Academic Emergency Medicine
Edited by George Jelinek
24.1 Research methodology
David M Taylor
Introduction
One important strategy in clinical research is to compare groups of people. These might be different groups or the same group pre- and post-intervention. The methods used are mainly non-experimental, that is observational. They are based on what we can observe and compare in groups of people within populations. By comparing the characteristics (such as behaviours and exposures) and the health experiences of these groups of people, it is possible to identify associations that might be responsible for the cause of a disease.
Initiating the research project
The research question
The research question forms the basis of every research study and is the reason that it is undertaken. It is the scientific, clinical, practical or hypothetical question that, when answered, will allow the researcher to apply newly found knowledge for some useful purpose.
The research question may be generated from many sources, including questions raised by clinical observations, the published medical literature, scientific conferences, seminars and discussions or the effectiveness of currently used or new treatment.
The study hypothesis
A hypothesis is a bold statement of what we think the answer to the research question is. Essentially, it is our best guess of what the underlying reality is. As such, it has a pivotal role in any study. The purpose of a research study is to weigh the evidence for and against the study hypothesis. Accordingly, the hypothesis is directly related to the research question.
In expressing a hypothesis, the researcher needs to be very specific about who or what is to be observed and under what conditions. A failure to define clearly the study groups and the study endpoints often leads to sloppy research.
The study aims
The aims of a study are a description of what the researcher hopes to do in order to weigh up the evidence for and against the study hypothesis.
Just as the research question begs the hypothesis, the hypothesis begs the study aims. The examples above demonstrate clearly the natural progression from research question through to the study aims. This is a simple, yet important, process and time spent defining these components will greatly assist in clarifying the study’s objectives. These concepts are discussed more fully elsewhere [1,2].
Assembling the research team
Most research projects are undertaken as collaborative efforts with the co-investigators each contributing in their area of expertise. Co-investigators should meet the criteria for co-authorship of the publication reporting the study’s findings [3].
Usually, the person who has developed the research question takes the role of principal investigator (team leader) for the project. Among the first tasks is to assemble the research team. Ideally, the principal investigator determines the areas of expertise required for successful completion of the project (e.g. biostatistics) and invites appropriately skilled personnel to join the team [1]. It is advisable to keep the numbers within the team to a minimum. In most cases, three or four people are adequate to provide a range of expertise without the team becoming cumbersome. It is recommended that nursing staff be invited to join the team, if this is appropriate. This may foster research interest among these staff, improve departmental morale and may greatly assist data collection and patient enrolment.
All co-investigators are expected to contribute time and effort to the project, although the extent of this contribution will vary. The temptation to include very senior staff or department heads simply to bolster the profile of the project should be avoided if possible. It is recommended that personality and track record for ‘pulling one’s weight’ be considered when assembling the team. There is little more frustrating than having poor contributors impede the progress of a study. Assigning specific responsibilities, in writing, to each member of the team is a useful tactic in preventing this potential problem. However, care should be taken to ensure that the timelines for assignment completion are reasonable.
The importance of good communication within the research team cannot be overemphasized. This is usually the responsibility of the principal investigator and may involve regular meetings or reports. At the risk of flooding each co-investigator with excessive or trivial communications (e.g. e-mail), selected important communications should be forwarded as they appear, for instance, notification of ethics committee approval and updates on enrolment.
Development of the study protocol
The protocol is the blue print or recipe of a research study. It is a document drawn up prior to commencement of data collection that is a complete description of the study to be undertaken [4]. Every member of the study team should be in possession of an up-to-date copy. Furthermore, an outside researcher should be able to pick up the protocol and successfully undertake the study without additional instruction.
Purpose of the study protocol
Protocol structure
The protocol should be structured largely in the style of a journal article’s Introduction and Methods sections [4]. Hence, the general structure is as follows:
Introduction
Methods
 Study setting and period – a description of where and when the study will take place.
Study setting and period – a description of where and when the study will take place.
 Data-collection instruments, e.g. questionnaires, proformas, equipment.
Data-collection instruments, e.g. questionnaires, proformas, equipment.
 Data-collection procedures including quality-control procedures to ensure integrity of data.
Data-collection procedures including quality-control procedures to ensure integrity of data.
 Ethical issues – subject confidentiality, safety, security and access to data.
Ethical issues – subject confidentiality, safety, security and access to data.

This general plan should be followed in the preparation of any study protocol. However, the final protocol will vary from study to study.
