CHAPTER 25
Paediatric clinical biochemistry
CHAPTER OUTLINE
POSTNATAL INVESTIGATION OF THE SMALL FOR GESTATIONAL AGE NEWBORN
Interpretation of renal function tests
CALCIUM AND PHOSPHORUS METABOLISM
Disorders of calcium and phosphorus metabolism
Unconjugated hyperbilirubinaemia: physiological jaundice
Unconjugated hyperbilirubinaemia: pathological causes
INTRODUCTION
National statistics for the UK record approximately 700 000 live births per year. In England and Wales in 2010, the infant mortality rate was 4.3 deaths per 1000 live births, the lowest ever recorded, which compares with an infant mortality rate of 12 deaths per 1000 live births in 1980. This considerable decrease in infant mortality can be partly explained by improvements in health care and more specifically, in midwifery and neonatal intensive care. The neonatal period, defined as the first four weeks of life, is associated with the highest mortality rate in childhood, with infants of very low (< 1500 g) and low (< 2000 g) birth weight accounting for a significant proportion of deaths. The mortality rate reflects the consequences of congenital malformation and disease and the difficulties of transition from intra- to extrauterine life, particularly in premature infants; such a high rate is not seen again until the seventh decade of life. For these reasons, the majority of this chapter is devoted to neonatal clinical biochemistry. Disorders of older children are only included where there are particular problems with interpretation of clinical biochemistry data.
The most common cause of admission to a neonatal intensive or special care unit is prematurity. ‘Pre-term’ in the practical sense rarely includes neonates of more than 32 weeks of gestation at birth. The problems of prematurity can be related to immaturity of vital systems, for example the lungs, kidneys and central nervous system, or inadequacy of energy stores or of nutrients such as calcium and iron that are accreted during the third trimester of pregnancy. Another high-risk group is those neonates with a birth weight inappropriate for their gestational age. In most cases, these babies are small for gestational age (SGA), defined as birth weight below the 10th centile for newborns of the same gestational age in the same population. However, neonates born to mothers with diabetes may be overweight as a result of hyperglycaemia in utero.
POSTNATAL INVESTIGATION OF THE SMALL FOR GESTATIONAL AGE NEWBORN
Inappropriate birth weight is often readily explicable from the clinical history. Common causes of growth retardation are multiparity, pre-eclampsia, infection and drug addiction. An uncommon example would be a first-born SGA infant with microcephaly, the mother of whom should be investigated for possible hyperphenylalaninaemia by analysis of maternal plasma amino acids.
Intrauterine infections
Antenatal infections can affect the developing fetus, leading to congenital malformations and/or low birth weight. Many viruses have teratogenic effects on the fetus, so maternal infection in the first trimester, when organogenesis is occurring, tends to cause the greatest effect. In the UK, universal screening for syphilis, hepatitis B, HIV and susceptibility to rubella infection is offered to all mothers in the early stages of pregnancy, with follow-up diagnosis and treatment available for both mother and infant as appropriate.
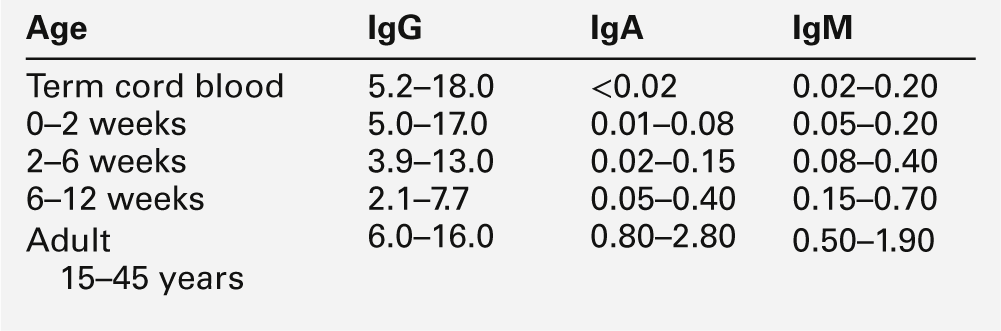
Intrauterine infection is indicated by an increase in the infant’s serum IgM concentration (Table 25.1) and is often associated with prolonged unconjugated hyperbilirubinaemia and abnormal liver function tests. Measurement of the serum IgM concentration is only useful as an indicator of intrauterine infection before six weeks of age. After this time, the normal rise in serum IgM concentration may mask any early increase, and investigation requires specific serological testing.
Maternal drug abuse
Two percent of women in urban areas of the UK have been reported to have had a positive screening test for at least one prohibited substance at the time they first attended for antenatal care. These positive tests included amphetamines, barbiturates, cannabinoids, cocaine metabolites, methadone and opiates.
The consequence of maternal drug abuse is dependent upon the intensity of use. Infants may be born prematurely and/or SGA, and a proportion exhibit withdrawal symptoms during the neonatal period. The appearance of these symptoms depends upon the rates of clearance of the drugs and their active metabolites. For example, the irritability, tremors and convulsions of heroin withdrawal appear during the first 24 h of life, whereas the same symptoms do not generally appear until after 24–48 h when methadone is the substance involved.
A urine drug screen of a symptomatic neonate is often negative. Poor drug penetration across the placenta means that there are higher concentrations in maternal tissues and, consequently, it is more rewarding to screen maternal urine. The likelihood of detecting maternal drug abuse by investigating the urine of the neonate follows a decreasing order of cocaine > methadone > heroin > benzodiazepines.
RESPIRATORY DISORDERS
Respiratory distress
Up to 55% of the fetal cardiac output goes to the placenta. At birth there is an increase in peripheral resistance and a reduction in pulmonary vascular resistance, both of which ensure the closure of the foramen ovale and constriction of the ductus arteriosus. Blood is then diverted through the pulmonary vessels and the adult type of circulation is established. In newborn infants, haemoglobin F (HbF) is the predominant haemoglobin, accounting for about 75% of the total. Haemoglobin F has a greater affinity for oxygen than haemoglobin A (HbA) and its oxygen dissociation curve is shifted to the left, ensuring adequate oxygen exchange at a significantly lower partial pressure of oxygen (PO2). As the partial pressure of arterial oxygen (PaO2) in blood rises with postnatal age, blood HbF concentrations fall, and by six months of age HbF accounts for only about 5% of the total haemoglobin.
Respiratory distress syndrome (RDS) may be caused by a variety of conditions (see Box 25.1). The most common cause in pre-term infants is hyaline membrane disease, primarily attributable to immature surfactant synthesis. Surfactant ensures patency of the alveoli by reducing the surface tension of the alveolar wall, and is made up of phosphatidylcholine (lecithin), phosphatidylglycerol and phosphatidylinositol. Surfactant synthesis begins by the 20th week of gestation. It increases slowly until the 34th week and then more rapidly as the type II alveolar cells mature. The rate of synthesis is sensitive to cold, hypoxia and acidaemia and may be halved by postnatal exposure to temperatures < 35°C or an arterial [H+] > 56 nmol/L (pH < 7.25). High intrauterine glucose concentrations, resulting from poorly controlled maternal diabetes mellitus, can also delay maturation of fetal surfactant synthesis. Fetal type II alveolar cell maturation may be enhanced by maternal steroid therapy. Corticosteroids are often given to women who are at risk of premature delivery at 24–34 weeks of gestation.
