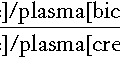CHAPTER 24
Inherited metabolic disease
Fiona Carragher; Mike Champion
CHAPTER OUTLINE
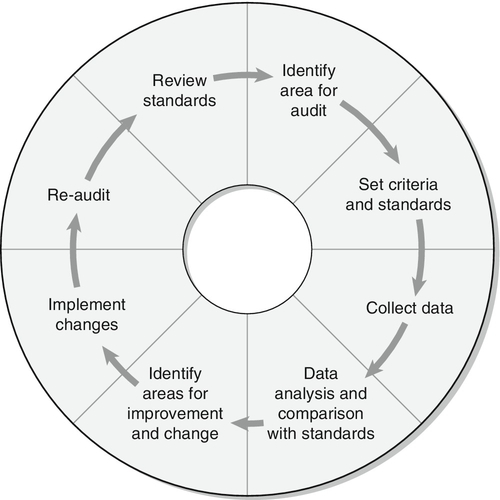
CLINICAL PRESENTATION AND PATHOPHYSIOLOGY
Autosomal recessive inheritance
Autosomal dominant inheritance
Essential laboratory investigations
Strategies to replace a missing product
Inhibition of product breakdown
Strategies to reduce the formation of toxic metabolites
Blockage of site of action of toxic metabolites
INTRODUCTION
Inherited metabolic diseases (IMDs), also known as inborn errors of metabolism, are inherited conditions that develop as a result of mutations that affect the function of proteins. The majority of IMDs are monogenic conditions and the mutant proteins are enzymes, but others involve structural proteins, receptors, hormones or transport proteins. Although they are inherited, not all IMDs present in the newborn period: some present later in childhood or not until adult life.
Inherited metabolic diseases are individually rare. Of those that present in childhood, the commonest, such as phenylketonuria (PKU) and medium chain acyl-CoA dehydrogenase deficiency (MCADD), have an incidence of 1 in 10 000. However, collectively, the IMDs that present in childhood are thought to occur with an incidence of 1 in 750 live births, although the true incidence is unknown, as no newborn screening programme is comprehensive, and many of these conditions go undiagnosed. More diagnoses are being secured with advancing diagnostic techniques, such as tandem mass spectrometry, and the growing awareness of these disorders by clinicians. Similarly, the therapeutic options are continuing to expand, increasing the pressure to detect cases at an earlier stage. Importantly, improving treatments leads to longer survival that, in turn, may present new clinical challenges such as the management of pregnancy in an affected mother. Altering the natural history of a condition may also reveal previously unknown long-term complications.
If IMDs that present in adult life (e.g. familial hypercholesterolaemia, genetic haemochromatosis) are included, at least 1 in 100 individuals has one of these conditions. If disorders such as the haemoglobinopathies are included (see Chapter 28), the prevalence is greater still.
CLINICAL PRESENTATION AND PATHOPHYSIOLOGY
Inherited metabolic diseases may present at any age. However, there are key times when presentation is more common, either owing to the individual having to survive biochemically without the support of the mother’s placenta, or to additional metabolic stresses at that particular time.
Neonatal presentation
Many IMDs present in the neonatal period. They may be considered in four broad categories: problems of synthesis and breakdown of complex molecules; intoxications; energy deficiency states, and seizure disorders.
Defects in synthesis and breakdown
Many complex molecules are integral to cell-to-cell communication and ordered patterning within the developing embryo. Failure to make these complex molecules can, therefore, result in disordered embryogenesis, presenting as a dysmorphic neonate at birth. An example is Zellweger syndrome, the most severe of the peroxisomal biogenesis defects. Affected neonates have a typical appearance, with a large fontanelle, prominent forehead and hypertelorism, hypotonia, hepatomegaly and calcific stippling, particularly of the knees and shoulders, on X-rays. The principle defect in peroxisomal disorders lies within the PEX genes, which encode the peroxin proteins critical for the targeting and importing of peroxisomal enzymes and proteins into peroxisomes, resulting in multiple enzyme deficiencies. This group of disorders is diagnosed by the analysis of very long chain fatty acids (VLCFAs), which are elevated in plasma owing to the block in their oxidation, which is a peroxisomal process. Management remains supportive, rather than curative.
Smith–Lemli–Opitz syndrome is another example of a synthetic defect. It results from a block in the penultimate step in cholesterol synthesis, 3β-hydroxysterol-Δ7-reductase. The production of cholesterol, an essential component of cell membranes, is decreased with marked elevation of its precursor 7-dehydrocholesterol (7-DHC) in body fluids and tissues. The characteristic dysmorphology includes anteverted nares, low-set ears, micrognathia, ptosis and microcephaly. Other features include syndactyly of the 2nd and 3rd toes, present in 98% of patients, genital and renal anomalies, learning difficulties and severe failure to thrive. Dietary cholesterol replacement and statin treatment have been used with the aim of inhibiting cholesterol synthesis and reducing accumulation of 7-DHC; no convincing effects have been seen with either approach, but this may be because the phenotype is determined by the in utero availability of cholesterol.
Problems with the breakdown of complex molecules result in storage disorders. These tend not to be apparent at birth, but rather become so with time as the substance(s) stored in excess begins to affect structure and function. For example, affected children with Hurler syndrome (mucopolysaccharidosis type I, MPSI) appear normal at birth, but the gradual accumulation of glycosaminoglycans over time produces the typical coarsening of the features, corneal clouding, organomegaly and dysostosis multiplex (distortion of the normal bony architecture secondary to storage material) that are characteristic of the condition. However, it is parental concerns about delayed development, rather than the coarse features, that usually bring these patients to medical attention. Storage disorders in which dysmorphology and organomegaly are often present in the first month of life include I-cell disease, infantile sialic acid storage disease and early infantile GM1 gangliosidosis.
Intoxications
Intoxication is the classic presentation of inherited metabolic disorders. An unremarkable period immediately after birth is followed by increasing clinical abnormality as the baby feeds, and toxic metabolites, which cannot be broken down because of the metabolic block, accumulate. Prior to birth, these toxic metabolites are cleared via the placenta. Typically, clinical features develop within 48–72 h after birth, but can take considerably longer. Phenylketonuria (phenylalanine hydroxylase deficiency) is an intoxication, but the first signs of the condition do not usually develop until 6–12 months after birth when motor developmental milestones are not met. Phenylketonuria (PKU) also illustrates two other features of inherited metabolic diseases affecting enzymes: accumulation of the substrate of the defective enzyme may lead to increased metabolism via alternative, normally minor, pathways by a mass action effect (phenylalanine is transaminated to phenylpyruvate and phenylketones), and there may be deficiency of the normal product of the enzyme (in this case, tyrosine). Both of these may contribute to the clinical presentation.
Urea cycle defects typically present with encephalopathy in the first days of life. As milk feeds are established, the block in the conversion of waste nitrogen, derived from the amine groups of amino acids, to urea produces hyperammonaemia and increases glutamine formation. Ammonia interferes with neurotransmission causing astrocyte swelling, and glutamine increases the risk of cerebral oedema, owing to the osmotic load as it accumulates in the brain. Ammonia is also a respiratory stimulant, acting on the respiratory centre in the brain stem to produce a respiratory alkalosis, which is an unusual finding in a sick neonate. Diagnosis relies on measuring plasma ammonia concentration in conjunction with plasma amino acids and urinary orotic acid to determine the location of the block (Fig. 24.1). Immediate management depends on attempting to increase the clearance of ammonia and supplementation of arginine, usually a non-essential amino acid, synthesized in the urea cycle, which becomes an essential amino acid in many of the urea cycle defects. Low arginine also contributes to neurotoxicity by reducing nitric oxide and creatine synthesis.
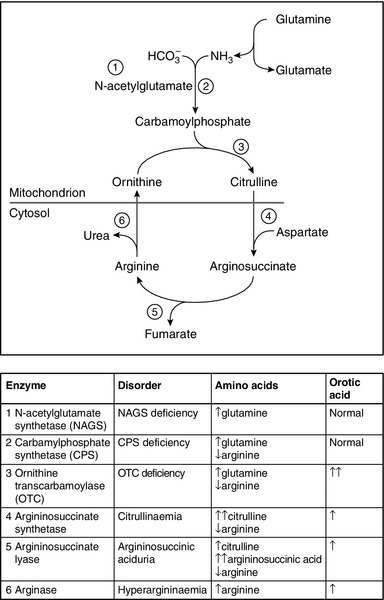
FIGURE 24.1 The urea cycle. The numbers refer to the enzymes listed in the table, deficiencies of which cause changes in the plasma concentrations of amino acids and increased urinary excretion of orotic acid as indicated.
Galactosaemia (galactose 1-phosphate uridyltransferase deficiency) usually presents a little later, at the end of the first week of life, with jaundice due to conjugated bilirubinaemia, hepatomegaly, coagulopathy and characteristic ‘oil drop’ cataracts. The diagnosis is made by measuring the enzyme activity in red cells. The exact pathogenesis is not fully elucidated, but restriction of galactose and lactose (a disaccharide of glucose and galactose) is effective in reversing the liver toxicity. However, the diet does not prevent all adverse effects of the disease and it is clear that endogenous galactose production is significant. As with many IMDs, some infants present later than the neonatal period, with the renal effects of galactosaemia, namely proximal tubulopathy and rickets, rather than the hepatic consequences. A similar difference in presentation is seen in tyrosinaemia type 1, with an early hepatotoxic picture not unlike galactosaemia and a later Fanconi-type presentation due to nephrotoxicity. The key diagnostic marker in tyrosinaemia type 1 is the detection of succinylacetone on urinary organic acid analysis.
Energy deficiency disorders
Energy deficiency disorders are the result of either a fundamental block in energy production, as seen in the congenital lactic acidoses, or a failure of adequate energy production in the absence of a regular food supply. The congenital lactic acidoses have a number of causes (Box 24.1), with a definitive diagnosis being made in only approximately half of the patients. Clinical features develop early, but significant problems due to metabolic decompensation may take longer to develop, in comparison with intoxications.
The key diagnostic marker is a raised plasma lactate concentration but, in practice, secondary hyperlactataemia (due, e.g. to hypoxia, hypovolaemia, hypotension etc.) is more common. The distinguishing diagnostic clue is the absence of ketones in the secondary elevations. Spurious elevation may result from squeezing the arm to elevate the vein prior to venepuncture. A free-flowing sample is required and an arterial stab should be used if a free-flowing sample cannot be obtained otherwise. Further evidence may be gleaned from measuring lactate in the cerebrospinal fluid (CSF), which reflects brain lactate over days rather than at that specific time. Secondary elevations of CSF lactate are seen in primary brain infections such as encephalitis and meningitis, and following seizures.
In certain disorders, energy deficiency only becomes apparent when feeding is interrupted. Neonates with fatty acid oxidation defects remain asymptomatic while fed, but can develop hypoketotic hypoglycaemia during prolonged fasting or intercurrent infections. The commonest fat oxidation defect, MCADD, usually presents much later, at around the age of one year, but a subgroup present in the first few days of life, before feeding becomes established, most commonly if they are breast fed. Neonatal glycogen stores are more rapidly exhausted than those in older infants and adults, requiring mobilization of fat stores for energy. Fats cannot be directly utilized by the brain, requiring conversion in the liver to ketones, which can be. Failure to produce ketones results in encephalopathy as a result of a combination of hypoglycaemia and the accumulation of acylcarnitines. Clinically evident hypoglycaemia is a late feature: treatment, either with oral glucose polymer or intravenous 10% dextrose, should be started before it occurs.
Seizure disorders
Inherited metabolic diseases are a rare cause of neonatal seizures compared with birth asphyxia and infections, and are usually a late, non-specific feature of blocks in intermediary metabolism. There are, however, a number of IMDs that typically present with seizures at this time that will not be detected on routine investigation and so need to be specifically excluded if no other cause is apparent (Table 24.1). Seizures may have been present in the antenatal period and been interpreted as the baby being particularly active because of increased fetal movements. Some mothers do note the rhythmic nature of these movements.
