CHAPTER 29
The haemoglobinopathies
CHAPTER OUTLINE
The structure and function of haemoglobin
The genetic control of haemoglobin synthesis
STRUCTURAL HAEMOGLOBIN VARIANTS
Other structural haemoglobin variants
INTRODUCTION
Since the discovery of its central role in oxygen transport in the 19th century, haemoglobin has proved to be a source of fascination to scientists and physicians alike, and is arguably the most widely studied protein in man. Its principal disorders, the thalassaemias and sickle cell disease, quantitative and qualitative defects of synthesis, respectively, are among the most common serious inherited diseases in man and have been pivotal in efforts to understand the molecular basis of human diseases.
It is estimated that about 7% of the world’s population carry the mutation for at least one type of haemoglobinopathy, and that 400 000 babies are born each year with a severe form. Approximately 230 000 babies are born each year with sickle cell disease in sub-Saharan Africa, and 120 000 are born with severe thalassaemia in southern and South-East Asia. There is good epidemiological and experimental evidence that the extraordinarily high frequency of haemoglobinopathies and their geographical distribution are due to the relative protection that they provide against malaria and its serious complications. This principally involves improved survival and reproductive success for heterozygotes compared with the normal population, which outweighs the reduced reproductive success of homozygotes. Thalassaemia reaches polymorphic frequencies (> 1%) in nearly all populations, except those of northern Europe, northern Asia and the indigenous peoples of Australia, the Americas and the Arctic. The common abnormal haemoglobins have more limited distributions (Fig. 29.1).
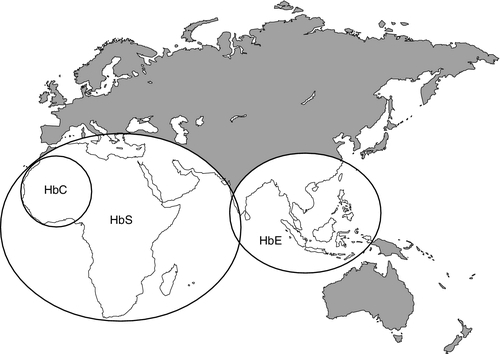
FIGURE 29.1 Primary geographic distribution of haemoglobins (Hb) S, C and E.
Since there is considerable overlap in the world distribution of thalassaemias as well as the common structural haemoglobin variants, co-inheritance of more than one haemoglobin abnormality is common. This generates an extremely diverse range of clinical phenotypes.
The structure and function of haemoglobin
Haemoglobin is a tetramer comprising two α and two β globin chains (α2β2). One molecule of haem, which contains iron and can bind to oxygen, is attached to each globin chain, lying in a hydrophobic cleft. The amino acid sequence of this haem pocket shows marked homology between different animal species, suggesting a specific and important function. Through its iron atom, each haem group is capable of binding one molecule of oxygen. The reversible binding of oxygen to haemoglobin is allosteric, giving rise to the characteristic shape of the oxygen dissociation curve (Fig. 29.2). On binding oxygen, conformational changes occur in both individual globin chains and the tetrameric structure of haemoglobin. The predominant change is closure of the gap between the two β-chains. The site at which this occurs is of critical functional importance. 2,3-Diphosphoglycerate (2,3-DPG), the principal regulator of oxygen affinity in red cells, binds to this part of the haemoglobin molecule. This serves to separate the two β-chains, favouring the deoxygenated conformation and thereby reducing the oxygen affinity of haemoglobin. The observation that oxygen affinity is often reduced in anaemic patients is explained by the adaptive increase in intracellular 2,3-DPG concentration. This enhances oxygen delivery to the tissues and is one of the important reasons why patients frequently tolerate chronic anaemia with very few symptoms. Conversely, the higher oxygen affinity of fetal blood necessary to maintain adequate maternal–fetal oxygen transport is, in large measure, due to the lower affinity of 2,3-DPG for fetal haemoglobin (HbF).
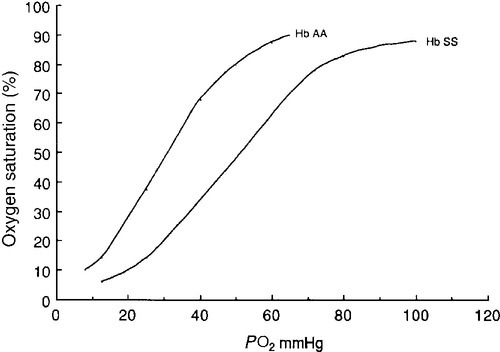
FIGURE 29.2 The oxygen dissociation curves of normal (AA) and sickle (SS) haemoglobin (Hb).
The other main physiological influence on haemoglobin oxygen affinity is the Bohr effect. First recognized at the turn of the century through the effect of carbon dioxide on lowering the oxygen affinity of whole blood, it is now known to reflect the sensitivity of oxygen binding by haemoglobin to changes in blood pH. An increase in hydrogen ion concentration stabilizes the deoxygenated conformation of the haemoglobin molecule, and so physiological acidosis leads to a reduction in the oxygen affinity. If acidosis is sustained, this effect may be counterbalanced by the reduction in intracellular 2,3-DPG concentration due to modulation of red cell glycolysis. Several inherited variants of haemoglobin have been described that stabilize the molecule in either the oxy or deoxy conformation. In some cases, the interaction with 2,3-DPG or the Bohr effect is modified.
A further factor affecting haemoglobin function is temperature. An increase in temperature reduces oxygen affinity, whereas a fall increases it. As the human is normally isothermic, this is usually of little consequence in terms of physiological adaptation, though during exercise, when oxygen consumption is increased, a raised temperature in the muscles together with an increase in hydrogen ion concentration favours release of oxygen by haemoglobin. Extreme hypothermia has many adverse consequences, including increased haemoglobin oxygen affinity causing reduced oxygen delivery to tissues.