The incidence of hyaline membrane disease is inversely related to postconceptional age. The signs of the disease, which occur within four hours of birth, include sternal retraction, intercostal and subcostal recession, expiratory grunt and tachypnoea with a respiratory rate > 60 per min. A characteristic ‘ground glass’ appearance is present on radiological examination of the chest. The appearance of these reticulogranular opacities allows diagnosis of hyaline membrane disease with 90% confidence. Other entities that may produce similar opacities include immature lung, wet lung disease, neonatal pneumonia, idiopathic hypoglycaemia, congestive heart disease, maternal diabetes and early pulmonary haemorrhage.
Group B streptococcal pneumonia may present as early as four hours after infection, during birth, from this common vaginal organism. It occurs more often in the pre-term than the full-term infant. Measurement of serum C-reactive protein and microbiological investigations can be used to distinguish bacterial pneumonia from hyaline membrane disease.
Meconium aspiration and transient tachypnoea of the newborn (TTN) occur most often in full- or post-term infants. The passage of meconium (the contents of the fetal bowel) in utero is associated with fetal hypoxia. In some cases, the contaminated liquor is aspirated. Clinical symptoms usually appear 12–24 h after birth. Transient tachypnoea of the newborn occurs in term neonates, usually those born by caesarean section, and appears to be due to incomplete stimulation of adrenergic mechanisms for lung clearance during birth. Pneumothorax may occur as a complication of either of these conditions or as a result of mechanical ventilation of pre-term infants.
In RDS, the neonate develops hypoxia and a respiratory acidosis, both of which cause an increase in pulmonary vascular resistance and thus pulmonary hypertension with a left-to-right shunt. Hypoxia enhances anaerobic glycolysis and may result in lactic acidosis. Additional complications include brain damage and oedema, and hypotension that may lead to renal failure, paralytic ileus and/or necrotizing enterocolitis (NEC).
Non-pulmonary causes of respiratory distress are usually self-evident and normally improve with treatment of the underlying conditions (see Box 25.1). Up to 20% of infants weighing < 1750 g at birth have a patent ductus arteriosus (PDA). Medical management of PDA includes fluid restriction and stimulation of diuresis. Pharmacological closure may be achieved using indometacin, an inhibitor of prostaglandin synthetase. Contraindications to this treatment include renal insufficiency (plasma creatinine concentration > 160 μmol/L) with or without oliguria, bleeding disorders and NEC.
Management of respiratory distress
This may involve assisted ventilation with oxygen, aiming to maintain the PaO2 in the range 6–12 kPa, PaCO2 5.5–8.0 kPa and arterial O2 saturation (SaO2) 88–95%. Although careful monitoring of blood gases is required, repeated blood sampling may cause anaemia. Anaemia may necessitate transfusion, usually with adult haemoglobin. Increased HbA may compromise oxygen uptake in the lungs in the presence of low alveolar PO2, thereby further aggravating tissue hypoxia. Correction of the metabolic acidosis with sodium bicarbonate can cause oedema and precipitate heart failure, owing to sodium and water overload. Too much oxygen may result in retrolental fibroplasia with retinal detachment and blindness. Long-term ventilation is associated with bronchopulmonary dysplasia, hyperinflated emphysematous lungs with extensive alveolar destruction and widespread fibrosis. The proposed role of free oxygen radicals in the development of these complications has led many paediatricians to administer vitamin E routinely to all infants on ventilators.
Transcutaneous PO2 (TcO2) polarographic electrodes can be used to monitor oxygen treatment in infants who have good skin perfusion; the results correlate well with arterial PO2 measurements. Transcutaneous PO2 is measured at a skin temperature of 44 °C so the electrodes require frequent re-siting with recalibration in order to prevent skin burns. These problems do not occur with pulse oximetry, which measures SaO2 of oxyhaemoglobin and reduced haemoglobin during an arterial pulse, by differential light absorption at 660 and 940 nm. The results correlate well with arterial PaO2 measurements at SaO2 values > 65%, but are falsely low in the presence of methaemoglobin because its molar extinction characteristics imitate reduced haemoglobin.
Surfactant administered through an endotracheal tube significantly reduces the incidence and complications of respiratory distress in newborns at risk of developing hyaline membrane disease.
Apnoea of prematurity
Apnoea of prematurity, defined as a cessation of breathing for > 20 s, with or without bradycardia and cyanosis, occurs in up to 85% of infants weighing < 1000 g at birth. The major cause of apnoea is immaturity of the central respiratory drive, with poor sensitivity to changes in PaCO2. This is compounded by the suppressed respiratory response to hypoxia, which serves to reduce oxygen requirements in utero and which persists in the pre-term infant. Poor coordination of the major respiratory muscles of the chest wall and the upper airways can lead to obstruction, usually at the level of the pharynx. Respiratory effort against a closed airway distorts the chest wall and activates the intercostal phrenic inhibitory reflex. Apnoea is worsened by infection, thermal instability and hypoglycaemia.
Apnoea of prematurity is treated with methylxanthines, which augment central respiratory drive and increase the sensitivity of chemoreceptors to changes in PaCO2.
RENAL FUNCTION
The investigation of renal disorders and the monitoring of fluid and electrolyte replacement in pre-term infants are complicated by immaturity of organ function, and by the difficulty in collecting accurately timed urine samples.
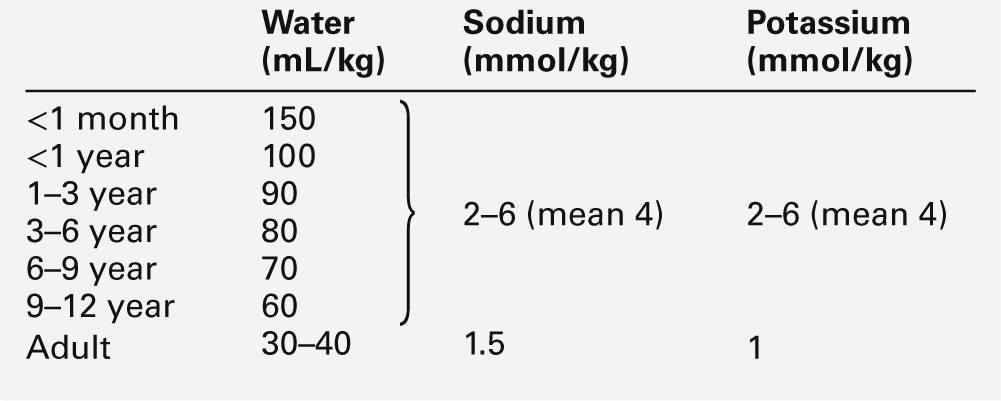
Nephrons develop from about the sixth week of gestation and start producing urine from about the tenth week. The full complement of nephrons is present by the 36th week of gestation. Glomerular function develops more rapidly than tubular function. At term, the tubules are relatively short and underdeveloped. They increase in length and develop increasing absorptive and secretory function during the neonatal period. Functional immaturity is characterized by an inappropriately high urinary sodium excretion for the glomerular filtration rate (GFR) and an impaired response to a sodium load. This is pertinent to the management of pre-term infants, in whom the sodium requirement per kg body weight may be higher than that for term or older infants, with more mature tubular function (Table 25.2). The loops of Henle are principally juxtamedullary in position: they, too, are relatively short compared with those in older infants and adults and do not penetrate deeply into the renal medulla, thereby limiting renal concentrating ability.