TABLE 24.1
Inherited metabolic disorders presenting with neonatal seizures and their diagnostic investigations
| Disorder | Investigation |
| Biotinidase deficiency | Plasma biotinidase Urine organic acids |
| Non-ketotic hyperglycinaemia (NKH) | CSF:plasma glycine ratio |
| 3-Phosphoglycerate dehydrogenase deficiency | CSF serine |
| Pyridoxine 5′ phosphate oxidase deficiency | CSF neurotransmitters Urine organic acids Trial of pyridoxal phosphate PNPO genotyping |
| Pyridoxine-dependent seizures | Trial of pyridoxine Plasma α-aminoadipic semialdehyde Antiquitin genotyping |
| Purine disorders | Urinary purine studies |
| Sulfite oxidase/molybdenum cofactor deficiency | Dip-stick test for sulphite on fresh urine Urinary purine studies |
| Peroxisomal disorders | Plasma very long chain fatty acids |
Presentation at weaning
Weaning is a time when new dietary components may first be encountered, and if any one of their metabolic pathways is blocked, clinical features may develop. Weaning may also result in greater intake of the particular substrate whose metabolism is compromised, e.g. protein. If the relevant metabolic pathway is unable to cope with the increased load, the threshold for the development of clinical features may be breached. An example of the latter is a partial urea cycle defect. Prior to weaning, protein intake may have been within the tolerance of the compromised pathway, but with the addition of solids, greater quantities of protein can be ingested. If, as a result, the pathway’s capacity is exceeded, hyperammonaemia will ensue. A similar effect is seen in children with hyperphenylalaninaemia detected on newborn screening. A decision is usually made to monitor concentrations to see if the phenylalanine increases with greater food intake, particularly around the time of weaning.
Hereditary fructose intolerance (aldolase B deficiency) does not present until an infant is exposed to fructose, which typically occurs around the time of weaning when pureed fruit is introduced; the sucrose in fruit is broken down to fructose and glucose. There is no fructose or sucrose in breast milk or formula milk. In the affected patient, fructose 1-phosphate accumulates, inhibiting glucose production, promoting hypoglycaemia and depleting inorganic phosphate, thereby reducing ATP production. Clinically, the first features are nausea and vomiting with postprandial hypoglycaemia. If the condition is not recognized, and fructose ingestion persists, the infant will fail to thrive and may develop liver and kidney failure. Confirmatory enzymology requires liver biopsy; however, in some patients a clinical diagnosis can be made on the basis of a history of exposure to fructose and reversal of symptoms after elimination of sources of fructose from the diet. The diagnosis may then be confirmed by genotyping.
Presentation in later infancy
Infancy is a time when there is a considerable risk of infections, as the body is exposed to numerous infectious agents for the first time and the immune system is still developing. Infection causes increased metabolic stress; patients with known IMDs may decompensate and others may present for the first time. Some IMDs show seasonal variation in presentation: for example, MCADD presents more frequently in the autumn and winter months, because of the increase in infections at this time of year.
Glutaric aciduria type I (GA-I) is an autosomal recessive defect in lysine catabolism. Affected infants may have large heads but minimal neurological signs prior to catastrophic metabolic decompensation around the end of the first year, usually precipitated by an infection. Such decompensation causes damage to the basal ganglia, with resultant irreversible dystonia and movement disorder. In presymptomatic siblings, prospective early aggressive management of intercurrent illnesses with antibiotics, protein (lysine) restriction, carnitine supplements and hyperalimentation with glucose polymer or intravenous dextrose, can reduce the incidence of decompensation and subsequent neurological sequelae. Measurement of urinary organic acids shows elevated glutarate and 3-hydroxyglutarate in this condition. Occasional patients have been described with classic histories and abnormal enzymology in fibroblast studies without the typical urine abnormalities; plasma acylcarnitine analysis may show reduced free carnitine and a glutarylcarnitine peak, but may be normal in such patients.
Infants may present with IMDs towards the end of the first year, when growth slows. On average, a baby will put on just under 7 kg in weight in the first year of life, compared with 2 kg each subsequent year during childhood. This means that for the same protein intake, more will need to be catabolized, as less is needed for growth. This increased pressure on an affected metabolic pathway may result in decompensation.
Presentation at puberty
Puberty is recognized as a difficult time for teenagers and rebellion affects all areas of life, including caring for their health. Patients with significant IMDs may break their diets or fail to take their medication in an attempt to be ‘normal’. Both can precipitate decompensation. However, new presentations are also seen at this time, probably as a result of changes in growth and the hormonal milieu. For girls with urea cycle defects, it is well recognized that following menarche, symptoms may fluctuate in time with the menstrual cycle, being worse in the few days leading up to, and including, the start of the period. The use of hormonal therapy to suppress ovulation and menstruation has proven helpful in some patients.
Presentation during adulthood
Inherited metabolic diseases are often considered to be paediatric conditions, but it must be remembered that they can present at any age. This may be owing to the defect being less severe so that an individual’s metabolism has not previously been sufficiently stressed to provoke decompensation. Patients with partial ornithine transcarbamoylase (OTC) deficiency, the commonest of the urea cycle defects, may remain asymptomatic throughout childhood and only present in adult life. Some adults may not have been exposed previously to sufficient quantities of metabolites they cannot deal with. Some adults with hereditary fructose intolerance will have learned at an early age that sweet, sugary foods make them feel unwell and will subconsciously avoid them. Many patients with this condition have perfect dentition owing to their self-imposed diet.
For some IMDs, presentation differs between adults and children. The severe form of X-linked adrenoleukodystrophy (ALD) usually presents at 5–10 years with progressive neurological deterioration, ultimately leading to spastic quadriplegia, seizures, vegetative state and death. More than 90% of affected boys have adrenal insufficiency. However, adrenal involvement may precede, or follow, neurological symptoms by years. There is wide phenotypic variability within families, and another family member may not present until adulthood. The cerebral form in adults is extremely rare, accounting for < 5% of all cases. The commoner adult presentation is adrenomyeloneuropathy (AMN), with a slowly progressive neurological picture mimicking spinal cord syndrome, with spastic gait and difficulty voiding urine. Some 10% of patients have only adrenal involvement, and 10% remain asymptomatic. Adrenoleukodystrophy is the commonest peroxisomal disorder. The defective adrenoleukodystrophy protein (ALDP) is thought to play a role in the uptake of VLCFAs across the peroxisomal membrane, but the exact pathophysiology is not completely understood. The use of Lorenzo’s oil (a 4:1 mixture of glyceryl trioleate and glyceryl trierucate) reduces the accumulation of the VLCFAs, but fails to prevent neurological decline in symptomatic patients. It appears to have a role in asymptomatic boys reducing the risk of developing abnormalities apparent on magnetic resonance imaging. Bone marrow transplantation can be performed to stabilize cerebral ALD in the very early stages of demyelination. The varied phenotype is not explained by differences in genotype, as family members with different manifestations of the condition will usually have the same mutation.
Adrenoleukodystrophy also demonstrates another variation in adult presentation, that of the manifesting heterozygote. This is seen in some X-linked conditions where a carrier female develops symptoms. Two-thirds of females carrying an ALD gene mutation have some degree of neurological involvement, ranging from brisk reflexes and mild abnormalities on clinical examination to a full-blown AMN-like picture. This may be misdiagnosed as multiple sclerosis.
A similar phenomenon is seen in Fabry disease (α-galactosidase deficiency). Deposition of glycosphingolipids in blood vessel walls, heart, kidneys, skin and autonomic ganglia produces cerebrovascular disease, cardiac disease, nephropathy, angiokeratoma, corneal dystrophy and acroparaesthesia (severe pain in the extremities). It was believed that the majority of female carriers were entirely asymptomatic throughout life, but it is now clear that one-third of female carriers have significant symptoms, which may be so severe as to warrant treatment. One explanation for the spectrum of severity is the random inactivation of the X chromosome in cells – lyonization. If sufficient numbers of the unaffected X chromosomes are suppressed, a significant number of cells will have a functioning, affected X chromosome. The resulting enzyme function may be low enough for symptoms to develop.
A significant number of IMDs present for the first time in adulthood. The classic mitochondrial syndromes that led to the recognition of mitochondrial DNA (mtDNA) mutations and their role in pathology are all primarily adult presentations: an example is Leber hereditary optic neuroretinopathy, presenting most commonly in the third decade of life with bilateral, painless central vision loss.
The IMDs that typically present only in adult life usually involve the accumulation of a toxic substance that, although it begins at birth, takes many years to become manifest. Important examples include heterozygous familial hypercholesterolaemia (homozygotes usually present with xanthomata or coronary disease in the second decade), familial combined hyperlipidaemia and primary (genetic) haemochromatosis (excepting the rare neonatal variant). All these conditions are discussed elsewhere in this book.
Presentation during pregnancy
The physiological stresses of pregnancy may precipitate crises in women with IMDs so meticulous care is required to ensure the best outcome for the mother and baby. Some women develop symptoms in pregnancy because they carry an affected child, although they do not have the condition themselves. The classic examples of this are the long chain fatty acid oxidation defects, long chain hydroxyl-acyl-CoA dehydrogenase deficiency (LCHADD) and very long chain acyl-CoA dehydrogenase deficiency (VLCADD). These conditions are recessively inherited and the mother, therefore, is an obligate carrier with 50% enzyme activity, which is compatible with normal life and function. Clinical manifestation of most IMDs only occurs if activity is < 5%. However, the mother also has to combat the metabolic stresses of pregnancy, which increase the load on the pathway. If the fetus is affected, she will also have to break down the long chain acylcarnitines that the fetus is producing owing to the block in its metabolism. Such women do not present with the typical hypoketotic hypoglycaemia seen in affected infants, but liver function is impaired, precipitating HELLP (hepatomegaly, elevated liver enzymes and low platelets) syndrome or the rarer, more severe acute fatty liver of pregnancy (AFLP). This may require intensive care and early delivery of the fetus. Only a small number of women with HELLP and AFLP carry affected fetuses, but it is imperative that their newborn infants are screened by measurement of plasma acylcarnitines to exclude a fat oxidation defect. Screening only for the associated dicarboxylic aciduria, using urinary organic acid analysis, is not sensitive enough and has led to the diagnosis being overlooked.
Presentation postpartum
Women with partial OTC deficiency may present for the first time a few days after delivery. They are able to withstand the increased metabolic stress of pregnancy and delivery, but the massive protein load presented by the involution of the uterus precipitates hyperammonaemia. Many of these women have never had previous symptoms that might alert their obstetrician to the cause of their illness, although some have an aversion to high protein diets.
Clinical abnormalities in the infant may also reveal an undetected IMD in the mother. Maternal PKU syndrome is a description of the clinical consequences to the fetus of in utero exposure to elevated plasma phenylalanine resulting from PKU in the mother. A mother not known to have PKU, usually because she was born in an area without a neonatal screening programme, may have had hyperphenylalaninaemia that was sufficiently mild for her not to have come to medical attention, but significant enough to affect the fetus. Typical features are low birth weight, microcephaly, cardiac abnormalities and developmental delay. The infant does not have PKU or hyperphenylalaninaemia, and therefore does not require dietary restriction. The diagnosis is usually made when the infant is investigated for microcephaly or developmental delay. Plasma phenylalanine needs to be measured in the mother for diagnosis; urinary amino acids may not be sufficiently sensitive and diagnoses have been missed when only this investigation has been used.
NEWBORN SCREENING
Newborn screening is used to detect conditions that have a presymptomatic period during which treatment can dramatically improve outcome. In the UK, conditions screened for at this time include congenital hypothyroidism (see below), PKU, MCADD, cystic fibrosis and sickle cell disorders. A pilot study is underway in the UK to examine an expanded neonatal screening programme including maple syrup urine disease, glutaric aciduria type 1, isovaleric acidaemia, LCHADD and pyridoxine unresponsive homocystinuria. This is modest compared to some countries, e.g. the USA where newborns are screened for over 30 conditions. However, for many of these conditions the natural history is not fully understood, treatments are only partially effective and infants may present and die prior to the screening result being available. The detection of infants who will never develop symptomatic disease remains a real concern. During the development of MCADD screening, mutations were identified in infants with raised octanoylcarnitine that have never been associated with clinical abnormalities.
Screening is also used to detect IMDs in populations with higher frequencies of a particular condition, usually secondary to a founder effect (a mutation occurring early in the settlement of a geographically isolated area or within a limited gene pool so that it occurs with high frequency), for example Tay–Sachs disease in the Ashkenazi Jewish population.
