Toxicology Emergencies
Edited by Mark Little
29.1 Approach to the poisoned patient
Lindsay Murray
Introduction
Drug overdose in adults usually occurs in the context of self-poisoning, which may be either recreational or an act of deliberate self-harm.
Deliberate self-poisoning accounts for 1–5% of all public hospital admissions in Australia [1,2]. The bulk of the medical management of cases presenting to hospital is carried out in the emergency department (ED) and the emergency physician is expected to be expert in the field. Although the management must vary considerably according to the nature and severity of the poisoning, some general principles apply.
Above all, it must be remembered that the acute overdose presentation is only a discrete time-limited event in the course of the underlying condition, which is usually psychiatric or social in origin.
Pathophysiology and clinical features
The effects of ingestion of pharmaceuticals or illicit drugs range from the non-toxic to the life threatening and may involve any system. Poisoning is a dynamic presentation and the patient may present at varying points in the time course of the poisoning. Consequently, rapid clinical deterioration or improvement may be observed after the initial presentation and assessment.
Acute morbidity and mortality from poisoning is usually a consequence of the cardiovascular, respiratory or central nervous system (CNS) complications of the poisoning. Less commonly, hepatic, renal or metabolic effects are potentially life threatening.
The most frequent life-threatening respiratory complication of poisoning is ventilatory failure, which is usually a consequence of CNS depression. Less commonly, it is secondary to ventilatory muscle paralysis. The frequency and depth of respirations are reduced. Respiratory failure may also be caused by direct pulmonary toxicity or complications, such as pulmonary aspiration or non-cardiogenic pulmonary oedema (Table 29.1.1).
Table 29.1.1
Toxic causes of respiratory failure

Cardiovascular manifestations of poisoning include tachycardia, bradycardia, hypertension, hypotension, conduction defects and arrhythmias (Table 29.1.2). Bradycardia is relatively rarely observed and is associated with a number of potentially life-threatening ingestions. Tachycardia is commonly observed and is usually benign. It may be due to intrinsic sympathomimetic or anticholinergic effects of a drug or a reflex response to hypotension or hypoxia. Hypotension is also commonly observed and may be due to a number of different causes (Table 29.1.2). Hypertension is unusual. Severe hypertension is usually associated with illicit drug use and is important because it may produce complications such as intracerebral haemorrhage.
Table 29.1.2
Cardiovascular effects of poisoning

CNS manifestations of poisoning include decreased level of consciousness, agitation or delirium, seizures and disordered temperature regulation. A decreased level of consciousness is a common presentation of poisoning and is associated with many drugs, some of which are listed in Table 29.1.1. Although usually a direct drug effect, CNS depression is occasionally secondary to hypoglycaemia, hypoxia or hypotension. Common causes of agitation or delirium following overdose are listed in Table 29.1.3. Toxic seizures are potentially life threatening and important causes are listed in Table 29.1.4.
Table 29.1.3
Toxic causes of agitation or delirium

Table 29.1.4
Amphetamines
Bupropion
Carbamazepine
Chloroquine
Cocaine
Isoniazid
Mefanamic acid
Theophylline
Tramadol
Tricyclic antidepressants
Venlafaxine
Hypothermia is usually a complication of environmental exposure secondary to a decreased level of consciousness or altered behaviour. Hyperthermia is a direct toxic effect and causes are listed in Table 29.1.5. Severe hyperthermia is rapidly lethal if not corrected.
Table 29.1.5
Amphetamines
Anticholinergics
Cocaine
MAO inhibitors
Salicylates
Serotonin syndrome
Metabolic and other manifestations of poisoning include hyper- and hypoglycaemia, hyper- and hyponatraemia, acidosis and alkalosis and hepatic failure.
Acute poisoning is distinguished from many other forms of acute illness in that, given appropriate supportive care over a relatively short period, a full recovery can usually be expected. A small number of potentially fatal poisonings may demonstrate progressive toxicity despite full supportive care. These are the so-called cellular toxins and include colchicine, iron, salicylate, cyanide, paracetamol, theophylline and digoxin. In some of these cases, early aggressive gastrointestinal decontamination, timely administration of antidotes or the institution of techniques of enhanced elimination may be life saving.
Mortality or morbidity may also result from specific complications of a poisoning. These include trauma, pulmonary aspiration, adult respiratory distress syndrome, rhabdomyolysis, renal failure and hypoxic encephalopathy. These complications usually occur prior to arrival in the ED.
Pulmonary aspiration frequently complicates a period of decreased level of consciousness or a seizure. It is a leading cause of in-hospital morbidity and mortality following overdose. This complication is characterized by rapid onset of dyspnoea, cough, fever, wheeze and cyanosis.
Rhabdomyolysis occurs as a direct toxic effect (rare) or secondary to excessive muscular hyperactivity, seizures, hyperthermia or prolonged coma with direct muscle compression. The urine is dark and acute renal failure can develop secondary to tubular deposition of myoglobin.
Assessment
Risk assessment
A risk assessment should be made as soon as possible in the management of the poisoned patient. Only resuscitation is a greater priority (Table 29.1.6). Risk assessment is a distinct quantitative cognitive step through which the clinician attempts to predict the likely clinical course and potential complications for the individual patient at that particular presentation [3]. An accurate risk assessment allows informed decision making in regard to all subsequent management steps including duration and intensity of supportive care and monitoring, screening and specialized testing, decontamination, enhanced elimination, antidotes and disposition. Factors that are taken into account when formulating this risk assessment include: the agent(s), the dose, the time since ingestion, the clinical features present and patient factors (Table 29.1.6). Specialized testing may refine risk assessment. Access to specialized poisons information in the form of a poisons information centre or in-house databases is often necessary to formulate an accurate risk assessment.
Table 29.1.6
Risk assessment-based approach to poisoning
| Resuscitation |
| Risk assessment |
| Supportive care and monitoring |
| Investigations |
| Decontamination |
| Enhanced elimination |
| Antidotes |
| Disposition |

Reproduced from Murray L, Daly F, Little M, Cadogan M. Toxicology handbook, 2nd edn. Sydney: Elsevier; 2011.
History
Every effort should be made to obtain information as to the type and dose of drug ingested, the time of ingestion and the progression of symptoms since ingestion. History provided by the patient, if they are awake, is usually reliable and should not be dismissed.
Physical examination
The focused physical examination of the poisoned patient aims to:
 identify any immediate threats to life and the need for intervention
identify any immediate threats to life and the need for intervention
 establish a baseline clinical status
establish a baseline clinical status
 identify intoxication syndromes
identify intoxication syndromes
The initial physical examination of the overdose patient in many ways parallels the primary survey of the trauma patient. The airway, breathing and circulation are assessed and stabilized as necessary. The level of consciousness should be assessed, the presence of seizure activity noted and the blood glucose and temperature measured.
A more complete examination is carried out when the patient is stable. This should include a full neurological examination, including assessment of the level of consciousness and mental status, pupil size, muscle tone and movements and the presence or absence of focal neurological signs. Poisoning normally causes global CNS depression and focal signs suggest an alternative diagnosis or a CNS complication, such as cerebral haemorrhage.
Other features that should be specifically sought are any evidence of associated trauma, the state of hydration, the condition of the skin, in particular the presence of pressure areas, the presence or absence of bowel sounds and the condition of the urine.
Several toxic autonomic syndromes, or ‘toxidromes’, have been described in relation to poisoning. The principal ones are listed in Table 29.1.7. Identification of these syndromes may narrow the differential diagnosis in cases of unknown poisoning.
Table 29.1.7
Toxic autonomic syndromes or ‘toxidromes’
| Toxidrome | Features | Common causes |
| Anticholinergic | Agitated delirium | Antihistamines |
| Tachycardia | Benztropine | |
| Hyperthermia | Carbamazepine | |
| Dilated pupils | Phenothiazines | |
| Dry flushed skin | Plant poisonings | |
| Urinary retention | Tricyclic antidepressants | |
| Ileus | ||
| Mixed cholinergic | Brady- or tachycardia | Organophosphates |
| Hypo- or hypertension | Carbamates | |
| Miosis or mydriasis | ||
| Sweating | ||
| Increased bronchial secretion | ||
| Gastrointestinal hyperactivity | ||
| Muscle weakness | ||
| Fasciculations | ||
| Mixed α- and β-adrenergic | Hypertension | Amphetamines |
| Tachycardia | Cocaine | |
| Mydriasis | ||
| Agitation | ||
| β-Adrenergic | Hypotension | Caffeine |
| Tachycardia | Salbutamol | |
| Hypokalaemia | Theophylline | |
| Hyperglycaemia | ||
| Serotonin | Altered mental status | Amphetamines |
| Autonomic dysfunction | Antihistamines | |
| Fever | Monoamine oxidase inhibitors | |
| Hypertension | NSSRIs | |
| Sweating | SSRIs | |
| Tachycardia | Tricyclic antidepressants | |
| Motor dysfunction | (Usually combined overdose) | |
| Hyperreflexia | ||
| Hypertonia (esp. lower limbs) | ||
| Myoclonus |
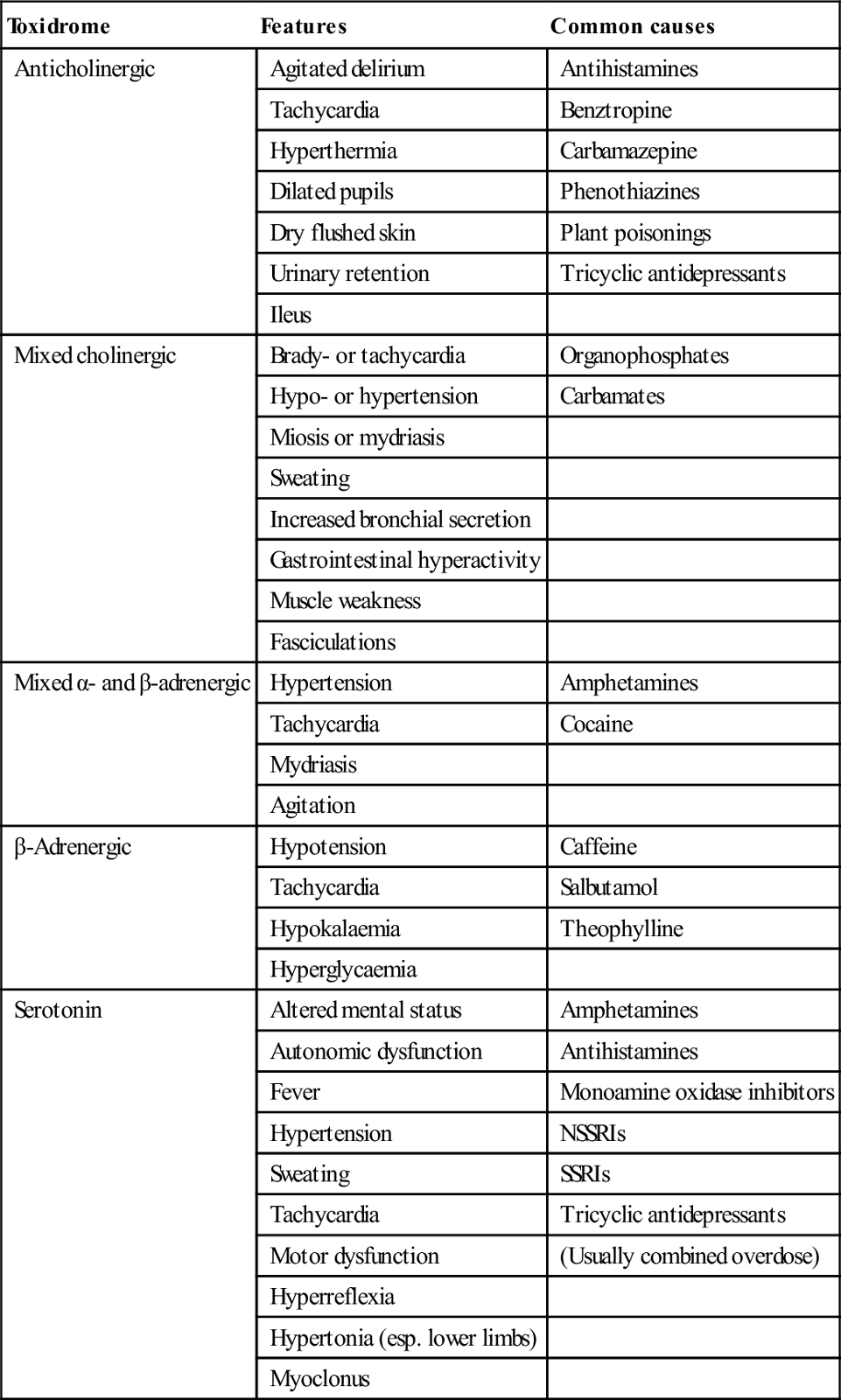
NSSRI: non-selective serotonin re-uptake inhibitor; SSRI: selective serotonin re-uptake inhibitor.
Poisons information
Information on the clinical course and toxic doses of specific pharmaceutical and non-pharmaceutical poisons is available on a 24 h basis throughout Australia by telephoning 131126. The poison information centres are staffed by trained poisons information specialists and are also able to refer cases to clinical toxicologists for consultation.
Treatment
The management of poisoning should be approached in a systematic way. Following initial resuscitation, further treatment is informed by the risk assessment (see Table 29.1.6).
Resuscitation, supportive care and monitoring
Supportive care is the key element in the management of poisoning. The vast majority of poisonings result in temporary dysfunction of one or more of the body systems. If appropriate support of the system in question is instituted in a timely fashion and continued until the toxic substance is metabolized or excreted, a good outcome can be anticipated. In severe poisonings, supportive care may be very aggressive and possible interventions are listed in Table 29.1.8.
