membrane attack complex). Activation can occur via two major pathways, the classical pathway and the alternative pathway. In addition, complement can be activated through the lectin pathway and may be recruited by proteolytic activity generated by other physiological pathways such as coagulation. The system is inherently labile and subject to regulation by various proteins (see below). A simple diagram of the complement system is shown in Figure 30.9. The role of the complement system as a whole is to destroy invading pathogens via the membrane attack complex and opsonization but there are many other effects of complement activation, as shown in Table 30.7. During activation of the earlier components (C2, C4, C3 and C5), smaller peptides are cleaved off the zymogens. These have powerful biological activities: they are able to initiate and potentiate inflammation by increasing blood vessel permeability and attracting inflammatory and immune cells to a site of injury via a process termed chemotaxis.
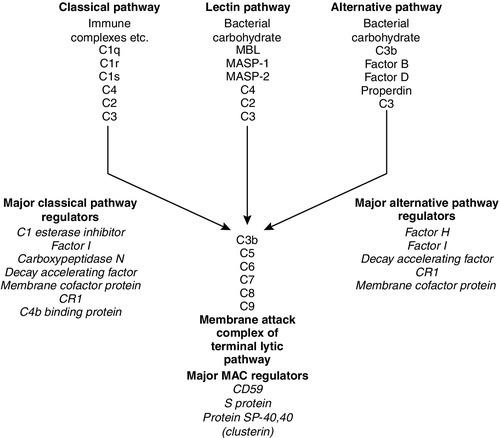
FIGURE 30.9 The complement system, showing the pathways for activation and their major regulatory proteins.
TABLE 30.7
The major actions of components of the complement system
| Complement components | Effect |
| C1q, C3, C4 | Clearance of immune complexes Clearance of cell debris |
| C3a, C4a, C5a (anaphylatoxins) | Histamine release from mast cells Increase smooth muscle contraction Increase vascular permeability Inflammation |
| C3b (opsonin) | Binding of antigen–antibody complex to receptors on phagocytes, enabling effective ingestion and destruction |
| C5a, C3a (chemotaxins) | Recruitment of phagocytes into areas of inflammation |
| C5–C9 (membrane attack complex) | Cell or bacterial lysis Cell signalling |
The larger fragment formed on activation usually has the alphabetical annotation ‘b’ (e.g. C3b) and the smaller fragment, ‘a’ (e.g. C3a). Some authors use a line over a component name (e.g. C1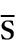 ) to denote an active enzyme that can activate the next component; this convention is unnecessary and has not been adopted in this chapter.
) to denote an active enzyme that can activate the next component; this convention is unnecessary and has not been adopted in this chapter.
Activation via the alternative pathway
The alternative pathway is initiated at the level of C3, bypassing the earlier components C1, C4 and C2, which are activated in the classical pathway. C3 spontaneously degrades in tissue fluids and is rapidly inactivated. However, on activating surfaces, including damaged tissue and components of bacterial cell walls such as lipopolysaccharides, degraded C3 is stabilized and can form an enzyme to activate further C3 and to generate an enzyme to cleave C5 and thus activate the terminal sequence of the pathway.
Activation via the classical pathway
Complexes of antigens with their antibodies (immune complexes) binding C1 activate the classical pathway. C1 is a trimolecular complex of C1s, C1r and C1q. Binding of C1 liberates an enzyme C1s (the C1 esterase), which is able to cleave C4 and C2, and an enzyme formed from a complex of the larger fragments of these two molecules can activate C3. The pathway beyond this is common with the alternative pathway.
Activation via the lectin pathway
Various components of bacterial cell walls are able to bind to a protein in the blood called mannose binding lectin (MBL); this complex is able to stimulate serine proteases that can directly activate component C4.
Regulation of the complement pathways
Many of the complement proteins have a natural cleavage site, making them inherently unstable; therefore the presence of a number of regulatory proteins is vital to stop uncontrolled activation of the cascade. Some of the control proteins circulate while others are membrane bound; some have enzyme activity, others are simple binding proteins. The important complement regulatory proteins are shown in Table 30.8.
TABLE 30.8
The major control mechanisms of the complement cascade
| Complement control mechanism | Activity |
| C1 esterase inhibitor | Protease inhibitor that blocks the activity of C1 esterase |
| Factor H | Binds C3b and enhances the degradative action of Factor I |
| Factor I | Enzyme that degrades C3b and C4b |
| C4b binding protein | Binds to C4 and enhances its destruction by Factor I |
| Protein S and SP-40,40 (clusterin) | Binds C5b67 and prevents formation of the membrane attack complex |
| Carboxypeptidase N | Enzyme that inactivates C3a, C4a and C5a |
| Decay accelerating factor | Transmembrane glycoprotein on most blood cells that binds C4b and inhibits C3 convertase |
| Membrane cofactor protein (CD46) | Membrane-bound C3 binding protein |
| Membrane attack complex inhibitory factor (CD59) | Membrane-bound protein that hinders insertion of the membrane attack complex |
| Complement receptor I (CRI, CD35) |
High affinity receptor for C3b and C4b on surface of erythrocytes |
Acute phase proteins
Inflammation is a fundamental pathological process that occurs in response to tissue injury. The inflammatory response has three main stages: local reactions; destruction and/or removal of the injurious material, and repair and healing of the damaged tissue. Stimulators of inflammation include trauma, infection, infarction and deposition of immune complexes; these result in the release of inflammatory mediators (see Table 30.9) and the classic signs of inflammation – redness, swelling, warmth or heat, pain and loss (or impairment) of function. These features may be localized, but in more severe inflammation there is a widespread response and the features also include fever, leukocytosis and the production of a variety of liver-derived acute phase proteins. The functions of these proteins are summarized in Table 30.10.
TABLE 30.9
Inflammatory mediators and their actions
| Mediator | Action |
| Platelet activating factor, histamine, prostaglandins | Vasodilatation |
| Platelet activating factor, histamine, complement C3a and C5a, bradykinin, leukotrienes | Increased vascular permeability |
| IL-8, IL-1, and TNFα, complement C5a, leukotrienes | Leukocyte adhesion and chemotaxis |
| Bradykinin and prostaglandins | Pain |
| Enzymes (proteases) and products of neutrophil respiratory burst (free radicals) | Tissue damage |
| Inflammatory cytokines IL-1, IL-6 and TNFα | Hepatic production of acute phase proteins |
TABLE 30.10
The functions of acute phase proteins
| Acute phase function | Examples |
| Mediating | Working as parts of networks of inflammatory mediators: C-reactive protein (CRP) binds a variety of ligands and activates complement; complement components are important opsonins and chemotactic factors; fibrinogen and the clotting components form clots and fibrin matrices as a basis for repair |
| Inhibiting | Inhibiting protease activity and controlling pathways: α1-antitrypsin and α1-antichymotrypsin inhibit the actions of enzymes released from leukocytes during phagocytosis; C1 esterase inhibitor inhibits part of the complement system |
| Scavenging | Inhibiting or eliminating noxious substances produced during the inflammatory process: haptoglobin combines with free haemoglobin to form a complex that is rapidly cleared by the liver; CRP may opsonize DNA and cell membrane debris |
| Regulating | Modulating the immune response: α1-acid glycoprotein is expressed on lymphocyte cell membranes |
| Repairing | Control and laying down of connective tissue elements: α1-antitrypsin and α1-antichymotrypsin are deposited in a sequential fashion on the surface of newly formed elastic fibres; α1-acid glycoprotein promotes fibroblast growth |
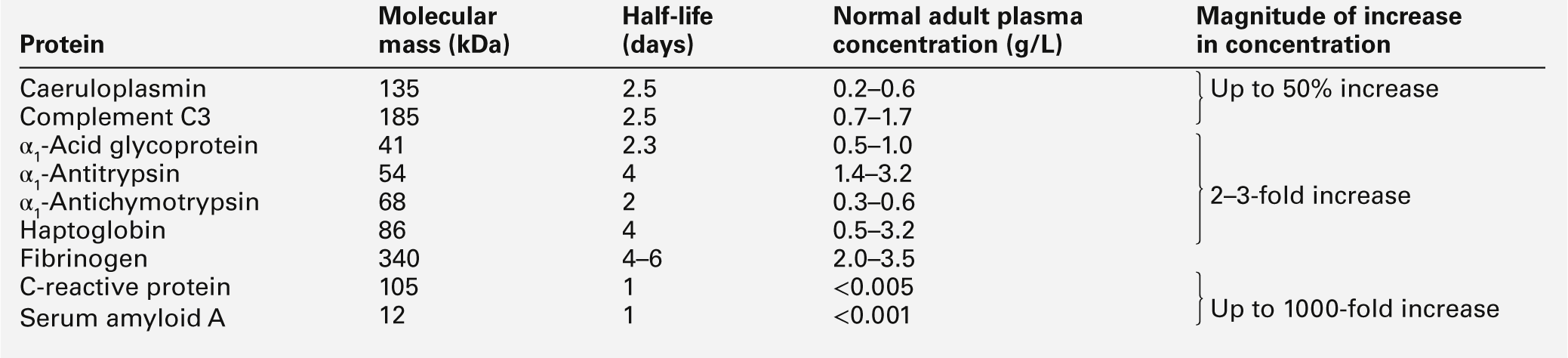
The acute phase proteins show diverse properties, particularly in the kinetics of their responses, as shown in Table 30.11. The magnitude of response is generally related to the degree of injury, although other factors such as catabolic rate, hormonal influences and genetic variability are also important.
Cytokines
These are biological signalling peptides, whose many actions include the control of the differentiation of white blood cells and the modulation of the actions of the cells of the immune system. They include the interleukins, interferons, tumour necrosis factors and various growth factors. They are summarized in Table 30.12.
TABLE 30.12
The major groups of cytokines
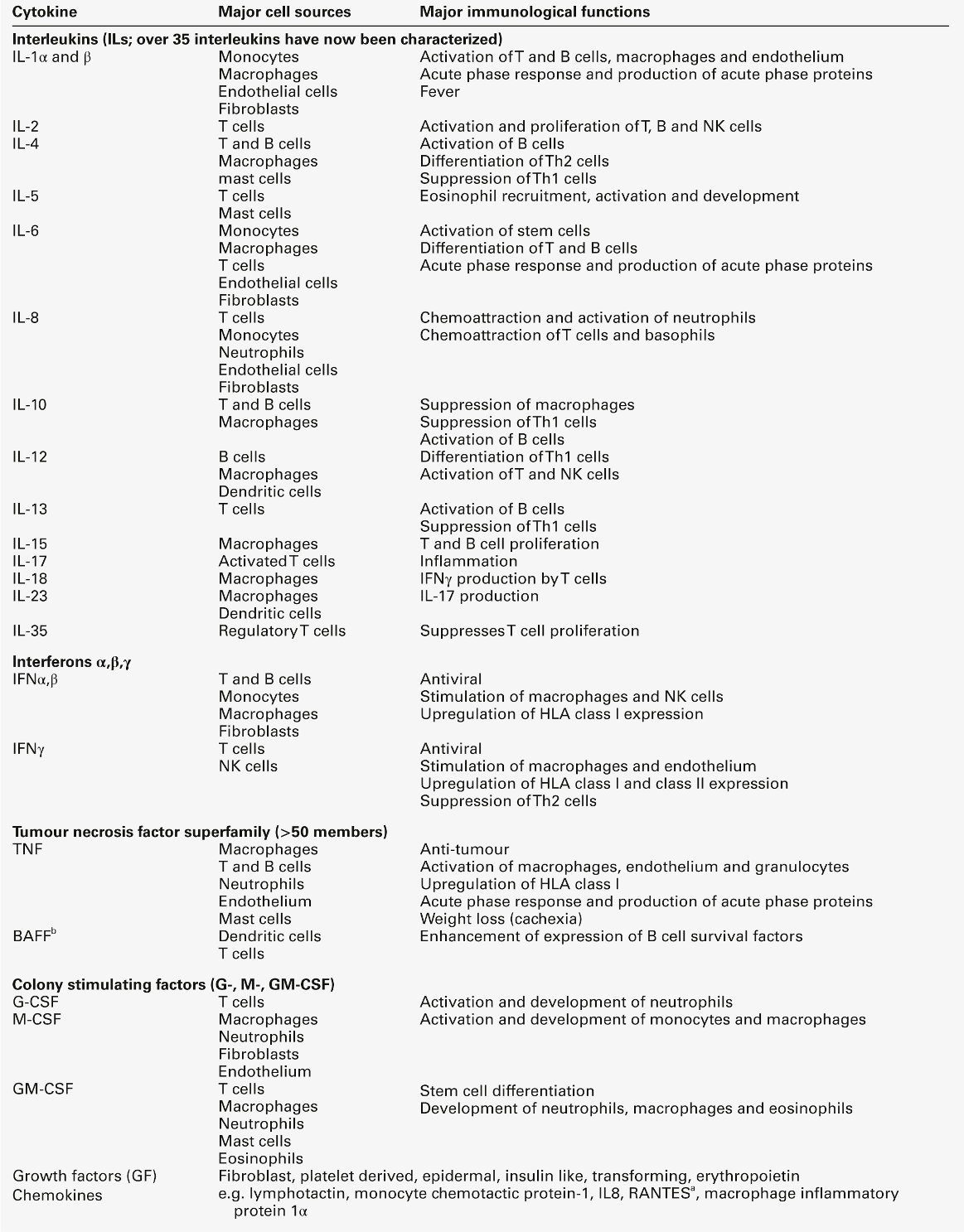
a RANTES: regulated on activation, normal T expressed and secreted; Th, T helper.
In general, cytokines, like hormones, act through high-affinity cell surface receptors that can be widely distributed on many cell types or can be restricted to one or two cell types. The receptors can be up- or downregulated or induced, depending upon the prevailing conditions. Most cytokines are glycated and their binding to the receptors may be influenced by the configuration of the sugar residues and by the other cytokines being released in the vicinity. Most cytokines are multifunctional molecules with diverse biological actions upon a wide variety of target cells. Cytokines, in general, show significant overlap in their functions, with any one function typically being shown by a number of cytokines (pleiotropism). They mostly act locally in either an autocrine or paracrine fashion; very few, for example IL-6, show a true endocrine function. The major differences between cytokines and hormones are shown in Table 30.13. Genetic deficiencies of cytokines are rare, possibly reflecting their vital role in the maintenance of health.
TABLE 30.13
Major differences between cytokines and endocrine hormones
| Property | Cytokine | Hormone |
| Sites of production | Many and varied | Many, but specific to each hormone |
| Cellular targets | Few | Many |
| Biological role | Fighting infection, tissue repair | Homoeostasis |
| Biological redundancy | High | Low |
| Biological pleiotropy | High | Low |
| Present in the circulation | Rarely | Yes |
| Sphere of influence | Predominantly autocrine/paracrine | Widespread and distant from site of production |
| Inducers | External insults | Primarily physiological changes (except stress hormones) |
Many cytokines interact with each other and the effect of a cytokine may change depending upon the prevailing micro-environment. It is important to stress that cytokine synthesis, including that of the proinflammatory cytokines, is a normal response to injury. Their functions should not be thought of as exclusively harmful, resulting in tissue damage or systemic shock. In the majority of instances, cytokines act to coordinate the elimination of invading organisms or removal of damaged tissue, thus avoiding excessive stimulation of specific immunity that could lead to hypersensitivity reactions.
Inflammatory cytokines
The cytokines interleukin (IL)-1β, IL-6 and tumour necrosis factor alpha (TNFα) are all central to the initiation of the inflammatory response. However, they do not function in isolation: other cytokines implicated in the inflammatory response include IL-8, 10, 11, 12, 17, 18, 23 and interferon γ. In addition, both defensive and injurious effects will involve the release of many mediators of inflammation such as peptides, for example the complement fragment C5a, and lipids, for example platelet activating factor. Many of these mediators act synergistically and also induce each other’s production and the production of other cytokines, resulting in both positive and negative feedback control pathways. Cytokines are also responsible, either directly or indirectly, for healing and successful resolution of the inflammatory insult. Severe inflammatory responses frequently overlap with specific immune responses, as they are often induced by infection and, furthermore, the tissue damage accompanying severe trauma may lead to secondary infection.
