[level-membership-for-emergency-medicine-category]
Environmental Emergencies
Edited by Mark Little
28.1 Heat-related illness
Ian Rogers
Introduction
Heat-related disorders have a broad range of potential aetiologies and manifestations. In some, the primary disorder is a failure of thermal homoeostasis whereas, in others, the hyperthermia is secondary to other processes. The major heat-related illnesses to consider are exercise-associated collapse (EAC), heatstroke, neuroleptic malignant syndrome, serotonin toxicity and malignant hyperthermia. Although of different aetiologies, the latter four, all associated with significantly elevated core temperatures, share much common ground with regard to complications and treatment.
Epidemiology and pathophysiology
EAC is the most common heat-related illness presenting either to medical tents at sporting events or to emergency departments. EAC manifests at the end of a race when muscle pump enhanced venous return ceases and cardiac output drops. This leads to collapse, often with a brief loss of consciousness. Despite the claims of the advocates of fluid loading and sports drinks in exercise, the primary mechanism is a failure of prompt baroreceptor responses and not haemodynamically significant dehydration. Severe heat-related dehydration is rare.
The other, more serious, heat-related disorders are all associated with, or the potential for, significant hyperthermia which, if not treated promptly, results in similar pathophysiology at a cellular and organ system level. A core body temperature around or greater than 41.5°C results in progressive denaturing of a number of vital cellular proteins, failure of vital energy-producing processes and loss of cell membrane function. At a cellular level, the exact mechanisms leading to loss of cell membrane function and cell death in heat illness remain uncertain. At an organ system level, these changes may manifest as rhabdomyolysis, acute pulmonary oedema, disseminated intravascular coagulation, cardiovascular dysfunction, electrolyte disturbance, renal failure, liver failure and permanent neurological damage [1,2]. Any or all of these complications must be expected in severe heat illness.
The hallmark of heatstroke is failure of the hypothalamic thermostat, leading to hyperthermia and the associated additional pathophysiological features described above. Clinically, heatstroke can be divided into ‘exertional heatstroke’ due to exercise in a thermally stressful environment, and ‘classic heatstroke’, which occurs in patients with impaired thermostatic mechanisms. Common risk factors for heatstroke are listed in Table 28.1.1.
Table 28.1.1
Behavioural
Army recruits
Athletes
Exertion
Inappropriate clothing
Elderly
Inappropriate exposure
Babies left in cars
Manual workers
Pilgrims
Drugs
Anticholinergics
Diuretics
Phenothiazines
Salicylates
Stimulants/hallucinogens
Illness
Delirium tremens
Dystonias
Infections
Seizures
Certain drugs produce hyperthermia by mechanisms in addition to interference with thermostatic function. In severe serotonin toxicity and neuroleptic malignant syndrome, increased motor activity and central resetting of the hypothalamic thermostat combine to produce hyperthermia. In the case of serotonin toxicity, these effects are a consequence of a relative excess of central nervous system serotonin whereas, in neuroleptic malignant syndrome, dopamine depletion or dopamine receptor blockade is responsible.
The elevation of central nervous system serotonin in serotonin toxicity is usually associated with combinations of serotoninergically active drugs, taken either therapeutically or in overdose. The incidence of serotonin toxicity when such combinations are taken is not known, but is low and there is much individual variation in susceptibility [3]. Less often the syndrome is precipitated by a single serotoninergic agent. Drugs associated with the serotonin syndrome are listed in Table 28.1.2. The most commonly implicated are combinations of serotinergically active drugs, such as selective serotonin reuptake inhibitors (SSRIs), lithium, pethidine, monoamine oxidase inhibotors (MAOIs) and amphetamines.
Table 28.1.2
Drugs causing severe serotonin toxicity
Antidepressants
Monoamine oxidase inhibitors (MAOIs)
Selective serotonin reuptake inhibitors (SSRIs)
Selective serotonin and noradrenaline reuptake inhibitors (SSNRIs)
St John’s wort
Tricyclics
Analgesics
Pethidine
Tramadol
Recreational drugs
Amphetamines
Methylenedioxymethamphetamine (MDMA, ‘ecstasy’)
Neuroleptic malignant syndrome (NMS) is a rare idiosyncratic reaction to neuroleptic agents with an incidence of between 0.02% and 3.0%, depending on the diagnostic criteria used. It occurs in response to a single agent, usually at therapeutic dosage. In individuals, the occurrence may be dose related. Certain at-risk groups have been identified and are listed in Table 28.1.3.
Table 28.1.3
Risk factors for neuroleptic malignant syndrome
Patient factors
Agitation
Dehydration
Male sex (male:female=2:1)
Organic brain disease
Drug dosing factors
Depot neuroleptics
High initial neuroleptic dose
High-potency neuroleptic (e.g. haloperidol)
Rapid dosage increase
NB: Duration of drug exposure and toxic overdose are not related to risk of developing NMS.
Malignant hyperthermia is a genetically inherited disorder in which triggering agents cause a release of sarcoplasmic Ca2+ stores. The resulting elevation of myoplasmic Ca2+ stimulates many intercellular processes, including glycolysis, muscle contraction and an uncoupling of oxidative phosphorylation. This leads to hyperthermia that, in contrast to neuroleptic malignant and serotonin syndromes, is purely peripheral in origin.
Prevention
Prevention of exertional heatstroke should focus on the education of at-risk groups. Dehydration is not as important aetiologically in heatstroke as once thought. Exertional heatstroke is most often reported in shorter, high intensity exercise where marked dehydration is unlikely. So, although adequate fluid intake is needed for prolonged exercise, it is not a key factor in heatstroke prevention. As high ambient temperatures and high humidity predispose to exertional heatstroke, exertion in these environments should be limited.
Clinical features
Exercise-associated collapse
The clinical presentation of EAC will be familiar to all emergency practitioners as it mirrors that of poor cerebral perfusion from any other cause. Patients complain of nausea, vomiting, malaise and dizziness. There may be a history of collapse and there is likely to be a tachycardia and (orthostatic) hypotension. The orthostatic hypotension may manifest at the end of physical exertion by collapse with brief loss of consciousness that is typical of EAC. In this syndrome, and in distinction to heatstroke, the core temperature will be less than 40°C and neurological function will rapidly return to normal once the patient is lying down.
Heatstroke
The classic clinical features of heatstroke are neurological dysfunction, core temperature above 41.5°C and hot, dry skin. However, relying on this classic triad to make the diagnosis will result in a number of cases being missed. Loss of consciousness is a constant feature of heatstroke [1] but, by the time of ED presentation, conscious state may be improving, although some neurological abnormality will persist. Temperature readings may be misleadingly low, due either to effective pre-hospital care or to measurements at inappropriate sites, such as the oral cavity or axilla. Profuse sweating is a much more common feature than previously thought [1]. Other clinical features may include tachycardia, hyperventilation, seizures, vomiting and hypotension.
Serotonin toxicity
Serotonin syndrome is characterized by CNS, autonomic and motor dysfunction (Table 28.1.4). It is a clinical diagnosis. It develops after a latent period, which is normally a few hours, but may be as long as several days. The spectrum of illness produced is broad. Most patients are only mildly affected and may escape clinical detection. Only the most serious develop hyperthermia (usually in the setting of muscular rigidity) severe enough to produce the complications of rhabdomyolysis, disseminated intravascular coagulation and renal failure. Most cases will resolve within 24–48 h once the precipitating agents are withdrawn. Even in severe cases, the underlying biochemical abnormality rapidly improves, usually with the institution of muscular paralysis. The morbidity and mortality in these cases is caused by the complications that develop while the syndrome is active.
Table 28.1.4
Features of the serotonin toxicity
Central nervous system
Agitation
Anxiety
Confusion
Decreased level of consciousness
Seizures
Motor
Clonus
Hyperreflexia
Hypertonia
Incoordination
Myoclonus
Tremor
Autonomic
Diaphoresis
Diarrhoea
Hypertension
Hyperthermia
Tachycardia
Neuroleptic malignant syndrome
This syndrome manifests in patients who have recently been started on neuroleptic treatment or in whom the dose of a neuroleptic agent has been increased. It has been associated with almost all antipsychotics (both first and second generation) and has also been reported in patients in whom a dopaminergic agent has been rapidly withdrawn (e.g. in parkinsonism). There is a latent period of many hours to several days. Characteristically, there are four classic signs: fever, rigidity, altered mental state and autonomic instability. In practice, it may be difficult to distinguish clinically from serotonin toxicity unless a good drug history is obtained. As in serotonin toxicity, the spectrum of illness is very broad, with only the more severe cases developing hyperthermia and its complications [5].
Malignant hyperthermia
This occurs when a triggering agent is given to a susceptible individual, usually in the context of an anaesthetic. Triggering agents identified include inhalational anaesthetic agents, such as halothane, isoflurane and enflurane, as well as succinylcholine and ketamine. The first signs are failure to achieve muscle relaxation following succinylcholine, tachypnoea and tachycardia. If not recognized and treated, acidosis, rhabdomyolysis and hyperthermia will ensue. In some cases, signs and symptoms may be delayed or even reappear after apparently successful treatment, so that malignant hyperthermia may even present as a postoperative fever. Untreated, the mortality is as high as 70%, but this can be reduced tenfold by appropriate management.
Clinical investigations
Diagnosis of the hyperthermic disorders is based on the history, clinical picture and exclusion of alternative diagnoses. Investigations are thus directed towards excluding other possible causes of temperature elevation (e.g. infection, metabolic disorders) and evaluation of the specific complications of hyperthermia.
Patients with a presumed clinical diagnosis of EAC should still have serum electrolytes and creatine kinase measured to exclude exercise associated hyponatraemia and rhabdomyolysis, respectively. Should mental state not rapidly normalize with supine posture, then an urgent finger prick or serum glucose estimation is needed. Collapsed athletes should also have an ECG to identify unrecognized cardiac abnormalities.
All other heat disorders warrant a far more extensive laboratory and radiological work-up, as multiorgan system dysfunction is the rule [2,6]. Tests must include an ECG, serum electrolytes, disseminated intravascular coagulation (DIC) screen, liver function tests, muscle enzyme assays, renal function and urinalysis, serum glucose and a chest X-ray.
Treatment
Exercise-associated collapse
EAC responds rapidly to supine posture (ideally with the legs elevated), rest and oral fluids. Intravenous normal saline is rarely required as few athletes will be profoundly dehydrated. The use of ‘routine’ IV normal saline in collapsed athletes should be actively discouraged as it will worsen exercise associated hyponatraemia where there are persistent, inappropriate antidiuretic hormone levels.
Heatstroke
This is a true medical emergency. Early recognition and aggressive therapy in the field and in hospital can prevent substantial morbidity and mortality. The key management is aggressive cooling. Cooling rates of at least 0.1°C/min should be achievable. Several cooling methods have been proposed, including evaporative cooling, iced water immersion, ice slush, cool water immersion, iced peritoneal lavage and pharmacological methods [7]. A combination of methods is most widely used in EDs. All of the patient’s clothing should be removed and the patient sprayed with a fine mist of tepid water while gentle fanning is commenced (a ceiling fan is ideal). At the same time, areas with vascular beds close to the surface (neck, axillae and groins) should be packed with ice bags. This technique facilitates patient access and monitoring when compared to methods, such as ice-bath immersion even though an iced bath may offer more rapid cooling. Although ice-cold IV fluids can also aid in rapid cooling (as used in therapeutic hypothermia post-cardiac arrest), fluid requirements in heatstroke can be difficult to estimate and balance.
In hospital, shivering, seizures and muscle activity may need to be controlled with pharmacological agents, such as chlorpromazine, benzodiazepines and paralysing agents. Aspirin and paracetamol are ineffective and should be avoided. Intravenous fluids need to be used cautiously and may need titrating to central venous or pulmonary capillary wedge pressures. Maintain adequate oxygenation but avoid hyperoxia. Ventilatory support may be required. Urine flow needs to be maintained with initial volume loading, and later with mannitol or furosemide, to prevent secondary renal injury, especially from rhabdomyolysis. Electrolyte, acid–base and clotting disturbances should be closely monitored and treated by standard measures.
Serotonin toxicity
Treatment of the drug-related hyperthermia involves both specific pharmacological therapy and full supportive and cooling measures, as described above. The objective is to recognize and treat before serious complications occur. In mild cases of the serotonin syndrome, no treatment or small doses of benzodiazepines may be all that is required while awaiting spontaneous resolution. In severe cases, neuromuscular paralysis should be considered early, especially in cases of markedly altered mental state. The duration of treatment used is partly judged on the half-life of the presumed causative agents [4]. Specific antiserotoninergic drugs that can be used include chlorpromazine (12.5–50 mg IM/IV) and cyproheptadine (4–8 mg orally 8-hourly).
Neuroleptic malignant syndrome
Again, early recognition and full supportive care, combined with specific therapy, is the mainstay of treatment. Dopamine agonists, such as bromocriptine, may reduce the duration of the syndrome. It can be administered orally or by nasogastric tube at an initial dose of 2.5–10 mg tds.
Malignant hyperthermia
Dantrolene acts by inhibiting the release of calcium from the sarcoplasmic reticulum and is the specific agent used in the treatment of malignant hyperthermia. It should be given in addition to full supportive care and discontinuing the triggering agents. The dose is 2.5 mg/kg IV initially, repeated every 15 min up to a maximum of 30 mg/kg if needed.
Prognosis and disposition
In heatstroke, both the maximum core temperature and the duration of temperature elevation are predictors of outcome. Prolonged coma and oliguric renal failure are poor prognostic signs [1]. Mortality is still of the order of 10%, but most survivors will not suffer long-term sequelae [1,2]. Any patient with suspected heatstroke should routinely be referred to the intensive care unit for ongoing care. Most cases of EAC will be suitable for short-stay ED treatment or, indeed, simply for treatment on-site in an event medical tent.
Prognosis in the drug-related group of hyperthermia is dependent largely on the degree to which the complications have progressed before definitive and aggressive treatment is begun. Again, early referral to intensive care is indicated. Even with appropriate treatment, mortality for malignant hyperthermia approaches 7%. After recovery, the patient’s medication regimen will need to be reassessed although, in the case of neuroleptic malignant syndrome, it may be possible slowly to reintroduce a neuroleptic agent at a lower dose. With malignant hyperthermia, future anaesthesia will need to be modified to avoid precipitating agents. In addition, family members should be tested for susceptibility.
Controversies
28.2 Hypothermia
Ian Rogers
Introduction
Hypothermia is defined as a core temperature of less than 35°C. This can be measured at a number of sites (including oesophageal, right heart, tympanic and bladder). Rectal remains the routine in most emergency departments (EDs), despite concerns at how rapidly it equilibrates to and reflects true core temperature. Conventionally, hypothermia is divided into three groups: mild (32–35°C), moderate (29–32°C) and severe (<29°C) on the basis of measured core temperature. In a field setting, where core temperature measurements may not be possible, moderate and severe are often grouped together as they typically share the clinical features of absence of shivering and altered mental state. These categorization systems can be used both out of and in hospital as a guide to selecting rewarming therapies and prognosis. Mild hypothermia is considered the stage where thermogenesis is still possible; moderate is characterized by a progressive failure of thermogenesis; and severe by adoption of the temperature of the surrounding environment (poikilothermia) and an increasing risk of malignant cardiac arrhythmia. Nevertheless, there are substantial differences between individuals in their response to hypothermia.
Epidemiology and pathophysiology
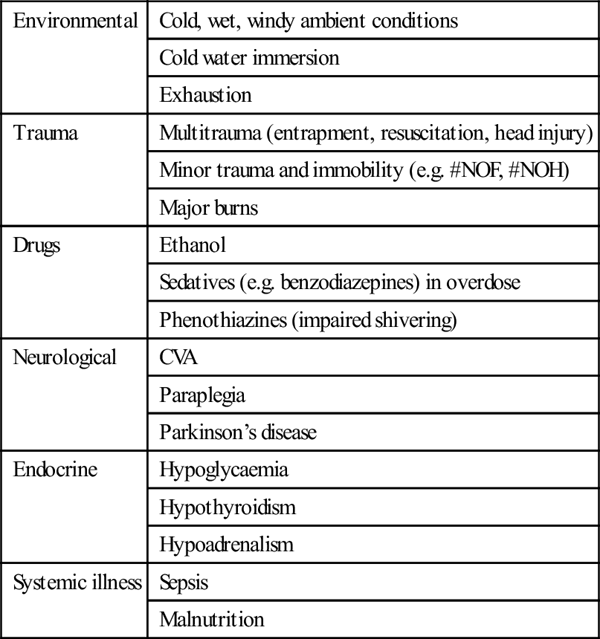
Hypothermia may occur in any setting or season [1]. True environmental hypothermia occurring in a healthy patient in an adverse physical environment is less common in clinical practice than that secondary to an underlying disorder. Common precipitants include injury, systemic illness, drug overdose and immersion and are outlined in more detail in Table 28.2.1. The elderly are at greater risk of hypothermia because of reduced metabolic heat production and impaired responses to a cold environment. Alcohol is a common aetiological factor and probably acts by a number of mechanisms, including cutaneous vasodilatation, altered behavioural responses, impaired shivering and hypothalamic dysfunction. Hypothermia in the ED setting is often associated with underlying infection [2].
Table 28.2.1
| Environmental | Cold, wet, windy ambient conditions |
| Cold water immersion | |
| Exhaustion | |
| Trauma | Multitrauma (entrapment, resuscitation, head injury) |
| Minor trauma and immobility (e.g. #NOF, #NOH) | |
| Major burns | |
| Drugs | Ethanol |
| Sedatives (e.g. benzodiazepines) in overdose | |
| Phenothiazines (impaired shivering) | |
| Neurological | CVA |
| Paraplegia | |
| Parkinson’s disease | |
| Endocrine | Hypoglycaemia |
| Hypothyroidism | |
| Hypoadrenalism | |
| Systemic illness | Sepsis |
| Malnutrition |
Clinical features
Despite substantial individual variations, it is still possible to describe the typical patient in each category of hypothermia. The clinical manifestations of hypothermia also depend on the features of the underlying aetiology.
Mild hypothermia manifests clinically as shivering, apathy, ataxia, dysarthria and tachycardia. Moderate hypothermia is typically marked by a loss of shivering, altered mental state, muscular rigidity, bradycardia and hypotension. In severe hypothermia, signs of life may become almost undetectable, with coma, fixed and dilated pupils, areflexia and profound bradycardia and hypotension. The typical cardiac rhythm of severe hypothermia is slow atrial fibrillation. This may degenerate spontaneously, or with rough handling, into ventricular fibrillation or asystole. In the field, moderate and severe hypothermia are often grouped together, with the key clinical feature of absent shivering suggesting the loss of the ability to rewarm without medical intervention.
Many complications may also manifest as part of a hypothermia presentation, although at times it may be difficult to separate cause from effect. These include cardiac arrhythmias, thromboembolism, rhabdomyolysis, renal failure, disseminated intravascular coagulation and pancreatitis.
Clinical investigations
Mild hypothermia with shivering and without apparent underlying illness needs no investigation in the ED.
Moderate or severe hypothermia mandates a comprehensive work-up to seek common precipitants and complications that may not be clinically apparent.
Biochemical and haematological abnormalities are frequently associated with hypothermia [1], although there is no consistent pattern. Blood tests that are indicated include sodium, potassium, glucose, renal function, calcium, phosphate, magnesium, amylase, creatine kinase, ethanol, full blood count and clotting profile. Arterial blood gases, if taken, should be accepted at face value, rather than adjusting for the patient’s temperature.
Impaired ciliary function, stasis of respiratory secretions or aspiration may be expected in moderate-to-severe hypothermia, so chest radiography should be routine. Other radiology may be indicated if a trauma-related aetiology is suspected.
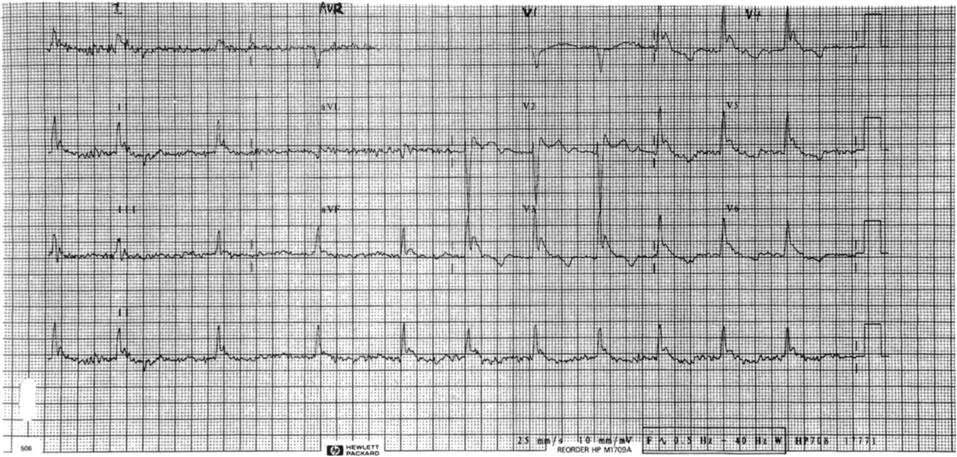
A 12-lead electrocardiograph and continuous ECG monitoring should be routine in moderate-to-severe hypothermia. The typical appearance is slow atrial fibrillation, with J or Osborn waves most prominent in leads II and V3–V6 (Fig. 28.2.1). The J wave is the extra positive deflection after the normal S wave and is more obvious and more commonly seen with increasing severity of hypothermia.
Treatment
General
The general and supportive management of hypothermia victims largely follows that of other critically ill patients. However, some syndrome-specific issues demand careful attention.
Muscle glycogen is the substrate preferentially used by the body to generate heat by shivering. All hypothermics, therefore, need glucose. In mild cases, this can be given orally as sweetened drinks or easily palatable food. With more severe hypothermia, gastric stasis and ileus are common and glucose should be given intravenously: 5% dextrose can be infused at 200 mL/h. Additional volume resuscitation with normal saline or colloid should be gentle, bearing in mind the contracted intravascular space in severe hypothermia and that hypotension that would be classified as severe at a core temperature of 37°C is a normal physiological state at 27°C. All intravenous fluids should be warmed to minimize ongoing cooling. Endotracheal intubation by a skilled operator is safe in severe hypothermia. Intubation is indicated as in any other clinical condition to provide airway protection or to assist in ventilation.
Ventilatory support and, where necessary, manipulation of acid–base status, should be titrated to maintain uncorrected blood gas pH and PCO2 within the normal range.
The slow atrial fibrillation so common in more severe hypothermia is a benign rhythm and requires no chemical or electrical correction. It will revert spontaneously with rewarming. Pulseless ventricular tachycardia and ventricular fibrillation should largely be managed along conventional lines. However, if initial DC shocks are unsuccessful, then others are unlikely to be so until the patient is warmer. Repeat countershocks are generally reapplied with every 1°C increase in core temperature. Magnesium may be the antiarrhythmic drug of choice in hypothermia.
The pharmacokinetics and dynamics of most drugs are substantially altered at low body core temperatures. Indeed, for many of the common drugs used in an ED they are unknown. Insulin is known to be inactive at<30°C. Hyperglycaemia, due in part to loss of insulin activity, is common in hypothermia, but should probably be managed expectantly until sufficient rewarming has occurred to ensure full endogenous insulin activity.
Rewarming therapies
Strategies for rewarming in hypothermia have only a limited evidence base on which to base recommendations. Although more invasive and rapid techniques are advocated for more severe hypothermia, there is little evidence to support this advice. The traditional concern of afterdrop (a paradoxical initial drop in core temperature with rewarming) is probably of little or no relevance in a clinical setting [3].
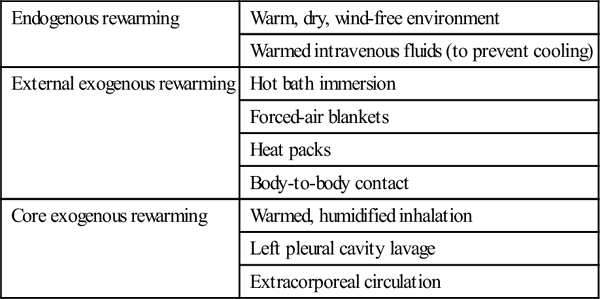
Rewarming therapies are broadly divided into three groups: endogenous rewarming, which is allowing the body to rewarm by its own endogenous heat production; external exogenous rewarming, which is supplying heat to the outside of the body; and core exogenous rewarming, which is applying the heat centrally. The classification of the commonly utilized rewarming therapies is outlined in Table 28.2.2.
