[level-membership-for-endocrinology-diabetes-and-metabolism-category]CHAPTER 27
Biochemical aspects of anaemia
CHAPTER OUTLINE
THE FORMATION, STRUCTURE AND FUNCTION OF THE NORMAL RED CELL
ANAEMIAS ASSOCIATED WITH A REDUCTION IN RED CELL PRODUCTION
The megaloblastic anaemias resulting from vitamin B12 and folate deficiency
Anaemias due to reduction in red cell production: inherited causes
Anaemias associated with reduction in red cell production: acquired causes
INTRODUCTION
Blood is often considered a liquid organ that acts as the transport system of the body. It is composed of a liquid (plasma) in which cells are suspended. There are three types of cells. Red cells are the most numerous; they play a vital role in the provision of oxygen to the organs and peripheral tissues. The white cells comprise the phagocytes, granulocytes and monocytes, which are mobilized in the blood to sites of infection and inflammation where they pass into the tissues to engulf and destroy bacteria and other objects deemed foreign by the body, and the lymphocytes which, via the secretion of antibodies and cytokines, coordinate defence against acute infection and maintain long term immune surveillance. Finally, the platelets, in conjunction with coagulation proteins in the plasma, allow prompt and effective haemostasis in the event of injury.
This chapter will focus on the structure and function of the red cells, or erythrocytes, and the pathophysiological mechanisms underlying the disorders of red cells that result in anaemia.
THE FORMATION, STRUCTURE AND FUNCTION OF THE NORMAL RED CELL
Formation
The first primitive blood cells develop in the embryo during the third week after conception and are seen in the yolk sac. These cells migrate to the liver and spleen, which become the major sites of blood formation (erythropoiesis) until after birth. From approximately five months’ gestation, erythropoiesis begins to occur in bone marrow. This is called medullary erythropoiesis (as opposed to extramedullary erythropoiesis, when cells are produced in other sites such as the liver and spleen). By birth, medullary erythropoiesis is occurring in the marrow cavity of nearly every bone in the body. With maturity, many of the marrow cavities become replaced with fat and, by adulthood, erythropoiesis is limited to the axial skeleton (sternum, vertebrae and pelvis only). The other sites retain the ability to produce blood cells if marrow function is compromised by various pathological processes.
Red cells survive for approximately 120 days and therefore the bone marrow needs to generate new haemopoietic cells continuously. It is estimated that the average adult requires synthesis of 1011 red cells per day. Red cells originate from pluripotent uncommitted stem cells that are capable of producing any type of blood cell. The earliest recognizable committed progenitor for erythroid cells is the colony forming unit – granulocyte, erythroid, megakaryocyte, macrophage (CFU-GEMM). This has a limited self-renewal capacity and may mature into the various haemopoietic lineages depending on various stimuli within the bone marrow microenvironment. The first unique erythroid progenitor is the burst forming unit – erythroid (BFU-E), from which further progeny develop into recognizable erythroid cells (see Fig. 27.1). Maturation of the erythroid cells involves primarily haemoglobin synthesis, which is vital for the subsequent function of the cell, and also condensation and eventual extrusion of the nucleus, removing the ability of the cell to proliferate. This process takes approximately seven days and results in the release of a reticulocyte, an immature red cell slightly larger than the mature red cell with a blue tinged cytoplasm owing to residual nuclear material. The reticulocyte then travels from the marrow to the spleen where the residual nuclear remnants are removed, producing the mature red cell.
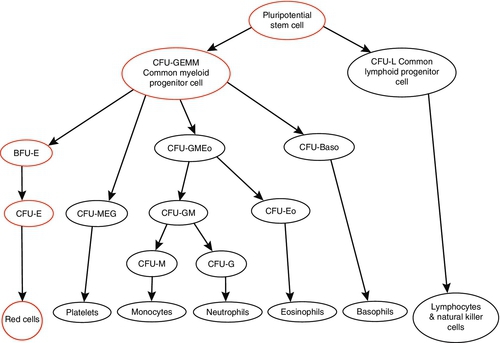
FIGURE 27.1 Schematic representation of haemopoiesis. BFU, burst forming unit; CFU, colony forming unit; E, erythroid; Eo, eosinophil; GEMM, granulocyte, erythroid, monocyte, megakaryocyte; GM, granulocyte, monocyte; L, lymphocyte; M, monocyte; MEG, megakaryocyte.
Thus, effective erythropoiesis requires a delicate balance between maintaining a pluripotent stem cell pool and allowing the development and terminal differentiation of the progeny to mature erythroid cells. The exact process by which this is achieved has not been elucidated but involves many transcription factors such as runt-related transcription factor 1 (RUNX1), acute myeloid leukaemia 1 (AML-1) protein, TEL oncogene (ETV6) and mixed lineage leukaemia (MLL) protein orchestrating, either alone or in conjunction with others, the intricate signalling processes required to produce mature erythroid cells. The dysregulation of these transcription factors, often via genetic mutations, results in a range of haematological malignancies that afford scientists a means of studying the role of these factors in normal erythropoiesis. GATA-1, with its cofactor FOG-1 (friend of GATA-1), is one of the most important transcription factors involved in the terminal differentiation of the erythroid cell. Its name is derived from its ability to bind to DNA sites with the consensus sequence (AT)GATA(AG) within the promoter regions of many of the genes encoding red cell membrane proteins and enzymes.
Erythropoiesis is the production of mature, haemoglobin-rich, red cells that carry oxygen to the tissues of the body. It is therefore not surprising that hypoxia is a major driver of erythropoiesis via the induction of hypoxia inducible factor (HIF), a transcription factor pivotal to coordinating the body’s response to hypoxia. This binds to hypoxia response elements (HREs), resulting in the activation of genes for proteins that play a vital role in oxygen delivery, such as vascular endothelial growth factor (leading to new blood vessel formation) and erythropoietin (a hormone which stimulates erythropoiesis).
Structure of the red cell
Red cells have a biconcave disc shape and a membrane structure that gives them particular strength and deformability to allow their passage through capillary networks, allowing the effective oxygenation of the tissues. The membrane consists of a phospholipid bilayer, with charged hydrophilic phosphatidyl groups forming the outer and inner surfaces, and a hydrophobic interior. It is stabilized by integral membrane proteins that span the lipid bilayer, providing numerous functions, including the transmembrane passage of ions and interaction with the proteins of the cytoskeleton below. The cytoskeletal protein spectrin forms tetramers that aggregate into a hexameric scaffolding that lies on the cytoplasmic surface of the cell membrane to preserve its shape and structure. The ankyrins anchor membrane proteins to spectrin and maintains correct orientation of membrane ion channels. Abnormalities in any of these proteins result in an alteration of shape and flexibility of the red cell with a consequent reduction in survival. This is the pathological basis of the inherited haemolytic anaemias, of which the commonest is hereditary spherocytosis.
Function of the red cell
The most important function of the red cells is to transport oxygen from the lungs to the tissues and to return the carbon dioxide they produce to the lungs. Normal adult haemoglobin consists of two α globin chains and two β globin chains; a haem prosthetic group is bound to each chain. Haem comprises a tetrapyrrole IXα ring bound to a single Fe2 + ion. The binding of a single molecule of oxygen to a haem group results in a change in conformation of the haemoglobin molecule that promotes further oxygen binding to the other subunits. These allosteric interactions allow for the efficient uptake and release of oxygen. In order to achieve this, the red cell requires a source of both ATP and reducing power. ATP generation is vital to maintain the deformability of the membrane and to regulate water and ion exchange. The reducing power of the red cell is required both to protect against the oxidation of the lipid bilayer and to reduce the methaemoglobin that is formed when ferrous iron (Fe2 +) is oxidized to its ferric form (Fe3 +) back to functional deoxyhaemoglobin. Anaerobic glycolysis is the main pathway responsible for supplying the cell with both ATP and nicotinamide adenine dinucleotide (reduced) (NADH), a cofactor for methaemoglobin reductase, the enzyme that catalyses the reduction of methaemoglobin to functional haemoglobin (see Fig. 27.2).
