[level-membership-for-endocrinology-diabetes-and-metabolism-category]CHAPTER 26
Introduction to haematology and transfusion science
David Ah-Moye; Ceinwen Davies; Joanne Goody; Peter Hayward; Rebecca Frewin
CHAPTER OUTLINE
Analysis of the full blood count
Erythrocyte sedimentation rate and plasma viscosity
Tests for infectious mononucleosis
Abnormal white cell morphology
Laboratory tests of coagulation
Interpretation of coagulation tests
Investigation of suspected transfusion reaction
INTRODUCTION
Haematology is the study of blood, its formation, composition, functions and diseases. Blood cell formation (haemopoiesis) is described in Chapter 27. Haematology laboratories perform a wide range of tests of the composition and function of the blood that assist the clinician in the diagnosis and management of disease. They are typically divided into three principal sections: general haematology, haemostasis and blood transfusion. This chapter provides an overview of the different diagnostic techniques offered in each.
GENERAL HAEMATOLOGY
The general haematology laboratory performs numerical, morphological and biochemical analysis of blood samples (now often as part of a combined blood sciences laboratory). Blood samples must be anticoagulated, for example with ethylenediamine tetra-acetic acid (EDTA) for determination of full blood count and reticulocyte count, blood film examination and plasma viscosity and tri-sodium citrate for coagulation tests and measurement of erythrocyte sedimentation rate.
Analysis of the full blood count
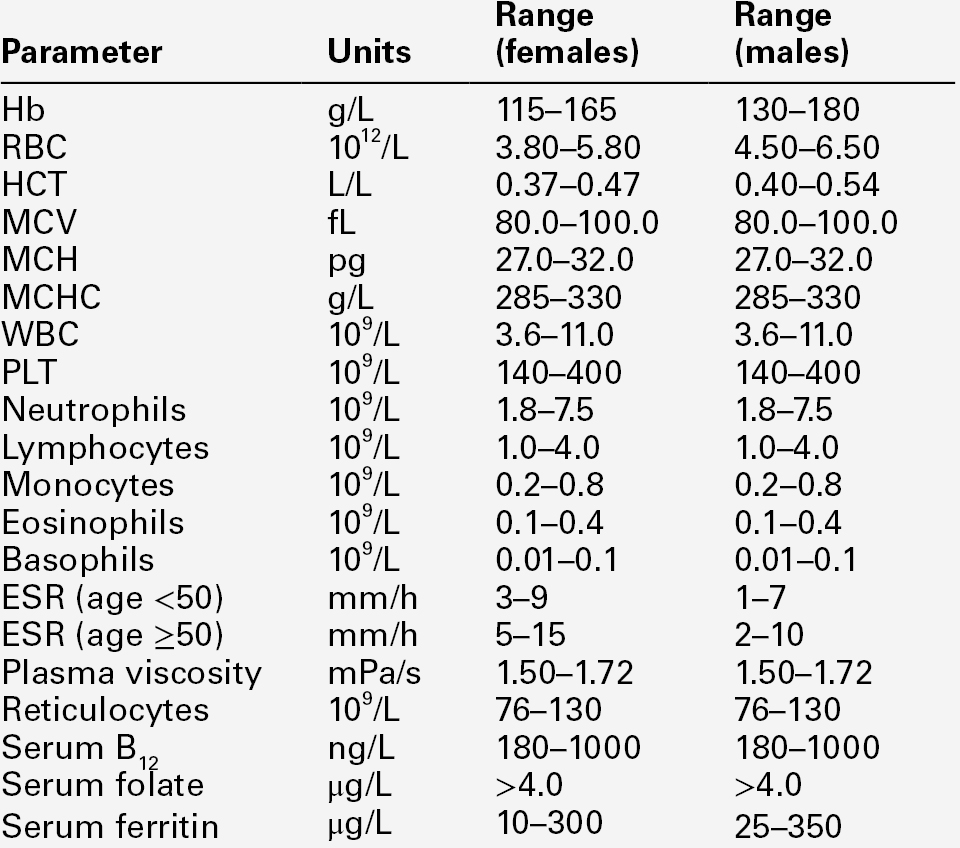
The full blood count (FBC) is the most requested test profile in haematology. It comprises:
• haemoglobin (Hb) concentration
• red blood cell (RBC) count
• red cell indices (volume and haemoglobin content)
• white blood cell (WBC) count
• white blood cell differential count (neutrophils, lymphocytes, monocytes, eosinophils and basophils)
• platelet count.
The FBC is a valuable first-line screening test from which further investigations may be generated (see Table 26.1). The clinician uses information from the FBC in conjunction with the clinical history, physical examination and other investigations to help formulate and distinguish between differential diagnoses.
Full blood counts are usually performed on fully automated analysers, which provide accurate quantitation of cell parameters and can generate alerts, commonly known as ‘flags’, to the presence of unusual characteristics that may warrant review of a blood film.
Haemoglobin
The main function of haemoglobin, which is present solely within the red cells, is to transport oxygen from the lungs to the tissues (see Chapter 5). Haemoglobin is measured by spectrometry. Diluted whole blood is mixed with a reagent (e.g. Stromatolyser™) that results in cell lysis and conjugates the haemoglobin with a compound such as potassium cyanide or sodium lauryl sulphate prior to measurement of its absorbance. The presence of high concentrations of substances that increase absorbance, e.g. triglyceride-rich lipoproteins, may cause a falsely raised haemoglobin result with some methods.
Cell counting
Blood cells are counted and sized either by electrical impedance or laser light scattering methods. The impedance method utilizes the fact that blood cells are very poor conductors of electricity. When a cell passes through a small aperture across which an electric current is applied, there is a measurable rise in the electrical impedance, with the pulse height being proportional to the volume of the cell. Each cell generates a separate impulse. Light scatter utilizes the fact that cells scatter light when passed through a laser beam: the number of cells can be counted and information about the cells can be obtained according to the pattern of scatter. White cell counting is performed after lysis of the red cells.
Clumping of cells results in underestimation of cell numbers. This may occur, for example, as a result of autoagglutination of red cells in cold agglutinin disease or in vitro clumping of platelets because of inadequate mixing with EDTA anticoagulant.
Red cell indices
The red cell indices are a combination of measured and derived red cell parameters. The red cell indices usually reported are:
• haematocrit (HCT) or packed cell volume (PCV), the percentage volume of red cells in the blood
• mean cell haemoglobin (MCH), the average mass of haemoglobin per red cell
• mean cell haemoglobin concentration (MCHC), the average concentration of haemoglobin per red cell.
Some analysers measure the MCV and calculate the HCT or PCV; others measure the HCT and calculate the MCV.
Mean cell volume
is useful when assessing anaemia. For example, it is reduced in iron deficiency anaemia and increased in megaloblastic anaemia.
Mean cell haemoglobin
is reduced in iron deficiency anaemia and thalassaemias; further tests such as ferritin and HbA2 measurement may be performed to differentiate between these two conditions.
Mean cell haemoglobin concentration
is reduced in iron deficiency but is probably a more useful tool within the laboratory than clinically. It is very stable in health, so it can be used as an internal quality control; a significant change in the daily mean MCHC indicates a drift in analyser calibration or a fault in its function. The MCHC is also useful for detecting anomalous results: a falsely raised Hb concentration, for example as a consequence of lipaemia, or a falsely reduced RBC count, for example owing to red cell autoagglutination, will result in a falsely raised MCHC.
The formulae for the derived parameters are as follows:
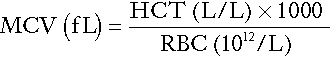
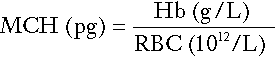
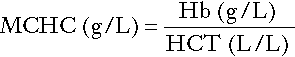
White cell differential
The white blood cells are involved in the cellular immunological response to infection and other causes of inflammation. The five types of white blood cell differ in volume, conductivity, light scattering properties, uptake of fluorescent dyes and cytochemical stains, and resistance to lytic agents. These properties, used in various combinations, enable their identification. Figure 26.1 shows typical white cell scattergrams for a normal blood sample.
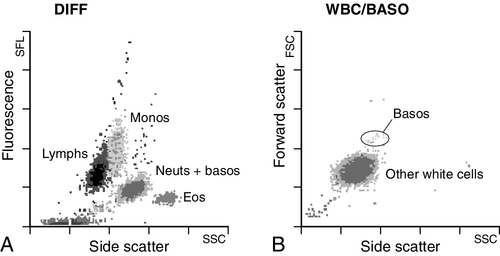
FIGURE 26.1 Five-part WBC differential from Sysmex XE2100TM analyser. Note that neutrophils and basophils cannot be separated in the side fluorescence (SFL) vs side scatter (SSC) population scattergram (A). The basophil count is obtained by treating the white cells with an acidic reagent that shrinks all white cells, leaving bare nuclei, except for basophils, which remain intact (B). The neutrophil count is then derived by subtracting the basophil count from the combined neutrophil and basophil count. Lymphs, lymphocytes; monos, monocytes; neuts, neutrophils; eos, eosinophils; basos, basophils.
Platelet count
The main function of platelets is to form a plug at sites of damage to vascular endothelium. A low platelet count (thrombocytopenia) may result from decreased production, increased consumption or abnormal pooling in the spleen. Decreased production is a consequence of either general marrow failure or selective depression, for example by viral infection, drugs and chemicals. Platelet consumption is a feature of certain types of autoimmune diseases and occurs in disseminated intravascular coagulation (which is often a result of septicaemia) and thrombotic thrombocytopenic purpura. A high platelet count (thrombocytosis) may be seen as part of a reactive process such as infection or in myeloproliferative neoplasms. Automated haematology analysers routinely measure the platelet count by impedance or laser light scattering; the reference method uses flow cytometry after labelling the platelets with CD41 or CD61 antibodies.
Reticulocyte count
Reticulocytes are immature red cells, which are released from the bone marrow into the circulation one to two days before maturation. An increase in the reticulocyte count is indicative of an increase in red cell production (erythropoiesis) to meet physiological demand, for example in haemolytic anaemia.
Reticulocytes contain ribosomal RNA, which can be detected by two main methods. When unfixed red cells are incubated with dyes such as new methylene blue, the ribosomal RNA precipitates out and appears as a blue reticular network within the cells, which can be visualised using a light microscope, allowing the reticulocytes to be counted. Most automated FBC analysers now perform reticulocyte counts by flow cytometry, using a fluorescent dye that binds to the ribosomal RNA; the number of reticulocytes can be expressed as a percentage of total red cells and as an absolute count.
Erythrocyte sedimentation rate and plasma viscosity
Erythrocyte sedimentation rate (ESR)
expressed in mm/h, is a measure of the rate at which red blood cells settle when an anticoagulated blood sample is left to stand in a column.
Plasma viscosity
is usually measured by an automated viscometer. The plasma is drawn through a capillary tube under constant pressure and temperature; the rate of flow is measured and expressed as viscosity of plasma relative to distilled water.
Measurement of ESR or plasma viscosity may be useful when assessing the acute phase response. Both are affected by the presence of large proteins such as fibrinogen, α2-macroglobulin and immunoglobulins, and are therefore usually raised during an acute phase response. They are non-specific screening tools for inflammation, and are useful in monitoring of diseases such as rheumatoid arthritis and response to treatment for example with non-steroidal anti-inflammatory drugs and anti-tumour necrosis factor.