In low birth weight infants, glomerular function is adequate for growth but may be inadequate to cope with the increased nitrogenous load during periods of catabolism, starvation, hypoxia, infection and infusion of nitrogen-containing solutions. Tubular function is adequate for the filtered load associated with a reduced GFR and can normally maintain an appropriate excretory function and systemic acid–base status during the anabolic growth phase.
The GFR increases with postconceptional age as renal blood flow increases and renal vascular resistance decreases; full functional maturity is not reached until about the second year of life.
Total body water constitutes about 85% of the body weight of pre-term infants weighing < 1.0 kg at birth, compared with about 75% in term infants and 60% in adults (Fig. 25.1). Relatively more water is in the extracellular compartment than in the intracellular compartment. Total body sodium is about 45 mmol and potassium 75 mmol, and blood volume about 70 mL. During the first few weeks of life, there is a contraction of the extracellular space associated with an increase in urinary sodium loss. This results in an initial 10–15% reduction in body weight. The onset of natriuresis coincides with improvement of lung function and is probably related to a reduction in pulmonary vascular resistance and the release of natriuretic peptides. This contributes to the contraction of the extracellular fluid space. Excess fluid and sodium administration, before the onset of the natriuretic phase, may worsen respiratory distress, delay the closure of the ductus arteriosus and precipitate oedema with hyponatraemia. It is not appropriate to rely on spot urinary sodium concentrations to monitor water and sodium requirements during the first few weeks of life in a premature infant.
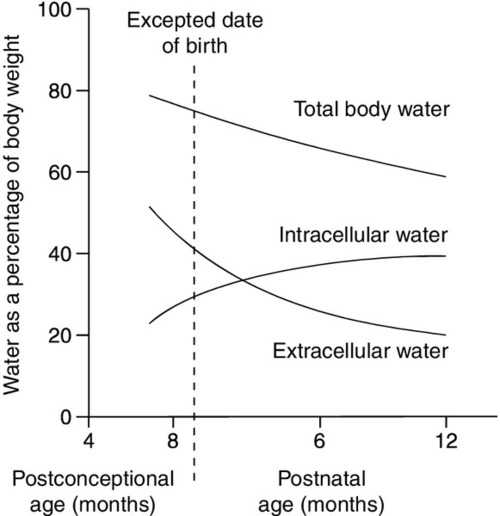
FIGURE 25.1 Changes in body water as a function of age for pre-term and term infants. The timescale indicates expected date of birth and postnatal age.
Antidiuretic hormone (ADH) secretion, in response to volume reduction and hyperosmolality, occurs from about the 25th week of gestation. The interstitial osmolality of the renal medulla, which is the main determinant of the urine concentrating ability in the presence of ADH, is dependent on the countercurrent multiplication mechanism in the loops of Henle and on the interstitial concentrations of sodium, chloride and urea. The reduced GFR and urea clearance decrease the tubular reabsorption of urea and the interstitial urea concentration; consequently, the concentrating ability is impaired, despite appropriate ADH output. Even full-term infants have a limited capacity to conserve water, and urine osmolality rarely rises > 700 mmol/kg. Approximate fluid requirements are shown in Table 25.2.
Hyponatraemia
In neonates, hyponatraemia may be caused by maternal fluid retention or overload during labour, or excess neonatal hypotonic fluid administration during the postnatal period. In addition, inappropriate antidiuresis may develop as a consequence of respiratory disease or intraventricular haemorrhage. In older infants, a dilutional hyponatraemia may be seen, caused by a combination of water retention and sodium depletion in response to increased intestinal or renal fluid losses. Signs of hyponatraemia are related to the rate of fall of plasma sodium concentration rather than to its actual value, and may include hypotension, drowsiness and convulsions. Congenital adrenal hyperplasia (CAH) must always be considered as a possible cause of hyponatraemia (see Chapter 21).
Hypernatraemia
As in adult patients, hypernatraemia may be caused either by water depletion or by excess sodium administration or retention. Insensible water loss is significantly greater in pre-term infants compared with children and adults. The reasons include:
• greater surface area to body volume ratio
• increased skin blood flow
• increased metabolic and respiratory rates
• lack of subcutaneous fat
• greater transepidermal fluid loss.
The epidermis of the skin matures by about the 28th week of gestation with keratinization of the stratum corneum. Consequently, infants born before 28 weeks are at greater risk of excess fluid loss and may lose up to 60 mL/kg/24 h of water through the skin, compared with about 10 mL/kg/24 h in term infants. Additional environmental factors, such as temperature and humidity, also affect transepidermal fluid loss.
Impaired responses to changes in blood volume and plasma osmolality make infants particularly vulnerable to developing hypernatraemia. Increased urinary free water loss may be caused by glycosuria secondary to either hyperglycaemia or to the low renal threshold for glucose. In pre-term infants, glycosuria may be present when the plasma glucose concentration is as low as 5.6 mmol/L. Diabetes insipidus, caused, for example, by intracranial injury at birth, may also result in an increase in free water clearance.
Although the major cause of sodium and water imbalance in premature infants is related to the functional immaturity of organs, it is important to be aware that some drugs may exacerbate the situation, e.g. sodium bicarbonate, indometacin and methylxanthine derivatives such as caffeine.
Morbidity and mortality from hypernatraemia are caused by the increased extracellular osmolality and its effects on fluid distribution between fluid compartments. Infants with hypernatraemia may present clinically with irritability and lethargy, convulsions and coma; lesions within the CNS include intracerebral and intraventricular haemorrhages, as well as sinus and small vessel thromboses. The majority of infants who survive suffer no long-term sequelae, although a minority develop persistent neurological abnormalities.
Hydrogen ions
Under normal circumstances, the kidneys can excrete the hydrogen ion load and generate sufficient buffering capacity to maintain normal systemic acid–base status, despite apparent immaturity of renal tubular function. However, if hydrogen ion production is increased, premature infants are prone to develop a metabolic acidosis in addition to the respiratory acidosis of respiratory distress. The proximal tubular threshold for bicarbonate reclamation is reduced, as is the distal tubular response to an ammonium chloride load: thus the generation of buffering capacity is reduced. In addition, urinary phosphate excretion, which is dependent on GFR, phosphorus intake and plasma phosphate concentration, may be significantly reduced in infants with phosphate depletion.
Metabolic acidosis is frequently related to disorders of respiratory and cardiac function. Echocardiography is invaluable in the investigation of congenital abnormalities of the heart and of left ventricular function. Inherited metabolic diseases, such as mitochondrial dysfunction with lactic acidosis and organic acidaemias, should be excluded early by appropriate investigations (see Chapter 24).
Interpretation of renal function tests
Renal function tests must be interpreted with caution in pre-term infants. Despite the low glomerular filtration rate (GFR), plasma urea concentrations are low in neonates compared with adults, because of increased utilization of nitrogen. Plasma concentrations fluctuate markedly in response to changes in the catabolic rate, GFR and nitrogen intake. Plasma creatinine concentrations are high at birth, being determined principally by maternal concentration: they fall initially and then gradually increase with respect to postnatal age and muscle mass. Measurements may be subject to analytical variation owing to negative interference by bilirubin, and reference intervals may therefore be method dependent.