Table 28.2.2
Rewarming therapy classification
| Endogenous rewarming | Warm, dry, wind-free environment |
| Warmed intravenous fluids (to prevent cooling) | |
| External exogenous rewarming | Hot bath immersion |
| Forced-air blankets | |
| Heat packs | |
| Body-to-body contact | |
| Core exogenous rewarming | Warmed, humidified inhalation |
| Left pleural cavity lavage | |
| Extracorporeal circulation |
Endogenous rewarming is a mandatory component of any emergency rewarming protocol. It consists of drying the patient, covering them with blankets, placing them in a warm and wind-free environment and warming any intravenous or oral fluids that are administered. Endogenous rewarming alone can be expected to rewarm at a rate of about 0.75°C/h. For most patients above 32°C (the level at which shivering thermogenesis is typically preserved), endogenous rewarming is the only therapy required. The exception is the exhausted patient in whom shivering has ceased at a core temperature higher than expected. Although more sophisticated techniques, such as bath immersion, will more rapidly rewarm a mildly hypothermic patient, there is no evidence that an increased rewarming rate improves prognosis in this group.
In moderate hypothermia, endogenous heat production is likely to fail progressively and more aggressive exogenous rewarming therapies are indicated. Hot-bath immersion has the theoretical disadvantage of causing peripheral vasodilatation, with shunting of cool blood to the core and convective heat loss. This might be expected to increase core afterdrop and produce circulatory collapse. In fact, rewarming rates of at least 2.5°C/h with minimal afterdrop have been achieved using baths at 43°C [4]. Nevertheless, substantial practical difficulties are obvious with monitoring a more seriously ill patient immersed in a bath. This method of rewarming can only be recommended for otherwise healthy patients who are expected to make a rapid recovery from accidental environmental hypothermia (e.g. immersion in very cold water).
The two therapies that have been best studied and are widely used in moderate hypothermia are forced-air rewarming and warm humidified inhalation [5]. Forced-air rewarming is achieved by covering the patient with a blanket filled with air at 43°C. These devices direct a continuous current of air over the patient’s skin through a series of slits in the patient surface of the blanket. This method produces minimal, if any, afterdrop, is apparently without complication and, combined with warm humidified inhalation, should produce rewarming at about 2.5°C/h. The value of warm humidified inhalation is probably by preventing ongoing respiratory heat loss. Given its widespread availability and lack of complications, it seems reasonable to combine it with forced-air rewarming in moderate hypothermia [6]. Body-to-body contact and chemical heat packs are often recommended as field treatments for all degrees of hypothermia. In mild hypothermia, it seems that the benefit of any heat they deliver is negated by an inhibition of shivering thermogenesis. In more severe cases, where shivering is absent, it may be that even the small amount of exogenous heat they deliver is beneficial, but this remains unproven.
In severe hypothermia, more aggressive exogenous rewarming therapies may be indicated in order rapidly to achieve core temperature above 30°C, the threshold below which malignant cardiac arrhythmias may occur spontaneously. Bladder, gastric or peritoneal lavage with warm fluids are all relatively ineffective methods of heat transfer and, as such, are not recommended for use in emergency situations. When available, full cardiopulmonary bypass achieves rewarming rates of about 7.5°C/h without core afterdrop. Pleural lavage using large volumes of fluid warmed to 40–45°C through an intercostal catheter may be nearly as effective. Both techniques have the advantage of delivering heat to the heart which acts as a heat pump to distribute rewarming to key core organs, but are clearly invasive and carry associated risks. These risks are certainly acceptable in a hypothermic arrest but, in the non-arrested patient, a slower rate of rewarming using forced-air and warm humidified inhalation may be more appropriate.
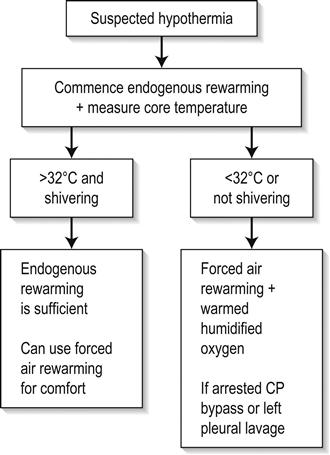
A suggested rewarming algorithm based on the evidence available to date is shown in Figure 28.2.2.
Prognosis and disposition
Attempts at developing a valid outcome prediction model for hypothermia are likely to be frustrated by its multifactorial aetiology. Recovery with appropriate treatment is likely from accidental environmental hypothermia when there is no associated trauma. To date, the coldest patient to survive accidental hypothermia neurologically intact had an initial measured temperature of 13.7°C [7]. Although increasing severity of hypothermia does worsen prognosis, the major determinant of outcome is the precipitating illness or injury. Reported mortality rates vary from 0 to 85%.
Mild hypothermics without associated illness or injury can be safely managed at home in the care of a responsible adult. Moderate hypothermia may be treatable in a short-stay observation ward, but often requires a longer inpatient stay to manage underlying illness or injury. Severe hypothermics are at risk of multiorgan system complications and should be considered for admission to an intensive care unit.
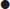
28.3 Dysbarism
David R Smart
Introduction
This chapter focuses on medical problems that develop secondary to breathing gases at higher than normal atmospheric pressure (dysbarism). This usually occurs in the context of scuba (self-contained underwater breathing apparatus) diving, a popular recreational activity in Australasia. Diving is generally very safe and serious decompression incidents occur approximately 1:10 000 dives. However, because of a high participation rate, between 200 and 300 cases of decompression illness are treated in Australia each year. It is estimated that 10 times that number of divers experience less serious health problems after diving. Emergency physicians are often the first medical staff to assess the diver after a diving accident and it is essential they understand the risks and potential injuries.
Diving physics and physiology
An understanding of pressure and some gas laws is essential to understand the pathophysiology of diving injuries. The air pressure at sea level is 1 atmosphere absolute (ATA). Multiple units are used to measure pressure (Table 28.3.1). For every 10 m a diver descends in sea-water, the pressure increases by 1 ATA. This pressure change impacts on gas spaces within the body according to Boyle’s law.
Table 28.3.1
Atmospheric pressure at sea level in various units
1 Atmosphere absolute (ATA)
101.3 kPa (SI units)
1.013 Bar
10 m of sea water (MSW)
760 mm of mercury (mmHg)
14.7 pounds per square inch (PSI)
Boyle’s law states that, at a constant temperature, the volume of a gas varies inversely to the pressure acting on it:
< ?xml:namespace prefix = "mml" />
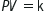
where P=pressure, V=volume and k=constant.
The proportionate change in volume is greatest near the surface (Table 28.3.2).
Table 28.3.2
Depth vs pressure and gas volume (Boyle’s law)
| Depth (m) | Absolute pressure (ATA) | Gas volume (%) |
| 0 | 1 | 100 |
| 10 | 2 | 50 |
| 20 | 3 | 33 |
| 30 | 4 | 25 |
| 40 | 5 | 20 |
Dalton’s law states that the total pressure (Pt) exerted by a mixture of gases is equal to the sum of the pressures of the constituent gases (Px, Py, Pz):
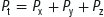
Therefore, as divers breathe air at increasing atmospheric pressure, the partial pressures of nitrogen and oxygen increase:

A diver breathing air at 40 m is inhaling a gas with a partial pressure of oxygen equivalent to breathing 100% oxygen at the surface. At partial pressures above 3 ATA, the PN2 affects coordination and judgement (‘nitrogen narcosis’). Oxygen may also become toxic at partial pressures greater than 1 ATA. Recreational scuba diving generally has a limit of 40 m because of these effects.
Henry’s law states that at a constant temperature the amount of a gas that will dissolve in a liquid is proportional to the partial pressure of the gas in contact with the liquid:
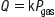
where Q=volume of gas dissolved in a liquid, k=constant and Pgas=partial pressure of the gas.
Henry’s law is relevant in diving illness because it is the basis of decompression illness (DCI). As the ambient pressure increases, the diver is exposed to increasing partial pressures of nitrogen, which dissolves in bodily fluids. The amount of nitrogen absorbed depends on both the depth (which determines the partial pressure of nitrogen) and the duration of the dive. Tissues also take up nitrogen at different rates depending on their blood supply and permeability. Eventually, the tissues become saturated with nitrogen and no further absorption occurs. As the diver ascends and ambient pressure decreases, the partial pressure of nitrogen in some tissues will exceed ambient pressure, resulting in tissue supersaturation. If the diver ascends slowly enough, nitrogen diffuses out of the tissues and is transported, safely dissolved in the blood, to the lungs for elimination. This is known as ‘off-gassing’.
If the diver ascends too rapidly, sufficient nitrogen bubbles will form in their body to cause decompression illness. Oxygen does not cause problems because it is rapidly metabolized by the tissues.
Barotrauma
Barotrauma occurs when changes in ambient pressure lead to expansion or contraction of gas within enclosed body cavities. The change in gas volume distorts or tears adjacent tissue. Injury by this mechanism may occur to the middle ear, inner ear, sinuses, lungs, eyes (via the diver’s mask) and, rarely, the gut. Different injury patterns occur in breath-hold divers (snorkellers) compared to those breathing compressed air. Both breath-hold and scuba divers may experience injury of the middle and inner ear, sinuses and eyes if they do not equalize pressures in the gas spaces as they descend. Breath-hold divers are unlikely to injure their lungs as their lung volumes reduce as they descend and return to their original volume as they ascend to the surface by the increasing ambient pressure.
Middle-ear barotrauma
Pathophysiology
Middle-ear barotrauma (MEBT), the most common medical disorder of diving, usually occurs during descent. Increased ambient pressure results in a reduction of middle-ear volume. If equalization of the volume via the eustachian tube is inadequate, a series of pathological changes results. The tympanic membrane (TM) is deformed inwards, causing inflammation and haemorrhage. Middle-ear mucosal oedema is followed by vascular engorgement, effusion, haemorrhage and, rarely, TM rupture.
Clinical features
Symptoms of middle-ear barotraumas include ear pain, tinnitus and conductive hearing loss. Mild vertigo may also be experienced. More severe vertigo and pain occur if water passes through a perforated TM. Severe vertigo and significant sensorineural hearing loss should alert the emergency physician to possible inner-ear barotrauma (IEBT) (see below). MEBT severity is graded by visual inspection of the TM (Table 28.3.3). An audiogram is useful to document any hearing loss.
Table 28.3.3
Grading of severity of middle-ear barotrauma
| Grade 0 | Symptoms without signs |
| Grade 1 | Injection of TM along handle of malleus |
| Grade 2 | Slight haemorrhage within the TM |
| Grade 3 | Gross haemorrhage within the TM |
| Grade 4 | Free blood in middle ear |
| Grade 5 | Perforation of TM |
Treatment
Treatment of MEBT consists of analgesia, decongestants and ear, nose and throat (ENT) referral if there is TM perforation or suspected IEBT. Antibiotics are indicated for TM rupture because of potential contamination with water. The patient should not dive again until symptoms and signs have resolved, any TM perforation has healed, and the eustachian tube is patent.
Inner-ear barotrauma
Pathophysiology
Sudden pressure changes between the middle and inner ears can cause rupture of the round or oval windows or a tear of Reissner’s membrane. This usually occurs during rapid descent without equalizing or forceful Valsalva manoeuvres.
Clinical features
Symptoms include sudden onset of tinnitus, vertigo, nausea and vomiting, vestibular symptoms and profound sensorineural hearing loss, which may not be apparent until the diver has left the water. Onset of symptoms after the dive while performing an activity that increases intracranial pressure (e.g. heavy lifting) suggests IEBT. Coexistent middle-ear barotrauma is absent in about one-third of cases.
The main differential diagnosis is DCI involving the inner ear or vestibular apparatus. Inner-ear DCI usually occurs on deep dives using helium and oxygen mixtures (heliox) and is typically accompanied by other symptoms or signs of DCI. Frequently it is difficult to distinguish between IEBT and vestibular DCI. Isolated inner-ear DCI has been reported in sports divers breathing air.
Treatment
Treatment of IEBT consists of avoidance of activities that increase intracranial pressure and urgent (same day) ENT referral for more detailed assessment and audiometry. Surgical repair may be undertaken when vertiginous symptoms are severe. Vomiting should be treated with antiemetics and the diver kept supine with their head on a pillow. If DCI is excluded, then a 45° semirecumbent position is preferred. If DCI cannot be excluded the diver should have a trial of recompression. In one series, exposure to pressure did not worsen the diver’s condition. The benefit of steroids in IEBT has not been confirmed.
It was thought that further diving was contraindicated after IEBT, but recent case data suggest that diving might be possible following full recovery of hearing.
External ear barotrauma
Ear-canal barotrauma is very rare and only occurs if there is a complete obstruction of the canal (usually by wax or ear plugs), creating a non-communicating gas cavity between the obstruction and the TM. Treatment is symptomatic. ENT specialist referral may be necessary if the TM cannot be visualized.
Sinus barotrauma
Pathophysiology
Mucosal swelling and haemorrhage occur if the communication of the sinuses with the nasopharynx is blocked and equalization of sinus pressure is not possible during descent. The frontal sinuses are most commonly involved.
Clinical features
Sinus pain usually develops during descent. Maxillary sinus involvement can refer pain to the upper teeth or cheek. There may be resolution of the pain at depth, due to mucosal oedema and blood filling the volume deficit left by gas compression. Pain and epistaxis may occur as the diver ascends. The pain usually persists after diving. Tenderness will be noted over the affected sinus. In doubtful cases, a sinus CT (computed tomography) scan will assist the diagnosis.
Treatment
Treatment includes analgesia, decongestants and recommendations to avoid diving until asymptomatic. Antibiotics may be required if secondary infection occurs.
Mask squeeze
If divers fail to exhale air into their masks on descent, the reduced volume inside the mask can cause pain, petechiae and conjunctival haemorrhage. In assessing these divers, it is important to confirm that they have normal visual acuity and fundi are normal. Treatment is with analgesia alone.
Gastrointestinal barotrauma
Expansion of gas within the gastrointestinal tract on ascent can occasionally cause colicky abdominal pain. Rupture of the stomach is rare but has occurred where panic or equipment failure has led to air swallowing and rapid ascent. Presentation is with abdominal pain and distension. Shoulder pain may be due to diaphragmatic irritation or coexisting DCI. Sub-diaphragmatic free air may be visible on an erect chest X-ray. The differential diagnosis includes pulmonary barotrauma, because air can enter the peritoneum via the mediastinum and oesophageal or aortic openings in the diaphragm. The diagnosis is confirmed with endoscopy and surgical repair is necessary.
Dental barotrauma
Severe tooth pain may occur with descent or ascent if air is trapped under a decaying tooth or recent filling. Percussion of the involved tooth is painful. Treatment is with analgesia and dental repair.
Pulmonary barotrauma
Pathophysiology
Breathing compressed air at depth, the diver’s lungs contain greater amounts of gas than they would on the surface. Divers are trained to breathe continuously during ascent or to exhale continuously if they have lost their air supply. Pulmonary barotrauma results when a diver ascends without exhaling adequately and the expanding gas in the lungs exceeds the lung’s elasticity, tearing alveoli. This occurs most commonly when a diver runs out of air, panics and ascends too rapidly. The change in pressure over 1 m near the surface is sufficient to cause lung barotrauma. It has been reported in student divers training in swimming pools and in helicopter escape training. It can also occur with a normal ascent if there is a localized area of lung that does not empty properly, as is possible in divers with asthma, reduced pulmonary compliance or air trapping.
The resultant clinical syndromes depend on the sites at which the air escapes and include arterial gas embolism (AGE), pneumomediastinum and pneumothorax.
Clinical features
Onset of symptoms is usually rapid. If pneumomediastinum or pneumothorax is detected after diving, it is essential to look for features consistent with associated gas embolism. These include impairment or loss of consciousness, cognition impairment including loss of memory, or neurological abnormalities. Sometimes the abnormalities are subtle and tests of cognition and memory should be performed in addition to a detailed history and thorough examination.
Treatment
If AGE is suspected, then the affected individual should be kept supine and urgent recompression treatment is required. Management of AGE is discussed under the heading of decompression illness. Lung barotrauma is regarded as the cause of AGE, however, in early studies, only about 5% of divers with AGE had radiographic evidence of a pneumothorax on plain chest X-ray. Subtle signs of extra-alveolar air suggesting pulmonary barotrauma are present in nearly half with more sophisticated imaging, such as CT.
The reverse also applies. If divers present with a pneumomediastinum or pneumothorax, then they may have up to 50% chance of AGE. The signs of AGE in these circumstances may be subtle with only a brief period of loss of memory or dizziness.
Pneumomediastinum and subcutaneous emphysema can usually be managed conservatively. If symptoms are severe, 100% oxygen can accelerate resolution of the trapped gas. If recompression is required for coexistent AGE, then the pneumomediastinum does not require any specific additional management unless a pneumothorax is present.
Isolated pneumothorax resulting from pulmonary barotrauma is very uncommon. Pneumothorax from pulmonary barotrauma should be managed in the same way as non-diving-related causes and recompression is not necessary. If recompression is required for coexisting AGE, a chest tube with a Heimlich valve should be placed before commencing treatment, because the size of any remaining pneumothorax will increase markedly on depressurization.
Once the acute management of pneumomediastinum and pneumothorax has occurred, the divers should be referred to a diving medical specialist for long-term follow up, because the conditions will impact upon their future diving fitness.
Decompression illness
Classification and criteria for diagnosis
Diving accidents involving bubbles were traditionally divided into decompression sickness (DCS; due to nitrogen bubbles coming out of tissue) and arterial gas embolism (AGE; due to pulmonary barotrauma releasing air into the circulation). DCS is then classified as type I or II. Type I DCS involves the joints or skin only; type II involves all other pain, neurological injury, vestibular and pulmonary symptoms.
In the 1990s, the term ‘decompression illness’ (DCI) was proposed to include both DCS and AGE, for the following reasons:
 AGE can be caused by arterialization of venous bubbles released from tissues
AGE can be caused by arterialization of venous bubbles released from tissues
 Pre-hospital and emergency management prior to recompression is identical
Pre-hospital and emergency management prior to recompression is identical
The current classification system describes DCI in terms of four components:

 evolution of symptoms (spontaneously resolving/static/progressive/relapsing)
evolution of symptoms (spontaneously resolving/static/progressive/relapsing)
 body system affected (musculoskeletal/cutaneous/lymphatic/neurological/vestibular/cardiorespiratory)
body system affected (musculoskeletal/cutaneous/lymphatic/neurological/vestibular/cardiorespiratory)
For example, a diver may be classified as having acute progressive neurological DCI with no evidence of barotrauma. The classification has been generally adopted in Australia and New Zealand, but not in North America. DCI is a satisfactory term from a management perspective but, from a scientific perspective, it does not describe differing aetiologies and pathophysiology.
Pathophysiology
DCI occurs if excessive nitrogen comes out of solution to form bubbles which gain access to the venous and lymphatic systems or if bubbles form within tissues themselves. The formation of bubbles requires tissues to be supersaturated with nitrogen and for ascent to be excessively rapid. As bubbles form in tissues, they distort tissue architecture, which results in impaired function, pain and inflammation and is probably responsible for most musculoskeletal symptoms.
Many bubbles entering the venous system do not cause symptoms. In fact, using ultrasonic detection methods, intravascular microbubbles are detected after approximately 60% of routine dives. It appears that these bubbles are safely filtered by the lung and diffuse into the alveoli.
Bubbles entering the arterial system are more likely to cause serious problems. This can occur under several circumstances. Large volumes of bubbles may overwhelm the pulmonary filter and arterialize. Bubbles may also bypass the lungs via a right-to-left shunt. Up to one-quarter of the population may have a patent foramen ovale (PFO). Under normal circumstances, the valve over the foramen is kept closed by the pressure difference between the left and right atria. However, during diving, the pressure differential may reverse during a Valsalva manoeuvre or with acute increases in right-sided pressures associated with a large pulmonary gas load.
Alternatively, gas can enter the circulation following pulmonary barotrauma. Air entering the pulmonary arterial system is carried to the pulmonary capillaries where it is trapped and reabsorbed by the alveoli. Air entering the pulmonary venous system, however, will pass through the heart and result in AGE.
Gas bubbles entering the circulation (either from tissues or barotrauma) cause both mechanical and biochemical abnormalities. Trapping in the pulmonary circulation may result in elevation of right heart and pulmonary pressures, leading to increased venous pressures, reduced cardiac output and impairment of tissue microcirculation. Arterial bubbles can cause end- organ ischaemia, although most pass through the capillaries and into the venous system. Most of the deleterious effects are a consequence of secondary inflammation of the vascular endothelium.
Bubble–endothelial interaction activates complement, kinin and coagulation systems and precipitates leucocyte adherence. This results in increased vascular permeability, interstitial oedema and microvascular sludging. The end result is ischaemia and haemoconcentration. Increased vascular permeability of the cerebral circulation will produce cerebral oedema. Vasospasm and reduced flow occurs approximately 1–2 h after bubbles have passed through the arterial tree. This explains the commonly observed clinical course of a diver with a cerebral AGE experiencing an initial deterioration (bubble emboli), followed by spontaneous improvement (bubbles pass through the cerebral capillaries) and then a subsequent secondary deterioration. Animal studies have demonstrated that bubbles travel against arterial flow because of their buoyancy and lodge in the highest point of the body (hence the logic of maintaining supine position after decompression accidents).
Prevention
A number of dive tables and computer algorithms have been developed in an attempt to avoid nitrogen supersaturation of tissues and improve diver safety. Limits are placed on depth, time and ascent rates to allow safe decompression after diving. However, as with all mathematical models which attempt to predict biological behaviour, the dive tables are far from perfect. One series has shown that 39% of DCI cases were within the limits of the table they were using and 24% within the limits of the conservative Canadian Defence and Civil Institute of Environmental Medicine (DCIEM) tables. Historically, it was assumed that DCI could not occur after dives shallower than 10 m, but it is now known that this can occur, particularly if there has been more than one dive per day or multiple ascents. The occurrence of DCI is a probabilistic event where risk increases with increasing depth, time, numbers of dives, numbers of ascents and rates of ascent.
Flying after diving can precipitate DCI. Even if there are no bubbles at the end of the dive, excess nitrogen remains in the tissues and is slowly off-gassed. Further reduction in ambient pressure at altitude can cause bubbles or enlarge pre-existing asymptomatic ones. Current guidelines advise against flying for 12 h after a single short no-decompression dive and 24 h following multiple or decompression dives.
Clinical features
Onset of any symptoms during or in the hours after diving should be regarded as DCI until proven otherwise. Failure to recognize and treat milder cases can lead to permanent morbidity because the disease can progress as the bubble load increases with time. Early onset of symptoms or signs (up to 1 h), especially those that are neurological in nature, indicates a serious decompression emergency and recompression is a time-critical treatment. Milder syndromes of decompression illness may develop up to 24 h after a dive or even later if there is a precipitant, such as heavy exercise or ascent to altitude (e.g. flying).
In general, pulmonary barotrauma that results in AGE has a dramatic clinical presentation and the onset of major neurological symptoms and signs occurs within seconds to minutes after the dive. DCI caused by intravascular bubbles from barotrauma can be rapidly fatal and has a mortality of 5% in sport divers who reach a recompression chamber alive. In Australia, it is the second most common cause of diving-related death after drowning. The brain is the organ most commonly affected, probably because of the vertical positioning of the diver on ascent. Cerebral gas emboli can cause sudden loss of consciousness, convulsions, visual disturbances, deafness, cranial nerve palsies, memory disturbance and asymmetric multiplegias. Hemiplegia is much less common than asymmetric multiplegias. Symptoms almost always begin within 10 min of surfacing. Sudden loss of consciousness on surfacing should be assumed to be due to cerebral gas emboli. Spontaneous improvement may occur with first aid measures, but relapse is common.