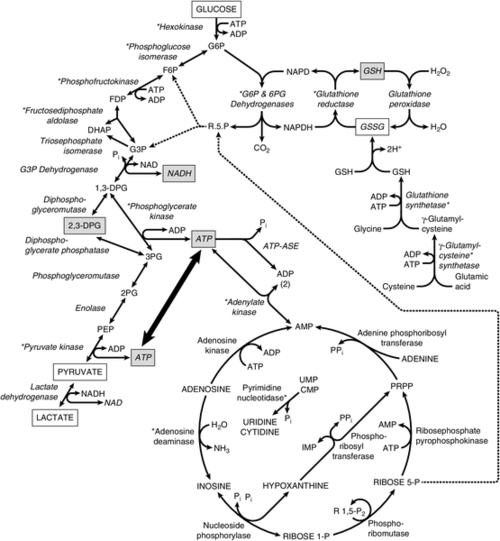
FIGURE 27.2 Pathways of energy metabolism in human erythrocytes. Glucose 6-phosphate (G6P) may be degraded anaerobically to two molecules of lactate via the Embden–Meyerhof (glycolytic) pathway (on the left) or oxidatively via the dehydrogenases of the pentose phosphate pathway. Ribose 5-phosphate (R5P) can re-enter anaerobic glycolysis after conversion to fructose 6-phosphate (F6P) and glyceraldehyde 3-phosphate by enzymes of the terminal pentose phosphate pathway and is also a product of adenosine or inosine degradation. 2,3-Diphosphoglycerate (2,3-DPG) may be generated instead of ATP by diversion of triose through the Rapoport–Luebering shunt. Glutathione may be directly synthesized from constituent amino acids, and its cycling from oxidized (GSSG) to reduced form (GSH) is dependent on generation of reduced pyridine cofactor (NADPH). Asterisks (*) indicate those enzymes found to be defective in association with hereditary haemolytic anaemia. Abbreviations: ADP, adenosine diphosphate; AMP, adenosine monophosphate; ATP, adenosine triphosphate; ATPase, adenosine triphosphatase; CMP, cytidine monophosphate; DHAP, dihydroxyacetone phosphate; 1,3-DPG, 1,3-diphospholycerate; 2,3-DPG, 2,3-diphosphoglycerate; FDP, fructose 1,6-diphosphate; F6P, fructose 6-phosphate; G3P, glyceraldehyde 3-phosphate; G6P, glucose 6-phosphate; GSH, reduced glutathione; GSSG, oxidized glutathione; IMP, inosine monophosphate; NAD, nicotinamide adenine dinucleotide (oxidized); NADH, nicotinamide adenine dinucleotide (reduced); NADP, nicotinamide adenine dinucleotide phosphate (oxidized); NADPH, nicotinamide adenine dinucleotide phosphate (reduced); PEP, phosphoenolpyruvate; 2PG, 2-phosphoglycerate; 3PG, 3-phosphoglycerate; Pi, inorganic phosphate; PPi, inorganic pyrophosphate; PRPP, phosphoribosyl pyrophosphate; RIP, ribose 1-phosphate; R1,5-P2, ribose 1,5-diphosphate; R5P, ribose 5-phosphate; UMP, uridine monophosphate.
The pentose phosphate pathway produces nicotinamide adenine dinucleotide phosphate (reduced) (NADPH) and pentose sugars. The latter are returned to the mainstream of glycolysis if necessary. Reduced nicotinamide adenine dinucleotide phosphate is essential for keeping glutathione in the reduced state, thus enabling it to perform its antioxidant functions. These consist mainly of eliminating hydrogen peroxide and free radicals and thus preventing oxidation of the red cell membrane or haemoglobin. The Rapoport–Luebering shunt is responsible for producing 2,3-diphosphoglycerate (2,3-DPG) from the 1,3-diphosphoglycerate that is produced in anaerobic glycolysis. 2,3-Diphosphoglycerate modulates the oxygen affinity of haemoglobin (see Chapter 5).
Various non-glycolytic enzymes contribute to red cell metabolism in a complementary fashion. Some of these are involved in glutathione metabolism as shown in Figure 27.2. Others catalyse nucleotide metabolism and participate in adenylate salvage pathways. During maturation of the reticulocyte, the ribosomal DNA is catabolized to its constituent nucleotides by the enzyme ribonuclease. The resulting nucleotides of adenine are useful to the cell and are processed through appropriate salvage pathways. Nucleotides of pyrimidine, however, can be harmful and must be eliminated. This is achieved through their dephosphorylation by a specific enzyme, pyrimidine 5′-nucleotidase, following which the resultant nucleosides leave the cell by passive diffusion.
As the red cell matures, it extrudes its nucleus and mitochondria, and becomes incapable of protein synthesis. Thus, none of the enzymes contained within the red cell can be replaced and the decline in metabolic activity results in a more rigid and fragile cell membrane. This becomes damaged during its passage through the capillaries and the red cell is eventually removed by cells of the reticuloendothelial system, particularly the macrophages of the spleen.
ANAEMIA
Anaemia is defined as a blood haemoglobin concentration below that which would be expected for a healthy individual of that age or sex. Regardless of its cause, the symptoms of anaemia are similar, reflecting the effects of poor oxygen delivery to tissues. These symptoms may often be vague and include tiredness, headaches and breathlessness.
Anaemia may be classified on the basis of the size of the red cells (microcytic, normocytic and macrocytic). This chapter, however, will discuss anaemias using a classification based on kinetic aspects of red cell production as follows:
• reduction in red cell production
• inherited causes
• acquired causes
• increased red cell loss
• haemolysis (inherited or acquired).
ANAEMIAS ASSOCIATED WITH A REDUCTION IN RED CELL PRODUCTION
Iron deficiency anaemia
Iron deficiency anaemia is one of the commonest causes of anaemia worldwide and has significant socioeconomic consequences. Other haematinics essential for adequate erythropoiesis are vitamin B12 and folate.
Iron physiology
Iron plays a vital role in many metabolic processes. It is an essential component of both haemoglobin and myoglobin and also plays an important part in electron transfer and the generation of energy (e.g. in cytochrome c oxidase and catalase). Non-haem iron is also important in DNA synthesis as it is essential for the function of ribonucleotide reductase. Many of these roles are aided by the fact that iron may exist in two stable oxidized states, as soluble ferrous (Fe2 +) iron and as insoluble ferric (Fe3 +) iron.
Iron requirements
The total body content of iron in normal adults is 3–4 g (see Table 27.1). Approximately two-thirds is contained in haemoglobin and the remainder in myoglobin, iron containing enzymes and the reticuloendothelial cells. Approximately 30 mg of iron is required per 24 h for haemoglobin in new red cells and the majority of this is supplied from the reticuloendothelial macrophages that recycle the iron from senescent red cells. However, approximately 1 mg of iron is lost from the body every 24 h and needs to be replaced from dietary sources. Women of childbearing age have additional blood loss from menses or pregnancy, resulting in a further 0.5 mg average loss of iron per 24 h. Infants and adolescents require additional iron during periods of rapid growth. A normal diet provides approximately 15 mg of iron but most is in the form of insoluble iron complexes and only 5–10% is absorbed.

Iron absorption
Absorption of iron occurs in the proximal duodenum. It is aided by the low pH of the hydrochloric acid secreted into the gastric lumen and the presence of reducing agents such as ascorbic acid, which help maintain the iron in the more soluble ferrous form. In contrast, other agents such as tannins, phytates and phosphates bind the iron within the intestinal lumen, inhibiting its absorption.
Although iron has an essential metabolic function, excess is toxic and therefore it is not surprising that iron metabolism is tightly regulated.
Hepcidin plays a critical role in iron metabolism. This 25 amino acid peptide is produced predominantly in the liver, from an 84 amino acid precursor. Its synthesis is stimulated by iron and inflammation (driven by IL-6) and inhibited by iron deficiency, hypoxia (via HIF) and conditions such as ineffective erythropoiesis where the concentration of growth differentiating factor 15 (GDF-15) in the developing erythroblasts is increased. Hepcidin negatively regulates iron homoeostasis by binding to ferroportin: the resultant hepcidin–ferroportin complex is phosphorylated, internalized by the cell, ubiquitinated and subsequently degraded in the lysosomes. Ferroportin is a transmembrane protein that functions as a basolateral exporter of iron, playing an essential role in the release of iron from macrophages, intestinal enterocytes and placental syncytiotrophoblasts, allowing the uptake and transfer of iron by transferrin to sites of need. Thus the stimulation of hepcidin production results in reduced iron absorption and mobilization (see Fig. 27.3). Genetic haemochromatosis (see Chapter 14), where there is significant iron accumulation and deposition in various body tissues resulting in cirrhosis, cardiac failure and diabetes, is a consequence of a defect in the hepcidin pathway.
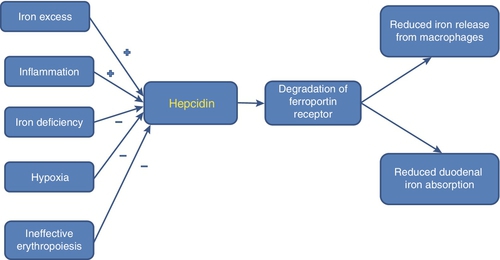
FIGURE 27.3 Factors affecting hepcidin production by the liver and consequent iron absorption and utilization. When hepcidin production is stimulated by conditions such as iron excess or inflammation, it binds to and results in the degradation of ferroportin in enterocytes and macrophages, preventing the absorption and utilization of iron. Conditions associated with iron deficiency or ineffective haemopoiesis inhibit hepcidin production, thereby resulting in iron accumulation.