Mechanisms of immunological damage
Immune responses are usually well controlled and host damage is minimal and reversible. Under some circumstances, resolution does not occur and an exaggerated or persistent response leads to irreversible host tissue damage and, in extreme situations, even death. These inappropriate reactions are termed hypersensitivity reactions. These are classified into four types (I-IV), each with a different mechanism. Type I hypersensitivity reactions, central to allergic responses, may occur without involvement of any other type of hypersensitivity reaction. Most reactions, however, involve more than one type of response and do not, alone, define a pathogenetic mechanism that underlies a group of diseases. There are also other reactions that do not fit neatly into this classification, for example stimulatory reactions, such as when specific immune cells are stimulated by external agents, and when autoantibodies have a stimulatory effect, for example as occurs with thyroid receptor antibodies in Graves disease.
Hypersensitivity reactions result in the release of inflammatory mediators. Some mediators have direct effects on local, or even distant, tissues; others recruit and activate effector cells that further contribute to tissue damage.
Type I hypersensitivity
Type I hypersensitivity reactions are IgE mediated. The IgE antibodies are formed to an antigen (or allergen), with an individual’s tendency towards making IgE being determined by many factors including genetic, T cell responsiveness and antigenic burden. The IgE binds to high-affinity IgE receptors on the surfaces of mast cells and basophils, and these cells are now primed to react the next time the cells come into close proximity with the allergen. The cross-linking of IgE on the cell surfaces causes rapid cellular degranulation and liberation of a number of chemical mediators. The mediators released by mast cell degranulation include the preformed molecules histamine, protease enzymes, proteoglycans (heparin) and chemotactic factors. Reaction of antigen with IgE on mast cells also stimulates synthesis and release of platelet activating factor (PAF), leukotrienes (B4, C4 and D4) and prostaglandins (mainly PGD2). The mediators of type I hypersensitivity reactions are shown in Table 30.14.
TABLE 30.14
Mediators of type I hypersensitivity reactions
| Mediator | Pharmacological effect |
| Histamine | Vasodilatation, capillary permeability, bronchoconstriction |
| Heparin | Control of histamine release |
| Leukotrienes (various) | Bronchoconstriction, airway tissue oedema |
| Prostaglandins (various) | Potent mediators of inflammatory response |
| Platelet activating factor | Platelet aggregation |
| Tryptase | Proteolytic enzyme activates C3 |
| Kininogenase | Kinins → vasodilatation → oedema |
| Cytokines (IL-5, IL-8, TNFs) | Chemoattractants |
The actions of histamine depend on the site of release. In the airways, it induces smooth muscle contraction; in the skin, it causes the hallmark wheal and flare response. Widespread activation of mast cells leads to systemic effects of circulatory shock, hypotension, collapse, chest tightness and, in the most severe cases, respiratory arrest and death: this is anaphylactic shock. Type I hypersensitivity reactions occur rapidly (within approximately 20 min of an insult) and are also called ‘immediate hypersensitivity reactions’.
Type II hypersensitivity
The important characteristic of type II hypersensitivity reactions is that the antigens involved are localized to the membranes of the target cells; they may be synthesized cell products, cell membrane components or bound foreign molecules (e.g. a drug). The circulating antibodies (IgG or IgM) bind to the cell-bound antigen and activate complement. Full activation of the complement pathway results in the membrane attack complex (C5–9) being assembled on the cell surface, resulting in target cell lysis. Partial activation via C3 will also render the cell a target for phagocytosis, via C3b and IgG that bind to appropriate receptors on phagocytic cells. Activation of phagocytic cells leads to release of further tissue-damaging enzymes. The tissue damage persists for as long as antibody and antigen are present.
Some of the more important type II responses involve red blood cell antigens (including transfusion reactions and haemolytic disease of the newborn) and autoantibodies to cell surface components, for example autoimmune haemolytic anaemia (in which the target is red blood cells), idiopathic thrombocytopenia purpura (in which the target is platelets) and Goodpasture syndrome (in which the target is the kidney glomerular basement membrane).
Type III hypersensitivity
Type III hypersensitivity reactions are also termed immune complex reactions. Complexes of antigen and antibody form in the circulation and are then deposited in susceptible tissues; they may also form directly in the tissue. The latter mechanism is termed the Arthus reaction, and is typically seen with repeated insect stings, where a red swollen lesion develops after a sting. The tissue damaging mechanisms are similar to those described for the antigen-antibody complexes that form in type II responses. The response times of types II and III hypersensitivity reactions are slower than that of type I reactions; they typically develop 3–6 h after exposure to antigen. The response can also become chronic, particularly in autoimmune reactions, where antigen persists.
The clinical manifestations of type III hypersensitivity reactions relate to the tissue deposition, for example vasculitic (skin), serum sickness (systemic), nephritis (kidneys) and extrinsic allergic alveolitis (lungs).
Type IV hypersensitivity
These reactions are cell mediated. Membrane receptors on sensitized T lymphocytes recognize antigen and the cells (particularly Th1) release cytokines that in turn activate macrophages. The major mediators of the hypersensitivity response are products of activated macrophages including enzymes, coagulation factors, complement, superoxide ions, leukotrienes, prostaglandins and cytokines. The hallmark of responses to persistent antigen is formation of granulomas that wall off the inflammatory focus.
Type IV reactions are also known as delayed type hypersensitivity reactions because symptoms occur 24–48 h after re-exposure to antigen. They are seen in response to mycobacteria (e.g. Mycobacterium tuberculosis) and are the mechanism underlying contact dermatitis.
Conclusion
This brief introduction has described the major components of the immune system and their interactions during immune responses. Antigens, typically encountered across mucosal surfaces, are taken up by antigen presenting cells (APCs) and processed and presented on the major histocompatibility antigen (MHC) molecules of these cells to the lymphocytes in lymphoid tissue. The lymphocytes, with their randomly rearranged receptors (TCR and Ig), traffic around the lymphoid tissue until activated by an antigen, whose three-dimensional shape can engage the receptor. Activated cells communicate via cytokines and various peptides and proteins to induce lymphocyte proliferation and maturation. Plasma cells of the B lymphocytic lineage secrete antibody that circulates, coats the mucous membranes and bathes the tissues. Re-exposure to a previously encountered antigen stimulates a rapid and specific response via antibody, complement, phagocytosis and T cell killing to provide long-lived immunity to infection. The complexity of the system makes it robust but also provides opportunities for something to go wrong and result in diseases of the immune system.
DISEASES OF THE IMMUNE SYSTEM
Introduction
Diseases of the immune system can be classified as follows:
• allergic reactions, which may result from any of the hypersensitivity reactions previously described or from direct activation of inflammatory mediators
• autoimmune disease resulting from immune reactions against body tissues
• malignancy of organs or cells of the immune system.
In addition, severe sepsis resulting from damage to the immune system rather than direct bacterial tissue damage may be considered as an immunological disease.
These disorders will be considered in more detail in the following sections.
Immune deficiency
Development of immunity in humans
The ability to recognize antigen is present at birth, but except on the rare occasions when there has been an intrauterine infection, antigens will not previously have been encountered. The newborn will not have memory responses so are potentially vulnerable to infection. In the first 3–6 months of life, during which many microbes will be met, immunity is provided by maternally derived IgG, giving time for a child’s own system to mature and to establish memory. Active transfer of IgG occurs from 12 weeks of gestation but the major portion is transferred after 32 weeks of gestation. A term baby should have adult plasma IgG concentrations. Premature babies will have reduced IgG concentrations and are more vulnerable to infection. Babies who grow poorly in utero may also have low concentrations. A diagram showing serum immunoglobulin concentrations from pre-term to adulthood is shown in Figure 30.10. Term babies have different immune responsiveness from adults, as shown in Table 30.15.
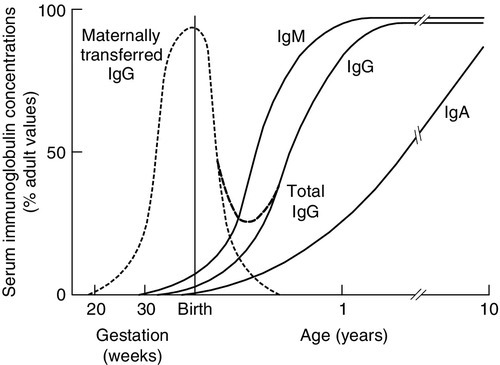
FIGURE 30.10 Serum immunoglobulin concentrations with age. IgG is actively transferred across the placenta to be gradually replaced by the IgG synthesized by the infant. The assays should be capable of reliably detecting concentrations for IgG of 1 g/L, IgA of 0.07 g/L and IgM of 0.1 g/L (after Adinolfi (ed). Immunology and development. Heinemann: London, 1969).
TABLE 30.15
Immune responsiveness in term neonates
| Components | Responsiveness |
| B cells | Normal numbers but immature (CD5+) |
| Antibodies | Able to make IgM, with good response to protein antigens but poor response to carbohydrates |
| Maternal IgG | |
| Complement | Classical pathway, 90% of adult; alternative pathway, 60% of adult; C8 and C9, only 20% of adult |
| T cells | Higher numbers than in adults and immature neonates |
| Cytokines | IL-2, as adult; IFNγ, 20% of adult; Th2 cytokines very low |
| Cytotoxicity | Tc only 30–60% of adult; NK 50% of adult |
Deficiencies of the immune system can be due to the lack of a component (or group of components) or defective (or absent) function of a component. Owing to immune interactions, defects in one component may affect another. For example, T cell defects may also result in marked antibody deficiency when T helper activity is compromised.
Infection and immune deficiency
The hallmark of immune deficiency syndromes is frequent, severe or unusual infection. The pattern of infections can give an indication of the type of deficiency, although in young children, this may be difficult to judge. It is estimated that children have eight minor infections per year and it is suggested that significant infection should be regarded as one requiring antibiotic treatment. Even this can be misleading, as the threshold for prescribing antibiotics varies between practitioners. The infections typically seen in specific and non-specific immune deficiencies are summarized in Table 30.16. In general, systemic viral and fungal infections are more typical of T cell deficiencies and common respiratory tract and gut infections are more typical of B cell deficiencies. With regard to the non-specific immune system, defects in complement tend to predispose to most common bacterial infections and impose an increased risk of autoimmune disease, while defects in phagocytes are linked with most common skin infections, cutaneous abscesses and lung abscesses.

In addition to the pattern of infections, there are other factors that can be used to build up an impression of whether immune deficiency should be considered. These are listed in Box 30.1.
Investigation of patients with suspected immune deficiency
The various categorizations of immune deficiency (primary or secondary, cell mediated or humoral etc.) are made at the end of the diagnostic process, not the beginning. The investigation of patients with recurrent, atypical or severe infections, irrespective of age, should follow the same basic pattern, but the detailed investigative process needs to be tailored to the individual patient. A careful history, including the type of symptoms, age of onset of symptoms, organisms responsible (where known) and family history (particularly of immune problems) is essential. Microbiological investigations and basic tests like a full blood count and white cell differential are vital. The process aims to determine the following:
• are all the components of the immune system present?
• are all the components functional (alone or in consort)?
• can the system respond to a defined challenge?
• what is the molecular defect?
As a general guide, the simple tests, and those that are important in the more common immune deficiencies, should be done first. The more complex, functional and genetic tests should be done later in the investigation process (unless there are strong indications from the clinical manifestations or family history). Figure 30.11 shows a basic plan for the immunological investigation of a patient with suspected immune deficiency. Following this plan should make it possible to classify the type of immune deficiency.
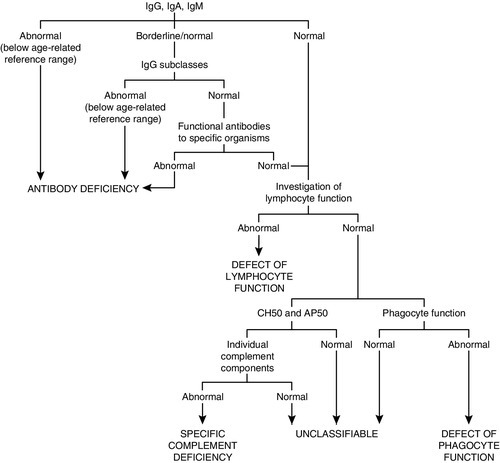
FIGURE 30.11 A basic plan of investigations in suspected immune deficiency (after IUIS/WHO 1981, 1988, 2009).
Primary immunodeficiencies
Clinically significant primary immune deficiency is rare. Many patients suffer years of ill health before they are appropriately investigated and a diagnosis made. The classification of these disorders is by an International System (see Further reading, below) based on the identified defect.
B Lymphocyte (humoral) system
IgA deficiency
This is the most common of the primary immune deficiencies, affecting approximately 1:700 of the population, and is characterized by low plasma IgA concentrations (< 0.07 g/L for total IgA deficiency). Patients are often asymptomatic, although they can suffer with repeated respiratory tract and sinus infections, especially if IgA deficiency is associated with IgG subclass deficiency. No particular treatment is indicated, although, if the patient is symptomatic, a low threshold for antibiotic treatment should be adopted. Salivary IgA concentration matures at approximately six weeks of age and can be measured to give an early indication of deficiency. Patients with IgA deficiency can develop antibodies to any IgA present in blood products and this can cause transfusion reactions.
Common variable immunodeficiency (CVID)
This is a primary immune deficiency, although the age of presentation can be any time from childhood to late in adulthood. It is not one clearly defined disease, rather a heterogeneous group of undifferentiated syndromes with the common feature of defective (quantitative and/or qualitative) antibody production. The most common presentation is with recurrent sinus and pulmonary infections. The critical laboratory test is quantification of immunoglobulin concentrations (with serum and urine electrophoresis to exclude myeloma). These are typically well below the lower limits of the age-related reference ranges. Patients can also have poor responses to vaccines, low CD4 counts, abnormal cell-mediated immunity and changes in the B cell subset profile. Treatment is with intravenous or subcutaneous immunoglobulin (IgG) replacement, with the dosage being calculated on the basis of body weight. Antibiotics can be given prophylactically or a low threshold adopted for their use in treatment. The IgG should be given every 2–3 weeks and trough immunoglobulin concentrations should be checked regularly, particularly in children.
X-linked agammaglobulinaemia
This condition, also known as Bruton agammaglobulinaemia, is caused by mutations in the gene for a B cell-specific tyrosine kinase (Btk). This results in no B cell development in the bone marrow, a lack of circulating B cells and profoundly low immunoglobulin concentrations. Patients present with recurrent bacterial infection from 4–5 months of age, once protective maternal IgG has disappeared. Treatment is with replacement immunoglobulin, as in patients with CVID.
T Lymphocyte (cell-mediated immunity) system
Severe combined immunodeficiencies (SCID)
This is a group of deficiencies with a variety of underlying molecular defects. They are the most profound of the immune deficiency diseases and patients typically present in early infancy. The most common of the diseases is the X-linked SCID that results from a defect in the common γ-chain of the IL-2 receptor. This affects the responsiveness to IL-2, IL-4, IL-7, IL-9, IL-15 and IL-21, so that there is no T cell or NK-cell function; although B cells can be detected, they have no T cell help to make antibodies. The lymphocyte count and T and B cell numbers are the most important investigations when considering SCID; immunoglobulin concentrations are not particularly helpful (owing to the presence of maternal IgG, and the normally low IgA and IgM concentrations expected in young babies). Any baby with suspected SCID should be referred to a paediatric immunologist for investigation. Treatment is with stem cell transplantation or, more recently, gene therapy.
DiGeorge syndrome
This condition is associated with deletions in the chromosomal region 22q11. It is a clinical spectrum of developmental defects including congenital cardiac defects, dysmorphic facies (including cleft palate), immune deficiency secondary to thymic hypoplasia and hypocalcaemia due to parathyroid aplasia or hypoplasia. The abnormal development of the thymus results in reduced T cell numbers and/or function and increased susceptibility to infection. The immunological abnormalities, which are seen in approximately 25% of patients, usually improve with age.