Study design
Study design, in its broadest sense, is the method used to obtain data to weigh up the evidence for and against the study hypothesis. Many factors influence the decision to use a particular study design and each design has advantages and disadvantages. For a more extensive discussion on study design the reader is referred elsewhere [1,5].
Observational studies
In general, research studies examine the relationship between an exposure or risk factor (e.g. smoking, obesity, vaccination) and an outcome of interest (e.g. lung cancer, cardiac disease, protection from infection).
In observational (non-experimental) studies, the principal challenge is to find a naturally occurring experiment, i.e. a comparison of two or more populations that enables the investigator to address a hypothesis about the outcome of interest.
Cross-sectional studies
Cross-sectional studies examine the present association between two variables. For example, within a population you could take a single random sample of all persons, measure some variable of interest (e.g. lung function) and then correlate that variable with the presence or absence of lung cancer. Data are often collected in surveys and the information on exposure and outcome of interest is collected from each subject at one point in time. The main outcome measure obtained from a cross-sectional study is prevalence.
Ecological studies
Ecological studies relate the rate of an outcome of interest to an average level of exposure that is presumed to apply to all persons in the population or group under investigation. So, for example, we could determine the association between the average amount smoked per capita in different countries and the incidence of lung cancer in each country.
Cohort studies
In a cohort study, a group of individuals, in whom the personal exposures to a risk factor have been documented, are followed over time. The rate of disease that subsequently occurs is examined in relation to the individuals’ exposure levels. For example, within a population you could take a sample (cohort) of healthy individuals, document their personal past and ongoing smoking history, and relate that to the subsequent occurrence of lung cancer in that same sample. Although not as powerful a study design as clinical trials (see below), cohort studies are able to provide valuable data relating to the causation of disease.
Case-control studies
Case-control studies involve a comparison between a representative sample of people with an outcome of interest (cases) and another sample of people without the outcome (controls). If an antecedent feature (exposure) is found to be more common in the cases than the controls, this suggests an association between that exposure and the development of the outcome. The frequencies of past exposures to risk factors of interest are compared in each group. Case-control studies provide only medium level evidence of an association between exposure and outcome of interest.
Case reports and case series
This study design is often employed in emergency medicine research. The clinical details (history, management, outcome) of interesting or similar patients are described. This study design provides weak evidence for an association between exposure and outcome of interest and is best employed for hypothesis generation. For example, a series of patients who all developed skin necrosis after being bitten by a certain spider would reasonably lead to the hypothesis that the venom of the spider of interest contained a particular tissue necrosis factor. However, this hypothesis would need to be proven by the isolation of the factor and experimental demonstration of its effects.
Data for case reports/series are often extracted from medical record reviews or existing databases. This is one reason for the weakness of this study design insofar as the data were most likely collected for purposes other than the research study. Accordingly, such data are often of low quality and may suffer from inaccuracies, incompleteness and measurement bias.
Experimental studies
In an experimental study, the researcher is more than a mere observer and actively manipulates the exposure of study subjects to an exposure of interest (risk) and measures the effects (outcomes) of this manipulation.
The preferred form of experimental study is currently the randomized, controlled trial, in which the intervention is randomly assigned at the level of the individual study subject. Although this is the most scientifically rigorous design, other study designs must often be used for a number of reasons including:
For ethical reasons, we cannot easily use experimental studies to study factors that are thought to increase the risk of disease in humans. For example, you could not do a study where you ask half of the group to smoke for 10 years and half of the group to remain non-smokers.
Main types of clinical trials
Key features of clinical trials
Randomization
This is a process by which patients are allocated to one of two or more study groups, purely by chance. Randomization prevents any manipulation by the investigators or treating doctors in the creation of the treatment groups. This prevents a situation whereby a doctor can, for example, allocate the sicker (or not so sick) patients to a new treatment. Randomization also helps to produce study groups comparable to one another with respect to known, as well as unknown, confounding variables (e.g. risk factors). The most convenient methods of randomizing patients are random number tables in statistical textbooks or computerized random-number-generating programs.
A fundamental aspect of randomization is that it must only take place after the commitment to participate has been made (enrolment has taken place). Another important principle is that randomized patients are irrevocably committed to follow up and must not be excluded from, or lost to, follow up, regardless of their subsequent compliance or progress (‘intention to treat analysis’).