Table 29.1.8
Supportive care measures for the poisoned patient
| Airway | Endotracheal intubation |
| Breathing | Supplemental oxygen |
| Ventilation | |
| Circulation | Intravenous fluids |
| Inotropes | |
| Antihypertensives | |
| Antiarrhythmics | |
| Defibrillation/cardioversion | |
| Cardiac pacing | |
| Cardiopulmonary bypass | |
| Metabolic | Hypertonic dextrose |
| Hypertonic saline | |
| Insulin/dextrose | |
| Calcium salts | |
| Sodium bicarbonate | |
| Agitation/delirium | Benzodiazepines |
| Butyrophenones | |
| Seizures | Benzodiazepines |
| Barbiturates | |
| Body | External rewarming |
| Temperature | External cooling |
| Impaired renal function | Rehydration |
| Haemodialysis |
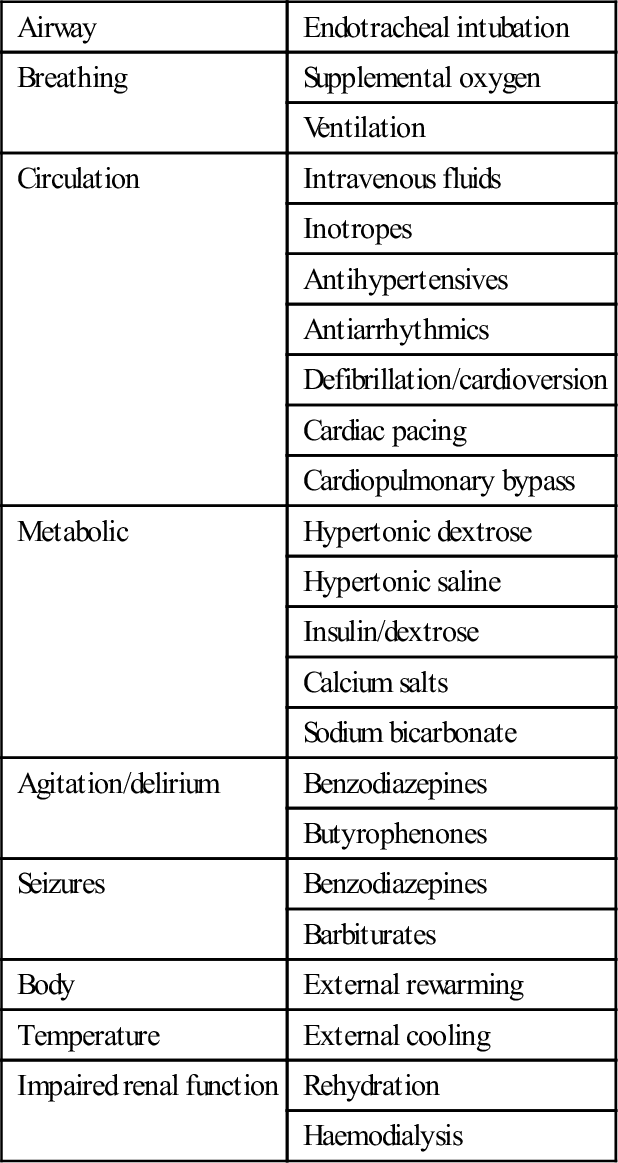
The specific supportive management of a number of manifestations or complications of poisoning warrants further mention insofar as it may differ from the standard management of such conditions with other aetiologies.
Cardiopulmonary arrest from poisoning should be aggressively resuscitated. Direct current cardioversion is rarely successful in terminating toxic arrhythmias and should not take precedence over establishing adequate ventilation and oxygenation, cardiac compressions, correction of acidosis or hypovolaemia and the administration of specific antidotes. Resuscitative efforts should be continued beyond the usual time frame. In cardiac arrest due to drugs with direct cardiac toxicity, the use of cardiopulmonary bypass or extracorporeal membrane oxygenation (ECMO) until the drug is metabolized may be life saving.
In general, intravenous benzodiazepines are the drugs of choice for control of toxic seizures. Large doses may be required. Hypoxia and hypoglycaemia must be corrected if they are contributory factors. Patients with toxic seizures do not generally need long-term anticonvulsant therapy. Isoniazid-induced seizures are difficult to control without administration of an adequate dose of the specific antidote, pyridoxine.
The management of pulmonary aspiration is essentially supportive, with supplemental oxygenation and intubation and mechanical ventilation if necessary. Neither prophylactic antibiotics nor corticosteroids have been shown to be helpful in the management of this condition, which is essentially a chemical pneumonitis.
Toxic hypertension rarely requires specific therapy. Most cases are mild and simple observation is sufficient. Agitation or delirium is a feature of many intoxications associated with hypertension and adequate sedation with benzodiazepines usually lowers the blood pressure. Severe toxic hypertension is most likely in toxicity from cocaine or amphetamine-type drugs and treatment may be indicated to avoid complications, such as cardiac failure or intracerebral haemorrhage. The drug of choice in this situation is sodium nitroprusside by intravenous infusion. The extremely short duration of action of this vasodilator allows accurate control of hypertension during the toxic phase and avoids the development of hypotension once toxicity begins to wear off.
Management of rhabdomyolysis consists of treatment of the causative factors, fluid resuscitation and careful monitoring of fluids and electrolytes. The role of mannitol and urinary alkalinization in reducing the risk of renal failure is not clear. Established acute renal failure requires haemodialysis, often for up to 6 weeks.
Decontamination
The aim of decontamination of the gastrointestinal tract is to bind or remove ingested material before it is absorbed into the circulation and able to exert its toxic effects. This is a very attractive concept and has long been considered one of the fundamental interventions in management of the overdose patient.
However, gastrointestinal decontamination should not be regarded as a routine procedure in the management of the patient presenting to the ED following an overdose. The decision to perform gastrointestinal decontamination and the choice of method should be based on an assessment of the likely benefit, the likely risk and the resources required. Gastrointestinal decontamination should only be considered where there is likely to be a significant amount of a significantly toxic material remaining in the gut. It is never indicated when the risk assessment predicts a benign course. Efforts at decontamination technique should never take precedence over the institution of appropriate supportive care.
Three basic approaches to gastrointestinal decontamination are available: gastric emptying, administration of an adsorbent and catharsis.
Gastric emptying can be attempted by the administration of an emetic, most commonly syrup of ipecac, or by gastric lavage. In volunteer studies, both of these techniques removed highly variable amounts of marker substances from the stomach even if performed immediately after ingestion and the effect diminished rapidly with time to the point of being negligible after 1 hour [4,5]. Clinical outcome trials have failed to demonstrate improved outcome as a result of routine gastric emptying in addition to administration of activated charcoal, except, perhaps, in patients presenting unconscious within 1 h of ingestion [6–8].
The principal adsorbent available to clinicians is activated charcoal (AC), which effectively binds most pharmaceuticals and chemicals, and is currently the decontamination method of choice for most poisonings. Materials that do not bind well to charcoal are listed in Table 29.1.9.
Table 29.1.9
Materials that do not bind well to activated charcoal

Charcoal is ‘activated’ by treatment in acid and steam at high temperature. This process removes impurities and greatly increases the surface area available for binding. Activated charcoal (AC) is packaged as a 50 g dose premixed with water or sorbitol, which is likely to be sufficient for the majority of ingestions. Adult patients are usually able to drink AC slurry from a cup. If the level of consciousness is too impaired to allow this, they should be intubated first. Administration of AC is absolutely contraindicated unless the patient has an intact or protected airway.
Volunteer studies demonstrate that the effect of AC diminishes rapidly with time and that the greatest benefit occurs if it is administered within 1 h. There is as yet no evidence that AC improves clinical outcome [9].
There is no evidence to suggest that the addition of a cathartic, such as sorbitol, to AC improves clinical outcome [10].
Apart from rarely employed endoscopic and surgical techniques, whole-bowel irrigation (WBI) is the most aggressive form of gastrointestinal decontamination. Polyethylene glycol solution (Golytely is administered via a nasogastric tube at a rate of 2 L/h until a clear rectal effluent is produced. This usually takes about 6 h and requires one-to-one nursing. In volunteer studies, this technique reduced the absorption of slow-release pharmaceuticals and so may be of benefit in life-threatening overdoses of these agents. Again, clinical benefit has not yet been conclusively demonstrated [11]. The use of WBI has also been reported in the management of potentially toxic ingestions of iron, lead and packets of illicit drugs. Whole-bowel irrigation is contraindicated if there is evidence of ileus or bowel obstruction and in patients who have an unprotected airway or haemodynamic compromise.
Enhanced elimination
A number of techniques are available to enhance the elimination of toxins from the body. Their use is rarely indicated, as only a very few drugs capable of causing severe poisoning have pharmacokinetic parameters that render them amenable to these techniques (Table 29.1.10).
Table 29.1.10
Techniques of enhanced elimination
| Technique | Suitable toxin |
| Multiple-dose activated charcoal | Carbamazepine |
| Dapsone | |
| Phenobarbitone | |
| Phenytoin | |
| Theophylline | |
| Urinary alkalinization | Phenobarbitone |
| Salicylate | |
| Haemodialysis | Ethylene glycol |
| Lithium | |
| Metformin lactic acidosis | |
| Methanol | |
| Salicylate | |
| Theophylline | |
| Valproic acid | |
| Charcoal haemoperfusion | Theophylline |
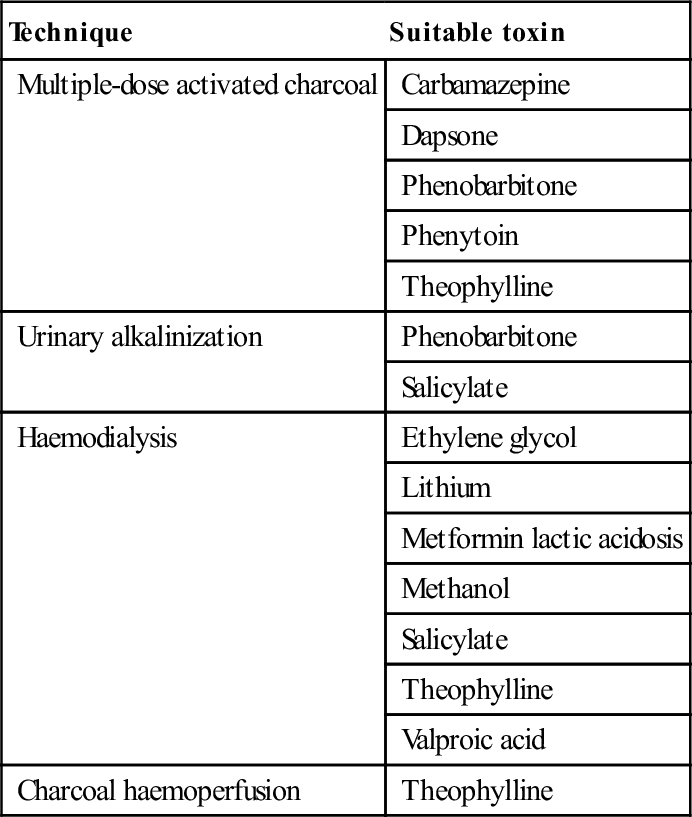
Multiple-dose AC (25–50 g every 3–4 h) may enhance drug elimination by interrupting the enterohepatic circulation or by ‘gastrointestinal dialysis’. Gastrointestinal dialysis is the movement of a toxin across the gastrointestinal wall from the circulation into the gut down a concentration gradient that is maintained by charcoal binding. For this technique to be effective, a drug must undergo considerable enterohepatic circulation or, in the case of ‘gastrointestinal dialysis’, have a small volume of distribution, small molecular weight, low protein binding, slow endogenous elimination and bind to charcoal [12]. The advantages of this technique are that it is non-invasive and simple to carry out.
Alkalinization of the urine enhances urinary excretion of drugs that are filtered at the glomerulus and are unable to be reabsorbed across the tubular epithelium when in an ionized form at alkaline pH. For elimination to be effectively enhanced by this method, the drug must be predominantly eliminated by the kidneys in the unchanged form, have a low pKa, be distributed mainly to the extracellular fluid compartment and be minimally protein bound.
Haemodialysis (HD) and haemoperfusion (HP) are both very invasive techniques and for that reason are reserved for potentially life-threatening intoxications. Only a small number of drugs that have small volumes of distribution, slow endogenous clearance rates, small molecular weights (HD) and bind to charcoal (HP) will have their rates of elimination significantly enhanced by these procedures.
Antidotes
Very few drugs have effective antidotes. Occasionally, however, timely use of an antidote may be life saving or substantially reduce morbidity, time in hospital or resource requirements. Antidotes that may be indicated in the ED setting are listed in Table 29.1.11. However, it must be remembered that antidotes are also drugs and are frequently associated with adverse effects of their own. An antidote should only be used where a specific indication exists and then only at the correct dose, by the correct route and with appropriate monitoring. Because many antidotes are so infrequently used, obtaining sufficient supplies when the need arises can be difficult. Every ED must review its stocking of antidotes and have a plan for obtaining further supplies should the need arise.
Table 29.1.11
| Poisoning | Antidote |
| Atropine | Physostigmine |
| Benzodiazepines | Flumazenil |
| Cyanide | Dicobalt edetate, hydroxocobalamin |
| Digoxin | Digoxin-specific Fab fragments |
| Insulin | Dextrose |
| Iron | Desferoxamine |
| Isoniazid | Pyridoxine |
| Methaemoglobinaemia | Methylene blue |
| Methanol and ethylene glycol | Ethanol, fomepizole |
| Organophosphates and carbamates | Atropine, oximes |
| Opioids | Naloxone |
| Paracetamol | N-acetyl cysteine |
| Sulphonylureas | Dextrose, octreotide |
| Tricyclic antidepressants | Sodium bicarbonate |
| Warfarin, brodifacoum | Vitamin K |
Differential diagnosis
It is essential to exclude important non-toxic diagnoses in the patient presenting with coma or altered mental status presumed to be due to drug overdose. These diagnoses include head injury, intracerebral haemorrhage or infarction, CNS infection, hyponatraemia, hypoglycaemia, hypo- or hyperthermia, post-ictal states and psychiatric disorders.