X-linked hyper-IgM syndrome/CD40 ligand deficiency
This immune deficiency is due to a defect in the gene for a cell surface receptor called CD40 ligand (CD154). The binding of CD40 (on antigen-presenting cells and B cells) to CD40 ligand (on T cells) is vital for B cell maturation and class switching. Presentation is similar to X-linked agammaglobulinaemia but these patients are also susceptible to opportunistic infections. They typically have low plasma concentrations of IgG, IgA and IgE and increased concentrations of IgM (as high as 10 g/L). Assessment of the expression of CD40 ligand on T cells is the most important investigation. The bacterial infections can be well controlled with replacement IgG, but patients may develop infections with Cryptosporidium, and can develop a sclerosing cholangitis and liver failure. Rare, autosomal recessive forms of hyper-IgM syndrome also exist.
Phagocytic (polymorphonuclear and mononuclear) system
Chronic granulomatous disease
This condition, which can be X-linked or autosomal recessive, is caused by abnormalities in the NADPH oxidase system. This is critical in the respiratory burst and the production of antimicrobial free radicals in neutrophils. Patients present with recurrent abscesses or infections and granulomata. An abnormally low respiratory burst in the neutrophils is the most significant finding; this property is increasingly being investigated by flow cytometric assays. Molecular testing is used to confirm the diagnosis.
Leukocyte adhesion defect types I and II
Recruitment of neutrophils, lymphocytes and monocytes into sites of infection relies on the presence of adhesion molecules, for example CD11a/18 and selectins. Absence or deficient expression of these molecules on cell surfaces results in poor cellular adherence, migration and phagocytosis. Patients present with recurrent, life-threatening bacterial infections and impaired wound healing.
Complement system
Deficiencies of all the complement components have been identified but have varied clinical consequences. Deficiencies of C3 and C6 are particularly associated with recurrent infections. The most important initial investigation is assessment of the integrity of the whole complement system, using haemolytic assays of the classical and alternative pathways or enzyme linked immunosorbent assay (ELISA)-based total complement pathway tests. If these are abnormal, individual components can be investigated: this usually requires referral to specialized centres.
C1 Esterase inhibitor deficiency
This inherited (autosomal dominant) deficiency of the control protein C1 esterase inhibitor results in hereditary angioedema – episodes of subepithelial swelling of the skin (especially of the face, limbs and trunk), gut, larynx etc. The major laboratory findings are low C4 and C1 esterase inhibitor concentrations but normal C3 concentrations. There are rare forms in which antigenic C1 esterase inhibitor concentrations are normal but function is impaired and C4 concentrations are usually low. Measurement of C1 esterase inhibitor should also be considered in patients with apparent anaphylaxis, as the two conditions have similar features in their early stages. However, it should be noted that C4 concentrations may also be low post-anaphylaxis. C1 esterase inhibitor deficiency can also be seen secondary to B cell malignancy when the monoclonal protein interferes with the normal activity of C1 esterase inhibitor.
Transient hypogammaglobulinaemia of infancy
This condition is not strictly a primary immune disorder but is mentioned here for completeness. Some babies show a very slow maturation of their own antibody production, resulting in a lengthening of the physiological trough in concentrations after maternal IgG has disappeared. These babies show normal responses to immunization and will eventually achieve normal antibody concentrations. No particular treatment is indicated, but patients should be monitored regularly in case antibody concentrations do not improve or they become symptomatic.
Secondary immune deficiency
Secondary immune deficiencies are significantly more common than primary immune deficiencies. They are the predominant form in adults, although they do occur in children. Investigation should concentrate on identifying the underlying process rather than identifying the nature of the defect in immunity. The causes of secondary immune deficiency are summarized in Table 30.17.
TABLE 30.17
Causes of secondary immune deficiency
| Cause | Examples |
| Immunosuppression | Corticosteroids |
| Chemotherapy | Cyclophosphamide, methotrexate, vincristine |
| Radiotherapy | Total body irradiation |
| Malnutrition | Protein malnutrition, vitamin deficiencies, trace element deficiencies |
| Infection | HIV, cytomegalovirus (CMV), Epstein–Barr virus, some bacteria, e.g. mycobacteria post-septicaemia |
| Malignancy | Myeloma, lymphoma, leukaemia |
| Loss of protein(or other components) | Nephrotic syndrome, protein losing enteropathy, loss from skin, e.g. burns |
| Miscellaneous | Trauma, type 1 diabetes, splenectomy, inflammatory bowel disease |
There are two broad mechanisms of secondary immune deficiency: inadequate production of a component or excessive loss of a component. Iatrogenic causes of immune deficiency rarely warrant investigation as they are accepted consequences of treatment for other (usually more severe) diseases. Malnutrition is the single most common cause of immune deficiency worldwide.
The most clinically important virally induced secondary immune deficiency is that related to HIV infection. The investigation of this relies particularly on measurements of CD4+ T cell numbers and viral load. Infections with other viruses, for example Epstein–Barr virus and cytomegalovirus, may be associated with some degree of secondary immune deficiency (usually of the cell-mediated responses).
Adults with symptoms of immune deficiency without an obvious cause should be investigated for lymphoid malignancy. These conditions can affect both the cell- and antibody-mediated immunity, making the patient particularly prone to infection. The investigation of lymphoid malignancies is discussed on p. 589.
Protein loss
The most frequent type of immune deficiency associated with excessive component loss is hypogammaglobulinaemia. The loss of immunoglobulins, if sufficiently marked to cause low plasma concentrations, is certain to be accompanied by the loss of albumin (a protein of lower molecular weight and higher plasma concentration than the immunoglobulins). It is useful, therefore, to check serum albumin concentrations together with immunoglobulin concentrations when investigating this group of patients. A normal albumin concentration with low immunoglobulins would suggest suppression of production rather than excessive loss. It is also vital to remember that protein loss via the kidneys can be seen with lymphoid malignancy so serum and urine electrophoresis must be performed. Protein loss through the gut can be difficult to prove, but a raised faecal α1-antitrypsin concentration is good evidence that protein is being lost into the gut. α1-Antitrypsin is used because, as a protease inhibitor, it is relatively resistant to proteolytic degradation in the gut; nevertheless, specimens must be frozen as soon as possible after collection to minimize degradation.
Splenectomy
Patients without a spleen, for example owing to surgical removal following trauma, have an increased susceptibility to overwhelming infection with bacteria such as Pneumococcus, Meningococcus and Haemophilus influenzae type B. Patients who are having elective splenectomy should be vaccinated with pneumococcal, haemophilus and meningococcal A and C vaccines; asplenic patients should take prophylactic antibiotics. The measurement of specific antibody titres (approximately six weeks post-vaccination) may be indicated to confirm adequate antibody responses. Asplenic patients may not maintain optimal antibody concentrations and these should be measured every 2–3 years, depending on the clinical findings. Patients found to have suboptimal concentrations should be re-immunized.
Allergies
Various mechanisms exist, whereby an individual can have an unexpected or inappropriate response to an agent that is not typically considered to be harmful. The mechanisms of such reactions include types I–IV hypersensitivity reactions, direct pharmacological effects (e.g. from biogenic amines in certain foods), biochemical effects (e.g. due to alcohol or other drugs) and intolerances due to enzyme defects (e.g. lactose intolerance). The term allergy is often used by lay people to describe such reactions but this term should only be applied to type I hypersensitivity reactions mediated by IgE.
The prevalence of allergies is increasing rapidly; it is estimated that 15–20% of the population have some type of allergy. The clinical features of allergic reactions include:
• anaphylaxis
• eczema and atopic dermatitis
• bronchospasm
• conjunctivitis
• diarrhoea
• headache
• malabsorption
• pneumonitis
• pruritus
• rhinitis
• urticaria
• vomiting.
Patients may have only one symptom or a mixture of symptoms. They can range in severity from mild and inconvenient to damaging to health and, in the extreme, to life-threatening anaphylactic shock.
Investigation of patients with allergies
It is important to investigate patients with suspected allergy to provide a logical basis for exclusion of the allergen, with the objective of improving well-being and preventing severe reactions. Investigation must start with the patient’s history and examination; the important points in the history are shown in Table 30.18. The exceptions are patients who complain of mild symptoms clearly related to eating one particular food, who often do not need investigation and should simply be advised to avoid that food, and patients with mild reactions to inhaled allergens, for example hay fever and seasonal rhinitis, who should attempt to avoid exposure and be treated symptomatically. However, if a patient has had a severe reaction, investigation is indicated to identify positively the causative agent. Accepted reasons for investigating patients with allergies are shown in Table 30.19.
TABLE 30.18
Important points in taking the history in a patient with suspected allergy
| Point | Notes |
| Age | Babies and young children should be investigated by people with appropriate training. Allergy can have a wider impact in children – loss of schooling, poor health, exclusion from normal activities |
| Family history | A positive family history increases the likelihood of allergy in an individual |
| Symptoms (what symptoms and where, e.g. gut, respiratory, skin etc.) | Can help to narrow down the allergen and direct investigation |
| Symptoms (severity) | Severe symptoms, e.g. anaphylaxis, need rapid investigation |
| Symptoms – all year or seasonal | Can help to identify the allergen and direct the testing, e.g. respiratory symptoms all year round are unlikely to be due to pollen allergy |
| Daytime or night-time, indoors or outdoors | Can help to identify the allergen and direct the testing, e.g. symptoms at night are more likely to be due to house dust mites, feathers etc. |
| Frequent animal contact | Household pets, but also animals of friends and relatives etc. |
| Does the patient know (or believe) what causes the symptoms? | Often the patient knows what precipitates the symptoms – but they may be wrong |
| Has exclusion been attempted? | Patients often spontaneously avoid something that makes them unwell |
TABLE 30.19
Indications for investigating patients with allergies
| Reason | Notes |
| Severe symptoms | Anaphylactic reaction or symptoms affecting patient’s overall health |
| Progressively worsening symptoms | Every time the patient encounters the allergen, the symptoms appear to be worse – next time it could be anaphylactic |
| Allergen is difficult to exclude or hidden | Foods, e.g. nuts, eggs, occupational allergens, particular tree pollens, favourite pet |
| Patient needs a diagnosis | Showing a patient test results may help them accept their condition and accept the reason for the proposed management, particularly if it involves dietary or other restrictions |
There are two general ways of investigating allergy but they are only useful as an adjunct to a good clinical history and examination. They are skin prick testing and measurement of IgE (total and specific). Both are widely used but have advantages and disadvantages, as shown in Table 30.20. In practice, both methods of investigation have a role in the allergy clinic and the allergist will use a combination depending upon a patient’s age, symptoms, the putative allergens and the overall clinical picture.
TABLE 30.20
Comparison of skin prick testing and specific IgE in the investigation of allergy
| Property | Skin prick testing | Specific IgE |
| Specificity | Atopic patients may show positive skin tests to several antigens, not all of which cause symptoms | Atopic patients may show positive specific IgE to several antigens, not all of which cause symptoms |
| Applicability | In vivo test, so assesses the patient’s response to the allergen | In vitro test, so more a test of the potential to react to an allergen |
| Use in children | Can be difficult in young children because they have to sit still for ~ 20 min | Requires a blood test but many allergens can be checked on a single specimen |
| Dermatological symptoms | Can be impossible in patients with extensive skin disease, e.g. eczema, dermatitis | Dermatological disease does not interfere |
| Risk | Must be done under medical supervision with resuscitation facilities available | As for a blood test |
| Convenience | Results available in the clinic within ~ 20 min | Samples need to be tested in the laboratory; results in days or weeks |
| Antihistamines | Limited use if the patient is taking antihistamines | Antihistamines do not interfere |
| Sensitivity | Can be insensitive for diagnosis of food allergy | Can give negative results even with highly suggestive history. Specific IgE may be negative in the first few weeks after a severe reaction |
| Allergens | Can be difficult if the allergen is toxic or insoluble test material often less purified and similar to native allergen | Can be used for toxic or insoluble allergens Allergens are highly purified and can show variability between different manufacturers |
| Standardization | Subjective with interoperator variability Saline and histamine must be used as negative and positive quality controls Results reported as wheal and flare diameter |
Objective with results reported in arbitrary kAU/L of IgE (or grades) related to the international reference preparation External QA schemes available to validate results |
Raised plasma total IgE concentrations are seen in allergic disease, parasitic infections, immune deficiencies, malignancies, liver disease and some viral infections. With regard to allergies, there are some important caveats. While a raised total IgE concentration suggests a high risk of allergic disease, it does not directly relate to the severity of disease. An IgE concentration within the reference range (particularly in children) does not exclude significant allergy and if there are strong clinical indications, further investigation is warranted.
The IgE that has activity against a particular defined antigen is called specific IgE. The term RAST refers to the technique that was first developed to detect and quantify IgE to specific allergens and, although now scientifically outdated, the term is still commonly used. Specific IgE is reported as kAU/L or converted to a grade based upon the concentration; both these results can be used as a guide to the patient’s potential reactivity to that allergen (or group of allergens). (The subscripted ‘A’ refers to ‘allergen’; however, the units are arbitrary in that they are calibrated against a total IgE standard rather than an allergen-specific IgE standard.) The grades corresponding to various ranges of specific IgE concentrations, and their significance, are shown in Table 30.21. Like total IgE, the specific IgE does not correlate with disease activity between patients. However, in individual patients (particularly children), there is some predictive value in specific IgE concentrations, for example to predict whether a challenge test would be appropriate, so reporting specific IgE in units rather than grades is now accepted.
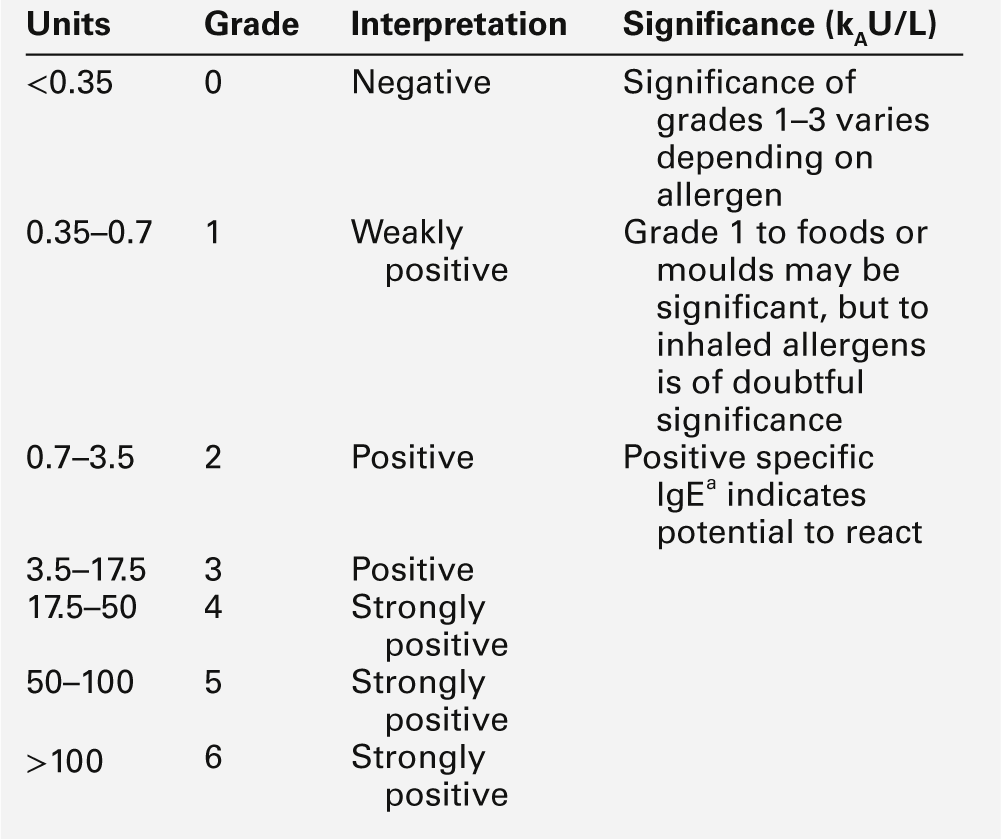
Anaphylaxis
Anaphylaxis is a medical emergency. It is the sudden, generalized shock and collapse that occurs when patients react to substances to which they are exquisitely sensitive. The clinical features (see Table 30.22) are induced via an IgE-mediated mechanism (anaphylaxis), but similar, non-IgE mediated (anaphylactoid) reactions also occur. The precipitating agents are varied, with drugs, bee and wasp venoms, peanuts, latex, fish and eggs being the most common. The mediators involved are shown in Table 30.9. Initial treatment should be aimed at protecting respiratory and cardiovascular function and tissue oxygenation. Adrenaline (epinephrine), antihistamines and steroids are the mainstays of treatment, together with general measures (e.g. oxygen and intravenous fluid). Long-term management must include avoidance of the precipitating allergen (which in practice may be difficult). Patients with a history of anaphylactic reactions should carry a self-injecting adrenaline device (and be instructed in its use) to administer if they inadvertently encounter the relevant allergen. In some situations, for example sensitivity to bee and wasp venoms, desensitization can be successful.