Newborn screening is widely practised for congenital hypothyroidism, but while some of these infants have inherited disorders of thyroid hormone synthesis, some have thyroidal agenesis or dysgenesis (that is, failure of development of the gland), for which a genetic basis has not been defined.
INHERITANCE
The molecular bases of IMDs are mutations in genes that adversely affect the functions of specific proteins. The patterns of inheritance vary, but the majority of these conditions are autosomal recessive.
Autosomal recessive inheritance
Autosomal recessive inheritance requires both parents to carry a mutation affecting the same gene. It is estimated that we each carry 250–300 loss of function mutations in our genes, but, as we also have a normal copy of the gene, the resultant 50% activity is more than sufficient for normal function: as a result, carriers of autosomal recessive conditions are generally not clinically affected (patients with IMDs typically have < 5% activity). Even if the parents are carriers for the same condition, both faulty copies of the gene need to be passed on to the embryo, resulting in a 1 in 4 risk of the infant being affected in each pregnancy (Fig. 24.2A). There is also a 1 in 4 chance that neither faulty gene will be inherited and a 2 in 4 (1 in 2) chance that the embryo will be a carrier.
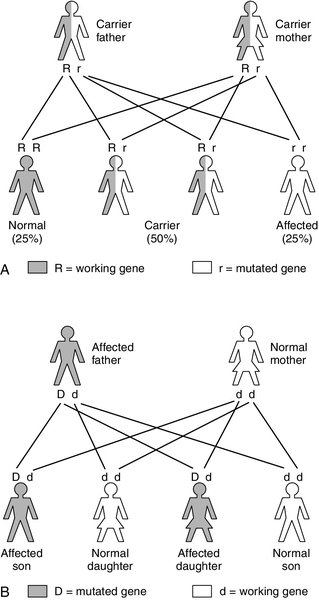
FIGURE 24.2 (A) Autosomal recessive inheritance. (B) Autosomal dominant inheritance.
If the frequency of the gene defect in the general population is known, the incidence can be calculated: for example the carrier frequency for PKU is 1 in 50, so the incidence equals (1/50 × 1/50) (the chance of two carriers having children together) multiplied by 1/4 (the chance of them having an affected child), that is, 1/10 000. If a certain condition is known to exist in a family, genetic counsellors can use calculations of this sort to inform individual couples of their risk of having an affected child. For example, if the sister of an individual PKU were to have a child with an unrelated man, the risk of the baby having the condition would be 1 in 300, which is considered negligible. (She is not affected: she has a 2 in 3 chance of being a carrier and 1 in 3 of being homozygous normal – the denominator is three rather than four because she is unaffected). The man’s chance of being a carrier is 1 in 50 (the carrier frequency in the general population), so the risk of two faulty genes being passed to an embryo is 2/3 × 1/50 × 1/4 = 1/300.)
Consanguinity within a family increases the risk of autosomal recessive conditions and there are some disorders with a high frequency within particular populations. There may be a founder effect within certain populations, for example the Pennsylvanian Mennonites, in whom the severe form of maple syrup urine disease has an incidence of 1:176. In these kindred, lineage can be traced to one couple who emigrated from Europe in the eighteenth century. The tradition of first cousin marriages in some immigrant groups is increasing the incidence of IMDs in some areas of the UK.
It should be noted that, although affected individuals with autosomal recessive IMDs are often classified as homozygotes, the existence of multiple mutations affecting individual genes means that some are, strictly speaking, compound heterozygotes.
Autosomal dominant inheritance
Autosomal dominant inheritance is rare in IMDs with the exception of some of the porphyrias, for example acute intermittent porphyria, hereditary coproporphyria and porphyria variegata (see Chapter 28). The risk of transmission in autosomal dominant disorders is 1 in 2, as only one faulty copy of the gene is required to cause disease (Fig. 24.2B), although variable penetrance and other factors may influence the degree to which the offspring are affected. Dominant inheritance is less common than recessive, as severe disease may result in death prior to an individual reaching reproductive maturity and so the defective gene fails to pass on to the next generation. However, in some cases, the mutation may have occurred de novo in the embryo. For some dominantly inherited conditions, the condition may provide some protection against another serious condition (e.g. the sickle haemoglobin trait provides protection against P. falciparum malaria, see Chapter 29).
X-linked inheritance
X-linked recessive inheritance occurs with a variety of IMDs, for example: OTC deficiency; pyruvate dehydrogenase complex deficiency; Hunter syndrome (mucopolysaccharidosis type II); Lesch–Nyhan syndrome (a purine disorder); Fabry disease (sphingolipidosis), and ALD. This pattern of inheritance is characterized by carrier females (unaffected, as they have a normal copy of the gene on their other X chromosome) passing the gene to their affected sons (the Y chromosome does not carry the gene so there is no normal copy) (Fig. 24.3A). The chance of a son being affected is 1 in 2 in each pregnancy, the same as the risk of a daughter being a carrier. As the affected gene lies on the X chromosome, affected fathers can only pass it to their daughters, who will be obligate carriers.
FIGURE 24.3 (A) X-linked recessive inheritance. (B) X-linked dominant inheritance.
Unlike autosomal recessive conditions, in X-linked disorders carriers may manifest the disorder clinically, for example in OTC deficiency. As discussed previously, in carrier females the variation in the degree of symptoms is due to lyonization, the random inactivation of one of the two X chromosomes in all cells, including hepatocytes. The severity of disease depends on the percentage of hepatocytes expressing the normal gene. This can lead to a varied clinical presentation within families, with some female carriers presenting with severe hyperammonaemia in the neonatal period and others being apparently unaffected.
X-linked dominant conditions require only one copy of the gene to be inherited to express the condition, hence males and females are equally affected, for example vitamin D-resistant rickets (Fig. 24.3B).
Mitochondrial inheritance
Mitochondria are unique intracellular organelles, in that they have their own genes. However, a fully functional mitochondrion is the product of both the nuclear and mitochondrial genomes with the vast majority of genes, in the order of 1300, being encoded in the nucleus. If disease-causing mutations arise in the nucleus, these can be inherited in the usual way, for example as autosomal recessive or dominant, or X-linked traits. Mitochondrial DNA (mtDNA) is inherited in a matrilineal fashion; that is, mtDNA is inherited exclusively from the mother. Paternal mitochondrial DNA is present in the mitochondria in the sperm’s tail to provide ATP for propulsion. On fertilization, the tail of the sperm is left outside and hence no paternal mtDNA enters the zygote. Mitochondrial DNA mutations can therefore be inherited only from females, but can affect males or females. Point mutations inherited in this fashion include those for: myoclonic epilepsy, ragged red fibres (MERRF); mitochondrial encephalomyopathy, lactic acidosis and stroke-like episodes (MELAS) and Leber hereditary optic neuropathy.
The degree to which the offspring will be affected in these conditions is influenced by the mutant load, in that each cell has many mitochondria and each mitochondrion has multiple copies of mtDNA, a mix of normal (wild type) and mutant. The presence of normal and mutant mtDNA within the same cell is called heteroplasmy. During cell division, the mtDNA becomes randomly divided between the daughter cells (Fig. 24.4). The proportion of mutant mtDNA may drift towards homoplasmy of either normal or mutant mtDNA. When the proportion of mutant mtDNA reaches a certain threshold within an individual cell, cellular function is compromised and, if sufficient cells are similarly affected, clinical features will develop. The distribution of the affected mitochondria within the body will determine the presentation. Mitochondrial disorders typically present with multiorgan involvement; however, the high energy-requiring organs are more frequently involved, especially brain, muscle, liver, heart, kidneys and eyes. Cells that divide rapidly may clear the mutant mtDNA because cells with normal function have reproductive advantage, e.g. refractory anaemias and enteropathy can resolve as the marrow or gastrointestinal tract recover, whereas brain and muscle involvement tend to be progressive.
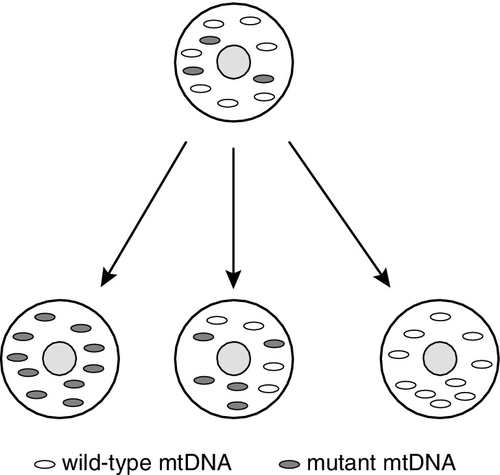
FIGURE 24.4 Mitochondrial DNA replication. Mitochondria randomly segregate to daughter cells following replication. Each cell may contain mitochondria with both mutant and wild-type DNA (heteroplasmy). Daughter cells may drift towards homoplasmy for either wild-type or mutant mtDNA. As the percentage mutant mtDNA load increases, the chance of developing clinical features increases.
Rearrangements (deletions/duplications) within the mitochondrial genome are usually sporadic and therefore the risk of recurrence is small. Examples include Kearns–Sayre syndrome (external ophthalmoplegia, pigmentary retinopathy, heart block) and Pearson syndrome (anaemia and pancreatic dysfunction, both endocrine and exocrine).
DIAGNOSTIC STRATEGIES
Close collaboration between the clinician and the metabolic laboratory is essential to establish the diagnosis of an IMD promptly. Links should be established between referring centres and specialist metabolic units, so that urgent cases can be discussed, investigations planned and a structured approach taken to the use of small samples. Details of the patient’s drug and feeding history, as well as transfusion status, will be needed for a full interpretation of results. Ideally, in the specialist unit, the clinical and laboratory services should be integrated with a multidisciplinary approach to the investigation and interpretation of results. This should lead to rapid diagnosis and initiation of appropriate treatment.
Essential laboratory investigations
When the suspicion of an IMD is raised, there are a number of basic laboratory investigations that can be performed immediately in the local hospital. These analyses will include full blood count and serum electrolytes as well as the more specific investigations discussed below. Their results may provide clues to the diagnosis and suggest further investigations.
Blood gas analysis
Blood gas analysis is a key investigation in a patient with a suspected IMD, particularly during acute episodes of illness. The most common acid–base imbalance seen in the paediatric population is a metabolic acidosis, secondary to infections, catabolic states or severe dehydration. The difficulty in interpretation in these circumstances is that these conditions are often the triggers of acute decompensation in patients with IMD. Calculation of the anion gap may assist in interpretation of the blood gas analysis: metabolic acidosis with an increased anion gap (> 16 mmol/L) is observed in many IMDs, whereas metabolic acidosis with a normal anion gap is more likely to be due to diarrhoea or renal tubular acidosis.
In patients with a metabolic acidosis and suspected IMD it is important to test the urine for ketones. Point-of-care testing for urine and blood ketones, using simple stick technology, is readily available in most neonatal and paediatric units and provides a rapid indication of the ketotic status of the patient. Ketosis is a physiological response to fasting, and is often seen in normoglycaemic children with severe nausea and vomiting. However, during the neonatal period, metabolic acidosis with ketonuria is nearly always pathological and should prompt consideration of branched chain amino acid disorders: propionic acidaemia, methylmalonic acidaemia, isovaleric acidaemia and maple syrup urine disease, as possible diagnoses. Urinary organic acids, blood acylcarnitine and amino acid analysis should be undertaken urgently to elucidate the underlying defect.
The detection of urine ketones, in addition to blood lactate and glucose measurement, provides the basis for the differential diagnosis of metabolic acidosis in the context of IMD (Box 24.2). Although diabetes mellitus is not an IMD, it is the most important diagnosis to consider when ketoacidosis is found associated with hyperglycaemia. In ketotic patients with hypoglycaemia, the gluconeogenetic and glycolytic defects should be considered first. The most suggestive features in this group are hepatomegaly and lactic acidaemia, although neither is consistently present. If the ketosis is sustained, the rare ketolysis defects (succinyl-CoA:3-oxoacid-CoA transferase deficiency and 3-oxothiolase deficiency) should be considered, as these can be easily missed. Urine organic acid profiling followed by enzyme and mutation analysis will be needed to confirm a diagnosis of these rare conditions.