Clinical investigations
Investigations should only be performed if they are likely to affect the management of the patient. They are employed as either screening tests or for specific purposes.
In poisoning, screening tests aim to identify occult toxic ingestions for which early specific treatment might improve outcome. The recommended screening tests for acute poisoning are the 12-lead ECG and the serum paracetamol level. The ECG is used to exclude conduction defects which may predict potentially life-threatening cardiotoxicity. The serum paracetamol is useful to ensure that paracetamol poisoning is diagnosed within the time available for effective antidotal treatment.
Other specific investigations may be indicated to exclude important differential diagnoses, confirm a specific poisoning for which significant complications might be anticipated, assess the severity of intoxication, assess response to treatment or assess the need for a specific antidote or enhanced elimination technique.
The patient with only minor manifestations of poisoning may require no other blood tests apart from a screening paracetamol level. Pregnancy should be excluded in women of childbearing age by serum or urine β-HCG if necessary. More seriously ill patients may require electrolyte, renal and liver function tests and a full blood count, creatine kinase and arterial blood gases. Urinalysis reveals myoglobinuria in significant rhabdomyolysis.
Routine qualitative drug screening of urine or blood in the overdose patient is rarely useful in planning management.
Measurement of serum drug concentrations is only useful if this provides important diagnostic or prognostic information or assists in planning management. Some drug levels that may be useful are listed in Table 29.1.12. For most cases, drug overdose management is guided by clinical findings and not by drug levels. Some drugs commonly taken in overdose for which serum concentrations are of no value in planning management are listed in Table 29.1.13.
Table 29.1.12
Drug levels that may be helpful in the management of selected cases of overdose
Carbamazepine
Digoxin
Dilantin
Lithium
Iron
Paracetamol
Phenobarbitone
Salicylate
Theophylline
Valproate
Table 29.1.13
Drug levels that are not helpful in the management of overdose
| CNS drugs | Cardiovascular drugs |
| Antidepressants | ACE inhibitors |
| Benzodiazepines | β-Blockers |
| Benztropine | Calcium channel blockers |
| Cocaine | Clonidine |
| Newer antipsychotics | |
| Opiates | |
| Phenothiazines |
Radiology has a limited role in the management of overdose. A chest X-ray is indicated in any patient with a significantly decreased level of consciousness, seizures or hypoxia. It may show evidence of pulmonary aspiration. A computed tomography scan of the head may be indicated to exclude other intracranial pathology in the patient with an altered mental status. The abdominal X-ray is useful in evaluating overdose of radiopaque metals including iron, lithium, potassium, lead and arsenic.
Disposition
Both the medical and the psychiatric disposition of the overdose patient must be considered. A good risk assessment is essential to determining timely and safe disposition.
The majority of overdose patients who remain stable at 4–6 h after the ingestion do not need further close monitoring and may be admitted to a non-monitored bed until manifestations of toxicity completely resolve. An emergency observation ward is ideal for this purpose.
Any patient who develops clinical manifestations of intoxication severe enough to require the institution of specific supportive care measures requires admission to an intensive care environment. A few patients will require admission for prolonged monitoring based on the history of the ingestion. For example, anyone with a history of ingestion of colchicine, organophosphates, slow-release theophylline or slow-release calcium channel blockers requires admission because of the possibility of delayed onset of severe toxicity.
Psychiatric evaluation of deliberate self- poisoning cases is indicated as soon as the patient’s medical condition permits. All such patients must be continuously supervised until the psychiatric evaluation has taken place.
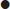
29.2 Cardiovascular drugs
Betty Shuk Han Chan and Angela Chiew
Calcium channel blockers and β-blockers
Introduction
The calcium channel blockers (CCBs) and β-blockers are widely prescribed in the community. In overdose, they present with similar clinical pictures of potentially life-threatening impairment of cardiac function. The management of both types of overdose is similar and they are discussed together.
Pharmacokinetics
Standard CCB preparations are rapidly absorbed from the gastrointestinal tract, with onset of action occurring within 30 min. Pharmacokinetic parameters are shown in Table 29.2.1. Verapamil and diltiazem undergo significant first-pass hepatic clearance. Verapamil is metabolized to norverapamil, which possesses 15–20% of verapamil’s pharmacological activity and is renally excreted. Diltiazem is metabolized to deacetyldiltiazem, which has half the potency of the parent compound and undergoes biliary excretion. The elimination half-lives of all CCBs may be prolonged following massive overdose. Amlodipine has a longer plasma half-life (30–50 h) than other CCBs.
Table 29.2.1
Pharmacological profiles of the calcium channel blockers
| Class | Phenylalkylamines | Benzothiazepines | Dihydropyridines |
| Prototype | Verapamil | Diltiazem | Nifedipine |
| Hours to peak plasma concentration (NR/SR) | 1.5/5–7 | 2.3/5–11 | 0.5/5 |
| Half-life (h) | 3–7/10–12 | 3–5/6–7 | 2–5–5–7 |
| Half-life in massive overdose (h) | 10–12 | 8–9 | 7–8 |
| Absorption (%) | >90 | >90 | >90 |
| Vd (L/kg) | 4 | 5 | 1.2 |
| Protein binding (%) | 90 | 80–90 | 90 |
| Predominant excretion route | (1) Hepatic; (2) renal | Hepatic | Renal |
| Active metabolite | Yes (20%) | Yes (25–50%) | No |
| Heart rate (%) | −10 | −15 | +10 |
| Systemic vascular resistance (%) | −10 | −10 | −20 |
| AV node conduction velocity (%) | −20 | −25 | +10 |
NR: normal release; SR: slow release.
Adapted from Kerns W II, Kline J, Ford MD. β-Blocker and calcium channel blocker toxicity. Emerg Med Clin N Am 1994;12:365–89 with permission.
Importantly, slow-release preparations of both verapamil and diltiazem are widely prescribed and are associated with much longer times to peak plasma concentration and clinical effect.
Absorption of β-blockers is rapid, with peak clinical effects occurring within 1–4 h. Pharmacokinetic parameters of the principal β-blockers are detailed in Table 29.2.2. Agents with high lipid solubility, such as propranolol, penetrate the blood–brain barrier better than the water-soluble agents and hence cause greater central nervous system (CNS) toxicity.
Table 29.2.2
Pharmacological profiles of the β-blockers
| Agent | β1-selective | Membrane stabilization | Absorption (%) | Protein binding (%) | Volume of distribution (L/kg) | Elimination/half-life (h) | Lipophilic |
| Atenolol | Yes | No | 50 | <5 | 0.6–1.1 | Renal/6–9 | Weak |
| Carvedilol | No | Yes | 25 | 98 | 2 | Hepatic/6 | Weak |
| Esmolol | Yes | No | NA | 55 | 3.4 | Blood esterase 9 min | Weak |
| Labetalol | No | No | 90 | 50 | 5.1–9.4 | Hepatic/3–4 | Weak |
| Metoprolol | Yes | No | 90 | 12 | 5.6 | Hepatic/3–4 | Moderate |
| Oxyprenolol | No | Yes | 90 | 80 | 1.2 | Hepatic/2–3 | Moderate |
| Pindolol | No | Yes | 90 | 57 | 1.2–2 | Renal/3–4 | Moderate |
| Propranolol | No | Yes | 90 | 93 | 3.4–6 | Hepatic/3–4 | High |
| Sotalol | No | No | 70 | 0 | 0.23–0.7 | Renal/9–10 | Weak |
| Timolol | No | No | 90 | 10 | 1.3–3.6 | Renal/4–5 | Weak |
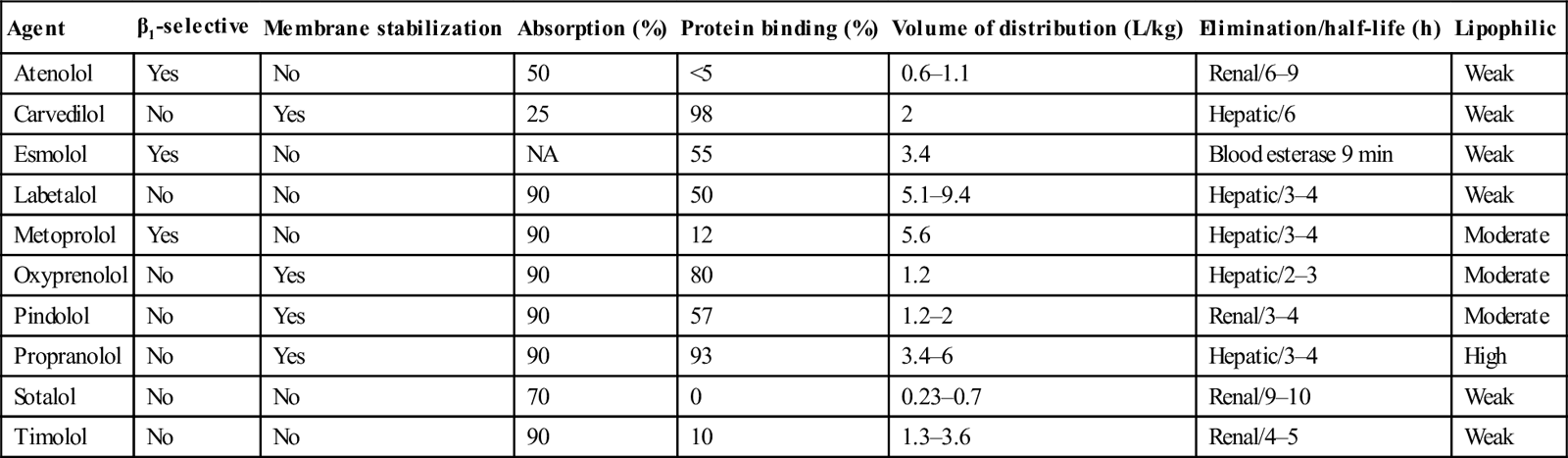
Adapted from Kerns W II, Kline J, Ford MD. β-Blocker and calcium channel blocker toxicity. Emerg Med Clin N Am 1994;12:365–89 with permission.
Pathophysiology
CCBs antagonize the entry of extracellular calcium into cardiac and smooth muscle, but not skeletal muscle. Upon entry into cells, calcium participates in mechanical, electrical and biochemical reactions. It is involved in excitation–contraction of cardiac and smooth muscles, as well as phase O depolarization in the sinus and atrioventricular (AV) nodes by calcium influx through channels. CCBs affect myocardial contractility and slow conduction through the sinus and AV nodes. Contraction of smooth muscle is mediated by calcium influx, which is inhibited by CCBs. This results in vasodilatation and secondary reflex tachycardia from an increase in sympathetic activity.
The different classes of CCB have somewhat different pharmacological and toxic effects, as a consequence of their different binding characteristics to the dihydropyridine (DHP) receptors. Verapamil, a phenylalkylamine, produces more profound cardiac conduction defects and equal reductions in systemic vascular resistance when compared with other CCBs on a mg/kg basis. Verapamil is more likely to produce symptomatic decreases in blood pressure, heart rate and cardiac output than diltiazem, a benzothiazepine. The DHPs, which include amlodipine, felodipine, lercanidipine, nifedipine and nimodipine, preferentially bind to vascular smooth muscle and predominantly decrease systemic and coronary vascular resistance. With the exception of felodipine, they also produce a reflex tachycardia by the unloading of baroreceptors.
β-Blockers prevent the binding of catecholamines to β-receptors (β1, β2). β1-Receptors are located in the myocardium, kidney and eye and β2-receptors in adipose tissue, pancreas, liver and both smooth and skeletal muscle. β1-Stimulation produces increased chronotropy and inotropy in the heart, increased renin secretion in the kidney and increased aqueous humor production. β2-Stimulation relaxes smooth muscle in the blood vessels, bronchial tree, intestinal tract and uterus.
Blockade of β-receptors results in blunting of the metabolic, chronotropic and inotropic effects of catecholamines. Some β-blockers, especially propranolol, may also impede sodium entry via myocardial fast inward sodium channels, thus slowing phase 0 of the action potential. This results in a prolonged QRS duration on the electrocardiogram and produces cardiotoxicity in overdose similar to that of the tricyclic antidepressants.
The different β-blockers have slightly differing pharmacological properties, including selectivity for β-adrenoreceptors, intrinsic sympathomimetic activity and membrane-stabilizing activity. The relative affinity for β-adrenoreceptors may influence expression of toxicity. Atenolol, esmolol and metoprolol are β1-selective agents and therapeutic use of these drugs is less likely to produce the peripheral vasoconstriction, bronchospasm and disturbances in glucose homoeostasis that result from β2 inhibition. However, pharmacological specificity decreases with increasing dose. Several β-blockers have partial agonist activity such that, although they block the β-receptor to catecholamines, they also weakly stimulate the receptor. This partial agonist activity may have a protective effect in overdose.