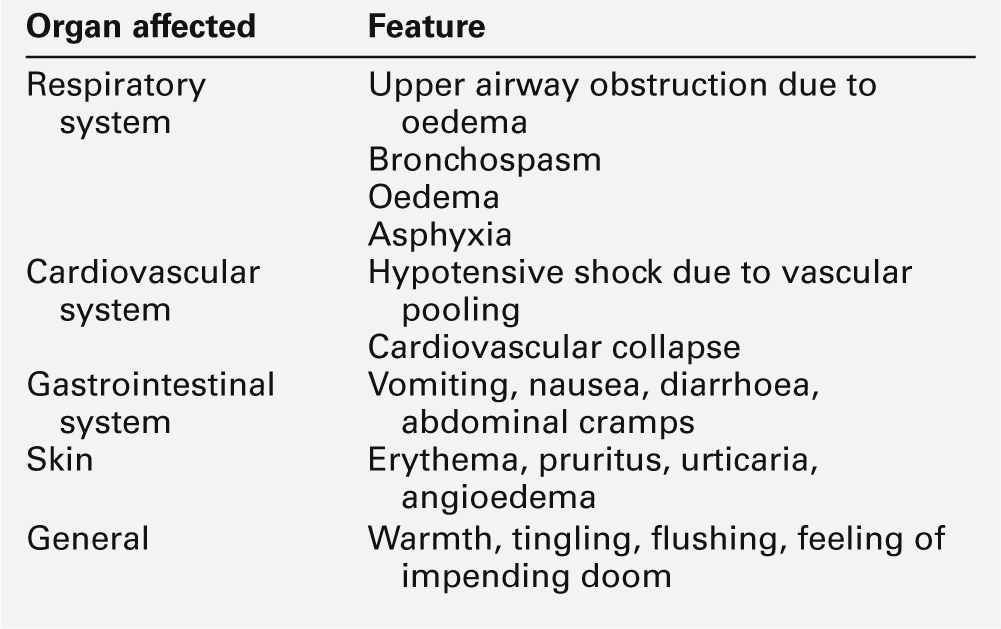
The investigation of anaphylaxis
It is important to confirm that an apparently anaphylactic reaction is a type I hypersensitivity reaction, and to try to identify the precipitating agent. Serum tryptase is a good marker of mast cell degranulation and can be used for this purpose; the minimum number of samples required is three: at times zero (as soon after the start of the reaction as possible and preferably within 30 min), at 1–2 h post reaction and a third sample 24 h post reaction or during convalescence. Ideally, additional samples at 3, 6 and 12 h should be requested to help interpretation. The peak concentration (> 50 μg/L) is reached 1–3 h after the start of the reaction; values gradually return to normal by 24 h. The timing for further investigation will be guided by the clinical situation but may involve closely supervised skin testing and/or total and specific IgE, generally with interpretation by an immunologist. Hereditary angioedema can cause symptoms very similar to those of a type I hypersensitivity reaction, so measurement of C3, C4 and C1 esterase inhibitor concentrations may be indicated.
Autoimmune diseases
The immune system is tolerant to self-antigens or makes only limited responses to them. Sometimes, this tolerance breaks down and the immune system makes a progressive response that attacks the body’s own tissue, resulting in autoimmune disease. The factors that trigger such responses are not well understood; many factors are likely to be involved, for example the patient’s immunological status, including their HLA types, any pre-existing inflammation and the presence of any infection. The initial damaging stage is usually cell mediated, and biopsies from affected tissue often show monocytic and lymphocytic infiltration. The production of autoantibodies can be part of the pathogenetic process or a result of a response to hidden antigens that are released as a consequence of cellular destruction. The general characteristics of pathogenetic antibodies and non-pathogenetic antibodies are shown in Table 30.23. The presence and the concentration of many of these antibodies are used to help diagnose and, occasionally, to monitor autoimmune diseases.
TABLE 30.23
Characteristics of pathogenetic and non-pathogenetic antibodies
| Characteristic | Pathogenetic antibodies | Non-pathogenetic antibodies |
| Antigen distribution | Cell surface antigens, e.g. glomerular basement membrane, acetylcholine receptor, thyroid stimulating hormone receptor | Antigens within the cell |
| Specificity | Antibodies rarely seen in other conditions | Antibodies often seen in the absence of clinical features |
| Concentration | Related to disease severity | Unrelated to disease severity |
| Mirrors disease activity | Unrelated to disease activity |
Autoantibodies can be detected by their reaction against tissue containing the relevant antigens, using subjective methods such as indirect immunofluorescence. Samples found to be positive by these sensitive (but not very specific) methods are then analysed, using more specific and quantitative methods, for example ELISA. Automated ELISA-based ‘screening’ techniques are increasingly being used, although the validity in a routine setting is questionable.
It is important that detection of these autoantibodies is not considered to be a ‘gold standard’ test. They are, at best, markers of disease: they have significant limitations and should be used as part of a diagnostic panel rather than the presence of individual autoantibodies being regarded as diagnostic of any particular disease.
An arbitrary distinction is often made between organ-specific and non-organ-specific autoimmune diseases, but in reality there is a spectrum of involvement ranging from only one or several organ(s) being affected to autoimmune diseases with systemic manifestations.
Autoimmune endocrine diseases
The autoimmune endocrine disorders include diseases of the thyroid, pancreatic islets, adrenal cortex, gonads, pituitary and parathyroids. More than one endocrine organ can be affected, either simultaneously or at different times during the patient’s life. Autoimmune disease affecting the parathyroids or pituitary is rare, and is not discussed further in this chapter.
Thyroid
Autoimmune thyroid disease is discussed in detail in Chapter 19. It includes primary myxoedema, Hashimoto thyroiditis and Graves disease. Measurement of thyroid antibodies is of limited use. Approximately 95% of patients with Hashimoto thyroiditis have antibodies to thyroid peroxidase. There is little point in measuring them in patients with obvious hypo- or hyperthyroidism. Measurement is best reserved for patients with equivocal thyroid function test results and when thyroid disease is suspected, but there are unusual clinical findings. Thyroid peroxidase antibody measurements are not required for monitoring patients on thyroid hormone replacement. Thyroid peroxidase antibodies are also found in Graves disease, but are neither of diagnostic value nor useful in monitoring the response to treatment. They are non-pathogenetic, so their concentration does not change in relation to disease activity.
Patients with untreated Graves disease have antibodies to the thyroid stimulating hormone (TSH) receptor molecule, to which they bind, stimulating the gland and causing hyperthyroidism. These antibodies are pathogenetic, but while the titre of antibodies correlates with thyroid gland hyperfunction, their measurement is not required routinely for diagnosis, nor is it used for monitoring treatment. The antibody is an IgG and capable of crossing the placenta and causing (transient) neonatal hyperthyroidism. A high maternal antibody titre in the third trimester indicates an increased risk of this condition. Less frequently, antibodies to the TSH receptor block the binding of TSH and cause hypothyroidism.
Pancreas
Type 1 diabetes is caused by progressive autoimmune destruction of pancreatic islet cells. Antibodies to islet cells and the enzyme glutamic acid decarboxylase can be found before clinical onset of disease and for a fairly short period early in its course, often disappearing later. There is considerable interest in how these antibodies participate in the immunological damage and whether the immune process may be susceptible to modulation. There is no clear indication for measuring these antibodies in most patients with diabetes, although they can sometimes be useful in patients with atypical clinical or laboratory findings. (More details can be found in Chapter 15.)
Adrenals
Approximately 60% of cases of Addison disease are due to autoimmune destruction of the adrenal cortex. Antibodies can be demonstrated to a number of adrenal cell components and these can show cross-reactivity to steroid-producing cells of the ovary, testis and placenta, potentially leading to gonadal failure.
Autoimmune polyendocrine syndromes (APS)
These are a well-recognized group of conditions where two or more endocrine organs are affected. Table 30.24 shows how the conditions may be classified.

Autoimmune diseases of the gut
Pernicious anaemia (see Chapter 27) causes impaired absorption of dietary vitamin B12. Patients develop a macrocytic anaemia and may develop neurological complications. Antibodies to gastric parietal cells and intrinsic factor are detectable in the majority of patients.
Coeliac disease is characterized by destruction of small intestinal villi, leading to malabsorption. It is triggered by an immunological response to the gliadin component of gluten (a protein in wheat and other cereals) but the endogenous antigen is endomysium, the tissue that surrounds the smooth muscle fibres of the gut. The initial response is T cell mediated but gradually antibodies (IgG and/or IgA) are made to gliadin, endomysium and tissue transglutaminase (the important antigen within the endomysium). Detection of antibodies is valuable in screening for the condition and may be sufficient for diagnosis in patients presenting with typical features (see Chapter 12, but the ‘gold standard’ diagnostic investigation is evidence of villous atrophy on jejunal biopsy. The UK National Institute for Health and Care Excellence (NICE) has issued a guideline on the diagnosis of coeliac disease (see: www.nice.org.uk/nicemedia/pdf/CG86FullGuideline.pdf).
Autoimmune liver diseases
The majority of cases of chronic hepatitis are due to viral infections (particularly with hepatitis B and C viruses) but approximately 20% of cases are autoimmune in origin. The distinction between autoimmune and infectious hepatitis is made mainly on the basis of the clinical features and the results of virological studies. It is important to identify the cause of chronic hepatitis, because interferon treatment (which is used for viral hepatitis) may worsen autoimmune liver disease and immune suppression (used for autoimmune liver disease) can worsen the viral disease. Unfortunately, patients with viral hepatitis sometimes make the very antibodies used in the investigation of autoimmune hepatitis.
Autoimmune hepatitis generally affects young females and is classified into types I and II. The typical age of presentation for type I is in the teenage years (∼10–20 years of age), while the peak age of presentation for type II is 7–8 years. Table 30.25 shows typical results of autoantibody measurements in autoimmune liver diseases. The antibodies are usually detected by indirect immunofluorescence. Considerable skill may be needed to interpret the immunofluorescence patterns and follow-up investigations, for example by ELISA, may sometimes be necessary. In general, high antibody titres are more closely associated with disease than low antibody titres. The hepatic autoantigens are often enzymes; for example, 2-oxoacid dehydrogenase is the specific antigen for the M2 subtype of mitochondrial antibodies that is the most specific for primary biliary cirrhosis.
TABLE 30.25
Investigations in liver disease, focusing particularly on the measurement of autoantibodies
b Antimitochondrial antibodies are virtually pathognomic of primary biliary cirrhosis.
Liver disease is not usually an indication for measurement of immunoglobulin concentrations, although abnormal immunoglobulin concentrations are common in liver disease, particularly when chronic. For example, patients with primary biliary cirrhosis usually have markedly raised polyclonal IgM concentrations; patients with alcoholic liver disease tend to have raised polyclonal IgA concentrations, and patients with persistent infections or inflammatory responses show raised polyclonal IgG and IgA concentrations. However, these patterns are non-specific and of negligible diagnostic value. Serial measurements of IgG may, however, be of value in monitoring patients with autoimmune chronic hepatitis.
Autoimmune skin diseases
The blistering skin diseases, pemphigus and pemphigoid, are caused by antibodies to components of the skin. These IgG antibodies bind to antigens in the skin, activate complement and disrupt the skin basement membrane (pemphigoid) or skin intercellular cement (pemphigus) causing intraepidermal blisters.
Autoimmune kidney diseases
Immunological damage due to the deposition on the glomerular basement membrane (GBM) of immune complexes (either to antigens of the GBM (as in Goodpasture syndrome) or formed in the circulation), is an important pathogenetic mechanism in renal disease. When the clinical features suggest a diagnosis of autoimmune kidney disease, evidence from renal biopsy often provides a definitive diagnosis. However, laboratory data can reduce the need for biopsy. The antibodies particularly implicated in autoimmune kidney disease are shown in Table 30.26. Laboratories should have protocols that have been agreed with the local renal physicians for investigating patients with renal disease, but detection of antinuclear antibody (ANA), antineutrophil cytoplasmic antibodies (ANCA) and GBM antibodies should ideally be available on an urgent basis (although, note that in immunological terms, this means results being available within 24 h of request). Patients with ANCA-associated vasculitis and GBM disease can also have pulmonary involvement (Goodpasture syndrome can manifest as haemoptysis). The vascularity of the lungs and kidneys makes them particular targets for vasculitic disease and the antibodies formed to either glomerular or alveolar basement membrane often show cross-reactivity with the other membrane. The concentrations of GBM antibodies and of antibodies to proteinase 3 (and myeloperoxidase) can be used to monitor patients’ responses to treatment and, in the vasculitides, to provide an early warning of exacerbations of disease.
TABLE 30.26
Antibody mediated renal disease
| Disorder | Serological findings | Biopsy findings |
| Antiglomerular basement disease (Goodpasture syndrome) | Serum antibodies that react with normal basement membrane (GBMa) | Linear IgG, C3 along basement membrane |
| SLE nephritis | Antinuclear antibodiesa (see text) | Granular IgG, IgM, C3, C4 along GBM |
| Vasculitis, e.g. Wegener granulomatosis Microscopic polyarteritis |
Anti-neutrophil cytoplasmic antibodiesa (ANCA) Staining in the cytoplasm hence c-ANCA and is associated with antibodies to proteinase 3 ANCA Staining in the perinuclear area hence p-ANCA and is associated with antibodies to myeloperoxidase |
Inflammatory – nothing specific |
| Cryoglobulinaemia | Cryoglobulins (proteins that form a precipitate on cooling of serum) | IgG, IgM, C3 along capillary wall |
a The indirect immunofluorescent staining patterns are shown in Figure 30.12.
Testing for renal autoantibodies is essential early in the investigation of unexplained renal impairment. Such patients should also have their serum and urine investigated for paraproteins (including Bence Jones proteinuria). The investigation of samples for cryoproteins is often forgotten but can be a vital test. The cryoproteins of relevance are not those that are seen with large monoclonal components that precipitate in the cold in vivo. Rather, they are high molecular weight immune complexes associated with infectious agents, for example hepatitis B and C viruses, which lodge in blood vessels, activating complement and causing vasculitis. This process can affect any organ, but the kidneys, with their large vascular network, are a particular target. This cryoprecipitation is a laboratory artefact, generated by storage of serum samples in the cold; the amount of precipitate can be very small, but careful isolation and investigation typically reveals monoclonal or polyclonal IgM showing rheumatoid factor activity.
Autoimmune articular diseases
These diseases are also referred to as connective tissue diseases. They are discussed in more detail in Chapter 32. There is considerable overlap between the clinical features seen in individual conditions. Autoantibodies are used as markers of the diseases; their presence contributes to, but does not establish, the diagnosis.
Rheumatoid arthritis (RA)
Rheumatoid factor (RF) is an autoantibody (usually IgM) directed against the Fc portion of IgG. It is not specific for rheumatoid arthritis, also occurring in infections, malignancies, persistent inflammatory conditions and in up to 10% of healthy adults, with increasing frequency in elderly patients. A raised RF is one of the diagnostic criteria for RA but is not essential for the diagnosis. There is a correlation between higher RF concentrations and more severe disease and poorer long-term prognosis in RA; less severe disease is seen in seronegative patients. Rheumatoid factor is not of value in monitoring RA: measurement of C-reactive protein (CRP), reflecting the severity of inflammation, is superior. Changes in CRP precede both radiological and clinical changes.
