[level-membership-for-endocrinology-diabetes-and-metabolism-category]CHAPTER 28
The porphyrias
inherited disorders of haem synthesis
Michael N. Badminton; George H. Elder
CHAPTER OUTLINE
Biochemistry of haem synthesis
Molecular genetics of the porphyrias
PORPHYRIAS PRESENTING WITH ACUTE ATTACKS
The autosomal dominant acute porphyrias
Erythropoietic protoporphyria and X-linked dominant protoporphyria
INTRODUCTION AND OVERVIEW
The porphyrias are a group of eight metabolic disorders that result from inherited or acquired functional abnormalities of enzymes of the haem biosynthetic pathway (Table 28.1 and see Fig. 28.1, below). No disease has yet been associated with defects in the 5-aminolaevulinic acid (ALA) synthase-1 (ALAS1) gene that encodes the ubiquitous isoform of ALAS, the first enzyme of the pathway. However, gain of function mutations in the erythroid-specific ALAS (ALAS2) gene cause X-linked dominant protoporphyria (XLDPP), whereas loss of function causes X-linked sideroblastic anaemia.
TABLE 28.1
Overview of the porphyrias indicating inheritance, prevalence and main clinical presentation

AR, autosomal recessive; AD, autosomal dominant.
a Also known as PBG deaminase.
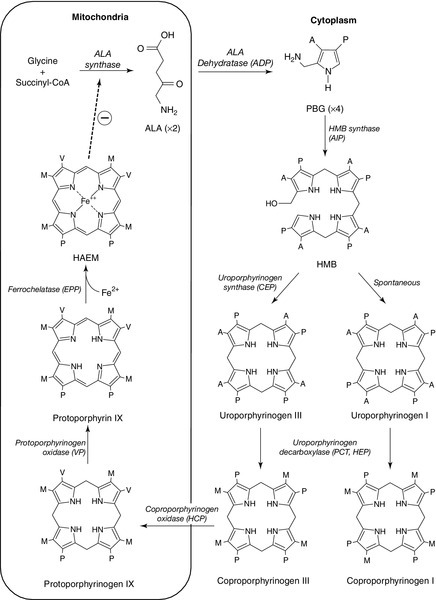
FIGURE 28.1 The haem biosynthetic pathway. The side chains are denoted by: A, acetic acid; M, methyl; P, propionic acid; V, vinyl. For other abbreviations, see Box 28.1.
The principal clinical features of the porphyrias are neurovisceral or cutaneous, or both. This chapter approaches the subject from a practical perspective, describing how and why patients present, how they are diagnosed and how patients and their families should be managed. A list of frequently used abbreviations is provided in Box 28.1.

Biochemistry of haem synthesis
Haem is essential for life and is synthesized in all cells, although the major sources are bone marrow (80%) and liver (15%). Haemoproteins include haemoglobin and myoglobin, which are the most abundant; the mitochondrial respiratory cytochromes; enzymes such as catalase and tryptophan pyrrolase, and the cytochrome P450 enzymes that are components of many essential metabolic processes, including the metabolism of xenobiotics.
The haem synthetic pathway comprises eight steps, each catalysed by a specific enzyme, of which the first and last three are mitochondrial and the remainder cytosolic (Fig. 28.1). The first enzyme, ALAS, catalyses the condensation of succinyl-CoA and glycine into 5-aminolaevulinic acid and is the rate-controlling step in all cells. Regulation of ALAS1 in the liver and other non-erythroid tissues is via inhibition by haem, the end product of the pathway, a characteristic that underlies both the pathogenesis and treatment of acute attacks of porphyria. In erythroid cells, regulation of haem synthesis is iron dependent. The second step is the synthesis of porphobilinogen (PBG), a watersoluble, colourless monopyrrole, catalysed by ALA dehydratase. The third step is the polymerization of four molecules of PBG to form the colourless linear tetrapyrrole, 1-hydroxymethylbilane (HMB), by the enzyme hydroxymethylbilane synthase (HMBS, also known as PBG deaminase). This linear molecule is cyclized into the tetrapyrrole ring structure, uroporphyrinogen III, by the enzyme uroporphyrinogen III synthase.
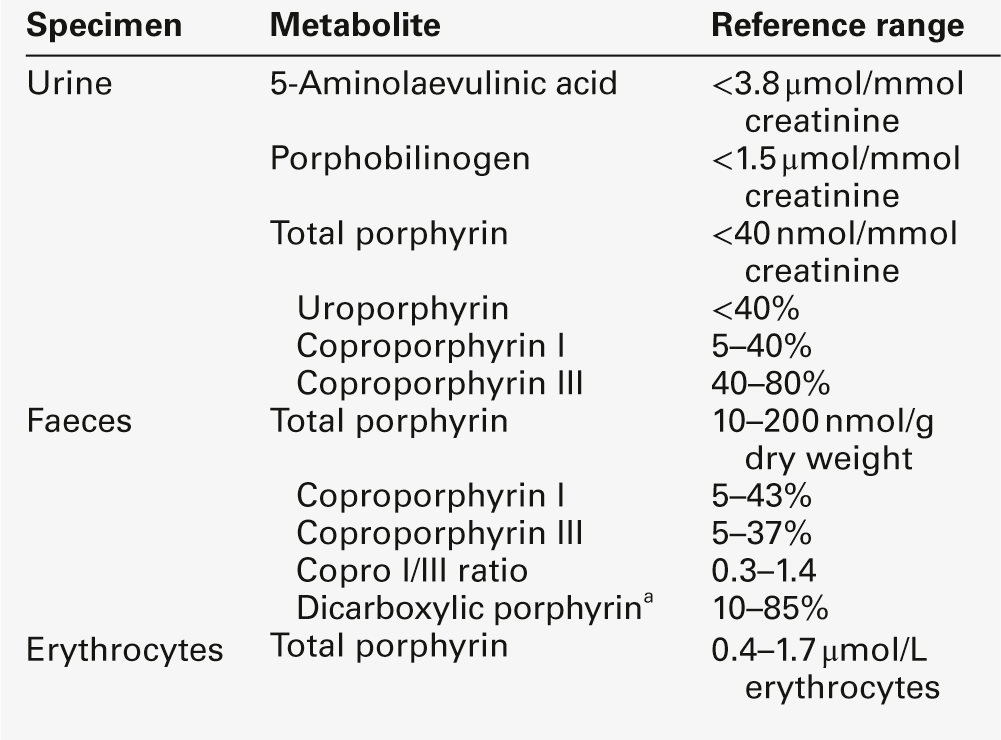
The porphyrinogens are colourless, non-fluorescent, unstable compounds, which rapidly oxidize to their red-purple porphyrin equivalents. Porphyrins absorb light, are fluorescent and therefore photosensitizing. These properties also make them relatively straightforward to measure in the clinical biochemistry laboratory. Uroporphyrinogen III, which is hydrophilic by virtue of its eight carboxy groups, is converted to coporphyrinogen III by uroporphyrinogen decarboxylase, which catalyses the sequential removal of four carboxyl residues. Two further carboxyl groups are removed in an oxygen-dependent dehydrogenation-decarboxylation reaction catalysed by the enzyme coproporphyrinogen oxidase (CPOX) to form protoporphyrinogen, which is then oxidized to protoporphyrin IX by protoporphyrinogen oxidase (PPOX). Progressive decarboxylation makes these precursors and the corresponding porphyrins increasingly hydrophobic, which determines their routes of excretion (see Fig. 28.2). The final step in the pathway is insertion of ferrous iron (Fe2 +) into protoporphyrin to form haem, catalysed by ferrochelatase (FECH). In the absence of iron, other divalent cations, such as zinc, may be inserted. Although only the III isomer can progress through the pathway to form protoporphyrin IX and haem, HMB may spontaneously cyclize into the uroporphyrinogen I isomer. This forms a substrate for uroporphyrinogen decarboxylase (UROD) and may be converted to coproporphyrinogen I, but is not metabolized further. Adult reference ranges for porphyrins and their precursors are shown in Table 28.2.
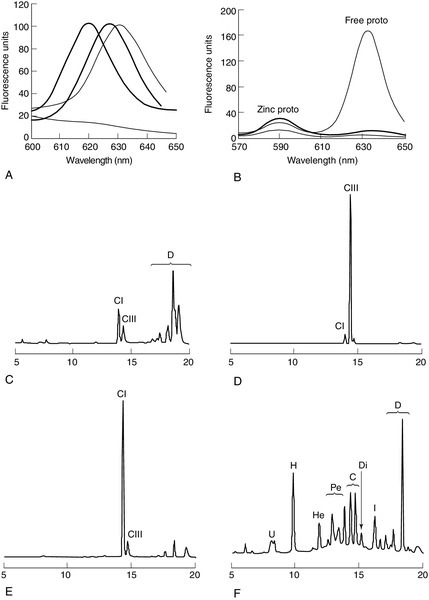
FIGURE 28.2 Examples of key laboratory findings that allow the porphyrias to be distinguished biochemically. (A) Plasma porphyrin fluorescence scans showing the three distinct emission peaks at 620 nm (AIP, CEP, PCT, HCP), 626 nm (VP) and 631 nm (EPP). A negative plasma scan (lower line) is also shown. (B) Fluorescence emission scan of whole blood allows zinc chelated protoporphyrin (proto) (increased in anaemia and lead poisoning) to be distinguished from free protoporphyrin, which is increased in EPP. Markedly increased zinc chelated and free protoporphyrins are characteristic for XLDPP. The lower line indicates a normal scan. High performance liquid chromatography (HPLC) analysis of faecal porphyrins allows three porphyrias with a plasma fluorescence emission peak at 620 nm to be distinguished (faecal porphyrin excretion is normal in AIP). (C) Normal faecal porphyrin HPLC trace indicating predominantly coproporphyrin (CI) isomer and dicarboxylic (D) porphyrins (protoporphyrin, pemptoporphyrin, deuteroporphyrin). (D) Faecal porphyrin pattern found in HCP indicating increased coproporphyrin, which is almost entirely the III isomer. (E) Faecal porphyrin trace seen in CEP indicating increased coproporphyrin isomer I. (F) Faecal porphyrin pattern seen in PCT patients indicating the increased excretion of partially decarboxylated intermediates; heptacarboxylic (H), hexacarboxylic (He) and pentacarboxylic (Pe) porphyrins as well as isocoproporphyrin (I) and dehydroisocoproporphyrin (Di), which are pathognomonic for PCT. U, uroporphyrin.