It will be apparent from Box 24.2 that the predominant causes of metabolic acidosis in IMDs are ketosis and lactic acidosis. Lactic acidosis is frequently present in the acutely ill, when circulatory collapse results in tissue hypoxia. It is also frequently associated with organic acid disorders, such as propionic acidaemia and methylmalonic aciduria, as a result of secondary interference with the metabolism of coenzyme A. Ketone analysis, again, provides clues to the differential diagnosis, since, although ketosis is a consistent finding in many IMDs, it is generally absent in lactic acidosis secondary to hypoxia. Secondary causes of lactate accumulation should be excluded before an inherited disease of lactate/pyruvate metabolism is sought.
Respiratory alkalosis is a far less frequent finding in IMDs and when it is present, plasma ammonia should be measured urgently. Ammonia stimulates the respiratory centre, causing hyperventilation, resulting in a decrease in pCO2. The finding of acute encephalopathy with respiratory alkalosis should raise the suspicion of hyperammonaemia that, if confirmed, should prompt immediate investigations to exclude urea cycle disorders. In hyperammonaemia due to organic acid disorders, the acid–base disturbance is typically a metabolic acidosis.
Blood glucose
Hypoglycaemia is a presenting feature of many IMDs, such as those in which there is a primary block in glucose metabolism, for example glycogen storage diseases, or those in which glucose metabolism is affected secondarily, for example tyrosinaemia type I. Further investigations to identify the underlying cause should ideally be carried out when the child is hypoglycaemic. If this window of opportunity is missed, it is often difficult to make the diagnosis, since biochemical abnormalities may not be present during normoglycaemia; the child may then have to be subjected to a controlled fast with all the risks associated with this procedure. It is best practice for any hospital with a paediatric workload to have an agreed protocol for the investigation of hypoglycaemia. This protocol should be instituted if a child has a laboratory blood glucose of < 2.6 mmol/L (or point-of-care testing glucose < 3 mmol/L). Best practice guidelines are available from the MetBioNet website: www.metbio.net
Plasma ammonia
It is essential to measure plasma ammonia in all children with acute or chronic encephalopathy, recurrent vomiting or hyperventilation. Suspicion of an IMD should be raised in an infant who is well at birth, with abnormal clinical features developing only after the first 24 h once feeds have been established. Clues may be gleaned from the family history: for example, where a background of early male deaths or females with episodic illness would suggest X-linked OTC deficiency.
Hyperammonaemia is often missed, particularly in the neonatal period, when the clinical presentation is non-specific and can mimic sepsis. All centres with neonatal and paediatric units should therefore be able to provide rapid analysis of plasma ammonia. Care should be taken in the sample handling, with ammonia-free tubes used for collection of blood and the separation of plasma from cells within 15 min.
Overwhelming hyperammonaemia in the neonatal period is usually due either to a urea cycle disorder (approximately 70%) or to an organic acid disorder (approximately 30%). The hyperammonaemia in organic acid disorders is a result of a deficiency in acetyl-CoA required for the synthesis of N-acetylglutamate, a preliminary step in urea synthesis. The finding of a metabolic acidosis in a patient with hyperammonaemia is suggestive of an organic acid disorder. Since the severity of hyperammonaemia does not allow a distinction between the causes, and treatment will differ depending on the underlying defect, rapid biochemical follow-up with second-line investigations is required. Analysis of urinary organic acids and orotic acid, in conjunction with plasma amino acids and acylcarnitines, is essential; results can usually be available the same day after discussion with the regional metabolic laboratory. Figure 24.5 provides a flowchart for the investigation and diagnosis of neonatal hyperammonaemia.
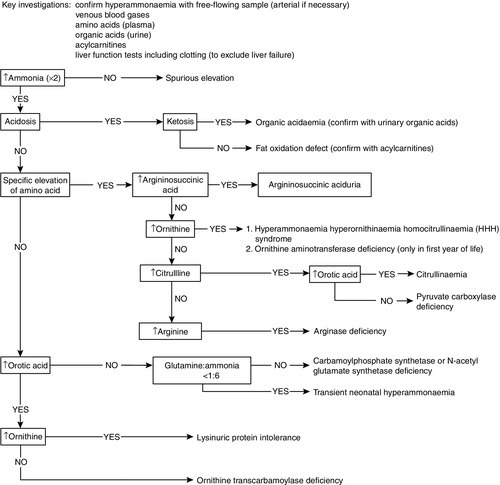
FIGURE 24.5 A flowchart for the investigation of hyperammonaemia.
In older children and adults, plasma ammonia should be measured in those with unexplained encephalopathy (progressive or chronic relapsing), vomiting or drowsiness, Reye-like syndrome (hyperammonaemia, hypoglycaemia and raised amino-transferases), neurological dysfunction or ataxia. Clinical illness is often episodic in the late onset group, presentation being associated with catabolism such as occurs during infections. If a complete dietary history is taken clues, such as self-selection of a low protein diet, may be revealed. In those patients with a Reye-like presentation, the possibility of a fatty acid oxidation disorder should be considered.
Liver function tests
Jaundice, accompanied by other features of liver dysfunction, is one of the most frequent presenting features of IMDs. This is not surprising, given the central role of the liver in metabolism. Liver function tests are therefore essential first-line investigations in suspected IMDs. They should comprise measurement of bilirubin (total and conjugated), aminotransferases, alkaline phosphatase and the prothrombin time (or international normalized ratio).
Many IMDs present with liver failure, often in the neonatal period. The liver function test findings are characterized by a severe conjugated hyperbilirubinaemia and raised serum aminotransferases. The most striking of this group of disorders is classic galactosaemia (galactose 1-phosphate uridyl transferase deficiency), which typically presents after introduction of milk feeding. The principal IMDs causing neonatal liver disease are listed in Box 24.3. This box does not include the inherited hyperbilirubinaemias, which are not associated with other features of hepatic dysfunction.
Measurement of ketones
Ketosis is a normal response to fasting and, when not associated with either acidosis or hypoglycaemia, should, in infancy and childhood, be considered as physiological. However, as previously mentioned, the presence of ketones in a neonate is abnormal and requires urgent follow-up. Although ketosis is a physiological response, it is likely to be of clinical significance when it is associated with acidosis. The detection of urine ketones, using a simple point-of-care testing device, is therefore the starting point for the investigation of metabolic acidosis (see Box 24.2).
The absence of ketones can also give a clue to the underlying IMD, the most classic example of which is hypoketonuria with hypoglycaemia due to a fatty acid oxidation defect such as MCADD. Patients with this group of disorders are able to mobilize fat stores during periods of fasting or catabolic stress but, owing to enzyme deficiencies, are unable to oxidize the fatty acids completely, leading to a relative deficiency of acyl-CoA required for ketogenesis. Although classically considered to lead to a complete absence of ketone production, it is more common to see some ketones present but at an inappropriately low concentration. The presence of ketonuria in hypoglycaemia should therefore not preclude investigation for fatty acid oxidation defects.
Urinary reducing substances
The detection of reducing substances in the urine, using simple but non-specific tablets, has historically been a mainstay of first-line testing for IMD. The withdrawal from the market of Clinitest® (Bayer) in 2011 has prompted a review of this practice and a streamlining of investigations of disorders of monosaccharide metabolism. Thin layer chromatography (TLC) may be useful to identify sugars that are present, but may give a negative result in children with IMD in whom the abnormal pathway is not stressed, for example if an infant with classic galactosaemia has been placed on a lactose-free milk formula. There are also analytical concerns with using sugar TLC, most notably a lack of a robust external quality assurance scheme and the reproducibility of results being highly operator dependent.
In the investigation of classic galactosaemia, where jaundice and liver dysfunction can rapidly deteriorate to hepatic failure, whole blood should be sent for urgent analysis of galactose 1-phosphate uridyl transferase activity. In older infants, with progressive liver dysfunction and hepatomegaly presenting after weaning, the diagnosis of hereditary fructose intolerance should be considered. Liver biopsy for aldolase activity and mutation analysis will be required for diagnosis.
Second-line investigations
Plasma and urinary amino acids
Amino acids are important intermediates in a number of metabolic pathways and are a significant source of energy, particularly during fasting. Their metabolism may involve potentially toxic intermediates that are normally further metabolized, but may accumulate in metabolic diseases involving amino acid metabolism and thus lead to organ damage. Amino acid analysis is therefore a key second-line investigation when an inherited metabolic disease is suspected (Box 24.4), and is frequently requested as part of a metabolic screen.
Individually, amino acid disorders are rare, with the most common (PKU) having an incidence of 1 in 10 000. However, the combined incidence of all disorders that can be diagnosed using amino acid analysis is in the order of 1 in 6000. As a group, they are heterogeneous, both in terms of clinical symptoms and age of presentation. The onset of symptoms may occur anywhere from the neonatal period to adulthood, but is classically associated with periods of protein catabolism, such as delayed feeding in a neonate, or intercurrent infections in an infant. Acute symptoms are associated with the accumulation of large amounts of amino acids that overwhelm a defective pathway, resulting in the production of toxic metabolites. There are certain presenting features in neonates, e.g. acute encephalopathy and ketosis, which should prompt consideration of amino acid analysis. However, the clinical presentation of many of these disorders is disease specific, e.g. gyrate atrophy in ornithine aminotransferase deficiency, and may warrant specific investigation. An understanding of the nuances of presentation in amino acid disorders is the key to making a diagnosis.
Amino acid disorders can be simply categorized as primary or renal disorders (Table 24.2). In primary disorders, there is a defective metabolic pathway with accumulation of specific amino acids or metabolites as a result of an enzyme deficiency. A classic example is maple syrup urine disease, caused by a deficiency in the enzyme branched chain oxoacid dehydrogenase, which leads to the accumulation of valine, leucine and isoleucine and the presence of the abnormal metabolite alloisoleucine. In primary amino acid disorders, the most informative sample is plasma, giving a snap shot of the metabolic pathway. Urine is less useful in this group since the excretion of amino acids is much more variable (especially in prematurity) and is significantly prone to interference from medication. Urine is, however, essential for the diagnosis of renal amino acid disorders such as cystinuria. In this group, amino acid metabolism is intact, but renal tubular reabsorption of specific amino acids is defective. The abnormal pattern is therefore only observed in urine.
TABLE 24.2
Renal amino acid disorders
| Disorder | Urinary amino acid pattern |
| Cystinuria | Cystine, ornithine, arginine, lysine |
| Hartnup disease | Neutral amino aciduria |
| Lysinuric protein intolerance | Lysine, ornithine, arginine |
| Iminoglycinuria | Proline, hydroxyproline, glycine |
It is important to be aware of the type of investigation undertaken when amino acid analysis is requested. Simple urine ‘spot tests’, such as the cyanide-nitroprusside test for cystine and homocystine, are often employed by local hospitals as a preliminary to referral to a specialist centre. These simple assays are becoming increasingly obsolete and are limited by a lack of sensitivity and specificity and should not be used in isolation. Other screening techniques may be employed, such as thin layer chromatography, but again are limited in the detection of anything but gross abnormalities: some disorders, particularly those with mild biochemical changes, may be missed by these methods. If these qualitative techniques are employed, their limitations must be understood, and conveyed to the requesting clinician, so that diagnoses are not missed.
Amino acids are best analysed using quantitative techniques such as ion exchange chromatography. These methods are sufficiently sensitive to detect the newly recognized amino acid disorders characterized by low concentrations of amino acids, such as 3-phosphoglycerate dehydrogenase deficiency, a disorder of serine synthesis.
Ion exchange chromatography is most suited to ‘routine’ analysis, but is limited by the long analysis time in the acute situation. To complement the approach, rapid tandem mass spectrometry (TMS), with analysis of a targeted list of amino acids and other metabolites (see section on acylcarnitines below), allows a diagnosis to be made swiftly and treatment to be instituted in an acutely sick child. Combining rapid targeted TMS testing in the acute situation, with a routine approach that clearly states which disorders are excluded, may provide a balanced solution to the provision of amino acid analysis.