Clinical features
Calcium channel blockers
The severity of toxicity is determined by a number of factors, including the amount and characteristics of the drug ingested, the underlying health of the patient, co-ingestants and delay until treatment. The majority of serious cases and deaths result from the ingestion of verapamil or diltiazem, the most toxic of the CCBs. Ingestion of as few as 10 tablets of the higher dose formulation of verapamil or diltiazem can cause severe toxicity. Elderly patients and those with congestive cardiac failure may develop toxicity with ingestions of two to three times their normal daily dose. The principal clinical features are shown in Table 29.2.3. Ingestion of toxic amounts of standard preparations typically produces symptoms within 2 h, although maximal toxicity may not occur for up to 6–8 h. The slow-release preparations can produce significant toxicity with onset of symptoms more than 6 h post-ingestion. The major threats to life are myocardial depression and hypotension. Overdose of DHPs often produces tachycardia with normal blood pressure during the first 30 min, followed later by hypotension and bradycardia in large ingestions (>10 mg/kg). Even though amlodipine has been reported to be less toxic than verapamil and diltiazem, it can cause severe shock in large overdoses. With verapamil and diltiazem poisoning, nausea, vomiting, hyperglycaemia and metabolic acidosis can develop. All CCBs can cause symptoms of cerebral hypoperfusion, such as syncope, lethargy, lightheadedness, dizziness, altered mental status, seizures and coma.
Table 29.2.3
Clinical features of CCB overdose
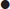

β-Blockers
In one large series of patients with β-blocker overdose, 30–40% of patients remained asymptomatic and only 20% developed severe toxicity. Most of the life-threatening presentations or deaths that have been reported in the literature are due to overdosage of propranolol or sototol. Significant toxicity is more likely to develop in patient ingestions with these β-blockers, in patients with pre-existing cardiac disease or where there is co-ingestion of other drugs with effects on the cardiovascular system, especially CCBs and cyclic antidepressants. Ingestion of more than 1.5 g propranolol is associated with severe toxicity. If β-blocker toxicity is to develop, it is usually observed within 6 h of ingestion.
Sinus node suppression, conduction abnormalities and decreased contractility are typical. First-degree AV block, AV dissociation, right bundle branch block and intraventricular conduction delay have been reported.
Propranolol in overdose, has sodium channel blocking effect, that is characterized by cardiotoxicity including prolongation of the QRS interval and ventricular arrhythmias that more closely resemble tricyclic antidepressant overdose. Sotalol has both β-blocker activity and class 3 antiarrhythmic properties. Class 3 drugs lengthen the duration of the QT interval owing to prolongation of the action potential in His–Purkinje tissue. Therefore, ventricular arrhythmias, such as torsades de pointes, are more common with sotalol.
Hypotension occurs as a result of negative inotropic effect. In addition, CNS effects, such as depressed conscious level and seizures, can occur, especially with the more lipid-soluble and membrane-depressant agents, such as propranolol. Hypoglycaemia is reported following atenolol overdose.
Clinical investigation
The ECG is essential in evaluating and monitoring toxic conduction defects. Serum drug levels are unhelpful in management. Patients with severe toxicity require monitoring of serum electrolytes and glucose. Serum calcium must be closely monitored if calcium salts are administered therapeutically.
Treatment
The primary aim in both β-blocker and CCB toxicity is to restore perfusion to vital organs by increasing cardiac output and the methods used are similar.
Supportive management may include airway and ventilatory support, intravenous fluid administration, early implemenation of hyperinsulinaemia euglycaemic therapy and administration of inotropes. Transcutaneous or transvenous pacing may be tried in cases with profound bradycardia, but often is of limited benefit. Severe cases may require studies on cardiac output and peripheral vascular resistance using either Swan–Ganz catheter or pulse contour cardiac output monitoring (PiCCO) and invasive blood pressure monitoring.
If safe to do so, oral-activated charcoal should be administered as soon as practicable to all those presenting after ingestion of slow-release preparations and may be considered for other β-blocker ingestions. More aggressive decontamination, with whole-bowel irrigation, is indicated following overdose with slow-release CCBs.
A number of drugs play a role in the management of significant CCB or β-blocker poisoning, although none is a completely effective antidote. Suggested doses are shown in Table 29.2.4.
Table 29.2.4
Useful drugs in the management of CCB and β-blocker toxicity
| CCBs | β-Blockers | |
| Calcium | Calcium gluconate 10% 30 mL (child 0.6 mL/kg) IV over 10 min OR calcium chloride 10% 10 mL (child 0.2 mL/kg) IV over 10 min. Repeat every 5 min as required. Further administration guided by serum calcium concentrations | |
| Catecholamines | Adrenaline (epinephrine) infusion started at 1 μg/kg/min and titrate to maintain organ perfusion | Isoprenaline or adrenaline (epinephrine) infusion titrated to maintain organ perfusion |
| Sodium bicarbonate | A bolus dose of sodium bicarbonate 8.4% 1–2 mmol/kg, every 3–5 min, to correct severe metabolic acidosis to a pH greater than 7.3 | A bolus dose of sodium bicarbonate 8.4% 1–2 mmol/kg, every 3–5 min, titrated to a narrowing of the QRS complex, resolution of arrhythmias |
| Hyperinsulinaemia euglycaemia | Actrapid 1 U/kg IV bolus followed by an infusion starting at 1 U/kg/h. Give with 50% dextrose 50 mL followed by infusion to maintain euglycaemia | Actrapid 1 U/kg IV bolus followed by an infusion commencing at 1 U/kg/h. Give with 50% dextrose 50 mL followed by infusion to maintain euglycaemia |
Calcium, an inotropic agent, is the initial drug of choice for CCB toxicity and has also been used successfully for β-blocker poisoning. Administration must be closely monitored, with ionized calcium measured 30 min after commencing the infusion and then second-hourly. Catecholamines are useful in attempting to restore adequate tissue perfusion.
Hyperinsulinaemic euglycaemia therapy (HIET) is increasingly advocated as therapy for hypotension unresponsive to fluids and calcium salts, with many toxicologists using HIET early in the management of these poisonings if inotropes are being considered. This therapy is supported by animal work and multiple human case reports, but a randomized controlled trial is lacking. Insulin administration switches cardiac cell metabolism from fatty acids to carbohydrates. It restores calcium fluxes and improves myocardial contractility. The recommended initial dose of actrapid is 1 U/kg IV followed by an infusion commencing at 1 U/kg/h. Although the optimal dose is still to be determined, there are some human case reports suggesting increasing doses up to 10 U/kg/h. This should be accompanied by an initial bolus dose of 50 mL 50% dextrose followed by an infusion of dextrose to maintain euglycaemia. Case reports often describe patients needing no more than 25 g/h of dextrose while poisoned (i.e. 50 mL/h of 50% dextrose).
The use of glucagon is supported only by case reports and some animal studies. There are no clinical trials supporting its efficacy in either calcium channel or β-blocker poisoning. Due to the significant doses often required, it is frequently difficult to source adequate stocks of glucagon for use as an inotropic agent. As such, its use in the treatment of calcium channel or β-blocker poisoning is not routinely recommended.
Severe propranolol toxicity is usually due to sodium channel blockade and treatment is similar to tricyclic antidepressant poisoning, including intubation, ventilation and sodium bicarbonate.
There are no clinically effective methods of enhancing the elimination of CCBs or β-blockers. When all else fails, extra-corporeal life support has been shown to allow organ perfusion until reversal of cardiac dysfunction and elimination of the drugs.
Disposition
Following overdose of β-blockers or standard CCBs, patients should be observed in a monitored environment for at least 6 h. Overdoses of slow-release CCBs require monitoring for at least 16 h from the time of ingestion. All symptomatic patients should be admitted to a monitored environment until toxicity resolves.
Digoxin
Introduction
Both acute and chronic digoxin toxicity are potentially life-threatening presentations to the emergency department (ED). Early recognition and administration of the specific Fab fragment antidote, if indicated, usually results in a good outcome.
Pharmacokinetics
Digoxin is moderately well absorbed following oral administration, with a bioavailability in the range of 50–80%. The initial volume of distribution is relatively small, but it is then slowly redistributed, predominantly to skeletal muscle, to give a relatively large volume of distribution of approximately 7 L/kg. Digoxin is excreted predominantly unchanged by the kidney, with an elimination half-life of about 36 h.
Pathophysiology
At a subcellular level, digoxin inhibits the function of Na–K ATPase, which leads to intracellular depletion of potassium and accumulation of sodium and calcium ions. Alteration of ionic fluxes affects cell membrane conduction. At toxic concentrations of digoxin, the effects on the cardiac conducting system produce decreased conduction velocity throughout the system, increased refractoriness at the AV node and enhanced automaticity of the Purkinje fibres. Vagal tone is also enhanced. In acute digoxin poisoning, the sudden loss of Na–K ATPase function produces hyperkalaemia.
Clinical features
Two distinct clinical presentations of digoxin toxicity are observed: acute and chronic. Both are characterized by cardiac arrhythmias and virtually all types of arrhythmia have been reported in the context of digoxin toxicity.
Acute digoxin overdose in adults is usually intentional. The therapeutic margin for digoxin is relatively narrow and any ingestion with suicidal intent is regarded as potentially life threatening.
The non-cardiac manifestations of toxicity are nausea and vomiting and hyperkalaemia. Nausea and vomiting occur early and may be the presenting complaint. The most common cardiac manifestations are sinus bradycardia, increased ventricular ectopy, sinoatrial node arrest and first-, second- or third-degree heart block. Ventricular tachycardia and fibrillation may occur. In significant acute overdose, progressive worsening of the conduction disturbance over a period of hours is usually observed.
Chronic digoxin toxicity may be precipitated by therapeutic errors, intercurrent illnesses that decrease renal elimination of digoxin or by drug interactions. Common drug interactions include those with quinidine, CCBs, amiodarone and indomethacin. The patient is commonly elderly. Reduced muscle mass and reduced renal function in the elderly mean that both the volume of distribution and rate of elimination of digoxin may be substantially reduced.
Nausea and vomiting are also common manifestations of chronic digoxin toxicity and are frequent presenting symptoms. Neurological manifestations are characteristic of chronic toxicity and include visual disturbances, weakness and fatigue. The most common cardiovascular manifestations of chronic digoxin toxicity are arrhythmias and these may be sinus bradycardia, atrial fibrillation with slowed ventricular response or a junctional escape rhythm, atrial tachycardia with block and ventricular tachycardia and fibrillation.
Death from digoxin toxicity results from pump failure, severe cardiac conduction impairment or ventricular arrhythmia.
Clinical investigations
The most important investigations are the ECG, serum electrolytes and creatinine and serum digoxin concentration.
The ECG is invaluable in documenting the type and severity of any cardiac conduction defect. Serial ECGs may demonstrate worsening of the cardiac conduction defects as toxicity progresses.
In acute poisoning, the serum potassium rises as Na–K ATPase function is progressively impaired. Hyperkalaemia denotes significant acute digoxin toxicity. Prior to the availability of a specific antidote for digoxin poisoning, a serum potassium concentration>5.5 mEq/L was associated with a high probability of lethal outcome. Hyperkalaemia seldom occurs in chronic digoxin poisoning, unless patient has acute renal failure. In fact, these patients are frequently hypokalaemic and hypomagnesaemic secondary to chronic diuretic use. Both these electrolyte disorders are important as they exacerbate digoxin toxicity.
Serum digoxin levels taken at 6 hours post ingestion are useful in assessing and confirming toxicity, but must be carefully interpreted in the context of the clinical presentation. They do not accurately correlate with clinical toxicity. Therapeutic concentrations are usually quoted as 0.6–1.0 nmol/L (0.5–0.8 mcg/L). Significant chronic toxicity may be associated with 0.6–1.0 nmol/L (0.5–0.8 mcg/L) elevations of the serum digoxin concentration. This is particularly the case in the presence of pre-existing cardiac disease, hypokalaemia or hypomagnesaemia. Following acute overdose, the serum digoxin concentration is relatively high compared to tissue concentrations, until distribution is completed by 6–12 h post-ingestion. However, early concentrations greater than 15 nmol/L indicate serious poisoning.
Treatment
The best outcome is associated with early recognition of digoxin toxicity.
For chronic toxicity with minimal symptoms, management may involve no more than observation, cessation of digoxin administration, correction of hypokalaemia and hypomagnesaemia and appropriate management of any factors that contributed to the development of toxicity. However, the presence of any cardiovascular system effects, particularly in elderly patients, is an indication for the administration of Fab fragments of digoxin-specific antibodies. Apart from brady-tachy arrhythmias that are associated with haemodynamic instability, patients who have increased automaticies with cardiac or gastrointestinal symptoms may be considered for digoxin specific antibody. This is especially for patients who have renal failure with a Creatinine clearance <30 ml/min. From an economic viewpoint, the potential reduction in length of stay of 1-2 days as a result of treatment with digoxin specific antibody needs to take into consieration of the increase in the cost of 1 vial of digoxin Fab to A$850 per 40 mg vial.
Following acute overdose, the patient should be initially managed in a monitored area with full resuscitative equipment available. Immediate attention to the airway, breathing and circulation may be required. Intravenous access should be established and blood sent for urgent electrolytes and serum digoxin concentration. Although digoxin is well bound by charcoal, administration is usually difficult because of repetitive vomiting and attempts should not detract from other interventions.
The specific antidote to digoxin poisoning is Fab fragments of digoxin-specific antibodies, which should be administered as soon as possible in any potentially life-threatening digoxin intoxication. Commonly accepted indications for the administration of Fab fragments are listed in Table 29.2.5.
Table 29.2.5
Hyperkalaemia (K>5.5 mmol/L) associated with digoxin toxicity
History of ingestion of more than 10 mg of digoxin
Haemodynamically unstable cardiac arrhythmia
Cardiac arrest from digoxin toxicity
Serum digoxin concentration greater than 15 nmol/L
Fab fragments of digoxin-specific antibodies
These are derived from IgG antidigoxin antibodies produced in sheep. Removal of the Fc fragments of the antibodies greatly reduces the potential for hypersensitivity reactions and contributes to the remarkable safety profile of the product. Intravenously administered Fab fragments bind digoxin in the intravascular space on a mole-for-mole basis. As binding continues, digoxin moves down a concentration gradient from the tissue compartments to the intravascular compartment. Bound digoxin is inactive. A clinical response is usually observed within 20–30 min of administration. The Fab–digoxin complexes are excreted in the urine.