Overview of the porphyrias
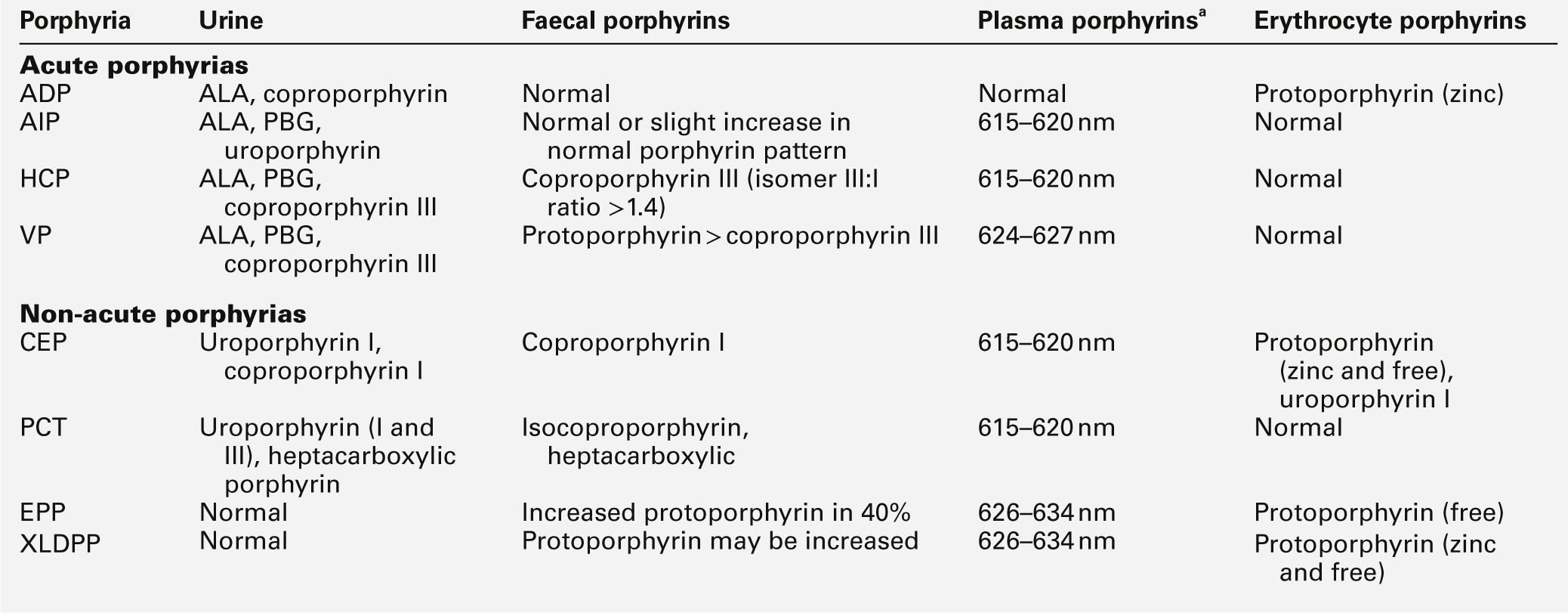
Clinically manifest porphyria is always associated with detectable overproduction of haem precursors. Each enzyme deficiency, and the increase in activity in XLDPP, gives rise to a specific pattern of overproduction, which defines the corresponding disease (Table 28.3). Three clinical manifestations may occur: acute neurovisceral attacks, skin lesions or both. Acute attacks of porphyria are always accompanied by overproduction of ALA and, in all but ALA dehydratase deficiency porphyria (ADP), of PBG. Porphyrias causing skin lesions are characterized by overproduction of porphyrins. Four of the eight porphyrias can present with acute neurovisceral attacks: the very rare autosomal recessive ADP and the three autosomal dominant acute porphyrias, acute intermittent porphyria (AIP), hereditary coproporphyria (HCP) and variegate porphyria (VP). Hereditary coproporphyria and VP can present with skin photosensitivity or acute attacks, or both. In the other four porphyrias, two types of photosensitization may occur: accumulation of hydrophobic free protoporphyrin in erythropoietic protoporphyria (EPP) and XLDPP is associated with acute photosensitivity, while accumulation of the more water-soluble porphyrins in porphyria cutanea tarda (PCT) and congenital erythropoietic porphyria (CEP) leads to fragile skin and bullae.
TABLE 28.3
The main biochemical findings in symptomatic porphyria
Porphyrin analyses on random urine, faeces and EDTA-preserved blood allow the individual porphyrias to be distinguished. Individual urine and faecal porphyrins are separated and measured by high-performance liquid chromatography with fluorescence detection. Note that EPP cannot be diagnosed by urine porphyrin analysis alone.
a Emission maximum of plasma fluorescence peak (for examples, see Fig. 28.2).
Molecular genetics of the porphyrias
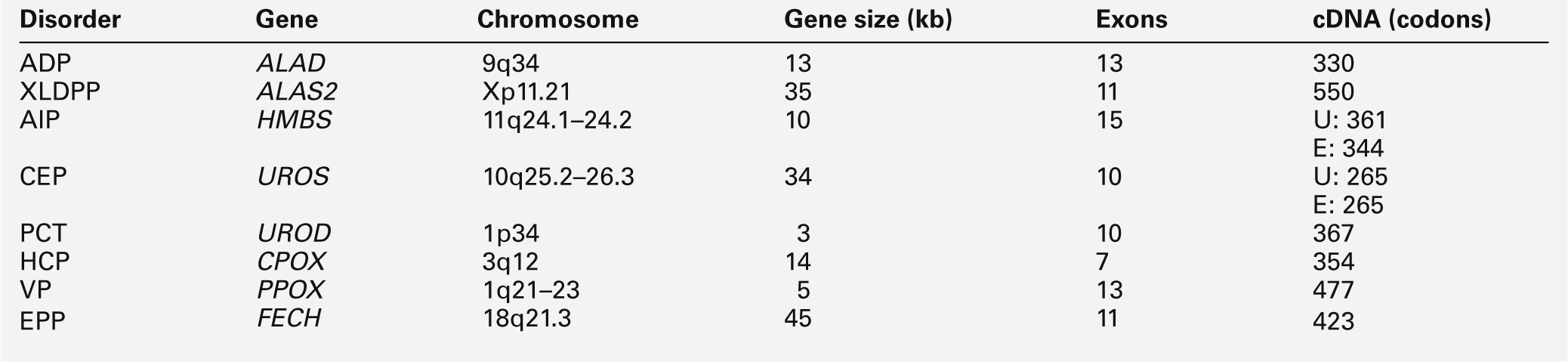
All the porphyrias, apart from the sporadic form of PCT, are single gene disorders that are inherited in autosomal dominant, recessive or X-linked patterns (see Table 28.1). Characteristics and chromosomal locations of individual genes are shown in Table 28.4. The HMBS and UROS genes are alternatively spliced to produce erythroid and ubiquitous isoforms. Disease-specific mutations that abolish or markedly decrease enzyme activity have now been identified in the genes for all the autosomal dominant porphyrias (Human Gene Mutation Database: www.hgmd.org). In most countries, mutational analysis has revealed extensive allelic heterogeneity, with large numbers of mutations identified in each gene, most of which are present in only one or a few families. The main exceptions are the W198X HMBS mutation in Sweden and the R59W PPOX mutation in South Africa. In both countries, these mutations have been multiplied by founder effects and account for the high prevalence of AIP in Sweden and of VP among individuals of Afrikaans ancestry in South Africa. The proportion of different types of mutation varies little between the diseases, with missense, nonsense, splice site and frameshift mutations contributing to the overall heterogeneity. Large deletions appear to be uncommon. Perhaps not surprisingly in view of the large number of different mutations, no clinically useful genotype-phenotype correlation has yet been identified in any autosomal dominant porphyria. With current methods of analysis, the sensitivity of mutation identification is over 95% in AIP and VP.
Incomplete penetrance is an important feature of all the autosomal dominant porphyrias; only a proportion of those who inherit a disease-specific mutation develop the disease. In this chapter, the terms latent or presymptomatic will be used to describe individuals who have inherited a porphyria gene but at the time of investigation have not had symptoms. Estimates of penetrance for the autosomal dominant acute porphyrias derived from family studies range from 10% to 40% and are influenced by age and rigour of phenotype definition. In the UK, about 80% of adults identified by family studies as carrying a gene for an acute porphyria never have an acute attack severe enough to require hospitalization. This figure is consistent with the observation that about 30% of patients with AIP present without a family history of the disease, yet family studies almost always reveal latent porphyria in their families, de novo mutation being uncommon. Studies of blood donors suggest that the gene for AIP may be present in about 0.06% of the general population. The low clinical penetrance of the acute porphyrias probably reflects a combination of environmental (see below) and genetic influences. The latter have not been identified but are likely to be at loci distant from the disease gene. Factors that determine clinical penetrance in familial PCT are described on page 544.
Clinical penetrance of the autosomal recessive porphyrias, ADP, CEP and EPP is close to 100%, although age at first presentation may be variable. They also show extensive allelic heterogeneity, with most patients whose parents are not consanguineous being compound heterozygotes. Other features of their genetics, and those of XLDPP, are discussed later in this chapter.
PORPHYRIAS PRESENTING WITH ACUTE ATTACKS
The autosomal dominant acute porphyrias
AIP, HCP and VP are autosomal dominant conditions that put the patient at risk of acute neurovisceral attacks, which may be life-threatening. In VP and HCP, skin lesions, indistinguishable from those of PCT, may occur either independently or in combination with acute attacks. In the UK, acute attacks of porphyria are estimated to affect about nine per million of the population, most of whom have AIP. Symptomatic VP is approximately half as common as AIP. HCP is the rarest of these acute porphyrias.
Pathophysiology of acute attacks
Acute neurovisceral attacks occur when hepatic haem synthesis is induced in the presence of deficient HMBS activity, resulting in an accumulation of the haem precursors ALA and PBG. In AIP, this is a primary deficiency, but in VP and HCP, it has been proposed that the deficiency is secondary to inhibition of HMBS by other metabolites in the haem pathway. The exact cause of neuronal damage has not been fully established, but current evidence from liver transplantation suggests that it is due to a hepatic neurotoxin, probably ALA. The neurological lesions comprise axonal degeneration and patchy demyelination of peripheral neurons with chromatolysis of anterior horn cells, brain stem nuclei and ganglia of the autonomic nervous system. There may also be diffuse neuronal loss and gliosis of the CNS. The result is damage to autonomic, motor and CNS neurons, giving rise to the characteristic clinical presentation, described below.
Clinical presentation of acute attacks
Acute attacks of porphyria are extremely rare before puberty and unusual after the menopause, having a peak occurrence in the third and fourth decades. Women are more frequently affected than men. In most patients, an attack will cease once the diagnosis has been made, appropriate treatment given and likely precipitants removed. However, particularly when there is delay in establishing the diagnosis, prolonged and very severe, life-threatening and sometimes fatal acute attacks may occur. Commonly ascribed precipitants include prescribed and illicit drugs, alcohol (particularly binge drinking), systemic infection and dieting either alone or in combination. In women, attacks may be related to the menstrual cycle, usually the luteal phase. However, in some cases no obvious aetiology is found.
An acute attack of porphyria normally starts with continuous abdominal pain, which becomes progressively more severe and is associated with nausea, vomiting and constipation. The patient may also complain of pain in the lower back, buttocks and inner thighs. The abdominal pain is typically diffuse, with no localizing signs or evidence of an acute abdomen on examination. The severity of pain appears out of keeping with the physical signs and often requires the administration of large parenteral doses of opiates. In the absence of an obvious cause, this may trigger concerns about opiate dependence among clinical staff who have no previous experience of the condition. Diminishing pain, particularly where no treatment has been given, does not always herald the end of an acute attack, and patients should be carefully monitored for the development of neuropathy. The autonomic neuropathy that results in the gastrointestinal symptoms also gives rise to tachycardia and hypertension in approximately two-thirds of patients. Patients frequently become dehydrated during an acute attack and may also develop hyponatraemia, which may worsen if inappropriate volumes of hypotonic intravenous fluid are infused. Plasma sodium concentration can fall quickly to extremely low values, precipitating convulsions, which occur in about 5% of patients. Seizures may also occur as a neurological manifestation of the acute attack, and present a particularly difficult management problem as many antiepileptic drugs are implicated as a cause of acute attacks. Rhabdomyolysis, which may lead to renal failure, is a recognized but rare complication of an acute attack.
Severe and untreated attacks can result in a predominantly motor peripheral neuropathy. In most cases, this is a symmetrical distal neuropathy resulting in wrist and foot drop, but occasionally there is progression to a flaccid paresis resembling Guillain–Barré syndrome and requiring prolonged ventilatory support. Prognosis for full recovery is excellent when attacks can be treated and halted. However, patients who require ventilatory support frequently experience a difficult recovery period, characterized by recurrent relapses triggered by the requirement for multiple prescribed drugs and complications such as infection. A small number of patients experience sensory changes such as dysaesthesia or hypoaesthesia in a similar distribution to the motor neuropathy. Other neurological signs reported include cranial nerve involvement including the optic nerve, which can lead to blindness.