Plasma viscosity has several advantages over ESR. For example, it is not affected by physiological and environmental factors. Measurement is rapid, reproducible and lends itself better to quality assurance procedures than measurement of ESR. Conversely, ESR is a cheap test, but is influenced by factors such as anaemia, sex, age and (in females) stage of the menstrual cycle and must be carried out within about six hours of venepuncture.
Flow cytometry
White cells express characteristic nuclear, cytoplasmic and surface antigens; this is referred to as the immunophenotype of the cell. Immunophenotyping has been made possible by the availability of a large number of antibodies, which target specific cellular antigens. For example, antibodies that are directed against the CD19 antigen will bind to B lymphocytes.
Immunofluorescence forms the basis of flow cytometric investigations. It uses antibodies conjugated to fluorescent dyes (fluorochromes) such as fluorescein isothiocyanate (FITC), phycoerythrin (PE) and phycoerythrin cyanin (PC). Whole blood or bone marrow is incubated with one or more antibodies, each conjugated to a different fluorochrome. The cells are then injected in single file into the path of a laser beam. As the cell passes through, the light is scattered in all directions. Forward scatter (FSC) is influenced by cell size whereas side scatter (SSC) is influenced by the cellular structure and contents. The different cell populations can be visualized by plotting SSC against FSC. As the cell–antibody complex passes through the laser beam it fluoresces. The different wavelengths of light emitted are filtered, enabling specific cell populations to be selected (gated) and the data can be displayed as dot plots or histograms. The dot plots show the intensity of fluorescence as well as the number of cells that are positive for the particular antigen; the intensity of fluorescence is proportional to the strength of antigen expression. In the example shown in Figure 26.2, whole blood was incubated with three antibody-dye conjugates, CD8-FITC, CD4-PE and CD3-PC5.
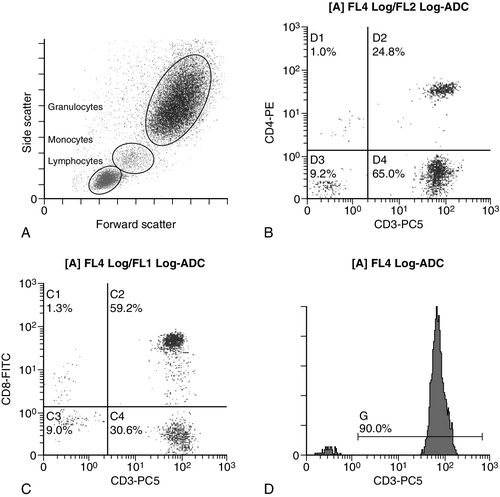
FIGURE 26.2 Scatter plots, dot plots and histogram printouts from Beckman Coulter FC500 flow cytometer. (A) Side scatter vs forward scatter, showing white cell populations. (B) CD4 vs CD3 dual dot plots. (C) CD8 vs CD3 dual dot plots. (D) Single histogram for CD3 gated for the lymphocyte population. Note: Granulocytes is the collective name for neutrophils, eosinophils and basophils.
Flow cytometry allows rapid immunophenotyping of cells. This is essential in the diagnosis and classification of leukaemias and lymphomas, but is also a useful tool in the assessment of non-malignant conditions and for monitoring the CD4 + lymphocyte count during the treatment of human immunodeficiency virus (HIV) infection. Flow cytometry can also be used to confirm feto–maternal haemorrhage (see p. 512), by the detection of Rh D positive fetal cells and measurement of haemoglobin F in the fetal cells.
Haematinic studies
These comprise direct measurement of the concentration of ferritin (a measure of iron status), vitamin B12 and folate. They are discussed further in Chapter 27.
Haemoglobinopathy screening
The haemoglobinopathies and the techniques used for their diagnosis are discussed in Chapter 29.
Tests for infectious mononucleosis
Infectious mononucleosis (IM), also known as glandular fever, is caused by the Epstein–Barr virus (EBV), which infects B lymphocytes. It is associated with a high titre of a heterophile antibody, so-called because it also reacts with red cells of other species, for example sheep, ox and horse. This antibody has been termed the Paul–Bunnell antibody, named after John Paul and Walls Bunnell who discovered them.
The Paul–Bunnell antibody is not adsorbed by guinea pig kidney, in contrast to other heterophile antibodies (Forssman antibodies). Differential adsorption tests utilize this principle; the patient’s serum is incubated with ox red cells and separately with guinea pig kidney antigen, after which horse red cells (reagent) are added to the mixtures. Red cell agglutination only in the latter mixture indicates the presence of the Paul–Bunnell antibody.
An alternative test is the latex agglutination test. A reagent containing latex microspheres coated with purified Paul–Bunnell antigen is mixed with the patient’s serum; agglutination indicates presence of the Paul–Bunnell antibody.
Definitive confirmation of EBV infection requires serological detection of a rise in the titre of specific anti-EBV capsid antigen IgM, which occurs in the first few weeks following infection.
MORPHOLOGY
Blood film examination
Microscopic examination of a well stained blood film is an essential part of many haematological investigations. The film may be made either manually or by an automated slide maker. After being air dried, it is stained with Romanowsky dyes such as May–Grünwald Giemsa or modified Wright stain, which contain azure B and eosin. This enables the assessment of the size, shape, maturity and contents of blood cells by light microscopy. Blood film examination is useful in the investigation of the causes of anaemia and to diagnose white cell disorders such as leukaemia and parasitic infections such as malaria. Less commonly, a blood film may be requested for specific reasons such as to look for acanthocytes (red cells with irregular spicules), which are associated with abetalipoproteinaemia, liver disease and several rare degenerative neurological diseases.
Normal red cell morphology
In health, there is little variation in the shape and size of red blood cells. On a well spread and stained blood film, red cells appear circular in outline, with a well stained outer rim and a paler central area that occupies approximately a third of the cell (see Fig. 26.3). The normal red cell appears slightly smaller than the nucleus of a small lymphocyte. It is referred to as normocytic (normal size) and normochromic (normal staining characteristics).
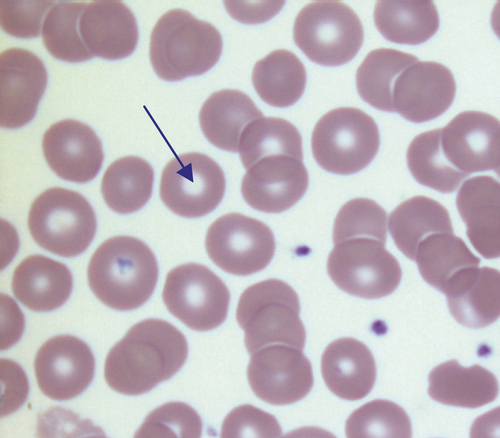
FIGURE 26.3 Normal red cells. Shows area of central pallor (arrow) occupying one-third the size of the cell.
Valuable information can be obtained from the shape of a red cell, as shown in Figure 26.4. In many haematological conditions there may be an increase in the proportion of non-specifically abnormal red cells, termed poikilocytosis. It is also good practice to check the patient’s biochemistry results, to corroborate the morphological features; for example, if acanthocytes are present, the liver function tests should be checked, as acanthocytes are commonly present in liver disease.
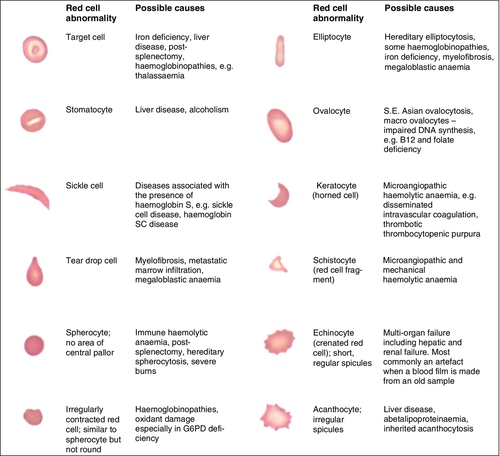
FIGURE 26.4 Red blood cells. Commonly observed morphological changes in red cells and the disorders in which they arise.
Morphology of the anaemias
Anaemia is a condition in which the number of red blood cells, or their oxygen-carrying capacity, is insufficient to meet physiological needs. This is conventionally defined as a reduction in haemoglobin concentration below the reference range, i.e. < 130 g/L in adult males and < 115 g/L in adult females. The causes and assessment of anaemia are discussed in Chapter 27.
Iron deficiency anaemia
In iron deficiency anaemia, the red cells are smaller than normal (microcytosis). This is because the maturing red cells undergo an extra cellular division before the critical haemoglobin concentration required to arrest mitosis is achieved. The cells are also hypochromic, with a larger area of central pallor (see Fig. 26.5). Polychromatic cells (immature red cells that have a bluish hue on routine staining because of retained ribosomal ribonucleic acid) may be present, indicating increased erythropoiesis.
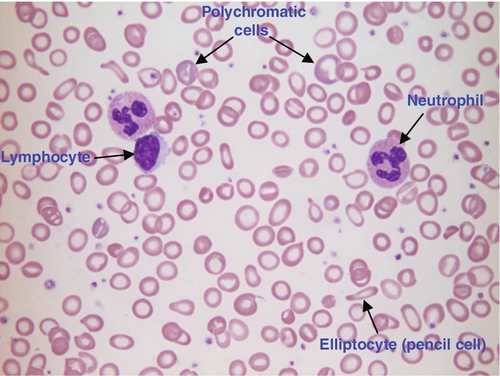
FIGURE 26.5 Iron deficiency anaemia (IDA). Hb 58 g/L, MCV 52.1 fL, MCH 12.7 pg, PLT 608 × 109/L, ferritin < 5 μg/L. Blood film shows microcytic hypochromic red cells, polychromatic red cells and pencil cells. The film also shows two neutrophils, a lymphocyte and an increase in platelet numbers.
Megaloblastic anaemia
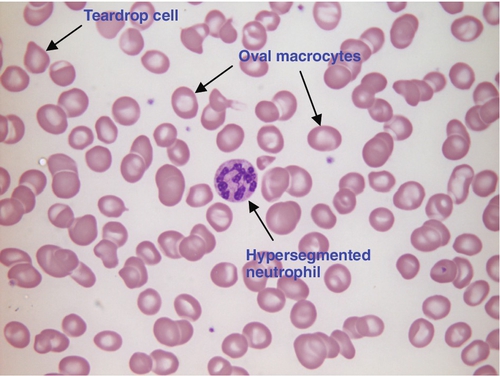
Megaloblastic anaemia is commonly caused by deficiency of vitamin B12 or folate, both of which are essential for DNA synthesis, or the administration of drugs that interfere with DNA synthesis (e.g. methotrexate). Defective DNA synthesis results in the nucleus maturing at a slower rate than the cytoplasm, thereby producing a red cell that is larger than normal (macrocyte). Teardrop cells and red cell fragments may also be seen, as a consequence of ineffective erythropoiesis (see Fig. 26.6). A common finding is the presence of hypersegmented neutrophils (defined as presence of a nucleus with more than five lobes).