The genetic control of haemoglobin synthesis
Human globins are encoded by the α and β gene clusters located on chromosomes 16 and 11, respectively (see Fig. 29.3). The α-like genes include an embryonic gene (ζ) and two adult genes (α1 and α2). The β-like genes comprise an embryonic gene (ε), duplicated fetal (γ) and adult (β and δ) genes. Expression of these genes is developmentally regulated during fetal life. The array of α- and β-like genes on their respective chromosomes reflects the order in which they are expressed. The control of the switch from embryonic, to fetal and then to adult globin synthesis, seems to be controlled by the interaction between several transcription factors, the locus control regions upstream of the gene cluster and the promoter regions of the individual genes.
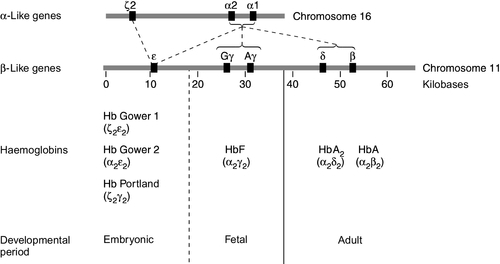
FIGURE 29.3 Organization of the human globin gene cluster and developmental changes in haemoglobin.
Normally, the synthesis of α- and β-like chains is carefully balanced to avoid the accumulation of free globin chains, although it is not clear how this balance is maintained. In the thalassaemias, where this balance is severely disturbed, the excess of one type of globin chain is central to the pathophysiology.
The switch in globin synthesis during development has important implications for clinical expression of the haemoglobinopathies. Complete failure of α-chain synthesis, which, since the α genes are duplicated on each chromosome, occurs only when there is loss of function involving all four α genes, becomes evident early in fetal life. By contrast, even if there is little or no normal β-chain synthesis, the effects will not be manifest until the switch from fetal γ to adult β gene expression is completed after birth. This switch is gradual, and abnormalities of β globin synthesis rarely result in clinical problems before three months of age. Reversing this switch, and reactivating fetal haemoglobin is also potentially curative for diseases caused by mutations of the β globin genes.
THE THALASSAEMIAS
The thalassaemias are one of the commonest human autosomal recessive disorders, with approximately 120 000 severely affected individuals born annually worldwide. The most common and clinically significant forms are α and β thalassaemia and the β thalassaemia-like structural variant, haemoglobin E. These may be further subdivided according to whether there is no output of globin (α0 or β0 thalassaemia) or some preservation of globin chain synthesis (α+ and β+ thalassaemia). In the case of α thalassaemia, this is complicated further by the duplication of expressed α genes. α Thalassaemia is clinically significant only when three or four α globin genes are lost. The clinical consequences of thalassaemia in the homozygous (or compound heterozygous) state may be understood in terms of the imbalance in globin chain synthesis that results from absent or reduced synthesis of either α or β globin. Unpaired α or β globin chains are toxic, and damage the developing red cell, causing it to die whilst still in the marrow, which is characteristic of thalassaemia and called ineffective erythropoiesis. Heterozygotes are unaffected clinically, and manifest the condition as slight anaemia with reduced red cell size and haemoglobin content. Increasing globin chain imbalance, as occurs in homozygous and compound heterozygous states, results in more marked anaemia and bone marrow expansion, with corresponding development of symptoms.
A characteristic of thalassaemia syndromes is their marked phenotypic heterogeneity. For over 20 years now, the molecular basis of thalassaemia has been studied and understood in great detail. The number of mutations identified as causing thalassaemia is large and continues to grow. However, in most populations, a small range of 5–10 different thalassaemia mutations accounts for about 90% of the mutant alleles found in that particular area.
α Thalassaemia
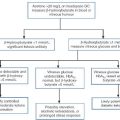
Though particularly common in South-East Asia, where carrier rates may reach 50%, α thalassaemia is widely distributed in all major populations apart from northern Europeans (see Fig. 29.1). The pathophysiological effects of α thalassaemia reflect the degree of impairment in α globin production (see Table 29.1). The majority of cases are due to deletions involving one or both α globin genes.
TABLE 29.1
The α thalassaemia syndromes
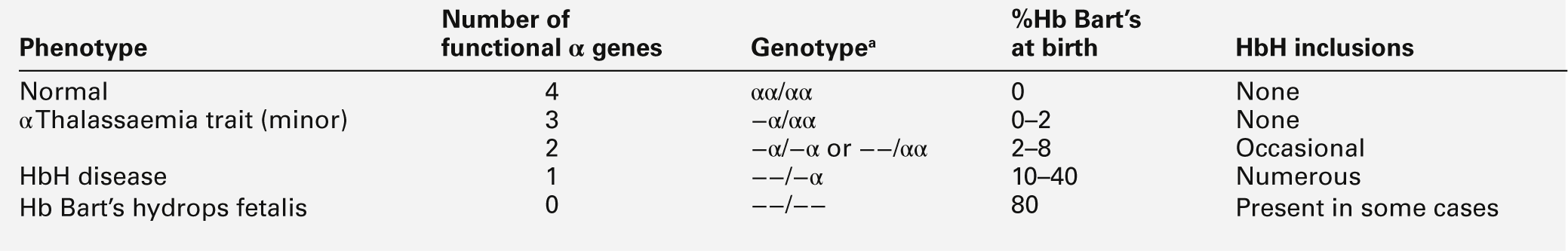
a − α, denotes loss of function of one α gene on a chromosome, i.e. α + thalassaemia.