The CNS is frequently involved. Acute mental changes are common and include anxiety, insomnia, confusion, hallucinations and paranoia, which resolve completely following the acute attack. There is no evidence that any form of chronic psychiatric illness is associated with any of the acute porphyrias. Less common CNS features are cerebellar syndrome, pyramidal signs, transient cortical blindness and altered consciousness.
Most patients have only one or a few attacks, with a major attack often being followed by two or three minor ones before full remission. However, about 5% of AIP patients suffer from frequent acute attacks, which in women may be premenstrual, and which may continue for several years (see p. 541). Obvious triggers are usually not found and the repeated admissions to hospital can severely affect the quality of life, particularly if patients have young families. Repeated admissions and high opiate requirement may also lead medical staff who are unfamiliar with the condition to question the diagnosis and the patient’s motives.
Chronic complications
Some patients develop a chronic pain syndrome. Pain is usually in the peripheries, often constant and may be triggered by minor stimuli. The cause of this is unknown; management is particularly difficult since it is important to avoid addictive analgesics. Treatment with haem arginate (p. 540) is usually of no benefit.
Studies from several countries have demonstrated an increased risk of hepatocellular carcinoma (HCC) in the absence of chronic liver disease in patients with an acute hepatic porphyria even when this is clinically latent. The risk is particularly high in Sweden, where a recent study found a standardized increased risk ratio of 64 for AIP gene carriers aged over 54 years and recommended that AIP patients over the age of 50 years should be screened annually for HCC by ultrasound examination. The benefit from screening in other countries has yet to be assessed. It has also been suggested that all HCC patients in whom no obvious cause is found should be screened for acute porphyria.
Renal impairment has been described as a complication of AIP, affecting particularly those patients who have previously suffered acute attacks. Hypertension is also common in these patients, but in many, the decline in renal function precedes the onset of hypertension, which may also be a consequence of the porphyria. Renal biopsies have shown glomerular sclerosis and interstitial fibrosis, but no evidence of inflammation or immunodeposits. A proportion of patients may progress to end-stage renal failure, requiring dialysis and/or renal transplantation. Patients who have had active disease should therefore have their renal function and blood pressure monitored regularly, and antihypertensive therapy should be instituted in an attempt to limit progression of renal impairment.
Diagnosis of acute porphyria
Diagnosis of an acute attack in a newly presenting patient requires the demonstration of increased excretion of urinary PBG in a fresh, random sample of urine that has been protected from light. An assay capable of quantitative or semiquantitative measurement of PBG should ideally be available in all acute hospitals. In practice, many non-specialist units use rapid qualitative screening tests, particularly out of hours, which have low sensitivity and poor specificity. All positive screening tests should be confirmed by a specific quantitative method with expression of results in relation to creatinine to correct for urine concentration. If urine PBG and ALA concentrations are normal during the early phase of an acute illness, all acute porphyrias, including ADP, are excluded as a cause of that illness. However, both PBG and ALA may return to within normal limits within a few days after the onset of symptoms in VP and HCP, and negative findings should be interpreted with caution when there has been a delay in collecting samples. However, in both these disorders, urinary and faecal porphyrins remain elevated for a prolonged period. Conversely, urinary PBG excretion usually remains elevated for many months or even years after an acute attack in AIP, and an increased PBG does not necessarily indicate an acute attack unless a marked increase from baseline can be shown to coincide with symptoms. Management of a clinically diagnosed acute attack should be started immediately, without waiting for confirmation of a positive screening test or determination of the type of acute porphyria.
Establishment of the type of acute porphyria requires analysis of plasma and faecal porphyrins (see Table 28.3; Fig. 28.2). Faecal porphyrin analysis is essential to distinguish HCP, in which coproporphyrin III accounts for most of the increased faecal porphyrin excretion, from AIP, in which faecal porphyrin excretion is normal or only slightly increased without any change in the coproporphyrin isomer ratio. Variegate porphyria can easily be identified by demonstrating a characteristic plasma porphyrin fluorescence emission peak at 625–628 nm. Enzyme analyses and genetic studies are not required for the diagnosis of new cases of clinically overt porphyria. Their use in family studies is detailed later in the chapter.
Laboratory monitoring of patients during and after acute attacks by regular measurement of porphyrin precursors is rarely indicated; treatment should be guided by clinical assessment. The one exception is where a severe attack has progressed to flaccid paresis and clinical assessment is difficult. In these circumstances, weekly monitoring may provide useful information on whether the condition is stable or deteriorating.
Management of an acute attack
Supportive treatment
As soon as the acute attack is confirmed, drugs or other recognized precipitants should be withdrawn. Safe and effective management of symptoms and complications, with support from an expert centre, is essential to minimize the stress; effective pain relief is a major component. This invariably requires opiates, and support from a specialist pain team can be very helpful as very high doses may be required. A phenothiazine may be used for anxiety and restlessness, and to decrease the opiate requirements. Antiemetics should be prescribed for nausea and vomiting. Adequate fluid and energy intake should be ensured, if necessary by intravenous infusions of 0.9% sodium chloride containing a minimum of 5% dextrose, with regular monitoring of electrolyte status in view of the risk of hyponatraemia. Where drug treatment of precipitants such as infection, coexisting conditions or other features of an acute attack, such as hypertension, tachycardia or convulsions, are required, care should be taken to select medications that are considered safe. Information on drug safety is continually under review and safe drug lists are likely to change regularly (see Welsh Medicines Information Centre: www.wmic.wales.nhs.uk). Where no safe alternative is on the list, an expert centre should be consulted for further advice on patient management. Information about specialist centres offering support in Europe is available from the European Porphyria Network (EPNET): www.porphyria-europe.org.
Specific treatment
Specific treatment is aimed at suppressing hepatic haem precursor production by administering intravenous haem, which binds to albumin and is transported to the liver, where it downregulates ALAS activity. Treatment is effective if given early, preferably within the first 24–48 h. It will not reverse established neuropathy due to axonal degeneration. Within Europe, haem is available as haem arginate (Normosang® Orphan Europe) and is provided as a concentrated stock solution (25 mg/mL), which should be diluted immediately before use in 100 mL 0.9% saline and administered at a dose of 3 mg/kg body weight in a single intravenous infusion over at least 30 min on each of four consecutive days. As there is no evidence for toxicity, it is practical in most adults to infuse the entire contents of the vial for each dose. However, in smaller patients it may be possible to obtain two doses from each vial, limiting the overall cost of treatment. The main side-effect of therapy is thrombophlebitis at the site of infusion. This can be minimized by careful flushing of the infusion site with normal saline, and re-siting the i.v. cannula after each infusion. Alternatively, the haem can be diluted in 20% human serum albumin, which has proved to be effective in preventing thrombophlebitis, particularly in patients who require regular haem arginate infusions. Lyophilized haem (Panhematin®, Abbott) for intravenous infusion is available in the USA, but is less stable than haem arginate, and has more potential side-effects, including thrombocytopenia and coagulopathy.
With the advent of intravenous haem therapy, carbohydrate loading, which also decreases hepatic ALAS1 activity, is no longer indicated as a specific treatment unless haem is unavailable or there is a delay in obtaining it. High-dose carbohydrate (300–500 g glucose/24 h) should be given intravenously via a central venous catheter.
Preventing acute attacks
All patients should be advised about avoiding factors that increase the risk of an acute attack. These include drugs, alcohol and fasting or dieting, particularly diets in which carbohydrate is avoided completely. Weight reducing diets should be undertaken under the supervision of a dietitian. Patients should also be encouraged to register with an organization such as the MedicAlert Foundation, which provides jewellery inscribed with appropriate information in case of emergencies. It is helpful to provide patients with written information explaining the disorder and, where applicable, they should be provided with details of existing patient support groups (e.g. the British Porphyria Association: www.porphyria.org.uk). They should also be offered the opportunity to see a clinician with a special interest in porphyria on at least one occasion. Patients in remission (i.e. those who have had an acute attack) should be followed-up annually, preferably at a specialist centre or in conjunction with one. Local follow-up is also important in order to ensure contact with clinical services should admission be required. Through such shared care arrangements, most intervening problems can be managed successfully through correspondence or over the telephone.
Severely affected patients
A minority of patients with acute porphyria suffer from repeated acute attacks, occasionally as often as every 3–4 weeks. The majority are women with AIP in their 20s and 30s and their management can present a major challenge to clinicians and the acute medical services. A careful history should be taken to ascertain any obvious provoking factor, such as smoking, alcohol, drugs or stage of the menstrual cycle. An individualized management protocol should be drawn up by the local physician with help from a recognized national porphyria service; these operate in many European countries, including the UK, where the National Acute Porphyria Service (NAPS) has been commissioned specifically to support the management of patients with active porphyria. The patient should be encouraged to seek early medical intervention as prompt treatment may abort an acute attack, limiting the length of inpatient stay. This is best achieved by rapid direct access to inpatient facilities and the support of a clinical team who knows the patient and is experienced in dealing with the treatment of acute attacks.
When attacks appear to be associated with the luteal phase of the menstrual cycle, they may be reduced in frequency and/or severity by suppression of ovulation using gonadotrophin releasing hormone agonists. If this succeeds and is to be continued for longer than six months, measures to prevent osteoporosis become essential. Oestrogen supplementation, preferably in the form of patches, is tolerated by some patients, and diminishes any menopausal symptoms, but carries a small risk of provoking further attacks. Progestogens should not be used and most women will therefore require regular endometrial monitoring under the supervision of a gynaecologist. Adequate vitamin D and calcium intake should be ensured, by supplementation if required. Bisphosphonates have also been used in this situation to reduce the risk of osteoporosis.
Regular infusion of haem arginate reduces the frequency and severity of acute attacks in many patients. The minimum effective dose frequency, varying between monthly and weekly single infusions, should be established for each individual patient. There is no typical regimen. Where this treatment is being considered, it is often helpful to contact an expert porphyria centre to discuss the individual details with a clinician with experience of its use. An indwelling central venous line is invariably required, although the specific type of line used should be dictated by local expertise and patient choice. Dilution of haem arginate in albumin solutions may reduce the need to replace blocked venous catheters.
Where these measures fail to control the attacks with resulting repeated life-threatening crises, problems with central venous access arise, progressive renal dysfunction occurs or quality of life is assessed as very poor, patients should be considered for liver transplantation, which is curative. Careful counselling and assessment is required, preferably in centres with experience with orthotopic liver transplantation (OLT) for this indication. Referral should ideally be before any chronic damage becomes irreversible. Liver transplantation has also confirmed that overproduction of haem precursors by the liver is central to the pathogenesis of acute attacks. This evidence has supported a project to develop gene therapy using a recombinant viral vector targeting the liver as a treatment for AIP.
Recurrent acute abdominal pain should not automatically be ascribed to the porphyria, particularly if the patient reports that the pain is not the same as previously experienced. Other conditions should be considered, as failure to diagnose these can have serious, possibly fatal consequences. In this situation, measuring urine PBG and/or ALA may be helpful in patients with VP or HCP. In AIP patients, a baseline measurement is required to aid interpretation of later results.