Iron transport and storage
Transferrin, a single chain polypeptide produced in the liver, acts as the plasma transporter of iron, binding two molecules of ferric iron per molecule. The transferrin–iron complex binds to transferrin receptors, cell surface glycoproteins consisting of two identical 95 kDa subunits linked by a disulphide bridge, localized on cells dependent on iron for function, e.g. for the production of haemoglobin, myoglobin or iron dependent enzymes. After binding, the complex is internalized and degraded, releasing the iron. Ferritin is the primary iron storage protein, found in all tissues but especially the reticuloendothelial system, with small amounts in the plasma. Each molecule of ferritin can store up to 4000 atoms of iron and consists of 24 subunits of two different types, H and L. Variation in the proportion of H and L subunits results in molecular heterogeneity, for example heart and red cell ferritin contains more H subunits than ferritin from liver and spleen, which are rich in L subunits.
The synthesis of all the proteins involved in iron metabolism is finely controlled at the molecular level by having iron response elements (IREs) within their mRNA. In iron replete situations, iron response proteins (IRPs), present in the cytoplasm of many cells, are non-functional by virtue of the binding of an iron–sulphur complex. However, when the cell is iron deplete, the IRPs bind to IREs to stimulate the gene transcription of proteins that enhance iron absorptive capacity.
Causes of iron deficiency anaemia
Iron deficiency is most commonly secondary to blood loss, usually from gastrointestinal or menstrual sources. In the developing world, hookworm infestation resulting in gastrointestinal blood loss is the commonest cause of iron deficiency anaemia; the resultant reduced working capacity has significant socioeconomic impact. Coeliac disease, which is associated with flattening of the duodenal mucosal villi with subsequent malabsorption, accounts for 5% of cases of iron deficiency. Other causes include inadequate dietary iron during periods of rapid growth such as puberty.
Clinical consequences of iron deficiency
Iron deficiency causes a hypochromic microcytic anaemia (see Chapter 26) with additional clinical features such as koilonychia (a spoon-shaped malformation of the nails), angular stomatitis, glossitis, alopecia and pica. There are concerns that significant iron deficiency in infants results in impaired mental development.
Laboratory determination of iron status
Red cell parameters
Iron deficiency is associated with a reduction in the red cell mean corpuscular volume (MCV) and mean cell haemoglobin concentration (MCHC).
Hypochromic red cells
Some automated analysers allow the determination of numbers of hypochromic red cells. Counts of > 6% are suggestive of iron deficiency.
Serum iron
Measurement of serum iron concentration alone provides little useful information of iron status as values show considerable variation within normal individuals. Low concentrations are seen in iron deficiency but are also seen in the anaemia of chronic disease and after surgery.
Serum ferritin
This parameter provides an accurate reflection of iron stores in healthy individuals with normal concentrations ranging from 15 μg/L to 300 μg/L. However, it is an acute phase reactant and its concentration may therefore be falsely normal in iron deficiency with coexistent inflammation.
Serum iron binding capacity, transferrin and transferrin saturation
In the plasma, each molecule of transferrin binds up to two iron ions. Thus, the total iron binding capacity (TIBC) of plasma reflects serum transferrin concentration. It is raised in iron deficiency and pregnancy. Transferrin saturation is a ratio of serum iron concentration to TIBC expressed as a percentage. A value of < 16% is suggestive of iron deficiency whereas elevated fasting transferrin saturations of > 55% in males and > 50% in females are suggestive of iron overload. In the anaemia of chronic disease, both serum iron and TIBC are reduced, thus the transferrin saturation is usually normal.
Serum transferrin receptor
As erythroblasts mature, cleavage releases the extracellular portion of the transferrin receptor into the plasma. The concentration of the transferrin receptor rises in iron deficiency but not in the anaemia of chronic disease. Transferrin receptor concentration can be measured by enzyme immunoassay, although the assay is only available in a few specialist centres.
Hepcidin
Assays for this protein are under development but as yet are poorly standardized. Controversy exists as to whether urinary or serum hepcidin measurements are superior.
Bone marrow aspiration
At present, staining bone marrow aspirates for iron is the ‘gold standard’ for assessing iron deficiency.
The megaloblastic anaemias resulting from vitamin B12 and folate deficiency
The megaloblastic anaemias give rise to characteristic morphological appearances: the red cells are macrocytic and hypersegmented neutrophils are present (see Chapter 26). Ineffective erythropoiesis is apparent, manifest in the serum by raised unconjugated bilirubin and reduced haptoglobin concentrations and markedly raised lactate dehydrogenase (LDH) activity. Megaloblastosis arises when either drugs or deficiencies of cobalamin or folate interfere with DNA synthesis.
Folate metabolism
Folates are a group of compounds derived from pteroylglutamic acid (see Fig. 27.4). Excellent dietary sources of folates include green leafy vegetables, liver, yeast and nuts (see Table 27.1).
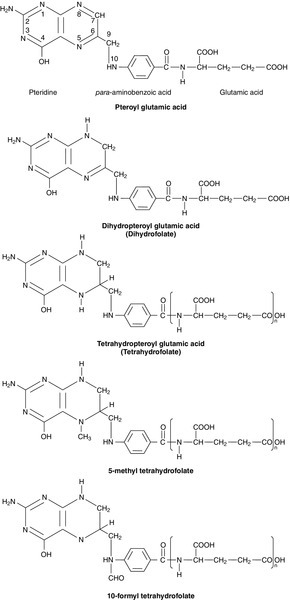
FIGURE 27.4 Pteroylglutamic acid and derivatives. The nitrogen atoms in the 5 and 10 positions are the sites involved in single carbon transfer reactions. Tetrahydrofolate and its 5-methyl and 10-formyl derivatives are shown as polyglutamates.
Folate requirements
A normal diet in the developed world contains about 250 μg of folates per day but < 1% of this is in the form of folic acid. The principal naturally occurring folates are tetrahydrofolate (THF), 5-methyl tetrahydrofolate (5-methyl THF) and 10-formyl tetrahydrofolate (10-formyl THF), which are polyglutamated. Total body folate in the adult is 10 mg, providing sufficient stores for four months given a daily metabolic requirement of 100 μg (see Table 27.1).
Absorption of folate
The majority of folate is absorbed in the upper small intestine, where pteroylpolyglutamate hydrolase on the mucosal brush border hydrolyses folate polyglutamates to the monoglutamate form. They are then converted to 5-methyl THF and transported by an active carrier mediated mechanism across the enterocytes and into the portal blood.
Within the plasma, one third of 5-methyl THF is loosely bound to albumin whilst the remainder is unbound. It is taken up by active transport into replicating cells. Here the folates play a vital role by acting as coenzymes in the transfer of single carbon groups in pathways involved in the synthesis of purines, pyrimidines (for DNA and RNA synthesis) and methionine (for methyl donor reactions) (see Table 27.2).
TABLE 27.2
Reactions in which pteroylglutamate acid derivatives are involved
| Folate derivative involved as: | Reaction |
| Single carbon donor | |
| 10-formyl tetrahydrofolate (THF) | Purine synthesis |
| 5,10-methylene THF | Thymine synthesis |
| 5,10-methylene THF | Synthesis of glycine from CO2 and 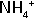 |
| 5-methyl THF | Homocysteine → methionine |
| Single carbon acceptor | |
| THF (forms 5,10-methylene THF) | Serine → glycine |
| THF (forms 5-formimino THF) | Breakdown of histidine |
Causes of folate deficiency
• Malabsorption: diseases of the small intestine, coeliac disease, tropical sprue.
• Excessive utilization or loss: pregnancy, prematurity, haemolytic anaemias, skin conditions, inflammatory conditions, haemodialysis.
• Antifolate drugs: anticonvulsants, trimethoprim, methotrexate.
Features of folate deficiency
Folate deficiency is thought to cause megaloblastic anaemia by inhibiting thymidylate synthesis, which is required for DNA production. It causes a macrocytic anaemia with hypersegmented neutrophils and a megaloblastic bone marrow. It may be associated with a pancytopenia. For further details on morphology, see Chapter 26.
Laboratory determination of folate status
Both serum and red cell folate concentrations can be measured by immunoassay. Serum folate concentration is affected by recent dietary intake and may be low after only a short period of inadequate diet. Conversely, serum folate concentration may rise in vitamin B12 deficiency owing to a blockage in the conversion of 5-methyl THF, the major circulating form, to THF.
Red cell folate may be a more accurate test of body folate status as its concentration is not affected by recent diet. However, red cell folate concentration is also reduced in vitamin B12 deficiency.
Vitamin B12 metabolism
Structure of vitamin B12
The vitamin B12 (cobalamin) molecule is centred on an atom of cobalt; this is the only known function of cobalt in humans. The surrounding corrin ring is made up of four pyrrole units in a similar manner to porphyrins (see Fig. 27.5), with variations in two substitutions above and below the ring giving rise to the various forms of the vitamin. Vitamin B12 is mainly contained within the mitochondria in the 5′-deoxyadenosyl form, where it plays a role in the conversion of L-methylmalonyl-CoA to succinyl-CoA. The other main form, methylcobalamin, is found within the cytoplasm and plasma where it is a cofactor for the conversion of homocysteine to methionine, a vital part of the pathway that creates a universal methyl donor (see Fig. 27.6).