−−, denotes loss of function of both α genes on a chromosome, i.e. α0 thalassaemia.
Haemoglobin Bart’s hydrops fetalis is the most severe form of α thalassaemia, where all four α genes are affected, abolishing or severely diminished α chain synthesis. The disease becomes manifest in fetal life. With the failure of normal production of fetal haemoglobin (α2 γ2), surplus fetal γ-chains combine to form tetramers (γ4) recognized electrophoretically as haemoglobin Bart’s. The fetus is only able to survive at all in utero because of the presence of increased amounts of the embryonic haemoglobin Hb Portland (ζ2γ2). Haemoglobin Bart’s possesses markedly increased oxygen affinity and is ineffective as an oxygen transporter. In an attempt to compensate for impaired tissue oxygen delivery, there is erythroblastosis with extramedullary haemopoiesis, leading to hepatic and splenic enlargement. Severe functional anaemia leads to tissue hypoxia, increased capillary permeability, and cardiac failure, which lead ultimately to fetal hydrops and death in utero or stillbirth in late pregnancy.
Haemoglobin H disease occurs when there is loss of function of three out of four α genes. There is sufficient residual α-chain synthesis to allow production of some normal fetal and adult haemoglobin, and fetal development is generally normal. A variable amount (10–40%) of Hb Bart’s is detectable at birth. After birth, the excess β-chains form tetramers detectable electrophoretically as the fast variant haemoglobin H (β4). Haemoglobin H (HbH) precipitates within red cells with the formation of inclusion bodies leading to shortened red cell survival. Staining of these inclusions with the redox dye brilliant cresyl blue serves as a useful diagnostic test. The clinical effects of HbH disease vary considerably. Most patients have a mild to moderate haemolytic anaemia, which may be exacerbated during pregnancy or parvovirus B19 infection, accompanied by splenomegaly. Growth and development are usually normal. As in other congenital haemolytic anaemias, there is an increased tendency to form pigment gallstones.
It follows that Hb Bart’s hydrops fetalis and HbH disease occur only when at least one parent carries the α0 genotype (–/αα). This provides an explanation for the observation that these disorders occur primarily in southern China, South-East Asia and the eastern Mediterranean where α0 thalassaemia is prevalent, and are not seen in other parts of the world, for example Africa and India, where α0 thalassaemia is very rare.
Individuals in whom one or two α genes are dysfunctional are clinically unaffected, although they show thalassaemic red cell indices and traces of Hb Bart’s at birth (see Table 29.1). This form of α thalassaemia trait is very common in most populations, with a 3.7 kb deletion accounting for most cases. Non-deletional forms of α thalassaemia are increasingly recognized as DNA analysis becomes more widespread and sophisticated.
β Thalassaemia
β Thalassaemia is prevalent throughout tropical and North Africa, the Mediterranean, Middle East and large parts of south and South-East Asia, including India (see Fig. 29.1). In some parts of the world, for example Cyprus, the heterozygote (carrier) frequency may reach 15%. Over 300 different mutations of the β globin gene or its promoter are known to cause β thalassaemia, the majority being single nucleotide substitutions (point mutations) or small deletions. Geographically, these segregate non-randomly, so that four or five specific mutations account for the majority of β thalassaemia genes in individual ethnic or geographic groups. Each mutation typically occurs on a single β globin gene haplotype, suggesting that in most instances individual β thalassaemia mutations have arisen historically on a single occasion.
Though both α and β thalassaemia share reduced globin chain synthesis, the pathogenesis of anaemia in severe forms of these conditions is distinct. In severe forms of β thalassaemia (β thalassaemia major), there is severe impairment or absence of β-chain production. As a consequence, excess α-chains accumulate and precipitate in red cell precursors leading to their destruction within the bone marrow. There is also a degree of shortened red cell survival, although ineffective erythropoiesis predominates. Red cells containing fetal haemoglobin (HbF) survive preferentially since there is less globin chain imbalance. The anaemia produces tissue hypoxia, which stimulates erythropoietin production and massive expansion of erythropoiesis in the bone marrow and extramedullary sites.
If untreated, β thalassaemia major leads to severe anaemia, failure to thrive, impaired growth and bony distortion, with death in early childhood. As a result of the expansion in erythropoiesis, there are bone deformities and enlargement of the liver and spleen. Medullary expansion of the facial bones and skull gives rise to a characteristic ‘thalassaemic’ facies, splaying of the teeth and frontal bossing. With the advent of effective therapy, this picture is now seldom seen in the developed world. The treatment of β thalassaemia major comprises regular blood transfusion to maintain the haemoglobin above 95 g/L. At this level of haemoglobin, bony deformity and hypersplenism due to anaemia are minimized. Leukocyte-depleted blood should be used to prevent alloimmunization against white cell antigens. The blood should be matched for an extended range of blood groups, including ABO, Rh and Kell, to reduce the frequency of alloantibody formation.