Coronary arterial emboli rarely may present as acute myocardial infarction or arrhythmia. Abdominal organs and skin may also be embolized. Elevation of serum creatine kinase (predominantly from skeletal muscle), serum transaminase and lactate dehydrogenase levels in divers with AGE suggests that emboli are distributed more extensively than previously recognized. Peak creatine kinase (CK) may be a marker of the degree and severity of AGE.
Onset of DCI due to gas bubbles coming out of solution can be equally as dramatic (especially after rapid ascents from deep dives), but frequently evolves over hours post-dive. DCI caused by bubbles released from tissues usually causes symptoms within 1 h of completing a dive and 90% of cases have symptoms within 6 h. Neurological symptoms occurring around 30 min after a dive have been associated with PFO. Common symptoms include profound fatigue, myalgia, periarticular pain and headache. Shoulders and elbows are the joints most commonly involved. The pain is usually a dull ache, which may initially be intermittent and migrate from joint to joint, but later becomes constant. Movement aggravates the pain, but local pressure with an inflated sphygmomanometer cuff may improve it. Paraesthesia and numbness may accompany the pain suggesting concomitant neurological disease.
Neurological DCI may present as personality change, headache, memory loss, visual defects, convulsions, confusion and altered level of consciousness. A flat affect may be the only symptom. The vestibular system can also be involved, with dizziness, vertigo, vomiting, nystagmus and ataxia.
Spinal-cord involvement occurs in up to 60% of cases of neurological DCI. The exact cause of spinal DCI is still debated. It may be a result of venous infarction of the cord due to obstruction of the epidural vertebral venous plexus. Other explanations include ischaemia and inflammation from bubble emboli or the formation of local bubbles within the spinal cord (autochthonous bubbles). Symptoms include back pain, paraesthesia and paraplegia, with bowel and bladder involvement. It is potentially disastrous to misdiagnose back pain coming on a few minutes after a dive as musculoskeletal pain and not consider spinal cord DCI.
If the bubble load overwhelms the pulmonary filter, a diver can present with a syndrome known as pulmonary DCI or ‘the chokes’. The symptoms of this syndrome include dyspnoea, pleuritic substernal chest pain, cough, pink frothy sputum, cyanosis and haemoptysis. It indicates the diver has sustained a large intravascular gas load, so a careful inquiry about other symptoms of DCI is mandatory and, if present, recompression is advised. Diving-related pulmonary oedema and salt-water aspiration syndrome are the major differential diagnoses.
A variety of rashes may be caused by cutaneous bubbles; however, these syndromes affect less than 10% of divers. The most common presentations are pruritus with no rash, a scarlatinaform rash with pruritus and cutis marmorata. Cutis marmorata begins as a spreading erythema but subsequently develops a marbled appearance of pale areas surrounded by cyanotic mottling.
Assessment of the injured diver
The injured diver requires simultaneous assessment and treatment. One hundred per cent oxygen treatment should be continued during the assessment. If the history suggests AGE, the patient should be kept in the horizontal position to avoid re-embolization. If symptoms are progressing rapidly, the examination should be brief but thorough so as to ensure rapid access to recompression. In serious cases, some of the historical information may be obtained once the diver is receiving treatment in the recompression chamber.
The diagnosis of DCI is made on history and examination. A full dive history must be obtained, in addition to the medical history. Important details include the number of dives over recent days, depth, bottom time (the time from beginning descent to beginning direct ascent), performance of any decompression or safety stops, dive complications, such as rapid ascents, surface interval between dives and the time interval between completing the dive and onset of symptoms. Previous dive experience, equipment used and gases breathed should also be recorded. A history of using surface supply equipment (the ‘Hookah’ apparatus) should alert the examining physician to the possibility of carbon monoxide poisoning and carboxyhaemoglobin measurement is required. Cold water, hard exercise during the dive, increasing age, multiple ascents and repetitive dives are predisposing factors in the development of DCI. Any exposure to altitude (>300 m) or heavy exercise post-dive should be recorded.
A thorough examination, particularly of the neurological system, to detect subtle abnormalities is required. It is also helpful to perform basic tests of cognitive function, such as the mini-mental state examination. For milder static DCI syndromes with delayed presentation, the sharpened Romberg test provides useful information. It is performed by asking the patient to stand heel-to-toe with open palms on opposite shoulders. The patient is stable. They are then asked to close their eyes and timed until they lose balance or achieve 60 s. A score of less than 60 s is suggestive of DCI in an injured diver. This test should not be performed if the history was suggestive of AGE or if there are neurological symptoms or signs.
Clinical investigations
Recompression should only be delayed for investigations if they will directly alter management. A full blood count and electrolytes are useful in that intravascular fluid depletion is common in severe DCI and the degree of haemoconcentration may correlate with eventual neurological outcome. Serum CK and liver function tests (LFTs) may be indicators of gas embolism; however, these do not influence clinical management. The blood glucose level should be checked in divers with impaired consciousness. A chest X-ray is indicated if pulmonary barotrauma is suspected, because a pneumothorax requires treatment before recompression. A dilemma occurs if the diver has a neurological presentation, because they should not be moved from the horizontal position until they are recompressed. If CAGE is suspected and CT is available, a supine CT scan of the thorax is preferable to a chest X-ray to diagnose pneumothorax or pneumomediastinum. Ultrasound in emergency may also be used to confirm a pneumothorax. Magnetic resonance imaging has no role in the acute investigation of DCI.
Treatment
First aid
One hundred per cent oxygen provides the maximum gradient for diffusion of nitrogen out of the bubbles. A large consecutive comparative series involving over 2000 divers has demonstrated that first aid oxygen significantly improves outcomes for divers with decompression illness. It should be administered in the pre-hospital setting and continue until and during recompression. Failure to improve on oxygen does not rule out DCI. Conversely, complete improvement on oxygen does not obviate the need for recompression. The diver should be supine or in the left lateral position if unable to protect their airway. Traditionally, the Trendelenburg position was advocated to reduce bubble embolization to the brain, but is now thought to increase the risk of cerebral oedema and should only be used if required to maintain blood pressure. The diver should be prevented from sitting or standing up, to avoid bubbles redistributing from the left ventricle to the brain. Initial resuscitation is along standard basic and advanced life support protocols. If intubation is required, the endotracheal tube cuff should be filled with saline prior to recompression to avoid a change in volume and a tube leak as ambient pressure increases.
Intravenous isotonic crystalloids should be commenced and titrated to response. Glucose-containing fluids are to be avoided because they may exacerbate CNS injury. Divers who present after several days with mild symptoms may be adequately managed with oral fluids. A urinary catheter should be inserted for spinal cord DCI with bladder involvement. Hypothermia should be corrected.
Retrieval
Long-distance retrieval can either be by air transport pressurized to 1 ATA or by portable recompression chambers. There is little debate that the longer the delay in recompression of severe DCI, the worse the outcome. However, Australian experience suggests that the number of cases where a portable chamber would have made a difference is so small that their use is unwarranted, largely because of the time required to prepare and transport portable chambers. Commercial aircraft are pressurized to 0.74 ATA (2440 m) and not appropriate to retrieve DCI patients, unless arrangements can be made to fly lower and pressurize to sea level. Road retrieval is not suitable over great distances or where an altitude of 300 m will be exceeded. Consultation with a hyperbaric physician should occur if retrieval is difficult.
Recompression
Recompression in a hyperbaric chamber is indicated even if the diver becomes asymptomatic with first aid, because otherwise many will relapse. The relapse may be more severe than the original presentation, due to the pathophysiological changes already initiated by bubbles in the microvasculature and tissues or redistribution of bubbles. Response to recompression is determined by time to recompression and the initial severity of injury. Recompression should always occur as soon as possible. It is particularly urgent for severe cases where treatment commenced later than 4 h after injury is associated with a poor response. Mild cases often respond despite longer delays to recompression.
Two types of hyperbaric chamber are available to administer recompression treatment:
Hyperbaric oxygen has the following beneficial effects:
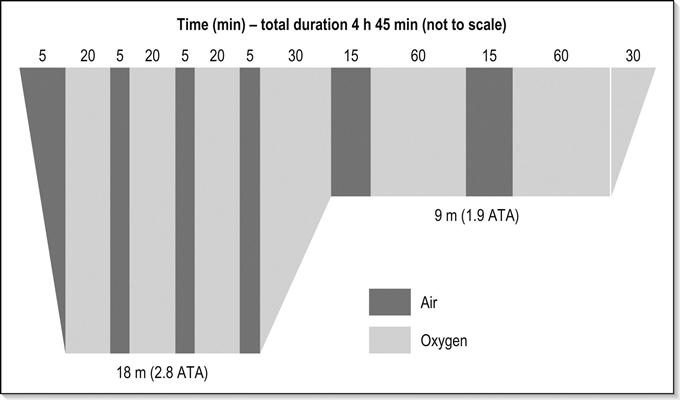
There are no published randomized trials comparing recompression protocols and hence no international agreement on how to manage DCI. The general consensus is that initial treatments should begin with a standard 18 m (2.8 ATA) table breathing 100% oxygen. Some studies have suggested a benefit from initially recompressing deeper, however, this procedure is not universally accepted and subject to considerable debate.
The identical Royal Navy 62 (RN62) and US Navy 6 recompression tables have become the standard of care for initial treatment of diving accidents in Australia and New Zealand. These are 18 m tables, lasting 4.75 h to 7.25 h (Fig. 28.3.1). Recompression is followed by gradual decompression. A response to treatment is usually evident by the second air break. If there is a partial response then there is the option of extending the table at 18 m. If there is minimal or no response and there is no doubt about the diagnosis, then it is reasonable to proceed to a deeper table (most units use the Comex 30 table). Because of the risks of oxygen toxicity at greater than 18 m, a combination of helium and oxygen (heliox) is used. Anecdotal evidence suggests that this technique is particularly effective for severe spinal cord DCI. In-water recompression is dangerous and difficult and should only be considered if retrieval is impossible. Hypothermia and oxygen toxicity pose serious risks during treatment and supervision by an experienced hyperbaric physician is essential.
Adverse effects of hyperbaric oxygen
Adverse effects of hyperbaric oxygen are uncommon. Even in non-divers, significant middle-ear barotrauma interrupting treatment occurs in 1/170 treatments. Claustrophobia is even rarer at 1/910 treatments.
The most serious adverse effect is oxygen toxicity and the attendant must continually watch for signs of its development. Toxicity is due to the formation of oxygen free radicals, which overwhelm the body’s antioxidants. It can affect the brain and the lung.
Cerebral oxygen toxicity can occur with brief exposures to 2 ATA oxygen and pulmonary toxicity may occur with prolonged exposure to 0.5 ATA or higher. The most common presentation of cerebral oxygen toxicity is muscle twitching, particularly of the lips and face. Other possible symptoms include apprehension, vertigo, visual disturbance, nausea, confusion and dizziness. If the oxygen is removed at this stage, progression to generalized convulsions may be avoided. Convulsions can, however, occur without premonitory symptoms. Treatment is as for any generalized convulsion, although removal of the oxygen will almost always stop it. Decompression should not be attempted during the convulsion as this may cause pulmonary barotrauma. Oxygen can be safely reinstituted 15 min after all symptoms have resolved. Predisposing factors to cerebral oxygen toxicity include fever, steroids, a past history of epilepsy and carbon monoxide poisoning. Incidence is directly proportional to time of exposure and inspired oxygen partial pressure. The incidence of convulsions in divers treated at 2.8 ATA on the RN62 is 0.56%.
Pulmonary oxygen toxicity manifests initially as an asymptomatic reduction in vital capacity, followed by cough and retrosternal pain. The symptoms usually abate when treatment is completed. Up to 10% reduction in vital capacity has been measured during extended treatments, which reverses within 24 h of completing treatment.
Adjuvant therapies for DCI
Recent research in the use of lignocaine infusions in patients undergoing open-heart surgery has demonstrated a significant benefit for the lignocaine group in terms of the incidence of postoperative neuropsychiatric abnormalities. The mechanism of injury in open-heart surgery is likely to be gas emboli and therefore provides a useful model for divers with AGE. There is now sufficient evidence to recommend a 48-h lignocaine infusion at standard antiarrhythmic doses to divers with unequivocal CAGE. A randomized clinical trial has demonstrated a reduction in symptoms after treatment for decompression illness if tenoxicam was administered to divers in the recovery phase. There was a reduction in total recompression requirements.
Prognosis after treatment
Relapses may occur after initial recompression and all neurological cases should be observed in hospital to allow immediate recompression if deterioration occurs. Further daily recompression is carried out until the patient stops improving or becomes asymptomatic and then one additional treatment is performed. Follow-up treatments are usually at 18 m, using either the RN61 table (18 m for 45 min, ascent to 9 m over 30 min, 9 m for 30 min then ascent over 30 min) or the 18:60:30 table (18 m for 60 min then ascent over 30 min).
Residual symptoms occur in up to 30% of cases and are more likely where recompression is delayed. Delays to treatment are not unusual with a mean time to recompression of 68 h in one series. There is no clear time interval after diving that recompression becomes ineffective. Many divers with DCI still respond to treatment even when delayed for 7–10 days and, therefore, any diver with unexplained symptoms after diving should be referred to a diving medicine specialist.
Flying after treatment and return to diving
Recommendations for flying and diving after treatment for DCI vary greatly and are not evidence based. Flying should be avoided for at least 1 week after treatment to avoid relapse. It is reasonable to permit resumption of diving after 4 weeks if there are no residual symptoms or signs. Because of the risk of recurrence, further diving is contraindicated if the DCI is thought to be due to pulmonary barotrauma, or where there are residual neurological signs or symptoms.
Other issues
Vertigo and headache in divers
These two symptom complexes are challenging to assess and diagnose. There are several possible causes of vertigo in divers. Vertigo developing while the diver is underwater is extremely dangerous: it can induce panic and lead to a rapid ascent. It can disorientate the diver so that they do not know which way the surface is and is often associated with vomiting.
The most common cause, alternobaric vertigo, begins just as divers commence their ascent and is caused by a unilateral pressure difference between the middle and inner ears. It usually lasts only a few minutes. Middle-ear barotrauma can also cause mild vertigo. Other causes include inner-ear DCI, inner-ear barotrauma and TM rupture. Any persistent vertiginous symptoms may indicate a more serious cause, such as neurological decompression illness or inner-ear barotrauma. Headache occurring during or after diving has a number of possible diving-related causes, such as sinus and mask squeeze, carbon dioxide accumulation, carbon monoxide toxicity, decompression illness, patent foramen ovale, ill-fitting wetsuits and marine envenomation. It is recommended that for all divers presenting with vertigo or headache, there is early consultation with a diving medicine specialist.
Oxygen toxicity
Cerebral oxygen toxicity in the diver underwater causes the same problems as in the hyperbaric chamber. Divers are more likely to develop toxicity underwater than in the chamber because immersion, exercise and carbon-dioxide retention increase the risk. The use of oxygen-enriched gases, such as nitrox, may increase the risk of cerebral oxygen toxicity. Enriched-air divers should ensure they stay at depths that maintain an oxygen partial pressure of less than 1.4 ATA.
Nitrogen narcosis
Described by Jacques Cousteau as ‘rapture of the deep’, nitrogen narcosis is due to the anaesthetic effect of nitrogen dissolved in lipid membranes. Symptoms are similar to those of alcohol intoxication. Some divers experience it at 30 m and almost all by 50. Loss of consciousness occurs at 90 m. This condition will not present to the emergency department because it is immediately reversible on ascent. However, it may result in other diving accidents, such as rapid ascent or near drowning. Divers planning to dive deeper than 50 m should use an alternative to air, such as heliox.
Gas contamination
Contaminants may be in the air before compression, added during compression or already in the tanks. Common contaminants include carbon dioxide, carbon monoxide and oil. Increasing partial pressures of the contaminant gases at depth may result in toxicity. Contamination is rare but must always be included in the differential diagnosis of injured divers, particularly those presenting with headache, shortness of breath or loss of consciousness at depth.
Diving-related pulmonary oedema
Pulmonary oedema in the diver may be caused by DCI, near drowning or immersion itself. Pre-existing cardiovascular disease, increasing age (>40), hypertension and beta blockade appear to be risk factors for immersion-induced pulmonary oedema. Symptoms often begin while the diver is still at depth, distinguishing it from DCI. There may also be episodes occurring when immersed but not diving (e.g. swimming). Treatment is supportive and recompression is not required provided DCI can be excluded. However, if detected, the occurrence of pulmonary oedema as a result of diving has long-term ramifications for future diving fitness.
Controversies
Important phone numbers
24-h services offering advice on management, retrieval and location of the nearest hyperbaric facility:
Australia
New Zealand
USA
UK
28.4 Radiation incidents
Paul D Mark
Introduction
In August 1945, the first atomic fission bombs were detonated above the Japanese cities of Hiroshima and Nagasaki with devastating effects. Most radiation incidents, however, have been accidental with the most serious occurring in 1986 at Chernobyl in the former Soviet Union when a nuclear reactor unit exploded, dispersing radioactive material over a wide area. One hundred and thirty six people developed the acute radiation syndrome, of which 28 died. A dramatic increase in thyroid cancers was observed among those who were either very young or in utero. The majority of incidents, however, have involved small numbers of people and many have occurred as a result of deliberate bypassing of safety procedures.
There were no deaths from exposure to radiation or cases of radiation sickness following the 2011 Fukushima accident but over 160 000 people had to be evacuated from their homes to ensure this.
Much of our knowledge of the long-term effects of ionizing radiation comes from the study of people who survived the Chernobyl incident and the atomic detonations over Japan.
In Australia, the Australian Radiation Incidence Registry records all accidents where exposures occur that are not ‘within the limits known to be normal for the particular source of radiation and for the particular use being made of it’. Very few accidents are recorded in which individuals received exposure or contamination significant enough to cause health concerns [1]. Strict licensing and control systems, coupled with improving technology and training, have helped to minimize the number of Australian radiation incidents.
The advent of terrorism might increase the risk of multiple casualty incidents, particularly from the use of radiation dispersal devices. Significant acute irradiation from such a device is unlikely, but victims may present with a variety of clinical syndromes and their management requires a multidisciplinary approach and close liaison between clinical staff and health physicists [2].
Decontamination is essential for all patients to avoid the delayed effects of continuing low-dose radiation.
Radiation sources and incidents
Worldwide, the most common radiation sources are:
With X-ray equipment and accelerators, the victim may be exposed to radiation but this does not make the tissues radioactive. These patients pose no threat to others, including medical attendants.
Unsealed radioactive material has the potential to cause radioactive contamination. This may be external on clothing or skin or internal following inhalation, ingestion or absorption through body orifices, mucous membranes and wounds. Following internal contamination, radioactive material may become incorporated into the patient’s tissues.
Other than for accidents involving nuclear processing and reactor plants, accidents usually lead to either exposure or contamination. Contaminated patients rarely suffer significant radiation exposure and exposed patients are seldom contaminated [1].
There are no nuclear reactors in Australia except for the occasional visiting nuclear powered warship. These vessels are closely monitored while in Australian ports.
The Fukushima reactors closed down when the earthquake struck in March 2011, however, the subsequent tsunami disabled the cooling systems. Following the approved release of a quantity of radioactively contaminated water, some seafood in the immediate vicinity of the power station has been found to exceed regulatory limits. The hydrogen explosion resulted in the release of a gaseous plume, which contaminated some milk and vegetable produce. The government enacted measures to prevent the distribution and sale of several foodstuffs and fish.
Terrorism
The most likely means for terrorist organizations to deploy radiation is a radiation dispersal device (RDD) or ‘dirty bomb’. These weapons use conventional explosives, such as trinitrotoluene, ammonium nitrate or other explosive material, to spread radioactive substances. A variety of substances could be used including americium, caesium, cobalt, iodine, phosphorus, plutonium, strontium, tritium and uranium. Only some of these are available in Australia.
RDDs are sometimes called ‘weapons of mass disruption’ because of the fear they engender in the population, multiple casualties, contamination of widespread areas and the economic cost [5]. Immediate injuries are generally the result of blast or thermal effects. Few contain sufficient material to cause acute radiation injury. Only those trapped near the site of detonation run this risk. However, radioactive material will be spread over a large area and many people might be exposed to the risks of low-dose radiation. Hospital staff treating the victims of RDD explosions are at negligible risk provided they wear appropriate protective equipment. Unlike surface burst nuclear weapons, RDDs do not cause fallout downwind of the detonation.
Radioactive material without the explosive component may constitute a radiation exposure device (RED) and could potentially be hidden in a crowded space, such as a theatre, where it could cause occult irradiation. Industrial sources are the most prevalent REDs in the civilian sector. An improvised nuclear device (IND), like a small nuclear weapon, produces blast, thermal and radiation energy; exposing people to high dose external radiation, inhalation of radioactive materials, particulate contamination and ingestion of radioactive materials in the food chain.
Measuring radioactivity
Radioactivity of an isotope is expressed as the average number of atoms that disintegrate per second. The Becquerel (Bq) is the SI unit for one nuclear disintegration per second. The activity of a given mass of a radioactive substance with a short half-life will decrease with time.
Ionization in air can be measured by portable dosimeters to give an estimate of the levels of radioactivity at the site of an incident. This is used to calculate the exposure level of a patient with acute radiation illness. The units used are Roentgens. Dosimeters are also used in hospitals to measure the level of radiation to which staff members have been exposed or to monitor patients during decontamination.
The absorbed dose of radiation is the amount of ionization energy deposited in matter by ionizing radiation. One Gray (Gy) is equivalent to one joule per kilogram. The effect of a given dose of radiation depends on the type of radiation emitted and the tissue type irradiated.
Type of radiation emitted
Different types of ionizing radiation transfer energy to tissue at different rates. The Sievert (Sv) is the international unit of effective radiation dose and is obtained by multiplying the absorbed dose measured in Gray (Gy) by a quality factor to reflect the different effects of each radiation type and their potential biological damage. For beta and gamma radiation 1 Sv=1 Gy. Alpha and neutron radiation deposit more energy in tissue so the quality factor is higher.
Alpha particles, composed of two protons and two neutrons, do not penetrate the dermis but may cause local damage if ingested, inhaled or absorbed through open wounds. Beta radiation, consisting of electron-like particles, travels about a metre through the air and is stopped by clothing. It often causes radiation injury to exposed skin. Gamma particles have no mass and are similar to X-rays, penetrating the body freely and causing the acute radiation syndrome if the trunk is involved. Neutrons are produced only during nuclear detonations and, while they can technically make an irradiated victim emit radiation, this is not clinically significant.
Grays are the preferred measure for determining acute effects while Sieverts are more useful in predicting chronic effects.
The average natural background radiation is 2 mSV per annum in Australia. The Australian National Occupational Health and Safety Commission’s standard for a worker is a maximum effective dose of 50 mSv in any year (or 20 mSv per year averaged over 5 years).
Pathophysiology
Radiation damages tissue both directly and indirectly by the production of free radicals from water molecules. Direct damage to cell membranes may cause changes in permeability and the release of lysosomes. Germinal, haemopoietic and gastrointestinal epithelial cells are relatively radiosensitive. The cells of bone, liver, kidney, cartilage, muscle and nerve tissue are relatively radioresistant. The delayed effects of radiation depend on whether the dose is lethal or sublethal to the tissue involved.
Lethal (deterministic) injuries are threshold dependent. Cells are killed when they receive a radiation dose, which varies with different tissues. Clinical expression occurs when the amount of cell killing cannot be compensated for by proliferation of viable cells. The acute and chronic radiation syndromes are deterministic. The earliest delayed effect of acute radiation injury, cataract formation at about 10 months, is an example of this type of injury.
For sublethal (stochastic) injuries there is no threshold level of radiation and the consequence is based on statistical probability. Sublethal injury to chromosomes is the most important effect of ionizing radiation. Double-strand breaks are not easily reparable, especially if the damage occurs simultaneously to both strands. This results in broken chromosomes with no template for repair. The exposed ends of chromosome fragments may join up at random, resulting in morphological chromosomal abnormalities. Sublethal damage to chromosomes is implicated in the development of tumours. Children are more prone to radiation-induced carcinogenesis. Although the incidence of malignancy in adults is increased by radiation exposure, the age at which malignancies are clinically expressed does not change. The estimated increase in lifetime risk of fatal cancer is 0.008% per millisievert of gamma radiation exposure [4]. Therefore, an individual who is exposed to 100 milligrays (twice the acceptable Australian occupational annual exposure) has a 0.8% increase in the lifetime risk of fatal cancer.