The interpretation of tubular function tests may be complicated in premature infants in whom the presence of a generalized aminoaciduria and glycosuria may be misinterpreted as a type 2 renal tubular acidosis (RTA). However, despite immature tubular function, maximum phosphate reabsorption occurs in premature infants when plasma phosphate concentrations fall below about 1.5 mmol/L. Impaired response to an ammonium chloride load in the presence of a systemic acidosis may be wrongly interpreted as a type 1 RTA, rather than as a result of immaturity of distal tubular function.
CARBOHYDRATE METABOLISM
The fetus receives a constant, transplacental supply of glucose and amino acids, maintained by maternal intermediary metabolism. Although free fatty acids are transported across the placenta, they are not used for oxidative metabolism but are stored in adipose tissue as triglycerides (triacylglycerols). At birth, the umbilical vein plasma glucose concentration is about 70% that of the maternal circulation. After birth, the plasma glucose concentration falls, resulting in an increase in the circulating concentrations of glucagon, catecholamines and growth hormone (GH), which, together with a fall in plasma insulin concentration, stimulate the release of glucose from hepatic glycogen and activate lipolysis and gluconeogenesis (Fig. 25.2). Increased hepatic cAMP synthesis is associated with glycogenolysis and, through induction of synthesis of phosphoenolpyruvate carboxykinase, gluconeogenesis. Substrates such as pyruvate, lactate and alanine, which are diverted to gluconeogenesis, are no longer available to form malonyl-CoA and hence fatty acids. As the cytosolic concentration of malonyl-CoA falls, its inhibitory effect on carnitine palmitoyltransferase I activity is reduced. Carnitine palmitoyltransferase I plays a role in transferring long chain free fatty acids, released from triglycerides through the action of hormone-sensitive lipase, into the mitochondria where they undergo β-oxidation. Activation of 3-hydroxy-3-methylglutaryl-CoA (HMG-CoA) synthase enhances the hepatic formation of ketones from acetyl-CoA.
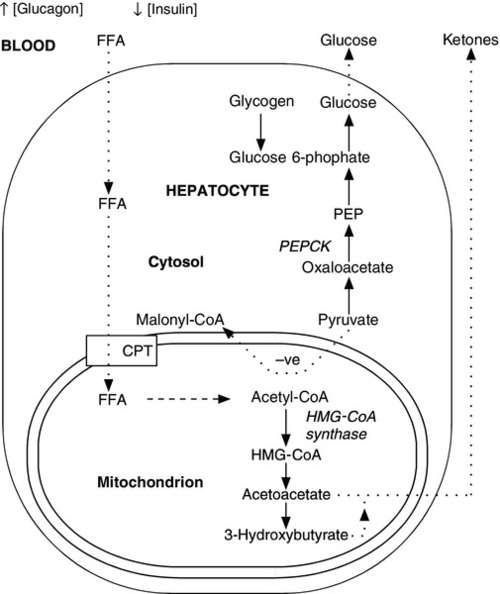
FIGURE 25.2 Changes in energy metabolism at birth. Plasma glucagon rises and insulin falls after birth, in response to falling plasma glucose concentrations. Hepatic glycogenolysis is stimulated. Increased synthesis of phosphoenolpyruvate carboxykinase (PEPCK) stimulates gluconeogenesis. The − ve sign indicates that pyruvate is no longer used to form malonyl-CoA. Malonyl-CoA-dependent inhibition of carnitine palmitoyltransferase (CPT) is reduced and free fatty acids (FFA) can be transferred into the mitochondria where they are subject to β-oxidation. Activation of 3-hydroxy-3-methylglutaryl-CoA (HMG-CoA) synthase permits hepatic ketone synthesis. The dotted lines indicate transfer (active or passive) across membranes.
Although glucose is the major source of energy for the brain, fetal and neonatal brains have an enhanced capacity to utilize ketones, lactate and amino acids as alternative fuels. Circulating free fatty acids and glycerol appear within 3 h after birth in the fasting full-term infant, but plasma concentrations of ketones begin to rise only after 12–48 h of fasting. This relative resistance to ketonaemia is caused by the high turnover of ketones in the neonate, for example ketones can be metabolized at a rate of 13–22 pmol/kg/min, after only 8 h of fasting and may account for up to 25% of the infant’s basal energy requirements. Such a high ketone turnover is observed only after several days of fasting in adults.
The maintenance of normal plasma glucose concentrations during the immediate postpartum period depends upon regulated insulin and glucagon secretion, adequate reserves of both glycogen and triglycerides and the possession of operational intermediary metabolism. When all these factors are present and functioning appropriately, glucose production can reach 5–8 mg/kg/min in full-term infants, compared with about 2 mg/kg/min in adults. This requirement for a high rate of glucose production is related to the higher brain to body mass ratio of infants.
Neonatal hypoglycaemia
Hypoglycaemia of the newborn is defined as a blood glucose concentration of < 2.5 mmol/L. It occurs in up to 0.4% of all newborns, but is much more common in infants of diabetic mothers (previously diagnosed as having diabetes or with gestational diabetes) and in low birth weight and premature infants. Signs of hypoglycaemia include:
• feeding problems
• abnormal cry
• floppiness
• pallor
• apnoea
• cyanosis
• convulsions.
The severity of symptoms will, in part, depend on the rate of fall of the plasma glucose concentration and on the infant’s ability to adapt to alternative sources of energy. In all cases, early diagnosis and the prompt restoration of normoglycaemia are critical in order to minimize damage to the developing CNS.
Infants at risk of developing hypoglycaemia should be monitored regularly. Hypoglycaemia detected by point-of-care testing should be confirmed by laboratory testing, as falsely low results may be obtained in the presence of a high packed cell volume and poor peripheral perfusion of the site of testing.
The aetiology of neonatal hypoglycaemia must be identified so that causes of persistent hypoglycaemia associated with rare disorders and inherited diseases of intermediary metabolism can be excluded (see below). Some causes of hypoglycaemia that may occur during the neonatal period or later in infancy are shown in Box 25.2.
The major cause of hypoglycaemia in pre-term infants is inadequacy of energy stores that would normally have accumulated during the last trimester of pregnancy. The glycogen content of fetal liver increases by 300%, from 0.9 to > 3.9 g/100 g, between 31 and 40 weeks of gestation. Small for gestational age infants also have reduced energy stores and they, too, are at risk of hypoglycaemia. Infants in shock, for example following perinatal asphyxia or because of neonatal sepsis, may develop hypoglycaemia owing to underperfusion of the liver.
Hyperinsulinism is the most common cause of severe and persistent hypoglycaemia in term infants. Hyperinsulinaemic infants are particularly prone to complications of hypoglycaemia, as the high insulin concentrations inhibit lipolysis and the generation of alternative energy sources, such as ketones.
Hyperinsulinism in infants born to diabetic mothers is due to in utero hyperglycaemia-induced hyperplasia of the islets of Langerhans. Idiopathic transient hyperinsulinism may occur in interuterine growth-retarded infants and those suffering from perinatal asphyxia.