An inevitable complication of multiple red cell transfusions is iron accumulation. If untreated, this leads to iron deposition throughout the body, which may cause endocrine dysfunction with diabetes, hypogonadotrophic hypogonadism and hypoparathyroidism, and, ultimately, death due to cardiac failure and liver damage. This may be prevented by the use of iron chelators. Desferrioxamine, which increases both urinary and faecal iron excretion, is administered parenterally, usually by continuous overnight subcutaneous infusion, since it is ineffective orally. Ascorbic acid may enhance urinary iron excretion when given with desferrioxamine. The clinical efficacy of desferrioxamine is limited by its subcutaneous route of administration, and two oral iron chelators are currently licensed for use. Deferiprone is probably less effective than desferrioxamine at removing hepatic iron, but may be particularly effective at removing cardiac iron, and can be used in combination with desferrioxamine. Side-effects include arthropathy and unpredictable neutropenia, and weekly full blood count monitoring is recommended for those taking it. Deferasirox is a newer oral chelator with similar efficacy to desferrioxamine and a good safety profile. The degree of iron overload and the efficacy of chelation can be monitored by measuring serum ferritin concentration, liver biopsy to quantitate iron and non-invasive measurements using magnetic resonance imaging (MRI) and the superconducting quantum interference device (SQUID). Cardiac and hepatic MRI using T2* and R2 protocols are increasingly used. Accurate quantitation of the volume of transfused blood is also valuable in assessing the degree of iron overload.
With adequate transfusion and chelation therapy, most children with β thalassaemia major attain normal growth and sexual development and survive well into adult life. Allogeneic bone marrow transplantation from human leukocyte antigen (HLA)-matched siblings has been used with considerable success for treatment of β thalassaemia major. The best results, with disease-free survival rates of up to 95%, have been obtained in younger children with little or no evidence of end-organ damage due to iron overload. Trials of gene therapy are taking place in France and the USA, with one patient to date successfully rendered transfusion-independent, although with one cell clone predominating, owing to the inadvertent activation of a potential oncogene.
As in other thalassaemia syndromes, there is considerable heterogeneity in the clinical expression of β globin gene mutations. Much of this heterogeneity is unexplained, but two factors have been identified: first, the extent to which β globin production is impaired, which reflects the nature of the underlying gene mutation, and second, co-inheritance of other interacting genetic determinants that modify clinical severity. Mutations that severely disrupt or abolish β globin synthesis (β0 thalassaemia) include those that prevent normal splicing of mRNA (splice junction or cryptic splice site mutations) or generate a non-functional mRNA by premature translation termination (nonsense mutations), and small nucleotide deletions or insertions that result in the normal triplet genetic code being read out of frame (frameshift mutations). Other mutations, for example those of the β globin chain promoter, which alter the level of gene expression (transcriptional mutations), usually result in reduced rather than absent β globin synthesis (β+ or β++ thalassaemia). Among the independent genetic factors that ameliorate the clinical effects of β thalassaemia are co-inheritance of α thalassaemia, which partially redresses the imbalance in globin chain synthesis and therefore results in less ineffective erythropoiesis, and hereditary persistence of fetal haemoglobin (HPFH). HPFH can be directly linked to the β globin cluster, and its causes include large deletions and point mutations in the promoter regions of the γ globin genes. Three other major loci which control HbF concentrations have now been identified: the Xmn1 polymorphism in promoter region of the Gγ globin genes, the HMIP locus on chromosome 6q23.3 and BCL11A on chromosome 2. These factors account for some cases of the clinical syndrome thalassemia intermedia, in which there is a variable degree of anaemia, without dependence on regular blood transfusions (Box 29.1).
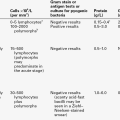
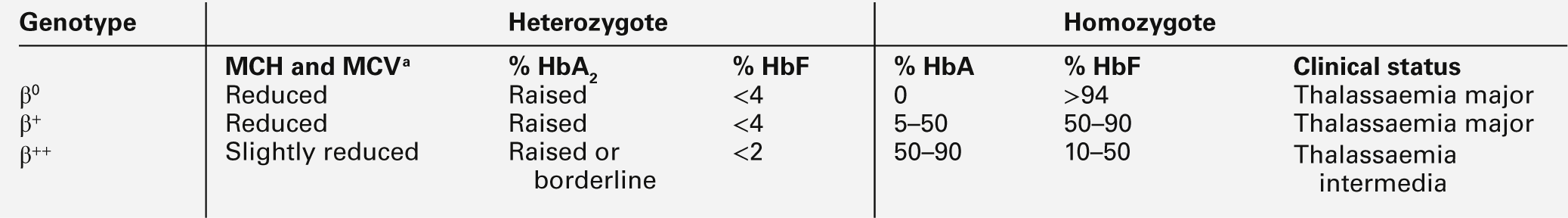
The heterozygous (carrier) state for β thalassaemia (β thalassaemia trait) is usually harmless. Some of the phenotype characteristics of β thalassaemia heterozygotes are shown in Table 29.2. Most individuals have slightly reduced haemoglobin concentrations and an elevated red cell count. A raised HbA2 (α2δ2) concentration is an important diagnostic marker and distinguishes β thalassaemia trait from α thalassaemia, in which the HbA2 is normal or low. This presumably reflects a higher output from the δ globin gene, which normally plays a minor role in adult haemoglobin synthesis (normal HbA2 < 3.2%) in the face of reduced β globin production. Similarly, HbF concentrations are frequently slightly raised in β thalassaemia heterozygotes. Rarely, dominant β thalassaemia occurs.
STRUCTURAL HAEMOGLOBIN VARIANTS
Over 700 qualitative variants have been identified, in which the haemoglobin molecule is structurally, and in some cases functionally, altered. These arise from diverse molecular defects, most commonly single amino acid substitutions, but also insertion or deletion of amino acids and polypeptide fusion as a result of recombination between globin genes, for example haemoglobin Lepore. The most common structural variants are haemoglobins S, C, DPunjab and E, which each affect many millions of people worldwide.