Chronic pain, which may be neuropathic, is a common problem in patients who have suffered regular acute attacks. Pain may be in the abdomen, limbs or lower back and may, in some cases, be present almost continuously. Although there may be an initial response to haem arginate, suppression of ovulation or other methods used to manage recurrent acute attacks, this is rarely sustained. Opiates are not a long-term solution in view of the risk of dependence, and should not be prescribed between acute attacks. Analgesia with a non-steroidal anti-inflammatory drug can, in some cases, be successful, and some patients may benefit from medication used to treat neuropathic pain such as gabapentin or pregabalin. Detailed discussion with the patient to explain that these symptoms are not due to an acute attack can be reassuring.
Managing asymptomatic relatives of patients
Once the diagnosis of a specific type of acute porphyria has been established in a new patient, screening should be offered to family members, so that those found to be affected, most of whom will have latent porphyria, can be offered specific advice as to how to limit the risk of suffering an acute attack.
Family studies
Metabolite measurements are highly specific but are almost always normal before puberty and have low sensitivity in adults (Table 28.5). Enzyme measurements are more sensitive but their sensitivity and specificity are limited by the overlap between activities in disease and in normal subjects. They have now largely been replaced by mutation detection by DNA analysis, which is specific and 100% sensitive if the mutation that causes porphyria in the family under investigation is known. It is essential, therefore, that children, and those family members whose specific porphyrin biochemistry is normal, be offered gene testing. Enzyme measurements, for example erythrocyte HMBS assay for detection of latent AIP, and gene tracking using intragenic single nucleotide polymorphisms, may be useful in the few families in which a disease-specific mutation cannot be identified.

Safe prescribing
Prescription drugs are an established precipitant of acute attacks and, even in known porphyria patients, careless prescribing has induced acute attacks or made evolving ones worse. It is therefore essential to provide information concerning the safe use of prescription medication to both those affected and their clinicians. A recent initiative in Europe has resulted in a review of the available evidence on a significant proportion of the current pharmacopoeia and provided an overall assessment of safety for individual drugs (www.drugs-porphyria.org).
Drugs can be classified into three broad groups, based on clinical experience, experimental evidence and an understanding of their metabolism and excretion. These are: Not Porphyrinogenic (safe); Porphyrinogenic (unsafe); and Of Uncertain Safety (i.e. should be used with caution), which the European system subclassifies into three groups: Probably Not Porphyrinogenic, Possibly Porphyrinogenic and Probably Porphyrinogenic. In the first instance, clinicians should be encouraged to prescribe from a list of drugs that are known to be safe and to avoid those that are definitely unsafe. However, no drug should be seen as completely unusable as there may be clinical situations where a safe alternative is either unsuitable or not available. In these circumstances, an assessment of benefit versus risk should be made, taking into account the severity of the medical condition and porphyria activity. Support from a national centre with expertise in managing porphyria, and access to the most up-to-date drugs information, should also be obtained. If an unsafe drug has to be prescribed, urine PBG should be measured before starting, and at regular intervals during treatment.
Specific situations
Pregnancy
Although acute attacks do occur during pregnancy, there does not appear to be a significantly increased risk and most patients tolerate pregnancy, delivery and the puerperium without any adverse consequence. However, it is usually recommended that pregnancy should be delayed until patients have been free of severe acute attacks for a year. Patients should not be allowed to experience prolonged periods of stress and fasting during labour. Effective pain relief, including spinal or epidural anaesthesia, should be administered and, where necessary, intravenous fluids containing glucose should be used to prevent the development of a catabolic state. Where an acute attack does occur during pregnancy, haem preparations have been used and there have been no reports of any adverse effects.
Anaesthesia
General anaesthesia can be safely undertaken in porphyria patients, provided care is taken to select drugs that are known to be safe. General measures to reduce stress and limit periods of fasting should also be instituted, and postoperative complications, such as infection treated aggressively with drugs chosen from the safe list. Regional and dental anaesthesia using local anaesthetics have been safely undertaken in many acute porphyria patients without problem.
Rare forms of acute porphyria
ALA dehydratase deficiency porphyria
5-Aminolaevulinic acid dehydratase deficiency porphyria (ADP) is an autosomal recessive acute porphyria, presenting with neurovisceral symptoms, that results from deficient ALAD activity; the biochemistry is characterized by marked increases in the plasma concentration and urinary excretion of ALA. The prevalence cannot be accurately assessed, but is extremely low with fewer than ten cases reported worldwide to date, and, as yet, none from the UK.
In addition to the very elevated plasma and urinary ALA, other biochemical features include a normal or slightly increased urine PBG, markedly elevated urine coproporphyrin excretion, normal faecal porphyrin and increased erythrocyte protoporphyrin. Confirmation of the diagnosis requires exclusion of lead poisoning, which gives rise to similar biochemical features, and demonstration of decreased ALAD activity in erythroid and non-erythroid cells that is not reversed by addition of excess zinc and a sulfhydryl group reducing agent. Mutational analysis may also be helpful in confirming the diagnosis, and may be required for genetic counselling.
Clinical presentation is variable with reported ages of presentation from birth through to 63 years. The predominant presentation is with acute attacks of abdominal pain and neuropathy that resemble those seen in the autosomal dominant acute porphyrias, and similarly may be triggered by factors such as prescribed drugs that induce ALAS activity. Heterozygote carriers are asymptomatic, but may be at increased risk from the effects of environmental toxins, such as lead, that inhibit ALAD activity. Treatment with hematin and glucose has not been effective in all cases. Liver transplantation in a patient with ADP did not protect the patient from further acute attacks, nor correct the biochemical abnormalities. The marked enzyme deficiency in other tissues, particularly nervous tissue, probably contributes to the poor clinical outcome in this autosomal recessive condition.
Homozygous acute porphyrias
Homozygosity for null mutations of haem biosynthetic genes is lethal in early embryonic development. However, very rare so-called homozygous variants of all the autosomal dominant acute porphyrias have been reported, in which patients are homozygous or compound heterozygous for mutations that have some residual activity. All present in childhood and have other phenotypic differences from their (heterozygous) autosomal dominant counterparts.
Homozygous AIP is clinically the most severe of these variants and is usually associated with a progressive leukodystrophy. Both homozygous HCP and homozygous VP present with skin lesions in childhood with, in the latter, clinodactyly (curving of the little finger towards the ring finger) and sometimes other abnormalities, including short stature and neurological defects. Acute attacks have been reported in homozygous HCP but not homozygous VP. Missense mutations in exon 6 of the CPOX gene cause harderoporphyria, a relatively benign condition characterized by neonatal jaundice, persistent mild haemolytic anaemia, skin lesions and excretion of the tricarboxylic porphyrin, harderoporphyrin, in faeces. All these variants can be distinguished from their (heterozygous) autosomal dominant counterparts by demonstrating very low activity of the relevant enzyme and by mutational analysis. Erythrocyte protoporphyrin concentrations are also increased in all these conditions and this may serve as a useful initial diagnostic indicator. With the exception of harderoporphyria, porphyrin excretion patterns in the homozygous acute porphyrias are indistinguishable from those of the autosomal dominant forms.
THE CUTANEOUS PORPHYRIAS
Bullous porphyrias
The bullous porphyrias all give rise to identical skin lesions, and so cannot be distinguished with certainty on clinical grounds alone. Although patients with CEP or hepatoerythropoietic porphyria (HEP, see p. 545) tend to present in infancy or childhood, and those with PCT, VP or HCP in adulthood, exceptions do occur: occasionally PCT presents in early childhood, while the skin lesions of late onset CEP may first appear in adults and be mistaken for PCT. Biochemical investigation is essential for accurate diagnosis (see Table 28.3). It is important to distinguish between the disorders in view of the differences in prognosis, treatment and susceptibility to acute attacks. In addition, clinically identical skin lesions, termed pseudoporphyria, can occur in the absence of any disturbance of porphyrin metabolism as a consequence of drug reactions, prolonged use of sun beds and in association with long-term haemodialysis.
Pathophysiology of skin lesions
Porphyrin-induced photosensitivity results from the absorption of visible light at the surface of the skin. Porphyrins enter the dermis from the plasma and interact with light of 400–410 nm wavelength, which is capable of penetrating to the level of the dermis and basement membrane. This photoactivation of the porphyrin ring gives rise to an unstable, excited-state molecule, which can release energy as light or react with oxygen by resonance energy transfer to form a singlet oxygen species or superoxide anion. These reactive oxygen species cause cellular damage via reactions with lipids, proteins and DNA, resulting in complement activation, degranulation of mast cells and damage to the dermis and basement membrane. This process gives rise to the characteristic features evident on histopathology: perivascular accumulation of amorphous hyaline material involving the small vessels of the dermis. This PAS-positive material includes deposits of immunoglobulin and complement and appears to emanate from the walls and lumens of the blood vessels. Bullae occur where the epidermis splits from chronically damaged, thickened basement membrane and forms a roof for a pocket of clear fluid. The dermal papillae are distorted and flattened by the accumulated amorphous hyaline material and the floors of the bullae have a characteristic festooned appearance.
Skin symptoms and signs
The skin lesions may occur in any sun-exposed area, most frequently the backs of the hands, the forearms, the face and the feet, particularly when open sandals are worn. Fragile skin that tears following minor mechanical stress is the commonest feature. Injury can occur in response to everyday household activities, and patients frequently wear gloves to avoid damaging their skin. Vesicles and bullae filled with clear fluid are commonly present, as are small white spots or milia (Fig. 28.3). Haemorrhagic bullae are uncommon. Bullae rupture easily and form crusted lesions that heal slowly, resulting in chronic scarring of the skin and areas of depigmentation. Secondary infection of open lesions may occur, which worsens the scarring and in severe cases, may result in photomutilation and disfiguration. Other features include hypertrichosis, usually on the forehead and temples; this is particularly noticeable and troubling in women. Less common clinical features include hyper- or hypopigmentary changes, onycholysis, conjunctival damage and scarring alopecia. Because of the chronic nature of the conditions, the relationship to sunlight is not always recognized by the patient.
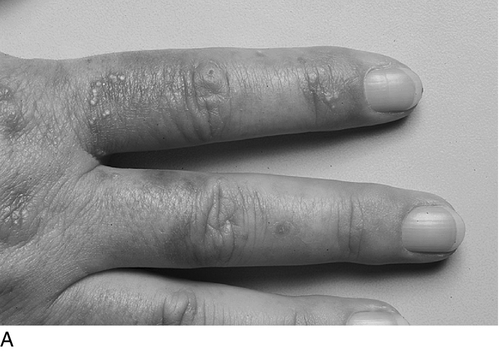
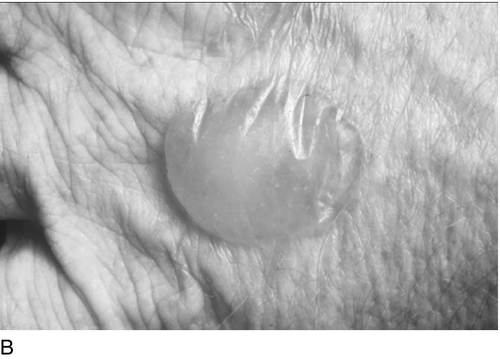
Biochemical features and diagnostic approach
Each of the bullous porphyrias has a characteristic pattern of porphyrin excretion during active disease that allows the conditions to be distinguished (see Table 28.3). Ideally, samples of urine, faeces and EDTA-anticoagulated blood should be sent for full porphyrin analysis on all patients. However, the single most useful analysis in patients with active skin lesions is a plasma porphyrin fluorescence emission scan, which, if normal, excludes active cutaneous porphyria (see Fig. 28.2). Many dermatologists now follow this approach and will send the other samples required for diagnosis only if the plasma porphyrin screen is abnormal. However, it is essential that the plasma scan be undertaken in a fluorometer that is sufficiently sensitive, which requires it to be fitted with a red-sensitive photomultiplier. An increased plasma porphyrin fluorescence emission peak with a maximum at 624–627 nm is diagnostic for VP. However, it does not necessarily indicate active porphyria, as positive plasma scans are found in approximately 60% of adults with latent VP and in most of those in remission. An emission maximum at 615–620 nm is consistent with skin lesions caused by porphyria but does not distinguish between PCT, CEP or HCP. However, these conditions can be unequivocally differentiated by urinary and faecal porphyrin analyses.