Persistent hyperinsulinaemic hypoglycaemia of infancy (PHHI), previously called familial hyperinsulinism, pancreatic nesidioblastosis or hyperinsulinaemic hypoglycaemia, causes recurrent severe hypoglycaemia that may present in term newborn infants or during the first few weeks of life. It can be subdivided into two types: focal adenomatous hyperplasia of the islets of Langerhans and diffuse (but discrete) hyperplasia. Mutations in several different genes that code for proteins involved with insulin release have been identified as being responsible. These include mutations associated with the two subunits of the β cell ATP-sensitive potassium channel – the sulphonylurea receptor proteins (SUR1) and the inward rectifier potassium channel proteins (Kir 6.2). Activating mutations in the glucokinase (GK) and glutamate dehydrogenase (GDH) genes have also been identified in this disorder. Glutamate dehydrogenase is a regulator of insulin secretion in pancreatic β cells and of ureagenesis in the liver, and defects in enzyme activity are characterized by hyperinsulinism with mild, asymptomatic hyperammonaemia. In many patients, the only therapeutic option is partial pancreatectomy, which carries the long-term risk of diabetes mellitus as apoptosis depletes the β-Cell mass. Those forms of PHHI resulting from mutations in GK and GDH are more amenable to pharmacological intervention, for example with diazoxide.
If the amount of glucose required to prevent recurrence of hypoglycaemia exceeds about 10 mg/kg/min, then hyperinsulinaemia is likely. If there is no known history of maternal diabetes mellitus or any of the other classic features (e.g. cherubic appearance and polycythaemia), blood samples taken during the hypoglycaemic episode should be assayed for insulin and 3-hydroxybutyrate. In hyperinsulinaemic hypoglycaemia, the plasma insulin concentration is inappropriately high for the low plasma glucose concentration; plasma C-peptide concentrations are not a reliable reflection of pancreatic β-Cell function in neonates because of renal immaturity and reduced GFR. A plasma 3-hydroxybutyrate concentration < 1.1 mmol/L during hypoglycaemia suggests that fatty acid oxidation and ketone synthesis are impaired. This can be due to hyperinsulinaemia, inadequate adipose stores or to a defect in β-oxidation of fatty acids.
The symptomatic treatment of hypoglycaemia is not without its complications, which include hyperglycaemia associated with hyperosmolality and cerebral hemorrhage, glycosuric osmotic diuresis and water overload.
CALCIUM AND PHOSPHORUS METABOLISM
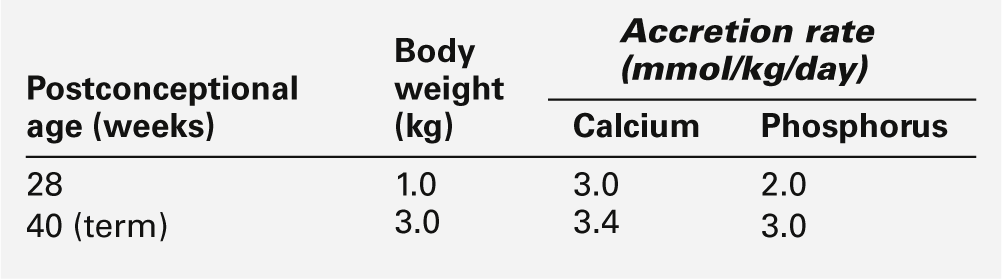
Disorders of calcium and phosphorus metabolism are relatively common in pre-term infants. Calcium and phosphorus are actively transported across the placenta with fetal plasma concentrations higher than those in the maternal circulation. The transplacental transport is probably enhanced by locally synthesized 1,25-dihydroxy vitamin D (1,25(OH)2D) and by parathyroid hormone-related peptide (PTHrP), for which receptors have been identified in the placenta. The high plasma calcium concentration suppresses fetal parathyroid gland activity and probably inhibits fetal 1,25(OH)2D synthesis. During the last trimester of pregnancy, there is a three-fold increase in fetal calcium and phosphorus uptake (Table 25.3): the changes in fetal hormone concentrations facilitate bone mineralization and growth. Consequently, infants born prematurely during the last trimester of pregnancy are significantly disadvantaged and are prone to develop complications of calcium and phosphorus metabolism.
Disorders of calcium and phosphorus metabolism
Hypercalcaemia
Hypercalcaemia is relatively uncommon during the neonatal period. The condition may occur in phosphorus-depleted premature infants receiving unsupplemented breast milk or parenteral nutrition: it is invariably associated with hypophosphataemia. Treatment with phosphorus supplementation alone may precipitate acute hypocalcaemia. Idiopathic hypercalcaemia, with a normal or high normal plasma phosphate concentration, is more common in mature infants but may occur in premature infants receiving high-dose vitamin D prophylaxis. It may also be associated with subcutaneous fat necrosis of infants. This disorder usually affects healthy full-term infants who have suffered perinatal asphyxia. These infants usually develop subcutaneous plaques over the buttocks and back. The condition is caused by excess production of 1,25(OH)2D by the granulomatous cells, similar to that observed in sarcoidosis in adults. Although very rare, it is the most common cause of hypercalcaemia in full-term neonates.
Hypercalcaemia during infancy and childhood is also rare but may be caused by immobilization, primary hyperparathyroidism, familial hypocalciuric hypercalcaemia or infantile hypercalcaemia (Williams syndrome). Familial hypocalciuric hypercalcaemia is a rare, autosomal dominant condition caused by mutations in the calcium sensing receptor (CASR). It is characterized by mild hypercalcaemia and a low urine calcium excretion and does not usually require treatment. Infants with mutations of the CASR gene may occasionally present in the neonatal period with severe hypercalcaemia (neonatal severe primary hyperparathyroidism). Features of Williams syndrome include failure to thrive, hypercalcaemia, elfin-like facial features, supravalvular aortic stenosis and learning disabilities. Microdeletion of the 7q11.23 region on chromosome 7, which contains the elastin gene, can be demonstrated by fluorescence in-situ hybridization (FISH).
Hypocalcaemia
Some causes of neonatal hypocalcaemia are shown in Box 25.3. After parturition, plasma total and ionized calcium concentrations fall, reaching a nadir between the 24th and 48th hour of life, then rising, such that the ionized calcium concentration is 1.10–1.40 mmol/L by about the fifth day of life. Plasma ionized calcium concentrations as low as 0.70 mmol/L have been observed in pre-term infants without apparent clinical abnormalities. The degree of fall is inversely related to gestational age and is more marked in infants born to diabetic mothers and those with perinatal asphyxia. These changes, which are usually self-limiting, are probably caused by transient suppression of fetal parathyroid gland activity.
Permanent causes of hypocalcaemia include hypoparathyroidism, either X-linked or autosomal recessively inherited, or as part of the DiGeorge syndrome and pseudohypoparathyroidism. DiGeorge syndrome is associated with hypoplasia or aplasia of the parathyroid glands and congenital heart anomalies with partial or complete absence of the thymus. Many infants have a partial deletion of chromosome 22q11.
Pseudohypoparathyroidism is characterized by hypocalcaemia and hyperphosphataemia with appropriately increased secretion of parathyroid hormone (PTH) but end-organ resistance to its effect: in the common form of the condition this is caused by mutations in the GNAS gene. Clinically, infants with this disorder present with a constellation of features known as Albright hereditary osteodystrophy. Signs include short stature, dysmorphism, obesity and shortening of the fourth and fifth metacarpals. There may be associated hypothyroidism; subcutaneous calcification may also occur.
Osteopenia of prematurity
Significant failure of bone mineralization, or osteopenia, occurs in > 50% of premature infants weighing < 1000 g at birth. Osteopenia has a reported incidence of up to 13% in infants weighing < 1500 g at birth.