Autoimmune haemolytic anaemia
Haemolytic anaemia is a term used to describe anaemia caused by a shortened red cell lifespan. In autoimmune haemolytic anaemia, the red cells are coated with antibody (usually IgG) that is directed against the patient’s own cells. When these cells circulate through the reticuloendothelial system, the surface of the red cells is eroded, leading to a change in their shape from a biconcave disc to a sphere (see Fig. 26.7).
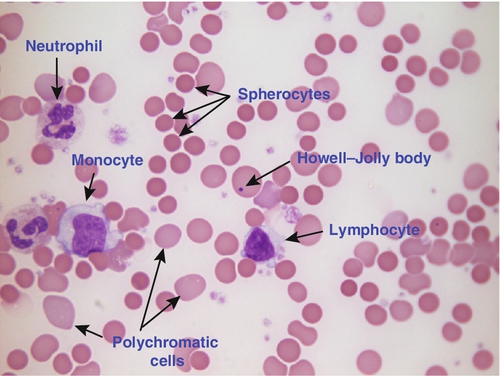
FIGURE 26.7 Warm autoimmune haemolytic anaemia. Hb 104 g/L, reticulocytes 604 × 109/L, haptoglobin < 0.2 g/L (reference range 0.7–3.2), direct antiglobulin test positive. Blood film shows numerous spherocytes, polychromatic cells and a red cell containing a Howell–Jolly body. Howell–Jolly bodies are typically found in patients who have undergone splenectomy. The film also shows two neutrophils, a monocyte and a lymphocyte.
Microangiopathic haemolytic anaemia
Microangiopathic haemolytic anaemia is a term that is used to describe the anaemia that results from physical damage to the red cells following the occlusion of arterioles and capillaries as a result of fibrin deposition or platelet aggregation. There are numerous causes, including infections (resulting, for example in disseminated intravascular coagulation (Fig. 26.8) or haemolytic uraemic syndrome), physical trauma (e.g. from mechanical heart valves) and autoimmune (e.g. thrombotic thrombocytopenic purpura). The blood film contains numerous fragmented cells (called schistocytes), and immature red cells (which have variable blue–grey colouration (polychromasia) owing to the presence of ribosomal material) released by the marrow in an attempt to compensate for the shortened red cell survival (see Fig. 26.9).
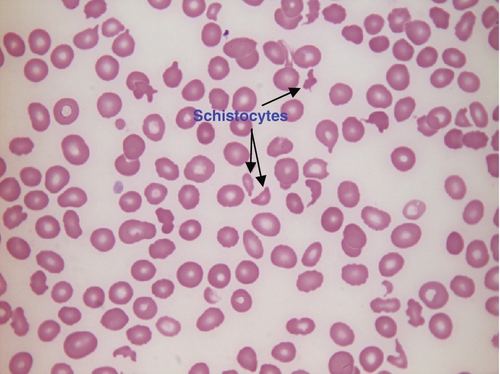
FIGURE 26.8 Disseminated intravascular coagulation (DIC). Hb 89 g/L, WBC 3.0 × 109/L, PLT 77 × 109/L. Blood film shows numerous schistocytes and reduced platelet numbers.
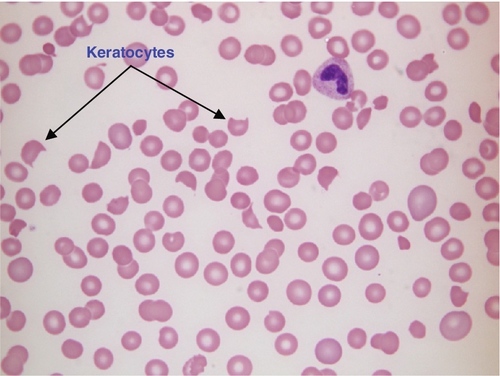
FIGURE 26.9 Haemolytic uraemic syndrome. Hb 84 g/L, WBC 24.3 × 109/L, PLT 23 × 109/L. Blood film shows numerous red cell fragments, some with the characteristic horn shape (keratocyte) and reduced platelet numbers. The WCC is increased because of neutrophilia as a result of bacterial infection with E. coli.
Malaria
Malaria is caused by infection with parasites of the Plasmodium species. It causes anaemia through marrow suppression, splenic pooling and sequestration of red cells. There is also haemolysis because of removal and destruction of parasitized red cells, and intravascular haemolysis when the sporozoites are released from the infected red cells. There are four Plasmodium species, each having distinct morphological features that can be recognized by microscopic examination of a stained thick blood film in the majority of infected patients (see Fig. 26.10).
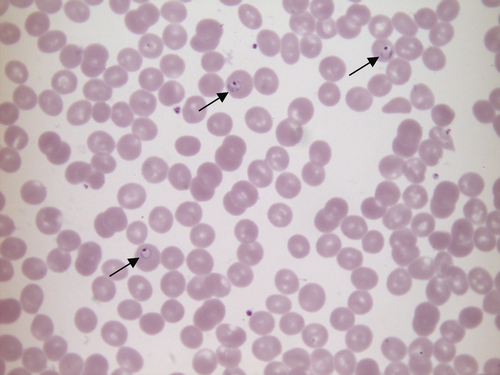
FIGURE 26.10 Plasmodium falciparum malaria. Hb 149 g/L, WBC 6.1 × 109/L, PLT 111 × 109/L. Blood film stained by the dilute Giemsa method, showing Plasmodium falciparum trophozoites (ring forms) in three red cells (arrows).
Normal white cell morphology
In health, five types of WBCs (also called leukocytes) are found in the peripheral blood (see Fig. 26.11). Their function is to protect the body against infection and other foreign invasion. This is facilitated by phagocytosis (neutrophils, eosinophils, basophils and monocytes) and humoral and cell mediated immunological activity (lymphocytes).
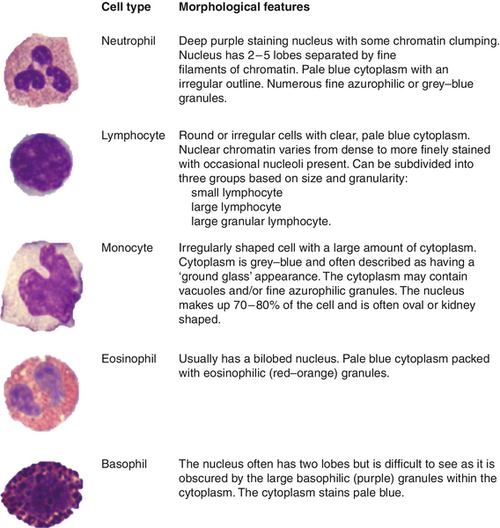
FIGURE 26.11 White blood cells. Normal morphological features of the myeloid and lymphoid white blood cells, shown in order of prevalence in the peripheral blood.
The most common cause of a raised WBC count (leukocytosis) is a neutrophil leukocytosis (neutrophilia), which may occur after severe exercise or tissue damage, and in bacterial infections, metabolic disorders and neoplasms of both haematological and non- haematological aetiology. The monocyte count may be raised in chronic bacterial infections and malignancies (again of both solid and haematological origin). A raised lymphocyte count (lymphocytosis) may be found in viral infections such as infectious mononucleosis and mumps, in chronic lymphocytic leukaemia and non-Hodgkin lymphoma. Allergic disorders, parasitic infections and Hodgkin lymphoma are associated with a raised eosinophil count.
A low WBC count (leukopenia) is usually a reflection of a reduction in the concentration of neutrophils (neutropenia) or of all blood cells (pancytopenia). Causes of neutropenia include therapeutic drugs, autoimmune diseases and viral infections, whereas pancytopenia is generally caused by marrow failure.
Abnormal white cell morphology
Morphological abnormalities may be divided into those resulting from benign and malignant disorders. Benign disorders include:
• toxic changes in neutrophils in bacterial infection; neutrophils often exhibit coarser granules in the cytoplasm, cytoplasmic vacuolation and Döhle bodies (Fig. 26.12)
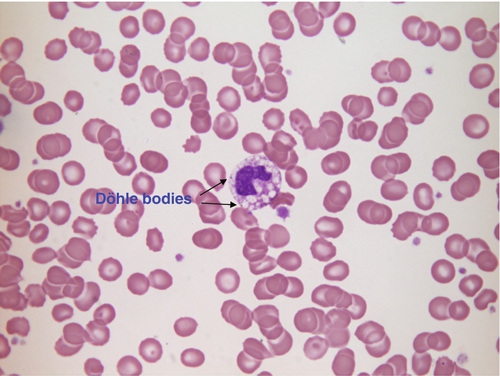
FIGURE 26.12 Bacterial infection. Hb 101 g/L, WBC 4.9 × 109/L, PLT 218 × 109/L.
Blood film shows toxic changes in a neutrophil: coarse granulation of the nucleus, cytoplasmic vacuolation and Döhle bodies (arrows) on the edge of the cytoplasm. Döhle bodies may also be found in myelodysplastic syndromes and acute myeloid leukaemia.
• neutrophil hypersegmentation in megaloblastic anaemia (Fig. 26.6)
• atypical lymphocytes in viral infection (Fig. 26.13)
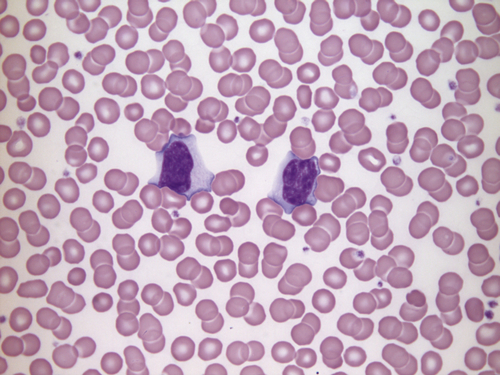
FIGURE 26.13 Infectious mononucleosis. Hb 165 g/L, WBC 23.9 × 109/L, lymphocytes 13.9 × 109/L, PLT 195 × 109/L. Blood film shows two atypical lymphocytes with basophilic cytoplasm. These are T lymphocytes that react against B lymphocytes infected with EBV. The film shows a typical appearance of red cells ‘wrapped’ around the lymphocytes.
• inherited disorders, for example Pelger–Huët anomaly and May–Hegglin anomaly.
Haematological malignancies
The haematological malignancies are clonal diseases originating from a single cell in the bone marrow or lymphoid tissue that has become genetically altered. They represent approximately 7% of all malignant diseases. They include acute leukaemias, chronic leukaemias, myeloproliferative neoplasms, myelodysplasia, non-Hodgkin lymphoma and multiple myeloma and its related disorders. The World Health Organization (WHO) classification of tumours of haematopoietic and lymphoid tissues is widely used to classify these malignancies. This system is based on morphologic, immunophenotypic, cytogenetic, molecular genetic and clinical features.