Blinding
Blinding is the most effective method of minimizing systematic error (bias) in clinical trials. In single-blinded studies, patients participating in the trial are unaware which treatment they are receiving but the investigators do know. In double-blind studies, neither the subjects nor the investigators know which patient is receiving which treatment. This type of study is usually only feasible with drug studies where it is possible to provide identically appearing medication. This is often achieved using the double dummy approach in which patients receive two medications, one active and the other placebo. The alternative treatment involves a swap-over of the active and placebo medications. Even in apparently blinded studies, there may be various indicators that allow the patient or investigator to determine which treatment they are receiving. In this circumstance, additional methods of bias control may be needed.
Concepts of methodology
Validity and repeatability of the study methods
It is essential that the study uses valid and repeatable methods, that is, measurements that measure what they purport to measure. Ideally, the validity of each of the measurements used in any study should be tested, during the design stage of the study, against another method of measuring the same thing that is known to be valid.
Two types of validity are described:
Response rate
Non-response is a problem for many types of observational study. Often people who participate in a study (responders) have different characteristics from those who do not (non-responders). This can introduce substantial selection bias into the prevalence estimates of a cross-sectional study. In order to minimize this bias, as large a sample as possible is required. To this end, investigators undertaking cross-sectional surveys aim for at least 70% of invited participants actually to respond. Unfortunately, a target response rate of 70% is often not met and low response rates are likely to impact significantly upon bias and validity of the study.
Study variables
A variable is a property or parameter that may vary from patient to patient. The framework for the study hypothesis is the independent variable. This variable is often the factor that is thought to affect the measurable endpoints, or dependent variables, in the study. For example, cigarette smoking causes lung cancer. In this example, cigarette smoking is the independent variable and lung cancer is the dependent variable, as its incidence and nature depends upon cigarette smoking.
Study endpoints
Study endpoints are variables that are impacted upon by the factors under investigation. It is the extent to which the endpoints are affected, as measured statistically, that will allow us to weigh up the evidence for and against the hypothesis. For example, a researcher wishes to examine the effects of a new anti-hypertensive drug. It is known that this drug has minor side effects of impotence and nightmares. A study of this new drug would have a primary endpoint of blood pressure drop and secondary endpoints of the incidence of the known side effects.
Essentially, all forms of investigation involve counting or measuring to quantify the study endpoints. In doing so, there is always the opportunity for error, either in the measurement itself or in the observer who makes the measurement. Such errors (measurement bias) can invalidate the study findings and render the conclusions worthless.
Sampling study subjects
There are several important principles in sampling study subjects:
Sampling frame
This is a list of all members (for instance persons, households, businesses) of the target population that can be used as a basis for selecting a sample. For example, a sampling frame might be the electoral roll, the membership list of a club or a register of schools. It is important to ensure that the sampling frame is complete, that all known deficiencies are identified and that flaws have been considered (omissions, duplications, incorrect entries).
Sampling methodology
Probability sampling
When every member of the population has some known probability of inclusion in the sample, we have probability sampling. There are several varieties:
Simple random sampling: in this type of sampling, every element has an equal chance of being selected and every possible sample has an equal chance of being selected. This technique is simple and easy to apply when small numbers are involved, but requires a complete list of members of the target population.
Systematic sampling: this employs a fixed interval to select members from a sampling frame. For example, every twentieth member can be chosen from the sampling frame. It is often used as an alternative to simple random sampling as it is easier to apply and less likely to make mistakes. Furthermore, the cost is less, its process can be easily checked and it can increase the accuracy and decrease the standard errors of the estimate.
Stratified sampling
A stratified sample is obtained by separating the population into non-overlapping groups or strata (e.g. males and females) and then selecting a single random (or systematic) sample from each stratum. This may be done to:
 obtain separate estimates for each stratum
obtain separate estimates for each stratum
 accommodate different sampling plans in different strata, e.g. over-sampling.
accommodate different sampling plans in different strata, e.g. over-sampling.
However, the strata should be designed so that they collectively include all members of the target population, each member must appear in only one stratum and the definitions or boundaries of the strata should be precise and unambiguous.
Non-probability sampling
Convenience sampling is an example of non-probability sampling. This technique is used when patients are sampled during periods convenient for the investigators. For example, patients presenting to an emergency department after midnight are much less likely to be sampled if research staff are not present. This technique is less preferred than probability sampling, as there is less confidence that a non-probability sample will be representative of the population of interest or can be generalized to it. However, it does have its uses, such as in in-depth interviews for groups difficult to find and for pilot studies.