The extraordinary clinical efficacy of digoxin-specific fragments has been well documented in a few multicentre studies. These studies demonstrated the safety of the product, with the adverse reactions reported being hypokalaemia (4%), rapid atrial fibrillation or worsening of congestive cardiac failure (3%) and allergic reaction (0.8%).
The correct dose of Fab fragments may be calculated on the basis that 40 mg (one vial) will bind 0.5 mg of digoxin. If the dose ingested is unknown and/or a steady-state serum digoxin concentration is not available, dosing of Fab fragments must be empiric. A reasonable empiric dosing is to give 2 vials initially and then check for a clinical response. Further digoxin specific antibody may be given to neutralise half of the body burden once digoxin concentration is available. If the digoxin ingested dose or serum concentration is known, give half of the equimolar dose is adequate to stablise patient with severe digoxin toxiicty. In cardiac arrest, give a bolus dose (5 vials), and this dose can be repeated after 30 to 60 minutes if there has been no clinical response. Smaller doses (two vials) are usually sufficient to reverse the effects of chronic toxicity.
It is important that ED staff are aware of the amount and location of supplies of Fab fragments within their own institution and know the most rapid way to acquire further stocks should the need arise.
Serum digoxin concentrations will be extremely high following the administration of Fab fragments because most assays measure both bound and unbound digoxin.
Disposition
Patients with mild, chronic digoxin toxicity (gastrointestinal symptoms only) may be discharged after cessation of digoxin therapy provided there are no significant electrolyte disturbances, renal failure or other precipitating medical conditions. Following administration of Fab fragments, cases of chronic toxicity with conduction defects usually require medical admission for observation and treatment of intercurrent illness.
Acute overdoses require close observation for at least 12 h. Those that develop toxicity require admission and an appropriate level of monitoring. Following successful administration of digoxin-specific Fab fragments, patients must be carefully monitored for hypokalaemia and worsening of any underlying medical conditions for which digoxin may have been prescribed therapeutically. All intentional ingestions require psychiatric evaluation prior to medical discharge.
Clonidine
Introduction
Clonidine, an imidazoline derivative, is a central α2-adrenergic agonist. It was first developed in the 1960s as a nasal decongestant. It is currently used for the management of hypertension, attention deficit hyperactivity disorder (ADHD) as well as withdrawal symptoms from drug and alcohol addiction, tobacco withdrawal and Tourette’s syndrome. Clonidine toxicity often mimics that of opioids.
Pharmacokinetics
Clonidine is well absorbed with a bioavailability of almost 100%. The peak concentration in plasma and effect is observed within 1–3 h. The elimination half-life is 6–24 h with a mean half-life of 12 h. Half of the administered dose is excreted unchanged by the kidney.
Pathophysiology
Clonidine activates central α2-receptors. This results in a reduction in CNS sympathetic outflow at the vasomotor centre in the medulla oblongata. Clonidine is thought to reduce blood pressure through a reduction in cardiac output as well as its weak peripheral α-adrenergic antagonist properties. Clonidine also stimulates parasympathetic outflow and this may contribute to the slowing of heart rate as a consequence of increased vagal tone. Paradoxically, clonidine overdose can result in an initial hypertension from its partial α1-adrenergic agonist effect. It is suggested that clonidine’s inhibition of sympathetic outflow is mediated through endogenous opiate release.
Clinical features
Clonidine can cause transient hypertension from initial vasoconstriction with parenteral administration followed by hypotension. In addition to bradycardia and conduction defects, it can cause a central chlorpromazine-like effect with sedation. Other CNS symptoms include coma, seizure, miosis, reduced respiration and hypothermia. The median onset of symptoms following clonidine ingestion is 30 min and patients are usually symptomatic on arrival at the ED. Symptoms usually resolve by 24 h.
Investigations
The ECG is essential in evaluating and monitoring for bradycardia and conduction defects.
Treatment
The management of clonidine poisoning is primarily supportive. Hypotension usually responds to intravenous fluids. Atropine has been shown to abolish bradycardia in some case reports. Occasionally, inotropes may be required to maintain haemodynamic stability. Hypertension is usually short lived and rarely requires treatment. Patients are usually symptomatic on arrival and the benefits of administering activated charcoal are unlikely to outweigh the risk of aspiration.
Disposition
Patients should be observed in hospital until they are asymptomatic and bradycardia has resolved. They do not require ongoing cardiac monitoring for a stable sinus bradycardia.
Class 1c Antiarrhythmics
Introduction
Apart from the class one antiarrhythmics, many drugs in overdose cause sodium channel blockade. Drugs highly associated with sodium channel blockade and QRS widening are shown in Table 29.2.6. Overdose with class 1c antiarrhythmic drugs is among the most serious ingestions and is associated with a high morbidity and mortality. Overdoses with these agents are rare but they may be rapidly lethal with profound cardiovascular collapse. Drugs in this class include flecainide and propafenone; they are used for the treatment of SVT and ventricular arrhythmias. Class 1c antiarrhythmics block the fast inward sodium channel during phase 0 of the action potential. They have slow offset kinetics and cause complete blockade of sodium channel for a much longer duration than class 1a and 1b antiarrhythmics.
Table 29.2.6
Drugs highly associated with QRS widening and sodium channel blockade
| Antidepressants | Tricyclic antidepressants |
| Venlafaxine | |
| Antiepileptic drugs | Carbamazepine |
| Antihistamines | Diphenhydramine |
| Antipsychotics* | Chlorpromazine |
| Cardiovascular drugs | Flecainide |
| Propranolol | |
| Local anaesthetics | Bupivacaine |
| Ropivacaine | |
| Others | Bupropion |
| Chloroquine, hydroxychloroquine and quinine | |
| Cocaine | |
| Dextropropoxyphene | |
| Dolasetron | |
| Orphenadrine |
*Antipsychotic drugs are not highly associated with QRS widening, but are commonly taken in overdose.
Reproduced with permission from Toxicology and Wilderness Expert Group. Drugs highly associated with QRS widening and sodium channel blockade (Table 17.1) [revised 2011 June]. In: Therapeutic guidelines eTG complete [Internet]. Melbourne: Therapeutic Guidelines Limited; 2012 Nov.
Pharmacokinetics
Flecainide has a high oral bioavailability and a rapid onset of action of 30 to 60 min. It has a long elimination half-life of 7–23 h. In adults, ingestions of 800 mg or more should be considered as life threatening. Similarly, propafenone has a long elimination half-life.
Clinical Features
In overdose, they have a rapid onset of clinical symptoms, typically with 30 min to 2 h. Overdose symptoms include nausea, vomiting, hypotension, bradycardia, varying degrees of atrioventricular block and tachyarrhythmia. In severe cases, coma and seizures may occur. Severe intoxication is frequently fatal because of the rapid onset of hypotension and ventricular arrhythmias.
Investigations
The ECG is essential in evaluating and monitoring the QRS duration, conduction defects and QT interval. The most common and important ECG change in overdose is QRS widening (more than 120 ms). Although QT prolongation occurs in flecainide overdose, torsades de pointes is rare.
Treatment
The mainstay of management of class 1c overdose is good supportive care including inotropic support, gastrointestinal decontamination and early and repeated doses of sodium bicarbonate to treat any broad complex arrhythmias and hypotension. Treatment is similar to other sodium channel blocking agents, such as the tricyclic antidepressants and includes plasma alkalinization to a pH of 7.5 with hyperventilation and repeated boluses of sodium bicarbonate. The use of antiarrhythmic drugs is problematic and are generally contraindicated and sufficient amounts of sodium bicarbonate should be used first.
Disposition
Asymptomatic patients with a normal ECG 4 h after ingestion of flecainide are unlikely to develop toxicity. Patients who are symptomatic should be admitted to a monitored area.
Cardiac arrest due to cardiovascularly active drugs
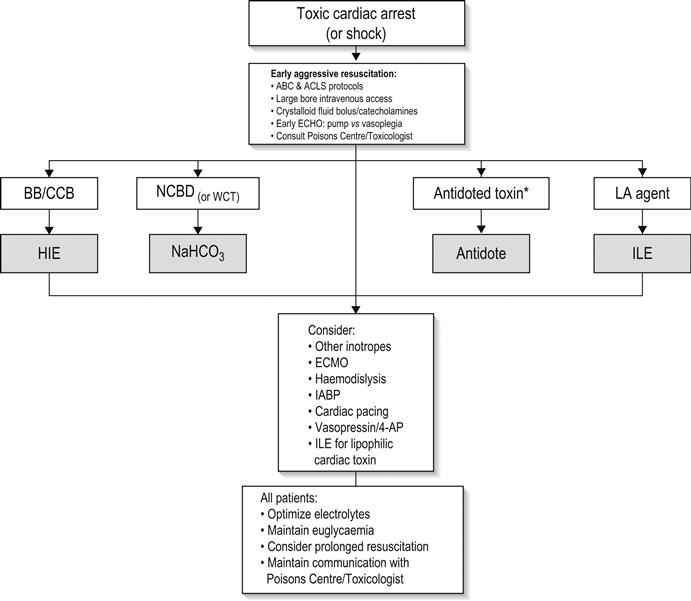
It is important for clinicians to be aware that cardiac arrest due to overdose of cardiovascularly active drugs may necessitate prolonged CPR and resuscitative manoeuvres and consideration of heroic measures, such as cardiopulmonary bypass (Fig. 29.2.1). Consultation with a clinical toxicologist is always recommended prior to cessation of resuscitative efforts in these cases.
Controversies
29.3 Antipsychotic drugs
Dino Druda and Shaun Greene
Introduction
The antipsychotics form a heterogeneous group of medications that has evolved since chlorpromazine was first used to treat schizophrenia in the 1950s. The first-generation or so-called ‘typical’ antipsychotics caused many adverse effects, especially movement disorders such as extrapyramidal symptoms (EPS) and tardive dyskinesia (TD). They also have very little efficacy in treating the negative symptoms of schizophrenia (social withdrawal, anhedonia, poverty of speech, etc.). This led to the development and marketing of the second-generation, or atypical, antipsychotics in the late 1980s. In general, these drugs have fewer tendencies to cause movement disorders and have efficacy in managing the negative symptoms of schizophrenia, while maintaining efficacy in the management of acute psychosis. For this reason, they have largely replaced the older antipsychotic as first-line therapy in the treatment of schizophrenia and psychotic disorders.
Pharmacology
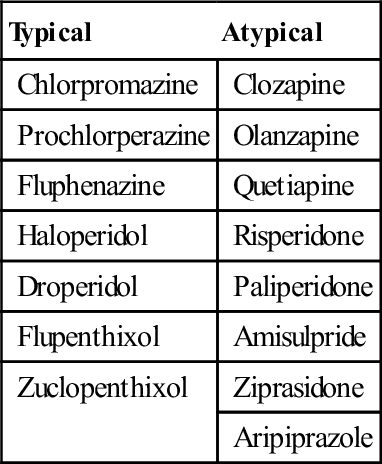
There are numerous ways of classifying the antipsychotic drugs; typical or atypical as described above (Table 29.3.1), by their chemical structure or according to neuroreceptor binding affinity.
Table 29.3.1
Typical and atypical antipsychotic drugs
| Typical | Atypical |
| Chlorpromazine | Clozapine |
| Prochlorperazine | Olanzapine |
| Fluphenazine | Quetiapine |
| Haloperidol | Risperidone |
| Droperidol | Paliperidone |
| Flupenthixol | Amisulpride |
| Zuclopenthixol | Ziprasidone |
| Aripiprazole |
All antipsychotic drugs produce their beneficial therapeutic effects by antagonizing the dopamine D2 receptors in the mesolimbic system. The first-generation antipsychotics were classified as high or low potency depending on their affinity for the D2 receptor. However, antagonism of the other D2 receptors leads to many of the adverse clinical effects. Antagonism of D2 receptors in the nigrostriatal pathway leads to movement disorders (EPS, akathisia, TD), antagonism of the D2 receptors in the mesocortical area can contribute to the negative symptoms and antagonism of D2 receptors in the anterior pituitary stimulates prolactin secretion which can lead to gynaecomastia and galactorrhoea. Blockade of D2 receptors in the anterior hypothalamus is associated with alterations in temperature regulation, which can lead to hypo- or hyperthermia and may be involved in the development of neuroleptic malignant syndrome, which is discussed later. The antiemetic effect of some of the antipsychotics is due to antagonism of the D2 receptors in the chemoreceptor trigger zone in the medulla.
The newer atypical antipsychotics also derive therapeutic efficacy from affinity and antagonism at various serotonin (5-HT) receptors. Antagonism at the 5-HT2A receptor is implicated both in increasing the efficacy of treating the negative symptoms of schizophrenia and also in reducing the incidence of EPS.
Agents with high antagonism of muscarinic M1 and M2 receptors (e.g. olanzapine, quetiapine) can cause an agitated delirium and peripheral features characteristic of anticholinergic toxicity (flushing, dry skin, tachycardia, urinary retention, etc.). Drugs that have a higher anticholinergic activity than dopaminergic tend to cause fewer extrapyramidal effects. High relative antagonism of histamine H1 receptors leads to sedation and, to a lesser extent, hypotension. Antagonism at the α1-adrenergic receptor can result in hypotension (e.g. quetiapine, clozapine) and clozapine also antagonizes the α2-receptor, although the clinical significance of this is uncertain.
Several first-generation antipsychotics can block voltage-gated fast sodium channels which, in overdose, can lead to slowing of cardiac conduction thus prolonging the QRS complex and impairing myocardial contractility. Blockade of the delayed rectifier potassium channel causes delayed repolarization and leads to prolongation of the QT interval.