Individual disorders
Porphyria cutanea tarda
Porphyria cutanea tarda (PCT) is the commonest of the porphyrias, with a prevalence of approximately 1:25 000. It results from partial deficiency of the enzyme uroporphyrinogen decarboxylase (UROD) in the liver and is characterized biochemically by overproduction of uroporphyrin, 7-, 6- and 5-carboxyl porphyrins and isocoproporphyrin. In many patients, there is evidence for underlying liver cell damage, and biochemical tests of liver function are often abnormal. Histopathological examination usually reveals only minor abnormalities: mild fatty infiltration with some hepatocyte necrosis and periportal inflammation. Cirrhosis is present in fewer than 20% of patients, but when it does occur, it carries a much greater risk of hepatocellular carcinoma than does comparable cirrhosis without PCT.
The majority of patients in the UK (80%) have sporadic or type I PCT, in which UROD deficiency is restricted to the liver. Most of the remaining 20% have familial or type II PCT, in which UROD deficiency is present in all tissues and is inherited in an autosomal dominant pattern. As with other autosomal dominant porphyrias, the clinical penetrance of familial PCT is low, with symptoms in fewer than 10% of those who inherit the condition. Porphyria cutanea tarda may also be caused by exposure to various halogenated aromatic hydrocarbons as in the outbreak of hexachlorobenzene poisoning in south-eastern Turkey in the late 1950s.
Whether the patient has familial or sporadic PCT, overproduction of sufficient porphyrin to produce symptoms appears to require a decrease in hepatic UROD activity to well below 50% of normal. This is brought about through the reversible inactivation of UROD by a porphomethene inhibitor formed from uroporphyrinogen by an iron-dependent oxidative mechanism, probably catalysed by hepatic cytochrome P450s. Less inactivation of UROD is required for symptoms to occur in familial PCT, where prior enzyme activity is half-normal and, although there is a wide overlap, patients tend to present at a younger age than those with sporadic PCT. Other risk factors for both types of PCT include alcohol misuse, conditions that increase hepatic iron content such as hereditary haemochromatosis, hepatotropic viruses (particularly hepatitis C virus, HCV) and HIV, and prescribed oestrogens: two or more risk factors are often present. Most patients have some hepatic iron overload but amounts are usually below those in overt haemochromatosis. Among patients of northern European descent, about 20% are homozygous for the haemochromatosis (HFE) gene C282Y mutation, irrespective of the type of PCT, but heterozygosity for this mutation is a less important risk factor. Prevalences for antibodies to HCV range from 8% to 79%, being highest in southern Europe and the USA and lowest in northern Europe. When PCT develops in HCV, it is usually an early manifestation; in contrast, it occurs as a late complication of HIV infection. Conditions less frequently associated with PCT include chronic renal disease, diabetes mellitus, systemic lupus erythematosus and various haematological malignancies.
Clinical management of patients with PCT should start with the identification of any predisposing factors that can be withdrawn or treated. Newly diagnosed patients should be investigated for underlying liver disease, tested for iron overload by measuring transferrin saturation and HFE genotyping, and screened for hepatitis viruses and, if indicated, HIV infection. Where there is evidence of chronic liver disease, referral to a hepatologist is advisable. All patients should be given general advice about avoidance of sunlight, encouraged to protect their hands from trauma by wearing gloves and advised to seek early treatment of any infected lesions. In contrast to patients with acute porphyrias, patients with PCT need not avoid any drugs, apart from antimalarial doses of chloroquine and its derivatives.
Two effective treatments for PCT are available. Venesection of a unit of blood at one- or two-weekly intervals to deplete hepatic iron stores prevents inactivation of hepatic UROD and eventually restores enzyme activity to basal levels. Treatment should be continued until transferrin saturation falls to 16% or less or the patient becomes anaemic (haemoglobin less than 120 g/L). Serum ferritin concentration is less satisfactory for monitoring, as values may be affected by concomitant liver disease. If it is used, treatment should continue until the concentration is 25 μg/L or less. Although resolution of skin lesions occurs within 3–9 months, biochemical remission usually takes longer as large quantities of porphyrins stored in the liver are released and excreted in the urine. Other methods used to reduce iron stores include subcutaneous desferrioxamine and, in patients with renal failure, erythropoietin without iron supplementation. Treatment with low-dose chloroquine (125 mg twice weekly) or hydroxychloroquine (100 mg twice weekly), which complexes with uroporphyrin and mobilizes it for excretion from the liver, is also effective. Larger doses will provoke an acute hepatotoxic reaction with systemic symptoms and should be avoided. Clinical improvement usually becomes evident within 3–4 months and treatment should be continued until total urinary porphyrin excretion falls to normal. Chloroquine treatment may be combined with venesection if response to monotherapy is slow. Choice between these two treatments is often determined by local preference. There is little evidence that chloroquine worsens liver disease, but it is potentially hepatotoxic in PCT and should probably be avoided in patients with severe liver disease. Iron depletion should be used for all HFE C282Y homozygotes; it is the only effective method in renal failure and ensures compliance, but is more expensive. Both treatments produce prolonged remission and, particularly in sporadic PCT, may prevent relapse. Nevertheless, long-term management should take into account the possibility of relapse, and patients in remission may benefit from an annual review. Patients with significant liver disease, familial haemochromatosis or other associated conditions should be managed by an appropriate specialist.
Determination of the type of PCT is best carried out by mutational analysis of the UROD gene. However, it is not essential for clinical management. Treatment is the same for both types, while the low clinical penetrance, absence of potentially life-threatening symptoms and availability of effective treatments means that mutational analysis for family studies is difficult to justify, except perhaps for those rare families in which more than one individual has overt PCT.
Hepatoerythropoietic porphyria
This is a rare variant of familial PCT in which patients are homo- or heteroallelic for UROD mutations. Uroporphyrinogen decarboxylase activity is markedly decreased in all tissues, although the liver appears to be the main source of excess porphyrins. Skin lesions usually appear in early childhood and are identical to those of PCT. They persist and may occasionally become as severe and disfiguring as in CEP. Other organs are rarely affected; although severe anaemia has been described in HEP, most patients have no significant haematological abnormalities. Differentiation from other early onset porphyrias requires analysis of urinary, faecal and erythrocyte porphyrins, with measurement of erythrocyte UROD activity and/or mutational analysis of the UROD gene. Photoprotection is the only effective treatment. There is no associated iron overload and patients do not respond to phlebotomy or chloroquine. For most patients, the disease does not appear to affect survival.
Congenital erythropoietic porphyria
Congenital erythropoietic porphyria (CEP), or Gunther disease, is a rare bullous porphyria, with a UK prevalence of 1 in 3 million. It is pan-ethnic, although like other autosomal recessive conditions prevalence is increased in communities where consanguinity is common. Marked deficiency of UROS results in the accumulation of large amounts of uroporphyrin I, formed by non-enzymatic cyclization of HMB, and, to a lesser extent, of coproporphyrin I. The main source of excess porphyrin production is the bone marrow, with less than 1% produced from other tissues. The severity of the condition varies from hydrops fetalis, resulting from in utero haemolytic anaemia, to the late onset forms in which the disease becomes manifest in late childhood and young adults. The severity appears to correlate with the level of residual enzyme activity, with the more marked deficiency resulting in very high plasma and red cell porphyrin concentrations, which result in two major pathophysiological consequences: marked skin photosensitivity and haemolytic anaemia with ineffective erythropoiesis and splenomegaly. Most porphyrin is released from cells destroyed in the bone marrow but some comes from red cells sequestrated and destroyed by the spleen. The haemolytic anaemia associated with CEP, which may be caused by porphyrin-induced damage to erythroid cell membranes, further stimulates erythropoiesis, increasing porphyrin overproduction.
The majority of patients present soon after birth with skin blisters, occasionally in response to phototherapy treatment for neonatal hyperbilirubinaemia, and red-brown staining of their nappies. The skin lesions are more severe and extensive than in other bullous porphyrias, and progressive damage can lead to photomutilation affecting the ears, nose, eyelids and fingers. Porphyrins also accumulate in bone and teeth, causing discolouration, evident as erythrodontia – reddish–brown teeth that fluoresce red on UV illumination. Bone marrow hyperplasia may lead to osteolytic lesions. Other skeletal abnormalities include decreased bone density, possibly resulting from low vitamin D due to strict avoidance of sunlight, leading to pathological fractures. The hands are frequently severely affected with contractures, atrophy and bone resorption of the terminal phalanges.
The variable phenotype dictates the treatment approach, and early on in the course of the disease, mutational analysis of the UROS gene can provide useful information as there is some correlation between genotype and phenotype. Homozygosity for a mutation present on about 30% of CEP alleles, C73R, carries a particularly poor prognosis. Depending on the combination of mutations, this information can be used to classify the likely clinical severity as mild, moderate or severe and predict likely treatment requirements. Mutational analysis may also be required for prenatal diagnosis.
As with other cutaneous porphyrias, the mainstay of treatment is avoidance of sunlight; special measures such as fitting filters to house and car windows may be required. Aggressive treatment of any infected skin lesions with antibiotics is essential. Regular blood transfusions aimed at maintaining the haematocrit above 33% suppresses erythropoiesis and can successfully decrease porphyrin production and improve photosensitivity. In patients with transfusion-dependent anaemia, it is essential to use desferrioxamine or other iron chelators to control iron accumulation, which has been an important cause of death in the past. If hypersplenism occurs, splenectomy may reduce haemolysis and transfusion requirements. Other treatment modalities reported on small numbers or individual patients, and shown to be of limited benefit, include β-carotene, oral activated charcoal, plasmasorbent therapy, hydroxyurea and haematin infusion. Bone disease can be managed with bisphosphonate therapy and vitamin D supplementation where deficiency is evident. Prophylactic vitamin D should also be considered.
Allogeneic bone marrow transplantation (BMT) has been undertaken successfully in more than ten patients with CEP, resulting in a marked decrease in porphyrin production and photosensitivity in all cases. The main limitation is the availability of suitable donors. Use of cells from a heterozygous donor does not appear to affect the outcome. The major risks are the morbidity and mortality associated with the procedure itself, and bone marrow transplantation should therefore be reserved for the most severely affected patients. Clinical care for patients with CEP should be provided by a multidisciplinary team, including a dermatologist, haematologist, ophthalmologist and a porphyria expert, in order to assist patients and their families make informed decisions about this form of treatment.
Gene therapy for the erythropoietic porphyrias, particularly CEP, is being actively pursued. The success of BMT indicates that introduction of active enzyme into a sufficient number of deficient cells by ex vivo transformation should be curative. Gene delivery methodology has been successfully developed for other bone marrow disorders, for example X-linked severe combined immunodeficiency.
Erythropoietic protoporphyria and X-linked dominant protoporphyria
Two clinically indistinguishable porphyrias present with acute photosensitivity without skin fragility. In both, the photosensitivity results from accumulation of protoporphyrin in the skin. The concentration of protoporphyrin is also increased in erythrocytes, plasma, liver and other tissues. Most patients with this presentation have erythropoietic protoporphyria (EPP) but about 5% have XLDPP.