Data-collection instruments
Surveys
Surveys are one of the most commonly used means of obtaining research data. While seemingly simple in concept, the execution of a well-designed, questionnaire-based survey can be difficult.
Designing a survey
From a practical point of view, the following points are suggested:
Before a survey
During the survey
After the survey
 Cross-check all the data again.
Cross-check all the data again.
 Perform the main data analysis.
Perform the main data analysis.
If possible, incorporate commonly asked questions into your questionnaire. One good source of such questions is standard surveys (such as Australian Bureau of Statistics). There are many other sources of pre-validated questions (for instance measures on quality of life, functional ability and disease-specific symptoms). The scientific literature, accessible through MEDLINE and other databases, is a good start. This is particularly important if you want to compare the sample with other surveys or, in general, if you want to be able to compare the sample’s responses to previously completed work.
Also, previously used questionnaires for similar topics are very helpful and often can be used directly. The advantage to doing this is that these questionnaires’ reliability and validity are established.
The wording of a question can affect its interpretation. Attitude questions with slightly different wordings can elicit differing responses, so several questions on the same topic may be helpful to be certain that the ‘true attitude’ of the respondent is obtained. This technique can enhance internal validity and consistency.
Pre-testing of a questionnaire is most important. Consider the following points:
 Assess face validity of all questions.
Assess face validity of all questions.
 Do different people have similar interpretations of questions?
Do different people have similar interpretations of questions?
 Do closed questions have appropriate possible answers?
Do closed questions have appropriate possible answers?
It is always worth checking with your colleagues to determine whether the questionnaire will answer the study question. Also, test the questionnaire on a cross-section of potential respondents of differing reading levels and background. There can be a few surprises and several revisions may be required before the final questionnaire is determined.
Data-collection proformas
These documents, also called case report forms, are generally used to record individual case data that are later transferred to electronic databases. These data may be obtained from the patient directly (e.g. vital sign measurements) or extracted from the medical records or similar source.
While simple in concept, careful design of a data-collection proforma should be undertaken. First, a list of the data required should be drafted and translated into data fields on the proforma. These fields should be clearly laid out and well separated. Prior to data collection, the proforma should be trialled on a small selection of subjects. In such an exercise, it is commonly found that the data fields are not adequate for the collection of the required data. Hence, revision of the proforma is often required.
Consideration should be given to the ease of data entry and extraction from the proforma. Data entry should progress logically from the top to the bottom of the document without interruption. This is particularly important for data extraction from medical records. Data extracted from the front of the record should be entered at the top of the proforma and so on. Consideration should also be given to later translation of the data to an electronic database. This should follow the same principles as described above. If possible, design a proforma that will allow data to be scanned directly into an electronic database.
Bias and confounding
Study design errors
In any study design, errors may occur. This is particularly so for observational studies. When interpreting findings from an observational study, it is essential to consider how much of the association between the exposure (risk factor) and the outcome may have resulted from errors in the design, conduct or analysis of the study [5]. The following questions should be addressed when considering the association between an exposure and outcome:
Systematic error (bias)
Bias resulting from the way a study is designed or carried out can result in an incorrect conclusion about the relationship between an exposure (risk factor) and an outcome (such as a disease) of interest [5]. Small degrees of systematic error may result in high degrees of inaccuracy. Many types of bias can be identified:
Confounding
This is not the same as bias. A confounding factor can be described as one that is associated with the exposure under study and independently affects the risk of developing the outcome [5]. Thus, it may offer an alternative explanation for an association that is found and, as such, must be taken into account when collecting and analysing the study results.
Confounding may be a very important problem in all study designs. Confounding factors themselves affect the risk of disease and, if they are unequally distributed between the groups of people being compared, a wrong conclusion about an association between a risk factor and a disease may be made. A lot of the effort put into designing non-experimental studies is in addressing potential bias and confounding. For example, in an often-cited case-control study on the relationship between coffee drinking and pancreatic cancer, the association between exposure and disease was found to be confounded by smoking. Smoking is a risk factor for pancreatic cancer; it is also known that coffee drinkers are more likely to smoke than non-coffee drinkers. These two points create a situation in which the proportion of smokers will be higher in those who drink coffee than in those who do not. The uneven distribution of smokers then creates the impression that coffee drinking is associated with an increased rate of pancreatic cancer when it is smoking (related to those who drink coffee and to pancreatic cancer) that underlies the apparent association.