The measurement of IgG antibodies to citrullinated peptides (anti-cyclic citrullinated peptide, CCP) is increasingly being used in investigation of patients with RA. It shows similar clinical specificity and sensitivity to RF but the presence of CCP antibodies can precede overt signs of RA. A combination of RF and CCP antibodies is becoming important to stratify patients with RA and to direct treatment, e.g. use of anti TNF. The UK National Institute for Health and Care Excellence has issued a guideline on the management of RA in adults (see: http://www.nice.org.uk/guidance/cg79).
Other connective tissue diseases
The detection of antibodies to nuclear components is one of the most important laboratory tests in the investigation of patients with suspected systemic lupus erythematosus (SLE), Sjögren syndrome and scleroderma. The term ‘antinuclear antibodies’ is used to describe this diverse group of autoantibodies that react with nuclear and cytoplasmic antigens that are common to all nucleated cells. The antigens typically have functions in the cell cycle, or in transcription and translation.
Antinuclear antibodies
Antinuclear patterns are detected by indirect immunofluorescence using either frozen sections of animal tissue or cultured human cancer cell lines, most commonly the epithelial cell line HEp-2. This is usually a very sensitive test and specimens that test positive for antinuclear antibodies then need testing for one of the nuclear antigens by specific methods. These specific tests use purified or partially purified antigens. There is a move towards using combined specific tests instead of the subjective immunofluorescence assays, but these methods are not yet widely accepted as potential replacements.
The pattern of the immunofluorescence reaction gives some indication of the antigen specificity because of the different distribution of antigens within the nucleus and cytoplasm. Figure 30.13 shows a diagram of a nucleus with the distribution of the important antigens. The more frequently identified and clinically useful antibodies are listed in Table 30.27. Specificities other than those listed rarely have clinical significance, are usually present at low concentrations and can be found non-specifically in many infectious and inflammatory conditions.
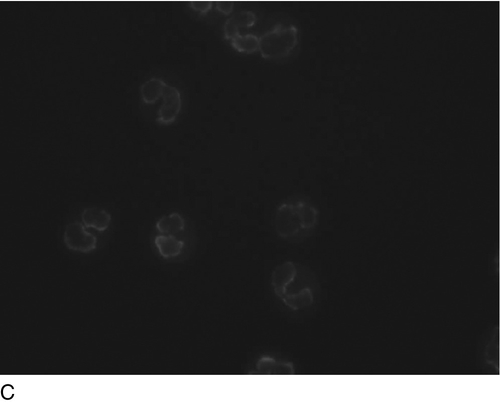
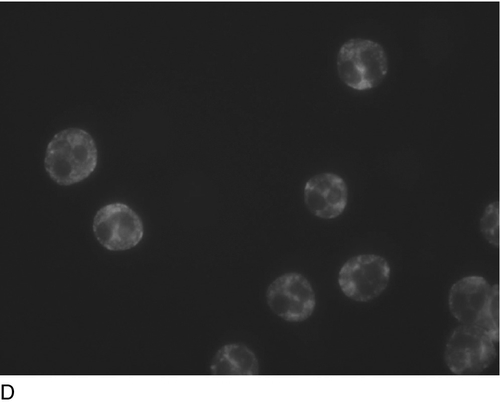
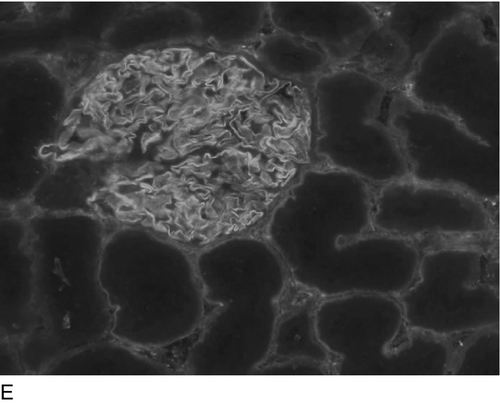
FIGURE 30.12 Indirect immunofluorescent patterns of: (A) homogenous and (B) speckled ANA; (C) pANCA and (D) cANCA and (E) anti-GBM antibodies.
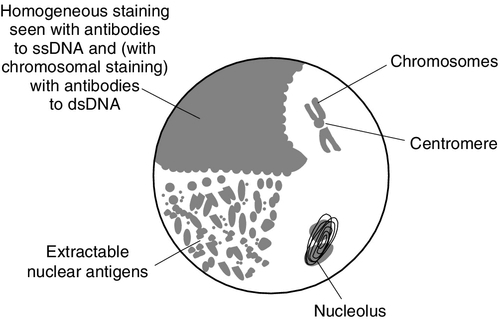
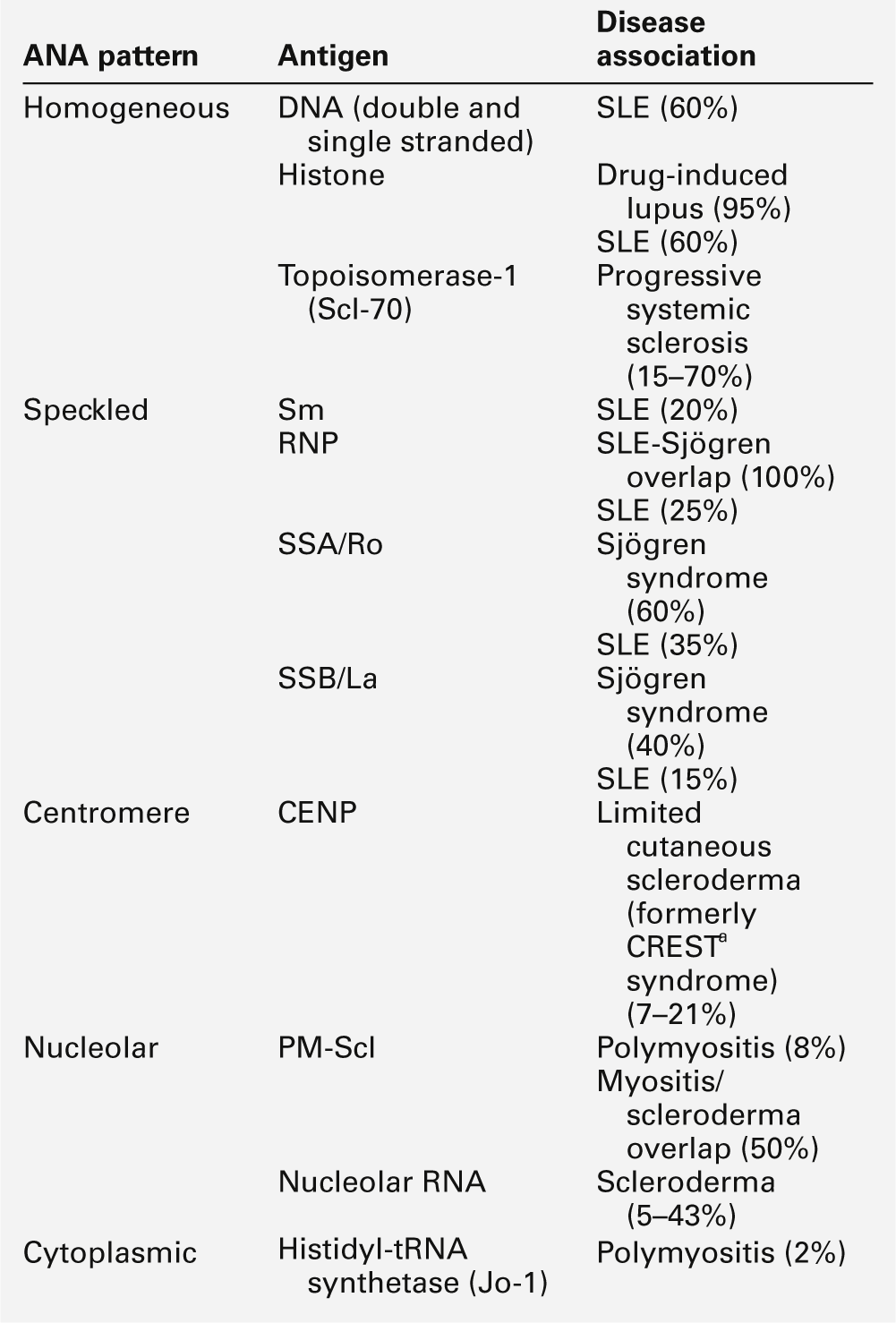
Antibodies to double-stranded DNA
Antibodies to single-stranded (ss) DNA are frequently present in SLE but can also be demonstrated in many other inflammatory conditions and in infections. Antibodies to double-stranded (ds) DNA are one of the important diagnostic criteria in SLE. The most frequently used methods for the quantification of anti-dsDNA are ELISAs, but the immunofluorescence assay using Crithidia luciliae is usually a more specific (although less sensitive) assay. High antibody concentrations to dsDNA are associated with renal complications of SLE and quantification of dsDNA antibodies is useful (together with C3 and C4 concentrations) to monitor the disease and response to treatment.
Antibodies to extractable nuclear antigens
Numerous antibodies to various nuclear or cytoplasmic protein antigens (commonly termed the extractable nuclear antigens or ENAs) are detectable in connective tissue disorders (see Table 30.27). They may be of diagnostic value but, in contrast to antibodies to dsDNA, there is negligible value in using these tests for monitoring disease activity.
Antinuclear antibodies in pregnancy
Antinuclear antibodies are of the IgG class and can therefore be transported across the placenta to the fetus. Antibodies to dsDNA can cause a transient, self-limiting, neonatal lupus syndrome. Antibodies to the Ro antigen cross-react with the fetal heart tissue and can cause neonatal heart block.
Antiphospholipid antibodies
These are a group of antibodies that are associated with an increased tendency for thrombosis. The most clinically relevant are antibodies to cardiolipin, β2-glycoprotein 1 and ‘lupus anticoagulant’. They may occur in some patients with SLE but can also be found in patients with recurrent miscarriages, unexplained transient ischaemic attacks, strokes or thrombotic events.
LYMPHOID MALIGNANCIES
Lymphocytes go through a complex process from the start of their lives as naïve lymphocytes to end-differentiated, functional lymphocytes. The complexity of the process means that there are many opportunities for a cell to undergo a malignant transformation that may result in malignant disease. The classification of lymphoid malignancies is based upon the normal physiological pathways and the place in this pathway where the malignant transformation has occurred. The leukaemias are malignancies of cells whose mature forms are normally found in the blood, while the lymphomas are tumours of the non-recirculating lymphoid cells. The diagnosis and management of lymphoid malignancy is predominantly the remit of haematologists. Flow cytometric analysis of peripheral blood for panels of CD markers is important as are cytological examinations of either lymph node or bone marrow biopsies. The detection of the products of monoclonal B cells, monoclonal proteins, is, however, a vital test in the investigation of patients with suspected B cell malignancy (the category that includes myeloma). There are no specific biochemical investigations for the diagnosis and management of malignancies of the T cell lineage, and the subsequent discussion therefore focuses on malignancies of B cells.
B lymphocytes and monoclonal proteins
Early B lymphocytes, with their randomly rearranged B cell receptors (IgM), leave the bone marrow and traffic around the lymphoid tissue, searching for appropriate antigens. Once a lymphocyte meets an antigen, there is a process of maturation to secrete high-affinity antibody (after class switching) and to maintain a memory response. Plasma cells are end-differentiated B cells that reside in the bone marrow and secrete immunoglobulins. There is normally considerable microheterogeneity in the immunoglobulin molecules. This results in polyclonal immunoglobulins, representing all classes and light chain types, with a large number of different idiotypes produced by a large number of different B cell clones. A monoclonal immunoglobulin of a single heavy chain class, light chain type and idiotype, results from the proliferation of a single clone of B cells. Monoclonal B cells, especially those that have undergone malignant transformation, do not necessarily produce normal, intact immunoglobulin: in approximately 20% of patients with myeloma, they produce only monoclonal free light chains (detectable in the urine as Bence Jones protein, BJP). Almost all variations on the normal immunoglobulin molecules are possible: monoclonal monomeric (7S), instead of the normal pentameric IgM, may be secreted, monoclonal fragments, for example free heavy chains, half molecules and truncated molecules can all occur; so, too, can combinations of immunoglobulin fragments and intact monoclonal immunoglobulins. The class of immunoglobulin produced by the tumour can give a clue to the place in the B cell development where the malignant transformation occurred. For example, tumours of early B cells often secrete monoclonal IgM, while tumours of end-differentiated B cells tend to secrete IgG or IgA.
Monoclonal proteins or fragments appear as homogeneous, compact bands on electrophoretic separations. These bands should be called monoclonal proteins but are often called paraproteins, M components or M proteins. Monoclonal proteins detectable in serum are most often intact immunoglobulins; if there is renal damage these proteins can leak into the urine. Low molecular weight fragments and monoclonal free light chains (BJPs) can pass readily through normal glomeruli and appear in the urine in the absence of renal damage but may also be detectable in the serum.
Clinical significance of monoclonal proteins
The finding of a monoclonal protein in the serum and/or urine can be associated with both malignant and benign conditions, as indicated in Table 30.28.
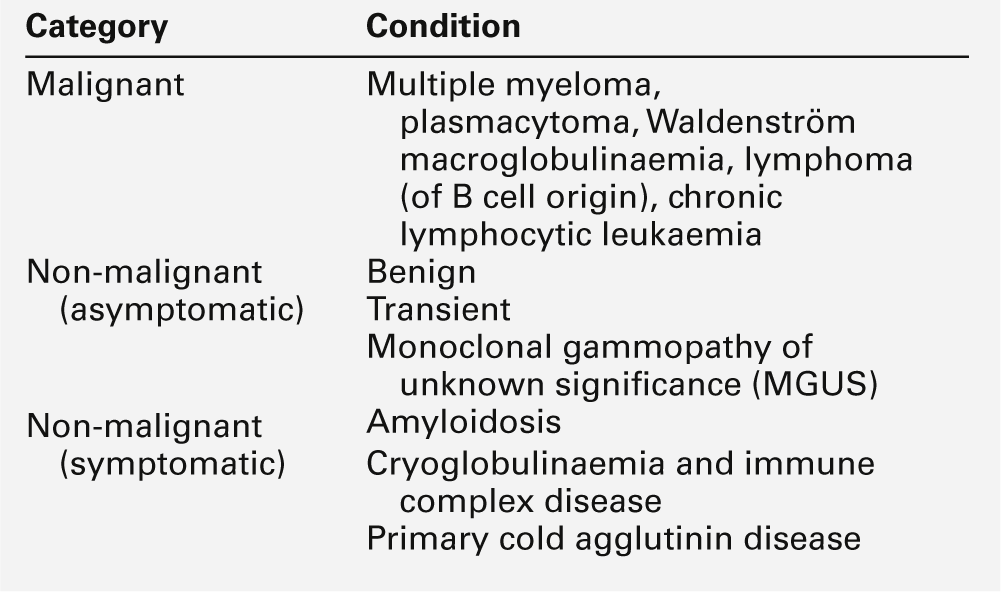
In malignant diseases, paraproteins are sensitive tumour markers (see below), but their presence in (apparently) non-malignant diseases limits their specificity. However, immunoglobulin fragments are rarely found in non-malignant monoclonal B cell expansions and their presence is thus highly suggestive of malignant B cell proliferation. The clinical features associated with paraproteinaemia can be due to the effects of the underlying tumour or to the properties of the monoclonal protein. Features related specifically to paraproteins are listed in Table 30.29. Some paraproteins also show autoantibody activity, for example against other immunoglobulins (e.g. rheumatoid factor) in immune complex disease and cryoglobulinaemia, against the I red cell antigen in primary cold agglutinin disease, and against myelin-associated glycoprotein in peripheral neuropathy.