Sickle cell anaemia
Sickle cell anaemia (HbSS), caused by homozygous inheritance of structurally abnormal haemoglobin, is one of the commonest genetic disorders with an estimated 60 million carriers worldwide and 230 000 affected infants born annually in Africa alone. The prototypic molecular disease, it was the first human disorder to be understood at a molecular level. The landmark discovery of sickle haemoglobin (HbS) more than half a century ago by Pauling and Itano, was succeeded by demonstration of the underlying single amino acid substitution and the gene mutation responsible. Sickle haemoglobin polymerizes on deoxygenation to produce long molecules that damage and distort the red cell, producing the classic sickle-shaped erythrocytes that give the disease its name. The resulting microvascular occlusion and shortened red cell survival combine to produce a clinical syndrome that comprises anaemia, vaso-occlusion, vasculopathy, haemolysis, inflammation, hypercoagulability, organ damage and susceptibility to infection due to hyposplenism.
A single base change (CAG→CTG) in the sixth codon of the β globin gene determines the substitution of valine for glutamic acid, which results in the structural variant sickle haemoglobin S. While the sickle mutation occurs predominantly in those of African origin, with smaller numbers affected in the countries of the Mediterranean littoral, Saudi Arabia and India, population migration has resulted in its spread globally. The allele frequency of βs is thought to be high because of protection against the complications of severe malaria in the heterozygous state, as described earlier for thalassaemia mutations.
Although HbSS is the commonest cause of sickle cell disease, this condition occurs with other genotypes, in which there is co-inheritance of a different β globin mutation. The most important of these compound heterozygous states are: HbSC disease, HbSβ+ thalassaemia and HbSβ0thalassaemia, although about 15 different genotypes have been identified as causing sickle cell disease. Among sickling disorders, HbSS and Sβ0 thalassaemia are the more severe clinically, with a greater degree of anaemia (haemoglobin 60–80 g/L) and more severe organ damage. While HbSC and Sβ+ thalassaemia are perceived as milder genotypes than HbSS, there is considerable overlap and some types of end-organ damage, particularly proliferative retinopathy, occur more frequently, particularly in HbSC disease. Other genetic factors that may influence disease expression include co-inheritance of α+ thalassaemia and high fetal haemoglobin (HbF) levels, both of which may ameliorate the severity of sickle cell disease.
The primary pathophysiological event in sickling is intracellular polymerization of deoxy HbS. The β6 valine substitution alters the surface charge of haemoglobin, resulting in interaction between haemoglobin tetramers and the formation of 14-stranded polymers. These are then ordered into parallel arrays of fibre bundles, which can be visualized by electron microscopy. The formation of polymer fibres (or gelation) is affected by four main variables: oxygen tension, HbS concentration, temperature and the presence of non-sickling haemoglobins. The mean corpuscular haemoglobin concentration (MCHC) has a profound effect on the kinetics of sickling through its effect on the delay period between deoxygenation and polymerization. Small increases in haemoglobin concentration, as occur with cellular dehydration, may therefore act as a trigger for the sickling process.
Polymer formation is accompanied by several changes in the membrane of sickle erythrocytes, making them less deformable, increasingly fragile and leaky to cations. These changes are thought to result not only from interactions between sickle haemoglobin and the membrane but also from oxidative damage caused by free radicals, concentrations of which have been shown to be increased in these cells. These membrane changes are initially reversible but become increasingly pronounced as the cell undergoes repeated cycles of sickling and unsickling, culminating in an irreversibly sickled cell (ISC). The percentage of ISCs has been used as a marker for the sickling process but correlates poorly with the clinical condition of the patient. Red cell membrane phospholipids are abnormally distributed in these cells, with the normal inner leaflet amino-phospholipids being rearranged on the outer leaflet of the lipid bilayer. Cation homoeostasis is also disordered, with potassium loss exceeding sodium gain, resulting in cellular dehydration and increased concentration of intracellular haemoglobin. This is accompanied by an up to four-fold increase in intracellular calcium. There is increased intravascular haemolysis, leading to a high plasma concentration of free haemoglobin, which binds nitric oxide avidly and results in a functional nitric oxide deficiency; this is implicated in the development of vasculopathy, which contributes to the complications of pulmonary hypertension, priapism and cerebrovascular disease. Other pathological processes include hypercoagulability, inflammation, oxidative stress and reperfusion injury, all of which contribute to the multisystem nature of the disease.
The polymerization of HbS and associated membrane changes cause a marked decrease in the ability of these cells to flow through the microvascular circulation, resulting in compromised oxygen delivery and setting up a vicious circle of worsening vaso-occlusion. There is also evidence of increased adherence of these red cells to vascular endothelium, which would increase deoxygenation and obstruct the flow of other red and white cells through the microvasculature.
The clinical manifestations of sickle cell disease are protean and include both acute and chronic complications. The majority of symptoms are attributable to the effects of vascular occlusion, since the anaemia is usually well tolerated. Sickle cell disease is a multisystem disease. Acute episodes or ‘crises’ can take several forms, including pain or severe anaemia; acute worsening of anaemia can be due to splenic or hepatic sequestration, or transient erythroid hypoplasia secondary to parvovirus B19 infection. The most common symptom is acute, severe pain, which may be preceded by infection, dehydration and cold exposure, though often no particular trigger is identified. Vaso-occlusion, which commonly involves the bones, results in avascular necrosis of the bone marrow with associated inflammation and increased intramedullary pressure. This results in painful swelling of the hands and feet (dactylitis) in early infancy. In older children and adults, the juxta-articular areas of long bones, flat bones like the ribs and pelvis and the vertebral column are most commonly affected. The management of acute pain is supportive, ensuring adequate analgesia, hydration and oxygenation. Other acute complications include priapism, retinal artery occlusion and sudden death.