Acute leukaemia
Acute leukaemias have a rapid onset and progression and are invariably fatal if left untreated. They are characterized by an accumulation of early haemopoietic cells, known as blast cells (see Fig. 26.14), in the bone marrow. Morphologically, acute leukaemia is defined as the presence of > 20% blast cells in the bone marrow or peripheral blood. The acute leukaemias are a heterogeneous group of conditions; the role of morphology in classifying them into subtypes of myeloid and lymphoid leukaemias has been superseded by the use of molecular genetics, cytogenetics and immunophenotyping. This allows classification of these complex disorders into prognostically relevant categories, and aids decision-making over the role of chemotherapy or the requirement for bone marrow transplantation after disease remission has been induced. The use of molecular genetics is becoming increasingly important in monitoring both the response of the disease to treatment and detecting patients at risk of relapse.
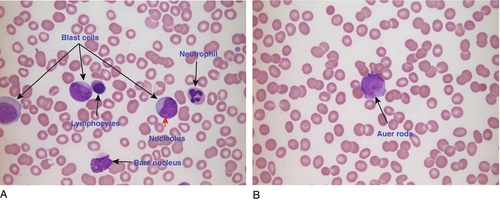
FIGURE 26.14 Two cases of acute myeloid leukaemia (AML). (A) Hb 111 g/L, WBC 1.0 × 109/L (3.6–11.0), blast cells 0.69 × 109/L, PLT 16 × 109/L. Blood film shows three myeloblasts containing two or more nucleoli. The film also shows a neutrophil and a lymphocyte. (B) Hb 127 g/L, WBC 30.3 × 109/L, blast cells 13.3 × 109/L, PLT 31 × 109/L. Blood film shows a myeloblast containing two Auer rods in the cytoplasm and reduced platelet numbers.
Chronic leukaemia
Chronic leukaemias are characterized by their slower disease progression and can broadly be divided into myeloid and lymphoid types.
Chronic myeloid leukaemia
is a clonal disorder of pluripotent stem cells, as shown by the presence of the Philadelphia (Ph) chromosome, an abnormal chromosome 22, which results from translocation t(9:22) (q34:q11) between chromosome 9 and 22. The fusion gene BCR-ABL1 is also created as a result of the translocation. The white cell count is usually > 50 × 109/L and the blood film shows all the stages of the myeloid series, from myeloblasts through to neutrophils, and an increase in basophils (see Fig. 26.15).
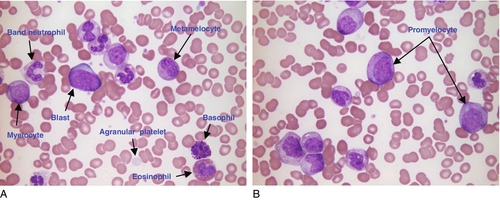
FIGURE 26.15 Chronic myeloid leukaemia. Hb 101 g/L, WBC 125.6 × 109/L, PLT 338 × 109/L. (A) and (B) are photographs of the same blood film showing all stages of differentiation of the myeloid lineage. Usually this is only seen in the bone marrow.
Chronic lymphoid leukaemias
are characterized by an accumulation of mature B or T lymphocytes. Included in this group are chronic lymphocytic leukaemia, prolymphocytic leukaemia, hairy cell leukaemia and plasma cell leukaemia. Chronic lymphocytic leukaemia is the most common of the chronic lymphoid leukaemias and occurs in older subjects, typically between 60 and 80 years of age. Immunophenotyping shows the cells to be of B-lineage. The lymphocyte count is usually > 5 × 109/L and the blood film shows presence of predominantly small lymphocytes and smear (smudge) cells (see Fig. 26.16).
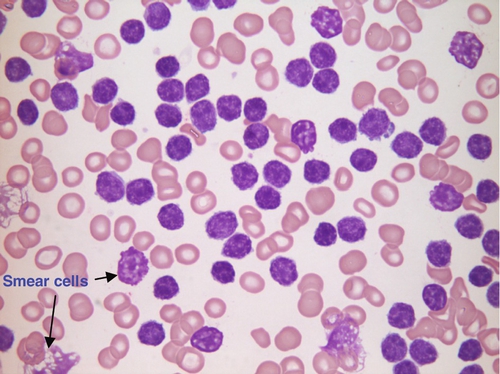
FIGURE 26.16 Chronic lymphocytic leukaemia. Hb 73 g/L, WBC 950 × 109/L (no neutrophils), PLT 85 × 109/L. Blood film shows numerous small lymphocytes and some smear cells (fragile lymphocytes damaged during spreading of blood film).
Myeloproliferative neoplasms
These are clonal disorders of haemopoietic stem cells, characterized by increased marrow proliferation of one or more myeloid cell lineages: granulocytic, erythroid, megakaryocytic and mast cells. The major myeloproliferative disorders are polycythaemia vera, essential thrombocythaemia and primary myelofibrosis (see Fig. 26.17). A single acquired mutation of the cytoplasmic kinase Janus-associated kinase 2 (JAK-2) is found in 90% of patients with polycythaemia vera and in 50% of those with essential thrombocythaemia and primary myelofibrosis.
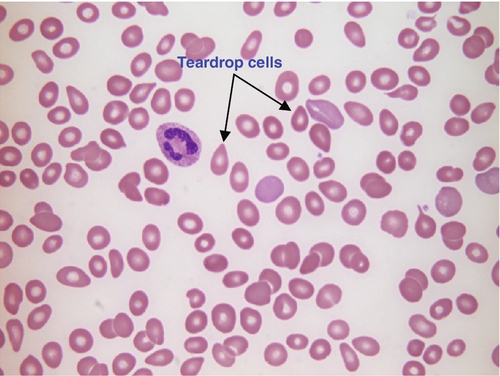
FIGURE 26.17 Myelofibrosis. Hb 86 g/L, WBC 4.3 × 109/L, PLT 56 × 109/L. Blood film shows numerous teardrop cells, some polychromatic cells, one neutrophil and few platelets.
Myelodysplasia
Myelodysplastic syndrome is a term used to describe a heterogeneous group of clonal disorders of haemopoietic stem cells. The bone marrow shows increased proliferation but, owing to ineffective haemopoiesis, there is a reduction in all myeloid cell lineages (pancytopenia) in the peripheral blood. There is dysplasia in at least one of the cell lineages. The blood film often shows hypogranular neutrophils, pseudo-Pelger neutrophils, large hypogranular platelets and a dimorphic red cell population (see Fig. 26.18). Myelodysplastic syndrome occurs in older subjects, with a median age of 69 years. The risk of developing acute leukaemia depends upon numerous prognostic factors, including the number of blasts present in the marrow, the number of blood cell types affected and cytogenetic changes.
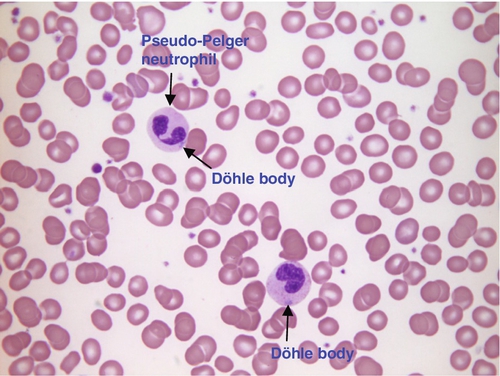
FIGURE 26.18 Myelodysplasia (MDS). Hb 110 g/L, WBC 33.6 × 109/L, PLT 209 × 109/L. Blood film shows two dysplastic neutrophils with few or no cytoplasmic granules. One of the neutrophils is a pseudo-Pelger cell (bi-lobed). Both neutrophils contain Döhle bodies.
Non-Hodgkin lymphoma
Non-Hodgkin lymphoma refers to a heterogeneous group of clonal lymphoid tumours. The term lymphoma is generally used to describe tumours arising in lymph nodes, the spleen and other solid organs, in contrast to leukaemias, in which the bone marrow is predominantly involved, but in some cases there is no clear distinction between the two. Characteristic lymphoma cells may be seen in the blood film, for example lymphocytes with a cleaved nucleus in follicular lymphoma (see Fig. 26.19).
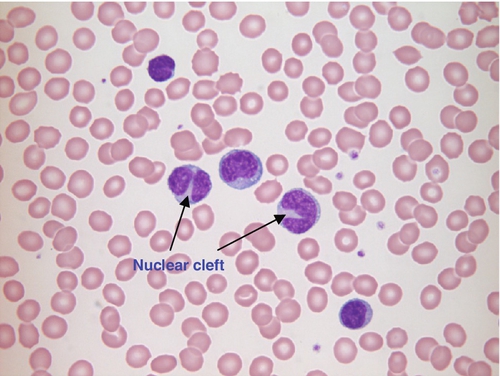
FIGURE 26.19 Follicular lymphoma Hb 101 g/L, WBC 38.5 × 109/L, lymphocytes 37.7 × 109/L, PLT 85 × 109/L. Blood film shows abnormal lymphocytes, two of which have deep nuclear clefts.
Multiple myeloma and related disorders are clonal malignancies of B lymphocytes. They are discussed in Chapter 30.
HAEMOSTASIS
Introduction
Haemostasis plays an essential role in preventing excess blood loss following injury. It is a complex process involving interactions between blood vessel walls, platelets and coagulation factors to form a haemostatic plug with the consequent activation of fibrinolysis, whereby the clot is gradually dissolved. Excessive activation of procoagulant mechanisms can lead to thrombosis and vascular occlusion; therefore a delicate balance is maintained via negative and positive feedback pathways.
Following blood vessel injury, the vessel constricts to reduce blood flow, and subendothelial collagen and tissue factor is exposed. Platelets adhere to the collagen and are activated, releasing procoagulant particles and undergoing a change in membrane configuration to expose negatively charged phospholipids and coagulation factor receptors. The initial platelet plug formed is unstable but is rapidly stabilized by activation of the coagulation cascade, resulting in the formation of fibrin.
The coagulation cascade
Historically, the coagulation process was divided into separate intrinsic and extrinsic pathways, terminating in the final common pathway with the formation of a stable fibrin clot. It is now established that the coagulation cascade is a single series of more complex interactions.
In vivo, blood coagulation is initiated by the exposure of tissue factor (TF) following endothelial cell damage. The TF binds to factor VII, and functions as a cofactor in the activation of factor VII, with the resulting TF:FVIIa complex activating factor X and factor IX, enabling the conversion of prothrombin to thrombin. The TF:FVIIa complex is quickly inhibited by tissue factor pathway inhibitor but the thrombin already generated enables the coagulation process to continue as it feeds back to activate coagulation factors V, VIII and XI, resulting in amplification of the brief initiator process. The ultimate goal of the coagulation process is the generation of thrombin, which converts fibrinogen to fibrin. Fibrin monomers spontaneously join together via hydrogen bonds, forming insoluble fibrin polymers; these are then cross linked by factor XIIIa, changing the relatively unstable primary platelet plug into a stable clot. In vitro, however, consideration of coagulation screen results in terms of the traditional pathways allows for the simpler interpretation of the common laboratory tests (see Fig. 26.20).
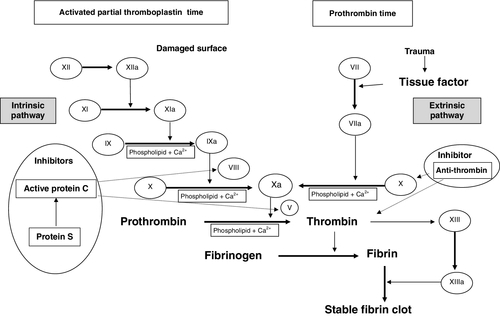
FIGURE 26.20 Coagulation cascade to illustrate use of laboratory tests and the function of naturally occurring inhibitors.