Osteopenia of prematurity is caused by impaired mineralization of normal osteoid of the epiphyseal growth plate with proliferation of unmineralized osteoid. The clinical condition usually presents between the 6th and 12th postnatal weeks, associated with a fall-off of longitudinal growth, with continued growth in head circumference and frontal bossing. Swelling of the costochondral junctions of the ribs may result in the classic ‘rachitic rosary’. Softening of the rib cage with impaired chest wall compliance and patchy lung atelectasis impairs ventilation and may cause late onset respiratory distress, so that ventilatory support may be required. Multiple non-traumatic fractures may also be present.
The aetiology of osteopenia of prematurity is multifactorial but the condition is primarily caused by deficient mineral substrate intake, particularly of phosphorus. Human breast milk is capable of providing up to 1.4 mmol/kg/24 h of calcium and 0.9 mmol/kg/24 h of phosphorus. Although these calcium and phosphorus intakes are adequate to enable normal bone mineralization and skeletal development in term infants, they fall far short of the mineral accretion that should have taken place during the last trimester of pregnancy and are not sufficient for a rapidly growing pre-term infant. Infants born as early as 26 weeks are able to hydroxylate vitamin D in the 25 position, but there may be maturational delay of the renal 1α-hydroxylase enzyme, and, consequently, concentrations of 1,25(OH)2D may be inappropriately low until about the third week of extrauterine life. This, coupled with the limited absorptive capacity of the pre-term intestinal tract, contributes to the mineral substrate deficiency.
The biochemical changes in plasma that may identify those infants at risk of developing clinically significant osteopenia of prematurity are shown in Table 25.4. Plasma alkaline phosphatase activity fluctuates considerably during the neonatal period, but rises rapidly to values above six times the upper adult reference limit in the majority of infants at risk. High plasma enzyme activities are inversely related to growth velocity, a fall in which is an early clinical manifestation of the disorder. Plasma phosphate concentrations are invariably low–normal or low and urinary phosphate excretion is significantly reduced. Plasma calcium concentrations are a poor indicator of impending rickets and may be low, normal or even raised, depending upon the aetiology and stage of the disorder. Urinary calcium excretion is variable and related to the plasma phosphate concentration: the lower the plasma phosphate, the greater the urinary calcium excretion.

The treatment for this common form of osteopenia of prematurity is to increase calcium and phosphorus intake in order to maintain a normal plasma calcium concentration, but, more importantly, to keep the plasma phosphate close to about 1.50 mmol/L. Vitamin D supplementation, in an active form such as 1-alphacalcidol, is often administered. Following successful treatment, plasma alkaline phosphatase activity may continue to rise for about two weeks before falling to expected pre-term values.
Rickets during childhood
Rickets occurs in growing children as a result of defective mineralization of growth plates: it is similar to osteomalacia in adults. The major cause of rickets during childhood is nutritional vitamin D deficiency. Other causes include malabsorption and anticonvulsant therapy. Hypocalcaemia with secondary hyperparathyroidism is invariably present and children may present with tetany and convulsions. Older infants may present with a characteristic waddling gait.
Inherited forms of rickets must be considered in the differential diagnosis. They can be divided into two main groups: those associated with hypophosphataemia, caused by an isolated or generalized renal tubular defect, and those in which the main defect is in vitamin D metabolism (Box 25.4). X-linked dominant hypophosphataemic rickets is the most common form and occurs in about one in 20 000 live births. Hypophosphataemia, with isolated hyperphosphaturia, usually presents during the first year of life and is associated with growth retardation. Remission may occur when the epiphyseal plates fuse and growth ceases, but the condition can relapse in later life. Female patients are less severely affected than males, the difference being caused by the random inactivation of one of the paired X chromosomes (lyonization). In females, presentation may be as osteomalacia in adulthood. A low–normal or low plasma concentration of 1,25(OH)2D in the presence of hypophosphataemia is suggestive of an altered feedback control on 1α-hydroxylase and impaired vitamin D metabolism.
Although rare, cystinosis is the most common cause of rickets in infants with type 2 RTA or Fanconi syndrome. Rickets develops by the second year of life and is probably caused by a combination of hypophosphataemia, systemic acidosis and impaired renal hydroxylation of vitamin D. In contrast, infants with type 1 RTA develop hypercalcuria and nephrocalcinosis and may present clinically with polyuria and polydipsia, renal stones or rickets, usually before the fifth year of life. The diagnosis of cystinosis is confirmed by the finding an increased concentration of white cell cystine.
Plasma alkaline phosphatase activity in infancy
Plasma alkaline phosphatase (ALP) fluctuates considerably in the pre-term infant, and activities up to six times the adult reference limit are considered appropriate for age.
Plasma ALP consists primarily of the bone isoform in infants, although fetal intestinal ALP, the gene for which is normally suppressed after the 30th week of gestation, may be present in the plasma of pre-term neonates receiving an oral fat intake. The hepatic isoform of ALP is rarely detected before one year of age, even in the presence of hepatic disease. Placental ALP is not present in the plasma of the newborn. Age and sex variation in plasma ALP during childhood and adolescence is shown in Figure 25.3.
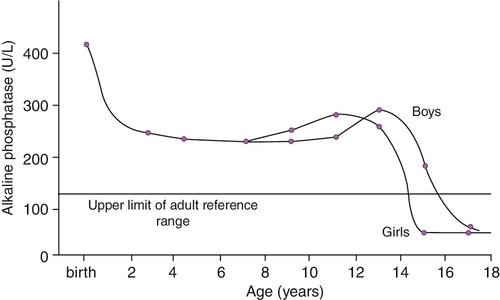
FIGURE 25.3 Age- and sex-related changes in plasma alkaline phosphatase activity during childhood and adolescence.
Transient hyperphosphatasaemia of infancy is a rare disorder of unknown aetiology that occurs in infants and children up to the age of about five years, although it may occasionally present in older children and adults. It is characterized by a rapid rise in plasma ALP activity to values > 20 times the upper adult reference limit and a subsequent return to normal, usually within three months. There is no associated bone or liver disease and the disorder has no proven long-term sequelae. Its early recognition is important as it prevents unnecessary investigations (see Chapter 13).
DISORDERS OF LIVER FUNCTION
Bilirubin metabolism
Bilirubin is formed in the reticuloendothelial system of the liver, spleen and bone marrow from the breakdown of haemoglobin. Unconjugated bilirubin is transported in plasma bound to albumin, which has two bilirubin binding sites, one high and one low affinity, with a maximum binding capacity of about 20 μmol bilirubin/g albumin.
Bilirubin crosses the sinusoidal membranes of hepatocytes by non-energy-dependent passive diffusion. It becomes bound to Y protein (ligandin) and is transported to the smooth endoplasmic reticulum where it is conjugated, by the enzyme uridine diphosphate (UDP)-glucuronosyl transferase, to form a water-soluble mono- and then diglucuronide. The monoglucuronide is the predominant conjugate of bilirubin in the newborn, but only accounts for about 15% of total bilirubin conjugates in adults. The ability of the neonatal liver to conjugate bilirubin is limited (see below), but by two years of age, the conjugating capacity greatly exceeds the rate of delivery of bilirubin to the liver and only 1% of the enzyme activity is required to clear bilirubin at the normal rate of production.