Sickle cell patients are at increased risk of bacterial infection due largely to loss of splenic function and, historically, overwhelming pneumococcal sepsis has been the major cause of early mortality. In developed countries, this risk has been significantly reduced by the introduction of pneumococcal prophylaxis in the form of pneumococcal vaccines and daily oral penicillin. The major cause of mortality after early childhood is the acute chest syndrome, which results from a combination of infection, infarction and fat embolism in the lungs, and is characterized by fever, severe chest pain, dyspnoea and pulmonary infiltrates. This complication needs prompt and vigorous management including oxygenation, hydration, intravenous antibiotics and blood transfusion. Increasing numbers of patients in developed countries are surviving to late middle and old age, and progressive multi-organ failure, particularly involving the kidneys, is an increasingly common form of death. From a very early age, most children with SCD display a number of renal abnormalities, including glomerular hyperfiltration and nephrogenic diabetes insipidus. Many go on to develop significant albuminuria in later childhood, with progressive renal failure contributing to death in about 30% of adults.
Stroke due to occlusion of the large cerebral vessels affects about 11% of patients by the age of 20, with the highest incidence between the ages of two and five years. Without treatment, there is a high rate of recurrence. Long-term transfusion significantly reduces the risk of a further stroke but carries with it the necessity for iron chelation to prevent siderosis. Transcranial Doppler scanning identifies children with early vasculopathy who are at high risk of stroke. Regular blood transfusion of children with such cerebral vasculopathy has been shown to be an effective form of primary stroke prevention, both in clinical trials and practice.
Pulmonary hypertension affects up to 5% of adults with sickle cell disease, and is thought to be associated with an increased risk of premature death. Increased rates of haemolysis have been linked to the pathology, and it may be part of a more general vasculopathy.
Chronic organ damage due to sickling may take several forms. Ischaemic damage to the bones and joints results in progressive destruction which, in the case of avascular necrosis of the hip, may lead to severe disability. Chronic restrictive lung disease may follow recurrent episodes of infection and infarction. As in other chronic haemolytic states, gallstones are common and found in nearly one-third of young adults with sickle cell disease. Proliferative retinopathy can result in bleeding, retinal detachment and blindness. Stasis and occlusion of the small vessels in the lower limbs may cause leg ulceration.
The prognosis for patients with sickle cell disease in developed countries has been transformed by early diagnosis, improved supportive care and, most importantly, prophylaxis against pneumococcal infection. At least 85% of HbSS patients and 95% of HbSC patients in the USA now survive to 20 years and 50% of patients survive beyond the fifth decade. Relatively little is known about the natural history of the condition in African countries, but the majority of children with sickle cell disease are thought to die before the age of five years. This markedly higher mortality in Africa reflects the importance of environmental factors, and is probably mainly related to malaria, pneumococcal and other infections. Sickle cell disease remains a disease without a cure. Haematopoietic stem cell transplantation has been successfully performed in a few patients but carries a procedure-related risk with 5% mortality. In the absence of reliable predictors of clinical severity, which varies widely among affected patients, this is difficult to justify in most cases. The role of transplantation is likely to increase as the procedure becomes safer and able to draw from a wider range of donors. Human trials of gene therapy are currently planned, but as yet, no patients have been successfully treated.
The search for effective anti-sickling agents has targeted the essential steps in the pathophysiology of sickle cell disease: polymer formation, membrane changes and interactions with the microvasculature. The first approach, aimed at combating polymer formation, either by altering oxygen affinity or increasing HbF concentrations, seems most likely to be successful. Hydroxycarbamide (hydroxyurea), which increases HbF concentrations, has been shown to reduce the frequency of acute pain and acute chest syndrome in randomized controlled trials, and is now an important therapeutic option in adults and children suffering frequent episodes of pain or severe chest problems.
Other structural haemoglobin variants
There are several hundred less common structural haemoglobin variants, most of which are very rare and have no clinical or functional consequences. An exception is the β globin variant haemoglobin E (β26 Glu→Lys), which is probably the most prevalent haemoglobin variant, being carried by an estimated 84 million individuals worldwide, mainly in South-East Asia. Haemoglobin E has an electrophoretic mobility similar to that of HbC and HbA2 at pH 8.9 but can be differentiated by citrate agar electrophoresis at acid pH. Unlike most other structural variants, inheritance of HbE is associated with a β thalassaemia phenotype with microcytosis and hypochromia. This is because the βE mutation activates a cryptic splice site that inhibits the normal splicing mechanism. Haemoglobin E is also unstable, which may contribute to the unexpectedly severe phenotype that occurs when HbE is co-inherited with β thalassaemia mutations. This compound heterozygous state results in thalassaemia major in about half of cases.
The other common abnormal haemoglobins are C and DPunjab, which affect millions, predominantly in West Africa and the Punjab region of India, respectively. In the homozygous state, there is mild haemolysis, with few if any symptoms. Co-inheritance of both of these variants with HbS results in sickle cell disease.
The unstable haemoglobin variants usually result from neutral substitutions affecting amino acid residues that contact the haem group and generally present as a congenital Heinz body haemolytic anaemia, for example Hb Köln and Hb Bristol. Heinz bodies are inclusion bodies seen in some red cells consisting of degraded haemoglobin. The diagnosis is made by the heat denaturation or isopropanol precipitation tests and identification of the globin mutation by protein or DNA analysis. Owing partly to the instability of the variant haemoglobin, only half of these variants are detectable by electrophoresis.