Once a stable clot has been produced and bleeding has ceased, the clot is reorganized and resorbed by a process called fibrinolysis. The main enzyme responsible is plasmin, which is formed from plasminogen.
In order to restrict coagulation factor activation to the site of injury, naturally occurring inhibitors including anti-thrombin, protein C and protein S are produced.
Laboratory tests of coagulation
Laboratory tests of coagulation attempt to mimic in vitro the complex in vivo coagulation processes, but cannot do so completely. The generation of an abnormal result does not therefore necessarily mean that the patient has a bleeding diathesis, and a normal result does not completely exclude a bleeding diathesis. It is important that the test results are interpreted in the clinical context; the indiscriminate use of coagulation screens is best avoided.
The standard tests for coagulation include prothrombin time (PT), activated partial thromboplastin time (APTT), fibrinogen concentration, D-dimer concentration and thrombin time (TT). The PT measures the coagulation factors involved in the classic extrinsic and common pathways and the APTT is used to test the coagulation factors involved in the classic intrinsic and common pathways.
Prothrombin time and APTT are measured by techniques that ultimately detect the formation of fibrin clot by either mechanical or optical methods.
Prothrombin time
Plasma is mixed with thromboplastin, which contains TF, phospholipids and calcium ions, all of which are required for the activation of the extrinsic pathway. This causes a fibrin clot to form via the extrinsic and common pathways. Different thromboplastin reagents result in differing prothrombin times but internationally standardized results can be generated by correlating the thromboplastin used with the WHO reference standard. This converts the PT result to an International Normalized Ratio (INR), which is used for monitoring patients on warfarin.
Activated partial thromboplastin time
Measurement of the APTT involves induction of the formation of a fibrin clot via the intrinsic and common pathways. Plasma is first incubated with partial thromboplastin, which contains a contact activator, e.g. micronized silica or kaolin, which activates factor XII and phospholipids, triggering the intrinsic pathway. The factor XIIa in turn activates factor XI; this reaction is not significant in vivo but is used in vitro to produce factor XIa independently of the extrinsic pathway. A second reagent containing calcium ions is then added, to enable the process to proceed to the formation of a fibrin clot.
Fibrinogen and thrombin time
Fibrinogen concentration can be estimated from the optical density changes observed when PT is measured using the optical method. A more accurate measurement can be performed using the thrombin clotting time. In this investigation, a thrombin-containing reagent is added to a dilution of the patient’s plasma: the resultant clotting time is proportional to fibrinogen concentration.
D-dimer concentration
D-dimers are cross-linked D fragments: they are only produced as a result of the breakdown of cross-linked fibrin and not from the degradation of fibrinogen and soluble fibrin. Around 2–3% of plasma fibrinogen is physiologically converted to cross-linked fibrin and then degraded. Thus a small quantity of D-dimer is present in the plasma of healthy individuals. Increased amounts of cross-linked fibrin degradation products are found in states of coagulation activation and increased plasma D-dimer concentration may indicate an increased rate of fibrinolysis; this may be a consequence of the presence of a deep venous thrombosis (DVT) or pulmonary embolus (PE), or a result of disseminated intravascular coagulation (DIC). It should be noted that the main diagnostic value of D-dimer measurement lies in its negative predictive value; raised concentrations are commonly found post surgery, in pregnancy, in malignancy and in other situations associated with activation of the fibrinolytic system, resulting in many false positives.
Plasma D-dimer concentration is usually measured by latex particle enhanced immunoassay. There are also many assays that measure fibrin degradation products (FDPs) often using latex agglutination techniques. The diagnostic value of FDPs is reduced by the fact that they may be generated from the breakdown of both fibrin and fibrinogen and thus their presence is not necessarily associated with clot formation.
Specific factor assays
Deficiency of individual clotting factors is detected by adding the patient’s plasma to artificially produced plasma that is deficient in one specific factor. Measurement of PT or APTT on this mixture enables calculation of factor concentration. Results are expressed as a percentage of normal activity. An abnormal result may reflect the presence of an inhibitor antibody as well as factor deficiency. This can be identified by performing a correction study where plasma from healthy subjects is added to the test plasma, in a 50:50 ratio. If correction is less than expected, the presence of an inhibitor of coagulation should be suspected.
Interpretation of coagulation tests
If a patient presents with a history of excessive bleeding or easy bruising, the use of the simple laboratory screening tests will guide the clinician to the likely underlying cause and what further investigations are appropriate. A guide to the interpretation of the results is given in Table 26.2.
TABLE 26.2
Screening tests used in the diagnosis of coagulation disorders
| Screening tests | Coagulation disorders indicated by abnormal result | Most common cause of coagulation disorder |
| Prothrombin time (PT) | Deficiency or inhibition of one or more of the following coagulation factors: VII, X, V, II or fibrinogen | Warfarin therapy Prematurity Disseminated intravascular coagulation (DIC) |
| Activated partial thromboplastin time (APTT) | Deficiency or inhibition of one or more of the following coagulation factors: XII, XI, IX VIII, X, V, II or fibrinogen | Haemophilia (factor VIII) Christmas disease (factor IX) Heparin therapy Liver disease DIC |
| Fibrinogen | Inflammatory or neoplastic conditions | Low concentrations: DIC Liver disease Fibrinolytic therapy High concentrations: Inflammation Neoplastic conditions |
| D-Dimer | Fibrin clot lysis | Pulmonary embolus Deep vein thrombosis DIC Postoperative Infection Cancer Pregnancy (raised in normal pregnancy) |
| Thrombin time | Deficiency or abnormality of fibrinogen or inhibition of thrombin by heparin or fibrinogen degradation products (FDPs) | Heparin therapy DIC Other thrombin inhibitors Hypofibrinogenaemia Dysfibrinogenaemia |
Haemophilia
The most common clotting factor deficiency associated with bleeding is factor VIII deficiency, also known as haemophilia A, followed by factor IX deficiency (haemophilia B). Inheritance of both conditions is sex-linked and can be caused by a number of different mutations. The severity of the bleeding disorder depends on the amount of active clotting factor: individuals with < 1% are classified as having severe haemophilia, those with 1–5% have moderate haemophilia and those with mild haemophilia have 5–40% of normal concentrations of active clotting factor. Patients with severe haemophilia suffer from recurrent severe and spontaneous bleeds. Bleeding may occur anywhere in the body but those that are associated with the most serious complications are joints, muscles, gastrointestinal tract and the brain. Most patients with severe haemophilia require regular prophylactic supplementation with recombinant factor concentrate and additional supplementation following episodes of bleeding or to cover surgery (see p. 513). Patients may develop antibodies (or inhibitors) to the factor infused, rendering it ineffective. In these situations, recombinant activated factor VII and activated prothrombin complex concentrates (factor VIII inhibitor bypassing activity, FEIBA) can be useful in the treatment of bleeding. Patients with mild haemophilia tend to bleed only after surgery or serious trauma and can often be managed successfully with desmopressin, which releases stored factor VIII from blood vessel walls.
Disseminated intravascular coagulation
Disseminated intravascular coagulation can become catastrophic if not treated rapidly. It may be triggered by numerous disorders, such as sepsis, disseminated malignancy and amniotic fluid embolism. These lead to the formation of small blood clots in blood vessels throughout the body, consuming coagulation factors and platelets and resulting in abnormal bleeding, e.g. from venesection sites, gastrointestinal tract and surgical wounds. Coagulation screen results will show prolonged PT and APTT, and low fibrinogen and high D-dimer concentrations. In these circumstances, the underlying cause should be corrected as soon as possible and platelets, fresh frozen plasma (FFP) and cryoprecipitate infused if bleeding is severe (see p. 513).
BLOOD TRANSFUSION
Introduction
In the UK, blood donation is entirely voluntary and open to healthy individuals aged 17–65 years at the time of first donation. Donors undergo rigorous health assessments; individuals with lifestyle risks or a recent history of infection are excluded from donating. Each donation is of 450 mL of whole blood and is tested for human immunodeficiency virus (HIV), hepatitis B and C, and syphilis, along with determination of the Rh and ABO blood group, before it is released to be transfused into a recipient.
Hospital blood transfusion laboratories perform a range of tests required for supply of patient-compatible blood and blood products and in the investigation of haemolysis resulting from blood incompatibility (including haemolytic disease of the newborn).
Blood group antigens
The red cell membrane carries a range of surface antigens. Those that differ between individuals of the same species as a result of genetic polymorphisms (alloantigens), and can therefore elicit an immune response between donor and recipient blood, are termed the blood group antigens. There are around 30 recognized blood group systems.
ABO blood group
This blood group system is considered the most important, because of the high risk of a severe haemolytic transfusion reaction if ABO incompatible blood is transfused.
There are three antigens in the ABO blood group system, A, B and H. The ABO gene is located on chromosome 9 and has three alleles, A, B and O. The A allele encodes a glycosyltransferase that adds N-acetylgalactosamine to the glycoprotein H antigen that is expressed on all normal red cells. The B allele encodes a different glycosyltransferase that adds D-galactose. The O allele is a deletion that results in loss of enzyme translation and therefore presence of unmodified H antigen. A and B are co-dominant alleles; AB individuals express both antigens.
Individuals produce antibodies to the antigen that they lack on their red cells. Landsteiner’s law states that, for whichever ABO antigen is not present on the red cells, the corresponding antibody is found in the plasma (see Table 26.3). Antibodies to A and B antigen develop in the first few months of life, without exposure to the red cell antigen, as a result of environmental exposure to bacteria. They are referred to as ‘naturally occurring’ antibodies. ABO antibodies are usually IgM but may be IgG.

It is essential that the ABO group of transfused red cells is compatible with the ABO blood group of the patient. Transfusion of even a few millilitres of red cells into patients who have the corresponding antibody results in a severe immune reaction, which may be fatal. Cells of blood group O are compatible with patients of all ABO blood groups so individuals of group O can be referred to as universal donors. Conversely, individuals of blood group AB can receive blood from any of the other groups and are referred to as universal recipients (see Table 26.3).
Rh blood group
The Rh (formerly rhesus) blood group consists of over 45 antigens, of which D, C, E, c and e antigen are usually typed in donor blood. All patient samples for blood grouping are routinely typed for their RhD status because the D antigen is clinically the most important after the ABO antigens. About 85% of the population express the D antigen and are called RhD positive; the remainder are termed RhD negative. The D antigen is highly immunogenic so individuals who are RhD negative but receive RhD positive blood are likely to produce anti-D antibodies. Anti-D has the potential to cause a severe haemolytic transfusion reaction and cause haemolytic disease of the newborn (HDN) (see below).
Expression of the D antigen is variable. Some patients express only small amounts of the D antigen; their RhD status is termed weak D. Other patients express only part of the D antigen; their RhD status is described as partial D. Partial RhD individuals can produce antibody to the part of the D antigen, which they lack, so they should receive RhD negative red cells if they require transfusion.