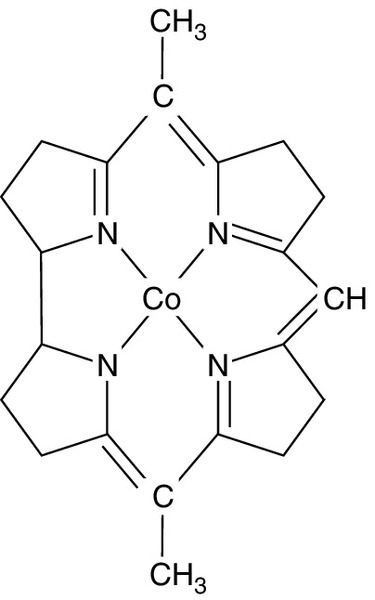
FIGURE 27.5 The corrin ring surrounding an atom of cobalt that forms the core of cobalamin. The other two ligands to cobalt (above and below the plane of the corrin ring), and substituents on the pyrrole units, are not shown.
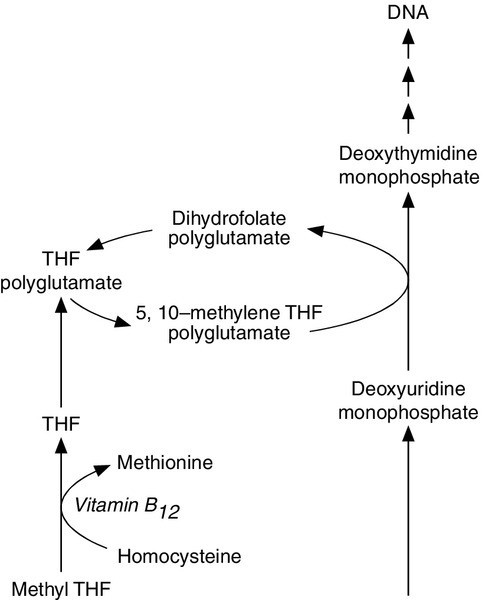
FIGURE 27.6 The link between folate and vitamin B12 deficiency in megaloblastic anaemia. 5,10-Methylene tetrahydrofolate polyglutamate is required for a rate-limiting step in DNA synthesis, the conversion of deoxyuridine monophosphate to deoxythymidine monophosphate. Vitamin B12 is required in one of the reactions converting the main circulating form of folate, 5-methyl THF, to 5,10-methylene THF.
Source of vitamin B12
Vitamin B12 is synthesized only by microorganisms and the only source for humans is food of animal origin. Liver is the richest source of vitamin B12, but it is present in almost all animal products, including milk. No vegetable food source contains significant amounts of vitamin B12 unless contaminated by bacteria (see Table 27.1).
Vitamin B12 requirements
An adult human requires only 1 μg of vitamin B12 per 24 h and has stores of 2–3 mg. Thus, it can take 3–4 years for vitamin B12 deficiency to develop (see Table 27.1).
Absorption of vitamin B12
Dietary vitamin B12 is bound to food proteins and must be freed by gastric acid. The parietal cells of the stomach secrete the glycoprotein intrinsic factor (IF), which combines with vitamin B12 to form a complex, which resists proteolytic digestion. The vitamin B12–IF complex passes through the small intestine until it binds to cubilin, a surface receptor on the enterocytes of the terminal ileum, where it is internalized. The enterocytes of the ileum have a limited capacity to absorb vitamin B12 because of a limited number of receptor sites; about half of a dose of 1 μg of vitamin B12 will be absorbed, the proportion falling markedly with higher doses. The enterocytes have a refractory period of about 6 h before they can absorb any more vitamin B12.
Within the enterocyte, the vitamin B12 is liberated and, after binding to the β-globulin carrier transcobalamin II (TCII), is released into the blood. The TCII–vitamin B12 complex is termed ‘holotranscobalamin’. Transcobalamin II readily releases the vitamin B12 to the bone marrow and other tissues, and, for this reason, holotranscobalamin is also described as ‘active B12’. Other transcobalamins also exist; transcobalamin I and III derive mainly from specific granules found in neutrophils and bind vitamin B12 tightly, in contrast to TCII, and do not release it into the tissues. Congenital TCII deficiency can occur, the affected infant presenting with megaloblastic anaemia a few weeks after birth.
Causes of vitamin B12 deficiency
• Nutritional: this is rare and is only seen in very strict vegans.
• Malabsorption: the commonest cause of vitamin B12 malabsorption is the autoimmune condition, pernicious anaemia, where gastric atrophy develops secondary to an inflammatory infiltrate. Autoantibodies to gastric parietal cells are seen in 90% of individuals with the condition and 50% develop IF autoantibodies, which either prevent vitamin B12–IF complex formation (binding antibodies) or the subsequent attachment of the vitamin B12 to the enterocyte mucosa (blocking antibodies).
• Gastric causes: such as total or partial gastrectomy.
• Intestinal causes: ileal resection and diseases of the terminal ileum such as Crohn disease or tropical sprue prevent vitamin B12 absorption. Deficiency is also associated with intestinal blind loop syndrome (because of metabolism of the vitamin B12 by the overgrowth of intestinal bacteria) and fish tapeworm (Diphyllobothrium latum), which binds cobalamin, preventing its absorption.
• Acquired: prolonged nitrous oxide exposure oxidizes methylcobalamin to an inactive state and results in functional vitamin B12 deficiency. This has been seen in dentists and anaesthetists. Metformin may be associated with low serum vitamin B12 concentrations. The exact mechanism for this is unknown but it has been postulated to be caused by metformin interfering with the calcium dependent channels responsible for the ileal absorption of the vitamin.
Features of vitamin B12 deficiency
Vitamin B12 is a coenzyme in the interconversion of the different forms of folate (see Fig. 27.6), so its deficiency results in a megaloblastic anaemia identical to that seen in folate deficiency. Deficiency may also result in neurological symptoms (e.g. peripheral neuropathy, dysfunction of the posterior columns of the spinal cord and sometimes psychotic illnesses and dementia). The precise mechanism for this has not been elucidated but may be linked with impaired conversion of homocysteine to methionine, resulting in either reduced availability of S-adenosylmethionine impairing sphingomyelin synthesis or in the toxic accumulation of the homocysteine metabolite S-adenosylhomocysteine.
Laboratory determination of vitamin B12 status
Serum vitamin B12
This is usually measured by automated immunoassay. Normal concentrations are 160–1000 ng/L. A low concentration is not specific for vitamin B12 deficiency and may be found in one-third of patients with folate deficiency and in normal pregnancy. Measurement holotranscobalamin (TCII – bound vitamin B12, or ‘active B12’) may be a more sensitive and specific indicator of physiologically relevant vitamin B12 deficiency.
Serum methylmalonate and homocysteine
Vitamin B12 deficiency results in the elevation of methylmalonate and homocysteine concentrations (see Fig. 27.6). However, concentrations of both compounds fluctuate and may be raised in renal impairment, smoking and (on single occasions) in up to 30% of normal volunteers, making definition of a specific cut-off level difficult.
Deoxyuridine suppression test
The conversion of deoxyuridine monophosphate (dUMP) to deoxythymidine monophosphate (dTMP) during DNA synthesis is by methyl group transfer, facilitated by both vitamin B12 and folate. The other source of dTMP is via phosphorylation of deoxythymidine, catalysed by thymidine kinase, which is subject to feedback inhibition by its product, dTMP. Normal marrow, pre-incubated with dUMP, successfully converts this to dTMP, and incorporates less subsequently added tritiated thymidine into DNA. If the marrow is deficient in either vitamin B12 or folate, methyl group transfer is reduced, so the cells have greater capacity to incorporate tritiated thymidine into DNA following pre-incubation with dUMP.
Antibody tests
Tests for the presence of antibodies to gastric parietal cells are positive in 90% of patients with pernicious anaemia; however, this test is not specific as it is also positive in around 15% of healthy elderly people. The presence of IF antibodies is more specific but is found in only 50% of patients with pernicious anaemia.
Schilling test
This test, which measured the 24-h urinary recovery of vitamin B12 following an oral radiolabelled dose of the vitamin, is now obsolete.
Anaemias due to reduction in red cell production: inherited causes
Inherited disorders of haemoglobin synthesis, such as sickle cell anaemia, result in ineffective erythropoiesis and a microcytic anaemia. These disorders are covered in Chapter 29.
Other inherited disorders of red cell production are rare and usually present in infancy. Fanconi anaemia is an autosomal recessively inherited disorder of a group of DNA repair proteins. Patients have various somatic deformities, a pancytopenia and markedly increased risk of both solid and haematological malignancies. Diamond–Blackfan syndrome is associated with an anaemia secondary to a reticulocytopenia and carries an increased risk of acute myeloid leukaemia.