Amino acid substitutions involving either α- or β-chains in the vicinity of the haem group can also result in altered oxygen affinity or a propensity to methaemoglobin formation. Haemoglobin variants in which oxygen affinity is significantly increased (e.g. Hb Chesapeake, Hb San Diego) are associated with erythrocytosis (polycythaemia). Low-affinity haemoglobins (e.g. Hb Kansas) and HbM variants (e.g. HbM Boston) cause cyanosis, usually with no associated signs or symptoms of disease. The possible consequences of abnormal haemoglobins are summarized in Table 29.3.
TABLE 29.3
The functional and clinical consequences of abnormal haemoglobins
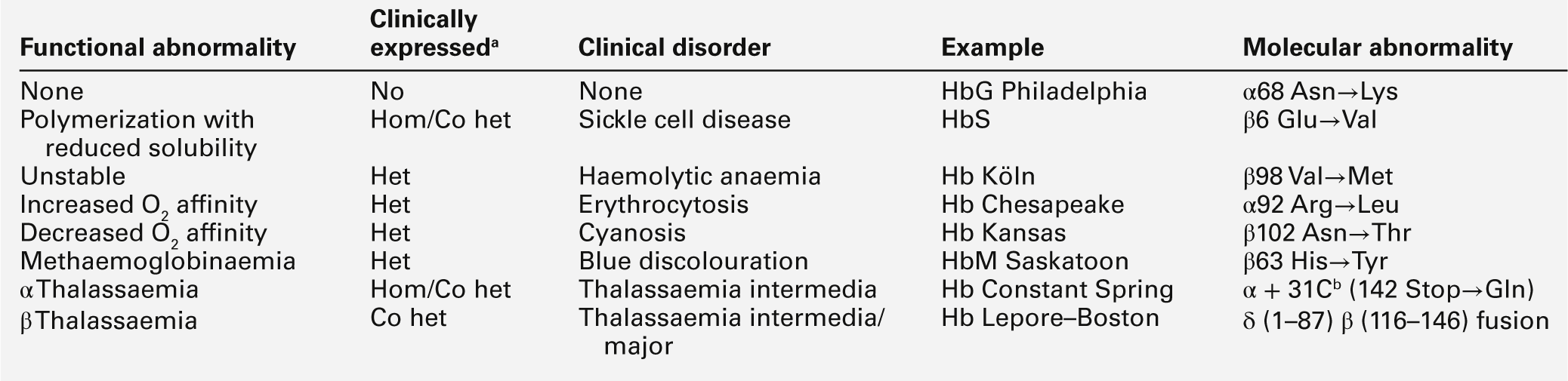
b This chain terminator mutation results in an α globin chain with an additional 31 amino acids.
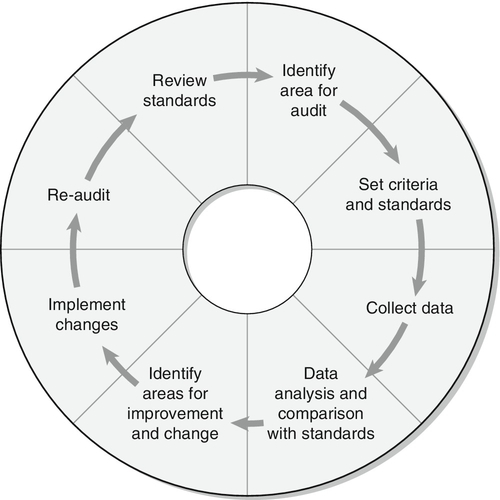
LABORATORY DIAGNOSIS OF HAEMOGLOBINOPATHIES
The primary investigation of a haemoglobinopathy should include a full blood count, peripheral blood film and haemoglobin electrophoresis (see Fig. 29.4). The full blood count allows assessment of haemoglobin formation, as judged by the red cell indices, the mean cell volume (MCV) and mean cell haemoglobin (MCH). Microcytosis (MCV < 76 fL) and hypochromia (MCH < 27 pg), in the face of a normal or raised red cell count (> 5.5 × 1012/L) in an iron-replete patient, suggest a diagnosis of thalassaemia. In the case of β thalassaemia major or intermedia, this is associated with a significant degree of anaemia, whereas in thalassaemia trait the haemoglobin is usually normal or only marginally reduced. Examination of a blood film stained by a Giemsa method may be helpful in confirming the diagnosis. However, definitive diagnosis often rests on electrophoretic or chromatographic analysis of haemoglobin in red cell haemolysates. Cellulose acetate electrophoresis at alkaline pH (8.9–9.1) is the most widely used method, being simple, rapid, inexpensive and effective in separating the common haemoglobin variants. In homozygous sickle cell anaemia, HbS predominates. A variable amount of HbF is present, higher proportions (> 10%) generally being associated with a milder clinical course. Haemoglobin A2 concentration is usually normal.
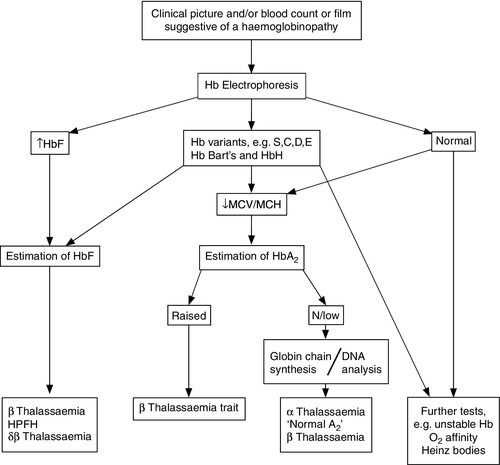
FIGURE 29.4 A simple algorithm for the diagnosis of haemoglobinopathies.