Other important blood groups and antibodies
Kell, Kidd, Duffy and MNS blood groups are clinically important because antibodies to antigens of these blood groups can cause either transfusion reactions or HDN or both. They are included routinely in antibody identification panels.
The Kell antibody (Anti-K) is the most commonly found antibody after those to the ABO and Rh blood group antigens. A third of all non-ABO, non-Rh alloantibodies are anti-K. It is good practice to give Kell-negative blood to patients who already have antibodies to other blood group antigens. Women of child-bearing age who require blood transfusion should also be transfused with Kell negative blood of the appropriate ABO and Rh grouping.
The Kidd antibodies (Anti-Jk(a) and Anti-Jk(b)) form another important antibody system that can cause severe acute and delayed transfusion reactions. Anti-Jk(a) is the most commonly implicated antibody in delayed transfusion reactions. Anti-Jk(a) may fall to low or undetectable concentrations in the patient’s plasma over time so can be difficult to detect.
Laboratory transfusion tests
Blood grouping and antibody screen
Commonly referred to as ‘group and save’, this is the most requested test and comprises two parts: ABO and RhD grouping, and an antibody screen.
ABO and D grouping
Antisera are mixed with the patient’s red cells; this is known as the forward group. In ABO grouping, the patient’s plasma is also mixed with known A and B cells: the reverse group. The positive and negative patterns of agglutination determine the patients ABO and RhD group (see Table 26.4). This can be performed manually or by automated methods but the principles remain the same.
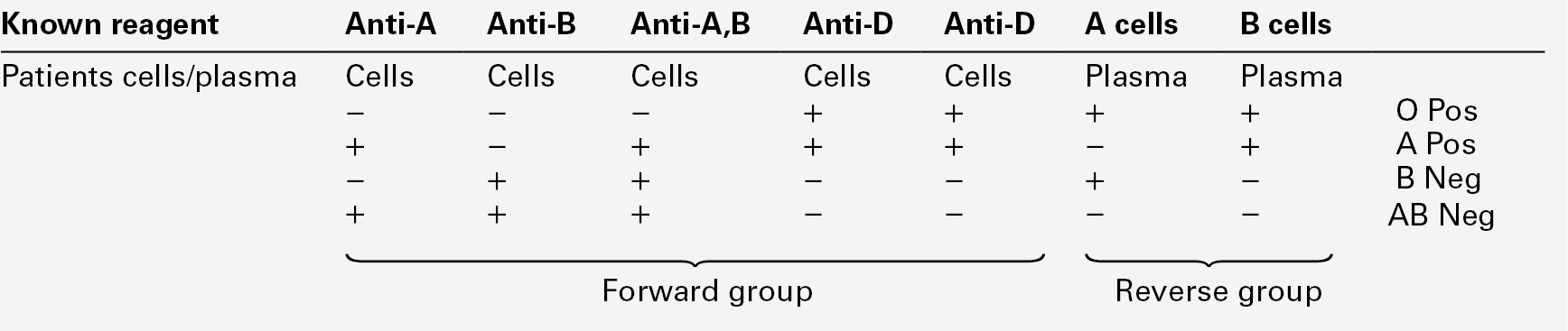
Antibody screening
This procedure is designed to detect clinically significant alloantibodies to blood group systems other than ABO. There are two stages to an antibody screen: sensitization and haemagglutination.
The patient’s plasma is incubated at 37 °C with two or three red cell samples of known blood group phenotype. These samples are always group O to avoid reactions that would result from ABO incompatibility. The antibody–antigen complex formation that occurs is termed sensitization. In the second stage, anti-human globulin (AHG) reagent is added, to encourage cross linking between sensitized red cells, as most antibodies are of the IgG class so cannot directly form cross links. If agglutination occurs, further tests are required to identify the antibody or antibodies present. This procedure is known as the indirect antiglobulin test (IAT).
The patient’s sample is retained and stored in accordance with national guidelines. It can subsequently be used in order to crossmatch blood for that patient, as described later. It is essential that, if a patient has recently been transfused, they are re-tested prior to any further transfusion in order to detect any new antibodies they may have developed in response to the previous transfusion.
Antibody identification panels
Antibodies that need to be identified are those that can cause transfusion reactions or HDN. Following a positive antibody screen, further tests are done to identify the antibody using the same theory as for antibody screening. The antibody panel usually contains at least ten different red cell reagents of known phenotype. There are usually two sets of red cell reagents in one panel set, one of which has been treated with an enzyme that cleaves off part of the red cell membrane, as a result of which some antigens are lost from the red cell membrane (MNS and Duffy systems) and others are exposed in order to produce stronger antibody binding (the Rh system). The use of both sets of cells is important as the differences in reaction for different blood group systems can help determine antibody identity, particularly when multiple antibodies are present.
Crossmatching (compatibility testing)
Crossmatching is designed to ensure compatibility of donor units with the recipient. The procedure can be either non-serological or serological. There are three main types of crossmatch:
• electronic crossmatch or blood issue (non-serological)
• immediate spin crossmatch (serological)
• full crossmatch (serological).
Electronic crossmatch
is the quickest to perform, enabling issue of blood within five minutes. A prerequisite is that a group and antibody screen has already been performed for that patient. In this non-serological process, the laboratory computer system checks for ABO and RhD compatibility. For a sample to be suitable for electronic crossmatching, a number of requirements must be met. There are some minor differences between guidelines issued by the British Committee for Standards in Haematology (BCSH) and the American Association of Blood Banks (AABB), but the following elements are generally considered essential in the UK:
• the ABO and RhD grouping procedure must have been performed on a fully automated grouping machine
• results must have been transferred to the laboratory computer system electronically, with no manual editing of results
• there must be two concordant ABO and RhD records on file for that patient
• no clinically significant antibodies have been detected presently or historically.
Transfusion laboratories may also adhere to additional locally determined rules, for example only donor units that have been electronically entered onto the computer system (via barcode scanning) being considered suitable for electronic issue. Not all laboratories perform electronic crossmatch, as some may not be equipped with automated analysers or do not have a laboratory computer system that is capable of processing the required algorithm.
Immediate spin crossmatch
is designed principally to detect ABO incompatibility. It may be used in an emergency when there is insufficient time to perform a full antibody screen. It may also be performed when the criteria for electronic issue are not fully met, for example if a sample has been run on an automated analyser that has not been able to interpret the group correctly. This could be caused by weak expression of anti-A, anti-B or RhD. It is also important that there is no record of the patient having clinically significant antibodies presently or historically. This is a manual technique performed in tubes: the patient’s plasma and donor red cells are incubated and then centrifuged to check for haemagglutination or haemolysis that would indicate ABO compatibility. Using this technique, it takes 10–15 min for blood to be issued from the laboratory.
Full crossmatch
is performed if time or equipment availability do not permit a sample to be processed on a blood grouping analyser, or following automatic processing if the patient is known to have clinically significant antibodies presently or historically. Some laboratories may not have adopted the immediate spin or the electronic crossmatch and therefore continue to use the full crossmatch routinely. In this procedure, the patient’s plasma is incubated with donor units and checked for compatibility using the IAT technique described in the previous section. Using this technique, it takes 45 min for blood to be issued from the laboratory.
Investigation of suspected transfusion reaction
Serious or life-threatening acute reactions are very rare. Acute haemolytic transfusion reactions are most commonly a consequence of errors in taking or labelling the sample, collecting wrong blood from the blood bank, inadequate checking of the product at the bedside or laboratory errors. Symptoms, such as the patient having a feeling of impending doom, agitation, flushing or pain at the venepuncture site or in the abdomen, flank or chest can occur after transfusion of only a few drops of blood. Clinical signs include fever and hypotension; generalized oozing from wounds or puncture sites can occur and there may be haemoglobinaemia or haemoglobinuria. Life-threatening transfusion reactions can also occur with platelet and FFP transfusions, for example owing to bacterial contamination or transfusion-related acute lung injury.
Following a suspected transfusion reaction, urgent blood tests (full blood count, assessment of kidney and liver function), along with assessment of the urine for haemoglobinuria, should be undertaken. The donor units and giving set should be returned to the laboratory, along with a fresh blood sample. The blood sample and donor units along with the original ‘group and save’ sample are then tested retrospectively for compatibility. At this point, it is also important to confirm the blood group of any units transfused and send the giving set and donor packs for culturing. Blood cultures should be taken if the patient has a persistent fever, and a coagulation screen performed to exclude developing DIC if the reaction is severe.
Haemolytic disease of the newborn
Haemolytic disease of the newborn (HDN) is a condition in which the neonatal red cells have a shortened lifespan because antibodies of maternal origin have crossed the placenta and coated them. These cells are then removed by the immune system. The mother will have produced the antibodies following fetal–maternal haemorrhage. It is not uncommon in pregnancy for small ‘silent’ bleeds to occur that are too small for detection. Antibodies that cause HDN are of the IgG class. The most common cause of HDN is ABO incompatibility, in which cases the haemolysis is usually mild. More severe cases of HDN can be caused by anti-D, anti-c and anti-K. Anti-K suppresses neonatal red cell production as the K antigen is one of the first antigens to be expressed on the red cells during red cell production.
Standard UK antenatal practice is that samples are taken for blood group and antibody screening when the patient is booked for antenatal care (8–12 weeks of gestation) and again at 28 weeks. If the mother is RhD negative, samples are also sent at delivery. Expectant mothers who have produced antibodies during pregnancy require close monitoring. Depending on when the antibody is first detected, BCSH guidelines state that grouping and antibody screening should be performed and antibody titre determined every four weeks up to the 28th week of pregnancy and then every two weeks thereafter. The antibody titre can be used as a guide to the potential severity of HDN, with the exception of anti-K for which there is no quantitative test and little correlation between concentration and the severity of HDN.
In the UK, the incidence of HDN caused by anti-D has been reduced following the implementation of national anti-D prophylaxis programmes for antenatal women. Pregnant women who are RhD-negative are given prophylactic anti-D sufficient to deal with any silent bleeds. Routine prophylaxis can either be given as two doses of 500 IU anti-D administered at 28 weeks and then 34 weeks of gestation or a single dose of 1500 IU administered at 28 weeks. At delivery, an RhD negative mother is given more anti-D if the baby is RhD positive. The amount they receive depends upon the results of a Kleihauer test, which is a slide test for the number of fetal cells in the maternal circulation (see Fig. 26.21). If any sensitizing events occur during the pregnancy, it will be necessary to administer a top-up dose of anti-D.
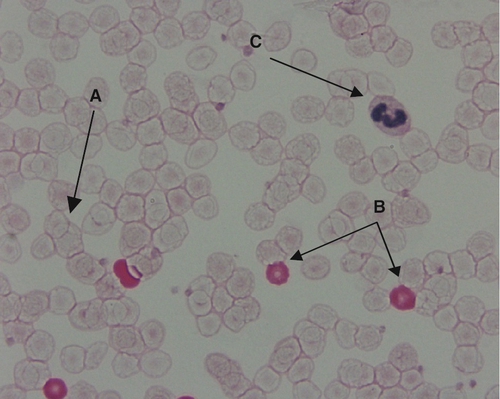
FIGURE 26.21 Kleihauer film. There is a good distinction between the three cell types: maternal cells (A), fetal cells (B) and white cells (C). The presence of RhD positive fetal cells is confirmed by flow cytometry.