Despite the heterogeneous nature of the antipsychotics, in general, they share similar pharmacokinetic properties. They are well absorbed after oral administration, with peak serum concentrations usually occurring within 2–6 hours of ingestion. This may be delayed following overdose of agents with significant anticholinergic properties. They are lipophilic, have large volumes of distribution and the majority of the agents are highly protein bound. They are extensively metabolized in the liver, with some having active metabolites.
Clinical effects
Adverse effects
Adverse effects at therapeutic doses may be dose related or idiosyncratic.
Extrapyramidal syndromes
These are a heterogeneous group of disorders characterized by abnormal neuromuscular activity. They can be particularly distressing to patients and may lead to difficulties with compliance and cessation of treatment. There are four well-recognized syndromes–acute dystonia, akathisia, parkinsonism and tardive dyskinesia. Of these, the first three are usually reversible, whereas tardive dyskinesia is irreversible, but occurs late, usually after months to years of treatment.
EPS are more common among the first-generation typical antipsychotics, especially those with high potency, such as haloperidol. Atypical antipsychotics are associated with a lower incidence of EPS, although it must be appreciated that EPS can occur with the use of any antipsychotic. Reactions are usually idiosyncratic, although can occur following overdose.
Acute dystonia is characterized by sustained involuntary muscle contraction, which commonly involves the face, head and neck, but can also involve the extremities. Rarely, the larynx can be involved, which may be life threatening. Risk factors for developing dystonia include male gender, young age and previous history of dystonic reaction. Onset is usually within a few hours of exposure, but may be delayed for several days.
Akathisia is characterized by an unpleasant sensation of restlessness or unease and, often, the patient is unable to remain still. It can be difficult to diagnose and can often be attributed to the underlying psychiatric condition rather than to the treatment.
Drug-induced parkinsonism is similar to idiopathic Parkinson’s disease, with rigidity and bradykinesia, although the characteristic tremor may be less pronounced. It is more common in older patients and patients on high potency agents.
Tardive dyskinesia is characterized by repetitive, involuntary, purposeless movements, classically involving the muscles of the face and mouth, although the limbs and trunk may be involved. It usually appears after months or years of therapy with antipsychotic medication and is usually resistant to treatment.
Cardiovascular effects
Cardiovascular effects of antipsychotics with therapeutic use include tachycardia, postural hypotension and ECG changes. Postural hypotension may be multifactorial, with α1-adrenergic blockade and direct myocardial depression playing a role. ECG changes may be diverse, with QRS prolongation, QT prolongation and non-specific ST-segment and T-wave changes being reported.
Seizures
All antipsychotics can lower the seizure threshold. However, seizures rarely complicate therapeutic use of antipsychotics, unless the patient has underlying risk factors, such as organic brain disease or epilepsy.
Metabolic syndromes
Chronic use of many of the antipsychotics is associated with the development of a metabolic syndrome, which can lead to weight gain, dyslipidaemia, hypertension and impaired glucose tolerance. These can be distressing and also contribute to the development of cardiovascular disease and type II diabetes. The development of these adverse effects can affect compliance with treatment. Metabolic affects are particularly associated with the use of olanzapine and clozapine.
Neuroleptic malignant syndrome (NMS)
NMS is a rare idiosyncratic adverse reaction, which can occur with any of the antipsychotic medications. Risk factors, diagnosis and management of NMS are described later in this chapter.
Clozapine
Clozapine is associated with a number of idiosyncratic effects that can occur with therapeutic use and requires more vigilant surveillance. These include agranulocytosis and myocarditis. These should be considered in the differential diagnosis if a patient on clozapine presents unwell.
Overdose
Following overdose of antipsychotics, the most common and significant manifestations involve the CNS and cardiovascular system.
Dose-dependent CNS depression occurs, ranging from lethargy and somnolence to coma and seizures. Airway protective reflexes may be impaired, requiring intensive care. Many of the agents can cause significant anticholinergic delirium with associated peripheral effects, such as urinary retention, flushed skin and reduction in sweat and saliva secretion.
The most common cardiovascular effects are tachycardia and hypotension. Tachycardia may be due to anticholinergic effects and also as a response to hypotension. Hypotension often occurs as a result of peripheral α1-receptor blockade, leading to vasodilatation.
ECG changes are often present after overdose and can include QRS prolongation and QT interval prolongation. Significant arrhythmias are uncommon, apart from following overdose with amisulpride, which can cause torsades des pointes.
Some of the specific clinical features following overdose of individual agents are described below.
Amisulpride
Overdose of amisulpride commonly causes QT prolongation, bradycardia and hypotension. Episodes of torsades de pointes have been reported and so amisulpride is considered to be particularly cardiotoxic. Onset of cardiotoxicity may be delayed for greater than 12 hours and QT prolongation can persist for many hours, with the potential to develop torsades de pointes abruptly. Ingestions greater than 4 g have been associated with development of prolonged QT and ingestions greater than 8 g can cause significant sedation and hypotension.
Chlorpromazine
Ingestions of greater than 15 mg/kg of chlorpromazine in children are associated with significant toxicity. Large overdoses>5 g, especially in drug-naïve patients, may lead to significant CNS depression, which may require intubation and intensive care due to loss of airway protective reflexes. Significant hypotension also occurs following overdose.
Clozapine
Acute overdose of clozapine leads to CNS depression, which is more pronounced in clozapine-naïve patients, who may require intubation. Seizures are reported in overdose. Despite the known anticholinergic properties of clozapine, hypersalivation is common. Miosis is classically described, but mydriasis can also occur. Agranulocytosis does not occur after a single overdose.
Haloperidol
Sedation and EPS are common following overdose of haloperidol. Haloperidol has also been associated with QT prolongation and arrhythmias following large ingestions or intravenous administration.
Olanzapine
Onset of clinical features following overdose is within 6 hours. Sedation and anticholinergic effects are the most common manifestations, leading to a combination of agitation and drowsiness, which may require intubation. Miosis may be noted. Tachycardia is common, but significant ECG abnormalities are rare.
Quetiapine
Quetiapine is available in immediate and extended release preparations. Overdose causes dose-related CNS depression and tachycardia. Hypotension and seizures are also reported after larger ingestions. Ingested doses of greater than 3 g are associated with increased length of stay and ICU admission. QTc prolongation is often reported, although the clinical significance of this is unclear, as there have been no reported cases of torsades de pointes. It may be that the prolongation of the QTc is as a result of overcorrection for tachycardia rather than intrinsic cardiotoxicity.
Risperidone
Risperidone is relatively benign following overdose, with tachycardia and dystonia being the most common effects. Onset of dystonia may be delayed and may recur after treatment.
Ziprasidone
Ziprasidone is associated with QT prolongation, both with therapeutic use and following overdose. Torsades de pointes has been reported after ziprasidone overdose with co-ingestants, but not in isolation.
Investigations
Antipsychotic toxicity is primarily diagnosed on history and examination for typical clinical features. Serum drug concentrations are not usually available in a clinically useful time frame or helpful in the management of acute overdose. In the case of patients on clozapine who present unwell to hospital, then a clozapine concentration can be measured. For these patients, a WCC is helpful when looking for agranulocytosis and troponin may be elevated in cases of clozapine-induced myocarditis. If this is suspected, then ECG and echocardiography may also be required.
Initial ECG evaluation should occur for all patients following antipsychotic overdose and any abnormality of the QRS or QT intervals warrants continuous cardiac monitoring. If initial ECG is normal, then it should be repeated after 6 hours in asymptomatic patients. More prolonged cardiac monitoring is required for patients following overdose with amisulpride or ziprasidone.
Treatment
The management of antipsychotic toxicity is primarily supportive. Attention to initial resuscitation should occur initially. Mild sedation requires no specific treatment. Patients with significantly decreased conscious state with loss of airway protective reflexes will require intubation, ventilation and intensive care.
Decontamination with activated charcoal can be considered if the presentation is within an hour and there is no clinical sign of CNS depression. Otherwise, administration of activated charcoal should be delayed until after the airway has been secured with intubation. There is no evidence for any benefit from enhanced elimination techniques, either with multidose activated charcoal or extracorporeal techniques and so they are not indicated.
Hypotension should initially be treated with an appropriate bolus of crystalloid solution. If there is no response to initial fluid bolus, then vasopressors may be required. There have been reports of worsening hypotension following the administration of adrenaline to patients who are hypotensive following quetiapine overdose, so noradrenaline is the preferred initial vasopressor. Patients with ventricular arrhythmias or with prolonged QRS should be managed with sodium bicarbonate in a similar fashion to patients with significant tricyclic antidepressant (TCA) toxicity. Intravenous bicarbonate 8.4% (1 mL=1 mmol) 1–2 mmol/kg boluses should be administered to obtain an arterial pH of 7.50–7.55. The pH may then be maintained in this range using hyperventilation in intubated patients. QT prolongation requires no specific management other than cardiac monitoring and correction of any potential contributing electrolyte abnormalities, such as hypokalaemia or hypomagnesaemia. Should torsades de pointes develop, it should be treated in the first instance with IV magnesium sulphate 50% 2–4 mL (1–2 g or 4–8 mmol) infusion over 10 minutes. Chemical or electrical overdrive pacing may also be required to avoid further instances.
Seizures are often self-limiting and require no treatment. However, should intervention be necessary, benzodiazepines should be used first line. Barbiturates and/or general anaesthesia are used for refractory seizures or status. There is no role for other anticonvulsants, such as phenytoin, in the management of drug-induced seizures.
Anticholinergic delirium should be managed initially with non-pharmacological measures, such as nursing in a quiet area and limiting stimulation. Urinary retention should be sought and relieved with a urinary catheter, as the distress caused by this may contribute to any agitation. Should medication be required, titrated doses of benzodiazepines (e.g. diazepam) should be used, although it must be appreciated that benzodiazepines may contribute to any CNS depressant effects of the ingested antipsychotic.
Acute dystonia is managed with an anticholinergic agent, such as benztropine (1–2 mg given IV or IM). Benzodiazepines may also be used. Repeat dosing may be required as the dystonia can recur.
Disposition
Patients should be observed for 6 hours after overdose of most antipsychotics. If they remain asymptomatic and have a normal ECG at this time, then they can be medically cleared for discharge. This observation period should be extended for 12 hours following significant ingestion of extended release preparations of quetiapine, ingestions of>4 g amisulpride and ingestions of ziprasidone.
Patients with significant symptoms should be admitted for observation until symptoms are resolving. Intensive care admission is required for patients requiring intubation and ventilation. In the event of any concerning ECG abnormalities, such as QT prolongation, then admission for cardiac monitoring is recommended until these changes are resolving.
Neuroleptic malignant syndrome
Neuroleptic malignant syndrome is a rare, idiosyncratic, potentially life-threatening adverse reaction that has been reported to occur with all the antipsychotics. There is a large variation in reported incidence, but recent pooled data suggest an incidence of 0.01–0.2%. The pathophysiology of NMS is not completely understood, but is thought to be due to central dopamine blockade, especially in the nigrostriatal and hypothalamic pathways. Risk factors for the development of NMS include male gender, young age, use of high potency antipsychotics, recent increase in dose, parenteral administration, dehydration and organic brain disease.
Clinical features
The onset of NMS occurs typically over 1–3 days. The typical clinical features are a combination of altered mental status, hyperthermia (temperature>38°C), autonomic dysfunction and muscular rigidity (‘lead pipe’ rigidity). The altered mental status ranges from delirium and confusion to stupor and coma. Autonomic dysfunction can manifest as tachycardia, cardiac arrhythmias, respiratory irregularities and hypo- or hypertension. The muscular rigidity is classically described as ‘lead-pipe’ rigidity, with increased tone and resistance to passive movement. There may be superimposed tremor leading to cogwheeling. Other neuromuscular abnormalities include bradykinesia, dystonia, mutism and dysarthria.
Differential diagnosis
There have been numerous diagnostic criteria proposed for NMS, but none are universally accepted. Alternate diagnoses must be considered and excluded, particularly CNS infection. Other conditions to be considered in the differential include heatstroke, thyrotoxicosis, serotonin toxicity, anticholinergic syndrome, malignant catatonia, non-convulsive status epilepticus, phaeochromocytoma and drug intoxication (MAOIs, sympathomimetics).
However, NMS must be considered in any patient on antipsychotic medication who is unwell, particularly when there is altered mental status, fever or muscle rigidity, especially if there has been a recent change in the antipsychotic regimen.
Investigations
There is no diagnostic test for NMS, although there are characteristic laboratory abnormalities in some cases. Increased muscle enzymes (CK, LDH, AST) are often present in cases of NMS. Leucocytosis is also frequently observed. There may be other electrolyte disturbance, metabolic acidosis and coagulation abnormalities.
Investigations to rule out alternate diagnoses need to be carried out, including CT scan of the brain and lumbar puncture to rule out CNS infection.
Treatment
Once NMS is diagnosed, it is essential to institute aggressive supportive care. The offending drug must be immediately withdrawn. Patients are often dehydrated, so fluid resuscitation should be commenced. Hyperthermia must be managed aggressively with passive and active cooling. If the temperature is>39.5°C, then intubation and neuromuscular paralysis should be considered. There is increased risk of thromboembolism, so prophylaxis should be commenced.
Benzodiazepines are often used early in the treatment of NMS and they may be effective in ameliorating symptoms in milder cases. Bromocriptine is a centrally acting dopamine agonist that can only be administered orally or via NG tube. The starting dose is 2.5 mg three times daily, increased to a daily maximum of 40 mg. It needs to be continued for 1–2 weeks, as premature discontinuation can lead to rebound symptoms. Potential adverse effects include vomiting and worsening of psychosis.