Radiation exposure to the gonads may produce temporary or permanent infertility in men depending on the dose. With temporary infertility, there is preservation of the secondary sexual characteristics. In the female, however, all ova are present at birth and larger radiation doses are required to produce sterility. Radiation-induced infertility in females is associated with premature menopause. Unlike animals, in humans, gonadal exposure to radiation does not affect future generations [8].
The fetus may receive less radiation than the mother when exposed to external radiation. However, when internal contamination occurs, it is possible for the fetus to receive a higher dose as material excreted in the urine collects in the maternal bladder. Exposures during organogenesis (weeks 3 to 7) may cause malformations. Exposure during weeks 8 to 25 causes decreasing IQ with increasing dose. There is a small increased risk of childhood cancers and possibly leukaemia.
Chronic radiation exposure
Chronic radiation exposure was first described in Russia following the exposure of workers in the plutonium enrichment programme to excessive doses of radiation over a period of time. Persons at risk have been exposed to radiation well above occupational health and safety standards for at least 3 years and have received a dose of 1 Gy or more to the bone marrow. Symptoms include sleep and appetite disturbance, easy fatiguability, impaired concentration and memory, vertigo, ataxia, paraesthesia, bone pain and hot flushes. Clinical findings include localized bone and muscle tenderness, tremor, hyperreflexia and underdeveloped secondary sexual characteristics. Investigations may reveal pancytopaenia and bone dysplasia. Following cessation of exposure, symptoms may slowly resolve.
Acute radiation exposure
Radiation exposure accidents usually involve penetrating radiation, such as high-energy X-rays or gamma rays. The effects are primarily due to the loss of cells in the body. Acute exposure is more dangerous than chronic, as it does not allow time for cell replacement or tissue recovery. Clinically, radiation exposure may produce a generalized acute radiation syndrome or a localized irradiation injury.
The acute radiation syndrome
The acute radiation syndrome refers to the effects of radiation on one or more body systems. The haemopoietic tissue alone is affected at doses of 1–4 Gy and produces pancytopaenia with its consequent risks of infection, bleeding and anaemia. Above 6 Gy, gastrointestinal effects are also manifest and the prognosis is poorer. The neurovascular syndrome occurs with doses above 20 Gy and is manifest by leaky capillaries, hypotension and a progressive decline in mental function with eventual death in weeks to months. The symptoms depend on the part of the body irradiated, the dose and the time over which it is delivered.
Clinical features
The course of the illness can be divided into four phases. The higher the dose, the shorter the duration of each phase and the more severe the symptoms:
The prodromal symptoms are due to the effects of radiation on cell membranes and the release of vasoactive amines. The symptoms are non-specific, with anorexia, nausea, vomiting, weakness, fever, conjunctivitis, erythema and hyperaesthesia. The time to emesis, presence of diarrhoea and duration of symptoms are markers of the severity of the exposure [5]. Vomiting, however, may be psychogenic.
The phase of manifest illness corresponds to the loss of cells. The haemopoietic syndrome occurs alone with whole-body radiation doses of between 1 and 4 Gy. It is due to loss of stem cells in the bone marrow. At these doses, some stem cells survive and recovery is therefore possible. The latent period lasts from 2 to 20 days and is followed by a rapid fall in the number of white blood cells and platelets. Recovery commences about 30 days after exposure, regardless of the exact dose.
The gastrointestinal syndrome predominates with radiation doses greater than 6 Gy. The prodromal symptoms are more severe. Early bloody diarrhoea suggests death within 2 weeks. The gastrointestinal symptoms recur during the manifest illness phase and can be very severe leading to dehydration and electrolyte imbalance. This syndrome is due to the loss of stem cells in the intestinal mucosal crypts. It is superimposed upon the haemopoietic syndrome with both occurring after a short latent period of under a week.
The neurovascular syndrome occurs with doses of greater than 20 Gy and is characterized by leakage of fluid into tissues and hypotension. The latent period is just a few days. Leakage into the brain causes neurological symptoms. These effects are superimposed on those due to gastrointestinal and haemopoietic damage. At very high doses, greater than 30 Gy, there is incapacitation usually within the first few minutes and certainly within 40 min. The effects are largely due to disruption of cell membranes and electrochemical inactivation of neurons. Death can be anticipated within hours. In a nuclear detonation, however, death from other injuries is more likely in those close enough to receive this level of exposure.
Whole body irradiation also produces visible changes in the skin. Hair epilation occurs at 3 Gy, erythema at 6 Gy, dry desquamation at 10 Gy and wet desquamation at 20 Gy. The erythema may come and go and occurs earlier with higher doses but rarely within 24 h.
Patients presenting after definite or presumed exposure to ionizing radiation can be triaged based on symptoms and lymphocyte counts. Less than 10% of people vomit if the radiation dose is less than 1 Gy, whereas most vomit if the dose is more than 2 Gy. Onset of emesis in less than 2 h suggests a dose of at least 3 Gy.
Treatment
The threshold for admission on initial presentation will depend on the number of casualties but, in general, patients who do not vomit within 6 h can be managed as outpatients. A useful triage tool for patients without other injuries or chronic illnesses utilizes a combination of the neutrophil/lymphocyte count and the presence of emesis at 4 or more hours post-exposure:
T=N/L+E, where E=0 if no emesis and E=2 if emesis.
If T is>3.7, the patient requires admission for further evaluation as the radiation dose is likely>1 Gy.
Supportive treatment includes maintenance of fluid and electrolyte balance, nutritional supplementation, antiemetics, such as ondansetron or granisetron, and antidiarrhoeals. Colony stimulating factors should be commenced as soon as possible if the radiation dose was>3 Gy and be continued until the lymphocyte count reaches 1000/mm3. Control of infection commences in the prodromal phase, with identification and aggressive treatment of any potential infection, so that the patient is in optimal condition to survive a period of manifest haemopoietic depression. To reduce the infection risk, patients may be kept home during the latent period and admitted to hospital when neutropaenia develops. Hospital management involves strict isolation and laminar airflow units. The prophylactic administration of antibacterial, antiviral, antifungal and antihelminthic therapy is reserved for the most severely neutropaenic. Non-absorbable agents are commonly used to sterilize the gastrointestinal tract. Anaerobic agents should be included if there is gut injury.
Management of neutropaenia follows the principles established in the management of bone marrow suppression secondary to chemotherapeutic agents. Fever is investigated and managed with empirical therapy in the first instance. If as many as 10% of the stem cells remain intact, the blood cells will repopulate. Platelet transfusion must be commenced early, especially if surgical procedures are required. The role of stem cell transplantation is evolving. Early reintroduction of enteral nutrition is important to maintain gastric acidity and prevent infectious organisms spreading from the gut to the respiratory system. Gastric acidity is maintained by avoiding antacids, H2 blockers and proton pump inhibitors: sucralfate is used for stress ulcer prophylaxis. Povodine-iodine or chlorhexidine is use for skin disinfection and shampoo. Meticulous oral hygiene must be maintained.
Clinical investigations
Acute radiation exposure is confirmed by laboratory investigation. A lymphocyte count of 1000/mm3 at 24 h suggests a dose of at least 2 Gy and the eventual development of the haemopoietic syndrome. A count of 500/mm3 suggests a radiation dose of 6 Gy and the subsequent development of both the gastrointestinal and haemopoietic syndromes. If lymphocytes disappear within 6 h, the dose is likely to be fatal.
Lymphocyte counts every 6–12 h for 48 h are useful for admitted patients further to refine the likely dose and clinical course [6].
The lymphocyte count may be less useful if there is significant concomitant trauma or at low levels of exposure. A dose-dependent increase in serum amylase is evident after 24 h.
Cytogenetic studies using blood collected at 48 h in a lithium heparin tube examine the number and structure of chromosomes. Radiation dose is reflected in the number of excess acentric and dicentric forms. T lymphocytes are relatively long-lived and reliable dose estimates can be made up to 5 weeks after collection of the sample. A newer method involves electron spin resonance of tooth enamel and can detect very low doses (0.1 Gy).
Prognosis
The LD50/60 is the dose at which half the victims succumb within 60 days. Without treatment, the LD50/60 is 4 Gy. With supportive care, antibiotics and colony stimulating factors, the LD50/60 is almost doubled up to around 7 Gy. Bone marrow transplantation may be used in patients exposed to 8–10 Gy.
Survival from the cardiovascular and neurovascular syndromes does not occur.
Combined injuries
Combined injury occurs when there is additional trauma, either physical or thermal, in addition to the radiation injury. The effects of the radiation exposure may become apparent earlier and may be more severe when other injuries are present. Healing of tissues, including callus formation at fracture sites, will be delayed even with subclinical radiation doses. Radiation exposure increases the probability of mortality when combined with other injuries or pre-existing conditions that result in immunosuppression, blood loss and danger of infectious complications. All administered blood products should be irradiated to remove the T-cell population and minimize graft-versus-host reactions. Platelets should be transfused if the platelet count falls below 20×109/L and, if surgery is anticipated, it should be maintained higher than 75×109/L. Emergency surgery, including the excision of dead tissue and the closure of wounds, should be completed within 48 h while some white blood cells remain. For thermal burns, early excision of potentially septic tissue and skin grafting are indicated. Wound closure is an important means of reducing vulnerability to infection. Non-urgent surgery should wait until any bone marrow suppression resolves.
Radiation pneumonitis may develop some time following the exposure and be confused with acute respiratory distress syndrome (ARDS).
Local irradiation injuries
The majority of local irradiation injuries occur when operators of X-ray diffraction units inadvertently place their fingers or hands in the direct X-ray beam. Other accidents have occurred when radioactive sources, often from industrial radiography equipment, are detached and then picked up and placed in the pockets of workers. There have been misadministrations of radiation to patients undergoing radiotherapy. The higher the dose, the greater the severity and the earlier the onset of the local injury. The smaller the area irradiated, the higher the dose required to produce a particular change.
Clinical features
Symptoms may include tenderness, itching, tingling and a changed sensitivity to heat and cold. Skin changes include epilation, erythema, dry desquamation, wet desquamation, blisters and radionecrotic lesions. If the area irradiated includes the epigastrium, nausea and vomiting may also occur. The degree of radiosensitivity of the skin depends on the thickness of the epidermis. The most sensitive areas are those that are also moist and subject to friction, such as the axillae, groins and skin folds. The least sensitive areas are the nape of the neck, scalp, palms and soles.
Erythema may not appear for some days. If it occurs within 48 h, the lesion will probably progress to ulceration. If irradiated skin appears normal at 72 h, the lesion is likely to be less severe but may still ulcerate in 1 or 2 weeks. Erythema may be delayed for up to 30 days. Pain is minimal unless ulceration occurs or the dose is extreme. Magnetic resonance imaging (MRI) and Doppler studies may help define the extent of the damage. Late effects include progressive tissue atrophy, fibrosis and chronic radiodermatitis with tissue breakdown. There may be stiffness and tenderness and decreased sensitivity to temperature change.
Treatment
Mild injuries may be simply observed. An effort should be made to protect the area from additional trauma. Topical corticosteroids may help. For more severe injuries, particularly with pain, local debridement and skin grafting may be necessary but should be delayed until the full extent of the lesion is known. Ideally, surgeons experienced in managing chronic vascular disease should be consulted. Amputation is reserved for gangrene. Skin grafts are indicated for areas of exposed cartilage or bone or for severe scarring. Topical antibiotics are often prescribed in an attempt to reduce infection. Vascular therapy with hyperbaric oxygen and pentoxifylline may be useful. In the long term, the irradiated area must be watched for the possible development of neoplastic change.
Occult radiation exposure
Occult exposure to radiation without an explosion might result in unsuspecting patients presenting with delayed symptoms. It should be in the differential diagnosis of the following, especially if associated with a 2- to 3-week prior history of nausea and vomiting:
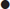
Contamination with radioactive material
The care of individuals who are contaminated with radioactive material requires similar preparation and precautions as for those contaminated with hazardous chemicals. Radioactive contamination has the advantage that it can be readily detected by instruments when on the skin. With the exception of Chernobyl, survivors of radiation accidents have not been sufficiently contaminated so as to pose a threat to emergency or hospital personnel using appropriate precautions and procedures.
Prevention
All staff using shielded or unshielded radiation sources in their daily work must be thoroughly trained in their safe use. Facilities using unshielded radioactive material must have procedures in place to deal with spillage and other accidents and all workers must be adequately trained in emergency procedures.
Preparedness
Emergency equipment must include appropriate monitors for detecting ionizing radiation or contamination, facilities for decontaminating victims and plastic bags for biological and other samples. Appropriate blocking or chelating agents should be stocked at the facility. Emergency planning must include early warning of the receiving hospital so that adequate preparations can be made prior to the arrival of patients.
Scene management
For incidents involving small numbers of patients, members of the rescue team should put on the protective clothing normally used by personnel working with radioactive material at that site. This includes gloves, facemask and cap. Gowns may be covered with large plastic aprons to make them waterproof. Additional measures, such as taping plastic bags over shoes, may be used if the normal protective clothing is judged inadequate. The implementation of life-saving procedures may make it necessary to forgo some of this protection. Contamination of the rescuer will be low and decontamination can be carried out later.
Serious illness or injury is not due to radiation per se and should be treated on its own merits. Unless the patient’s condition is serious, external decontamination begins at the scene so as to minimize internal contamination and incorporation of the radionuclide into the body tissues and to reduce the risk of contaminating other persons and the hospital environment. As much as 80% of contaminating material may be on the clothing [7]. Accordingly, the victim’s outer clothing should be removed at the earliest practicable stage. If monitoring is not available, it should be assumed that all outer clothing is contaminated. Clothing is cut from head to toe and down the sleeves, folded back over itself as it is cut and then rolled up. The person removing the contaminated clothing must wear protective clothing and limit contact with the outside of the victim’s clothing. The victim is then wrapped in plain sheets and transferred to hospital. If small contamination spots on the skin cannot be easily removed at the scene, they should be dressed and the victim transported to hospital.
At larger incidents, it may also be necessary to establish a controlled area, the periphery of which is located just beyond the region where contamination is detected above background levels. Rescue team members should wear the maximum level of personal protective equipment available. This should be removed at the perimeter of this area prior to both patient and rescuers leaving. Monitoring of all personnel leaving the area should be undertaken if facilities are available.
Portable vacuum units with high efficiency particulate air filters have reportedly been used to facilitate rapid decontamination outdoors.
Emergency department
The elements of planning for the management of radiation accident patients are similar to those for other types of emergencies, namely prevention, preparedness, response and recovery.
Facilities using unsealed radioactive sources should be identified in advance. These include nuclear medicine departments, scientific laboratories and nuclear facilities. An emergency department (ED) response plan should be developed and emergency response team membership designated. Equipment for monitoring, decontamination and contamination control should be in place. Regular practice is essential [8].
A decontamination area must be designated and be itself capable of adequate decontamination. Ambulant patients and lower acuity stretcher bound patients should be decontaminated outside the ED. Waste water may be legally discharged into normal draining systems if it does not exceed specified limits. In the clinical setting of a few patients, this is unlikely. Incidents involving contaminated or possibly contaminated patients rapidly deplete a receiving hospital’s emergency response. If multiple patients with possible contamination are being managed, the hospital may need to defer where possible the arrival of other patients.
Hospital protocols should include plans for dealing with relatives, the press and the public. The timely release of appropriate information is important. Persons issuing this information should be well versed in radiation medicine, as the avoidance of questions and confusion in answers may generate public uncertainty and panic. Security personnel will be required to restrict the entry of unauthorized persons to the treatment area.
Decontamination process
Life-saving procedures resulting from trauma or burns should take priority over consideration of the radiation aspects of the patient’s condition, even if preparations to minimize the spread of contamination have not been completed. A radiation physicist with appropriate monitoring equipment should be present in the ED. However, if patients arrive before monitoring is available, treatment of severe injury should proceed immediately and subsequent decisions regarding decontamination should be based on the patient’s likely exposure.
In the ideal situation, all patients should be monitored at triage and, if found to be contaminated, those without severe injury should be showered and re-monitored prior to admission to the ED. This is especially so if whole-body contamination has occurred, for example from a gaseous plume from a reactor accident [1]. Washing starts with the hair and works downwards. Patients should bend forward while washing their hair so that any contamination is not washed into their eyes, nose or mouth. Wounds should be covered with a waterproof dressing before showering to avoid washing contaminated water into them.
Because some patients with severe injury will require immediate admission to the ED, adequate preparations are necessary. The floor of the entry and some treatment cubicles should be covered with plastic and any non-essential items removed. Access to this controlled area must be strictly supervised and there should preferably be a buffer zone. Disposable fluid-repellent gowns are ideal but surgical gowns covered by plastic aprons are satisfactory. Lead aprons as used in X-ray departments are not satisfactory; these prevent exposure but not contamination and are heavy and hot to wear. Plastic bags are taped over the shoes and the cuffs of overalls should be taped and secured to the outsides of overshoes. Face-masks are required to protect against airborne contamination but they do not protect the face from being touched by contaminated hands. N95 masks may be superior to standard surgical masks [9]. Trauma masks with clear plastic visors are the best option. Two pairs of gloves should be worn. The inner ones should be surgical gloves taped to the sleeves. The outer gloves are not taped down and should be changed frequently. Hair cover is desirable. Rubbish bins lined with garbage bags serve as waste receptacles and should be emptied promptly to minimize the amount of radiation in the department.
Once the patient is in the controlled area, all clothing should be removed and other medical conditions assessed and treated. Blocking agents can be administered if they have not already been given. All mucosal surfaces should be swabbed to aid in the assessment of likely internal contamination. These include nostrils and ears, the mouth and rectum. The swabs should be placed in sealed labelled plastic bags and sent for radiation assessment and identification of the chemicals involved. Blood samples should be drawn for a baseline complete blood count, differential and absolute lymphocyte counts and later cytogenic analysis. A serum amylase is also important as the parotid is very sensitive to radiation.
External decontamination utilizes the principles of barrier nursing and contamination control. Staff should stand back from the patient except when actually examining them or performing procedures. Radiation exposure is inversely proportional to the distance from the source squared. Hospital personnel should be rotated during the decontamination procedure to minimize the perceived risk to any one individual. Pregnant staff should not be involved. Each staff member should shower following completion of their turn in decontamination.
The priority areas for external decontamination are wounds and orifices, as it is through these that the risk of subsequent internal contamination is greatest. Other priority areas include the hands, face and head, as early contamination removal reduces spread. Decontamination of intact skin is the last priority.
Wounds are decontaminated in the same manner as when removing dirt or bacteria. Deeper wounds should be opened up and thoroughly irrigated. Burnt areas also should be carefully irrigated. Metal fragments should be removed with forceps. Deep debridement and excision of a wound is rarely necessary in extreme cases where highly toxic material is embedded in the tissues. Decontamination efforts should continue until the radiation level is at background levels or there is minimal reduction with further washing.
The mouth is decontaminated by gentle irrigation and frequent rinsing with 30% hydrogen peroxide solution. Brushing of the teeth with toothpaste is helpful, as toothpaste contains chelating agents. External ear canals should be irrigated and nasal douches can be effective. The eyes are rinsed by directing a stream of water or saline from the inner canthus to the outer canthus, so that material is not forced into the lacrimal duct. Hair should be shampooed several times with the head deflected backwards over a basin to keep water from the eyes and ears. A hair dryer is used to dry the hair. Clipping of hair may occasionally be necessary.
The skin is washed initially with warm water and mild soap. If this is ineffective, 0.5% hypochlorite or stronger detergents can be used. If the skin becomes damaged or red and sore, cleansing should be discontinued. If contamination is only discovered after patients are admitted to an ED, the entire area through which they have passed should be taped off, surveyed with the help of a radiation physicist and, if necessary, decontaminated. Staff should put on protective clothing and remove nearby patients so as to create a spacious treatment area. Following a radiation incident, all equipment, instruments and work areas used in treating contaminated patients must be thoroughly cleaned.
Monitoring decontamination
Radiation physicists should check the background level of radiation in the ED from time to time so that they have a baseline from which to assess each patient’s exposure. Scanning should occur slowly to avoid missing radiation. Headphones should be used or the sound turned off to avoid alarming patients.
Internal contamination
Internal contamination causes no acute clinical effects and it is usually not feasible to confirm its presence before commencing treatment directed at the reduction of absorption, prevention of incorporation into tissues and promotion of elimination. Significant internal contamination has traditionally occurred through wounds or body orifices in small-scale accidents. It could readily occur on a wider scale following the explosion of an RDD, a reactor accident or a nuclear detonation. Absorption would be by inhalation of contaminated air and/or ingestion of foodstuffs contaminated by fallout. Radionuclides which have short effective half-lives, such as technetium used in nuclear medicine (t1/2=5 h), pose no danger. For isotopes with effective half-lives measured in days, the decision to treat will depend on the likely intake especially via the lungs, whether the drug is concentrated in tissue, such as iodine in the thyroid or uranium or americium in bone, whether the emission is high energy as with cobalt and whether the chemical itself is toxic. The effective half-life combines radioactive and chemical properties and describes the rate of elimination without decontamination.
Table 28.4.1 describes the radioisotopes most likely to be available in Australia, their common uses, emissions, toxicity, effective half-life and treatment.
Table 28.4.1
Isotopes likely to cause internal contamination in Australia
| Element | Emissions | Primary toxicity | Effective t1/2a | Common use | Detectionb | Absorption | Treatment |
| Americium241 | Alpha Gamma | Marrow suppression | Years | Smoke detector | Yes | Lung, skin | DTPA or EDTA IV |
| Caesium137 | Beta Gamma |
Whole body irradiation | 70 days | Medical radiology | Yes | Lung. GI tract, wounds | Prussian blue orally |
| Cobalt60 | Gamma Beta |
Whole body irradiation | 10 days | Medical radiology Commercial food irradiation | Yes | Lung | Penicillamine orally |
| Iodine131 | Beta Some gamma |
Thyroid | 8 days | Nuclear medicine therapy | Yes | Lung | Iodine orallyc |
| Tritium3 | Beta | No significant hazard | 12 days | Signs | No | Lungd | Increase fluids |
| Uranium235/238 | Alpha | Kidney | Can be permanent in bone | Fuel rods for reactorse | Yes | Lung | NaHCO3 Tubular diuretics |
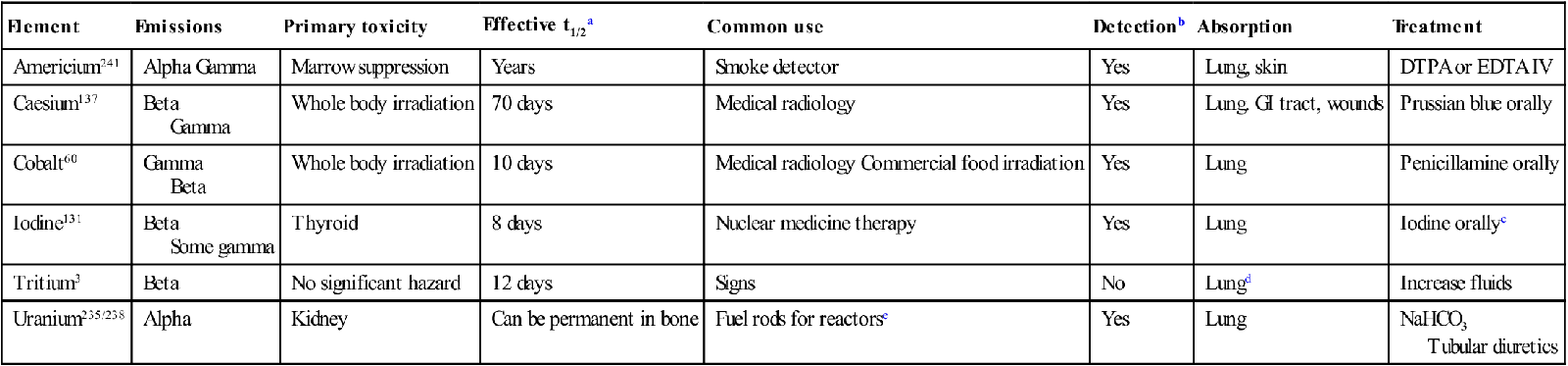
bDetection by standard radiation detection equipment.
cIodine dose in adults 130 mg daily.
dTritium is not a significant radiation hazard except perhaps in closed spaces.
eNatural and depleted uranium are not serious irradiation threats. GI: gastrointestinal.