Blood products
Red cells
Red cells are the most common blood product issued by a transfusion laboratory. Indications for red cell transfusion include active bleeding, acute anaemia caused by trauma or surgery, and chronic anaemia secondary to malignancy. For surgery in which blood loss is expected, red cells are crossmatched in advance in accordance with a maximum surgical blood ordering schedule (a schedule of the number of red cell units routinely crossmatched for each type of elective surgical procedure). Red cell concentrates have a maximum shelf-life of 35 days and must be stored at 4 ± 2 °C.
Transfusion of emergency group O RhD negative blood is an important option in life-threatening situations when there is insufficient time to provide crossmatched blood. These units must be easily available especially to the emergency department and theatres. The criteria for the selection of donor units should be determined locally, for example negative for the C and E Rh antigens and the Kell antigen, low titre of ABO antibodies and cytomegalovirus negative.
Platelets
Platelet transfusions are indicated for the prevention and treatment of haemorrhage in patients with thrombocytopenia or defective platelet function. They are, however, contraindicated in post transfusion purpura, thrombotic thrombocytopenia purpura and heparin induced thrombocytopenia as they would increase the risk of thrombosis. Platelet concentrates can be produced by pooling together platelets from whole blood donations or by obtaining platelets by apheresis. Platelets must be stored at 22 ± 2 °C and have a shelf-life of up to seven days with continuous gentle agitation.
Fresh frozen plasma
Fresh frozen plasma (FFP) contains all clotting factors found in plasma. Indications for its use are limited but it is most often used in patients with massive bleeding where clotting factors are being consumed or are diluted owing to the administration of large volumes of red cells or crystalloids. It must be stored at < − 25 °C and for a maximum of 24 months. It is most effective if used within four hours of thawing but can be stored at 4 °C post thawing for up to 24 h.
Cryoprecipitate
Cryoprecipitate is a plasma fraction rich in fibronectin, fibrinogen, factor VIII and von Willebrand factor. It is used particularly in patients who have low plasma fibrinogen concentrations such as during massive transfusions and in DIC. Like FFP, it must be stored at < − 25 °C and for a maximum of 24 months.
Factor concentrates
Factor concentrates can be either fractionated or recombinant. Fractionated products are produced by pooling multiple donations, which increases the risk of viral transmission to the recipient, making recombinant products the preferred option. Recombinant factor concentrates are used in patients with inherited deficiencies of clotting factors, either prophylactically or during bleeding episodes (see p. 509). Prothrombin complex concentrate, rich in factors II, VII, IX and X, is used for the emergency correction of warfarin-induced bleeding. Factor VII concentrate, although only licensed for use in inherited or acquired haemophilia with significant inhibitors or Glanzmann thrombasthenia (a platelet function disorder), is being increasingly used if life-threatening bleeding persists despite optimization of laboratory measured clotting and platelet parameters.
Risks of transfusion
The incidence of adverse events caused by blood transfusion is low, although the consequences are potentially severe. The main adverse risk is that of receiving the incorrect blood component. Strict procedures are in place to ensure vein to vein traceability of all blood products and to reduce the risks of misidentification of either the patient or donor. The very rare but fatal complication of transfusion-associated graft versus host disease, whereby immunocompetent donor lymphocytes are infused into an immunocompromised patient (from bone marrow transplantation or other defects in T cell immunity), is prevented by the irradiation of blood products to inactivate any residual white cells. The risk of transmitting infections is very small provided that appropriate screening and selection of donors is in place.
Regulations
In the UK, blood transfusion laboratories have to comply with a range of specific regulations and standards of practice, which are monitored by the Medicines and Healthcare Regulatory Authority (MHRA). The key standards are incorporated into UK law, making infringement a criminal offence. Adverse incidents associated with blood transfusion must be reported to the MHRA.
CONCLUSION
• Blood film examination is an essential part of a haematological investigation, as valuable information can be obtained from cell morphology.
• Prothrombin time, activated partial thromboplastin time, thrombin time, D-dimer and fibrinogen measurements are useful screening tests for coagulation disorders but should be interpreted within the clinical context.
• The blood transfusion laboratory performs a vital role in the provision of safe and effective blood product support.
• Antibody identification is an essential procedure in the prevention of transfusion reactions and haemolytic disease of the newborn.
ACKNOWLEDGEMENTS
We would like to thank our colleagues Rebecca Pleasance, Gemma Greenall and Colin Greig, who critically reviewed the material in this chapter and made many helpful suggestions. Rebecca Pleasance also provided the photographs and some of the text used in Figures 26.4 and 26.11.
Further reading
Bain BJ. Blood cells. 4th revised ed. Chichester: Wiley–Blackwell; 2006.
British Committee for Standards in Haematology Blood Transfusion Task Force. Guidelines for pre-transfusion compatibility procedures in blood transfusion laboratories. 2012a. www.bcshguidelines.com/documents/Compat_Guideline_for_submission_to_TTF_011012.pdf [Accessed 23 January 2013].
British Committee for Standards in Haematology Blood Transfusion Task Force. Guidelines on the investigation and management of acute transfusion reactions. 2012b. www.bcshguidelines.com/documents/ATR_final_version_to_pdf [Accessed 23 January 2013].
Daniels G. Human blood groups. 3rd ed. Chichester: Wiley–Blackwell; 2002.
Hoffbrand AV, Moss P, eds. Essential haematology. 6th ed. Oxford: Wiley-Blackwell; 2011.
[/level-membership-for-endocrinology-diabetes-and-metabolism-category][not-level-membership-for-endocrinology-diabetes-and-metabolism-category]CHAPTER 26
Introduction to haematology and transfusion science
David Ah-Moye; Ceinwen Davies; Joanne Goody; Peter Hayward; Rebecca Frewin
CHAPTER OUTLINE
Analysis of the full blood count
Erythrocyte sedimentation rate and plasma viscosity
Tests for infectious mononucleosis
Abnormal white cell morphology
Laboratory tests of coagulation
Interpretation of coagulation tests
Investigation of suspected transfusion reaction
INTRODUCTION
Haematology is the study of blood, its formation, composition, functions and diseases. Blood cell formation (haemopoiesis) is described in Chapter 27. Haematology laboratories perform a wide range of tests of the composition and function of the blood that assist the clinician in the diagnosis and management of disease. They are typically divided into three principal sections: general haematology, haemostasis and blood transfusion. This chapter provides an overview of the different diagnostic techniques offered in each.
GENERAL HAEMATOLOGY
The general haematology laboratory performs numerical, morphological and biochemical analysis of blood samples (now often as part of a combined blood sciences laboratory). Blood samples must be anticoagulated, for example with ethylenediamine tetra-acetic acid (EDTA) for determination of full blood count and reticulocyte count, blood film examination and plasma viscosity and tri-sodium citrate for coagulation tests and measurement of erythrocyte sedimentation rate.
Analysis of the full blood count
The full blood count (FBC) is the most requested test profile in haematology. It comprises:
• haemoglobin (Hb) concentration
• red blood cell (RBC) count
• red cell indices (volume and haemoglobin content)
• white blood cell (WBC) count
• white blood cell differential count (neutrophils, lymphocytes, monocytes, eosinophils and basophils)
• platelet count.
The FBC is a valuable first-line screening test from which further investigations may be generated (see Table 26.1). The clinician uses information from the FBC in conjunction with the clinical history, physical examination and other investigations to help formulate and distinguish between differential diagnoses.
Full blood counts are usually performed on fully automated analysers, which provide accurate quantitation of cell parameters and can generate alerts, commonly known as ‘flags’, to the presence of unusual characteristics that may warrant review of a blood film.
Haemoglobin
The main function of haemoglobin, which is present solely within the red cells, is to transport oxygen from the lungs to the tissues (see Chapter 5). Haemoglobin is measured by spectrometry. Diluted whole blood is mixed with a reagent (e.g. Stromatolyser™) that results in cell lysis and conjugates the haemoglobin with a compound such as potassium cyanide or sodium lauryl sulphate prior to measurement of its absorbance. The presence of high concentrations of substances that increase absorbance, e.g. triglyceride-rich lipoproteins, may cause a falsely raised haemoglobin result with some methods.
Cell counting
Blood cells are counted and sized either by electrical impedance or laser light scattering methods. The impedance method utilizes the fact that blood cells are very poor conductors of electricity. When a cell passes through a small aperture across which an electric current is applied, there is a measurable rise in the electrical impedance, with the pulse height being proportional to the volume of the cell. Each cell generates a separate impulse. Light scatter utilizes the fact that cells scatter light when passed through a laser beam: the number of cells can be counted and information about the cells can be obtained according to the pattern of scatter. White cell counting is performed after lysis of the red cells.
Clumping of cells results in underestimation of cell numbers. This may occur, for example, as a result of autoagglutination of red cells in cold agglutinin disease or in vitro clumping of platelets because of inadequate mixing with EDTA anticoagulant.
Red cell indices
The red cell indices are a combination of measured and derived red cell parameters. The red cell indices usually reported are:
• haematocrit (HCT) or packed cell volume (PCV), the percentage volume of red cells in the blood
• mean cell haemoglobin (MCH), the average mass of haemoglobin per red cell
• mean cell haemoglobin concentration (MCHC), the average concentration of haemoglobin per red cell.
Some analysers measure the MCV and calculate the HCT or PCV; others measure the HCT and calculate the MCV.
Mean cell volume
is useful when assessing anaemia. For example, it is reduced in iron deficiency anaemia and increased in megaloblastic anaemia.
Mean cell haemoglobin
is reduced in iron deficiency anaemia and thalassaemias; further tests such as ferritin and HbA2 measurement may be performed to differentiate between these two conditions.
Mean cell haemoglobin concentration
is reduced in iron deficiency but is probably a more useful tool within the laboratory than clinically. It is very stable in health, so it can be used as an internal quality control; a significant change in the daily mean MCHC indicates a drift in analyser calibration or a fault in its function. The MCHC is also useful for detecting anomalous results: a falsely raised Hb concentration, for example as a consequence of lipaemia, or a falsely reduced RBC count, for example owing to red cell autoagglutination, will result in a falsely raised MCHC.
The formulae for the derived parameters are as follows:
White cell differential
The white blood cells are involved in the cellular immunological response to infection and other causes of inflammation. The five types of white blood cell differ in volume, conductivity, light scattering properties, uptake of fluorescent dyes and cytochemical stains, and resistance to lytic agents. These properties, used in various combinations, enable their identification. Figure 26.1 shows typical white cell scattergrams for a normal blood sample.
FIGURE 26.1 Five-part WBC differential from Sysmex XE2100TM analyser. Note that neutrophils and basophils cannot be separated in the side fluorescence (SFL) vs side scatter (SSC) population scattergram (A). The basophil count is obtained by treating the white cells with an acidic reagent that shrinks all white cells, leaving bare nuclei, except for basophils, which remain intact (B). The neutrophil count is then derived by subtracting the basophil count from the combined neutrophil and basophil count. Lymphs, lymphocytes; monos, monocytes; neuts, neutrophils; eos, eosinophils; basos, basophils.