Bilirubin conjugates are secreted across the canalicular membrane into the bile by a multispecific anion transporter. The rate of secretion of bilirubin into the biliary canaliculi is dependent on the active secretion of bile salts and the rate of bile flow. Up to 25% of the conjugated bilirubin may be hydrolysed within the intestinal lumen, either non-enzymatically, under alkaline conditions, or by the mucosal enzyme β-glucuronidase, to unconjugated bilirubin, which is reabsorbed and returned to the liver via the enterohepatic circulation. Conjugated bilirubin is converted to bile pigments (urobilinoids), primarily by intestinal bacteria in the distal ileum and colon.
Unconjugated hyperbilirubinaemia: physiological jaundice
Bilirubin bound to albumin in the blood may be displaced by drugs, such as antibiotics, and by hormones and free fatty acids. Free bilirubin is lipid soluble and can cross the blood–brain barrier, where its toxic affects result in irreversible brain damage (kernicterus). This is thought to be caused by uncoupling of oxidative phosphorylation. Significant free bilirubin occurs when the plasma total bilirubin concentrations exceed 340 μmol/L in term infants, with a conjugated bilirubin < 25 μmol/L. The absence of a clear relationship between circulating bilirubin concentrations and the development of kernicterus in pre-term infants has led to the ‘action level’ for total bilirubin being reduced to as low as 170 μmol/L. Putative additional risk factors for the development of kernicterus include acidosis, sepsis and hypothermia. National guidelines have been adopted for the detection and management of neonatal hyperbilirubinaemia to prevent the potential devastating effects. However, given that the cause may be a sudden and unpredictable haemolytic crisis, kernicterus has yet to be completely eliminated.
Hyperbilirubinaemia is usually treated with phototherapy, which converts the bilirubin to water soluble isomers but, if severe, may require treatment by exchange transfusion. This latter procedure may be complicated by a fall in plasma ionized calcium concentration and there is an associated risk of cardiac arrest.
Transient unconjugated hyperbilirubinaemia, presenting with jaundice on the second or third day of extrauterine life and persisting for about two weeks, is known as ‘physiological jaundice of the newborn’. In premature infants, such jaundice may last for up to four weeks. During the first week of life, plasma bilirubin concentrations rise to > 220 μmol/L in 6% of healthy full-term neonates and a greater proportion of pre-term neonates. The cause is controversial and almost every phase of bilirubin metabolism and transport has been implicated. Bilirubin synthesis is increased during the first three weeks of life, particularly in premature infants, because of the reduced erythrocyte half-life. The high bilirubin production (100–140 μmol/kg body weight/24 h), almost three times the normal adult production, exceeds the capacity of the neonatal liver to clear unconjugated bilirubin from the plasma, owing to reduced membrane uptake and ligandin production, the low activity of UDP-glucuronosyl transferase and inefficient bile acid metabolism and transport.
After the third week of life, the increased bilirubin load is derived principally from the enhanced reabsorption of unconjugated bilirubin from the gut. The reduced uptake and conjugation of the increased load by the liver of the premature infant contributes to the prolonged ‘physiological’ jaundice in this age group. It has been suggested that bilirubin, through its oxidation to biliverdin, provides a scavenging mechanism for free radicals produced during disease and that physiological jaundice may have a beneficial role in the neonate.
Unconjugated hyperbilirubinaemia: pathological causes
Unconjugated hyperbilirubinaemia that occurs within 24 h of birth, which rises rapidly (> 85 μmol/L/24 h) or which is persistent, should prompt investigation for the secondary conditions shown in Box 25.5. Haemolytic disease of the newborn is now uncommon following the widespread use of prophylactic rhesus immune globulin: when it occurs, jaundice develops within the first six hours of life. Congenital spherocytosis and ABO haemolytic disease are usually associated with less severe jaundice. The latter condition resolves as the circulating erythrocytes coated with anti-A and anti-B antibodies are broken down.
Erythrocyte glucose 6-phosphate dehydrogenase deficiency is a common inherited cause of haemolytic disease and should be ruled out in neonates with persistent hyperbilirubinaemia. Haemolysis in this condition may be precipitated by exposure to chemicals, drugs and infection and may be more severe in the Mediterranean and Oriental type than in that seen in African-Americans.
Congenital infections such as syphilis, rubella and toxoplasmosis may cause increased erythrocyte turnover and haemolysis, which can contribute to unconjugated hyperbilirubinaemia.
Inherited disorders of bilirubin metabolism presenting in childhood
Inherited glucuronosyl transferase deficiency (Crigler–Najjar syndrome) is a rare disorder of impaired bilirubin conjugation that presents with progressive unconjugated hyperbilirubinaemia from birth. In type I, there is complete absence of hepatic glucuronosyl transferase activity. The disorder is inherited as an autosomal recessive trait. With modern management of phototherapy (10–12 h/day) and exchange transfusions, kernicterus is now less common, but affected individuals still go on to develop severe neurological complications during adolescence. Many individuals are treated by liver transplantation by the age of five years. Heterozygotes have normal plasma bilirubin concentrations but hepatic enzyme activity is reduced by about 50%. Some heterozygotes may present in the neonatal period with unconjugated hyperbilirubinaemia.
In type II, glucuronosyl transferase activity is detectable but markedly reduced. Unconjugated hyperbilirubinaemia presents at birth and persists into adult life. Milder forms of the disorder are indistinguishable from Gilbert syndrome, a benign autosomal dominant disorder characterized by mild fluctuating unconjugated hyperbilirubinaemia. Type II glucuronosyltransferase deficiency is inherited as an autosomal dominant trait with variable penetrance. As hepatic enzyme activity normally exceeds the rate of bilirubin delivery, small changes in enzyme activity can significantly alter the expression of the disorder. Treatment with phenobarbital, which induces enzyme activity, results in a rapid decline in plasma bilirubin concentration, maximal by the sixth day, in infants with the type II disorder but not with type I.
Conjugated hyperbilirubinaemia and hepatocellular disease
Conjugated hyperbilirubinaemia, defined in infants as a conjugated plasma bilirubin concentration of > 25 μmol/L, is always pathological. As in adults, the cause may be extra- or intrahepatic cholestasis or hepatocellular necrosis (Box 25.6). Routine liver function tests are often poor discriminators of the aetiology during the neonatal period, but the presence of pale stools is suggestive of cholestasis.
The biliary atresias comprise a group of disorders in which all grades of bile duct obstruction may be present, from a marked reduction in bile duct numbers to complete absence. Conjugated hyperbilirubinaemia develops during the first week of life and continues to increase, causing a fluctuating jaundice and failure to thrive. Early diagnosis, by abdominal ultrasound and isotope studies, is important, as extrahepatic atresia can be successfully treated surgically with the Kasai procedure (hepatic portoenterostomy). Liver transplantation is undertaken in some cases.
α1-Antitrypsin deficiency is probably the most common inherited metabolic disorder to cause neonatal conjugated hyperbilirubinaemia sufficient to mimic severe biliary atresia. Liver disease occurs in those patients with the PiZZ phenotype, of whom 10–20% present with neonatal cholestasis. The diagnosis must be confirmed by determining the phenotype/genotype.
Defects in the synthesis of bile acids (cholic acid and chenodeoxycholic acid) from cholesterol are rare inherited causes of cholestatic jaundice. The jaundice is usually associated with steatorrhoea and malabsorption of the fat-soluble vitamins. Defining defects of bile acid synthesis depends upon the identification of unusual profiles of bile acids or alcohols in urine.