Urinary organic acids
Urine organic acid profiling is an important second-line investigation in which up to 150 different IMDs can be identified from a single analysis. Organic acid analysis can identify intermediary metabolites from most amino acid, carbohydrate, purine and pyrimidine, neurotransmitter, cholesterol and fatty acid pathways, making it a powerful investigative tool. Using organic acid analysis, metabolic disorders can be identified by the presence of pathological concentrations of normal metabolites, for example fumaric acid in fumarase deficiency, or the presence of pathological (and in some cases pathognomic) metabolites such as succinylacetone in tyrosinaemia type I. Most laboratories use gas chromatography-mass spectrometry (GCMS) to undertake organic acid analysis, providing a qualitative interpretation of the profile. However, the use of quantitative analysis by stable isotope dilution should be considered when small concentrations of critical metabolites may lead to a diagnosis.
Many disorders can be detected by organic acid analysis regardless of whether the sample is collected when the child is acutely ill or not. However, some IMDs are clearly identifiable only during episodes of acute decompensation. Medium chain acyl-CoA dehydrogenase deficiency is one such disorder where the classic pattern of a medium chain dicarboxylic aciduria (adipic, suberic and sebacic acids), with an inappropriately low excretion of ketones in the presence of the pathognomonic metabolite hexanoylglycine, may only be apparent when the child is acutely unwell. Samples collected during episodes of acute decompensation are therefore vital for investigation of IMDs. The indications for urinary organic acid analysis are summarized in Box 24.5.
Urinary orotic acid
Increased urinary excretion of orotic acid is a feature of four IMDs involving the urea cycle (ornithine transcarbamoylase deficiency, citrullinaemia, argininosuccinic aciduria and argininaemia) (see Fig. 24.1) and is helpful in their diagnosis. There are a number of analytical techniques that can be used for identification of this key metabolite, for example GCMS or TMS, and it is often required as part of the rapid investigation of a sick infant.
Blood acylcarnitines
With the advent of TMS technology, acylcarnitine profiling has become a crucial investigation in the field of IMDs. The use of TMS for acylcarnitine profiling, combined with simultaneous analysis of other intermediary metabolites, enables the diagnosis of many of the treatable amino acid, organic acid, fatty acid oxidation and urea cycle disorders. In a clinical emergency, if one of these disorders is considered, then discussion with a specialist metabolic service to arrange urgent analysis should be a priority.
Acylcarnitine profiling allows the quantitation of saturated and unsaturated acylcarnitine species required for the diagnosis of a number of IMDs. In addition, the determination of both free and total carnitine (i.e. overall carnitine status) is required for the investigation of suspected carnitine transporter defects. Typically, acylcarnitine analysis is undertaken on plasma or blood spot samples, with only a small sample usually being necessary. Bile samples, collected post-mortem, on to newborn screening cards, can be used if the suspicion of IMD is raised at autopsy.
It should be noted that some conditions can only be diagnosed confidently with the use of acylcarnitine profiling: an example is VLCADD. Acylcarnitine analysis should therefore be undertaken if a fatty acid oxidation defect is suspected; these disorders typically present with hypoketotic hypoglycaemia, but may be associated with cardiomyopathy, rhabdomyolysis or hepatomegaly. In a pregnant woman with AFLP or HELLP syndrome, the possibility of LCHADD in her infant should be considered. Rapid acylcarnitine profiling is required for the newborn infant of a mother with a history of either condition.
Blood lactate and pyruvate
Lactate and pyruvate are important intermediaries in a number of metabolic pathways, particularly in the production of ATP under anaerobic conditions. It is therefore not surprising that assessment of these pathways, by analysis of blood lactate, is an essential measurement in the investigation of a suspected IMD. In the absence of tissue hypoxia, and assuming the sample was collected properly, using an appropriate antiglycolytic agent, a blood lactate of > 3 mmol/L should be considered abnormal and further investigations for an IMD initiated.
Lactic acidaemia is found in many groups of IMDs (see Box 24.1) and may lead to a metabolic acidosis when the lactate concentration exceeds 5 mmol/L. It is a frequent finding in organic acidaemias, urea cycle disorders and fatty acid oxidation disorders, as a result of secondary interference with the metabolism of coenzyme A, and is often accompanied by ketosis. These groups of IMDs can be rapidly distinguished with follow-up investigations such as organic acid, amino acid and acylcarnitine analysis.
The primary lactic acidaemias, for example defects in pyruvate metabolism or respiratory chain function, often present with severe metabolic acidosis in the neonatal period. A distinguishing feature of this group, as opposed to the secondary IMD groups noted above, is that the clinical picture is unrelated to intake of protein. In the primary lactic acidaemias, analysis of both blood lactate and pyruvate, calculation of the lactate: pyruvate (L:P) ratio and measurement of cerebrospinal fluid (CSF) lactate are of assistance in directing further investigations. It must be noted that pyruvate is an extremely unstable analyte and needs careful sample handling with immediate precipitation of whole blood. However, if accurate measurement of lactate and pyruvate is undertaken, the L:P ratio can give valuable insight into the cytoplasmic redox status. An elevated L:P ratio (> 25) is suggestive of a respiratory chain defect or pyruvate carboxylase deficiency, with a normal ratio (< 25) suggesting a disorder of gluconeogenesis or pyruvate dehydrogenase deficiency.
Further information can be gained from assessing plasma lactate concentrations in relation to the fed and fasted states. This is of particular use in the investigation of glycogen storage disorders (GSDs). Lactic acidaemia in the fasted state is a key finding in GSD type I (glucose 6-phosphatase deficiency), in contrast to GSD type III (amylo-1,6-glucosidase deficiency), in which the increase in lactate is most noticeable after feeding. In investigating these conditions, it is important that samples are collected both pre- and postprandially so that lactic acidaemia is not missed. It is for this reason that lactate is a key measurement in many diagnostic fasts and 24 h metabolite profiles (see below).
Urinary glycosaminoglycans
The mucopolysaccharidoses are a group of lysosomal storage disorders characterized biochemically by the accumulation of glycosaminoglycans (GAGs) in the urine. Clinically, they present with coarsening of the facies, in conjunction with features including skeletal abnormalities, hepatosplenomegaly and hearing loss. The majority of these features are not present at birth but develop progressively with age. If this group of disorders is suspected, a random urine sample should be collected for analysis of GAGs. In the first instance, quantitative analysis is usually undertaken with interpretation against age-related reference ranges. This is then followed-up by electrophoresis to identify the individual GAGs from the typical patterns characteristic of each disorder (see Table 24.3). Definitive diagnosis is by enzyme analysis.

Plasma very long chain fatty acids
Very long chain fatty acids, with carbon chain length ≥ 22, are exclusively metabolized by β-oxidation in peroxisomes. The analysis of plasma VLCFAs is therefore an important second-line investigation in suspected peroxisomal disorders. These are a genetically heterogeneous group but, in disorders both of peroxisomal biogenesis (such as Zellweger syndrome), and single peroxisomal protein defects (the most common of which is X-linked ALD), β-oxidation is impaired, leading to the accumulation of saturated VLCFAs. The finding of an elevated ratio of C26:C22 fatty acids is a clear indication of peroxisomal dysfunction. However, it should be noted that although analysis of VLCFAs identifies the presence of a peroxisomal disorder, results should be viewed in conjunction with the clinical findings, imaging and further laboratory analysis, such as erythrocyte plasmalogen, bile acid intermediates and pristanic acid to enable a precise diagnosis.
Functional and loading tests
The aim of functional or loading tests is to unmask IMDs that may be difficult to diagnose by conventional techniques. They are most often used in situations where metabolic findings are not clearly abnormal and the diagnosis is uncertain, for example many of the fatty acid oxidation disorders, where biochemical findings may be subtle, or absent, when the patient is well. If the window of opportunity to collect samples when the patient is hypoglycaemic or acutely unwell is missed, then a controlled diagnostic fast may be carried out to aid the diagnosis.
Diagnostic fast
During a diagnostic fast, blood samples are collected hourly. Investigations should include markers of intermediary metabolism (non-esterified fatty acids, hydroxybutyrate and lactate), acylcarnitines and counter-regulatory hormones (insulin, growth hormone and cortisol). Urine organic acids and blood acylcarnitine should be measured at the beginning and end of the fast. The blood glucose concentration is monitored using a point-of-care testing device appropriate for the detection of hypoglycaemia; such results must be confirmed in the laboratory. Given the risks associated with this procedure, diagnostic fasts must be performed under medical supervision in a specialist unit. In addition to providing valuable diagnostic information, a fast allows the determination of the maximum safe fasting period before the onset of hypoglycaemia, and is often undertaken on a yearly basis in patients with glycogen storage disease.
Allopurinol loading test
Loading tests can be used to expose an enzyme defect, where there is residual activity. The allopurinol test can be used in this way to confirm heterozygote status in female carriers of OTC deficiency. Urine is collected for measurement of orotidine and orotic acid, before and after administration of a dose of allopurinol. Although this is a safe and noninvasive approach, not all at-risk females will be detected. This strategy, although still in use, is now being superseded by the use of mutation analysis.
Confirmatory investigations
Enzyme analysis: general principles
Final confirmation of the diagnosis of a suspected IMD often requires enzyme analysis. In some cases, confirmatory testing is not undertaken, because the diagnosis can be inferred from a typical clinical history and a characteristic pattern of abnormal metabolites. In conditions such as PKU, the use of an invasive procedure, such as liver biopsy, to obtain tissue for enzyme analysis would not contribute further to the management of the patient. However, the majority of patients with a suspected IMD will have enzyme analysis to confirm the diagnosis.
Enzyme analysis can be undertaken in various tissue types, ranging from blood cells to liver and muscle samples, obtained by biopsy. It is of particular importance that for IMDs that are tissue specific, an appropriate specimen is used, e.g. liver biopsy samples in the urea cycle disorders and muscle or liver in the glycogen storage disorders that are either muscle or liver specific. If a patient with a suspected IMD is not expected to survive, plasma, urine, skin and, if possible, muscle and liver biopsies should be collected. Skin biopsies are of particular use since fibroblast cultures can be established and DNA harvested if required.
Red cell galactose 1-phosphate uridyltransferase
Classic galactosaemia, due to a deficiency in the enzyme galactose 1-phosphate uridyltransferase (Gal-1-PUT or GALT), leads to the accumulation of galactose and other toxic metabolites on introduction of lactose into the diet. In the past, testing for urine reducing substances provided an easily available screening test for galactosaemia. Now, clinical suspicion should be urgently followed-up with confirmatory testing of red cell GALT activity. The requesting clinician must state clearly the transfusion status of the child, since previous blood transfusion will invalidate the result. If a child has been transfused, it may be possible to infer the diagnosis by analysing parental blood and demonstrating that they are heterozygotes.
Lysosomal enzyme screening
The lysosomal storage disorders are a group of approximately 40 different IMDs characterized by the accumulation of macromolecules in lysosomes, as a result of specific enzyme or protein transporter defects. The accumulated material results in an increase in the size and number of these organelles, leading to cellular dysfunction and subsequent pathological features. The lysosomal storage disorders are generally classified on the basis of the accumulated material, for example sphingolipidoses, mucolipidoses and mucopolysaccharidoses. These conditions typically present in a chronic and progressive manner, in many cases with neurological degeneration. The principal clinical features are summarized in Box 24.6.
Diagnosis of this group of disorders is often difficult, as there is an overlap in the clinical presentation of each disease. A ‘screening’ approach is, therefore, adopted by specialist laboratories: this ensures the maximum possibility of making a diagnosis. Groups of lysosomal enzymes in plasma and leukocytes are analysed depending on the clinical features, for example whether the patient has neurodegeneration or the presence or absence of hepatosplenomegaly. The choice of enzymes is tailored to exclude the majority of lysosomal storage disorders. This approach requires good communication between the local hospital and specialist enzyme laboratory so that a systematic approach to the investigation is undertaken.
Complementation studies
Although in the vast majority of IMDs the enzyme deficiency is well characterized, there are still a number of disorders in which the protein or gene defect has not been identified. In these conditions, such as those that lead to decreased synthesis of adenosylcobalamin and the clinical features of cobalamin-responsive methylmalonic aciduria (classified as Cbla and Cblb), a different approach to confirm the diagnosis may be taken. To distinguish these different groups, complementation studies can be undertaken in cultured fibroblasts. The principle of the investigation is to compare the incorporation of a marker substance, in this case propionate, in parallel fused and unfused cell cultures. The classification of the patient in this way is of use in assessing the possible prognosis and excluding disease in other family members. However, it is likely that this approach will be superseded once the enzyme or gene locus is identified.