Platelet count
The main function of platelets is to form a plug at sites of damage to vascular endothelium. A low platelet count (thrombocytopenia) may result from decreased production, increased consumption or abnormal pooling in the spleen. Decreased production is a consequence of either general marrow failure or selective depression, for example by viral infection, drugs and chemicals. Platelet consumption is a feature of certain types of autoimmune diseases and occurs in disseminated intravascular coagulation (which is often a result of septicaemia) and thrombotic thrombocytopenic purpura. A high platelet count (thrombocytosis) may be seen as part of a reactive process such as infection or in myeloproliferative neoplasms. Automated haematology analysers routinely measure the platelet count by impedance or laser light scattering; the reference method uses flow cytometry after labelling the platelets with CD41 or CD61 antibodies.
Reticulocyte count
Reticulocytes are immature red cells, which are released from the bone marrow into the circulation one to two days before maturation. An increase in the reticulocyte count is indicative of an increase in red cell production (erythropoiesis) to meet physiological demand, for example in haemolytic anaemia.
Reticulocytes contain ribosomal RNA, which can be detected by two main methods. When unfixed red cells are incubated with dyes such as new methylene blue, the ribosomal RNA precipitates out and appears as a blue reticular network within the cells, which can be visualised using a light microscope, allowing the reticulocytes to be counted. Most automated FBC analysers now perform reticulocyte counts by flow cytometry, using a fluorescent dye that binds to the ribosomal RNA; the number of reticulocytes can be expressed as a percentage of total red cells and as an absolute count.
Erythrocyte sedimentation rate and plasma viscosity
Erythrocyte sedimentation rate (ESR)
expressed in mm/h, is a measure of the rate at which red blood cells settle when an anticoagulated blood sample is left to stand in a column.
Plasma viscosity
is usually measured by an automated viscometer. The plasma is drawn through a capillary tube under constant pressure and temperature; the rate of flow is measured and expressed as viscosity of plasma relative to distilled water.
Measurement of ESR or plasma viscosity may be useful when assessing the acute phase response. Both are affected by the presence of large proteins such as fibrinogen, α2-macroglobulin and immunoglobulins, and are therefore usually raised during an acute phase response. They are non-specific screening tools for inflammation, and are useful in monitoring of diseases such as rheumatoid arthritis and response to treatment for example with non-steroidal anti-inflammatory drugs and anti-tumour necrosis factor.
Plasma viscosity has several advantages over ESR. For example, it is not affected by physiological and environmental factors. Measurement is rapid, reproducible and lends itself better to quality assurance procedures than measurement of ESR. Conversely, ESR is a cheap test, but is influenced by factors such as anaemia, sex, age and (in females) stage of the menstrual cycle and must be carried out within about six hours of venepuncture.
Flow cytometry
White cells express characteristic nuclear, cytoplasmic and surface antigens; this is referred to as the immunophenotype of the cell. Immunophenotyping has been made possible by the availability of a large number of antibodies, which target specific cellular antigens. For example, antibodies that are directed against the CD19 antigen will bind to B lymphocytes.
Immunofluorescence forms the basis of flow cytometric investigations. It uses antibodies conjugated to fluorescent dyes (fluorochromes) such as fluorescein isothiocyanate (FITC), phycoerythrin (PE) and phycoerythrin cyanin (PC). Whole blood or bone marrow is incubated with one or more antibodies, each conjugated to a different fluorochrome. The cells are then injected in single file into the path of a laser beam. As the cell passes through, the light is scattered in all directions. Forward scatter (FSC) is influenced by cell size whereas side scatter (SSC) is influenced by the cellular structure and contents. The different cell populations can be visualized by plotting SSC against FSC. As the cell–antibody complex passes through the laser beam it fluoresces. The different wavelengths of light emitted are filtered, enabling specific cell populations to be selected (gated) and the data can be displayed as dot plots or histograms. The dot plots show the intensity of fluorescence as well as the number of cells that are positive for the particular antigen; the intensity of fluorescence is proportional to the strength of antigen expression. In the example shown in Figure 26.2, whole blood was incubated with three antibody-dye conjugates, CD8-FITC, CD4-PE and CD3-PC5.
FIGURE 26.2 Scatter plots, dot plots and histogram printouts from Beckman Coulter FC500 flow cytometer. (A) Side scatter vs forward scatter, showing white cell populations. (B) CD4 vs CD3 dual dot plots. (C) CD8 vs CD3 dual dot plots. (D) Single histogram for CD3 gated for the lymphocyte population. Note: Granulocytes is the collective name for neutrophils, eosinophils and basophils.
Flow cytometry allows rapid immunophenotyping of cells. This is essential in the diagnosis and classification of leukaemias and lymphomas, but is also a useful tool in the assessment of non-malignant conditions and for monitoring the CD4 + lymphocyte count during the treatment of human immunodeficiency virus (HIV) infection. Flow cytometry can also be used to confirm feto–maternal haemorrhage (see p. 512), by the detection of Rh D positive fetal cells and measurement of haemoglobin F in the fetal cells.
Haematinic studies
These comprise direct measurement of the concentration of ferritin (a measure of iron status), vitamin B12 and folate. They are discussed further in Chapter 27.
Haemoglobinopathy screening
The haemoglobinopathies and the techniques used for their diagnosis are discussed in Chapter 29.
Tests for infectious mononucleosis
Infectious mononucleosis (IM), also known as glandular fever, is caused by the Epstein–Barr virus (EBV), which infects B lymphocytes. It is associated with a high titre of a heterophile antibody, so-called because it also reacts with red cells of other species, for example sheep, ox and horse. This antibody has been termed the Paul–Bunnell antibody, named after John Paul and Walls Bunnell who discovered them.
The Paul–Bunnell antibody is not adsorbed by guinea pig kidney, in contrast to other heterophile antibodies (Forssman antibodies). Differential adsorption tests utilize this principle; the patient’s serum is incubated with ox red cells and separately with guinea pig kidney antigen, after which horse red cells (reagent) are added to the mixtures. Red cell agglutination only in the latter mixture indicates the presence of the Paul–Bunnell antibody.
An alternative test is the latex agglutination test. A reagent containing latex microspheres coated with purified Paul–Bunnell antigen is mixed with the patient’s serum; agglutination indicates presence of the Paul–Bunnell antibody.
Definitive confirmation of EBV infection requires serological detection of a rise in the titre of specific anti-EBV capsid antigen IgM, which occurs in the first few weeks following infection.
MORPHOLOGY
Blood film examination
Microscopic examination of a well stained blood film is an essential part of many haematological investigations. The film may be made either manually or by an automated slide maker. After being air dried, it is stained with Romanowsky dyes such as May–Grünwald Giemsa or modified Wright stain, which contain azure B and eosin. This enables the assessment of the size, shape, maturity and contents of blood cells by light microscopy. Blood film examination is useful in the investigation of the causes of anaemia and to diagnose white cell disorders such as leukaemia and parasitic infections such as malaria. Less commonly, a blood film may be requested for specific reasons such as to look for acanthocytes (red cells with irregular spicules), which are associated with abetalipoproteinaemia, liver disease and several rare degenerative neurological diseases.
Normal red cell morphology
In health, there is little variation in the shape and size of red blood cells. On a well spread and stained blood film, red cells appear circular in outline, with a well stained outer rim and a paler central area that occupies approximately a third of the cell (see Fig. 26.3). The normal red cell appears slightly smaller than the nucleus of a small lymphocyte. It is referred to as normocytic (normal size) and normochromic (normal staining characteristics).
FIGURE 26.3 Normal red cells. Shows area of central pallor (arrow) occupying one-third the size of the cell.
Valuable information can be obtained from the shape of a red cell, as shown in Figure 26.4. In many haematological conditions there may be an increase in the proportion of non-specifically abnormal red cells, termed poikilocytosis. It is also good practice to check the patient’s biochemistry results, to corroborate the morphological features; for example, if acanthocytes are present, the liver function tests should be checked, as acanthocytes are commonly present in liver disease.
FIGURE 26.4 Red blood cells. Commonly observed morphological changes in red cells and the disorders in which they arise.
Morphology of the anaemias
Anaemia is a condition in which the number of red blood cells, or their oxygen-carrying capacity, is insufficient to meet physiological needs. This is conventionally defined as a reduction in haemoglobin concentration below the reference range, i.e. < 130 g/L in adult males and < 115 g/L in adult females. The causes and assessment of anaemia are discussed in Chapter 27.
Iron deficiency anaemia
In iron deficiency anaemia, the red cells are smaller than normal (microcytosis). This is because the maturing red cells undergo an extra cellular division before the critical haemoglobin concentration required to arrest mitosis is achieved. The cells are also hypochromic, with a larger area of central pallor (see Fig. 26.5). Polychromatic cells (immature red cells that have a bluish hue on routine staining because of retained ribosomal ribonucleic acid) may be present, indicating increased erythropoiesis.
FIGURE 26.5 Iron deficiency anaemia (IDA). Hb 58 g/L, MCV 52.1 fL, MCH 12.7 pg, PLT 608 × 109/L, ferritin < 5 μg/L. Blood film shows microcytic hypochromic red cells, polychromatic red cells and pencil cells. The film also shows two neutrophils, a lymphocyte and an increase in platelet numbers.
Megaloblastic anaemia
Megaloblastic anaemia is commonly caused by deficiency of vitamin B12 or folate, both of which are essential for DNA synthesis, or the administration of drugs that interfere with DNA synthesis (e.g. methotrexate). Defective DNA synthesis results in the nucleus maturing at a slower rate than the cytoplasm, thereby producing a red cell that is larger than normal (macrocyte). Teardrop cells and red cell fragments may also be seen, as a consequence of ineffective erythropoiesis (see Fig. 26.6). A common finding is the presence of hypersegmented neutrophils (defined as presence of a nucleus with more than five lobes).
Autoimmune haemolytic anaemia
Haemolytic anaemia is a term used to describe anaemia caused by a shortened red cell lifespan. In autoimmune haemolytic anaemia, the red cells are coated with antibody (usually IgG) that is directed against the patient’s own cells. When these cells circulate through the reticuloendothelial system, the surface of the red cells is eroded, leading to a change in their shape from a biconcave disc to a sphere (see Fig. 26.7).
FIGURE 26.7 Warm autoimmune haemolytic anaemia. Hb 104 g/L, reticulocytes 604 × 109/L, haptoglobin < 0.2 g/L (reference range 0.7–3.2), direct antiglobulin test positive. Blood film shows numerous spherocytes, polychromatic cells and a red cell containing a Howell–Jolly body. Howell–Jolly bodies are typically found in patients who have undergone splenectomy. The film also shows two neutrophils, a monocyte and a lymphocyte.
Microangiopathic haemolytic anaemia
Microangiopathic haemolytic anaemia is a term that is used to describe the anaemia that results from physical damage to the red cells following the occlusion of arterioles and capillaries as a result of fibrin deposition or platelet aggregation. There are numerous causes, including infections (resulting, for example in disseminated intravascular coagulation (Fig. 26.8
[/not-level-membership-for-endocrinology-diabetes-and-metabolism-category]