Dantrolene interferes with calcium release in skeletal muscle cells and therefore reduces skeletal muscle activity. It may be useful in cases of NMS with prominent hyperthermia and muscle rigidity. It can be given by IV infusion at 2–3 mg/kg/day. However, there are conflicting reports regarding the efficacy and outcome benefit of dantrolene in the management of NMS. Electroconvulsive therapy (ECT) has been advocated for cases of NMS refractory to pharmacological treatments, although its efficacy is unclear.
Controversies
29.4 Antidepressant drugs
Shaun Greene and Dino Druda
Introduction
Severity of clinical toxicity following overdose (OD) of antidepressant drugs available in Australia varies according to the class of drug. Toxicity is dose dependent and produces clinical manifestations affecting multiple organ systems. Cardiovascular and neurological features can be life threatening. Early risk assessment and aggressive supportive care are essential in ensuring a good outcome.
Tricyclic antidepressants
Although efficacious in treating depression, tricyclic antidepressants (TCAs) are relatively more toxic than other classes of antidepressants in OD. Significant toxicity including death is associated with ingested doses of more than 10 mg/kg in adults and 5 mg/kg in children. Of the tricyclic antidepressants available in Australia (Table 29.4.1), dothiepin is associated with the greatest toxicity. Cardiovascular system dysfunction and coma typically manifest rapidly following significant ingestion. Good outcome is dependent on aggressive airway management, utilization of sodium bicarbonate and provision of supportive care in a critical care environment.
Table 29.4.1
Tricyclic antidepressants available in Australia
Amitriptyline
Clomipramine
Dothiepin
Doxepin
Imipramine
Nortriptyline
Trimipramine
Pharmacology
The tertiary amine structure of TCAs non-selectively interacts with multiple receptors throughout the body, most of which are not implicated in positive antidepressant effects. Pharmacodynamic interactions include:
 Stimulation of central postsynaptic histamine receptors producing CNS depression, sedation and coma.
Stimulation of central postsynaptic histamine receptors producing CNS depression, sedation and coma.
 Antagonism of peripheral α1-adrenergic receptors producing peripheral vasodilatation.
Antagonism of peripheral α1-adrenergic receptors producing peripheral vasodilatation.
 Varying degrees of antagonism at potassium, chloride and γ-aminobutyric acid (GABA) receptors.
Varying degrees of antagonism at potassium, chloride and γ-aminobutyric acid (GABA) receptors.
Tricyclic antidepressants are well absorbed following ingestion, undergo extensive first pass metabolism, are hepatically metabolized (often producing active metabolites), are highly protein bound and are lipophilic and therefore widely distributed throughout the body. Half-lives are relatively long (10–81 hours) and often observed to be longer following OD.
Clinical features
The most common clinical features of significant TCA overdose are CNS depression (varying from agitated delirium to coma) and sinus tachycardia; these manifest rapidly within 1–2 hours of exposure. Risk assessment based on dose ingested and associated anticholinergic, CNS and cardiovascular clinical effects are described in Table 29.4.2. Agitated delirium secondary to anticholinergic receptor agonism is not always evident in more severe cases as coma predominates. Sodium channel blockade and α-receptor mediated peripheral vasodilatation lead to supraventricular and ventricular arrhythmias, hypotension and asystole in a dose-dependent manner. Anticholinergic features may become more apparent during the recovery phase as histamine receptor-induced sedation resolves.
Table 29.4.2
Tricyclic antidepressants: dose-related risk assessment and clinical effects
| Dose | Effect |
| <5 mg/kg | Minimal symptoms |
| 5–10 mg/kg | Anticholinergic effects, mild sedation |
| Major toxicity not expected | |
| >10 mg/kg | Significant clinical toxicity likely to occur within 2–4 hours of ingestion |
| Anticholinergic effects | |
| Coma, myoclonic jerks, seizures (early in course, 3–4% of cases) | |
| Sinus tachycardia, supraventricular tachycardia, torsades de pointes, ventricular fibrillation, idioventricular rhythm, 2nd/3rd degree heart block, asystole, hypotension (myocardial dysfunction+peripheral vasodilation) | |
| >20 mg/kg | Coma, hypotension, potential for seizures and arrhythmias. Duration toxicity potentially>24 hours |
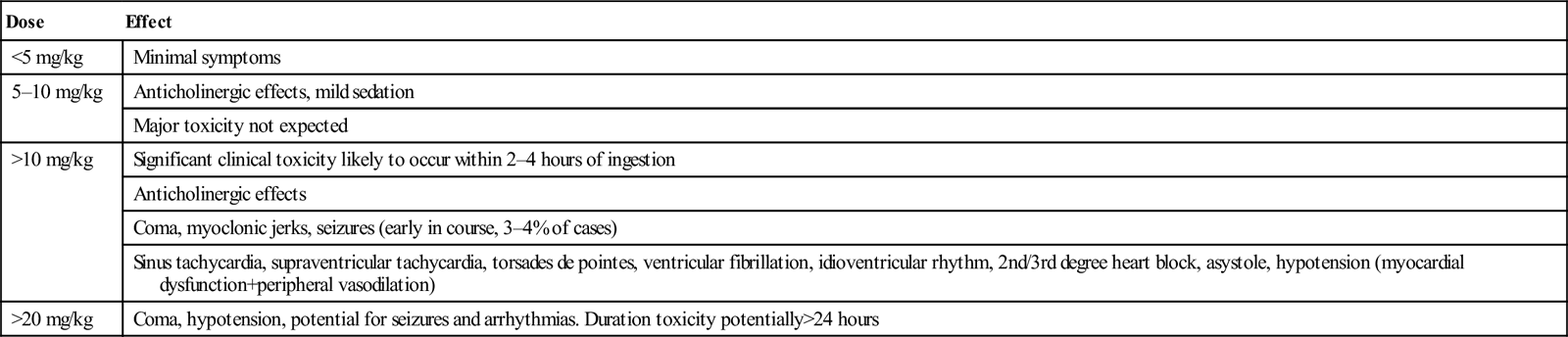
Clinical investigations
A 12-lead ECG is the most valuable prognostic investigation following TCA overdose. A terminal 40 ms axis between 120 and 270 degrees is a sensitive indicator of TCA presence but this is difficult to measure at the bedside. Measurement of maximal limb lead QRS duration is a useful predictor of toxicity. Prolongation of greater than 100 ms is associated with an increased incidence of coma, need for intubation, seizures, hypotension and arrhythmias. One study demonstrated no seizures or arrhythmias in patients with a QRS duration that remained<100 ms. Ventricular arrhythmias were predicted in one study by a QRS duration>160 ms. The finding of a positive R wave of greater than 3 mm in amplitude in lead aVR or a ratio of>0.7 between the amplitude of R and S waves in aVR are sensitive markers for seizures and arrhythmias. A rightward frontal plane QRS vector (indicated by an S wave in lead I and an R wave in aVR) is associated with TCA toxicity. Although QT prolongation is observed in TCA therapy and toxicity, this finding is not predictive of clinical toxicity.
TCA blood concentrations can be measured, but are poorly correlated with degree of clinical toxicity.
Treatment
Patients with significant clinical toxicity or those with a recent (previous 3–4 hours) reported ingestion of a potentially toxic amount of a TCA receive aggressive supportive care in a resuscitation area. Early securing of the airway via endotracheal intubation is indicated when there is any decrease in conscious state. Poor respiratory function and secondary hypoxia potentially worsen TCA toxicity.
Administration of activated charcoal should be considered within 1 hour of ingestion, provided facilities exist to protect the airway if decreased consciousness or seizures occur. Activated charcoal may reduce TCA absorption if administered to intubated patients via a nasogastric tube up to 4 hours post-ingestion.
Early aggressive use of sodium bicarbonate in conjunction with hyperventilation is indicated where there is any cardiovascular dysfunction in conjunction with QRS prolongation (>100 ms), hypotension unresponsive to initial intravenous fluid (see further discussion below) or in the presence of any arrhythmia. Intravenous bicarbonate 8.4% (1 mL=1 mmol) 1–2 mmol/kg boluses should be administered to obtain an arterial pH of 7.50–7.55. pH should then be maintained in this range using hyperventilation. Sodium bicarbonate provides hypertonic sodium, competitively overcoming sodium channel blockade. Alkalization improves sodium channel function and reduces the free concentration of TCA available to produce toxicity. Acid–base manipulation using sodium bicarbonate is more effective than hyperventilation alone in TCA toxicity. Sodium bicarbonate may be prophylactically beneficial in cases where there is a significant history of TCA ingestion and a QRS duration of>100 ms.
Other therapies, including concentrated hypertonic saline (3% sodium chloride) and lignocaine, may be beneficial in treating resistant arrhythmias. Class 1a antiarrhythmics, including procainamide, quinidine and phenytoin, are contraindicated.
Hypotension is treated with intravenous crystalloid (up to 20–30 mL/kg). Administration of sodium bicarbonate is indicated if hypotension persists. Inotropes are indicated for resistant hypotension despite intravenous fluid administration and normalization of acid–base status.
Disposition
Patients who are well with no CNS depression and a normal ECG 6 hours post-reported TCA ingestion are safe for medical discharge. Those with any signs of significant toxicity require admission to a critical care environment.
Bupropion
Pharmacology
Bupropion is a drug with a unicyclic structure used as a smoking cessation aid in Australia and as an antidepressant in a number of other countries. It has a structure similar to amphetamine and inhibits reuptake of dopamine, with a lesser effect on noradrenaline and serotonin. Bupropion is licensed for use as an antidepressant in some countries. It is only available as an extended release preparation in Australia. Bupropion and its active metabolite hydroxyl-bupropion have half-lives of approximately 20 hours. Both are relatively highly protein bound (84% and 77%). Bupropion has a volume of distribution of 2000 L. Mild anticholinergic properties may be evident following OD. Bupropion OD is characterized by a high risk of seizures; studies suggest hydroxyl-bupropion is responsible, although the exact mechanism is unclear.
Clinical features
Bupropion is only available as a modified release preparation in Australia. In therapeutic doses, bupropion may cause mild hypertension, postural hypotension in sporadic cases, headache, agitation, seizures and gastrointestinal irritation. Cardiac conduction abnormalities are not reported in therapeutic dosing.
Tachycardia, hypertension, nausea, vomiting, tremor and hallucinations may be observed following bupropion OD; however, severe agitation and seizures are the most significant clinical features; seizures are typically delayed up to 6–8 hours (in some cases up 16 hours) post-exposure and are dose dependent. In a series of 59 patients presenting following OD of modified release bupropion, seizures occurred in 30% of those ingesting<4.5 g, 50% with 4.5–9 g ingested and 100% of individuals who ingested>9 g.
QT and QRS prolongation, arrhythmias and hypotension have been reported following ingestions of>9 g of bupropion. QT prolongation is likely to be related to coexisting tachycardia. A QT nomogram should be utilized in the risk assessment of any measured QT prolongation related to bupropion toxicity. Deaths have been reported following massive ingestions.
Treatment
Provision of meticulous supportive care is the mainstay of management following bupropion OD. Patients should be managed in a monitored area with resuscitation facilities available in case of seizures or cardiovascular deteriorations. Activated charcoal should be considered in patients who are cooperative and not agitated within 4 hours of ingestion. Intravenous benzodiazepines should be administered in incremental doses to control agitation at an early stage as they may increase the threshold for seizure activity. Resistant agitation may require sedation and intubation. Seizures are managed using benzodiazepines.
Hypotension is initially treated with intravenous crystalloid. QRS prolongation is treated with intravenous sodium bicarbonate (see tricyclic antidepressant section).
Disposition
Patients who are well with a normal ECG and no agitation or seizures 16 hours post-bupropion exposure are safe for medial discharge. Patients with significant agitation, ongoing seizures, or CVS instability require admission to a critical care environment.
Monoamine oxidase inhibitors
Pharmacology
Monoamine oxidase inhibitors (MAOIs) either reversibly or irreversibly inhibit function of the enzymes monoamine oxidase A and B (MAO-A, MAO-B), leading to increased CNS concentrations of adrenaline, noradrenaline, serotonin and dopamine. The pharmacodynamic effects of non-selective irreversible MAOIs (including phenelzine and tranylcypromine) are not overcome until MAO is resynthesized, resulting in dose-dependent toxicity that may last for days. Moclobemide, a selective reversible inhibitor of MAO-A is more benign in OD, but may cause severe serotonin toxicity when combined with other serotonergic agents.
The MAOIs are well absorbed orally, undergo extensive first-pass metabolism, readily cross the blood–brain barrier and have moderate volumes of distribution. Peak concentrations occur within 2 or 3 hours of ingestion. Metabolites are renally eliminated.
Clinical features
Overdose of irreversible MAOIs is characterized initially by peripheral sympathomimetic stimulation and central nervous system excitation. Symptoms do not usually manifest until 6–12 hours post-overdose. Initial symptoms include tachycardia, agitation, restlessness, hyperreflexia and voluntary movements. As toxicity progresses, there is progressive muscle rigidity, respiratory failure, decreasing conscious state, hyperthermia, rhabdomyolysis, coma and cardiovascular collapse. Clinical toxicity may last for days.
Lone overdose of the reversible selective MAOI moclobemide generally produces only mild symptoms including tachycardia, nausea and anxiety. Overdose of moclobemide with another serotonergic agent may lead to significant serotonin toxicity.
MAOI adverse effects in therapeutic doses include the development of serotonin toxicity when combined with other serotonergic agents. The tyramine reaction may occur following ingestion of tyramine-containing foods. Tyramine is an indirect acting sympathomimetic. MAOI inhibition of tyramine metabolism can lead to a hyperadrenergic crisis with severe hypertension, intracranial haemorrhage, renal failure, disseminated intravascular coagulopathy (DIC) and rhabdomyolysis.