Anaemias associated with reduction in red cell production: acquired causes
Anaemia of chronic disease
Chronic diseases are often associated with an elevation of proinflammatory cytokines such as TNF-α and IL-6. These cytokines are potent inducers of hepcidin, which acts to inhibit intestinal iron absorption and the release of iron from macrophages. Thus iron studies in chronic diseases reveal a low serum iron and iron binding capacity with a raised ferritin. The anaemia of chronic disease is usually normocytic, but in approximately 20% of cases, is microcytic.
Aplastic anaemia
Stem cell failure can be caused by toxins affecting cell production. This may be predictable dose dependent toxicity, such as following exposure to high dose irradiation, cytotoxic drugs and benzene. Rarely, the aplasia occurs as an idiosyncratic reaction to drugs such as non-steroidal anti–inflammatories and chloramphenicol, or as a reaction to viruses such as hepatitis, which is probably immune mediated. It is sometimes associated with an acquired defect in the glycosylphosphatidylinositol (GPI) anchor, which plays an important role in the attachment of a number of cell surface antigens, protecting the cells from complement mediated premature destruction. This rare disorder is called paroxysmal nocturnal haemoglobinuria (PNH) (see p. 529).
Myelodysplasia
The myelodysplastic syndromes are a heterogenous group of disorders where there is ineffective haemopoiesis, resulting in progressive multilineage cytopenia and an increased propensity to transform to an acute leukaemia. The incidence of these conditions rises with age, with median onset in the seventh decade.
Malignant infiltration of the bone marrow
Infiltration of the marrow causes loss of normal haemopoietic tissue. This gives rise to a characteristic leukoerythroblastic appearance on a blood film. Immature white cells, nucleated red cells and red cells with a teardrop shape appear and the platelet count falls. Leukoerythroblastic anaemia is a characteristic finding when a malignancy has metastasized to the marrow. It also occurs in myelofibrosis, where there is an increase in reticulin fibres in the bone marrow and splenomegaly.
ANAEMIAS ASSOCIATED WITH INCREASED RED CELL LOSS
Bleeding
Acute bleeding, from any source, results in a reticulocytosis and thus, the anaemia may be normocytic or even macrocytic if the reticulocyte count is high, as these cells are larger than mature red cells. With time, iron stores become diminished and a microcytic anaemia develops.
Haemolysis
Haemolysis may be defined as the premature destruction of red cells, that is, the shortening of their normal life span of approximately 120 days. The destruction of the red cells may be effected by the macrophages in the reticuloendothelial system, for example in the spleen, liver or bone marrow; such destruction is said to be extravascular. Alternatively, destruction of red cells may take place within the circulation, i.e. intravascular haemolysis. Often there is a combination of both mechanisms. Normally, the bone marrow responds to the anaemia by increasing red cell production, and where this balances the loss of red cells, there is said to be a compensated haemolytic state. If the rate of the destruction exceeds the capacity of the marrow’s compensatory increase in erythropoiesis, a haemolytic anaemia ensues.
Thus, haemolytic anaemias are usually characterized by a triad of pathological features:
• increased red cell destruction resulting in anaemia
• increased red cell production resulting in reticulocytosis
• increased haemoglobin catabolism resulting in jaundice.
Laboratory features of haemolysis
In extravascular haemolysis, the haem moiety released from haemoglobin as a consequence of increased red cell destruction is broken down to protoporphyrin, with the liberation of the iron, which enters the plasma pool and is bound to transferrin, increasing its saturation. Protoporphyrin is metabolized to carbon monoxide and biliverdin, which is subsequently further reduced to bilirubin. An increased plasma unconjugated bilirubin concentration to > ~ 50 μmol/L results in jaundice. As unconjugated bilirubin is bound to albumin, it is not excreted in the urine, but there is increased urinary excretion of its metabolic end-product, urobilinogen (see Chapter 13).
In contrast, in intravascular haemolysis, there is release of haemoglobin into the plasma (haemoglobinaemia) and, when the renal threshold for haemoglobin is exceeded, haemoglobin is excreted in the urine (haemoglobinuria). Some of the haemoglobin in the glomerular filtrate is reabsorbed and degraded by the proximal tubular cells, with the iron released being deposited as haemosiderin. As the tubular cells desquamate, the presence of the intracellular haemosiderin iron can be detected in the urinary sediment by Perls reaction, which is positive for up to six weeks after the episode of intravascular haemolysis. The loss of iron may be sufficiently great to result in overt iron deficiency anaemia, in contrast to the iron accumulation seen in extravascular haemolysis.
Free haemoglobin in the plasma readily dissociates from its tetrameric structure to dimeric units consisting of α- and β-subunits. The haemoglobin α chain binds readily to the β chains of haptoglobin, a glycoprotein produced by the liver. The haemoglobin–haptoglobin complex is rapidly cleared by the reticuloendothelial system. In chronic haemolytic states, there is an increased removal of haptoglobin at a rate that exceeds the synthetic capacity of the liver, resulting in low or undetectable plasma haptoglobin concentrations. The return to a normal concentration takes approximately one week following cessation of the haemolytic process.
Any free plasma haem resulting from haemolysis forms complexes with either haemopexin or albumin. The haem–haemopexin complex is taken up and metabolized by the liver, resulting in a decrease in plasma haemopexin concentration. The haem–albumin complex may be oxidized to methaemalbumin, which has a characteristic absorption spectrum and gives a brownish colour to the patient’s plasma. Acute kidney injury is often seen in intravascular haemolysis. Studies have revealed that haem is directly toxic to renal tubular cells and may form intratubular casts with Tamm–Horsfall protein. The mitochondria of renal cells appear particularly susceptible to haem-mediated damage, with resultant impaired mitochondrial oxygen consumption and autophagocytosis. The kidney protects itself during intravascular haemolysis by inducing haemoxygenase-1 and ferritin production, which act to degrade the haem and bind any resultant free iron, respectively (see Fig. 27.7).
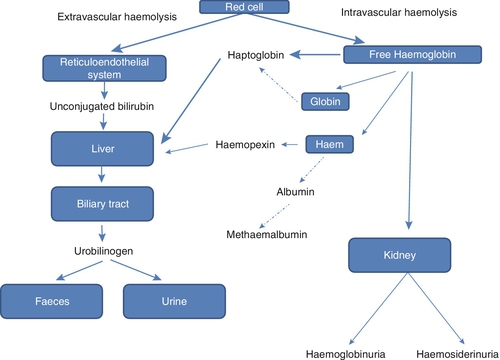
FIGURE 27.7 The pathway of haemoglobin metabolism in haemolytic anaemias. Haemoglobin is liberated from the breakdown of the red cells and becomes bound to haptoglobin. Any free haem binds to haemopexin. Both scavenger proteins are taken up by macrophages within the reticuloendothelial system. When these plasma proteins are consumed, the haem may be either free in the plasma or bound to albumin.
Causes of haemolytic anaemias
The disorders that cause haemolysis form a diverse group. Some of them are very common, for example sickle cell anaemia and thalassaemia, which affect millions worldwide, while some are very rare, for example aldolase deficiency, with only a few families reported in the world literature. The clinical features are equally diverse, varying from little or no clinically evident haemolysis as, for example, with hereditary elliptocytosis, to life-threatening acute intravascular haemolysis associated with Plasmodium falciparum malaria (blackwater fever). Clinical manifestations other than haemolysis may complicate the situation further; for example, sickle cell anaemia is associated with vaso-occlusive episodes that may result in life-threatening problems such as pulmonary crises, while some of the inherited deficiencies of enzymes such as triose phosphate isomerase and phosphoglycerate kinase are associated with severe neurological disease.
A classification of haemolytic disorders based on the site of haemolysis would identify very few disorders causing only intravascular haemolysis, as the vast majority are associated with predominantly extravascular haemolysis. Therefore, the best classification of haemolytic disorders is based on whether the haemolysis is inherited or acquired, as summarized in Box 27.1.
Inherited haemolytic anaemia
It is convenient to consider inherited red cell defects under three headings: membrane defects, enzymes defects and haemoglobinopathies. The haemoglobinopathies are considered further in Chapter 29.
Membrane defects
The commonest haemolytic anaemia caused by a membrane defect is hereditary spherocytosis. It is usually inherited in an autosomal dominant manner, resulting in haemolysis of varying severity; the majority of patients have a mild anaemia, although severe haemolysis resulting in neonatal kernicterus has been reported. It is characterized by spherocytic, osmotically fragile cells and in most cases is caused by a defect in the ankyrin–spectrin complex, although defects in other membrane proteins can also cause the condition. Extravascular haemolysis occurs predominantly in the spleen because of the poor deformability of the cells. Splenectomy is sometimes considered in patients, as prolongation of the red cell life span results in amelioration of the clinical features.