TABLE 30.29
Clinical manifestations of the physicochemical properties of paraproteins
| Condition | % of patients with paraproteins | Main Ig classes |
| Hyperviscosity syndrome | 8 | IgG3, IgA and, particularly, IgM |
| Amyloid deposition | 8 | Bence Jones |
| Renal tubular obstruction | 6 | Bence Jones |
| Thromboembolism | 5 | IgG |
| Immune complex disease | 4 | IgM, IgG |
| Monoclonal cryoglobulinaemia | 2 | IgM, IgG |
| Fanconi syndrome | 1 | Bence Jones |
Monoclonal proteins can also occur secondarily to infection and as part of immune reconstitution post-transplantation; in these situations, they are generally transient, disappearing over weeks or months. Monoclonal proteins may also be found in patients without any overt clinical signs of malignancy; this is particularly true in the elderly.
Prevalence of monoclonal proteins
The prevalence of monoclonal proteins is dependent on the population being considered. In symptomatic populations (e.g. hospital patients), paraproteins are predominantly associated with B cell malignancy, of which myeloma is the most common. In asymptomatic populations, the prevalence depends on the sensitivity of the electrophoresis used to detect the paraprotein and the characteristics of the group that is screened. The incidence of paraproteins rises with age; a survey of over 9000 apparently healthy individuals showed paraproteins in 0.1% of those aged 20–30 years, in 0.6% by age 60 years and in 6% of subjects aged 90–99 years.
Laboratory investigation of paraproteins
The detection, typing and quantification of monoclonal components in serum and urine are essential laboratory investigations in the diagnosis and monitoring of both malignant and non-malignant monoclonal B cell expansions. A serum paraprotein and/or urine BJP is a highly sensitive tumour marker for certain B cell malignancies, particularly myeloma (> 95%) and Waldenström macroglobulinaemia (100%). In these conditions, the monoclonal component is indeed a diagnostic feature. However, they are not entirely specific for these conditions and can be found in other B cell malignancies and non-malignant diseases (see Table 30.28). It is important to note that the detection of a monoclonal protein does not necessarily indicate that the patient has myeloma or, indeed, any malignant transformation of B cells, nor does the absence of a paraprotein completely exclude the presence of myeloma or B cell malignancy.
The presence of a monoclonal immunoglobulin may be suspected from the patient’s clinical presentation or from results of other investigations. The findings that should, in the absence of another explanation, prompt investigation for paraproteinaemia, are shown in Box 30.2.
Identification of paraproteins
The detection of monoclonal proteins is one of the two clear indications for the measurement of serum IgG, IgA and IgM concentrations and protein electrophoresis; these investigations should be done as a logical group of tests for optimum interpretation; this approach is in line with the consensus recommendations of the International Myeloma Workshop Consensus Panel (see Further reading, below). Protein electrophoresis is the only reliable method for the detection of paraproteins in serum and urine. Abnormal bands (in the serum), once detected by electrophoresis, can be quantified by scanning densitometry or directly from the capillary zone electrophoresis trace. Immunofixation should be used to type the monoclonal band; this will confirm clonality and identify the heavy and light chains.
Automated capillary zone electrophoresis (CZE) systems are becoming the most common method for serum protein electrophoresis, although agarose gel based methods are still in frequent use. Capillary zone electrophoresis techniques for urine protein electrophoresis are available but sample preparation is complex and the methods not sufficiently robust for routine practice. Serum proteins are separated by electrophoresis at pH 8.6; both agarose and CZE systems generate five major zones. Figure 30.14 shows the typical mobility of the major serum proteins. Agarose separations should be long enough (3–4 cm) to allow good separation of zones and show a good spread of the β-γ zone, which is achieved by properties of the agarose gel and buffer system that produce high endosmotic flow. The majority of serum paraproteins migrate in the β or γ zones, although they may be found anywhere on the electrophoretic separation between the α1 zone and the post-γ region. Monoclonal heavy chains may show as diffuse zones running very anodally in the α2-β region of the electrophoretic separation; IgD paraproteins can also form a diffuse zone in the β-γ region. Representative examples of serum protein electrophoretic patterns are shown in Figure 30.14.
FIGURE 30.14 Representative serum electrophoresis patterns. Tracks 1 and 10 are dilutions of normal serum. Dots to the right of some of the tracks indicate bands that require immunofixation for further investigation.
Paraproteins may be missed if they are present at low serum concentrations (< 5.0 g/L), when their mobility coincides with other bands such as β-globulins, or where there is no suppression of normal background immunoglobulin concentrations. This is most often seen with low-concentration IgM or IgA paraproteins. Therefore, whenever there is a raised concentration of IgA or IgM with no clear increased staining in the β-γ region that would indicate a polyclonal increase, immunofixation should be done, as it should be also if there is a high clinical index of suspicion of B cell malignancy but an apparently normal electrophoresis. IgD paraproteins and free heavy chains are susceptible to post-synthetic degradation, which results in diffuse paraprotein bands on electrophoresis, and these may be missed if present at low concentrations or if the operator is expecting to see a narrow band rather than a diffuse zone. Precipitation of paraproteins on cooling of blood samples (monoclonal cryoglobulins) and subsequent removal with the clot, can also result in failure to detect a significant paraprotein. There are a number of proteins that can be mistaken for monoclonal proteins in electrophoresis separations.
These include:
• a haptoglobin–haemoglobin complex as a result of in vitro haemolysis, causing splitting of the normally homogeneous α2 zone
• acute phase proteins, e.g. C-reactive protein, increases, which may result in additional bands being present
• fibrinogen in plasma or inadequately clotted samples, which results in an additional band in the fast γ region
• lipoproteins, which may give distinct bands in the β region in agarose systems
• haemoglobin, which migrates in the β region.
Urine should be examined in every patient in whom B cell malignancy is being considered. This is especially important if there is suppression of the serum immunoglobulins or if there is renal impairment, and it should be done even where no serum monoclonal component is detected. It is also important to monitor the amount of BJP during a patient’s follow-up, even when BJP is not detected at presentation, because it can appear later in the course of the disease (‘Bence Jones escape’).
The presence of BJP provides strong evidence of malignancy, although it can occur in apparently benign conditions. The International Federation for Clinical Chemistry (IFCC) recommends a sensitivity for the detection of BJP of 10 mg/L: high-sensitivity electrophoresis of urine is therefore essential. This can be achieved by using very sensitive stains when electrophoresis is performed on unconcentrated urines, or by concentrating urines at least 100-fold. The concentration devices separate the water, ions and low molecular weight peptides from higher molecular weight constituents across a semipermeable membrane. The molecular exclusion of these membranes should be between 11.5 kDa and 13.0 kDa. Some concentration systems are static; others require centrifugation to achieve separation. Urine concentrates should then be analysed alongside serum in the agarose systems. Ideally, a trace of albumin should be visible in every urine sample; failure to detect any albumin on the electrophoretic separation should prompt further concentration and re-analysis. The renal damage associated with Bence Jones proteinuria often results in complex patterns; immunofixation improves both specificity and sensitivity, so is very useful to help resolve these patterns. Representative examples of urine protein electrophoretic patterns are shown in Figure 30.15. Low concentrations of BJP can be seen with a significant glomerular proteinuria in patients with light chain renal amyloidosis. Any urine with a marked glomerular proteinuria should be investigated by immunofixation, even in the absence of a band suggestive of BJP.
FIGURE 30.15 Representative urine electrophoresis patterns. Tracks 1 and 10 are dilutions of normal serum; all urine samples contain a trace of albumin. Significant bands that require immunofixation for further investigation are indicated by dots to the right of the respective tracks.
Several proteins can be mistaken for monoclonal protein in urine electrophoresis separations, particularly where there is an element of tubular proteinuria. They include the following:
• lysozyme (migrating in the slow γ region)
• degraded fragments of protein of glomerular origin
• seminal fluid proteins (very rare)
• β2-microglobulin (can produce a prominent band when present in high concentrations).
Measurement of serum free light chains is now routinely available. A combination of serum protein electrophoresis and free light chain measurement has been suggested as a suitable screening protocol for B cell malignancy. This approach is now being used in some laboratories and is further discussed on p. 596.
Typing of monoclonal immunoglobulins in serum and urine
Immunofixation
Once a paraprotein band has been detected, it is important to identify the heavy and light chain components. The major indications for immunofixation are to:
• confirm clonality of a band detected by electrophoresis
• test for α, γ or μ heavy chains and κ and λ light chains (routinely)
• test for δ and ε heavy chains when a serum shows monoclonal light chains without a corresponding α, γ or μ heavy chain
• exclude monoclonal components being present at low concentrations, even where no band is apparent on electrophoresis but when there are appropriate clinical indications, e.g. amyloid
• exclude the presence of monoclonal IGA or IGM if there are raised concentrations without β-γ fusion (the appearance when these bands lose their separate identities)
• investigate the possibility of an apparent paraprotein in serum or urine being caused by a high concentration of another protein (e.g. fibrinogen, complement components, β2-microglobulin) (using antisera specific to these proteins)
• detect minimal residual disease or complete remission following haematopoietic stem cell transplantation for myeloma when no monoclonal component is seen on the electrophoretic separation.
When monoclonality has been confirmed, determination of the paraprotein type may provide useful additional information about the underlying tumour and prognosis. The distribution of heavy chain classes in B cell dyscrasias is shown in Table 30.30. The light chain distribution of the common paraproteins is similar to that of normal polyclonal immunoglobulins, with a predominance of κ over γ (about 2:1). IgD paraproteins are usually of γ light chain type, although κ may be found, and the few IgE paraproteins that have been reported have all been of κ light chain type.
TABLE 30.30
Paraprotein types in B cell dyscrasias

a Values in brackets = typical serum concentrations in g/L (except BJP);
b NHL, non-Hodgkin lymphoma; CLL, chronic lymphatic leukaemia: both are tumours of B cell origin.
c Can also show Bence Jones protein;
f does not include intact monoclonal Ig with BJP or multiple bands of the same paraprotein type.
The electrophoresis pattern may change during the course of a patient’s disease or treatment. For example, some patients may start producing BJP after having not done so at the time of diagnosis. Complete disappearance of a paraprotein is rare but is occurring increasingly with treatment regimens using high-dose chemotherapy and bone marrow transplantation. An oligoclonal banding pattern is often seen in patients after stem cell transplantation and it is important to distinguish this from the original paraproteinaemia.
Immunofixation enhances the sensitivity of gel electrophoresis both by removing background staining and selectively increasing the amount of protein (by adding antibodies) in the band of interest. Representative examples of serum and urine immunofixation patterns are shown in Figures 30.16 and 30.17, respectively. The choice of antisera is paramount: problems with this technique are often related to the use of poor antisera. This is especially true for the antisera to free κ and λ light chains. Such antisera are available but often react poorly. The antisera most widely used for immunofixation react with both free and bound immunoglobulin heavy and light chains. The presence of free light chains can be inferred if no heavy chain reaction is present. Immunofixation is not a quantitative method; antisera often show greater binding to free than to bound light chains.
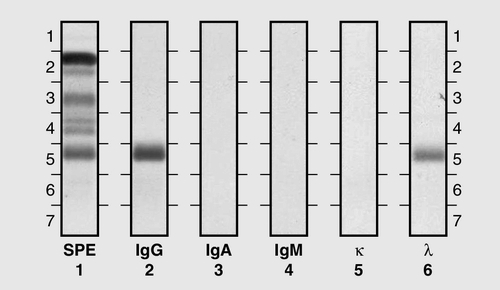
FIGURE 30.16 Representative immunofixation of a serum sample containing an IgGλ paraprotein.
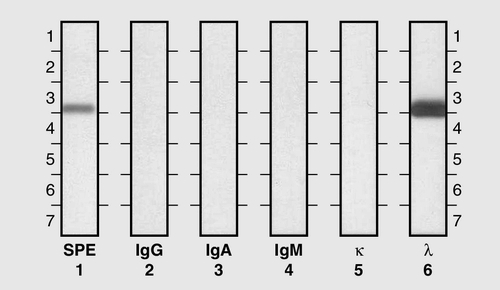
FIGURE 30.17 Representative immunofixation of a urine specimen containing a λ Bence Jones protein.
A major problem with immunofixation of urine is distinguishing between monoclonal light chains and light chain fragments generated from normal immunoglobulin catabolism, which result in a ‘ladder-like’ pattern, particularly in the immunofixation reaction with antiserum to κ light chains. This ‘κ banding’ pattern is most often seen in elderly patients with inflammatory conditions. Examples of BJP and κ banding are shown in Figures 30.17 and 30.18.
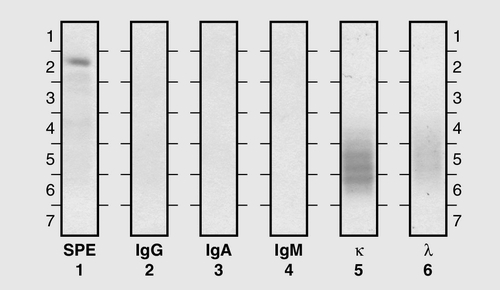
FIGURE 30.18 Representative immunofixation of a urine specimen showing κ banding (lane 5) (immunofixation will vary between manufacturers, in particular, the extent of separation).
Immunofixation techniques are not applicable in automated CZE systems; therefore, the technique of immunosubtraction has been developed for these systems. Immunosubtraction techniques work well for large monoclonal proteins but can lack sensitivity for small paraproteins and are inflexible; for example no reagents are available for typing IgD or IgE paraproteins or other non-immunoglobulin proteins.
In rare circumstances, the presence of free heavy chains may need to be confirmed. This is done by an adaptation of gel electrophoresis called immunoselection, or by molecular weight and immunoblotting methods that are best referred to expert laboratories.
Quantitation of monoclonal components
The concentration of a monoclonal component does not, in general, predict tumour mass. However, in individual patients, concentration does reflect tumour mass and is therefore an essential component in both the initial investigation and the assessment of the response to treatment of patients with B cell malignancies. Table 30.30 shows the average paraprotein concentration at presentation with myeloma with respect to the paraprotein type.
The only reliable method of paraprotein quantitation is to measure the percentage of the monoclonal protein (by scanning densitometry or from CZE read-out), with respect to the total electrophoretic separation or to the globulin fraction. The stains used in protein electrophoresis show differential dye binding between albumin and globulin; the more precise estimation of paraprotein is therefore derived from the percentage of relative dye binding of the paraprotein band compared with the total globulin fraction rather than the total protein. The CZE measuring system uses a UV detector system that overcomes the differential dye binding and also limits problems of non-linear dye binding at high paraprotein concentrations. Serum total protein is most often measured by biuret-based methods. A useful check is to take the sum of the paraprotein and albumin concentrations and add 10–15 g/L (to account for the other serum proteins): the result should be approximately equal to the measured serum total protein.
It is possible to quantitate reliably a serum paraprotein down to a concentration of 1 g/L, provided there is suppression of the background immunoglobulins. Monoclonal proteins with electrophoretic mobility in the β region can be difficult to measure. However, the concentration of the β-globulins is relatively constant. Therefore, when a paraprotein band migrates with β-globulins, the total band can be measured: significant changes in that concentration are likely to be due to changes only in the concentration of the monoclonal component. When it is possible to delineate a monoclonal peak for the purposes of measurement, densitometric quantitation should be reported; however, once the concentration has fallen to an extent that the peak cannot be clearly seen, immunochemical quantitation should be used. The concentration of monoclonal components can reach values in excess of 140 g/L, although this is rare. In this situation, falsely low concentrations can be seen because the paraprotein concentration has exceeded the capacity of the detection system; the monoclonal peak on the CZE tracing may appear with a flat or split top. The frequency of monitoring the concentration of monoclonal proteins will depend upon a patient’s clinical condition and treatment regimen. Cumulative reporting of paraprotein studies is invaluable in monitoring patients with myeloma and other paraproteinaemias.