Genetic mutation analysis
Mutation analysis is playing an increasingly important role in the diagnosis of IMDs The use of next generation sequencing has enabled a rapid and more cost-effective approach and is becoming a mainstay of diagnostic pathways.
Mutation analysis has many benefits in the diagnosis of IMDs. It is of particular use in prenatal diagnosis, where mutation analysis in chorionic villus samples (CVS) can be performed much earlier than enzyme diagnosis in cultured amniocytes. Once the mutation has been defined in the proband, testing of family members may overcome the need for invasive sampling, such as liver biopsies or functional tests that may give ambiguous results.
Where mutations are classified and the genotype/phenotype correlation is well recognized, this may provide useful information regarding prognosis and possible treatment options. In some disorders, such as MCADD, a common disease-causing mutation (A985G) is present in many cases, allowing the use of mutation analysis to secure the diagnosis. The approach of combining metabolite assays with mutation analysis confirms the diagnosis rapidly and may obviate the need for fibroblast culture. Problems may arise, however, when compound heterozygotes are identified: in these instances it might not be clear what the clinical outcome of a combination of potentially less severe mutations would be.
PRENATAL DIAGNOSIS
The availability of prenatal diagnosis gives a number of options to a family in whom a serious genetic disorder is suspected. In the field of IMDs, prenatal diagnosis is often requested if a previously affected child has a poor prognosis, particularly where the options for treatment are limited. Inherited metabolic diseases are generally diagnosed prenatally using either enzyme analysis in cultured amniocytes or mutation analysis in CVS.
Amniocentesis, undertaken in either the first or second trimester, provides fetal amniocytes for culture. Enzyme analysis can be undertaken in these cultures and the activity compared with appropriate reference ranges to confirm the diagnosis. The limitation of this approach is the time scale: amniocyte culture typically takes 4–6 weeks to establish, leading to additional anxiety for the family and limited time for decision-making. Since only a small number of analyses are undertaken each year, prenatal diagnosis of IMDs using these techniques is undertaken at only a few specialized centres.
Chorionic villus sampling is generally undertaken at 9–12 weeks of gestation, providing the opportunity for earlier diagnosis and giving additional time for counselling and safer termination, if this option is chosen. In this technique, fetal villus cells obtained by amnioscopy are dissected from the maternal decidual tissue, and DNA extracted and used for mutation analysis. Adherence to strict techniques is paramount, to avoid maternal cell contamination that may result in diagnostic ambiguity.
MANAGEMENT
Many treatment modalities are adopted to manage IMDs, dependent on whether pathology results from deficiency of the product, the build up of a toxic metabolite or a combination of the two. At present, most treatments remain symptomatic rather than curative.
Strategies to replace a missing product
Supply of precursor
Enzymes catalyse reactions that would proceed at a much reduced rate in their absence. Provision of large doses of precursor can shift the reaction in the direction of making the product by a mass action effect: in some cases this is sufficient to ameliorate symptoms.
Hydroxycobalamin is converted in the body via a number of intracellular conversions from its dietary hydroxycobalamin form to its two active forms, adenosylcobalamin and methylcobalamin. In the cobalamin disorders, blocks in these conversions lead to accumulation of homocysteine, methylmalonate or both, dependent on the position of the block (Fig. 24.6). Cobalamin C disorder is the commonest of these rare IMDs. Patients present in the neonatal period and first year with neurological deterioration, poor feeding and hypotonia, and often develop seizures. Anaemia is common, as is multiorgan involvement including the kidneys, liver and heart. The diagnosis is suggested by elevated plasma homocysteine and urinary methylmalonate concentrations, in the absence of hydroxycobalamin deficiency. Diagnosis is confirmed by fibroblast incorporation studies and decreased synthesis of adenosyl- and methylcobalamin. Treatment with regular intramuscular hydroxycobalamin leads to a marked reduction in homocysteine and methylmalonate, but not complete biochemical normalization. Clinical improvement is limited even in prospectively treated affected siblings.
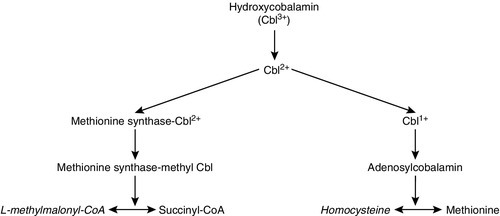
FIGURE 24.6 Cobalamin metabolism.
Replacement of product
Replacement of the deficient product of a blocked pathway is another solution to the treatment of some IMDs: it effectively bypasses the block. Its efficacy depends on the product being available and its potential to be given in a form that can be taken by the patient and then reach its site of action in a usable form.
Such an approach is used in the congenital disorder of glycosylation (CDG) Ib (phosphomannose isomerase deficiency). Glycosylation is an essential process in the body, as almost all proteins that are secreted or membrane bound are dependent on their glycans (carbohydrate side chains) for normal activity. The production of glycoproteins begins with the manufacture of the oligosaccharide precursor and its transfer to the nascent polypeptide chain in the endoplasmic reticulum, with subsequent modification within the Golgi apparatus to produce the desired glycan. Defects of the pathway were first established when examining transferrin, which has two glycans with four sialic acid residues. Defective glycosylation results in variation in the number of sialic acid residues, which affects the charge on the protein. This can be detected using isoelectric focusing, which shows additional bands. Children with CDG Ib present with recurrent vomiting and diarrhoea, coagulopathy, protein-losing enteropathy, hypoalbuminaemia, delayed growth and hyperinsulinaemic hypoglycaemia. Liver fibrosis has also been noted. Treatment with oral mannose effectively bypasses the block by replacing the missing product (Fig. 24.7). Clinically, there is a reduction in diarrhoea and vomiting with improved growth. Biochemically, the isoelectric focusing pattern normalizes as glycan production beyond the block is normal. Plasma concentrations of antithrombin III, the cause of the coagulopathy, also return to normal.
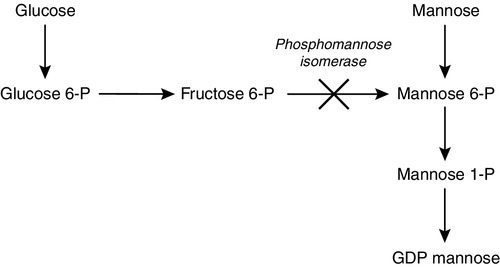
FIGURE 24.7 Mannose therapy for CDG Ib (phosphomannose isomerase deficiency). Mannose is phosphorylated by hexokinase to mannose 6-phosphate, thereby bypassing the block. GDP, guanosine 5′-diphosphate; P, phosphate.
In dihydropteridine reductase deficiency, there is decreased recycling of biopterin, the essential cofactor for pheknylalanine hydroxylase, the enzyme responsible for the conversion of phenylalanine into tyrosine, with resultant phenylketonuria. In addition, biopterin is the essential cofactor for two other hydroxylases in the body, namely tyrosine and tryptophan hydroxylases, required for the production of L-DOPA and 5-hydroxytryptophan (5-HT), respectively. These are, in turn, converted to dopamine and serotonin, which are two of the major neurotransmitters in the brain. Dihydropteridine reductase deficiency was originally termed malignant PKU, as following the introduction of the PKU screening in the 1970s it became apparent that there were very rare patients who still went on to have marked developmental delay and characteristic axial hypotonia and limb dystonia, in spite of excellent phenylalanine control by diet. Now, all neonates with hyperphenylalaninaemia detected on newborn screening are screened for biopterin defects, usually performed on dried blood spots by measuring total biopterins. Dihydropteridine reductase deficiency is managed with a phenylalanine restricted diet and supplementation with 5-HT and L-DOPA. The latter is given in combination with carbidopa in the ratio 4:1. Carbidopa is a peripheral decarboxylase inhibitor, which reduces peripheral conversion of L-DOPA to dopamine, thereby increasing the amount of L-DOPA reaching the brain and reducing side-effects, which include nausea, vomiting, postural hypertension, dystonic reactions and psychological disturbance. Dose increments should be small, as initially patients are very sensitive to the effect of the treatment owing to upregulation of receptors. Dosage requirements are monitored clinically and by measuring the breakdown products of dopamine and serotonin, namely homovanillic acid (HVA) and 5-hydroxyindoleacetic acid (5-HIAA), in the CSF. A slightly greater dose of L-DOPA is given compared with 5-HT, to mimic the naturally slightly higher amounts within the brain. In addition, folinic acid is required to combat the associated central folate deficiency seen in this condition.
Replacement of the missing product is also used in pyridoxal phosphate-dependent seizures in pyridoxamine 5-phosphate oxidase deficiency. Pyridoxal phosphate is the active form of pyridoxine, required for a number of essential reactions in the body and particularly in the brain. Pyridoxamine 5-phosphate oxidase deficiency presents with devastating neonatal seizures, which can be effectively treated by giving pyridoxal phosphate orally. The cessation of seizures is immediate and dramatic but associated with transient hypotonia and apnoea. Treatment should therefore be initiated with full resuscitation facilities available.
Replacement of the missing product in utero has been successful in 3-phosphoglycerate dehydrogenase deficiency, a serine biosynthesis disorder, characterized by congenital microcephaly, severe psychomotor retardation and intractable seizures. The treatment of affected children with oral L-serine has proved effective in preventing the seizures: however, development is not normalized and the congenital microcephaly suggests impaired fetal brain growth in utero. It has now been shown that giving L-serine to the mother during pregnancy can reverse the decrease in fetal head growth in utero with resultant normal neurological development. The key diagnostic investigation for this condition is analysis of serine concentration in CSF.
Synthetic analogues
It may not be possible to replace the product directly, owing to difficulties with absorption and denaturation when exposed to the acidity of the stomach and digestive enzymes. Synthetic analogues of the missing product are, however, used in some circumstances. N-acetylglutamate synthetase (NAGS) deficiency is a rare urea cycle defect that may present with devastating hyperammonaemic crises. N-acetylglutamate is an essential activator for carbamoylphosphate synthetase, the second enzyme in the urea cycle pathway (see Fig. 24.1). Carbamylglutamate, a synthetic analogue of N-acetylglutamate, is an effective treatment for NAGS deficiency. It allows activation of carbamoylphosphate synthetase, in the absence of a working NAGS enzyme, reducing the risk of hyperammonaemia and improving neurological outcome. In organic acidaemias, hyperammonaemia can occur as a result of secondary inhibition of NAGS; carbamylglutamate is used in their management.
Receptor agonists can be used where the site of action of a deficient product is a receptor. Aromatic amino acid decarboxylase (AADC) converts 5-HT into serotonin and L-DOPA into dopamine. Clinical features of AADC deficiency include intermittent oculogyric crises, limb dystonia, generalized hypotonia, developmental delay and autonomic dysfunction. The diagnosis is indicated by low concentrations of the neurotransmitter breakdown products, HVA and 5-HIAA, in CSF. Dopamine receptor agonists, such as pramipexole and rotigotine, are being used to reduce the frequency of crises, and also improve voluntary movements, but the benefits have so far been variable.
Alternate product
A further strategy employed in the treatment of IMDs is to use an alternate product to bypass the block. The classic example is the use of a ketogenic diet in deficiency of glucose transporter type 1 (GLUT1), a membrane transporter that transports glucose across the blood–brain barrier: a defective protein results in hypoglycaemia in the brain, with a normal plasma glucose concentration. Presentation is in infancy with epilepsy, global developmental delay and complex movement disorders. Diagnosis requires the simultaneous measurement of glucose in the plasma and CSF which reveals a decreased CSF:plasma glucose ratio. Although glucose is the preferred fuel for the brain, ketones may be used for energy and gain entry to the brain via the monocarboxylate cotransporter (MCT1) transporter, thus avoiding the block (Fig. 24.8). The ketogenic diet promotes ketogenesis by restricting carbohydrate (typically a 4:1 ratio of fat to combined protein and carbohydrate), with clinical improvement in seizure control, development and movements. The condition tends to stabilize after puberty, but the degree of residual impairment is highly variable.