The excess protoporphyrin is produced mainly or, in XLDPP, exclusively by erythroid cells in bone marrow. In EPP, protoporphyrin accumulation results from a decrease in ferrochelatase (FECH) activity to less than 35% of normal. Almost all patients are compound heterozygotes for one of a large number of FECH mutations that abolish or severely decrease FECH activity and a hypomorphic allele (FECH IVS3–48C) that has about three-quarters of the activity of the normal allele and is present in around 10% of white Europeans, 30–45% of Asians and < 3% of Africans. About 3% of patients lack the hypomorphic allele but have FECH mutations on both alleles, at least one of which retains some FECH activity. Within families, EPP segregates as an autosomal recessive disorder that may show pseudo-dominant inheritance in populations where the frequency of the hypomorphic allele is high. A late onset form of EPP associated with myelodysplasia or myeloproliferative disease and caused by acquired somatic FECH mutation has also been described.
X-linked dominant protoporphyria is caused by mutations in ALAS2 that disrupt the C-terminal region of erythroid ALAS2, thereby increasing its activity and leading to increased formation of protoporphyrin. Unlike in EPP, FECH activity is normal, which enables conversion of some of the protoporphyrin that accumulates to zinc- protoporphyrin. Families normally show inheritance through several generations. This pattern is unusual in EPP and is an important indicator of the possible presence of XLDPP. As might be expected for a gain in function mutation on an X chromosome, both sexes are similarly affected, though skewed inactivation of the mutant chromosome may lead to mild or no disease in a small number of women.
The diagnosis of both conditions can be established by demonstrating an increase in erythrocyte protoporphyrin using a method that distinguishes between free protoporphyrin and its zinc chelate, which is the predominant form in normal and iron-deficient erythrocytes. In EPP, almost all the protoporphyrin is in the free form; more than 15% of zinc-protoporphyrin in a patient with a markedly increased total protoporphyrin indicates possible XLDPP, in which zinc-protoporphyrin forms from 20% to 70% of the total (see Fig. 28.2). Mutational analysis of ALAS2 is required to confirm a diagnosis of XLDPP. Protoporphyrin is increased in plasma in EPP and XLDPP and has a fluorescence emission peak at 626–635 nm. Faecal protoporphyrin excretion is increased in about 40% of EPP patients, and some of those with XLDPP, but is not specific for either condition and is of limited value diagnostically. Protoporphyrin is hydrophobic and is not excreted in urine, which therefore cannot be used to diagnose or exclude EPP or XLDPP.
Skin symptoms and signs
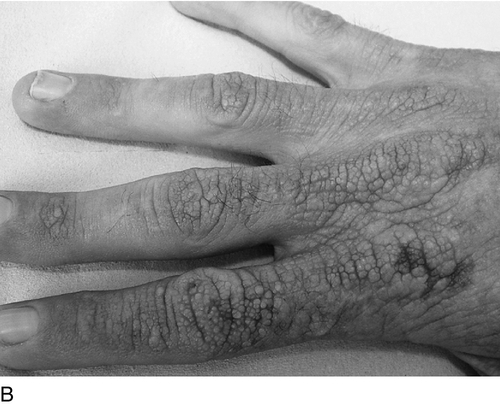
Both conditions are characterized by life-long acute photosensitivity with pain and swelling of sun-exposed skin, usually becoming evident within the first two years of life. Although symptoms typically appear in early childhood, diagnosis may be delayed for many years unless there is a family history. Patients give a characteristic history of burning pain within minutes of exposure to sunlight, which may only be relieved by cold water or wet towels. There may also be priming phenomena, whereby exposure to sunlight sensitizes the skin, lowering tolerance to sunlight on subsequent occasions. The oedema evident immediately after exposure has usually resolved by the time a doctor sees the child, hence the difficulty in diagnosis. With prolonged exposure, an erythematous reaction or even blistering can occur. Over time, patients develop chronic skin lesions with a waxy, thickened appearance. Chronic exposure of the face leads to linear scarring of the forehead and cheeks, with furrowing around the mouth (Fig. 28.4). Occasionally, marked thickening of the skin develops over the joints of the hands and fingers. Severe acute photosensitivity has a serious adverse effect on quality of life, sometimes worsened by delayed or incorrect diagnosis. Childhood can be a particularly difficult period and adult patients frequently state that they do not want to have children if they have to go through a similar experience.
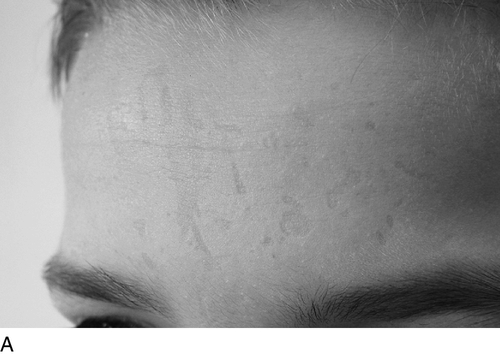
Treatment
Avoidance of sunlight is the mainstay of management of acute protoporphyrin-induced photosensitivity. Outdoor clothing should include full-length garments to cover the arms and legs, a wide brimmed hat (or peaked hat with a cloth covering the neck), closed shoes and gloves if possible. Sunscreen creams must be formulated to block wavelengths around 400–420 nm. The most effective are those based on titanium dioxide, which acts as a reflectant, and a specially formulated product available in three tinted versions (coral, beige and coffee) is available in the UK (Tayside Pharmaceuticals, Ninewells, Dundee). Where prevention fails, cold compresses and application of local corticosteroid creams to the affected area can provide symptomatic relief. Antihistamines may also provide some relief by reducing the immediate release of histamine by mast cells.
Several specific treatments have been proposed, although none has proved to be completely reliable. The most promising is narrow-band UV therapy, which induces sunlight tolerance equivalent to a sun protection factor (SPF) of 8. Effective treatment is limited by availability of equipment and the requirement for regular therapy. More traditionally, patients should be offered a trial of β-carotene therapy, which is believed to work by quenching oxygen radicals and/or interfering with the absorbance of light by protoporphyrin. Efficacy is variable, with a significant proportion of patients reporting no benefit. However, it is important to ensure an adequate dose of between 100 and 300 mg/day, sufficient to achieve plasma concentrations of 11–15 μmol/L (6–8 mg/L). Hormonal tanning agents based on α-melanocyte stimulating hormone (α-MSH) analogues are currently being investigated as a treatment and have shown promise in early trials.
Bone marrow transplantation (BMT) has also been used successfully in several EPP patients, either to treat a coexistent haematological malignancy or to protect a transplanted liver from recurrence of protoporphyric hepatopathy (see below). Although in most EPP patients, the risk of BMT outweighs the benefits, if it becomes possible to identify patients with a high risk of protoporphyric liver disease, BMT could be used to cure the condition and eliminate the need for liver transplantation.
Chronic complications and their management
Mild microcytic, normochromic anaemia, which is associated with reduced iron indices, is common in both conditions, affecting up to a third of EPP patients. Bone marrow studies show scattered sideroblasts and perimitochondrial iron accumulation, suggesting defective utilization of iron for haem synthesis. Iron therapy is, therefore, unlikely to resolve the anaemia and, in some cases, has been reported to worsen photosensitivity.
The most serious complication of EPP and XLDPP is progressive protoporphyric liver disease, which affects 1–2% of patients and may lead to severe acute liver failure. Excess protoporphyrin is excreted via the biliary tract and deposits of protoporphyrin are present in the hepatocytes of many patients. In a minority, this leads to liver damage that, although initially mild as indicated by small increases in plasma transaminase activities, will in some cases progress to cirrhosis. Further deterioration leads to cholestatic jaundice and abdominal pain that may be similar to the neurovisceral pain experienced in the acute porphyrias. Hypersplenism and the accompanying haemolysis stimulate erythropoiesis and, in combination with the decreased biliary protoporphyrin excretion, result in increased red blood cell protoporphyrin concentrations and worsening photosensitivity. This fulminant or decompensated liver failure is usually fatal unless the patient receives a liver transplant. Perioperative risks include severe phototoxic burns as a result high levels of circulating protoporphyrin. This can be reduced by preoperative treatments that reduce protoporphyrin concentrations; it is essential that the theatre lights be filtered. In addition, patients are at risk of neuronal damage leading to abdominal pain and profound motor neuropathy requiring ventilation. Finally, protoporphyric liver disease can recur in the transplanted liver and, in these circumstances, bone marrow transplantation to correct the metabolic defect and prevent further hepatic damage is the only viable option.
There is no reliable way of predicting which patients are at risk of severe liver disease, although there appears to be an increased risk in patients with XLDPP and those with true autosomal recessive EPP (mutations on both FECH alleles). All patients should therefore be seen regularly throughout their lives by a porphyria specialist to monitor both erythrocyte protoporphyrin concentrations and liver function. Patients in whom liver function is found to be abnormal, or whose protoporphyrin concentrations are markedly increased, should be referred to a hepatologist for full assessment and liver biopsy. At this stage, treatment to halt the progressive liver damage caused by the increasing protoporphyrin concentrations should be instituted. This should include avoiding alcohol, measures to enhance protoporphyrin excretion such as oral cholestyramine, activated charcoal or bile acids (ursodeoxycholic or chenodeoxycholic acid). In patients with incipient liver failure, particularly those awaiting liver transplantation, this should be combined with measures to reduce protoporphyrin production using hypertransfusion or haematin infusion to suppress erythropoiesis. Finally, exchange transfusion and plasmapheresis can be used to reduce circulating protoporphyrin concentrations immediately prior to transplantation.
SECONDARY DISORDERS OF PORPHYRIN METABOLISM
Abnormalities of porphyrin production and excretion may occur as a secondary manifestation of a large number of disorders other than the porphyrias (Table 28.6). These secondary disturbances of porphyrin metabolism are usually mild, but together are far more common than the inherited porphyrias and may cause clinical and diagnostic confusion unless recognized for what they are.
TABLE 28.6
Secondary disorders of porphyrin metabolism
| Porphyrin abnormalities | Condition | Differentiation from porphyria |
| Increased coproporphyrin I and III in urine | Hepatobiliary dysfunction Chronic liver disease Alcohol, drugs Severe illness, infections |
Normal urinary PBG/ALA Normal plasma porphyrin Normal faecal porphyrins |
| Increased dicarboxylic porphyrins in faeces | Increased haem in gut: diet or bleeding | Normal faecal coproporphyrin Normal plasma and erythrocyte porphyrin Normal urine porphyrins |
| Increased erythrocyte zinc protoporphyrin | Iron deficiency Haemolytic and other anaemias Lead poisoning |
Normal or slightly increased erythrocyte free protoporphyrin Normal plasma porphyrin |
| Increased urinary ALA and coproporphyrin III | Lead poisoning | PBG normal or increased much less than ALA Increased blood lead |
| Increased plasma porphyrin | Chronic renal failure (particularly long-term haemodialysis) Cholestatic jaundice |
Normal faecal porphyrins |
Urinary coproporphyrin excretion may be increased in any condition that impairs hepatobiliary excretion, by ingestion of alcohol and drugs that induce cytochrome P450 isoforms, in poisoning by lead, other heavy metals and halogenated hydrocarbons, and in many severe illnesses, including infections. In normal individuals, coproporphyrin I predominates in bile, while the III isomer is excreted mainly in urine. As biliary excretion declines, the proportion of coproporphyrin I increases in urine. In alcoholism uncomplicated by liver disease, and in lead poisoning, urinary excretion of coproporphyrin III is increased. In Dubin–Johnson syndrome, coproporphyrin I comprises 80% or more of the total urinary coproporphyrin excretion, which is usually normal or near normal: this pattern has been regarded as diagnostic and distinct from the patterns found in Rotor syndrome and primary biliary cholestasis, which resemble those of other hepatobiliary disorders.