Hereditary elliptocytosis is a more heterogeneous disorder characterized by large numbers of elliptically shaped red cells in the peripheral blood. It is caused by defects in the spectrin tetramers (see p. 517). There is an increased incidence in West Africa, possibly because of protection from infection by Plasmodium falciparum; in vitro studies have demonstrated reduced ability of this organism to parasitize red cells with the defects seen in hereditary elliptocytosis.
Hereditary stomatocytosis (HS) is rare and is associated with the appearance of ‘mouth like’ red cells on blood film. These cells are leaky to cations: hence the condition is associated with pseudohyperkalaemia. Splenectomy should be avoided, owing to the risk of thrombosis in these individuals. The variant termed dehydrated HS, or hereditary xerocytosis, may be associated with perinatal ascites, which resolves spontaneously within the first year of life.
Enzyme defects
These may be grouped under three headings:
• disorders of the pentose phosphate pathway and related enzymes of glutathione metabolism
• disorders of anaerobic glycolysis
• disorders of nucleotide metabolism.
Disorders of the pentose phosphate pathway and related enzymes of glutathione metabolism
The pentose phosphate shunt provides the reducing power of the red cell in the form of NADPH, which maintains glutathione in the reduced form (GSH) via the closely linked glutathione pathway. The GSH protects the red cells from oxidative damage; inadequate supplies result in peroxidation of the red cell membrane, denaturation of haemoglobin and its precipitation as Heinz bodies, resulting in reduced cell deformability and intravascular haemolysis.
The commonest disorder of the pentose phosphate pathway is a defect of the enzyme glucose 6-phosphate dehydrogenase (G6PD), affecting 400 million people worldwide. The gene for the enzyme is located on the X chromosome. The wild type is G6PD(B). More than 300 variants have been described with differences in maximum enzyme activity and/or gene expression. The best known of these affects people of African origin, G6PD(A-), or from Mediterranean countries, G6PD(Med). Most individuals with G6PD deficiency are asymptomatic but liable to have acute haemolytic crises under oxidative stress from certain drugs (see Table 27.3), ingestion of fava beans or acute infection. Favism has been described in Mediterranean countries since classical times.
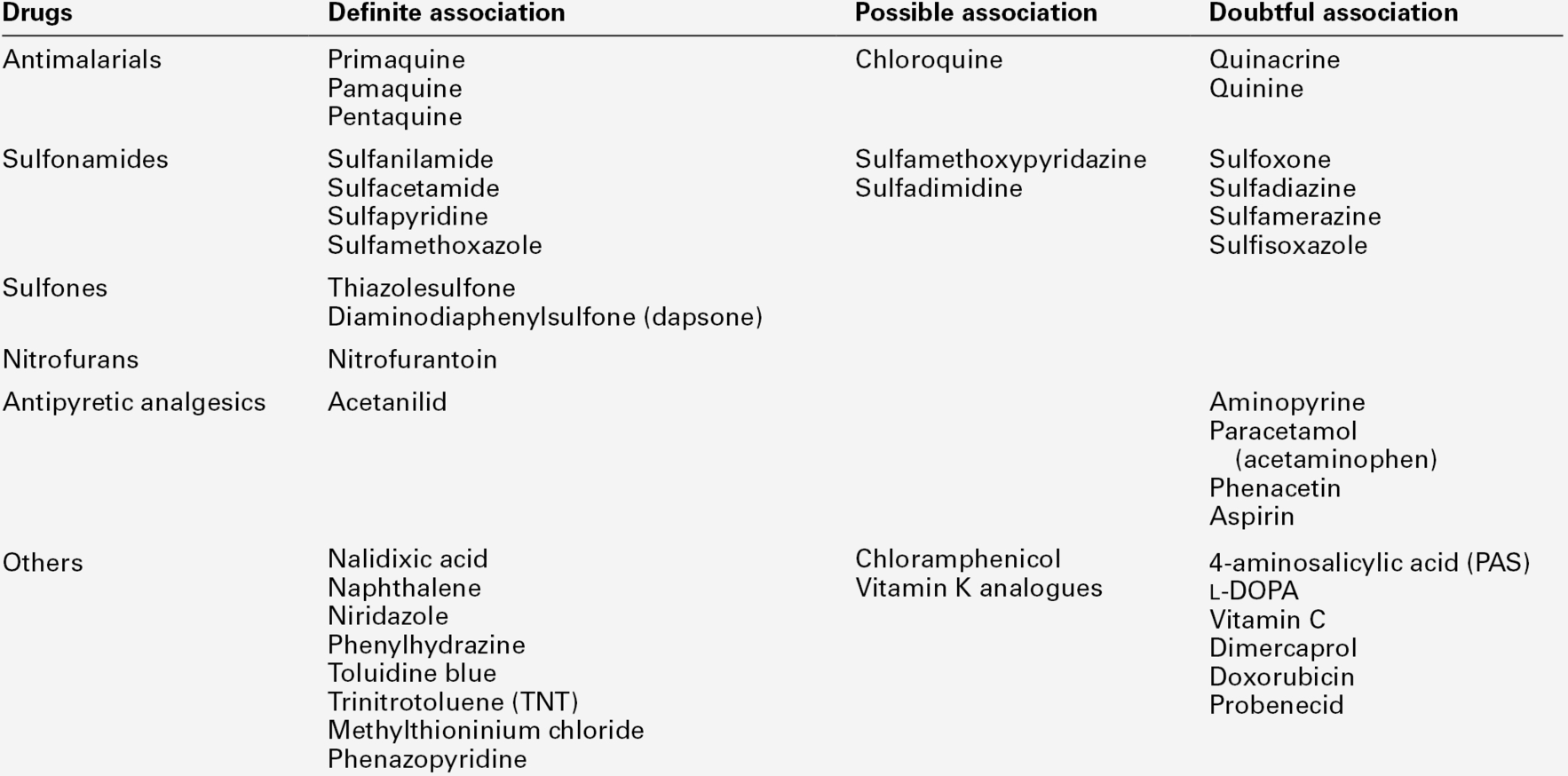
The cause of the haemolysis is a failure to produce enough GSH to protect the red cell membrane from oxidative stress. Glucose 6-phosphate dehydrogenase is the first and most important enzyme in the pentose phosphate pathway that generates the NADPH that is required for glutathione reduction. Normal in vivo flux uses only a small percentage of the maximum enzyme capacity. This explains why G6PD variants with low residual activities are compatible with almost normal function of the red cell. Only under oxidative stress is the protective capacity overwhelmed, leading to predominantly extravascular haemolysis.
In a relatively small number of sporadic cases, the G6PD variant is functionally so abnormal that there is chronic extravascular haemolysis even without any additional oxidative stress.
Disorders of anaerobic glycolysis
Deficiencies of the glycolytic enzymes hexokinase (HK), phosphoglucose isomerase (PGI), phosphofructokinase (PFK), aldolase (ALD), triose phosphate isomerase, phosphoglycerate kinase (PGK), enolase and pyruvate kinase (PK) have all been reported in association with chronic haemolytic anaemia. Almost all cases reported are inherited in an autosomal recessive mode except for PGK deficiency, which is X-linked. All are relatively rare; the commonest in this group is PK deficiency, which has a prevalence of ~ 1:20 000.
Because the glycolytic pathway is mainly concerned with the generation of ATP, it has been proposed that the primary pathogenic defect is low red cell ATP concentration causing rigidity of the red cells. However, the expected reduction in ATP concentration is not a constant finding. This may be partly explained by the presence of increased numbers of reticulocytes, which have higher concentrations of ATP, and/or the preferential destruction of the most metabolically affected red cells, giving a distorted picture of the concentration of the ATP at the time of investigation. It is likely, however, that a number of different factors contribute to the haemolytic process depending on the various metabolic abnormalities resulting from the enzyme deficiency.
Disorders of nucleotide metabolism
The most important abnormalities of nucleotide metabolism that are clearly associated with chronic haemolytic anaemia are deficiency of pyrimidine 5′-nucleotidase (P5N) and hyperactivity of adenosine deaminase (ADA).
The mechanisms of haemolysis in these conditions have not been established, although some interesting hypotheses have been proposed as logical explanations of the biochemical findings. In patients who are homozygous for P5N deficiency, there is an accumulation of pyrimidine nucleotides in the red cells. This could affect the metabolism of the cells by competing with adenine nucleotides, which are the normal cofactors of a number of important enzyme reactions, and by interfering with the function of the pentose phosphate pathway.
In some cases of ADA hyperactivity, there is a low red cell concentration of ATP. It has been hypothesized that this leads to depletion of adenosine through irreversible deamination, so that insufficient adenosine is available for the salvage pathway via adenosine kinase to replenish normal losses from the adenine nucleotide pool.
Acquired haemolytic anaemias
The acquired haemolytic anaemias can be divided into two main groups:
• immune haemolytic anaemias, where the production of antibodies mediates red cell destruction
• non-immune haemolytic anaemias, which are of numerous diverse causes.