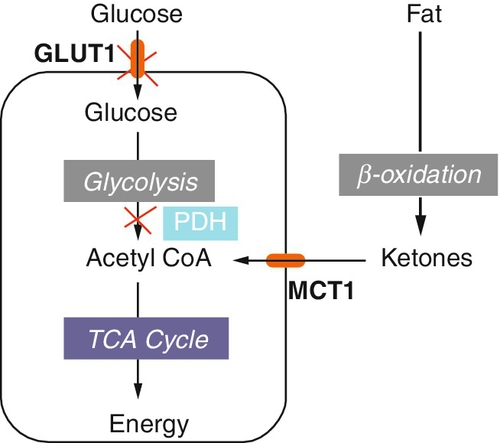
FIGURE 24.8 Providing alternate fuel utilizing ketones in the management of GLUT1 deficiency and pyruvate dehydrogenase deficiency. PDH, pyruvate dehydrogenase; GLUT1, glucose transporter protein 1; MCT1, monocarboxylate transporter 1; TCA cycle, transcarboxylic acid cycle.
Ketogenic diets are similarly used in pyruvate dehydrogenase deficiency, bypassing glycolysis.
Inhibition of product breakdown
Enzyme blocks in IMDs are not usually complete, and usually some product is made. Preventing or delaying breakdown of the product can have useful therapeutic effects. In AADC deficiency described above, a combination of a dopamine agonist and a monoamine oxidase inhibitor, such as tranylcypromine, can be used in combination. The latter prevents the natural breakdown of dopamine and serotonin and therefore boosts their concentrations. This has the positive effect of reducing the frequency of oculogyric crises and improving muscular tone.
Enzyme replacement therapy
Replacement of the missing enzyme is an obvious potential solution to the management of IMDs, but many obstacles have to be overcome, including production, purification and targeting of the enzyme. Access to the CNS across the blood–brain barrier remains a technical challenge. Nearly 40 years ago, it was noted that co-culture of fibroblasts from Hurler and Hunter syndrome patients mutually corrected the defects in vitro, suggesting the existence of a diffusible substance that could correct the block. Twenty years later, the first enzyme replacement therapy (ERT) became commercially available. Gaucher disease (glucocerebrosidase deficiency) has been the forerunner of the ever expanding number of conditions considered for ERT. Storage of glucocerebroside occurs in liver, spleen and bone, and, in two out of the three forms, brain. Type 1 is the most common, presenting with hepatosplenomegaly, anaemia, lung infiltration and bony changes: the brain is not involved. Type 1 is prevalent in the Ashkenazi Jewish population. Type 2 is rapidly progressive with severe neurological involvement and hepatosplenomegaly. Type 3 has brain involvement, but a much milder course. Inheritance of all three forms is autosomal recessive. The diagnosis is made by measuring the glucocerebrosidase activity in leukocytes.
The absence of brain involvement in type 1 made it an obvious target for ERT as the enzyme does not cross the blood–brain barrier. The enzyme used is a recombinant modified human placental glucocerebrosidase, with an exposed mannose terminus to enhance lysosomal targeting, following uptake by macrophages. Uptake of glycoproteins is mediated via carbohydrate-specific receptors, which are present on macrophages. Enzyme replacement therapy has revolutionized the management of type 1 Gaucher disease, with improvement in haematological parameters, regression of organomegaly and reduction in bone pain. The enzyme is given fortnightly as an intravenous infusion, initially in hospital, but later at home, utilizing a home-care nursing team. Some individuals produce antibodies to the enzyme protein, but therapy can usually continue.
Fabry disease (α-galactosidase deficiency) is another condition for which ERT has been developed. Previously, treatment was supportive, including dialysis and the use of analgesics. Enzyme replacement therapy is effective in reducing plasma glycosphingolipid concentrations, preserving renal function, improving cardiac conduction and reducing pain.
Replacement enzymes have been developed for other lysosomal storage disorders, including: Hurler syndrome (α-iduronidase deficiency); Hunter syndrome (iduronate-2-sulphatase deficiency); Morquio syndrome Type A (N-acetylgalactosamine-6-sulfatasedeficiency); Maroteaux Lamy syndrome (N-acetylgalactosamine-4-sulfatase deficiency); Pompe disease (acid maltase deficiency), and Wolman disease (lysosomal acid lipase deficiency). At present, relatively small numbers of patients have been treated, for relatively short time periods. It is hoped that early treatment will prevent storage occurring and therefore give optimal outcomes, but, for those conditions with neurological involvement, the lack of brain penetrance remains a concern.
Cofactor supplementation
Many enzymes have vitamin cofactors that are essential for normal function. Cofactor supplementation may increase residual enzyme activity in deficient patients. Although the absolute response may be quite small, because clinical features often only develop at extremely low levels of enzyme activity (< 5%), even a small increase (e.g. by 1% of normal activity) may prove to be clinically significant.
Patients may have sensitive deficiencies that are totally correctable by giving the enzyme cofactor. Some conditions can be characterized by their response to cofactor supplementation. An example is homocystinuria (cystathionine β-synthase deficiency), which is classified as being either pyridoxine responsive or pyridoxine unresponsive. In patients who respond to pyridoxine, there is a reduction in fasting plasma homocysteine concentration and characteristic hypermethioninaemia, on pyridoxine supplementation. A pyridoxine challenge should therefore be undertaken in all new patients, with measurements of plasma homocysteine and methionine concentrations before, during and after supplementation. Some patients have a partial response to pyridoxine.
Once a diagnosis has been established, cofactors are routinely given in a number of IMDs, to assess whether any clinical benefit can be gained by supplementation (Table 24.4).
TABLE 24.4
Vitamin cofactors, supplementation of which may be beneficial in inherited metabolic disorders
| Cofactor | Disorder |
| Ascorbic acid | Tyrosinaemia type III Transient tyrosinaemia of the newborn Glutathione synthase deficiency Congenital lactic acidosis |
| Biotin | Biotinidase deficiency Multiple carboxylase deficiency Propionic acidaemia Pyruvate carboxylase deficiency |
| Biopterin | Phenylalanine hydroxylase deficiency |
| Cobalamin | Methylmalonic aciduria |
| Folate (given as folinic acid) | Dihydropteridine reductase deficiency Uridine monophosphate synthase deficiency Methylene tetrahydrofolate reductase deficiency Methionine synthase deficiency Primary hyperoxaluria type 1 |
| Pyridoxine | Homocystinuria (cystathionine β-synthase deficiency) Ornithine aminotransferase deficiency Pyridoxine-responsive seizures |
| Pyridoxal phosphate | Pyridox(am)ine 5′-phosphate oxidase deficiency |
| Riboflavin | Glutaric aciduria type I Multiple acyl-CoA dehydrogenase deficiency Congenital lactic acidosis |
| Thiamin | Congenital lactic acidosis Maple syrup urine disease Pyruvate dehydrogenase deficiency |
| Tocopherol | Glutathione synthetase deficiency |
| Ubiquinone | Mitochondrial disorders |
Organ transplantation
Purified enzymes for replacement therapy are available for only a very small number of IMDs. A more crude form of enzyme replacement is whole organ transplantation. This most frequently involves the liver, as this is the principal site of many metabolic pathways, for example the urea cycle. It is important to examine whether there are significant extrahepatic manifestations of the disorder for which transplantation is being considered. If there is significant enzyme expression in other tissues, then it may be that liver transplantation, although improving hepatic enzyme function, will not counteract these other harmful effects. This importance of extrahepatic morbidity is demonstrated by methylmalonic acidaemia, where liver transplantation fails to protect the patient from further neurological decompensation. In contrast, the risk of metabolic stroke post liver transplantation in propionic acidaemia appears much lower, even though the enzyme deficiencies affect the same catabolic pathway (see Fig. 24.9). Correcting the defect in the liver abolishes metabolic decompensation, thereby permitting a free diet. Cerebrospinal fluid propionate concentrations do not completely normalize and therefore it has been suggested that relaxing the diet post transplant, but continuing modest protein restriction, may abolish the risk of further neurological sequelae.
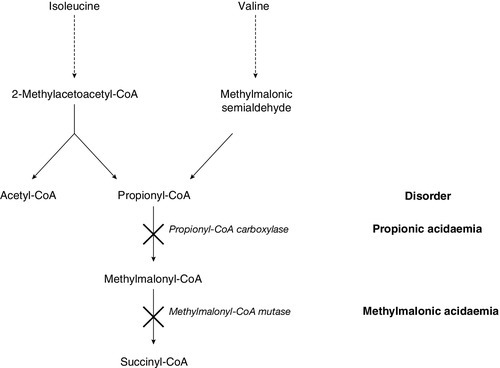
FIGURE 24.9 Isoleucine and valine metabolism, showing the position of enzyme blocks in propionic acidaemia and methylmalonic acidaemia.
Liver transplantation can also be used in urea cycle defects, but the decision as to whether to transplant depends on the severity of the defect, the extent of pre-existing damage from previous hyperammonaemic crises and the future risk of longer term complications of the disease. The survival rate for liver transplantation has improved dramatically and, in children, is quoted as being > 95% at five years. However, after transplantation, patients will require lifelong immunosuppressant therapy to prevent organ rejection, which increases the chance of significant sepsis and the development of lymphoproliferative disease. The use of partial liver transplantation, that is, transplanting a donor lobe without removing the native liver, has the added advantage of correcting the metabolic defect (100% liver activity is not required for normal function), while leaving the native liver (which is otherwise healthy) in situ. A partial liver transplant also has the potential, if gene therapy becomes standard practice, to allow the anti-rejection drugs to be stopped and the transplanted lobe to be rejected.
An alternative to transplantation of a whole liver, or a lobe of a liver, is hepatocyte transplantation. Neonates who present with OTC deficiency have a very poor prognosis. Mortality is extremely high and, even in prospectively treated patients (including those receiving a liver transplant in the first six months of life), neurological damage remains common. Hepatocyte transplantation has been used as a bridge to organ transplantation. Donor hepatocytes are infused into the hepatic circulation to seed in the native liver. Some of these cells engraft and replicate, thus increasing enzyme activity within the liver. This technique has been used to stabilize babies, and to avoid hyperammonaemic crises, before liver transplantation, which is usually carried out at about six months of age. Neurological outcome has been good. It is hoped that further refinement of this technique may allow longer lasting effects that would obviate the requirement for later surgical transplantation. Conditions in which there is liver destruction are likely to benefit the most, as this provides a stimulus for hepatic regeneration to which the transfused hepatocytes respond. Hepatocyte transplantation would be appropriate for several inherited disorders including Crigler–Najjar syndrome, glycogen storage disease type Ia and infantile Refsum disease. Immunosuppression is still required, but, as with auxiliary liver transplantation, there is the potential for withdrawing immunosuppressants when gene therapy becomes feasible, as the native liver remains in situ. A further advantage of hepatocyte transplantation is that livers that might otherwise be considered unsuitable for liver transplantation can be used as a source of healthy hepatocytes, as can redundant liver lobes following the transplant of livers from adults into children.
Bone marrow transplantation has been used in a number of lysosomal disorders, fundamentally changing the natural history of these conditions. Bone marrow transplantation has an advantage over ERT, in that a small number of stem cells do pass beyond the blood–brain barrier and therefore have the ability to improve neurological outcome. Bone marrow transplantation has been attempted in nearly all lysosomal disorders, but has proven ineffective in some, particularly those in which there is severe rapid neurodegeneration, such as San Filippo syndrome (MPS III). Bone marrow transplantation has proven successful in MPS type I, which is the model lysosomal condition for bone marrow transplantation, but it needs to be performed early, ideally within the first 12–18 months of life. However, bone marrow transplantation does not ameliorate all the problems of this condition, including articular disease and spinal problems.
Gene therapy
Correction of single gene disorders by gene therapy (see also Chapter 43), replacing the faulty copy of the gene with a fully working copy, remains the ultimate goal for the treatment of IMDs. The technical challenges have proven considerable; for example the cystic fibrosis gene was first cloned at the end of the 1980s, and nearly 20 years later gene therapy remains in development.