To assist in the determination of the extent of internal contamination a 24-h urine sample should be collected. If gastrointestinal contamination is suspected a 24-h stool sample should also be collected.
Selection of the appropriate technique or drug depends on knowledge of the radionuclide involved and its physical form [10]. For example, uranium is found in order of increasing radioactivity in depleted uranium used in artillery shells, natural uranium, fuel rods and weapons grade enriched uranium. The first two are not significant radiation hazards but the latter two can emit significant levels of gamma radiation if sufficient quantity is present.
Uptake by the various organs can be reduced by the use of blocking agents, dilution techniques or chelating agents.
Administration of stable iodine in the form of potassium iodate or potassium iodide tablets will reduce uptake by the thyroid gland by up to 90% if given less than 2 h after intake and by about 50% if in less than 3 h. Chelating agents and mobilizing agents may be useful for up to 2 weeks. Mobilizing agents, such as antithyroid drugs, increase the natural rate of turnover of a biological molecule and thereby increase excretion. Gastrointestinal decontamination is unusual but an enema might be used to empty the bowel. In the absence of external contamination, this would be the only circumstance in which internal contamination posed any risk to hospital staff.
Likely developments over the next 5–10 years
28.5 Drowning
David Mountain
Introduction
Australia, the driest inhabited continent, has one of the highest reported incidences of drowning in the developed world. It is a major cause of death in those under 30 years of age with peaks in young children and young adult males. Nomenclature and definitions are now generally agreed with all respiratory distress (of any level, e.g. cough, wheeze, etc.) from immersion or submersion defined as drowning (fatal or non-fatal).
Good outcomes are mainly determined by pre-hospital factors, particularly witnessed drowning, early cardiopulmonary resuscitation (CPR) (and patient response to CPR) and early access to emergency services. However, an accurate history, well-run resuscitation and informed judgement on prognosis will optimize outcomes, resource use and aid management of the patient and their family. Patients with spontaneous respiration and/or neurological responsiveness on arrival in the emergency department (ED) are expected to recover unless severe lung injury/acute respiratory distress syndrome (ARDS) supervenes. Treatment after recovering from a non-fatal drowning is mainly supportive, although therapeutic hypothermia and possibly extracoporeal membrane oxygenation (ECMO) may potentially improve outcomes in the future.
In many groups/regions, preventative and educative measures have reduced fatality rates dramatically in the last 20 years. Emergency physicians should be strong advocates of these initiatives.
Epidemiology
Overall, there is a marked preponderance of male over female deaths from drowning and, in adults, the ratio has been reported as high as 9:1. This ratio seems to have declined in recent years with fewer male deaths and drownings being reported. Groups with high rates of drowning include: infants (particularly males), young adult males (15–30 years), epileptics (up to 20×higher), overseas visitors, the mentally retarded and those from deprived/underresourced communities with poor public health initiatives. In young adult males, bravado, inexperience and alcohol lead to many deaths. Alcohol is found in 14–50% of adult drownings and the majority of male adult drownings are related to recreational activities in some series. In the elderly, underlying medical illnesses and suicide attempts are common causative factors. Most of these factors (except age) are associated with worse outcomes. Cold water is associated with worse outcomes overall (shorter time to submersion in icy waters), although some younger patients may survive very prolonged immersions when they rapidly cool their brains.
The ratio of those who initially survive (but require medical attention) to fatal drownings is not accurately known because of differences in nomenclature, definitions and the inability to collect all attendances related to drowning, but is estimated at between 2 and 20:1. In a well-conducted observational study from the Netherlands, the ratio of patients admitted to the intensive care unit (ICU) following drowning compared to those who died before admission was 2:1.
Prevention
Prevention of drowning is a major area for ongoing research and it is important that emergency physicians act as advocates for preventative strategies of proven benefit.
Patrolled beaches and early, good quality CPR with early assisted breathing are associated with better outcomes. Important public educational initiatives include early swimming lessons/survival techniques, beach safety and patrolled water areas and beaches, CPR training, protective fencing, parental supervision of children, raising public awareness of the dangers of mixing alcohol and water activities and wearing of life vests and appropriate safety equipment. Enforcement of alcohol laws on the water and safety regulations pertaining to providers of water activities are also important.
Definitions and terminology
Much confusion has been caused in research and management by imprecise drowning definitions. Phrases commonly used have been near-drowning, dry, wet, active, passive or silent, late or secondary drowning, immersion, submersion, suffocation and asphyxia. Modell historically gave succinct definitions with drowning defined as death due to suffocation (asphyxia) after submersion in a liquid medium then further divided into ‘dry’ or ‘wet’, depending on the presence or absence of aspirated fluid in the lungs. Near-drowning was defined as survival of any length after suffocation (asphyxia) due to submersion in a liquid medium.
In 2002, ILCOR (International Liaison Committee on Resuscitation) provided updated and internationally agreed Utstein style nomenclature for drowning. The system simplifies the definition of drowning to ‘… a process resulting in primary respiratory impairment from submersion/immersion in a liquid medium’. Implicit in this definition is that a liquid/air interface is present at the entrance of the victim’s airway, preventing the victim from freely breathing.
The distinction between ‘near-drowning’ and ‘drowning’ is redundant as all are drowning events, irrespective of the outcome Similarly, there is no distinction between ‘wet’ and ‘dry’ drownings; all drowning is wet by definition and just have differing degrees of aspiration. Descriptions of ‘active’ and ‘passive’ or ‘silent’ drowning (determined by bystander descriptions of activity) have been replaced by ‘witnessed’ and ‘unwitnessed’ drowning, defined according to whether or not entry to the water was observed. The term ‘secondary drowning’ was used to describe both problems causing drowning (e.g. intoxication, injury, illness, etc.) or death after drowning due to secondary problems (e.g. lung problems, hypoxic encephalopathy, etc.) and this description was inherently confusing. Therefore, associated precipitating factors and sequelae should be specifically described as such. Immersion describes any situation when the patient is unable to maintain a fluid-free air interface while submersion implies the whole airway is underwater.
Pathophysiology
The sequential pathophysiology of drowning is well described:
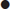
Clinical features and organ-specific effects
Lungs/airways
The major features are intense laryngospasm, bronchospasm, pulmonary hypertension and marked V/Q mismatch with physiological shunt. Even in patients without overt respiratory embarrassment after near drowning, shunts of up to 70% may occur and take up to a week to resolve. In the alveoli, there is surfactant loss, formation of protein-rich exudate and alveolar cell injury, often exacerbated by pneumonitis from gastric aspiration, chemicals and secondary infection (seen in up to 15% of intubated patients). These changes markedly reduce pulmonary compliance and oxygen transfer. The importance of the pulmonary insult in determining outcomes are seen by the fact that level of lung involvement from full cardiopulmonary arrest through different amounts of respiratory distress, down to cough or asymptomatic patients clearly stratify death and morbidity (Table 28.5.1).
Table 28.5.1
Grading of drowning severity–pre-hospital based on cardiorespiratory status
| Drowning grade | Dead | Grade 6 | Grade 5 | Grade 4 | Grade 3 | Grade 2 | Grade 1 | Rescue |
| Submersion time | >1 h/unknown | <1 h | ||||||
| Signs at scene/rescue | Clearly dead | No pulse No breaths |
Pulse No breaths |
Rales–all fields Hypotension | Rales–all fields BP normal | Rales–some BP normal |
Cough only | No signs |
| Mortality rate (%) | 100 | 88–93 | 31–44 | 18–22 | 4–5 | 1 | 0 | 0% |
| Management | Transport | CPR–ABC resus | Rescue ventilation | O2–prob ETT | O2–poss ETT | O2 | Check nil other probs | Nil required |
| Expected level of care | Forensic | ICU | ICU | ICU | HDU–ICU | ED review | Scene first aid | Nil required |
CPR: cardiopulmonary resuscitation; ICU: intensive care unit; ETT: ; HDU: high-dependency unit.
Modified from Szpilman D, Bierens JJLM, Handley AJ, Orlowski JP. Drowning. N Engl J Med 2012;366:2102–10 with permission.
Brain
The major effects on the brain are secondary to hypoxia and are the major cause of death in drownings. Cerebral oedema, convulsions and persistent vegetative states are all observed all too frequently. The possibility of trauma or an underlying medical complaint should be considered in the differential diagnosis of an altered mental state especially in unwitnessed events, drowning involving water transport or motor vehicles, the intoxicated and the elderly. The severity of hypoxic brain injury is a major determinant predicting survival from out of hospital arrest from drowning (Table 28.5.2).
Table 28.5.2
Modell/Conn classification of mental status following drowning
| Grade | Description of mental status | Equivalent GCS | Expected Likelihood of good outcome (neurologically intact) (%) |
| A | Awake/alert | 14–15 | 100 |
| B | Blunted | 8–13 | 100 |
| C | Comatose | 6–7 | >90 |
| C1 | Decerebrate | 5 | >90 |
| C2 | Decorticate | 4 | >90 |
| C3/4 | Flaccid coma or arrest | 3 | <20 |
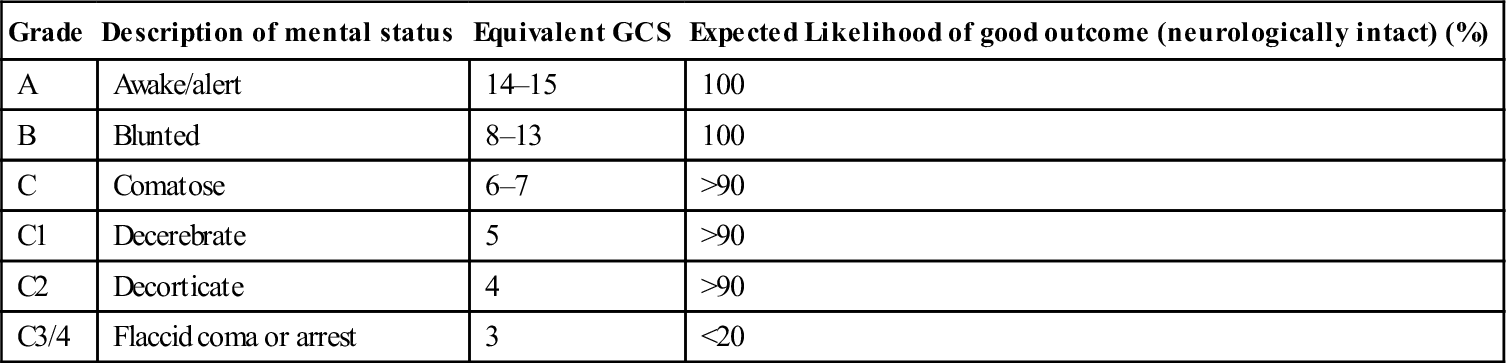
Cardiovascular
Most drowning patients are haemodynamically stable after resuscitation. Hypothermic patients may develop any arrhythmia and should be gently handled and aggressively rewarmed (see Chapter 28.2 Hypothermia). In older patients, underlying ischaemic heart disease should be considered. Congenital long QT syndrome may be associated with arrhythmia in some cold-water immersions.
Haematological
Haemolysis occurs occasionally in fresh-water drownings [8].
Renal
Acute tubular necrosis or tubular injury from hypoxia may occur. Electrolyte disturbances are rarely significant.
Gastrointestinal
Vomiting is frequently observed (up to 80% in some series). It is secondary to ingestion of large volumes of water, potentially aggravated by poor resuscitation techniques and positioning, often leading to aspiration. Diarrhoea is less frequent except with grossly polluted water. Hypoxic gut injury may contribute to late multiorgan failure, ARDS and potential sepsis.
Orthopaedic
Cervical spine injury should always be considered and excluded in drownings related to diving injuries. Coexistent trauma may complicate recreational drowning particularly if alcohol, water sports or boating related.
Treatment
Pre-hospital
Hypoxia (particularly brain hypoxia) is the major cause of almost all early mortality and morbidity and many late problems. Rapid institution of effective pre-hospital care, particularly supplemented (or exhaled responder) breathing/oxygenation (potentially in water if expert providers, e.g. surf lifesavers) and rapid emergency service activation are the most important factors in determining good outcome following drowning. All patients seen alive within 1 h of removal from cold water (<5–10°C) should be transported for definitive care. The level of pre-hospital care varies with the clinical severity of the case (see Table 25.8.1), ranging from asymptomatic (the majority) to cardiopulmonary arrest. Initial assessment of the ABCs may be done with the patient on their side to assist airway fluid drainage followed by institution of cardiopulmonary resuscitation (with an ABC emphasis) if respirations or pulse are absent. There is little role for in-water resuscitation except in deepwater retrievals on patrolled beaches with properly equipped expert retrievers who can get to shore easily. Lung drainage procedures (e.g. abdominal compressions) and the Heimlich manoeuvre are dangerous as they increase gastric aspiration. The Heimlich manoeuvre is only indicated for removal of a clearly inhaled foreign body and has no real place in drownings. The priority in drowning is to re-initiate breathing and, in drowning arrests, A and B are priorities. In particular, rescue breathing (or bagging if available) are obligatory (2 breaths with good chest movement before CPR). Additional breaths, up to 5, may be required if there is initial inadequate chest movement because of poor lung compliance with large fluid aspiration. Victims often vomit upon resumption of spontaneous respiration and obtunded, spontaneously breathing patients should be transported on their side to minimize the risk of aspiration. Wet clothing should be removed and the patient wrapped/covered to minimize further heat loss. If associated neck trauma is likely, the cervical spine should be immobilized. All symptomatic patients should be given supplemental high-flow oxygen. Early access to emergency medical systems is essential to minimize time to definitive care. A person with knowledge of the patient or witness to the drowning should be encouraged to go directly to the hospital or travel in the ambulance and, if not, a clear history taken and documented from bystanders.
Emergency department
History
Important factors in the history of arrested drowning include, environment of drowning and potential associated factors, e.g. water temperature, duration of submersion (or the time since last seen), time to institution of CPR, quality of CPR and response to first assisted breaths and/or CPR, time of first spontaneous breath or return of spontaneous cardiac output, initial Glasgow coma scale (GCS) and GCS after resuscitation recorded in Utstein style if possible. A collateral history regarding previous health problems (including psychiatric issues), use of alcohol and drugs, occurrence of vomiting and likelihood of associated trauma is also useful.
Initial resuscitation
Initial assessment and resuscitation, continuing the priorities established in the pre-hospital setting, is directed towards the assessment and maintenance of ABCs. Monitoring should include oximetry, capnometry, cardiac rhythm, blood pressure (BP) and core temperature (urinary or rectal probes are fine).
Airway management may initially simply involve clearing and positioning the airway and the provision of supplemental oxygen via a non-rebreathing mask. Endotracheal intubation is indicated if respiration is ineffective, saturations poor or lung fields are full of rales or the patient is comatose. Patients who cannot maintain a PaO2 greater than 90 mmHg on a non-rebreathing mask should be considered for early intubation, although continuous positive airway pressure (CPAP) ventilation is a potential alternative in the cooperative patient. Persistent rales in significant portions of the lung fields are a marker of a high risk for later severe respiratory compromise and potential ARDS. Patients with bronchospasm should be treated with nebulized β-agonists, with the use of steroids controversial unless the patient is a known asthmatic. In the unconscious patient, a nasogastric tube should be placed early after intubation to minimize the risk of pulmonary aspiration. All intubated patients require early positive end-expiratory pressure (PEEP) and end-tidal CO2 monitoring.
Cardiac complications should be managed according to standard treatment regimens except in patients with core temperatures less than 33°C. Hypothermic patients (see Chapter 28.2 Hypothermia) must be handled gently and antiarrhythmic drugs avoided if possible until rewarming has occurred. All rhythms without output require CPR. In general, asystole following drowning has the same dire prognosis as from other causes, particularly if still present after adequate resuscitation in the field. However, cold-water arrests (particularly in younger patients) do have occasional very good outcomes after very prolonged (60+ min) resuscitations. Hypotension is managed with early inotropes and judicious (e.g. small bolus) fluids, together with invasive monitoring if required. This is particularly important in patients with pulmonary oedema.
The management of hypothermia is described elsewhere in this book (see Chapter 28.2). Where cervical spine injury is a distinct possibility (especially following diving and water/motor vehicle accidents), cervical spine immobilization should be maintained until the injury can be excluded radiologically or clinically.
Ongoing management
Patients who require intubation, especially if pulmonary changes are present on chest X-ray, should be given PEEP. Commence with low pressures (5–7.5 cm H2O) and then increase until adequate oxygenation is achieved or hypotension or high airway pressures prevent further increases. Pressure-controlled ventilation may be added but may increase barotrauma and alveolar injury. Use should be discussed with the intensive care unit (ICU) that will be managing the ongoing care. These modalities improve outcome for near-drowning patients with secondary lung injury. Ventilatory weaning should begin as soon as possible after 24 h in order to minimize the risk of barotrauma. Maintenance of normoglycaemia, normovolaemia, normocarbia, seizure control and avoiding hyperthermia, hypoxia and hypotension are important in optimizing cerebral outcomes. Dehydration and prolonged hyperventilation are dangerous. Induced hypothermia post-arrest from drowning has been an area of some controversy (see below), but is recommended by expert consensus.
Experimental therapies
A number of other therapeutic modalities have been trialled in an effort to improve the outcome of lung and brain injuries caused by near-drowning. These include the following:
Clinical investigations
Ordering of investigations in the ED is guided by the clinical status of the patient, in particular, mental and cardiorespiratory status. All patients require continuous pulse oximtery and a chest X-ray which should be repeated at 4–6 h. Using the Modell/Conn classification of mental status (see Table 28.5.2), patients in group A only require a chest X-ray and oximetry. Patients in group B may also require a full blood count, electrolytes and creatinine, blood sugar, arterial blood gases and an ECG. If they do not improve rapidly after arrival and supplemental oxygen, they should be investigated and managed like group C patients. Patients in group C should also have liver function tests, creatine kinase and troponin at 6 h, coagulation profile, alcohol levels and possibly a drug screen, urine dipstick and microscopy, along with a computed tomography (CT) scan and possibly other imaging of the head if coma persists. Cervical spine X-rays and other trauma films are indicated if trauma is likely. Intracranial pressure monitoring, EEGs, MRI and brain injury markers are not really part of ED care and should be left to ICU discretion.
Prognosis
Mortality rates of 15–30% and persistent severe neurological deficit rates of up to 25% are reported in series of patients admitted to hospital following drowning events (but these studies are highly selected). Patients with prolonged cardiac arrests and poor initial response have a very high rate of mortality and persistent vegetative states (see Tables 28.5.1 and 28.5.2). Patients with relatively good GCS but with persistent hypoxia, early recovery from arrest and/or persistent rales are at high risk of secondary lung injury and multiorgan failure and have relatively high mortality rates (persistent rales 4–5%–recovered arrest 40%).
Potential prognostic features in drowning have been extensively evaluated in an effort to reduce the number of neurovegetative survivors, avoid prolonged CPR and to provide relatives and medical personnel with early accurate prognostic information. The most useful predictors of neurological outcome relate to the initial resuscitation (field predictors). Factors associated with good outcome include witnessed drowning, time to retrieval of less than 5 min, good-quality CPR provided within 10 min, a first spontaneous breath within 30 min of retrieval from the water and early return of spontaneous circulation before arrival at hospital. The last two are often associated with good neurological outcome, provided secondary lung injury does not supervene. Pre-hospital factors associated with poor outcome include male sex, unwitnessed or prolonged submersion, prolonged arrests (particularly asystole), fresh-water drownings, cold- water submersions and prolonged resuscitation before arrival at hospital. However, absolute field predictors of poor outcome have not been identified and all patients who arrive in the ED following drowning deserve full resuscitation efforts, short of clear signs of prolonged death (e.g. lividity, rigor) or clearly unsurvivable combination of prognostic features.
Emergency department prognostic factors have also been identified, but again no combination of factors reliably predicts all patients who will do badly. Features suggesting good outcomes in ED are pupillary response on arrival, perfusing cardiac rhythm on arrival or any motor response to pain on arrival. Asystole (particularly if the initial rhythm in the field and a significant pre-hospital time) is predictive of very poor outcome and, except in paediatric/young adults in ice-water drownings, should lead to early cessation of CPR in the ED. Hypothermia per se, has been described as a favourable prognostic indicator but is debatable. Although this may be true following near drowning of children or younger adults in ice-cold water with shorter immersion times (e.g. rapid brain cooling has occurred), hypothermia is generally a marker of prolonged submersion and, as such, is associated with a poor prognosis in most cases.
In-hospital factors associated with poor outcome include Glasgow coma score less than 5 on transfer to intensive care (less than 20% intact survival–see Table 28.5.2), fixed dilated pupils at 24 h (if not hypothermic) and any abnormality on CT scan in the first 36 h. However, a normal CT scan is of no prognostic value.
Disposition
All drowning victims requiring ED assessment should be carefully observed for a minimum of 6 h. Monitoring during that time should include pulse oximetry. Any patient with an abnormal chest X-ray or widespread respiratory rales or significant hypoxaemia after 6 h should be admitted with strong consideration for HDU/ICU environments. Those requiring intubation, with a history of cardiorespiratory arrest, persistently altered mental status or significant hypoxaemia require intensive care. Truly asymptomatic patients or non-progressive rales with stable oximetry, may be discharged home after 6 h of observation, but should be instructed to return to hospital if they develop worsening respiratory symptoms.
Controversies
28.6 Electric shock and lightning injury
Daniel Fatovich
Electric shock
Introduction and epidemiology
Electricity is an integral part of our everyday world and electric shock is common. Patients may present to the emergency department (ED) with resulting injuries that range from trivial to fatal (termed electrocution). Although permanent disability can occur, it is reassuring to note that if the initial exposure is survived, subsequent death is unlikely. For each death caused by electricity, there are two serious injuries and 36 reported electric shocks.
There are approximately 20–30 electrical fatalities each year in Australia. Victims are predominantly male and relatively young. Death is just as likely to occur at home as in the workplace, most often in summer. Electricians and linesmen are most at risk. The ratio of low-to-high-voltage deaths ranges from 3:1 to 7:1. The presence of water is associated with fatality. Electrical burns represent 3–5% of admissions to burns units.
Physics of electricity and pathophysiology of electrical injury
Electrical current passing through the body can cause damage in two ways:

The threshold for perception of an electrical current is 1 mA, which results in a tingling sensation. Current greater than 10 mA can induce muscular tetany and prevent the patient letting go of the current source. Paralysis of respiratory muscles occurs at 20 mA. The threshold for ventricular fibrillation is 100 mA (Fig. 28.6.1). Cardiac standstill and internal organ damage occurs at 2 A. The maximum ‘safe’ current tolerable for 1 s is 50 mA.
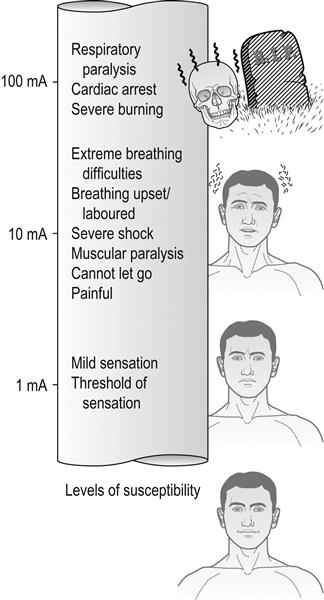
Ohm’s law is fundamental to the understanding of the physics of electricity. This states that:
The amount of current passing through the body is directly proportional to voltage and inversely proportional to resistance (current [amperes]=voltage [volts]/resistance [ohms]).