Immune haemolytic anaemias
These may be caused by autoantibodies produced against epitopes on the surface of the patient’s own normal red cells or by the development of alloantibodies, antibodies produced by the patient against foreign antigens introduced through blood transfusions or neoantigens that develop on red cells during drug induced haemolysis.
The autoimmune haemolytic anaemias are characterized by a positive Coombs test, which detects antibodies, with or without complement, bound to the surface of the red cell. These antibodies are often directed against the rhesus antigen system; they may be of any immunoglobulin subclass and display different thermal activities, hence the term ‘warm’ and ‘cold’ acting antibodies. The resulting haemolysis is usually extravascular in nature because of the ability of the autoantibody to fix complement, allowing opsonization and subsequent uptake by the macrophages of the spleen and liver. However, some autoantibodies produced are able to activate the complement pathway through to cell lysis, resulting in a more severe intravascular haemolysis.
Autoimmune haemolytic anaemias may occur without an obvious underlying cause (primary autoimmune haemolytic anaemia) or may be secondary to other conditions such as lymphoma, chronic lymphocytic leukaemia and autoimmune diseases including systemic lupus erythematosus and rheumatoid arthritis.
Non-immune haemolytic anaemias
The main causes of non-immune haemolysis are infections, traumatic and microangiopathic disorders, chemical and physical agents and acquired disorders of the red cell membrane.
Infections
The most important infection associated with haemolytic anaemia is Plasmodium falciparum, in which both extravascular haemolysis, from destruction of parasitized erythrocytes in the reticuloendothelial system, and intravascular haemolysis, when the parasites break out of cells, are seen. Rarely, the latter may be extreme enough to produce the dramatic picture known as blackwater fever, with severe intravascular haemolysis associated with a high percentage of parasitized erythrocytes, haemoglobinuria and acute kidney injury.
Other protozoal and bacterial infections that can be associated with haemolysis are listed in Box 27.1.
Traumatic and microangiopathic disorders
The common mechanism in these conditions is contact between red cells and an abnormal surface in the circulation. Cardiac prosthetic valves and intracardiac patch repairs have been associated with haemolytic anaemia, which is intravascular, though usually mild.
Microangiopathic haemolytic anaemia is a term used to describe a variety of conditions in which there is increased fibrin deposition in the microcirculation. This results in increased red cell fragmentation either directly by damage to red cells by fibrin strands or indirectly by the stress imposed by other cells flowing past cells trapped on fibrin strands. The end-result is intravascular haemolysis. Such conditions include haemolytic uraemic syndrome, which is seen mainly in childhood and in which renal failure is a prominent feature, and thrombotic thrombocytopenic purpura (TTP), which is associated with the diagnostic pentad of various neurological signs, red cell fragmentation, thrombocytopenia, purpura and renal failure. In haemolytic anaemia associated with hypertension (including pregnancy-associated hypertension), there is little fibrin deposition and it is postulated that there is increased fragmentation of red cells trapped at the endothelial surface as a result of the shearing stress of the flow of arterial blood. March haemoglobinuria is a rare and benign condition in which there is red cell destruction in the feet caused by mechanical trauma from walking or running over long distances.
Acquired disorders of the red cell membrane
Paroxysmal nocturnal haemoglobinuria (PNH) is a rare acquired disorder of the red cell membrane. It is associated with an acquired somatic mutation in the PIGA gene on the X chromosome. This encodes the enzyme complex phosphatidylinositol N-acetyl-aminyl-transferase subunit A (PIGA), which plays a pivotal role in the biosynthesis of glycosylphosphatidylinositol (GPI) anchors. These are responsible for the attachment of a large number of surface antigens to the cell membrane, including proteins that protect the cell membrane from complement-mediated attack. Thus GPI anchor deficiency renders the red cells susceptible to complement mediated cell lysis and intravascular haemolysis ensues. The condition is associated with the clinical triad of intravascular haemolysis, bone marrow failure and thrombosis.
Zieve syndrome is an uncommon disorder seen in alcoholics, in which intravascular haemolysis is possibly a consequence of changes in the lipid composition of the cell membrane. Vitamin E deficiency in neonates results in the loss of the protective antioxidant effect of this vitamin and an oxidative haemolysis ensues.
DIAGNOSIS OF HAEMOLYSIS
The search for evidence supporting the presence and cause of haemolysis can be considered in terms of clinical evidence and the results of laboratory investigations.
Clinical evidence
The clinical history should pay particular attention to the onset of jaundice and to any precipitating factors. The colour of the urine may suggest the presence of haemoglobinuria, suggesting intravascular haemolysis. Anaemia is often asymptomatic, either because of its mild degree or because of its chronicity. However, severe symptomatic anaemia is likely to be of recent onset and suggests an acquired aetiology. The general medical history, including previous surgery, is important but particular attention should be paid to travel, drug history and exposure to chemicals at work or recreation.
A family history may help to establish an inheritance pattern. The majority of families with congenital non-spherocytic haemolytic anaemias caused by enzymopathies show an autosomal recessive transmission, while most non-spherocytic anaemias with unstable haemoglobin or red cell membrane defects show an autosomal dominant pattern. The most important exceptions are the X-linked deficiencies of G6PD and PGK. In these, the affected individuals are hemizygous males or, very infrequently, homozygous females. The vast majority of heterozygous females are clinically unaffected and difficult to detect.
General physical examination provides additional information. In thalassaemia major, frontal bossing and prominence of the maxillae produce a characteristic facial appearance (see p. 553). Splenomegaly is often a prominent finding, particularly in the congenital haemolytic anaemias.
Laboratory investigations
These should be directed to answering two basic questions: is there any haemolysis and if so, what is the mechanism and cause?
Laboratory investigations for the presence of haemolysis
Red cell morphology
In haemolytic anaemias, there is a varying degree of anaemia accompanied by a reticulocytosis. The underlying cause of haemolysis may be suggested by the presence of specific morphological abnormalities (see Chapter 26), for example, spherocytes in both hereditary spherocytosis and autoimmune haemolytic anaemia, and red cell fragmentation in microangiopathic haemolytic anaemia. Heinz bodies, which are composed of denatured haemoglobin, are seen in G6PD deficiency, and some unstable haemoglobins, formed after exposure to oxidant drugs and chemicals, are revealed using the supravital stain crystal violet. Haemoglobin H inclusions (seen in α thalassaemia) can be demonstrated using brilliant cresyl blue. Excessive basophilic stippling, indicating the presence of undegraded RNA, suggests a block in the catabolism of RNA during reticulocyte maturation and is a constant finding in pyrimidine 5′-nucleotidase deficiency.
Total and unconjugated bilirubin
Haem breakdown results in the release of hydrophobic unconjugated bilirubin that is transported, bound to albumin, to the liver where it is conjugated. Therefore, haemolysis results in an increase in plasma unconjugated bilirubin concentration.
Haptoglobin
Any free haemoglobin released from red cell breakdown complexes with haptoglobin and is then cleared by the reticuloendothelial system. A decrease in the plasma haptoglobin concentration occurs when the daily haemoglobin turnover is doubled, irrespective of whether the haemolysis is extravascular or intravascular. Haptoglobin is more rapidly depleted in intravascular haemolysis. However, congenital ahaptoglobinaemia is seen in 2% of the Caucasian population and concentrations may also be lowered in megaloblastic anaemia (because of ineffective erythropoiesis) and in liver disease. Haptoglobin is an acute phase protein, its concentration rising in many inflammatory conditions, in pregnancy and during use of oral contraceptives and corticosteroids. Under these circumstances, a normal haptoglobin concentration does not exclude haemolysis.
Haemopexin
Haemopexin binds free haem in plasma and in severe intravascular haemolysis, when haptoglobin is depleted, plasma haemopexin falls to very low concentrations or becomes undetectable. However, in mild haemolysis, plasma haptoglobin concentration may be reduced or undetectable but plasma haemopexin concentration normal or only slightly reduced. Low concentrations of haemopexin may be found in renal or liver disease without any evidence of haemolysis and high concentrations are seen in diabetes mellitus, infection or carcinoma. This assay is only available at a few specialized centres.
Methaemalbumin
This is found in plasma when haptoglobin is depleted in the more severe intravascular haemolytic anaemias. It may be detected by the Schumm test: methaemalbumin reduced by the addition of ammonium sulfide shows an intense absorption band in the green spectrum.
Free haemoglobin
Free haemoglobin does not appear in the plasma until all available haptoglobin has been depleted, so it may be undetectable in mild degrees of haemolysis. Therefore, an increased plasma free haemoglobin concentration indicates a significant degree of intravascular haemolysis, provided that the possibility of in vitro lysis of red cells during red cell sampling or processing has been excluded. Free haemoglobin in the urine (haemoglobinuria) is also a good indicator of intravascular haemolysis. False positives may occur owing to haematuria (intact red cells in urine) or myoglobinuria, which must be excluded.