Quantitation of BJP in urine is generally more difficult than quantitation of monoclonal proteins in serum. Urine volume can be very variable, being influenced by factors such as fluid intake, renal function, state of hydration and time of day. Quantitation of BJP should start with the densitometric scan of the stained electrophoretic separation in a similar way to that used for serum paraproteins. The band can be expressed as a percentage of the total protein, but this can give misleading information, especially if the general background proteinuria is increased, making the percentage of BJP relatively lower. This percentage BJP can be used with the urine albumin or creatinine concentration to give the quantity of BJP expressed per gram of albumin or millimole of creatinine. It can also be used with the total protein concentration to generate a concentration in g/L and this result can be used with the 24 h urine volume to generate a 24 h excretion. There are both practical and analytical issues, with no one method being ideal for all situations. Most importantly, a consistent approach should be adopted so that it is reliable to compare results on the same patient during the course of the disease.
Cryoproteins
Cryoproteins are proteins that aggregate on cooling of the serum or plasma and usually redissolve on warming to 37 °C; immunoglobulins and fibrinogen can all show this property.
Cryofibrinogen is an unusual finding resulting from misdirected synthesis or by post-synthetic modifications of fibrinogen. It can also be (artefactually) induced by heparin; thus collection into EDTA, citrate or oxalate is essential. Cryofibrinogen will only be visible in plasma samples while cryoprecipitating immunoglobulin should be visible in both serum and plasma stored at 4 °C. Cryofibrinogenaemia typically produces skin lesions or thrombophlebitis migrans and patients are managed by dermatologists or haematologists.
Cryoprecipitating immunoglobulins are classified into types I, II and III, based on whether they are mono- or polyclonal, their approximate concentrations and other properties; details of this classification are shown in Box 30.3. Individual cryoproteins show particular thermal profile characteristics that influence their clinical significance. Monoclonal proteins that precipitate at temperatures close to 4 °C may not cause any significant problems, but those that precipitate close to room temperature frequently precipitate in the plasma in vivo and are more likely to become clinically manifest, irrespective of relative concentrations.
The clinical features associated with types II and III cryoproteins are linked more to their ability to deposit in tissue as immune complexes rather than to their tendency to precipitate in the cold. Types II and III cryoproteins are both associated with neuropathy, vasculitis and nephropathy.
Immune complex or cryoprotein deposition in tissues is likely to cause activation of the classical complement cascade. Low concentrations of complement (particularly C4) are a good marker of active immune complex disease; measurement of these components is an important part of the investigation of possible cryoproteinaemia. Careful specimen collection and handling are vital. Both serum and plasma samples should be collected, transported and separated at 37 °C. Failure to do this may result in significant cryoproteins being lost in the blood clot during centrifugation.
β2-Microglobulin
In conditions in which there is increased cell turnover, for example malignancy (particularly lymphoid), acquired immune deficiency syndromes and inflammatory conditions, plasma β2-microglobulin concentrations increase. This protein is cleared from the plasma by glomerular filtration followed by proximal tubular reabsorption and catabolism; plasma concentrations therefore reflect cell turnover and renal function. They have been shown to be an important prognostic indicator in myeloma.
α-Interferon, used in maintenance treatment of some β-Cell malignancies, induces marked increases in plasma β2-microglobulin concentrations, and this should be taken into consideration when using β2-microglobulin for the assessment of tumour response during α-interferon therapy.
B cell malignancies
Myeloma
Plasma cell disorders have been classified into monoclonal gammopathy of unknown significance (MGUS), smouldering (asymptomatic) multiple myeloma and symptomatic multiple myeloma, based on the presence and concentration of a monoclonal protein, number and morphology of bone marrow plasma cells and end organ damage (hypercalcaemia, renal insufficiency, anaemia, bone lesions attributable to the plasma cell proliferation disorder). Myeloma is the commonest of the malignant causes of paraproteinaemia. The most frequent presenting features are bone pain (in 70% of patients), hypercalcaemia (30%), fever (15%), renal failure (10%) and infection (10%). It is predominantly a disease of the elderly, with a peak incidence between 75 and 80 years.
The three major diagnostic features are the identification of a monoclonal protein in serum or urine, the presence of neoplastic cells in the bone marrow and the destruction of bone. The significance of these and other laboratory investigations is shown in Table 30.31.

As indicated in Table 30.30, IgG, IgA and BJP account for the majority of paraproteins in myeloma. IgD paraproteins occur occasionally, usually with λ light chains. IgM paraproteins are rare in myeloma; IgE paraproteins are extremely rare in any B cell malignancy. The paraprotein concentration at presentation does not predict survival; patients with Bence Jones and IgD paraproteins often present when the paraproteins are present only at relatively low concentrations (they also tend to present at an earlier age) but have shorter survivals than myeloma patients with IgG or IgA paraproteins, even though the concentrations at presentation are often higher in the latter. Bence Jones protein (with or without intact monoclonal immunoglobulin) is seen in over 80% of patients with myeloma; it is rarely found in benign conditions.
The natural history of myeloma is generally one of progression. No treatment can be regarded as curative, although significant remission may be obtained with newer treatments regimens that may include chemotherapy, steroids, autologous stem cell transplant, bisphosphonates and the newer biologics, lenalidomide, bortezomib and thalidomide. Hypercalcaemia can be a particular problem and cause severe dehydration and/or exacerbate renal failure; it is treated with rehydration and bisphosphonates. Monoclonal protein concentrations that exceed 30 g/L can cause hyperviscosity: this is seen particularly with IgA and IgG3 paraprotein which have a tendency to aggregate. The effects of hyperviscosity can be ameliorated by plasmapheresis. Spinal cord compression requires urgent treatment with dexamethasone, followed by radiotherapy; the latter can also be helpful when localized bone pain is a problem. Infections (often associated with secondary immune deficiency) must be treated promptly. The anaemia may respond to erythropoietin.
Solitary plasmacytoma
In addition to the disseminated form in multiple myeloma, plasma cell tumours may occur as apparently solitary lesions in either bone or soft tissue. Paraprotein concentrations may be very low and intensive urine concentration (× 300) may be necessary to reveal small traces of BJP. Immunofixation of serum, even in the absence of a visible band on electrophoresis, is also worthwhile as the high sensitivity of this technique may reveal a low concentration of paraprotein, which then acts as a valuable marker. Localized treatment by surgery or radiotherapy will frequently result in disappearance of the paraprotein. Patients should be followed up at three-monthly intervals for the first year, six-monthly for the second year and then annually, as there is always a risk that progression to disseminated disease will occur and this may take as long as 20 years from detection of the solitary tumour. Extramedullary deposits may also occur in myeloma, particularly with IgD and Bence Jones type.
Waldenström macroglobulinaemia
IgM myeloma is very rare; IgM paraproteins are more often seen with a heterogeneous group of lymphoproliferative diseases of the B cell lineage such as Waldenström macroglobulinaemia (WM), lymphoma and chronic lymphocytic leukaemia. Waldenström macroglobulinaemia is characterized by the presence of an IgM paraprotein and pleiotropic lymphoid proliferation in the bone marrow. It is generally a disease of elderly men. The presenting symptoms and laboratory investigations in WM are shown in Table 30.32. The malignant cells proliferate only slowly and are relatively non-invasive; it tends to be the high paraprotein concentration and its effects that account for the presenting features. Hyperviscosity (responsible for the headaches and visual disturbance) is associated with paraprotein concentrations over 30 g/L. Bence Jones protein does occur in WM patients but tends to be at lower concentrations and less damaging than in myeloma.
TABLE 30.32
Clinical features and results of investigations in Waldenström macroglobulinaemia
| Clinical feature | % of patients |
| Weakness and fatigue | 41 |
| Hepatosplenomegaly | 40 |
| Lymphadenopathy | 36 |
| Bleeding tendency | 31 |
| Weight loss | 18 |
| Neurological | 12 |
| Visual disturbances | 10 |
| Infection | 8 |
| Bone pain and arthralgia | 4 |
| Raynaud phenomenon | 3 |
| Investigation | Typical results |
| Serum and urine for presence of paraprotein | Narrow band on electrophoresis shown to be monoclonal on immunofixation |
| Bone marrow biopsy | Increased number of lymphocytoid cells |
| Plasma viscosity | Increased |
| Lymph node biopsy | Well preserved architecture with lymphocytoid infiltrate |
| Haematology investigations | Anaemia, clotting disorders, rouleaux formation, high ESR |
Lymphomas, chronic lymphocytic leukaemia and heavy chain diseases
A paraprotein (most often IgM) is found in 10–20% of patients with non-Hodgkin lymphoma (NHL) and 5–15% of patients with chronic lymphocytic leukaemia (CLL). These tumours arise from cells early in the B cell maturation and the monoclonal protein concentration is usually low. The tumours are diagnosed by biopsy and histological examination of lymphoid tissue and by immunophenotyping of blood and lymph node biopsy material.
Certain B-lymphoproliferative disorders are associated with synthesis of heavy chains or heavy chain fragments in the absence of light chain synthesis; most often the fragments consist of heavy chains with deletion of the variable and first constant region domain. Free heavy chains are more susceptible to enzymatic degradation than intact immunoglobulin; they undergo considerable post-synthetic modification and migrate in electrophoretic separations as rather diffuse zones, anywhere between the α2 and fast γ zone. Low concentrations of the heavy chain paraprotein may be missed on examination of the electrophoretic separation. The presence of heavy chains can be indicated by immunofixation when reaction with heavy chain, but not light chain, antiserum is seen. Identity is confirmed by an immunoselection technique whereby the serum is electrophoresed through an agarose gel layer containing antiserum to light chains where any complete immunoglobulin will be trapped; free heavy chains pass through the light chain ‘trapping’ layer then move into a second layer containing antiserum to the appropriate heavy chain (α, γ or μ) and precipitate as they travel through this layer to form a ‘rocket’. The rocket is usually directly visible but can be stained with protein stains if necessary. It is essential that a positive heavy chain sample and a myeloma protein of the same class are included as controls in the heavy chain immunoselection technique. Bence Jones protein is unusual in α- and γ-chain diseases and is more often seen with μ-chain disease. Immunosuppression of non-paraprotein immunoglobulin is frequent in the heavy chain diseases.
α-Chain disease represents the most distinct clinical entity among the heavy chain diseases. The disease is a lymphoma, usually of the gut, and is alternatively known as Mediterranean type lymphoma. It is interesting in that the initial diffuse infiltration appears benign and complete remission can be achieved in some patients with oral antibiotics. However, untreated, the condition progresses to frank malignancy. The diagnosis depends on the finding of α-chains in the serum. Gut biopsies show villus atrophy and plasma cell infiltration of the lamina propria. Clinical features include a malabsorption syndrome and diarrhoea.
γ-Chain disease is not characterized as a distinct clinical entity but has a varied clinical and pathological picture ranging from apparently non-malignant proliferation, for example systemic lupus erythematosus, to aggressive lymphoma. An association with autoimmune diseases has been recorded in about 25% of published cases, most frequently with rheumatoid arthritis and autoimmune haemolytic anaemia and, in a few cases, malignant NHL that was not diagnosed until many years after the autoimmune disorder.
μ-Chain disease is extremely rare and most often a variant of CLL.
Monoclonal gammopathy of unknown significance (MGUS)
A clearer definition of MGUS is gradually emerging as a serum monoclonal protein present at a concentration < 30 g/L, clonal bone marrow plasma cells < 10% and no end-organ damage (e.g. hypercalcemia, renal insufficiency, anaemia or bone lesions). Nevertheless, the finding of a paraprotein without any symptoms poses a problem, and a relatively frequent one, given that sensitive electrophoretic techniques are so readily available and there are few constraints on requesting such investigations. Benign disease can be indistinguishable from malignant disease early in its course. No single feature can help differentiate these two entities, but monoclonal protein concentration, the presence and degree of any immune suppression and the presence of BJP can give some guide. Thus, in general, benign disease is seen with serum paraprotein concentrations below < 10 g/L (not applicable for IgD or BJP only): higher concentrations, and particularly values > 30 g/L, suggest malignancy. Non-paraprotein immunoglobulins are suppressed in 75% of myeloma patients at presentation. The presence or absence of BJP is the most important distinguishing feature; myeloma should be considered in any patient with BJP in the urine at a concentration of > 10 mg/L. The International Myeloma Working Group suggests that the measurement of serum free light chains can help to stratify patients with MGUS. The risk of progression of MGUS to an overt malignancy has a cumulative probability of 10% at 10 years (approximately 1% per year). The follow-up time in patients with MGUS should be determined by the clinical findings and the patient’s age. A general guide would be to examine the paraprotein in serum and urine at three-monthly intervals for the first year, six-monthly for the second year and, if no progression occurs, annually thereafter. If any other features of B cell malignancy occur, full investigation is required.
Transient paraproteinaemia
Transient paraproteins are usually small IgG or IgM bands (~ 1–2 g/L); however, they may be > 10 g/L. They typically occur during infections, often in patients with compromised immune systems, for example patients with CLL or with iatrogenic immune suppression for organ transplantation. Transient paraproteins may persist for over one year but most disappear within a few months.
Serum free light chains (SFLC)
Methods for the quantification of serum free light chains are now widely available, with more manufacturers producing reagents. The assays use antibodies directed against the ‘hidden’ epitopes of free light chain molecules at the interface between the light and heavy chains of intact immunoglobulins. The general concerns about the immunochemical quantification of a monoclonal protein are as applicable to SFLC as they are to the quantification of intact monoclonal IgG, IgA or IgM. However, there are additional concerns about the FLC assays with respect to their linearity, performance in external quality assurance schemes, reproducibility between laboratories and quantification of the proteins. Nevertheless, there is clear value to these measurements in patients with light chain amyloidosis and in non-secretory myeloma where the serum monoclonal protein may be undetectable by other methods. They are being used as ’stringent’ criteria for remission in some trials investigating new treatment regimes in myeloma. There are also publications promoting their use to stratify risk in myeloma, other B cell dyscrasias and MGUS.
Amyloidosis
Amyloidosis is a heterogeneous group of clinical conditions characterized by the systemic or localized deposition of fibrils derived from a variety of protein precursors and having characteristic histological appearance. It can be systemic or localized, and both types can be acquired or hereditary (Table 30.33). This table also indicates the nature of the deposit and precursor proteins, where known. In addition to the precursor proteins, amyloid deposits can contain sulphated glycosaminoglycans and serum amyloid P component, which is a normal circulating protein in human plasma.