Factors that determine the effects of an electrical current passing through the body are:
Type of current
The vast majority of serious electrical injuries result from alternating current (AC), which is approximately three times as dangerous as direct current (DC). Alternating current can produce tetanic contraction of muscle such that the victim may not be able to let go of the current source. This is not a feature of direct current shock.
Human muscular tissue is sensitive to frequencies between 40 and 150 Hz. As the frequency increases beyond 150 Hz, the response decreases and the current is less dangerous. In Australia, a frequency of 50 Hz is used for household current because this is optimal for the transmission and use of electricity and also has advantages in terms of generation. As such, household current lies directly within the dangerous frequency range. It also spans the vulnerable period of the cardiac electrical potential and is thus capable of causing ventricular fibrillation.
Voltage
Voltage is the electromotive force in the system. In general terms, the greater the voltage the more extensive the injury, but it must be remembered that the amount of current passing through the body will also be determined by resistance (Ohm’s law). High voltage is defined as greater than 1000 V. Household voltage in Australia is 240 V. Voltages less than 50 V (50 Hz) have not been proved hazardous. Survival has been reported following shocks of greater than 50 000 V.
Resistance
Different tissues provide differing resistances to the passage of electrical current. Bone has the highest resistance, followed by, in decreasing order, fat, tendon, skin, muscle, blood vessels and nerves. Importantly, however, skin resistance varies greatly according to moisture, cleanliness, thickness and vascularity. Moist skin may have a resistance of 1000 Ω and dry, thick, calloused skin a resistance of 100 000 Ω. By Ohm’s law, dry skin resistance to a contact with a 240 V potential results in a current of about 2.4 mA, which is just above the threshold for perception. However, the resistance of wet or sweat-soaked skin drops to 1000 Ω, increasing the current flow to 240 mA, which is easily enough to induce ventricular fibrillation. Not surprisingly, moisture has been identified as a key factor in over half of electrocutions.
Current path
Prediction of injuries from knowledge of the current path is unreliable. Mortalities of 60% for hand-to-hand (transthoracic) and 20% for head-to-foot passage of current are quoted, but have not been verified. When current passes hand-to-hand (or hand-to-foot), only about 5% of the total current passes through the heart. If current passes leg-to-leg, no current traverses the heart.
Contact duration
The longer the duration of contact, the greater the potential for injury. Fortunately, most contacts are brief and frequently result in the victim being thrown back from the current source. This may result in a secondary injury, especially if the victim falls from a height.
Unfortunately, exposures to more than 10 mA of alternating current can induce sweating. Moisture decreases skin resistance and increases current flow, thereby reducing the ability to release the current source. This can progress to a fatal exposure.
Prevention
All members of the community must be encouraged to treat electricity with respect and to practise electrical safety. Licensed electrical contractors should be used to carry out any electrical repairs or installations. Water and electricity should never be mixed.
Residual current devices are useful in providing an additional level of personal protection from electric shock. These devices continuously compare current flow in both active and neutral conductors of an electrical circuit. If current flow becomes sufficiently unbalanced, then some of the current in the active conductor is not returned through the neutral conductor and leaks to earth. These devices operate within 10–50 ms and disconnect the electricity supply when they sense harmful leakage, typically 30 mA.
Clinical features
Electrical injury resembles a crush injury more than a burn. Invariably, the damage below skin level is more severe than the cutaneous wound suggests. The current passing through low-resistance structures produces massive necrosis of muscles, vessels, nerves and subcutaneous tissues.
The clinical manifestations differ from thermal burns in the following ways:
Burns
As electricity traverses the skin, energy is converted to heat. The smaller the area of contact, the greater the current density, heat production and the consequent skin and adjacent tissue destruction.
Electrothermal burns are best characterized by arc burns, which result from the external passage of current from the contact point to the ground. These may be associated with extensive damage to skin and underlying tissue. Secondary flame burns may occur when the current arc ignites clothing or nearby combustibles.
Electrical burns may range from first degree to third degree. The typical appearance is of a central depressed charred black area surrounded by oedema and erythema. Single or multiple exit wounds may be present.
Cardiac
Ventricular fibrillation is the usual cause of immediate death from electric shock and occurs at the time of the shock. Delayed arrhythmia resulting in death is exceptionally rare. Sinus tachycardia is common and non-specific ST- and T-wave changes may be observed. Atrial fibrillation occurs infrequently and usually resolves spontaneously. Acute myocardial infarction following electric shock has been reported.
Nervous system
Both acute and delayed neurological sequelae have been described following electric shock. Acute complications include respiratory arrest, seizures, altered mental state, amnesia, coma, expressive dysphasia and motor deficits. Reported delayed complications include spinal cord injury (myelopathy) with local amyotrophy and long tract signs, and reflex sympathetic dystrophy.
Peripheral nerve injury is usually associated with significant soft-tissue injury. It has also been reported in the absence of soft-tissue injury and such cases appear to have a good prognosis.
Renal
Acute renal failure may occur secondary to myoglobinuria. Electric shock results in disruption of muscle cells with the release of myoglobin and creatine phosphokinase, similar to a crush injury. Transient oliguria, albuminuria, haemoglobinuria and renal casts are common and there have been reports of high-output renal failure.
Vascular
Large and small vessel arterial and venous thrombosis are responsible for the tissue damage in electrical injury. Vascular complications have included immediate and delayed major vessel haemorrhage, arterial thrombosis and deep vein thrombosis.
Musculoskeletal
Tetanic muscle contractures can result in compression fractures of vertebral bodies, fractures of long bones and dislocations of joints. Injuries may also result from a secondary fall, rather than from the electric shock.
Other
Numerous complications involving other systems, including the eye (especially cataracts) have been reported following electric shock.
Electric shock in pregnancy
Reports of electric shock in pregnancy are rare and the true incidence is unknown. A high mortality has been reported in the literature. However, this may represent publication bias and a prospective cohort study concluded that, in most cases, accidental electric shocks during pregnancy do not pose a major fetal risk.
If there was an immediate problem, the mother may notice a sudden cessation of fetal movements. However, there is no preventative action possible in the ED. Other reported fetal complications of electric shock include intrauterine growth retardation, oligohydramnios and abortion.
Fortunately, therapeutic electric shocks, such as DC cardioversion and electroconvulsive therapy, are known to be safe in pregnancy. The critical factor is current path: accidental electric shocks include the uterus, whereas therapeutic shocks do not.
Treatment
Pre-hospital
Everyone should be aware of the pre-hospital management of electric shock. Most importantly, the rescuer should avoid becoming a further victim. The victim can be separated from the electrical source by using rubber, a wooden handle, a mat or any other non-conductive substance or, if possible, by turning off the electricity supply. Cardiopulmonary resuscitation (CPR) should begin immediately, if indicated, and help summoned. CPR may need to be prolonged. Ventricular fibrillation is the most common lethal arrhythmia after electric shock and early defibrillation provides the greatest chance for survival.
Emergency department
The majority of patients who present to the ED after electric shock are relatively well. Following appropriate assessment to exclude primary or secondary injury, an ECG should be performed. Cardiac monitoring is not indicated if the patient is asymptomatic and has a normal ECG. Most patients are able to be reassured and discharged directly from the ED. Measurement of creatine phosphokinase levels is not required. It should be remembered that exposure to an electric shock is an unpleasant experience and this should be acknowledged. Tetanus status should be checked.
Many patients have a degree of muscle pain following electric shock owing to the tetanic nature of alternating current. Simple analgesia is appropriate. Any secondary injury, such as fractures or loss of consciousness, should be treated as dictated by the injury.
If an arrhythmia is present it will usually resolve spontaneously and not require specific treatment. Delayed lethal arrhythmias have not been reported in patients without initial arrhythmias.
Severe electrical injury with extensive soft-tissue damage should be managed as a crush injury. This is more likely following high-voltage exposure, which results in a large exudation and sequestration of fluids in the damaged area. Emergency management includes adequate volume replacement and treatment of acidosis and myoglobinuria.
Emergency physicians should be aware of the low potential for fetal harm following electric shock in pregnancy. Publication bias suggests that apparently minor exposures can have profound effects. It would be prudent to adopt a conservative approach of performing a fetal heart Doppler assessment with obstetric follow up including ultrasound.
Prognosis
The prognosis for the majority of patients surviving the initial shock is excellent. Those with significant soft-tissue injury or secondary injury may be left with long-term deficits.
Disposition
The majority of patients presenting to the ED following an electric shock will be suitable for discharge home following assessment and reassurance as detailed above. Those suffering muscle pain secondary to tetanic contractions should be given simple analgesia and instructed to follow up with their general practitioner.
Patients with cardiac arrhythmias require admission for observation until the arrhythmia resolves. Those with evidence of neuropathy should be referred to a neurologist, as nerve conduction studies may be required.
Severe electrical injuries with extensive soft-tissue damage require admission to hospital and, sometimes, to an intensive care unit. All patients with electrical burns should be reviewed by a burns specialist and referral to a specialist burns unit may be indicated. Minor burns may be suitable for elective review.
Secondary injuries, such as loss of consciousness or fractures, should be admitted or referred on their merits.
The Taser
The Taser is a development of the stun gun. It is used by the police service to fill the operational gap between the baton and the gun for controlling potentially dangerous and violent suspects. ‘tasered’ victims are occasionally brought to the ED for assessment.
The device is a battery operated unit resembling a hand gun that fires two barbed electrodes on 7 m long copper wires at 60 m/s. The barbs attach to the subject’s skin or clothing and deliver up to 50 000 V of electricity in rapid pulses over 5 s. The current can cross up to 5 cm of clothing.
Electricity delivered by a taser is neither pure AC nor pure DC and is probably akin to rapid-fire low-amplitude DC shocks. The output is believed to stay near the surface of the body in the skin and muscles and does not penetrate into the internal organs. There is no evidence to date that this form of electrical delivery interfered with cardiac or neurological function in the 30 000 volunteers or in the reported operational uses.
One author concluded that the pre-existing injuries and toxic conditions leading to the patient being tasered are the most important problems requiring medical treatment after Taser use. It seems that the device is essentially safe on healthy people. However, there is limited evidence to base recommendations for the assessment and management of patients that are brought to the ED after being ‘tasered’. Suggestions for management of these patients attending EDs are:
 Any patient with chest pain or abnormal ECG should be assessed as per routine clinical practice.
Any patient with chest pain or abnormal ECG should be assessed as per routine clinical practice.
 Pregnant women>24 weeks’ gestation should be considered for cardiotocograpic monitoring.
Pregnant women>24 weeks’ gestation should be considered for cardiotocograpic monitoring.
It is clear that, properly used as a method of restraining violent people, Tasers are less likely than guns to cause injury and death of the target (and of the police officer). They are also generally more effective than other methods of restraint. The deaths that have followed taser use have occurred in people who were out of control and who had taken potentially fatal drugs. It is likely that the deaths would have occurred whether or not the Taser was used. However, the medical effects of multiple shocks on such persons is unknown.
Lightning injury
Introduction and epidemiology
There are several deaths each year in Australia from lightning. For each death, there are five injuries. These events are always prominent and emergency physicians should be familiar with the pathophysiology. In addition, about 60 people each year report injuries caused by lightning surges while using the telephone during thunderstorms.
Physics
Lightning occurs most commonly during thunderstorms. Particles moving up and down in a thunderstorm create static electricity, with a large negative charge building up at the bottom of clouds. Electrical discharge (lightning) occurs as a result of the great charge difference between the negatively-charged thundercloud underside and the positively charged ground. The duration of the lightning stroke is between 1 and 100 ms.
Lightning strike is very different from high-voltage electric shock (Table 28.6.1) and produces different clinical effects, requiring a different management approach.
Table 28.6.1
Lightning versus high-voltage injury
| Factor | Lightning | High voltage |
| Time of exposure | Brief instantaneous | Prolonged tetanic |
| Energy level | 100 million V 200 000 A |
Usually much lower |
| Type of current | Direct | Alternating |
| Shock wave | Yes | No |
| Flashover | Yes | No |
Adapted from Cooper MA. Lightning injuries. In: Auerbach P, et al (eds). Management of wilderness and environmental emergencies. New York: Macmillan; 1983: 500–21.
An interesting phenomenon called ‘flashover’ seems to save many victims from death by lightning. Current passes around and over, but not through the body. The victim’s clothing and shoes may be blasted apart. Only cutaneous flame-type burns result.
Clinical features
Immediate
Delayed
Other
Reports of lightning strike in pregnancy reveal a high rate of fetal death in utero, despite maternal survival.
Treatment
Pre-hospital
The important principle is that those who appear dead should be resuscitated first. Immediate institution of basic cardiopulmonary resuscitation in the field for those in asystole prevents secondary hypoxic cardiac arrest during the interval until cardiac function resumes spontaneously. Fixed dilated pupils should not be taken as an indicator of death after lightning strike.
Emergency department
Most lightning strikes are unwitnessed and diagnosis may be difficult in the unconscious or confused patient. The diagnosis should be considered where such patients were found outdoors in stormy weather. The presence of multiple victims, exploded clothing, linear or punctuate burns, keraunic markings or tympanic membrane rupture all add weight to the diagnosis. The differential diagnosis includes cerebrovascular event, seizure disorder, spinal cord injury, closed-head injury, Stokes–Adams attack, myocardial infarction and toxin effects.
Standard trauma resuscitation measures should be adopted. Examination of the ears for tympanic rupture and eyes for lens/corneal defects, retinal detachment and optic nerve injury is especially important. If the conscious state deteriorates after arrival, cranial computed tomography scan is indicated. Examination of the cardiovascular system should include an ECG.
Burns are rarely more than superficial and are managed expectantly using standard treatments. Tetanus prophylaxis should be arranged.
Treatment of lightning-induced limb paralysis is expectant. If it does not resolve within a few hours, other causes should be considered. Fasciotomy is unnecessary.
Standard therapy for ocular complications, such as retinal detachment or cataracts, is indicated. Baseline visual acuity should be documented for future reference.
Prognosis and disposition
For survivors of the initial strike the prognosis is excellent unless significant secondary injury has occurred. Admission for observation is indicated for those with abnormal mental status or ECG, or with significant burns or traumatic complications. The burns usually heal well and grafting is rarely required. For those with ocular complications, long-term ophthalmic follow up is necessary.
28.7 Altitude illness
Ian Rogers
Introduction
Altitude illness comprises a number of syndromes that can occur on exposure to the hypobaric hypoxic environment of high altitude. At any altitude, the partial pressure of inspired oxygen (PiO2) is equal to 0.21 times the barometric pressure minus water vapour pressure of 47 mmHg. At an altitude of 5500 m, barometric pressure is halved. On the summit of Mount Everest (8850 m), the PiO2 is only 43 mmHg and a typical climber without oxygen can be expected to have a PaO2 of<30 mmHg and a PaCO2 of about 13 mmHg [1]. In addition to the hypoxic stress of altitude, a subject may also be exposed to cold, low humidity, fatigue, poor diet and increased ultraviolet radiation. For the emergency physician, the unique feature of altitude illness is that it requires recognition and treatment in the field, frequently without access to sophisticated diagnostic and imaging techniques and often without access to rapid evacuation.
Epidemiology and pathophysiology
The human body has the capacity to acclimatize to hypoxic environments. This is principally achieved by increasing ventilation (the hypoxic ventilatory response effected by the carotid body), increasing numbers of red blood cells (via stimulation of erythropoietin), increasing the diffusing capacity of the lungs (resulting from increased lung volume and pulmonary capillary blood volume), increasing vascularity of the tissues and increasing the tissues’ ability to use oxygen (possibly owing to increased numbers of mitochondria and oxidative enzyme systems).
In some individuals, exposure to low PO2 initiates a sequence of pathophysiological changes, which result in oedema formation in the brain and lungs. The altitude illness syndromes, acute mountain sickness (AMS), high-altitude cerebral oedema (HACE) and high-altitude pulmonary oedema (HAPE), are the result of this oedema formation. The exact mechanism of these pathophysiological changes is still debated but vasodilatation is a key part.
In the brain, the development of oedema causes intracranial pressure (ICP) to rise. Initially, this is partially compensated for by displacement of cerebrospinal fluid (CSF) into the spinal space and adjustment of the balance between production and absorption of CSF. However, once these compensatory mechanisms are overwhelmed, ICP can rise beyond the cerebral perfusion pressure. Without intervention, cerebral blood flow ceases and the patient dies.
In the lung, non-cardiogenic pulmonary oedema develops. A significant rise in pulmonary artery pressure appears to be a crucial pathophysiological factor [2]. Impaired sodium driven clearance of alveolar fluid may contribute to HAPE [3]. It has been postulated that uneven pulmonary vasoconstriction increases the filtration pressure in non-vasoconstricted lung areas, worsening the interstitial and alveolar oedema.
The tendency to develop altitude illness is idiosyncratic. The major predisposing factors are the rate of ascent and the altitude reached. It is not related to physical fitness or gender. Individuals vary in their ability to compensate for changes in ICP and in their pressor responses to hypoxia. This may explain the reproducibility of AMS, HACE and HAPE in susceptible individuals and why some, and not others, develop symptoms at the same altitude. The risk is higher in those who have an impaired ventilatory response to hypoxia in normobaric conditions and with dehydration, vigorous exercise and the use of depressant drugs.
Prevention
The best form of prevention is gradual ascent to allow sufficient time for acclimatization. Although individuals vary in how quickly they acclimatize, a sensible recommendation is sleeping no more than 500 m higher than the previous day once above 2500 m. Keeping warm, avoiding alcohol, maintaining hydration and eating a high-carbohydrate diet to improve the respiratory quotient, may all decrease the incidence of altitude illness. Modest exercise on acclimatization days should be encouraged.
Acclimatization is not always practical or possible and so pharmacological agents may be required [4]. Acetazolamide reduces the incidence and severity of AMS/HACE when used prophylactically in subjects experiencing rapid ascent [5]. Doses recommended have decreased as a result of ongoing research [6]. Chemoprophylaxis can be achieved with 125 mg bd, starting the day before ascent and continued for 2 days after reaching high altitude. Dexamethasone 4 mg bd may be equally effective and may be more so when a rapid onset is required, such as in unacclimatized personnel involved in high-altitude rescue missions.
Nifedipine 20 mg slow-release tds or 30 mg bd provides protection against HAPE in susceptible individuals. More recent research suggests that other drugs, such as sildenafil, tadalafil and salmeterol, may have a role in HAPE prevention, but it is generally advised that vasodilators not be combined.
Clinical features
AMS is common, occurring in about 30% of subjects exposed to moderate altitude (3500 m). HACE and HAPE are less common, but a study in pilgrims at 4300 m reported AMS in 68%, HACE in 31% and HAPE in 5% of subjects [7]. The diagnosis is usually made purely on clinical assessment.
Acute mountain sickness
AMS is primarily a neurological syndrome, associated with some degree of respiratory compromise. The onset is usually 6–24 h after arrival at high altitude. The majority of patients present in the early stages when the symptoms are like those of a hangover and include headache, nausea, anorexia, weakness and lassitude. In the early stage of AMS, there are no abnormalities on physical examination and the oxygen saturation, if measured, should be no lower than that expected for a given altitude. Mild AMS is usually benign and self-limiting.
If the illness progresses, the more severe form of AMS is characterized by dyspnoea at rest, nausea and vomiting, altered mental state, headache and ataxia. Ataxia is the most useful sign of progression to serious illness. Left untreated, severe AMS may progress to life-threatening HACE or HAPE.
AMS can be scored using the Lake Louise AMS score [8]. This consists of five symptom groups: headache, gastrointestinal distress, fatigue or weakness, dizziness or light headedness and difficulty sleeping. Each symptom is scored on a scale from 0 (not present) to 3 (severe or incapacitating) and the totals of the five symptom groups are summed. A total score of 3 or more is considered diagnostic of AMS.
High-altitude cerebral oedema
HACE is the progression of neurological signs and symptoms in the setting of AMS. There is a progressive decline in mental status and truncal ataxia is a prominent physical finding. Focal neurological signs, such as third and sixth cranial nerve palsies, may develop as a result of raised intracranial pressure. Unrecognized and untreated, there may be rapid progression to coma and death due to raised intracranial pressure.
High-altitude pulmonary oedema
HAPE occurs in susceptible individuals who may have no underlying pulmonary or cardiac disease. It most commonly manifests on the second night at high altitude. In the early stages, the oedema is interstitial and the patient may only have a dry cough and decreased exercise tolerance. Few abnormalities will be seen on examination at this stage. As more fluid accumulates, the patient develops tachycardia, increasing dyspnoea, marked weakness, cough productive of frothy sputum and cyanosis. Pulse oximetry, if available, confirms profound hypoxia. A chest X-ray will demonstrate widespread interstitial and alveolar infiltrates. It may occur in conjunction with AMS/HACE or as an isolated clinical syndrome.
Treatment
Early recognition is an essential component of the management of all acute altitude syndromes. Developing symptoms in a party member may have substantial impact on route planning choices, particularly whether to halt ascent or descend. The goal is to stop the pathophysiological process with the key interventions summarized in Table 28.7.1.
Table 28.7.1
Key treatments in severe altitude syndromes
| HACE/severe AMS | HAPE |
| Descent | Descent |
| Oxygen | Oxygen |
| Simulated descent (e.g. Gamow bag) | Simulated descent (e.g. Gamow bag) |
| Dexamethasone 8 mg stat then 4 mg 6-hourly | Nifedipine SR 20 mg 8-hourly |
Acute mountain sickness and high-altitude cerebral oedema
A patient presenting with symptoms of mild AMS should be advised to halt ascent to allow time for acclimatization. They should rest, as physical exertion aggravates symptoms, and take simple analgesics and antiemetics if desired. It is important that the patient be closely observed for progression of symptoms.
With moderate symptoms, the management is the same as for mild AMS, with the addition of oxygen 2–4 L/min and, possibly, pharmacological agents. Acetazolamide, a carbonic anhydrase inhibitor, aids the normal process of ventilatory acclimatization by reducing the renal reabsorption of bicarbonate, resulting in metabolic acidosis and compensatory hyperventilation. It relieves symptoms, improves arterial oxygenation and prevents further impairment of pulmonary gas exchange. It also helps to maintain cerebral blood flow despite hypocapnia and opposes the fluid retention of AMS. The recommended treatment dose is 250 mg orally bd. Acetazolamide is a sulpha drug and contraindicated in those with known allergy. Dexamethasone is also an effective agent in this condition [9], presumably by reducing capillary permeability and ICP. It does not aid in acclimatization. It may be given as an alternative, or in addition to, acetazolamide. The recommended dose is 8 mg stat then 4 mg every 6 h.
If a patient shows signs of severe AMS progressing to HACE, then rapid and controlled descent is the highest priority. Oxygen 2–4 L/min should be administered. Additional therapy may be required if the illness is severe, the patient’s condition must be improved to allow descent or where immediate descent is not possible. Additional therapeutic options include dexamethasone 8 mg stat then 4 g every 6 hours and hyperbaric therapy using a portable fabric hyperbaric chamber (e.g. Gamow bag) [4]. The bags are expensive and need to be pumped continuously, but have the advantage of using air rather than oxygen.
High-altitude pulmonary oedema
Rapid and controlled descent, with oxygen, is the mainstay of management in a patient suffering from HAPE, although milder cases may be managed with oxygen without altitude change. In a large proportion of cases this is sufficient. Oxygen flow should be titrated to maintain adequate oxygen saturation. Continuous positive airway pressure may be required. The patient should be rested and kept warm, as cold may further increase pulmonary hypertension through sympathetic stimulation.
Nifedipine should be considered as adjunctive therapy to oxygen and descent. It lowers the raised pulmonary artery pressure that characterizes HAPE and results in clinical improvement, better oxygenation and progressive clearing of alveolar oedema on chest X-ray. The recommended dosage is 20 mg of the slow release formulation 8-hourly or 30 mg 12-hourly [4].
Controversies
[/level-membership-for-emergency-medicine-category][not-level-membership-for-emergency-medicine-category]
Environmental Emergencies
Edited by Mark Little
28.1 Heat-related illness
Ian Rogers
Introduction
Heat-related disorders have a broad range of potential aetiologies and manifestations. In some, the primary disorder is a failure of thermal homoeostasis whereas, in others, the hyperthermia is secondary to other processes. The major heat-related illnesses to consider are exercise-associated collapse (EAC), heatstroke, neuroleptic malignant syndrome, serotonin toxicity and malignant hyperthermia. Although of different aetiologies, the latter four, all associated with significantly elevated core temperatures, share much common ground with regard to complications and treatment.