Haemosiderinuria
After glomerular filtration, some free haemoglobin is absorbed by renal tubular cells and then broken down. The released iron is deposited as insoluble haemosiderin and is ultimately excreted in the urine when desquamation of the tubular cell occurs. Haemosiderin can be demonstrated within these cells by staining a cytospin preparation of the urine by the Perls technique. Haemosiderinuria can be detected for several weeks after a haemolytic episode, even when there has not been overt haemoglobinuria. Thus, the demonstration of intracellular haemosiderin in the urine is a good sign of mild intravascular haemolysis.
Red cell survival
This can be measured using radiolabelled (usually with 51Cr) autologous red cells. Daily measurements can allow for the calculation of red cell survival, usually combined with surface counting over the liver and spleen to estimate the relative contribution of each reticuloendothelial organ to the red cell destruction. If uptake of radiolabelled red cells is seen predominantly in the spleen, removal of this organ should ameliorate the haemolysis. This test is performed only in specialist centres.
Laboratory investigations for the cause of haemolysis
After the diagnosis of haemolysis has been made, further investigations are required to establish the precise cause of the reduced red cell survival. These should be directed along a logical pathway to avoid requests for inappropriate tests.
Coombs test (direct antiglobulin test)
This is used to detect the presence of red cells coated with immunoglobulin or complement, which is the hallmark of immune haemolysis. The test can be modified to allow the detection of specific immunoglobulin subclasses or complement fractions by the use of specific antisera.
Tests for abnormal haemoglobin
The screening tests for haemoglobinopathy may be described under three main headings, indicating the main haemoglobin property being investigated:
• functional abnormalities, e.g. sickling test, demonstration of Heinz bodies
• unbalanced haemoglobin synthesis, e.g. quantitation of haemoglobin A2 and haemoglobin F.
The detailed investigation of haemoglobinopathies is discussed in Chapter 29.
Osmotic fragility tests
These tests are no longer recommended in the routine assessment of haemolytic anaemias, having been replaced by flow cytometry techniques (see below). They are based on spectrophotometric assessment of the lysis of red cells after incubation in water or hypotonic saline for variable periods of time. Normal red cells, as a consequence of their biconcave shape, have an ability to take up more water before they lyse. Spherocytes, because of their high volume to surface area, have a very limited ability to take in water and thus lyse more readily. Conversely, cells with a reduced volume to surface area ratio, such as the cells in thalassaemia or iron deficiency, and reticulocytes, are relatively resistant to lysis. The tests are usually normal in enzyme defects apart from a tail of reduced fragility from the reticulocytes. A normal osmotic fragility does not exclude a diagnosis of hereditary spherocytosis as it may be normal in 10–20% of cases.
The autohaemolysis test
This measures the spontaneous haemolysis of blood incubated at 37 °C for 24 h. It is a useful screening test in cases of suspected haemolytic anaemia if the blood film is not morphologically suggestive of hereditary spherocytosis. If the test is entirely normal, then an intrinsic red cell defect is unlikely. Correction of the autohaemolysis by the addition of glucose (as an energy source) suggests a membrane defect, as these are associated with increased glucose consumption because of the increased cation leak through the membrane. In enzyme defects, the addition of glucose has no effect as the cells cannot use this energy source.
Flow cytometry
The use of the eosin 5-maleimide (EMA) binding test is now the recommended first-line investigation for the diagnosis of hereditary spherocytosis, where there is no family history or the blood film appearances are atypical. Eosin 5-maleimide binds to band 3 protein (so named on the basis of its electrophoretic mobility). This protein interacts with both ankyrin and protein 4.2 and, in turn, with the spectrin cytoskeleton, which is disrupted in hereditary spherocytosis, resulting in a decreased flow cytometric fluorescence signal. The test has a high level of specificity (99.1%) and sensitivity (92.7%) and can be performed rapidly.
Flow cytometry has also become the gold standard for the diagnosis of PNH, replacing the acid haemolysis (Ham) test. It uses fluorescent-labelled monoclonal antibodies to detect various GPI-anchored surface antigens, principally CD55 (decay accelerating factor) and CD59 (membrane inhibitor of reactive lysis), whose absence on the surface of red cells is compatible with a diagnosis of PNH.
Tests for enzyme deficiencies
It is useful for laboratories to be able to screen for the common red cell enzyme deficiencies, such G6PD and PK, and to indicate where the defect lies in the less common disorders, but further detailed investigations are best performed in specialist laboratories. It is important that leukocytes and platelets are removed from the samples as they generally have higher enzyme concentrations than the red cells. Also, it should be remembered that reticulocytes have increased concentrations of many ‘age-dependent’ enzymes, particularly HK, PK, ALD and pyrimidine 5′-nucleotidase. Therefore, if possible all samples should be tested either alongside a control sample with a similar reticulocyte count or the activity of the enzyme under investigation compared with that of a second ‘age-dependent’ enzyme.
Glucose 6-phosphate dehydrogenase
Screening tests for this enzyme deficiency depend on the inability of the cells to convert an oxidized substrate, such as NADP, to a reduced form that fluoresces under long wavelength UV light. Red cells with < 20% of normal G6PD activity do not fluoresce. Problems in interpreting this test are common, particularly in heterozygous women and affected males with G6PD(A-) deficiency and reticulocytosis. In the latter situation, the presence of increased numbers of reticulocytes, which contain increased amounts of G6PD(A-), may give false negative results. Thus negative results do not always exclude enzyme deficiency, while positive results should be confirmed by quantitative assay.
Pyrimidine 5′-nucleotidase
Screening for this deficiency is based on the different spectrophotometric properties of cytidine nucleotides (pyrimidine nucleotides), which absorb maximally in acidic solutions at 280 nm, whilst adenine and guanine (purine nucleotides) and uridine absorb at 260 nm. Normally, > 96% of nucleotides in red cells are purine analogues, but this falls to < 50% in cases of pyrimidine 5′-nucleotidase deficiency, where the pyrimidine nucleotides accumulate, causing the absorbance ratio to fall.
Red cell metabolites
2,3-Diphosphoglycerate (2,3-DPG), ATP and GSH are present in red cells in sufficiently high concentrations (millimolar) to be readily measurable by spectrophotometric techniques in most laboratories, providing valuable information on red cell enzymopathies. An increase in 2,3-DPG occurs in most anaemias. Furthermore, hypoxaemia, alkalosis and hyperphosphataemia all lead to increases in 2,3-DPG, irrespective of anaemia. A low concentration of 2,3-DPG is a useful indicator of an enzymopathy early in the glycolytic pathway. Hexokinase, PFK and PGI deficiencies have been frequently, but not invariably, associated with low concentrations of this metabolite. Extremely low 2,3-DPG concentrations have also been reported in a case of complete diphosphoglyceromutase deficiency, in which there is a high haemoglobin concentration because of an increased oxygen affinity in the absence of haemolysis.
A moderate reduction in GSH concentration has been reported in several patients with congenital non-spherocytic haemolytic anaemia including G6PD deficiency. However, very low concentrations of GSH could also be an indication of deficiency in either of the two enzymes of GSH biosynthesis: glutathione synthetase and γ-glutamylcysteine synthetase. In both enzymopathies, some patients have neurological disease in addition to the haemolytic anaemia, which is exacerbated by oxidative stress. For unknown reasons, high concentrations of GSH are present as an epiphenomenon in pyrimidine 5′-nucleotidase deficiency and several syndromes of dyserythropoietic anaemia and ineffective erythropoiesis. Neonates have higher red cell of GSH concentrations than adults.
Glycolytic intermediates
The in vivo concentrations of glycolytic intermediates are probably the best available measure of the functional status of the glycolytic enzymes. Theoretically, when a given enzyme is functionally defective, it should cause a metabolic block leading to an accumulation of its precursor metabolites and a relative reduction in the concentration of those after it. However, this is not always seen for various reasons, including the increased activity of the enzymes in reticulocytes, but measurement of the glycolytic metabolites may still provide a useful screening test for a metabolic block in some circumstances. For example, PK deficiency may be suspected because of relatively high concentrations of metabolites preceding the PK step, i.e. phosphoenolpyruvate, 2-phosphoglycerate, 3-phosphoglycerate and 2,3-DPG.
CONCLUSION
In the accurate assessment of a patient presenting with anaemia, the clinical history plays a vital role in distinguishing between congenital and acquired causes. This is further supplemented by utilizing the following investigations:
• examination of blood film morphology
• serum ferritin, vitamin B12 and folate concentations
• coombs test and reticulocyte count
• haemoglobin electrophoresis.
This strategy should result in a specific diagnosis for the majority of individuals, with only a minority (< 5% in the UK) requiring more specialized investigations.
ACKNOWLEDGEMENTS
The author is deeply indebted to Dr Pamela A. Gover, who prepared the chapter on Biochemical aspects of anaemia for earlier editions of this book.
Further reading
[/level-membership-for-endocrinology-diabetes-and-metabolism-category][not-level-membership-for-endocrinology-diabetes-and-metabolism-category]C
[/not-level-membership-for-endocrinology-diabetes-and-metabolism-category]