Solubility testing based on the reduced solubility of deoxy-HbS in the presence of reducing agents, for example sodium dithionite, has a limited role in the diagnosis of sickle cell disease since it does not differentiate the homozygous disease and carrier states. In an emergency situation, a positive solubility test taken in conjunction with a significantly reduced haemoglobin and typical red cell morphology points strongly towards a diagnosis of sickle cell disease. This should be confirmed by haemoglobin analysis at the earliest opportunity. Several other variants, including HbD, HbG and Hb Lepore, have an electrophoretic mobility identical to that of HbS on cellulose acetate but may be distinguished by the negative sickle solubility test and citrate agar gel electrophoresis at acid pH (6.0). Similarly, haemoglobins C, E and O, which co-migrate on cellulose acetate at alkaline pH, can be differentiated by citrate agar electrophoresis. Both HbE and Hb Lepore are associated with thalassaemic red cell indices, which further aids their distinction from electrophoretically similar variants. Isoelectric focusing improves the resolution of some structural variants and can also be used for neonatal screening of eluates from Guthrie (dried blood spot) cards, since it reduces interference from methaemoglobin present in such samples.
High performance liquid chromatography (HPLC) is a fast and sensitive method for separation and quantitation of haemoglobins that, in some cases, allows identification of variants not possible by other techniques. Since it is largely automated and requires minute quantities of sample, HPLC has become the method of choice for large-scale population testing such as neonatal screening programmes. Universal neonatal screening for sickle cell disease has been used in the USA and England for some time, and similar programmes are being introduced into other European, Middle Eastern and African countries, depending on the prevalence of the conditions and the resources available. Such programmes have resulted in significant benefits in terms of reduced mortality and morbidity due to improved care, early implementation of prophylaxis against pneumococcal infection and parental education. In β thalassaemia, the proportion of individual haemoglobins varies with the underlying genotype. Homozygous β0 thalassaemia is associated with a predominance of HbF, absence of HbA and variable amounts of HbA2 (range 1.0–6.0%, mean 1.7%). In individuals with homozygous β+ thalassaemia or compound heterozygous β0/β+ thalassaemia, a variable amount of HbA is present. Haemoglobin F is increased and distributed heterogeneously among red cells.
Accurate quantitation of HbA2 by HPLC or microcolumn chromatography is essential for the diagnosis of β thalassaemia trait, in which the HbA2 is elevated, typically > 3.5%. Carriers of ‘normal A2’ or ‘silent’ β thalassaemia due to mild β gene defects or co-inheritance of a δ gene mutation in cis or trans cannot easily be distinguished from α thalassaemia by conventional screening methods and require investigation by specialized techniques, including in vitro globin chain synthesis and DNA analysis. Analysis of globin chain synthetic ratios by tritiated leucine incorporation and carboxymethylcellulose chromatography is the definitive way of identifying individuals with thalassaemia, although it is rarely used because of its laborious nature.
The α thalassaemias are characterized electrophoretically by the presence of the fast moving variants, Hb Bart’s (γ4) and HbH (β4), which are most obvious in neonatal samples. In hydrops fetalis owing to homozygous α0 thalassaemia, Hb Bart’s predominates and is found in smaller amounts in other α thalassaemia syndromes in the neonatal period. Haemoglobin H may also be detected by staining for HbH inclusion bodies. The diagnosis of clinically silent forms of α thalassaemia is often one of exclusion, being made on the basis of the subject’s ethnic origin, microcytic hypochromic red cell indices and a normal or low HbA2 concentration in the presence of normal iron status. Definitive diagnosis can be made by DNA analysis, which can also often distinguish α0 and α+ thalassaemia.
While the majority of haemoglobinopathies can be diagnosed by haemoglobin electrophoresis, variants caused by amino acid substitutions that do not alter charge, such as those found in some unstable haemoglobins or haemoglobins with altered oxygen affinity, may escape detection. Further investigations that may be helpful in this context include assessment of haemoglobin instability and oxygen affinity. High throughput DNA analysis has made this a feasible way of screening for globin mutations in richer countries, with techniques such as multiplex ligation-dependent probe amplification allowing identification of large gene mutations which were previously only detectable using Southern blotting. Haemoglobin mass spectrometry can also be used to identify abnormal globins by measuring their mass accurately, with particular potential use in screening programmes which already use mass spectrometry.
The identification of couples at risk for major haemoglobinopathies by antenatal or preconceptional screening permits informed reproductive choice with the option of prenatal diagnosis. In most cases, this can now be accomplished in the first trimester by detection of mutant globin genes in chorionic villous DNA. In several countries, notably Cyprus where the carrier rate for β thalassaemia reaches 12%, this has led to a marked decline in the birth incidence of haemoglobin disorders. Preimplantation genetic diagnosis is increasingly used to allow selection of unaffected embryos, although it continues to be a demanding and expensive process that is not applicable to most couples. Attempts continue to develop non-invasive prenatal diagnosis using fetal cells and DNA in maternal blood, although this is currently not technically feasible as a routine procedure for the haemoglobinopathies.
CONCLUSION
Haemoglobin is arguably the best studied of all human proteins. It is a tetramer of two pairs of globin chains. Each chain binds a molecule of haem, to which a molecule of oxygen can become reversibly bound. The affinity of haemoglobin for oxygen can be modified by various physiological factors in ways that respond to the requirements of tissues for oxygen under different circumstances.
Inherited disorders of haemoglobin synthesis can be qualitative or quantitative. More than 400 structural variants have been described, of which the most important is haemoglobin S, the haemoglobin of sickle cell disease. Quantitative abnormalities of globin chain synthesis cause the thalassaemias. Advances in diagnosis and treatment have dramatically altered the prognosis for many affected patients, although there is still a limited range of treatments available.