The first condition for which gene therapy was used clinically was adenosine deaminase (ADA) deficiency, one of the causes of severe combined immunodeficiency disease (SCID). This is a combined profound impairment of both humoral and cellular immunity, presenting with multiple life-threatening recurrent infections. Bone marrow transplantation gives a good chance of complete cure; however, if no histocompatible donor is identified, ERT with polyethylene glycol-modified ADA (PEG-ADA) is given every one to two weeks, by intramuscular injection. Gene therapy trials started in the early 1990s, and involved the culture and collection of peripheral blood T cells of patients, insertion of a fully functional ADA gene, by means of a retroviral vector, and reinfusion into the patient. This improved patients’ immune function, but required repeated treatments as lymphocytes have a finite life span of a few months. The technique was further developed using gene transfer into stem cells, which have an unlimited life span. However, the retroviral vector randomly inserted the gene into the cells’ DNA. A total of four children subsequently developed leukaemia owing to the gene being inserted into an oncogene and therefore altering the control of this gene. Subsequent development and refinement of gene transfer technologies have allowed safer and more effective gene transfer and expression with 70% having sufficient improvement in immune function to remain off PEG-ADA. No serious adverse events have been reported with the refined vectors. Gene therapy trials are now under way for a number of IMDs.
Other molecular therapies
Nonsense mutations create a premature stop codon thus producing a truncated, usually non-functioning, protein. It is estimated that nonsense mutations account for 5–15% of disease-causing mutations. It was recognized that aminoglycosides could influence the ability to read through the premature stop codon and so produce a normal length functioning protein. Importantly, this affects only premature, and not normal, stop codons. Subsequent development of small read-through molecules has shown that boosting the affected enzyme by small amounts can significantly reduce disease severity. Clinical trials in cystic fibrosis have been promising and potential trials for patients with IMDs secondary to nonsense mutations are currently being planned.
Exon skipping is another technique to boost functioning protein production, by masking the faulty exon harboring a frameshift mutation, using small pieces of DNA (antisense oligonucleotides). Bypassing the frameshift mutation facilitates reading of the remaining exons. Animal work confirmed the principal in mouse models and now clinical trials have started in Duchenne muscular dystrophy and Huntington disease.
Strategies to reduce the formation of toxic metabolites
Reduction of metabolic load
For IMDs in which the pathological consequences are related to the accumulation of toxic substances, one of the simplest and most broadly used therapeutic strategies is to reduce the load on the block in the pathway, by reducing the amount of substrate to be processed by that enzyme. Phenylketonuria is the classic example: blood phenylalanine concentrations are reduced by limiting phenylalanine, by strict restriction of normal dietary protein intake. Phenylalanine is an essential amino acid and must not be removed from the diet completely so a small amount of natural protein is eaten to supply phenylalanine requirements. Measurement of blood phenylalanine concentrations is performed to monitor dietary control, usually by taking fingerprick samples onto Guthrie cards, which are sent to the laboratory by the family. Phenylalanine free amino acid supplements are prescribed to prevent dietary deficiency from the severe protein restriction. The availability of artificial low-phenylalanine foods on prescription has greatly improved the tolerability of the diet, owing to their variety and greater acceptability to the patient.
Reduction in metabolic load is also used acutely in a number of conditions that present with decompensations. In urea cycle defects, protein intake is stopped during the initial presentation, while other treatment strategies that help reduce the hyperammonaemia take effect. A similar approach is used in organic acidaemias, to reduce organic acid production. Once recovery is apparent, protein is reintroduced and intake is gradually increased. Removal of protein for longer than 72 h risks exacerbating catabolism, including that of muscle protein, with further production of organic acids (or of ammonia in urea cycle defects). It is therefore usual practice to introduce some protein after 48–72 h to promote anabolism. Patients with the severest blocks will still be able to tolerate 0.5 g/kg/24 h of protein for synthetic requirements and repair, without precipitating further decompensation.
Blockage of formation of toxic metabolites
In tyrosinaemia type I, the deficiency of fumarylacetoacetase results in the accumulation of fumarylacetoacetate, which causes hepatorenal toxicity. Nitisinone (NTBC) is a drug used to prevent the production of these toxic metabolites, by blocking the catabolic pathway of tyrosine at an earlier stage (4-hydroxyphenylpyruvate dioxygenase) (Fig. 24.10). This means that the patient has, in effect, been converted from having the severe, tyrosinaemia type I, phenotype to the milder, tyrosinaemia type III, phenotype. Clinically, tyrosinaemia type I presents with acute metabolic decompensation, associated with severe liver compromise and renal impairment and, in the longer term, the development of hepatocellular carcinoma. The clinical picture in type III is much milder, with some neurological involvement reported in some patients: liver and renal disease are not features. In view of the continued block in tyrosine catabolism, albeit at an earlier stage, a low tyrosine diet is still required, but the need for liver transplantation for hepatocellular carcinoma is dramatically reduced. Paradoxically, patients with milder tyrosinaemia type I that presents later, now do less well than those that present as neonates, as more hepatic fibrosis occurs before presentation and the introduction of NTBC.
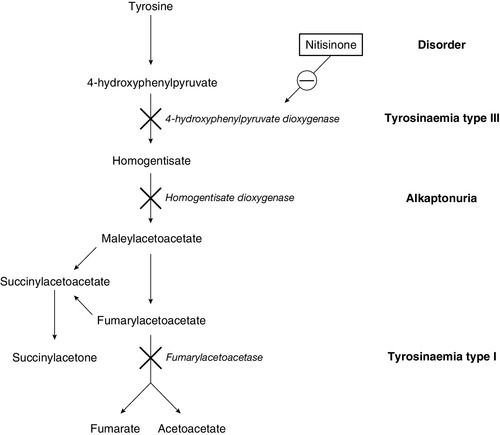
FIGURE 24.10 Blocking the production of toxic metabolites: nitisinone blocks tyrosine catabolism at the level of 4-hydroxyphenylpyruvate dioxygenase, thus preventing the production of the toxic metabolites maleylacetoacetate and fumarylacetoacetate; this modifies the clinical phenotype to that of the milder tyrosinaemia type III from the more severe type I.
Similarly, tyrosinaemia type III has a milder phenotype than alkaptonuria, caused by deficiency of homogentisate dioxygenase, the third enzyme in the pathway (see Fig. 24.10). This recessive condition, one of the conditions on which Garrod based his inherited metabolic traits theory, is manifest clinically by darkening of the urine on standing (usually presenting as staining of nappies). In the longer term, darkening of the sclerae and ear cartilage is observed, with subsequent darkening of the skin, especially over the nose, cheeks and axillae. In alkaptonuria, the joint involvement leads to arthritis, which can be severely debilitating. Low dose NTBC is now being evaluated in the management of this disorder.
Blockage of site of action of toxic metabolites
The effects of toxic metabolites can be limited by blocking their site of action. An example of this strategy is the use of dextromethorphan in the management of non-ketotic hyperglycinaemia, a glycine cleavage defect which results in high plasma concentrations of glycine. Glycine is a neurotransmitter with a central excitatory effect and inhibitory peripheral effect. Excessive concentrations, therefore, result in seizures with hypotonia. Infants classically present with increased fetal movements (seizures in utero), hiccups and progressive apnoeas in the first days after birth, before developing encephalopathy and profound global developmental delay in the longer term. Sodium benzoate is used to reduce glycine levels, but this has little effect on long-term outcome. The use of partial antagonists of the N-methyl-D-aspartate (NMDA) receptor, the site of glycine binding, e.g. dextromethorphan and ketamine, improve seizure control, but do little to improve developmental outcomes.
Strategies to remove toxic substances
It may not be possible to reduce toxin production, and in acute decompensation, rapid clearance of toxic metabolites is required. This can be achieved either with the use of drugs that permit alternate pathways of excretion, or with dialysis or haemofiltration.
Drugs
Carnitine therapy is widely used in the management of IMDs for conjugating toxic metabolites and facilitating their urinary clearance as carnitine conjugates. This therapeutic approach is particularly used in organic acidaemias, e.g. propionic acidaemia and glutaric aciduria type I. Glycine is used in a similar way in isovaleric acidaemia. In urea cycle defects, sodium benzoate and sodium phenylbutyrate are used to conjugate with glycine and glutamine, respectively, which can then be excreted in the urine, bypassing the urea cycle and reducing its metabolic load (Fig. 24.11). However, as conjugation may be immature in neonates, these drugs may prove less effective in the newborn period. In organic acidaemias, this treatment strategy is used to help control hyperammonaemia, which may cause sufficient vomiting to perpetuate the catabolic state driving metabolic decompensation.
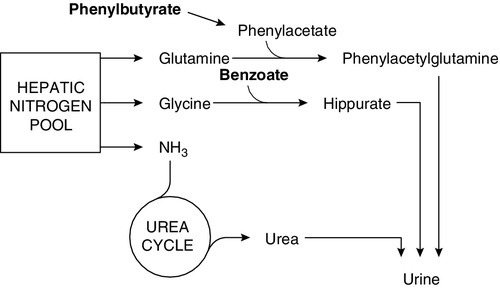
FIGURE 24.11 Alternate pathway therapy in urea cycle defects; phenylbutyrate is metabolized to phenylacetate, which can combine with glutamine, allowing the latter to be excreted as phenylacetylglutamine; benzoate can combine with glycine to form hippurate, which can be excreted in the urine.
Dialysis and haemofiltration
These techniques can be used effectively in acute settings, to clear toxic metabolites from the bloodstream. Originally, exchange transfusion was used for this purpose, but this procedure is inefficient and not without complications. Continuous venovenous haemofiltration, although not as efficient as haemodialysis at clearing toxic metabolites, has less impact on haemodynamic status. It is very effective at clearing lactate, ammonia, leucine and organic acids. Peritoneal dialysis is used in infants < 2.5 kg, owing to the technical difficulties of performing haemodialysis on small babies.
Additional treatments
Appropriate nutritional support is essential to promote anabolism and prevent the catabolic drive to decompensation. To ensure that adequate calories are provided, when patients with IMDs require intravenous infusions, they are given fluids containing higher concentrations of glucose than are used in intravenous fluids administered to most other patients. A minimum of 10% dextrose (with appropriate electrolyte additives) is used in all age groups. Patients with congenital lactic acidosis are an exception as, in this group, the use of high concentrations of dextrose may exacerbate hyperlactataemia; in these patients, 5% dextrose-based solutions are used. Fat emulsions are used to provide additional calories, provided that a fat oxidation defect has been excluded. Patients known to have IMDs, are instructed to use an emergency regimen to help to reduce catabolism, during episodes of intercurrent infection. This comprises a glucose polymer drink that has the advantage of easy absorption without a great osmotic load, thus reducing the risk of diarrhoea. These drinks can be administered at home during intercurrent illnesses, but if they are not tolerated, owing to vomiting or diarrhoea, admission to hospital is required, for intravenous dextrose therapy.
Substrate depletion
Different strategies are needed to manage storage disorders. Cystinosis is an autosomal recessive disorder presenting with a severe renal Fanconi syndrome and growth failure. The biochemical basis is a defective lysosomal membrane cystine transporter, leading to intracellular cystine accumulation. Cysteamine has been used successfully to reduce the progression of renal glomerular damage and improve growth. Cysteamine can combine with cystine to produce cysteine and a mixed disulphide cysteine-cysteamine, which can be exported by the cysteine and the lysine transporters, respectively.
Substrate deprivation
Substrate deprivation therapy is used in the management of lysosomal storage disorders. The principle is to reduce the amount of substrate synthesized so that it matches the impaired rate of catabolism. In this way, substrate does not accumulate. Miglustat is an inhibitor of glucosylceramide synthase, an essential enzyme in glycosphingolipid synthesis. It is used in the management of Gaucher disease type I and Niemann Pick C and is being investigated for the management of late onset Tay–Sachs disease.
CONCLUSION
Although the IMDs are individually rare, collectively they are an important cause of morbidity and mortality, particularly in the newborn, but in some cases in later life. Although the presentation of some IMDs is characteristic, for many it is non-specific, for example with growth failure, acidosis or hypoglycaemia. Nevertheless, the systematic use of investigations, starting with the simple and proceeding to more complex, will usually allow a diagnosis to be established and, when available, treatment to be commenced. Treatment is usually directed at overcoming the effect of the defective enzyme, but strategies for enzyme replacement are being developed and gene therapy may become more widely available in the future.