When total urinary porphyrin alone is measured, secondary coproporphyrinuria may suggest the presence of a porphyria but can readily be distinguished by demonstrating normal urinary PBG and ALA and faecal porphyrin excretion, with measurement of blood lead if increased coproporphyrin III or ALA suggests lead poisoning.
Haem is metabolized by the gut flora to protoporphyrin and related dicarboxylic porphyrins. Thus, even small increases in the haem content of the gut, which may not be large enough or occur sufficiently distally in the gut to produce a positive test for occult blood, may increase the total porphyrin content of faeces. Fractionation by HPLC shows that the increase is caused entirely by dicarboxylic porphyrins. Provided a dietary source of extra haem can be excluded, this finding may indicate alimentary tract bleeding of pathological significance.
Erythrocyte porphyrin concentrations are increased in iron deficiency, some other anaemias and in lead poisoning. In these conditions, the erythrocyte porphyrin is almost entirely zinc protoporphyrin and plasma porphyrin concentrations are normal. In some patients with iron deficiency, faecal porphyrin content may also be increased by gastrointestinal bleeding.
Plasma porphyrin concentrations are increased in cholestatic jaundice and in chronic kidney disease. Patients on long-term haemodialysis may develop a bullous dermatosis on sun-exposed skin that resembles PCT. Since PCT is also associated with end-stage kidney disease, and can be successfully treated by decreasing hepatic iron stores, it is important to distinguish between the two conditions. Plasma uroporphyrins and heptacarboxylic porphyrins are increased in both, though usually to a greater extent in PCT. Faecal isocoproporphyrin is increased only in PCT and its measurement is more reliable than plasma porphyrin analysis for identifying PCT in such patients.
CONCLUSION
The porphyrias are metabolic diseases characterized biochemically by the excessive production of porphyrins and their precursors as a result of defects in the enzymes leading to haem synthesis. Decreased haem synthesis leads to increased activity of the rate-limiting enzyme, 5-aminolaevulinic acid synthetase and accumulation of the substances involved in the pathway before the block. Porphyrias can be classified as acute (characterized by attacks of a predominantly neurovisceral nature) and chronic (characterized by photosensitivity). Some exhibit both characteristics. They can be distinguished by the pattern of porphyrin excretion, which is unique to each condition.
Further reading
Internet resources
British Porphyria Association. www.porphyria.org.uk
British patient support group website with links to other patient associations.
Cardiff Porphyria Service. www.cardiff-porphyria.org.uk
Website of the authors’ department with information about their services and a link to annually updated safe drug list.
The European Porphyria Network (EPNET). www.porphyria-europe.org
Website with details of specialist porphyria centres in Europe, information for patients in 12 different languages, information on diagnosis and treatment and regularly updated information on safe prescribing in the acute porphyrias.
University of Cape Town Porphyria Service. www.porphyria.uct.ac.za
Website of an expert group dealing with all porphyrias, with information for patients and professionals. Particularly useful section on patient management.
[/level-membership-for-endocrinology-diabetes-and-metabolism-category][not-level-membership-for-endocrinology-diabetes-and-metabolism-category]CHAPTER 28
The porphyrias
inherited disorders of haem synthesis
Michael N. Badminton; George H. Elder
CHAPTER OUTLINE
Biochemistry of haem synthesis
Molecular genetics of the porphyrias
PORPHYRIAS PRESENTING WITH ACUTE ATTACKS
The autosomal dominant acute porphyrias
Erythropoietic protoporphyria and X-linked dominant protoporphyria
INTRODUCTION AND OVERVIEW
The porphyrias are a group of eight metabolic disorders that result from inherited or acquired functional abnormalities of enzymes of the haem biosynthetic pathway (Table 28.1 and see Fig. 28.1, below). No disease has yet been associated with defects in the 5-aminolaevulinic acid (ALA) synthase-1 (ALAS1) gene that encodes the ubiquitous isoform of ALAS, the first enzyme of the pathway. However, gain of function mutations in the erythroid-specific ALAS (ALAS2) gene cause X-linked dominant protoporphyria (XLDPP), whereas loss of function causes X-linked sideroblastic anaemia.
TABLE 28.1
Overview of the porphyrias indicating inheritance, prevalence and main clinical presentation
AR, autosomal recessive; AD, autosomal dominant.
a Also known as PBG deaminase.
FIGURE 28.1 The haem biosynthetic pathway. The side chains are denoted by: A, acetic acid; M, methyl; P, propionic acid; V, vinyl. For other abbreviations, see Box 28.1.
The principal clinical features of the porphyrias are neurovisceral or cutaneous, or both. This chapter approaches the subject from a practical perspective, describing how and why patients present, how they are diagnosed and how patients and their families should be managed. A list of frequently used abbreviations is provided in Box 28.1.
Biochemistry of haem synthesis
Haem is essential for life and is synthesized in all cells, although the major sources are bone marrow (80%) and liver (15%). Haemoproteins include haemoglobin and myoglobin, which are the most abundant; the mitochondrial respiratory cytochromes; enzymes such as catalase and tryptophan pyrrolase, and the cytochrome P450 enzymes that are components of many essential metabolic processes, including the metabolism of xenobiotics.
The haem synthetic pathway comprises eight steps, each catalysed by a specific enzyme, of which the first and last three are mitochondrial and the remainder cytosolic (Fig. 28.1). The first enzyme, ALAS, catalyses the condensation of succinyl-CoA and glycine into 5-aminolaevulinic acid and is the rate-controlling step in all cells. Regulation of ALAS1 in the liver and other non-erythroid tissues is via inhibition by haem, the end product of the pathway, a characteristic that underlies both the pathogenesis and treatment of acute attacks of porphyria. In erythroid cells, regulation of haem synthesis is iron dependent. The second step is the synthesis of porphobilinogen (PBG), a watersoluble, colourless monopyrrole, catalysed by ALA dehydratase. The third step is the polymerization of four molecules of PBG to form the colourless linear tetrapyrrole, 1-hydroxymethylbilane (HMB), by the enzyme hydroxymethylbilane synthase (HMBS, also known as PBG deaminase). This linear molecule is cyclized into the tetrapyrrole ring structure, uroporphyrinogen III, by the enzyme uroporphyrinogen III synthase.
The porphyrinogens are colourless, non-fluorescent, unstable compounds, which rapidly oxidize to their red-purple porphyrin equivalents. Porphyrins absorb light, are fluorescent and therefore photosensitizing. These properties also make them relatively straightforward to measure in the clinical biochemistry laboratory. Uroporphyrinogen III, which is hydrophilic by virtue of its eight carboxy groups, is converted to coporphyrinogen III by uroporphyrinogen decarboxylase, which catalyses the sequential removal of four carboxyl residues. Two further carboxyl groups are removed in an oxygen-dependent dehydrogenation-decarboxylation reaction catalysed by the enzyme coproporphyrinogen oxidase (CPOX) to form protoporphyrinogen, which is then oxidized to protoporphyrin IX by protoporphyrinogen oxidase (PPOX). Progressive decarboxylation makes these precursors and the corresponding porphyrins increasingly hydrophobic, which determines their routes of excretion (see Fig. 28.2). The final step in the pathway is insertion of ferrous iron (Fe2 +) into protoporphyrin to form haem, catalysed by ferrochelatase (FECH). In the absence of iron, other divalent cations, such as zinc, may be inserted. Although only the III isomer can progress through the pathway to form protoporphyrin IX and haem, HMB may spontaneously cyclize into the uroporphyrinogen I isomer. This forms a substrate for uroporphyrinogen decarboxylase (UROD) and may be converted to coproporphyrinogen I, but is not metabolized further. Adult reference ranges for porphyrins and their precursors are shown in Table 28.2.
FIGURE 28.2 Examples of key laboratory findings that allow the porphyrias to be distinguished biochemically. (A) Plasma porphyrin fluorescence scans showing the three distinct emission peaks at 620 nm (AIP, CEP, PCT, HCP), 626 nm (VP) and 631 nm (EPP). A negative plasma scan (lower line) is also shown. (B) Fluorescence emission scan of whole blood allows zinc chelated protoporphyrin (proto) (increased in anaemia and lead poisoning) to be distinguished from free protoporphyrin, which is increased in EPP. Markedly increased zinc chelated and free protoporphyrins are characteristic for XLDPP. The lower line indicates a normal scan. High performance liquid chromatography (HPLC) analysis of faecal porphyrins allows three porphyrias with a plasma fluorescence emission peak at 620 nm to be distinguished (faecal porphyrin excretion is normal in AIP). (C) Normal faecal porphyrin HPLC trace indicating predominantly coproporphyrin (CI) isomer and dicarboxylic (D) porphyrins (protoporphyrin, pemptoporphyrin, deuteroporphyrin). (D) Faecal porphyrin pattern found in HCP indicating increased coproporphyrin, which is almost entirely the III isomer. (E) Faecal porphyrin trace seen in CEP indicating increased coproporphyrin isomer I. (F) Faecal porphyrin pattern seen in PCT patients indicating the increased excretion of partially decarboxylated intermediates; heptacarboxylic (H), hexacarboxylic (He) and pentacarboxylic (Pe) porphyrins as well as isocoproporphyrin (I) and dehydroisocoproporphyrin (Di), which are pathognomonic for PCT. U, uroporphyrin.
Overview of the porphyrias
Clinically manifest porphyria is always associated with detectable overproduction of haem precursors. Each enzyme deficiency, and the increase in activity in XLDPP, gives rise to a specific pattern of overproduction, which defines the corresponding disease (Table 28.3). Three clinical manifestations may occur: acute neurovisceral attacks, skin lesions or both. Acute attacks of porphyria are always accompanied by overproduction of ALA and, in all but ALA dehydratase deficiency porphyria (ADP), of PBG. Porphyrias causing skin lesions are characterized by overproduction of porphyrins. Four of the eight porphyrias can present with acute neurovisceral attacks: the very rare autosomal recessive ADP and the three autosomal dominant acute porphyrias, acute intermittent porphyria (AIP), hereditary coproporphyria (HCP) and variegate porphyria (VP). Hereditary coproporphyria and VP can present with skin photosensitivity or acute attacks, or both. In the other four porphyrias, two types of photosensitization may occur: accumulation of hydrophobic free protoporphyrin in erythropoietic protoporphyria (EPP) and XLDPP is associated with acute photosensitivity, while accumulation of the more water-soluble porphyrins in porphyria cutanea tarda (PCT) and congenital erythropoietic porphyria (CEP) leads to fragile skin and bullae.
TABLE 28.3
The main biochemical findings in symptomatic porphyria
Porphyrin analyses on random urine, faeces and EDTA-preserved blood allow the individual porphyrias to be distinguished. Individual urine and faecal porphyrins are separated and measured by high-performance liquid chromatography with fluorescence detection. Note that EPP cannot be diagnosed by urine porphyrin analysis alone.
a Emission maximum of plasma fluorescence peak (for examples, see Fig. 28.2).
Molecular genetics of the porphyrias
All the porphyrias, apart from the sporadic form of PCT, are single gene disorders that are inherited in autosomal dominant, recessive or X-linked patterns (see Table 28.1). Characteristics and chromosomal locations of individual genes are shown in Table 28.4. The HMBS and UROS genes are alternatively spliced to produce erythroid and ubiquitous isoforms. Disease-specific mutations that abolish or markedly decrease enzyme activity have now been identified in the genes for all the autosomal dominant porphyrias (Human Gene Mutation Database: www.hgmd.org). In most countries, mutational analysis has revealed extensive allelic heterogeneity, with large numbers of mutations identified in each gene, most of which are present in only one or a few families. The main exceptions are the W198X HMBS mutation in Sweden and the R59W PPOX mutation in South Africa. In both countries, these mutations have been multiplied by founder effects and account for the high prevalence of AIP in Sweden and of VP among individuals of Afrikaans ancestry in South Africa. The proportion of different types of mutation varies little between the diseases, with missense, nonsense, splice site and frameshift mutations contributing to the overall heterogeneity. Large deletions appear to be uncommon. Perhaps not surprisingly in view of the large number of different mutations, no clinically useful genotype-phenotype correlation has yet been identified in any autosomal dominant porphyria. With current methods of analysis, the sensitivity of mutation identification is over 95% in AIP and VP.