Epidemiology and pathophysiology
EAC is the most common heat-related illness presenting either to medical tents at sporting events or to emergency departments. EAC manifests at the end of a race when muscle pump enhanced venous return ceases and cardiac output drops. This leads to collapse, often with a brief loss of consciousness. Despite the claims of the advocates of fluid loading and sports drinks in exercise, the primary mechanism is a failure of prompt baroreceptor responses and not haemodynamically significant dehydration. Severe heat-related dehydration is rare.
The other, more serious, heat-related disorders are all associated with, or the potential for, significant hyperthermia which, if not treated promptly, results in similar pathophysiology at a cellular and organ system level. A core body temperature around or greater than 41.5°C results in progressive denaturing of a number of vital cellular proteins, failure of vital energy-producing processes and loss of cell membrane function. At a cellular level, the exact mechanisms leading to loss of cell membrane function and cell death in heat illness remain uncertain. At an organ system level, these changes may manifest as rhabdomyolysis, acute pulmonary oedema, disseminated intravascular coagulation, cardiovascular dysfunction, electrolyte disturbance, renal failure, liver failure and permanent neurological damage [1,2]. Any or all of these complications must be expected in severe heat illness.
The hallmark of heatstroke is failure of the hypothalamic thermostat, leading to hyperthermia and the associated additional pathophysiological features described above. Clinically, heatstroke can be divided into ‘exertional heatstroke’ due to exercise in a thermally stressful environment, and ‘classic heatstroke’, which occurs in patients with impaired thermostatic mechanisms. Common risk factors for heatstroke are listed in Table 28.1.1.
Table 28.1.1
Behavioural
Army recruits
Athletes
Exertion
Inappropriate clothing
Elderly
Inappropriate exposure
Babies left in cars
Manual workers
Pilgrims
Drugs
Anticholinergics
Diuretics
Phenothiazines
Salicylates
Stimulants/hallucinogens
Illness
Delirium tremens
Dystonias
Infections
Seizures
Certain drugs produce hyperthermia by mechanisms in addition to interference with thermostatic function. In severe serotonin toxicity and neuroleptic malignant syndrome, increased motor activity and central resetting of the hypothalamic thermostat combine to produce hyperthermia. In the case of serotonin toxicity, these effects are a consequence of a relative excess of central nervous system serotonin whereas, in neuroleptic malignant syndrome, dopamine depletion or dopamine receptor blockade is responsible.
The elevation of central nervous system serotonin in serotonin toxicity is usually associated with combinations of serotoninergically active drugs, taken either therapeutically or in overdose. The incidence of serotonin toxicity when such combinations are taken is not known, but is low and there is much individual variation in susceptibility [3]. Less often the syndrome is precipitated by a single serotoninergic agent. Drugs associated with the serotonin syndrome are listed in Table 28.1.2. The most commonly implicated are combinations of serotinergically active drugs, such as selective serotonin reuptake inhibitors (SSRIs), lithium, pethidine, monoamine oxidase inhibotors (MAOIs) and amphetamines.
Table 28.1.2
Drugs causing severe serotonin toxicity
Antidepressants
Monoamine oxidase inhibitors (MAOIs)
Selective serotonin reuptake inhibitors (SSRIs)
Selective serotonin and noradrenaline reuptake inhibitors (SSNRIs)
St John’s wort
Tricyclics
Analgesics
Pethidine
Tramadol
Recreational drugs
Amphetamines
Methylenedioxymethamphetamine (MDMA, ‘ecstasy’)
Neuroleptic malignant syndrome (NMS) is a rare idiosyncratic reaction to neuroleptic agents with an incidence of between 0.02% and 3.0%, depending on the diagnostic criteria used. It occurs in response to a single agent, usually at therapeutic dosage. In individuals, the occurrence may be dose related. Certain at-risk groups have been identified and are listed in Table 28.1.3.
Table 28.1.3
Risk factors for neuroleptic malignant syndrome
Patient factors
Agitation
Dehydration
Male sex (male:female=2:1)
Organic brain disease
Drug dosing factors
Depot neuroleptics
High initial neuroleptic dose
High-potency neuroleptic (e.g. haloperidol)
Rapid dosage increase
NB: Duration of drug exposure and toxic overdose are not related to risk of developing NMS.
Malignant hyperthermia is a genetically inherited disorder in which triggering agents cause a release of sarcoplasmic Ca2+ stores. The resulting elevation of myoplasmic Ca2+ stimulates many intercellular processes, including glycolysis, muscle contraction and an uncoupling of oxidative phosphorylation. This leads to hyperthermia that, in contrast to neuroleptic malignant and serotonin syndromes, is purely peripheral in origin.
Prevention
Prevention of exertional heatstroke should focus on the education of at-risk groups. Dehydration is not as important aetiologically in heatstroke as once thought. Exertional heatstroke is most often reported in shorter, high intensity exercise where marked dehydration is unlikely. So, although adequate fluid intake is needed for prolonged exercise, it is not a key factor in heatstroke prevention. As high ambient temperatures and high humidity predispose to exertional heatstroke, exertion in these environments should be limited.
Clinical features
Exercise-associated collapse
The clinical presentation of EAC will be familiar to all emergency practitioners as it mirrors that of poor cerebral perfusion from any other cause. Patients complain of nausea, vomiting, malaise and dizziness. There may be a history of collapse and there is likely to be a tachycardia and (orthostatic) hypotension. The orthostatic hypotension may manifest at the end of physical exertion by collapse with brief loss of consciousness that is typical of EAC. In this syndrome, and in distinction to heatstroke, the core temperature will be less than 40°C and neurological function will rapidly return to normal once the patient is lying down.
Heatstroke
The classic clinical features of heatstroke are neurological dysfunction, core temperature above 41.5°C and hot, dry skin. However, relying on this classic triad to make the diagnosis will result in a number of cases being missed. Loss of consciousness is a constant feature of heatstroke [1] but, by the time of ED presentation, conscious state may be improving, although some neurological abnormality will persist. Temperature readings may be misleadingly low, due either to effective pre-hospital care or to measurements at inappropriate sites, such as the oral cavity or axilla. Profuse sweating is a much more common feature than previously thought [1]. Other clinical features may include tachycardia, hyperventilation, seizures, vomiting and hypotension.
Serotonin toxicity
Serotonin syndrome is characterized by CNS, autonomic and motor dysfunction (Table 28.1.4). It is a clinical diagnosis. It develops after a latent period, which is normally a few hours, but may be as long as several days. The spectrum of illness produced is broad. Most patients are only mildly affected and may escape clinical detection. Only the most serious develop hyperthermia (usually in the setting of muscular rigidity) severe enough to produce the complications of rhabdomyolysis, disseminated intravascular coagulation and renal failure. Most cases will resolve within 24–48 h once the precipitating agents are withdrawn. Even in severe cases, the underlying biochemical abnormality rapidly improves, usually with the institution of muscular paralysis. The morbidity and mortality in these cases is caused by the complications that develop while the syndrome is active.
Table 28.1.4
Features of the serotonin toxicity
Central nervous system
Agitation
Anxiety
Confusion
Decreased level of consciousness
Seizures
Motor
Clonus
Hyperreflexia
Hypertonia
Incoordination
Myoclonus
Tremor
Autonomic
Diaphoresis
Diarrhoea
Hypertension
Hyperthermia
Tachycardia
Neuroleptic malignant syndrome
This syndrome manifests in patients who have recently been started on neuroleptic treatment or in whom the dose of a neuroleptic agent has been increased. It has been associated with almost all antipsychotics (both first and second generation) and has also been reported in patients in whom a dopaminergic agent has been rapidly withdrawn (e.g. in parkinsonism). There is a latent period of many hours to several days. Characteristically, there are four classic signs: fever, rigidity, altered mental state and autonomic instability. In practice, it may be difficult to distinguish clinically from serotonin toxicity unless a good drug history is obtained. As in serotonin toxicity, the spectrum of illness is very broad, with only the more severe cases developing hyperthermia and its complications [5].
Malignant hyperthermia
This occurs when a triggering agent is given to a susceptible individual, usually in the context of an anaesthetic. Triggering agents identified include inhalational anaesthetic agents, such as halothane, isoflurane and enflurane, as well as succinylcholine and ketamine. The first signs are failure to achieve muscle relaxation following succinylcholine, tachypnoea and tachycardia. If not recognized and treated, acidosis, rhabdomyolysis and hyperthermia will ensue. In some cases, signs and symptoms may be delayed or even reappear after apparently successful treatment, so that malignant hyperthermia may even present as a postoperative fever. Untreated, the mortality is as high as 70%, but this can be reduced tenfold by appropriate management.
Clinical investigations
Diagnosis of the hyperthermic disorders is based on the history, clinical picture and exclusion of alternative diagnoses. Investigations are thus directed towards excluding other possible causes of temperature elevation (e.g. infection, metabolic disorders) and evaluation of the specific complications of hyperthermia.
Patients with a presumed clinical diagnosis of EAC should still have serum electrolytes and creatine kinase measured to exclude exercise associated hyponatraemia and rhabdomyolysis, respectively. Should mental state not rapidly normalize with supine posture, then an urgent finger prick or serum glucose estimation is needed. Collapsed athletes should also have an ECG to identify unrecognized cardiac abnormalities.
All other heat disorders warrant a far more extensive laboratory and radiological work-up, as multiorgan system dysfunction is the rule [2,6]. Tests must include an ECG, serum electrolytes, disseminated intravascular coagulation (DIC) screen, liver function tests, muscle enzyme assays, renal function and urinalysis, serum glucose and a chest X-ray.
Treatment
Exercise-associated collapse
EAC responds rapidly to supine posture (ideally with the legs elevated), rest and oral fluids. Intravenous normal saline is rarely required as few athletes will be profoundly dehydrated. The use of ‘routine’ IV normal saline in collapsed athletes should be actively discouraged as it will worsen exercise associated hyponatraemia where there are persistent, inappropriate antidiuretic hormone levels.
Heatstroke
This is a true medical emergency. Early recognition and aggressive therapy in the field and in hospital can prevent substantial morbidity and mortality. The key management is aggressive cooling. Cooling rates of at least 0.1°C/min should be achievable. Several cooling methods have been proposed, including evaporative cooling, iced water immersion, ice slush, cool water immersion, iced peritoneal lavage and pharmacological methods [7]. A combination of methods is most widely used in EDs. All of the patient’s clothing should be removed and the patient sprayed with a fine mist of tepid water while gentle fanning is commenced (a ceiling fan is ideal). At the same time, areas with vascular beds close to the surface (neck, axillae and groins) should be packed with ice bags. This technique facilitates patient access and monitoring when compared to methods, such as ice-bath immersion even though an iced bath may offer more rapid cooling. Although ice-cold IV fluids can also aid in rapid cooling (as used in therapeutic hypothermia post-cardiac arrest), fluid requirements in heatstroke can be difficult to estimate and balance.
In hospital, shivering, seizures and muscle activity may need to be controlled with pharmacological agents, such as chlorpromazine, benzodiazepines and paralysing agents. Aspirin and paracetamol are ineffective and should be avoided. Intravenous fluids need to be used cautiously and may need titrating to central venous or pulmonary capillary wedge pressures. Maintain adequate oxygenation but avoid hyperoxia. Ventilatory support may be required. Urine flow needs to be maintained with initial volume loading, and later with mannitol or furosemide, to prevent secondary renal injury, especially from rhabdomyolysis. Electrolyte, acid–base and clotting disturbances should be closely monitored and treated by standard measures.
Serotonin toxicity
Treatment of the drug-related hyperthermia involves both specific pharmacological therapy and full supportive and cooling measures, as described above. The objective is to recognize and treat before serious complications occur. In mild cases of the serotonin syndrome, no treatment or small doses of benzodiazepines may be all that is required while awaiting spontaneous resolution. In severe cases, neuromuscular paralysis should be considered early, especially in cases of markedly altered mental state. The duration of treatment used is partly judged on the half-life of the presumed causative agents [4]. Specific antiserotoninergic drugs that can be used include chlorpromazine (12.5–50 mg IM/IV) and cyproheptadine (4–8 mg orally 8-hourly).
Neuroleptic malignant syndrome
Again, early recognition and full supportive care, combined with specific therapy, is the mainstay of treatment. Dopamine agonists, such as bromocriptine, may reduce the duration of the syndrome. It can be administered orally or by nasogastric tube at an initial dose of 2.5–10 mg tds.
Malignant hyperthermia
Dantrolene acts by inhibiting the release of calcium from the sarcoplasmic reticulum and is the specific agent used in the treatment of malignant hyperthermia. It should be given in addition to full supportive care and discontinuing the triggering agents. The dose is 2.5 mg/kg IV initially, repeated every 15 min up to a maximum of 30 mg/kg if needed.
Prognosis and disposition
In heatstroke, both the maximum core temperature and the duration of temperature elevation are predictors of outcome. Prolonged coma and oliguric renal failure are poor prognostic signs [1]. Mortality is still of the order of 10%, but most survivors will not suffer long-term sequelae [1,2]. Any patient with suspected heatstroke should routinely be referred to the intensive care unit for ongoing care. Most cases of EAC will be suitable for short-stay ED treatment or, indeed, simply for treatment on-site in an event medical tent.
Prognosis in the drug-related group of hyperthermia is dependent largely on the degree to which the complications have progressed before definitive and aggressive treatment is begun. Again, early referral to intensive care is indicated. Even with appropriate treatment, mortality for malignant hyperthermia approaches 7%. After recovery, the patient’s medication regimen will need to be reassessed although, in the case of neuroleptic malignant syndrome, it may be possible slowly to reintroduce a neuroleptic agent at a lower dose. With malignant hyperthermia, future anaesthesia will need to be modified to avoid precipitating agents. In addition, family members should be tested for susceptibility.
Controversies
28.2 Hypothermia
Ian Rogers
Introduction
Hypothermia is defined as a core temperature of less than 35°C. This can be measured at a number of sites (including oesophageal, right heart, tympanic and bladder). Rectal remains the routine in most emergency departments (EDs), despite concerns at how rapidly it equilibrates to and reflects true core temperature. Conventionally, hypothermia is divided into three groups: mild (32–35°C), moderate (29–32°C) and severe (<29°C) on the basis of measured core temperature. In a field setting, where core temperature measurements may not be possible, moderate and severe are often grouped together as they typically share the clinical features of absence of shivering and altered mental state. These categorization systems can be used both out of and in hospital as a guide to selecting rewarming therapies and prognosis. Mild hypothermia is considered the stage where thermogenesis is still possible; moderate is characterized by a progressive failure of thermogenesis; and severe by adoption of the temperature of the surrounding environment (poikilothermia) and an increasing risk of malignant cardiac arrhythmia. Nevertheless, there are substantial differences between individuals in their response to hypothermia.
Epidemiology and pathophysiology
Hypothermia may occur in any setting or season [1]. True environmental hypothermia occurring in a healthy patient in an adverse physical environment is less common in clinical practice than that secondary to an underlying disorder. Common precipitants include injury, systemic illness, drug overdose and immersion and are outlined in more detail in Table 28.2.1. The elderly are at greater risk of hypothermia because of reduced metabolic heat production and impaired responses to a cold environment. Alcohol is a common aetiological factor and probably acts by a number of mechanisms, including cutaneous vasodilatation, altered behavioural responses, impaired shivering and hypothalamic dysfunction. Hypothermia in the ED setting is often associated with underlying infection [2].
Table 28.2.1
| Environmental | Cold, wet, windy ambient conditions |
| Cold water immersion | |
| Exhaustion | |
| Trauma | Multitrauma (entrapment, resuscitation, head injury) |
| Minor trauma and immobility (e.g. #NOF, #NOH) | |
| Major burns | |
| Drugs | Ethanol |
| Sedatives (e.g. benzodiazepines) in overdose | |
| Phenothiazines (impaired shivering) | |
| Neurological | CVA |
| Paraplegia | |
| Parkinson’s disease | |
| Endocrine | Hypoglycaemia |
| Hypothyroidism | |
| Hypoadrenalism | |
| Systemic illness | Sepsis |
| Malnutrition |
Clinical features
Despite substantial individual variations, it is still possible to describe the typical patient in each category of hypothermia. The clinical manifestations of hypothermia also depend on the features of the underlying aetiology.
Mild hypothermia manifests clinically as shivering, apathy, ataxia, dysarthria and tachycardia. Moderate hypothermia is typically marked by a loss of shivering, altered mental state, muscular rigidity, bradycardia and hypotension. In severe hypothermia, signs of life may become almost undetectable, with coma, fixed and dilated pupils, areflexia and profound bradycardia and hypotension. The typical cardiac rhythm of severe hypothermia is slow atrial fibrillation. This may degenerate spontaneously, or with rough handling, into ventricular fibrillation or asystole. In the field, moderate and severe hypothermia are often grouped together, with the key clinical feature of absent shivering suggesting the loss of the ability to rewarm without medical intervention.
Many complications may also manifest as part of a hypothermia presentation, although at times it may be difficult to separate cause from effect. These include cardiac arrhythmias, thromboembolism, rhabdomyolysis, renal failure, disseminated intravascular coagulation and pancreatitis.
Clinical investigations
Mild hypothermia with shivering and without apparent underlying illness needs no investigation in the ED.
Moderate or severe hypothermia mandates a comprehensive work-up to seek common precipitants and complications that may not be clinically apparent.
Biochemical and haematological abnormalities are frequently associated with hypothermia [1], although there is no consistent pattern. Blood tests that are indicated include sodium, potassium, glucose, renal function, calcium, phosphate, magnesium, amylase, creatine kinase, ethanol, full blood count and clotting profile. Arterial blood gases, if taken, should be accepted at face value, rather than adjusting for the patient’s temperature.
Impaired ciliary function, stasis of respiratory secretions or aspiration may be expected in moderate-to-severe hypothermia, so chest radiography should be routine. Other radiology may be indicated if a trauma-related aetiology is suspected.
A 12-lead electrocardiograph and continuous ECG monitoring should be routine in moderate-to-severe hypothermia. The typical appearance is slow atrial fibrillation, with J or Osborn waves most prominent in leads II and V3–V6 (Fig. 28.2.1). The J wave is the extra positive deflection after the normal S wave and is more obvious and more commonly seen with increasing severity of hypothermia.
Treatment
General
The general and supportive management of hypothermia victims largely follows that of other critically ill patients. However, some syndrome-specific issues demand careful attention.
Muscle glycogen is the substrate preferentially used by the body to generate heat by shivering. All hypothermics, therefore, need glucose. In mild cases, this can be given orally as sweetened drinks or easily palatable food. With more severe hypothermia, gastric stasis and ileus are common and glucose should be given intravenously: 5% dextrose can be infused at 200 mL/h. Additional volume resuscitation with normal saline or colloid should be gentle, bearing in mind the contracted intravascular space in severe hypothermia and that hypotension that would be classified as severe at a core temperature of 37°C is a normal physiological state at 27°C. All intravenous fluids should be warmed to minimize ongoing cooling. Endotracheal intubation by a skilled operator is safe in severe hypothermia. Intubation is indicated as in any other clinical condition to provide airway protection or to assist in ventilation.
Ventilatory support and, where necessary, manipulation of acid–base status, should be titrated to maintain uncorrected blood gas pH and PCO2 within the normal range.
The slow atrial fibrillation so common in more severe hypothermia is a benign rhythm and requires no chemical or electrical correction. It will revert spontaneously with rewarming. Pulseless ventricular tachycardia and ventricular fibrillation should largely be managed along conventional lines. However, if initial DC shocks are unsuccessful, then others are unlikely to be so until the patient is warmer. Repeat countershocks are generally reapplied with every 1°C increase in core temperature. Magnesium may be the antiarrhythmic drug of choice in hypothermia.
The pharmacokinetics and dynamics of most drugs are substantially altered at low body core temperatures. Indeed, for many of the common drugs used in an ED they are unknown. Insulin is known to be inactive at<30°C. Hyperglycaemia, due in part to loss of insulin activity, is common in hypothermia, but should probably be managed expectantly until sufficient rewarming has occurred to ensure full endogenous insulin activity.
Rewarming therapies
Strategies for rewarming in hypothermia have only a limited evidence base on which to base recommendations. Although more invasive and rapid techniques are advocated for more severe hypothermia, there is little evidence to support this advice. The traditional concern of afterdrop (a paradoxical initial drop in core temperature with rewarming) is probably of little or no relevance in a clinical setting [3].
Rewarming therapies are broadly divided into three groups: endogenous rewarming, which is allowing the body to rewarm by its own endogenous heat production; external exogenous rewarming, which is supplying heat to the outside of the body; and core exogenous rewarming, which is applying the heat centrally. The classification of the commonly utilized rewarming therapies is outlined in Table 28.2.2.
Table 28.2.2
Rewarming therapy classification
| Endogenous rewarming | Warm, dry, wind-free environment |
| Warmed intravenous fluids (to prevent cooling) | |
| External exogenous rewarming | Hot bath immersion |
| Forced-air blankets | |
| Heat packs | |
| Body-to-body contact | |
| Core exogenous rewarming | Warmed, humidified inhalation |
| Left pleural cavity lavage | |
| Extracorporeal circulation |
Endogenous rewarming is a mandatory component of any emergency rewarming protocol. It consists of drying the patient, covering them with blankets, placing them in a warm and wind-free environment and warming any intravenous or oral fluids that are administered. Endogenous rewarming alone can be expected to rewarm at a rate of about 0.75°C/h. For most patients above 32°C (the level at which shivering thermogenesis is typically preserved), endogenous rewarming is the only therapy required. The exception is the exhausted patient in whom shivering has ceased at a core temperature higher than expected. Although more sophisticated techniques, such as bath immersion, will more rapidly rewarm a mildly hypothermic patient, there is no evidence that an increased rewarming rate improves prognosis in this group.
In moderate hypothermia, endogenous heat production is likely to fail progressively and more aggressive exogenous rewarming therapies are indicated. Hot-bath immersion has the theoretical disadvantage of causing peripheral vasodilatation, with shunting of cool blood to the core and convective heat loss. This might be expected to increase core afterdrop and produce circulatory collapse. In fact, rewarming rates of at least 2.5°C/h with minimal afterdrop have been achieved using baths at 43°C [4]. Nevertheless, substantial practical difficulties are obvious with monitoring a more seriously ill patient immersed in a bath. This method of rewarming can only be recommended for otherwise healthy patients who are expected to make a rapid recovery from accidental environmental hypothermia (e.g. immersion in very cold water).
The two therapies that have been best studied and are widely used in moderate hypothermia are forced-air rewarming and warm humidified inhalation [5]. Forced-air rewarming is achieved by covering the patient with a blanket filled with air at 43°C. These devices direct a continuous current of air over the patient’s skin through a series of slits in the patient surface of the blanket. This method produces minimal, if any, afterdrop, is apparently without complication and, combined with warm humidified inhalation, should produce rewarming at about 2.5°C/h. The value of warm humidified inhalation is probably by preventing ongoing respiratory heat loss. Given its widespread availability and lack of complications, it seems reasonable to combine it with forced-air rewarming in moderate hypothermia [6]. Body-to-body contact and chemical heat packs are often recommended as field treatments for all degrees of hypothermia. In mild hypothermia, it seems that the benefit of any heat they deliver is negated by an inhibition of shivering thermogenesis. In more severe cases, where shivering is absent, it may be that even the small amount of exogenous heat they deliver is beneficial, but this remains unproven.
In severe hypothermia, more aggressive exogenous rewarming therapies may be indicated in order rapidly to achieve core temperature above 30°C, the threshold below which malignant cardiac arrhythmias may occur spontaneously. Bladder, gastric or peritoneal lavage with warm fluids are all relatively ineffective methods of heat transfer and, as such, are not recommended for use in emergency situations. When available, full cardiopulmonary bypass achieves rewarming rates of about 7.5°C/h without core afterdrop. Pleural lavage using large volumes of fluid warmed to 40–45°C through an intercostal catheter may be nearly as effective. Both techniques have the advantage of delivering heat to the heart which acts as a heat pump to distribute rewarming to key core organs, but are clearly invasive and carry associated risks. These risks are certainly acceptable in a hypothermic arrest but, in the non-arrested patient, a slower rate of rewarming using forced-air and warm humidified inhalation may be more appropriate.