Prolonged parenteral nutrition may cause cholestatic liver disease with a progressive rise in plasma alkaline phosphatase activity and a later rise in the plasma aminotransferases. The aetiology is unclear but it may be caused by an ascending cholangitis, secondary to intestinal stasis. The condition usually resolves when parenteral feeding is stopped and enteral feeding is started or resumed but, in some children, the abnormalities persist and chronic liver disease may supervene. Some children have been successfully treated with combined liver and small bowel transplantation.
Hepatocellular disease in neonates is associated with raised plasma aminotransferase activities, reduced plasma albumin and prolonged prothrombin time. Clinically, there may be oedema and ascites. When liver damage has occurred in utero, for instance in congenital infections and some inherited metabolic diseases, infants may present soon after birth with the neurological effects of intracranial bleeding before liver disease becomes apparent clinically.
Postnatally acquired infection may be superimposed on an inherited metabolic disease, such as tyrosinaemia type 1 or classic galactosaemia.
Neonatal haemochromatosis is a rare inherited disorder characterized by jaundice, hepatic failure and hypoglycaemia in the newborn period. Although the distribution of iron overload in the liver and in extrahepatic tissues, such as the heart and pancreas, is similar to that of hereditary haemochromatosis, the two conditions are distinct. The aetiology of neonatal haemochromatosis is unclear; it is probably inherited as an autosomal recessive trait. The transferrin saturation (80–90%) and serum ferritin concentration are both markedly raised.
In many infants, it is impossible to differentiate cholestasis and hepatocellular disease. Several inherited metabolic diseases are associated with neonatal hepatocellular disease. Sometimes there are clinical pointers to a diagnosis, such as the craniofacial dysmorphia of the peroxisomal disorder Zellweger syndrome (p. 462). In the absence of any specific clinical signs, the following investigations should be performed:
• plasma: lactate, glucose, ammonia, amino acids, α1-antitrypsin, prothrombin time (international normalized ratio).
Given that the liver has significant metabolic turnover, it is not surprising that inherited metabolic diseases, such as fatty acid oxidation defects, glycogen storage disease type 1 and urea cycle disorders, are associated with early liver disease. Affected infants are acutely ill and the diagnosis may be indicated by hypoglycaemia or hyperammonaemia, in addition to deranged liver function tests.
Disorders in which the major initial biochemical findings relate to the liver include the acute form of tyrosinaemia type I (fumarylacetoacetate hydrolase deficiency). Signs and symptoms of the acute neonatal onset form include acute liver failure with hepatomegaly, a tubulopathy, hypoglycaemia, failure to thrive, vomiting, diarrhoea and a cabbage-like odour; some infants may present with a neurological crisis. Plasma tyrosine and methionine concentrations are markedly raised. Urine organic acid analysis shows raised concentrations of the tyrosine metabolites, 4-hydroxyphenyllactic and 4-hydroxyphenylpyruvic acids, and succinylacetone. Of these biochemical findings, only the latter, succinylacetone, is specific to the defect, and allows differentiation from transient tyrosinaemia of infancy. Tyrosinaemia type I can now be successfully treated using 2-(2-nitro-4-trifluoromethylbenzoyl)-1,3-cyclohexanedione (NTBC), which inhibits the enzyme 4-hydroxyphenylpyruvate dioxygenase and thus the formation of fumarylacetoacetate and succinylacetone. Succinylacetone inhibits the enzyme porphobilinogen synthase; consequently, older individuals with tyrosinaemia type I may present with features resembling those of acute intermittent porphyria. Hepatocellular carcinoma is a significant complication, and patients should be monitored regularly by measuring plasma α-fetoprotein.
There are three separate inherited disorders of galactose metabolism. The most common and severe of these (classic galactosaemia) is galactose 1-phosphate uridyltransferase (GALT) deficiency. The acute clinical presentation of galactosaemia includes vomiting and diarrhoea, hepatic dysfunction, with a pronounced coagulopathy, an Escherichia coli septicaemia due to impaired leukocyte bactericidal activity, and cataracts. Galactose 1-phosphate uridyltransferase activity should be measured in red blood cells.
Dietary galactose restriction can reverse these acute problems, but the development of long-term complications, such as developmental delay, speech abnormalities and premature ovarian failure, may not be affected by the early institution of therapy. The poor treatment outcome in classic galactosaemia is due either to endogenous production of galactose 1-phosphate through the action of epimerase or to the low intracellular availability of UDP-galactose, a necessary component of some glycolipids and glycoproteins.
Liver disease in older children
The most common causes of liver disease in childhood are viral, autoimmune and drug-induced hepatitis. However, some metabolic diseases, such as α1-antitrypsin deficiency, tyrosinaemia, Wilson disease or cystic fibrosis should be excluded. A Fanconi-like picture with osteopenia and rickets may indicate the presence of renal tubular damage secondary to tyrosinaemia type I, Wilson disease or hereditary fructose intolerance. Tyrosinaemia type I may also present with hypertension and a porphyria-like crisis, owing to succinylacetone-induced inhibition of 5-aminolaevulinic acid dehydratase (ALAD), the rate-limiting enzyme in the porphyrin metabolic pathway.
Wilson disease
This recessively inherited disorder is caused by the impaired hepatic incorporation of copper into caeruloplasmin and by reduced biliary excretion of copper. Copper accumulates in the liver and later in the brain and kidneys. The disorder may present with acute liver failure, cirrhosis or chronic hepatitis after four years of age but asymptomatic hepatocellular damage has been found earlier in life. A more detailed account of Wilson disease is given in Chapter 14.
Reye syndrome or Reye-like illness
Hepatitis associated with acute encephalopathy and fatty infiltration of tissues is known as Reye syndrome. The onset of the disorder may be precipitated by a variety of conditions, including viral illness, especially varicella or influenza A or B, drugs, such as salicylates or sodium valproate, and toxins, including insecticides and aflatoxins.
The condition usually presents in children aged 3–12 years. Following a viral-like illness, the child develops persistent vomiting and a progressive encephalopathy, with irritability, confusion and, in its most severe form, coma. Jaundice is rarely present. Biochemical features include hypoglycaemia, raised plasma ammonia concentration and aminotransferase activity and a prolonged prothrombin time.
Several inherited metabolic disorders present clinically and biochemically in a manner similar to Reye syndrome. These include disorders of medium and long chain fatty acid oxidation and some organic acidaemias. Reye syndrome should not be diagnosed in a child < 3 years of age until a genetic defect has been excluded. Given the possible link between Reye syndrome and salicylates it is recommended that they are not given to children under 16 years. This has led to a significant drop in the number of cases recorded.
Further reading
Internet resources [All Accessed October 2013]
Congenital disorders of glycosylation, http://www.euroglycanet.org
Mitochondrial disorders, http://www.neuro.wustl.edu/neuromuscular/mitosyn.html
Neuromuscular Disease Center, Washington University, St Louis, MO, USA. http://neuromuscular.wustl.edu/
Online Mendelian Inheritance in Man. http://www.ncbi.nlm.nih.gov/omim
Catalogue of human genes and genetic disorders maintained by the National Centre for Biotechnology InformationUSA
UK Metabolic Biochemistry Laboratory Network. http://metbio.net
Assay directory for specialist metabolites and enzymes for inherited metabolic disordersThere is also an active training and education initiative and best practice guidelines aimed at helping local non–specialist laboratories and clinical teams
UK Newborn Screening Programme Centre. http://newbornscreening-bloodspot.org.uk
Website includes details of the UK screening programme and UK standards