Treatment
Management of MAOI toxicity is primarily supportive. Asymptomatic patients presenting within 2 hours of ingestion of tranylcypromine or phenelzine may benefit from administration of activated charcoal. Patients with evidence of severe toxicity are managed in a resuscitation area with particular attention to supporting organ function and limiting complications. Hypertension is initially treated using titrated doses of intravenous benzodiazepines. Refractory hypotension requires treatment with intravenous nitrates, sodium nitroprusside or phentolamine. Beta-adrenergic blockers are contraindicated; unopposed α-receptor stimulation may worsen toxicity. Hypothermia unresponsive to sedation with benzodiazepines must be treated aggressively along conventional lines. Serotonin toxicity requires specific care as outlined below. Moclobemide toxicity usually only requires basic supportive care.
Disposition
Patients who remain clinically well 6 hours post-ingestion of moclobemide or 12 hours post-ingestion of phenelzine or tranylcypromine are safe for medical discharge. Patients who are symptomatic following overdose of an irreversible MAOI require admission to an intensive care unit.
Selective serotonin reuptake inhibitors
Selective serotonin reuptake inhibitors (SRRIs) are associated with fewer adverse effects both in therapeutic use and OD compared to TCAs. They are utilized in treating depression, obsessive–compulsive disorder, panic/anxiety disorders, eating disorders and chronic pain syndromes. Table 29.4.3 lists SSRIs available in Australia.
Table 29.4.3
Serotonin reuptake inhibitors available in Australia
Selective serotonin reuptake inhibitors (SSRIs)
Citalopram, escitalopram, fluoxetine, fluvoxamine, paroxetine, sertraline
Combined selective serotonin and noradrenaline reuptake inhibitors (SNRIs)
Venlafaxine, desvenlafaxine, duloxetine, reboxetine
Serotonin reuptake inhibition with α-adrenergic antagonism
Mirtazepine
Pharmacology
SSRIs increase synaptic concentrations of serotonin in the CNS via interaction with G-protein coupled serotonin receptors (14 different receptors have been described currently). With extended use, downregulation of serotonin inhibitory autoreceptors occurs, leading to increased serotonin synthesis and decreased reuptake.
SSRIs are well absorbed post-ingestion. SSRIs and metabolites are substrates for and inhibitors of numerous hepatic microsomal enzymes, the most important being CYP2D6. Hepatic metabolism produces various metabolites; many are active and capable of prolonging therapeutic effect and also increasing the probability of adverse drug interactions.
Clinical features
Therapeutic use of SSRIs is associated with headache, insomnia, sexual dysfunction, gastrointestinal symptoms, dizziness and fatigue. Serotonin toxicity can occur in therapeutic use if exposure to another serotonergic agent occurs.
OD is rarely associated with severe adverse effects. Mild CNS depression may occur, but is not severe. Nausea, vomiting, tachycardia, diaphoresis and dizziness are reported, but are generally mild and self-limiting.
QT prolongation and torsades des pointes have been reported following citalopram and escitalopram OD. Fluoxetine, citalopram and escitalopram may cause mild bradycardia.
Treatment
Activated charcoal is only indicated following massive ingestion of an SSRI within the previous hour. There is evidence that cardiovascular toxicity may be limited if activated charcoal is administered to patients who have taken>300 mg escitalopram or>600 mg citalopram within the previous 4 hours.
Treatment is primarily supportive. Regular ECGs and cardiac monitoring are indicated following ingestion>1000 mg citalopram (or 600 mg if activated charcoal has not been given within 4 hours of ingestion) or>400 mg escitalopram (or 400 mg if activated charcoal has not been given within 4 hours of ingestion). Cardiac monitoring is continued for at least 13 hours post-exposure in these cases or until the QT interval has normalized.
Combined serotonin and noradrenaline reuptake inhibitors
Duloxetine, reboxetine, venlafaxine and desvenlafaxine are the combined selective serotonin and noradrenaline reuptake inhibitor antidepressants available in Australia.
Pharmacology
Selective serotonin and noradrenaline reuptake inhibitor (SNRIs) inhibit both serotonin and noradrenaline reuptake in the CNS. Rapid downregulation of β-adrenergic receptors may increase the speed of onset of therapeutic effect compared to SSRIs.
SNRIs are rapidly absorbed after oral administration and are hepatically metabolized. Desvenlafaxine and venlafaxine are only available in Australia in modified release formulations.
Clinical features
Overdose produces a noradrenergic mediated sympathomimetic toxidrome characterized by tachycardia, tremor, nausea, vomiting, dizziness and agitation. Venlafaxine is associated with seizures following ingestions of>5 g. Increasing agitation may herald onset of seizures. Hyperthermia and rhabdomyolysis may occur with large ingestions. Cardiovascular toxicity may occur following ingestions of>8 g of venlafaxine. QRS prolongation, QT prolongation, ventricular arrhythmias and hypotension have been reported. Desvenlafaxine is more potent than venlafaxine therapeutically (50 mg desvenlafaxine is equivalent to 75 mg venlafaxine) and therefore thresholds for toxic effects are likely to be lower with desvenlafaxine.
Treatment
Following large ingestions of SNRIs, patients should be managed in an area equipped with cardiac monitoring and facilities to manage seizures. Activated charcoal may be beneficial if administered within an hour of a large ingestion. Intubation and administration of activated charcoal within 4 hours of ingestion of>5 g of venlafaxine should be considered.
Agitation should be controlled using incremental doses of benzodiazepines. Seizures are normally self-limiting, but may require treatment with intravenous benzodiazepines.
Hypotension is treated initially with intravenous fluid. Arrhythmias associated with QRS prolongation should be treated with sodium bicarbonate (see TCA section).
Disposition
Patients should be observed for at least 6 hours post-ingestion of standard release preparations and 16 hours post-ingestion of modified release preparations or until clinically well. Cardiac monitoring is indicated following large ingestions (>5 g venlafaxine). Ingestion of>5 g of venlafaxine mandates 24 hours of observation due to the risk of delayed seizures.
Mirtazapine
Mirtazapine is a unique antidepressant; in addition to blocking the reuptake of serotonin, mirtazapine is a centrally acting α2-antagonist (increasing concentrations of serotonin and noradrenaline) and serotonin receptor (5-HT2 and 5-HT3) agonist.
Overdose of mirtazapine typically only causes minor effects including minor sedation. Tachycardia, more significant CNS sedation and seizures have been reported following large ingestions. Treatment is supportive.
Serotonin toxicity
Pharmacology
Serotonin toxicity (traditionally described as serotonin syndrome) is a consequence of excess serotonin acting at CNS and peripheral receptor sites. Table 29.4.4 lists drugs associated with serotonin toxicity. Although the exact pathophysiological mechanism is not fully elucidated, 5-HT1A and 5-HT2A receptors appear to be predominantly involved.
Table 29.4.4
Selected drugs associated with serotonin toxicity
Increased serotonin production and release
Tryptophan, lysergic acid diethylamide (LSD)
Increased release of stored serotonin
Amphetamines (including MDMA), cocaine, lithium, mirtazapine
Impaired reuptake of serotonin into presynaptic nerve
SSRIs (fluoxetine, citalopram, sertraline, fluvoxamine, paroxetine), bupropion, fentanyl, cocaine, tramadol, venlafaxine
Inhibition of serotonin metabolism
Monoamine oxidase inhibitors (moclobemide, phenelzine, tranylcypromine), methylene blue, linezolid
Although serotonin toxicity may occur following lone therapeutic or excess exposure to a serotonergic agent, it most commonly occurs in the context of combination use of serotonergic drugs or in instances where there has been an inadequate ‘wash-out’ time between substitution of one serotonergic agent for another.
Clinical features
Serotonin toxicity normally develops within hours of exposure to the offending agent or agents. A number of diagnostic criteria have been suggested, but none widely validated. Most commonly, the diagnosis has been defined as the presence of clinical findings affecting three distinct systems (in the context of exposure to a serotonergic agent):
 autonomic system: tachycardia, flushing, diaphoresis, mydriasis, hyperthermia, tachypnoea, diarrhoea
autonomic system: tachycardia, flushing, diaphoresis, mydriasis, hyperthermia, tachypnoea, diarrhoea
Toxicity is seen along a spectrum and varies from subclinical non-specific feelings of anxiety and apprehension, through to life-threatening hyperthermia, autonomic instability, cardiovascular dysfunction, multiorgan failure, seizures and rigidity. Most patients recover, but deaths have been reported. The variety and variable severity of symptoms may make the diagnosis challenging and it is not uncommon for the differential diagnosis to include neuromuscular malignant syndrome (NMS). Distinguishing features include the slower onset of NMS and the typical associated ‘bradykinetic’ clinical picture and severe muscle rigidity. Serotonergic toxicity is characterized by hyperexcitability with hyperreflexia and clonus. Other differential diagnoses include CNS infection, anticholinergic syndrome, sympathomimetic syndrome, acute dystonia, malignant hyperthermia and non-convulsive seizures. Blood concentrations of serotonergic drugs do not correlate with clinical toxicity.
Treatment
Cessation of the serotonergic agents implicated in toxicity and avoidance of further serotonergic agents are mandatory in all cases. Benzodiazepines are first-line treatment to reduce muscle rigidity or to treat seizures. Cases of severe hyperthermia or muscle rigidity should be treated aggressively: sedation with neuromuscular paralysis and admission to a critical care environment.
The use of a number of antidotes known to antagonize 5-HT1 and 5-HT2 receptors has been reported. No clinical trials have examined the safety or efficacy of these drugs. They include cyproheptadine, olanzapine, chlorpromazine and propranolol. Of these, cyproheptadine appears to be the most widely utilized. The dose is 12 mg orally (via a nasogastric tube in the intubated patient) as a single dose followed by 4–8 mg orally 8-hourly. Chlorpromazine may be useful in cases where additional sedation is required, but the possibility of dehydration may combine with the peripheral vasodilating effects of chlorpromazine to produce significant hypotension.
29.5 Lithium
Mark Monaghan
Introduction
Lithium, the metal with the lowest molecular weight, is usually dispensed as the carbonate salt. It is widely used in the therapy of bipolar disorder and a number of other conditions. Both immediate-release and sustained-release preparations are available. This drug has a relatively narrow therapeutic index and chronic intoxication develops relatively frequently. Acute overdose is less common.
Pharmacokinetics
Standard lithium preparations are rapidly and completely absorbed after oral administration with peak serum levels occurring at 2–4 h. Absorption and time to peak level is delayed after administration of sustained-release preparations and following overdose. Once absorbed, lithium is slowly redistributed from the intravascular space to the total body water. Lithium is not metabolized and its elimination is almost exclusively renal. Lithium is freely filtered at the glomerulus but, under normal circumstances, approximately 80% of filtered ions are reabsorbed in the proximal tubule and only 20% are excreted in the urine. Under these circumstances, renal clearance of lithium is approximately 10–40 mL/min and its elimination half-life is 20–24 h. The renal elimination of lithium is greatly affected by sodium and water balance and by the presence of drugs that affect renal tubular reabsorption of sodium. In the early stages following acute overdose, renal elimination is much greater because lithium is relatively concentrated in the intravascular compartment and available for filtration at the glomerulus.
Clinical features
Acute lithium overdose
Patients who take a significant overdose of lithium carbonate as with any other metal salt, develop rapid onset of gastrointestinal toxicity characterized by nausea, vomiting, abdominal pain and diarrhoea. This gastrointestinal disturbance can be very severe and may result in significant fluid and electrolyte losses. It is usually observed where more than 25 g are ingested, but can occur following smaller doses. Gastrointestinal upset is not a prominent feature of chronic lithium toxicity.
Acute lithium overdose is much less likely to result in significant neurotoxicity than is chronic lithium toxicity. Neurotoxicity can develop following massive acute overdose if renal clearance is sufficiently impaired so as to allow redistribution of sufficient lithium from the intravascular compartment to tissue compartments before it could be excreted. This situation may develop if there is pre-existing renal failure or if inadequate fluid resuscitation leads to dehydration, sodium depletion or renal impairment as a consequence of the fluid losses from gastrointestinal toxicity.
Chronic lithium toxicity
Chronic lithium toxicity may develop in association with prolonged excessive dosing or, more commonly, as a result of impaired lithium excretion due to intercurrent illness or a drug interaction. Lithium excretion is impaired in renal failure and congestive cardiac failure because of reduced filtration at the glomerulus and also in water or sodium depletion states because of increased reabsorption of sodium (and lithium) in the proximal tubule. A number of drugs including non-steroidal anti-inflammatory drugs (NSAIDs), selective serotonin reuptake inhibitors (SSRIs), neuroleptics, angiotesin converting enzyme (ACE) inhibitors, thiazide diuretics and topiramate may either impair lithium excretion or exacerbate toxicity.
The clinical features of chronic lithium toxicity are almost exclusively neurological and the following severity grading system is widely used:
 Grade I (mild): nausea, vomiting, tremor, hyperreflexia, agitation, muscle weakness, ataxia
Grade I (mild): nausea, vomiting, tremor, hyperreflexia, agitation, muscle weakness, ataxia
 Grade II (serious): stupor, rigidity, hypotonia, hypotension
Grade II (serious): stupor, rigidity, hypotonia, hypotension
 Grade III (life threatening): coma, seizures, myoclonia, cardiovascular collapse.
Grade III (life threatening): coma, seizures, myoclonia, cardiovascular collapse.
The differential diagnosis for this presentation is broad and includes non-convulsive status epilepticus, serotonin and neuroleptic malignant syndromes, electrolyte abnormalities and CNS pathologies, such as sepsis.
While minor benign ECG changes may be observed, Lithium toxicity is generally not associated with significant cardiovascular effects, although delayed bradycardia has been reported.
Chronic lithium therapy is also associated with nephrogenic diabetes insipidus and thyroid dysfunction, which may complicate the clinical presentation of toxicity.