A suggested rewarming algorithm based on the evidence available to date is shown in Figure 28.2.2.
Prognosis and disposition
Attempts at developing a valid outcome prediction model for hypothermia are likely to be frustrated by its multifactorial aetiology. Recovery with appropriate treatment is likely from accidental environmental hypothermia when there is no associated trauma. To date, the coldest patient to survive accidental hypothermia neurologically intact had an initial measured temperature of 13.7°C [7]. Although increasing severity of hypothermia does worsen prognosis, the major determinant of outcome is the precipitating illness or injury. Reported mortality rates vary from 0 to 85%.
Mild hypothermics without associated illness or injury can be safely managed at home in the care of a responsible adult. Moderate hypothermia may be treatable in a short-stay observation ward, but often requires a longer inpatient stay to manage underlying illness or injury. Severe hypothermics are at risk of multiorgan system complications and should be considered for admission to an intensive care unit.
28.3 Dysbarism
David R Smart
Introduction
This chapter focuses on medical problems that develop secondary to breathing gases at higher than normal atmospheric pressure (dysbarism). This usually occurs in the context of scuba (self-contained underwater breathing apparatus) diving, a popular recreational activity in Australasia. Diving is generally very safe and serious decompression incidents occur approximately 1:10 000 dives. However, because of a high participation rate, between 200 and 300 cases of decompression illness are treated in Australia each year. It is estimated that 10 times that number of divers experience less serious health problems after diving. Emergency physicians are often the first medical staff to assess the diver after a diving accident and it is essential they understand the risks and potential injuries.
Diving physics and physiology
An understanding of pressure and some gas laws is essential to understand the pathophysiology of diving injuries. The air pressure at sea level is 1 atmosphere absolute (ATA). Multiple units are used to measure pressure (Table 28.3.1). For every 10 m a diver descends in sea-water, the pressure increases by 1 ATA. This pressure change impacts on gas spaces within the body according to Boyle’s law.
Table 28.3.1
Atmospheric pressure at sea level in various units
1 Atmosphere absolute (ATA)
101.3 kPa (SI units)
1.013 Bar
10 m of sea water (MSW)
760 mm of mercury (mmHg)
14.7 pounds per square inch (PSI)
Boyle’s law states that, at a constant temperature, the volume of a gas varies inversely to the pressure acting on it:
< ?xml:namespace prefix = "mml" />
where P=pressure, V=volume and k=constant.
The proportionate change in volume is greatest near the surface (Table 28.3.2).
Table 28.3.2
Depth vs pressure and gas volume (Boyle’s law)
| Depth (m) | Absolute pressure (ATA) | Gas volume (%) |
| 0 | 1 | 100 |
| 10 | 2 | 50 |
| 20 | 3 | 33 |
| 30 | 4 | 25 |
| 40 | 5 | 20 |
Dalton’s law states that the total pressure (Pt) exerted by a mixture of gases is equal to the sum of the pressures of the constituent gases (Px, Py, Pz):
Therefore, as divers breathe air at increasing atmospheric pressure, the partial pressures of nitrogen and oxygen increase:
A diver breathing air at 40 m is inhaling a gas with a partial pressure of oxygen equivalent to breathing 100% oxygen at the surface. At partial pressures above 3 ATA, the PN2 affects coordination and judgement (‘nitrogen narcosis’). Oxygen may also become toxic at partial pressures greater than 1 ATA. Recreational scuba diving generally has a limit of 40 m because of these effects.
Henry’s law states that at a constant temperature the amount of a gas that will dissolve in a liquid is proportional to the partial pressure of the gas in contact with the liquid:
where Q=volume of gas dissolved in a liquid, k=constant and Pgas=partial pressure of the gas.
Henry’s law is relevant in diving illness because it is the basis of decompression illness (DCI). As the ambient pressure increases, the diver is exposed to increasing partial pressures of nitrogen, which dissolves in bodily fluids. The amount of nitrogen absorbed depends on both the depth (which determines the partial pressure of nitrogen) and the duration of the dive. Tissues also take up nitrogen at different rates depending on their blood supply and permeability. Eventually, the tissues become saturated with nitrogen and no further absorption occurs. As the diver ascends and ambient pressure decreases, the partial pressure of nitrogen in some tissues will exceed ambient pressure, resulting in tissue supersaturation. If the diver ascends slowly enough, nitrogen diffuses out of the tissues and is transported, safely dissolved in the blood, to the lungs for elimination. This is known as ‘off-gassing’.
If the diver ascends too rapidly, sufficient nitrogen bubbles will form in their body to cause decompression illness. Oxygen does not cause problems because it is rapidly metabolized by the tissues.
Barotrauma
Barotrauma occurs when changes in ambient pressure lead to expansion or contraction of gas within enclosed body cavities. The change in gas volume distorts or tears adjacent tissue. Injury by this mechanism may occur to the middle ear, inner ear, sinuses, lungs, eyes (via the diver’s mask) and, rarely, the gut. Different injury patterns occur in breath-hold divers (snorkellers) compared to those breathing compressed air. Both breath-hold and scuba divers may experience injury of the middle and inner ear, sinuses and eyes if they do not equalize pressures in the gas spaces as they descend. Breath-hold divers are unlikely to injure their lungs as their lung volumes reduce as they descend and return to their original volume as they ascend to the surface by the increasing ambient pressure.
Middle-ear barotrauma
Pathophysiology
Middle-ear barotrauma (MEBT), the most common medical disorder of diving, usually occurs during descent. Increased ambient pressure results in a reduction of middle-ear volume. If equalization of the volume via the eustachian tube is inadequate, a series of pathological changes results. The tympanic membrane (TM) is deformed inwards, causing inflammation and haemorrhage. Middle-ear mucosal oedema is followed by vascular engorgement, effusion, haemorrhage and, rarely, TM rupture.
Clinical features
Symptoms of middle-ear barotraumas include ear pain, tinnitus and conductive hearing loss. Mild vertigo may also be experienced. More severe vertigo and pain occur if water passes through a perforated TM. Severe vertigo and significant sensorineural hearing loss should alert the emergency physician to possible inner-ear barotrauma (IEBT) (see below). MEBT severity is graded by visual inspection of the TM (Table 28.3.3). An audiogram is useful to document any hearing loss.
Table 28.3.3
Grading of severity of middle-ear barotrauma
| Grade 0 | Symptoms without signs |
| Grade 1 | Injection of TM along handle of malleus |
| Grade 2 | Slight haemorrhage within the TM |
| Grade 3 | Gross haemorrhage within the TM |
| Grade 4 | Free blood in middle ear |
| Grade 5 | Perforation of TM |
Treatment
Treatment of MEBT consists of analgesia, decongestants and ear, nose and throat (ENT) referral if there is TM perforation or suspected IEBT. Antibiotics are indicated for TM rupture because of potential contamination with water. The patient should not dive again until symptoms and signs have resolved, any TM perforation has healed, and the eustachian tube is patent.
Inner-ear barotrauma
Pathophysiology
Sudden pressure changes between the middle and inner ears can cause rupture of the round or oval windows or a tear of Reissner’s membrane. This usually occurs during rapid descent without equalizing or forceful Valsalva manoeuvres.
Clinical features
Symptoms include sudden onset of tinnitus, vertigo, nausea and vomiting, vestibular symptoms and profound sensorineural hearing loss, which may not be apparent until the diver has left the water. Onset of symptoms after the dive while performing an activity that increases intracranial pressure (e.g. heavy lifting) suggests IEBT. Coexistent middle-ear barotrauma is absent in about one-third of cases.
The main differential diagnosis is DCI involving the inner ear or vestibular apparatus. Inner-ear DCI usually occurs on deep dives using helium and oxygen mixtures (heliox) and is typically accompanied by other symptoms or signs of DCI. Frequently it is difficult to distinguish between IEBT and vestibular DCI. Isolated inner-ear DCI has been reported in sports divers breathing air.
Treatment
Treatment of IEBT consists of avoidance of activities that increase intracranial pressure and urgent (same day) ENT referral for more detailed assessment and audiometry. Surgical repair may be undertaken when vertiginous symptoms are severe. Vomiting should be treated with antiemetics and the diver kept supine with their head on a pillow. If DCI is excluded, then a 45° semirecumbent position is preferred. If DCI cannot be excluded the diver should have a trial of recompression. In one series, exposure to pressure did not worsen the diver’s condition. The benefit of steroids in IEBT has not been confirmed.
It was thought that further diving was contraindicated after IEBT, but recent case data suggest that diving might be possible following full recovery of hearing.
External ear barotrauma
Ear-canal barotrauma is very rare and only occurs if there is a complete obstruction of the canal (usually by wax or ear plugs), creating a non-communicating gas cavity between the obstruction and the TM. Treatment is symptomatic. ENT specialist referral may be necessary if the TM cannot be visualized.
Sinus barotrauma
Pathophysiology
Mucosal swelling and haemorrhage occur if the communication of the sinuses with the nasopharynx is blocked and equalization of sinus pressure is not possible during descent. The frontal sinuses are most commonly involved.
Clinical features
Sinus pain usually develops during descent. Maxillary sinus involvement can refer pain to the upper teeth or cheek. There may be resolution of the pain at depth, due to mucosal oedema and blood filling the volume deficit left by gas compression. Pain and epistaxis may occur as the diver ascends. The pain usually persists after diving. Tenderness will be noted over the affected sinus. In doubtful cases, a sinus CT (computed tomography) scan will assist the diagnosis.
Treatment
Treatment includes analgesia, decongestants and recommendations to avoid diving until asymptomatic. Antibiotics may be required if secondary infection occurs.
Mask squeeze
If divers fail to exhale air into their masks on descent, the reduced volume inside the mask can cause pain, petechiae and conjunctival haemorrhage. In assessing these divers, it is important to confirm that they have normal visual acuity and fundi are normal. Treatment is with analgesia alone.
Gastrointestinal barotrauma
Expansion of gas within the gastrointestinal tract on ascent can occasionally cause colicky abdominal pain. Rupture of the stomach is rare but has occurred where panic or equipment failure has led to air swallowing and rapid ascent. Presentation is with abdominal pain and distension. Shoulder pain may be due to diaphragmatic irritation or coexisting DCI. Sub-diaphragmatic free air may be visible on an erect chest X-ray. The differential diagnosis includes pulmonary barotrauma, because air can enter the peritoneum via the mediastinum and oesophageal or aortic openings in the diaphragm. The diagnosis is confirmed with endoscopy and surgical repair is necessary.
Dental barotrauma
Severe tooth pain may occur with descent or ascent if air is trapped under a decaying tooth or recent filling. Percussion of the involved tooth is painful. Treatment is with analgesia and dental repair.
Pulmonary barotrauma
Pathophysiology
Breathing compressed air at depth, the diver’s lungs contain greater amounts of gas than they would on the surface. Divers are trained to breathe continuously during ascent or to exhale continuously if they have lost their air supply. Pulmonary barotrauma results when a diver ascends without exhaling adequately and the expanding gas in the lungs exceeds the lung’s elasticity, tearing alveoli. This occurs most commonly when a diver runs out of air, panics and ascends too rapidly. The change in pressure over 1 m near the surface is sufficient to cause lung barotrauma. It has been reported in student divers training in swimming pools and in helicopter escape training. It can also occur with a normal ascent if there is a localized area of lung that does not empty properly, as is possible in divers with asthma, reduced pulmonary compliance or air trapping.
The resultant clinical syndromes depend on the sites at which the air escapes and include arterial gas embolism (AGE), pneumomediastinum and pneumothorax.
Clinical features
Onset of symptoms is usually rapid. If pneumomediastinum or pneumothorax is detected after diving, it is essential to look for features consistent with associated gas embolism. These include impairment or loss of consciousness, cognition impairment including loss of memory, or neurological abnormalities. Sometimes the abnormalities are subtle and tests of cognition and memory should be performed in addition to a detailed history and thorough examination.
Treatment
If AGE is suspected, then the affected individual should be kept supine and urgent recompression treatment is required. Management of AGE is discussed under the heading of decompression illness. Lung barotrauma is regarded as the cause of AGE, however, in early studies, only about 5% of divers with AGE had radiographic evidence of a pneumothorax on plain chest X-ray. Subtle signs of extra-alveolar air suggesting pulmonary barotrauma are present in nearly half with more sophisticated imaging, such as CT.
The reverse also applies. If divers present with a pneumomediastinum or pneumothorax, then they may have up to 50% chance of AGE. The signs of AGE in these circumstances may be subtle with only a brief period of loss of memory or dizziness.
Pneumomediastinum and subcutaneous emphysema can usually be managed conservatively. If symptoms are severe, 100% oxygen can accelerate resolution of the trapped gas. If recompression is required for coexistent AGE, then the pneumomediastinum does not require any specific additional management unless a pneumothorax is present.
Isolated pneumothorax resulting from pulmonary barotrauma is very uncommon. Pneumothorax from pulmonary barotrauma should be managed in the same way as non-diving-related causes and recompression is not necessary. If recompression is required for coexisting AGE, a chest tube with a Heimlich valve should be placed before commencing treatment, because the size of any remaining pneumothorax will increase markedly on depressurization.
Once the acute management of pneumomediastinum and pneumothorax has occurred, the divers should be referred to a diving medical specialist for long-term follow up, because the conditions will impact upon their future diving fitness.
Decompression illness
Classification and criteria for diagnosis
Diving accidents involving bubbles were traditionally divided into decompression sickness (DCS; due to nitrogen bubbles coming out of tissue) and arterial gas embolism (AGE; due to pulmonary barotrauma releasing air into the circulation). DCS is then classified as type I or II. Type I DCS involves the joints or skin only; type II involves all other pain, neurological injury, vestibular and pulmonary symptoms.
In the 1990s, the term ‘decompression illness’ (DCI) was proposed to include both DCS and AGE, for the following reasons:
AGE can be caused by arterialization of venous bubbles released from tissues
Pre-hospital and emergency management prior to recompression is identical
The current classification system describes DCI in terms of four components:
evolution of symptoms (spontaneously resolving/static/progressive/relapsing)
body system affected (musculoskeletal/cutaneous/lymphatic/neurological/vestibular/cardiorespiratory)
For example, a diver may be classified as having acute progressive neurological DCI with no evidence of barotrauma. The classification has been generally adopted in Australia and New Zealand, but not in North America. DCI is a satisfactory term from a management perspective but, from a scientific perspective, it does not describe differing aetiologies and pathophysiology.
Pathophysiology
DCI occurs if excessive nitrogen comes out of solution to form bubbles which gain access to the venous and lymphatic systems or if bubbles form within tissues themselves. The formation of bubbles requires tissues to be supersaturated with nitrogen and for ascent to be excessively rapid. As bubbles form in tissues, they distort tissue architecture, which results in impaired function, pain and inflammation and is probably responsible for most musculoskeletal symptoms.
Many bubbles entering the venous system do not cause symptoms. In fact, using ultrasonic detection methods, intravascular microbubbles are detected after approximately 60% of routine dives. It appears that these bubbles are safely filtered by the lung and diffuse into the alveoli.
Bubbles entering the arterial system are more likely to cause serious problems. This can occur under several circumstances. Large volumes of bubbles may overwhelm the pulmonary filter and arterialize. Bubbles may also bypass the lungs via a right-to-left shunt. Up to one-quarter of the population may have a patent foramen ovale (PFO). Under normal circumstances, the valve over the foramen is kept closed by the pressure difference between the left and right atria. However, during diving, the pressure differential may reverse during a Valsalva manoeuvre or with acute increases in right-sided pressures associated with a large pulmonary gas load.
Alternatively, gas can enter the circulation following pulmonary barotrauma. Air entering the pulmonary arterial system is carried to the pulmonary capillaries where it is trapped and reabsorbed by the alveoli. Air entering the pulmonary venous system, however, will pass through the heart and result in AGE.
Gas bubbles entering the circulation (either from tissues or barotrauma) cause both mechanical and biochemical abnormalities. Trapping in the pulmonary circulation may result in elevation of right heart and pulmonary pressures, leading to increased venous pressures, reduced cardiac output and impairment of tissue microcirculation. Arterial bubbles can cause end- organ ischaemia, although most pass through the capillaries and into the venous system. Most of the deleterious effects are a consequence of secondary inflammation of the vascular endothelium.
Bubble–endothelial interaction activates complement, kinin and coagulation systems and precipitates leucocyte adherence. This results in increased vascular permeability, interstitial oedema and microvascular sludging. The end result is ischaemia and haemoconcentration. Increased vascular permeability of the cerebral circulation will produce cerebral oedema. Vasospasm and reduced flow occurs approximately 1–2 h after bubbles have passed through the arterial tree. This explains the commonly observed clinical course of a diver with a cerebral AGE experiencing an initial deterioration (bubble emboli), followed by spontaneous improvement (bubbles pass through the cerebral capillaries) and then a subsequent secondary deterioration. Animal studies have demonstrated that bubbles travel against arterial flow because of their buoyancy and lodge in the highest point of the body (hence the logic of maintaining supine position after decompression accidents).
Prevention
A number of dive tables and computer algorithms have been developed in an attempt to avoid nitrogen supersaturation of tissues and improve diver safety. Limits are placed on depth, time and ascent rates to allow safe decompression after diving. However, as with all mathematical models which attempt to predict biological behaviour, the dive tables are far from perfect. One series has shown that 39% of DCI cases were within the limits of the table they were using and 24% within the limits of the conservative Canadian Defence and Civil Institute of Environmental Medicine (DCIEM) tables. Historically, it was assumed that DCI could not occur after dives shallower than 10 m, but it is now known that this can occur, particularly if there has been more than one dive per day or multiple ascents. The occurrence of DCI is a probabilistic event where risk increases with increasing depth, time, numbers of dives, numbers of ascents and rates of ascent.
Flying after diving can precipitate DCI. Even if there are no bubbles at the end of the dive, excess nitrogen remains in the tissues and is slowly off-gassed. Further reduction in ambient pressure at altitude can cause bubbles or enlarge pre-existing asymptomatic ones. Current guidelines advise against flying for 12 h after a single short no-decompression dive and 24 h following multiple or decompression dives.
Clinical features
Onset of any symptoms during or in the hours after diving should be regarded as DCI until proven otherwise. Failure to recognize and treat milder cases can lead to permanent morbidity because the disease can progress as the bubble load increases with time. Early onset of symptoms or signs (up to 1 h), especially those that are neurological in nature, indicates a serious decompression emergency and recompression is a time-critical treatment. Milder syndromes of decompression illness may develop up to 24 h after a dive or even later if there is a precipitant, such as heavy exercise or ascent to altitude (e.g. flying).
In general, pulmonary barotrauma that results in AGE has a dramatic clinical presentation and the onset of major neurological symptoms and signs occurs within seconds to minutes after the dive. DCI caused by intravascular bubbles from barotrauma can be rapidly fatal and has a mortality of 5% in sport divers who reach a recompression chamber alive. In Australia, it is the second most common cause of diving-related death after drowning. The brain is the organ most commonly affected, probably because of the vertical positioning of the diver on ascent. Cerebral gas emboli can cause sudden loss of consciousness, convulsions, visual disturbances, deafness, cranial nerve palsies, memory disturbance and asymmetric multiplegias. Hemiplegia is much less common than asymmetric multiplegias. Symptoms almost always begin within 10 min of surfacing. Sudden loss of consciousness on surfacing should be assumed to be due to cerebral gas emboli. Spontaneous improvement may occur with first aid measures, but relapse is common.
Coronary arterial emboli rarely may present as acute myocardial infarction or arrhythmia. Abdominal organs and skin may also be embolized. Elevation of serum creatine kinase (predominantly from skeletal muscle), serum transaminase and lactate dehydrogenase levels in divers with AGE suggests that emboli are distributed more extensively than previously recognized. Peak creatine kinase (CK) may be a marker of the degree and severity of AGE.
Onset of DCI due to gas bubbles coming out of solution can be equally as dramatic (especially after rapid ascents from deep dives), but frequently evolves over hours post-dive. DCI caused by bubbles released from tissues usually causes symptoms within 1 h of completing a dive and 90% of cases have symptoms within 6 h. Neurological symptoms occurring around 30 min after a dive have been associated with PFO. Common symptoms include profound fatigue, myalgia, periarticular pain and headache. Shoulders and elbows are the joints most commonly involved. The pain is usually a dull ache, which may initially be intermittent and migrate from joint to joint, but later becomes constant. Movement aggravates the pain, but local pressure with an inflated sphygmomanometer cuff may improve it. Paraesthesia and numbness may accompany the pain suggesting concomitant neurological disease.
Neurological DCI may present as personality change, headache, memory loss, visual defects, convulsions, confusion and altered level of consciousness. A flat affect may be the only symptom. The vestibular system can also be involved, with dizziness, vertigo, vomiting, nystagmus and ataxia.
Spinal-cord involvement occurs in up to 60% of cases of neurological DCI. The exact cause of spinal DCI is still debated. It may be a result of venous infarction of the cord due to obstruction of the epidural vertebral venous plexus. Other explanations include ischaemia and inflammation from bubble emboli or the formation of local bubbles within the spinal cord (autochthonous bubbles). Symptoms include back pain, paraesthesia and paraplegia, with bowel and bladder involvement. It is potentially disastrous to misdiagnose back pain coming on a few minutes after a dive as musculoskeletal pain and not consider spinal cord DCI.
If the bubble load overwhelms the pulmonary filter, a diver can present with a syndrome known as pulmonary DCI or ‘the chokes’. The symptoms of this syndrome include dyspnoea, pleuritic substernal chest pain, cough, pink frothy sputum, cyanosis and haemoptysis. It indicates the diver has sustained a large intravascular gas load, so a careful inquiry about other symptoms of DCI is mandatory and, if present, recompression is advised. Diving-related pulmonary oedema and salt-water aspiration syndrome are the major differential diagnoses.
A variety of rashes may be caused by cutaneous bubbles; however, these syndromes affect less than 10% of divers. The most common presentations are pruritus with no rash, a scarlatinaform rash with pruritus and cutis marmorata. Cutis marmorata begins as a spreading erythema but subsequently develops a marbled appearance of pale areas surrounded by cyanotic mottling.
Assessment of the injured diver
The injured diver requires simultaneous assessment and treatment. One hundred per cent oxygen treatment should be continued during the assessment. If the history suggests AGE, the patient should be kept in the horizontal position to avoid re-embolization. If symptoms are progressing rapidly, the examination should be brief but thorough so as to ensure rapid access to recompression. In serious cases, some of the historical information may be obtained once the diver is receiving treatment in the recompression chamber.
The diagnosis of DCI is made on history and examination. A full dive history must be obtained, in addition to the medical history. Important details include the number of dives over recent days, depth, bottom time (the time from beginning descent to beginning direct ascent), performance of any decompression or safety stops, dive complications, such as rapid ascents, surface interval between dives and the time interval between completing the dive and onset of symptoms. Previous dive experience, equipment used and gases breathed should also be recorded. A history of using surface supply equipment (the ‘Hookah’ apparatus) should alert the examining physician to the possibility of carbon monoxide poisoning and carboxyhaemoglobin measurement is required. Cold water, hard exercise during the dive, increasing age, multiple ascents and repetitive dives are predisposing factors in the development of DCI. Any exposure to altitude (>300 m) or heavy exercise post-dive should be recorded.
A thorough examination, particularly of the neurological system, to detect subtle abnormalities is required. It is also helpful to perform basic tests of cognitive function, such as the mini-mental state examination. For milder static DCI syndromes with delayed presentation, the sharpened Romberg test provides useful information. It is performed by asking the patient to stand heel-to-toe with open palms on opposite shoulders. The patient is stable. They are then asked to close their eyes and timed until they lose balance or achieve 60 s. A score of less than 60 s is suggestive of DCI in an injured diver. This test should not be performed if the history was suggestive of AGE or if there are neurological symptoms or signs.
Clinical investigations
[/not-level-membership-for-emergency-medicine-category]