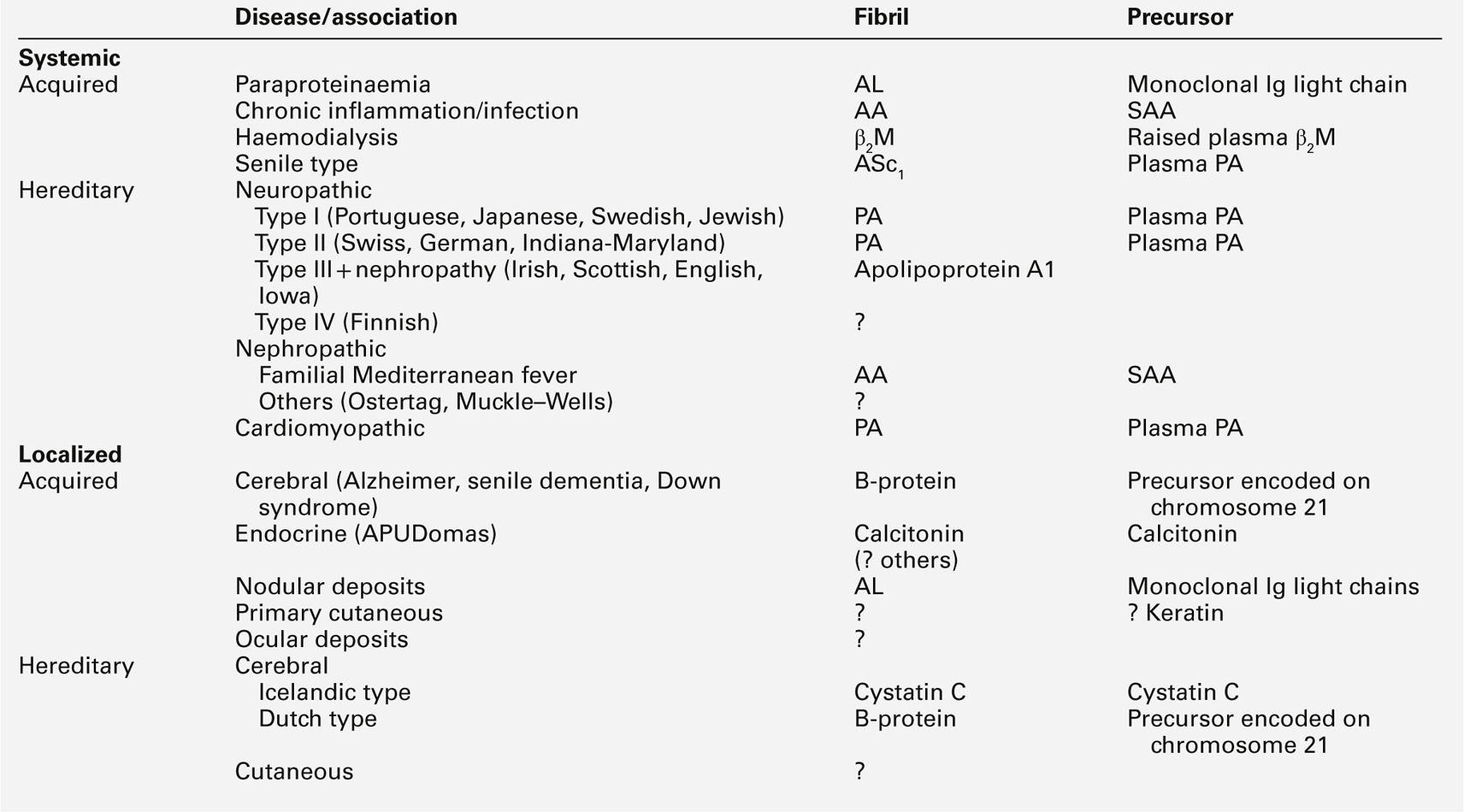
The tendency for monoclonal immunoglobulin to form amyloid fibrils (AL) can occur as a secondary complication of B cell malignancies. Amyloidosis occurs in 6–15% of patients with myeloma and is more common when the major paraprotein is free light chains. When amyloid fibrils are found in the absence of myeloma (previously classified as primary idiopathic amyloidosis), 90% of patients have a demonstrable serum paraprotein or Bence Jones proteinuria, albeit often at low concentrations; in addition, there is frequently a degree of hypogammaglobulinaemia and a slight increase in plasma cells in the bone marrow. Progressive amyloid deposition is the most serious clinical complication in these patients as they do not tend to progress to overt malignancy.
The clinical features of peripheral neuropathy, cardiomyopathy, purpura, macroglossia and carpal tunnel syndrome are similar in both the malignant and benign conditions and both have similar age distribution (mean 55–62 years), with males more frequently affected than females.
While the detection of a serum or urine paraprotein is an important finding in AL amyloidosis, the diagnosis depends on demonstration of amyloid fibrils in biopsy material. Tissue, usually rectal mucosa or abdominal fat, is stained with Congo red: the presence of characteristic apple green birefringence under polarized light is diagnostic. The National Amyloidosis Centre can localize amyloid deposits using gamma camera photographs after injection of radiolabelled serum amyloid P material. Increasingly, serum free light chain measurements are used to monitor patients but other biochemical markers (e.g. the cardiac biomarkers troponin-T (cTnT) and N-terminal pro–B-type natriuretic peptide (NT-proBNP) in cardiac amyloid) are equally important.
INFECTION AND SEPSIS
All bacteria are recognized as foreign and cause inflammation on entry into tissues. This is achieved by various mechanisms, many of which are innate. In addition, many components of bacteria are able to stimulate inflammatory or immune responses, and some of these are shown in Table 30.34.

Infection with a small bacterial load, particularly if confined to a local site, will cause cytokine production, a controlled inflammatory response and recruitment of immune cells. The outcome will be the resolution of the infection, the repair of any damage to surrounding tissue and wound healing. If the bacterial load is greater, the bacteria and/or the cytokine response may not be confined and may extend to produce a generalized systemic response. If this is unabated, it will eventually lead to septic shock, the condition in which vital tissues are inadequately perfused with blood, with progressive deterioration of circulation and falling blood pressure leading inevitably to death if untreated. Because the inflammation progresses through cascades of host-mediated reactions, once the inflammatory response has been initiated, stopping it becomes increasingly difficult – even removal of the bacteria may not affect the outcome.
Diagnosis and monitoring of infections
The diagnosis of infection is based on clinical findings, but microbiological investigation is often required to identify the organism responsible and information about antibiotic resistance may help guide treatment. However, there are situations when the clinical signs of infection may be masked, for example in patients who are immunosuppressed. In these situations, markers of acute phase response and, in particular, measurement of C-reactive protein (CRP) concentrations may be useful to give an independent marker of the presence of infection or inflammation and to monitor the response to treatment.
C-reactive protein and markers of the acute phase response
Measurement of the erythrocyte sedimentation rate (ESR) is one of the oldest ways of detecting the acute phase response. It is still frequently requested but has important limitations, including its slow response and insensitivity to small changes in disease activity. Plasma viscosity measurements have also been used to monitor the acute phase response but suffer from the same limitations as the ESR.
C-reactive protein is produced by the liver on stimulation with the inflammatory cytokines IL-6, IL-1 and TNFα. There is a rapid increase in CRP concentrations, with changes detectable as quickly as 6 h after the inflammatory insult, with a half-life of 19 h. C-reactive protein is the most widely measured acute phase protein, with raised concentrations occurring in both bacterial infections and inflammation. A raised CRP concentration is unequivocal evidence of an inflammatory response. However, viral infections do not usually cause a raised CRP and neither do some autoimmune diseases, for example SLE and scleroderma. Minimal inflammation causes a significant increase in CRP concentration but most analytical methods lack the sensitivity to detect this. There are also marked variations in quoted reference ranges, for example, < 0.1 mg/L, < 1 mg/L and < 10 mg/L. The concentration of CRP is related to the extent and severity of inflammation, with concentrations of 10–40 mg/L occurring in mild inflammation, 40–200 mg/L in acute inflammation and bacterial infections, and > 300 mg/L in extensive trauma and severe sepsis. In patients with bacterial infections, daily measurement of CRP may be useful to monitor response to treatment. Appropriate antibiotic therapy should result in the CRP falling by approximately 50% every 24 h. If the fall in CRP is slower than this, it suggests that the antibiotic regimen may not be appropriate. Patients with severe systemic inflammatory responses, for example the critically ill, often have infection on top of a marked inflammatory response, and in these situations, a fall in CRP of > 20% in 24 h is consistent with successful treatment of the infection, whereas a rise in CRP of > 20% in 24 h is consistent with a new infection. In chronic inflammatory diseases, for example rheumatoid arthritis, a falling CRP concentration precedes clinical and radiological improvements in disease.
Some laboratories are measuring CRP using assays with improved sensitivity (0.1–5 mg/L, so-called ‘highly-sensitive CRP’) for use as a marker of chronic low-grade inflammation to predict risk of coronary heart disease and progression of unstable angina. The combination of high analytical imprecision and high biological variation at low concentrations makes this difficult in a ‘one-off’ analysis, but if the analytical systems improve, CRP may become more widely applicable in predicting risk of cardiovascular disease. The major clinical applications of CRP measurement are shown in Table 30.35.

Procalcitonin (PCT) is a peptide that undergoes post-translational proteolysis into the mature hormone calcitonin. High plasma PCT concentrations are seen in patients with severe bacterial and fungal infections but also during the acute phase response post surgery. There is considerable interest in the possible use of this protein as a marker of acute sepsis, particularly in neonates, but it has not been shown to be clearly superior to CRP.
TRANSPLANTATION
Transplantation of organs or tissues is a complex procedure undertaken to restore a damaged or missing function, or to replace a lost, removed or absent ‘tissue’. Transplants may be autologous (within same person, e.g. stem cell transplant post-chemotherapy), syngeneic (identical genetics, i.e. from a twin) or allogeneic (from a different person). Table 30.36 shows the major types of transplantation currently performed in the UK, and some of the requirements applicable to each type of transplant. (General information about transplantation can be found at: www.histocompatibilityandimmunogenetics.com)
TABLE 30.36
Organ transplantation in the UK, April 2011–March 2012
| Organ/tissue | Number in UK |
| Kidney | 2801 |
| Heart/lung | 321 |
| Liver | 791 |
| Pancreas | 249 |
| Cornea | 3520 |
| Intestine | 22 |
| Stem cell transplantation (prepared from peripheral blood, umbilical cord or bone marrow) | Not available |
Taken from NHS Blood and Transplant Activity Report 2011–2012: www.organdonation.nhs.uk
Immunological issues in transplantation include rejection due to the graft being recognized by the immune system as foreign, graft-versus-host disease (GvHD) and infection secondary to the immune suppression required to prevent or control rejection or GvHD (see Box 30.4). The key issue, however, remains rejection of the graft. This can happen by three different mechanisms, shown in Box 30.5. The risk of rejection or GvHD can be reduced by matching the donor and recipient for ABO blood group and HLA type. This is particularly important for renal and stem cell transplants. A longer-term complication of modulating the immune system to accommodate transplanted tissue is an increased risk of malignancy.
Organ transplantation
Patients waiting for an organ transplant are registered, together with their HLA type, on a waiting list. As potential donors become available their HLA type is compared with those of the patients on the waiting list. Potential donor–recipient matches need to be further investigated using a specific test of the reactivity of the recipient’s serum against the donor. This is particularly to check whether the recipient has any pre-formed antibodies to the donor that could cause hyperacute rejection. Transplant patients usually need long-term immunosuppression to prevent graft rejection; the most common immunosuppressants with their modes of action are shown in Table 30.37.

Stem cell transplantation
This is the re-population of the bone marrow to rescue and restore an immune system ablated as a result of high dose chemo- or radiotherapy or to treat genetic diseases. Stem cells are undifferentiated cells that are capable of self-renewal and differentiation into more specialized cells. Stem cells can be harvested for transplant from cord blood or bone marrow but are now most commonly obtained from peripheral blood (see Table 30.38). The relative ease with which stem cells can be collected from peripheral blood and safely stored has made autologous (from the patient) peripheral blood stem cell transplant (PBSCT) a common ‘rescue’ treatment following high dose chemotherapy, e.g. for malignancy. Allogeneic (from a donor) stem cell transplants may be required to treat haematological malignancies, e.g. leukaemias, and bone marrow disorders such as thalassaemias, aplastic anaemia and severe combined immunodeficiency.

There are registers for people willing to be bone marrow or stem cell donors, which contain details of their HLA types. When a patient requires an allogeneic stem cell transplant (and a suitable family member is not available), their HLA type is used to search donor databases to try to find an appropriate match. If a donor is identified, their HLA type is double-checked and the potential recipient’s sample cross-matched with the potential donor. The transplant typically involves ‘conditioning’ with chemotherapy or radiotherapy to eradicate disease (depending on the reason for transplant) and the recipient’s functioning bone marrow. The donor stem cells are infused like a blood transfusion; they ‘home in’ to the bone marrow where they start to develop and multiply. The patients are immunosuppressed to prevent rejection and GvHD. The main immunological complications of PBSCT are shown in Box 30.5.
CONCLUSION
The immune system is complex and the diseases associated with abnormalities of the immune system can affect almost every part of the body. The common symptoms where the immune system is involved are shown in Table 30.39, together with the appropriate investigations and relevant disease. The list is not exhaustive but covers the most important immunological investigations.

Further reading
The following texts provide good accounts of more specialized areas of immunology.
Gahrton G, Durie BGM, Samsom DM, eds. Multiple myeloma and related disorders. London: Arnold; 2004.
Keren DF. Protein electrophoresis in clinical diagnosis. London: Arnold; 2003.
Rose NR, Mackay IR, eds. The autoimmune diseases. London: Academic Press; 1998.
APPENDIX 30.1 IMMUNOLOGICAL INVESTIGATIONS
Quantitative assays should be calibrated against available international reference preparations; be sensitive, specific, precise and where available, validated by adequate performance in external quality assurance (QA) schemes. It is important to mention these factors because there is a trend towards moving immunoglobulins and complement measurements from dedicated immunoassay analysers to main clinical chemistry analysers and we must not compromise our ability to detect immune deficiencies or the overall quality of the assays by losing precision and sensitivity. It is also worth remembering that babies and children will be a group of patients often investigated for immune deficiency and saving any ‘left-over’ samples for further analysis can be very useful.
Quantification of total immunoglobulin concentrations
Methods for the quantification of serum IgG, IgA and IgM concentrations are readily available. Quantification of serum immunoglobulin concentrations should be accompanied by serum protein electrophoresis and, in adults, by urine protein electrophoresis.
There is marked variation in immunoglobulin concentrations with age, and results must be interpreted using appropriate age-related reference ranges and the potential impact of transfusion of any blood products, maternal IgG and prematurity.
IgG subclasses
The IgG1 and IgG3 subclass concentrations mature first, with IgG2 and IgG4 concentrations maturing more slowly. There is considerable debate among immunologists about the value of IgG subclass measurements. It is likely that many IgG subclass requests are of negligible value; e.g. a patient with a low total serum IgG will have low IgG concentrations of subclasses. There may be a small number of patients where the subclass measurements may be useful but increasingly, quantitative specific antibody responses are being used.
Quantification of specific antibody responses
These assays quantify the antigen-specific IgG. Ideally, every individual should be vaccinated against tetanus and should have measurable IgG concentrations to tetanus. There are assays for IgG to Haemophilus influenza B, Pneumococcus and tetanus but the assays can be variable and the external QA programmes show marked variability in performance. Analysis of pre- and post-vaccination samples helps to limit the effect of inter-assay variability. Patients with a low specific antibody concentration should be re-vaccinated and the concentrations re-checked 4–6 weeks later. Unfortunately, antibody production and concentration are not necessarily good correlates of immunological protection so the interpretation of the results needs particular care.
Quantification of IgE
Reliable assays for IgE are becoming increasingly available on immunoassay analysers. It is interesting to note that in the investigation of immune deficiency (e.g. hyper IgE syndrome), immunologists now tend to look for raised IgE concentrations (into many thousands of kU/L) rather than low concentrations.
Complement
It is possible to quantify all the complement components but only C3 and C4 assays are readily available. Concentrations of other proteins, e.g. C1 esterase inhibitor can be easily measured but tend only to be available in more specialized centres. The functional assays are also vital in the investigation of immune deficiencies; these are variations on total haemolytic complement assays (for the classical and alternative pathways).
Enumeration of cell numbers
The total white cell count and differential is a vital investigation that will include total lymphocyte count and total neutrophil count but it cannot, for example, distinguish T lymphocytes from B lymphocytes. The various white cell types can be defined using flow cytometric assays that look at the size and granularity of the cells and the binding of fluorescent conjugated antibodies to the cell surface markers. The assays are usually done in immunology or haematology departments. Table 30.3 shows the common CD antigens, many of which are used in the investigation of suspected immune deficiency. Reference ranges are age-related and the results reported as either percentages or as absolute numbers.
Functional assays
There are immune deficiencies, where the various cell types are present but they lack any functional ability. Assays of lymphocyte function are laborious and complex and typically rely on the proliferation of cells after stimulation with mitogens, antigens or stimulation via specific receptors. These assays are often run over 3–5 days, so need careful planning, and the interpretation of the results needs great expertise, therefore they are only done in specialized centres.
Neutrophil function tests
Neutrophil functions tests are also complex and like lymphocyte function tests, the cells need to be alive for them to work. The most common analysis is for detection of the neutrophil respiratory burst, either by a flow cytometric assay (using dihydrorhodamine) or by looking at neutrophils under the microscope after being incubated with the redox dye, nitroblue tetrazolium.
Autoantibodies
The analytical problems with autoantibody testing are many and include the source, type and preparation of antigen, the method used and variation between each patient’s antibodies. Autoimmune serology, in comparison with general clinical biochemistry, is poorly standardized and this is compounded by large numbers of companies producing kit reagents for these tests. External quality assurance schemes are in place but they can make scary reading for a laboratory scientist!