Incomplete penetrance is an important feature of all the autosomal dominant porphyrias; only a proportion of those who inherit a disease-specific mutation develop the disease. In this chapter, the terms latent or presymptomatic will be used to describe individuals who have inherited a porphyria gene but at the time of investigation have not had symptoms. Estimates of penetrance for the autosomal dominant acute porphyrias derived from family studies range from 10% to 40% and are influenced by age and rigour of phenotype definition. In the UK, about 80% of adults identified by family studies as carrying a gene for an acute porphyria never have an acute attack severe enough to require hospitalization. This figure is consistent with the observation that about 30% of patients with AIP present without a family history of the disease, yet family studies almost always reveal latent porphyria in their families, de novo mutation being uncommon. Studies of blood donors suggest that the gene for AIP may be present in about 0.06% of the general population. The low clinical penetrance of the acute porphyrias probably reflects a combination of environmental (see below) and genetic influences. The latter have not been identified but are likely to be at loci distant from the disease gene. Factors that determine clinical penetrance in familial PCT are described on page 544.
Clinical penetrance of the autosomal recessive porphyrias, ADP, CEP and EPP is close to 100%, although age at first presentation may be variable. They also show extensive allelic heterogeneity, with most patients whose parents are not consanguineous being compound heterozygotes. Other features of their genetics, and those of XLDPP, are discussed later in this chapter.
PORPHYRIAS PRESENTING WITH ACUTE ATTACKS
The autosomal dominant acute porphyrias
AIP, HCP and VP are autosomal dominant conditions that put the patient at risk of acute neurovisceral attacks, which may be life-threatening. In VP and HCP, skin lesions, indistinguishable from those of PCT, may occur either independently or in combination with acute attacks. In the UK, acute attacks of porphyria are estimated to affect about nine per million of the population, most of whom have AIP. Symptomatic VP is approximately half as common as AIP. HCP is the rarest of these acute porphyrias.
Pathophysiology of acute attacks
Acute neurovisceral attacks occur when hepatic haem synthesis is induced in the presence of deficient HMBS activity, resulting in an accumulation of the haem precursors ALA and PBG. In AIP, this is a primary deficiency, but in VP and HCP, it has been proposed that the deficiency is secondary to inhibition of HMBS by other metabolites in the haem pathway. The exact cause of neuronal damage has not been fully established, but current evidence from liver transplantation suggests that it is due to a hepatic neurotoxin, probably ALA. The neurological lesions comprise axonal degeneration and patchy demyelination of peripheral neurons with chromatolysis of anterior horn cells, brain stem nuclei and ganglia of the autonomic nervous system. There may also be diffuse neuronal loss and gliosis of the CNS. The result is damage to autonomic, motor and CNS neurons, giving rise to the characteristic clinical presentation, described below.
Clinical presentation of acute attacks
Acute attacks of porphyria are extremely rare before puberty and unusual after the menopause, having a peak occurrence in the third and fourth decades. Women are more frequently affected than men. In most patients, an attack will cease once the diagnosis has been made, appropriate treatment given and likely precipitants removed. However, particularly when there is delay in establishing the diagnosis, prolonged and very severe, life-threatening and sometimes fatal acute attacks may occur. Commonly ascribed precipitants include prescribed and illicit drugs, alcohol (particularly binge drinking), systemic infection and dieting either alone or in combination. In women, attacks may be related to the menstrual cycle, usually the luteal phase. However, in some cases no obvious aetiology is found.
An acute attack of porphyria normally starts with continuous abdominal pain, which becomes progressively more severe and is associated with nausea, vomiting and constipation. The patient may also complain of pain in the lower back, buttocks and inner thighs. The abdominal pain is typically diffuse, with no localizing signs or evidence of an acute abdomen on examination. The severity of pain appears out of keeping with the physical signs and often requires the administration of large parenteral doses of opiates. In the absence of an obvious cause, this may trigger concerns about opiate dependence among clinical staff who have no previous experience of the condition. Diminishing pain, particularly where no treatment has been given, does not always herald the end of an acute attack, and patients should be carefully monitored for the development of neuropathy. The autonomic neuropathy that results in the gastrointestinal symptoms also gives rise to tachycardia and hypertension in approximately two-thirds of patients. Patients frequently become dehydrated during an acute attack and may also develop hyponatraemia, which may worsen if inappropriate volumes of hypotonic intravenous fluid are infused. Plasma sodium concentration can fall quickly to extremely low values, precipitating convulsions, which occur in about 5% of patients. Seizures may also occur as a neurological manifestation of the acute attack, and present a particularly difficult management problem as many antiepileptic drugs are implicated as a cause of acute attacks. Rhabdomyolysis, which may lead to renal failure, is a recognized but rare complication of an acute attack.
Severe and untreated attacks can result in a predominantly motor peripheral neuropathy. In most cases, this is a symmetrical distal neuropathy resulting in wrist and foot drop, but occasionally there is progression to a flaccid paresis resembling Guillain–Barré syndrome and requiring prolonged ventilatory support. Prognosis for full recovery is excellent when attacks can be treated and halted. However, patients who require ventilatory support frequently experience a difficult recovery period, characterized by recurrent relapses triggered by the requirement for multiple prescribed drugs and complications such as infection. A small number of patients experience sensory changes such as dysaesthesia or hypoaesthesia in a similar distribution to the motor neuropathy. Other neurological signs reported include cranial nerve involvement including the optic nerve, which can lead to blindness.
The CNS is frequently involved. Acute mental changes are common and include anxiety, insomnia, confusion, hallucinations and paranoia, which resolve completely following the acute attack. There is no evidence that any form of chronic psychiatric illness is associated with any of the acute porphyrias. Less common CNS features are cerebellar syndrome, pyramidal signs, transient cortical blindness and altered consciousness.
Most patients have only one or a few attacks, with a major attack often being followed by two or three minor ones before full remission. However, about 5% of AIP patients suffer from frequent acute attacks, which in women may be premenstrual, and which may continue for several years (see p. 541). Obvious triggers are usually not found and the repeated admissions to hospital can severely affect the quality of life, particularly if patients have young families. Repeated admissions and high opiate requirement may also lead medical staff who are unfamiliar with the condition to question the diagnosis and the patient’s motives.
Chronic complications
Some patients develop a chronic pain syndrome. Pain is usually in the peripheries, often constant and may be triggered by minor stimuli. The cause of this is unknown; management is particularly difficult since it is important to avoid addictive analgesics. Treatment with haem arginate (p. 540) is usually of no benefit.
Studies from several countries have demonstrated an increased risk of hepatocellular carcinoma (HCC) in the absence of chronic liver disease in patients with an acute hepatic porphyria even when this is clinically latent. The risk is particularly high in Sweden, where a recent study found a standardized increased risk ratio of 64 for AIP gene carriers aged over 54 years and recommended that AIP patients over the age of 50 years should be screened annually for HCC by ultrasound examination. The benefit from screening in other countries has yet to be assessed. It has also been suggested that all HCC patients in whom no obvious cause is found should be screened for acute porphyria.
Renal impairment has been described as a complication of AIP, affecting particularly those patients who have previously suffered acute attacks. Hypertension is also common in these patients, but in many, the decline in renal function precedes the onset of hypertension, which may also be a consequence of the porphyria. Renal biopsies have shown glomerular sclerosis and interstitial fibrosis, but no evidence of inflammation or immunodeposits. A proportion of patients may progress to end-stage renal failure, requiring dialysis and/or renal transplantation. Patients who have had active disease should therefore have their renal function and blood pressure monitored regularly, and antihypertensive therapy should be instituted in an attempt to limit progression of renal impairment.
Diagnosis of acute porphyria
Diagnosis of an acute attack in a newly presenting patient requires the demonstration of increased excretion of urinary PBG in a fresh, random sample of urine that has been protected from light. An assay capable of quantitative or semiquantitative measurement of PBG should ideally be available in all acute hospitals. In practice, many non-specialist units use rapid qualitative screening tests, particularly out of hours, which have low sensitivity and poor specificity. All positive screening tests should be confirmed by a specific quantitative method with expression of results in relation to creatinine to correct for urine concentration. If urine PBG and ALA concentrations are normal during the early phase of an acute illness, all acute porphyrias, including ADP, are excluded as a cause of that illness. However, both PBG and ALA may return to within normal limits within a few days after the onset of symptoms in VP and HCP, and negative findings should be interpreted with caution when there has been a delay in collecting samples. However, in both these disorders, urinary and faecal porphyrins remain elevated for a prolonged period. Conversely, urinary PBG excretion usually remains elevated for many months or even years after an acute attack in AIP, and an increased PBG does not necessarily indicate an acute attack unless a marked increase from baseline can be shown to coincide with symptoms. Management of a clinically diagnosed acute attack should be started immediately, without waiting for confirmation of a positive screening test or determination of the type of acute porphyria.
Establishment of the type of acute porphyria requires analysis of plasma and faecal porphyrins (see Table 28.3; Fig. 28.2). Faecal porphyrin analysis is essential to distinguish HCP, in which coproporphyrin III accounts for most of the increased faecal porphyrin excretion, from AIP, in which faecal porphyrin excretion is normal or only slightly increased without any change in the coproporphyrin isomer ratio. Variegate porphyria can easily be identified by demonstrating a characteristic plasma porphyrin fluorescence emission peak at 625–628 nm. Enzyme analyses and genetic studies are not required for the diagnosis of new cases of clinically overt porphyria. Their use in family studies is detailed later in the chapter.
Laboratory monitoring of patients during and after acute attacks by regular measurement of porphyrin precursors is rarely indicated; treatment should be guided by clinical assessment. The one exception is where a severe attack has progressed to flaccid paresis and clinical assessment is difficult. In these circumstances, weekly monitoring may provide useful information on whether the condition is stable or deteriorating.
Management of an acute attack
Supportive treatment
As soon as the acute attack is confirmed, drugs or other recognized precipitants should be withdrawn. Safe and effective management of symptoms and complications, with support from an expert centre, is essential to minimize the stress; effective pain relief is a major component. This invariably requires opiates, and support from a specialist pain team can be very helpful as very high doses may be required. A phenothiazine may be used for anxiety and restlessness, and to decrease the opiate requirements. Antiemetics should be prescribed for nausea and vomiting. Adequate fluid and energy intake should be ensured, if necessary by intravenous infusions of 0.9% sodium chloride containing a minimum of 5% dextrose, with regular monitoring of electrolyte status in view of the risk of hyponatraemia. Where drug treatment of precipitants such as infection, coexisting conditions or other features of an acute attack, such as hypertension, tachycardia or convulsions, are required, care should be taken to select medications that are considered safe. Information on drug safety is continually under review and safe drug lists are likely to change regularly (see Welsh Medicines Information Centre: www.wmic.wales.nhs.uk). Where no safe alternative is on the list, an expert centre should be consulted for further advice on patient management. Information about specialist centres offering support in Europe is available from the European Porphyria Network (EPNET): www.porphyria-europe.org.
Specific treatment
[/not-level-membership-for-endocrinology-diabetes-and-metabolism-category]