Common confounders
Common confounders that need to be considered in almost every study include age, gender, ethnicity and socioeconomic status. Age is associated with increased rates of many diseases. If the age distribution in the exposure groups differs (such as where the exposed group is older than the non-exposed group) then the exposed group will appear to be at increased risk for the disease. However, this relationship would be confounded by age. Age would be the factor that underlies the apparent, observed, association between the exposure and disease. Although age is a common confounder, it is the biological and perhaps social changes that occur with age that may be the true causes that increase the rate of disease.
There are several ways to control for the effect of confounding. To control for confounding during the design of the study, there are several possible alternatives:
Principles of clinical research statistics
Sample size
The sample must be sufficiently large to give adequate precision in the prevalence estimates obtained by the study for the purposes required. The most common mistake made by inexperienced researchers is to underestimate the sample size required. As a result, the sample size may be too small and not representative of the population that the sample is meant to represent. This usually leads to outcome measures that have very wide 95% confidence intervals and, hence, statistically significant differences between study groups may not be found.
To ensure that a study has adequate sample sizes to show statistically significant differences, if they are there, sample sizes should be calculated prior to the study commencement. In reality, sample size is often determined by logistic and financial considerations, that is to say a trade-off between sample size and costs.
Study power
The power of a study is the chance of correctly identifying, as statistically significant, an effect that truly exists. If we increase the sample size, we increase the power. As a general rule, the closer the power of a study is to 1.0, the better. This means that the type II error will be small, that is there will be only a small chance of not finding a statistical difference when there really is one. Usually, a power of 0.8 or more is sufficient.
Statistical versus clinical significance
To determine statistical significance, we can obtain a P value, relative risk or some other statistical parameter that is indicative of a difference between study groups. However, a statistical difference (e.g. P<0.05) between groups may be found if the study is highly powered (many subjects), even though the absolute difference between the groups is very small and not a clinically significant (meaningful) difference.
This difference is important for two reasons. First, it forms the basis of sample size calculations. These calculations include consideration of what is thought to be a clinically significant difference between study groups. The resulting sample sizes adequately power the study to demonstrate a statistically and clinically significant difference between the study groups, if one exists. Second, when reviewing a research report, the absolute differences between the study groups should be compared. Whether or not these differences are statistically significant is of little importance if the difference is not clinically relevant. For example, a study might find an absolute difference in blood pressure between two groups of 3 mmHg. This difference may be statistically significant, but too small to be clinically relevant.
Databases and principles of data management
The fundamental objective of any research project is to collect information (data) to analyse statistically and, eventually, produce a result. Data can come in many forms (laboratory results, personal details) and are the raw material from which information is generated. Therefore, how data are managed is an essential part of any research project [4].
Defining data to be collected
Many a study has foundered because the wrong data were collected or important data were not collected. Generally, data fall into the following groups:
 identification data: personal information needed to link to an individual patient
identification data: personal information needed to link to an individual patient
 administrative data: initials of the data collector, the study centre if a multicentred trial.
administrative data: initials of the data collector, the study centre if a multicentred trial.
Collect only the research data that are essential to answer the study question. Collection of data that will not be of use is time-consuming, expensive and may detract from the quality of the remaining data. However, there will usually be a minimum of data that must be collected. If these data are not collected, then the remaining data may not be analysed adequately. This relates particularly to data on confounding factors.
Database design
A database is a specific collection of data that is organized in a structured fashion. In other words, database software provides us with a way of organizing the data we collect from a research project in a systematic way.
Data entry
This refers to the entry of data into the electronic database, e.g. Access, Excel. Even if the study design and the data collection have been well done, the final data set may contain inaccurate data if the data-entry process is inadequate. This relates particularly to manually entered data where mistakes are bound to happen.
Data entry can be achieved in many ways:
Data validation
Effectively, this is a quality assurance process that confirms the accuracy of the data and can be done in the following ways:
Research ethics
Participation in a clinical trial may involve a sacrifice by the participant of some of the privileges of normal medical care for the benefit of other individuals with the same illness. The privileges forgone might include:
Participation may also require the discomfort and inconvenience associated with additional investigations and the potential incursion on privacy. Without the willingness of some individuals to make these sacrifices, progress in clinical medicine would be greatly impaired. Most individuals who now expect to receive safe and effective medical care are benefiting by the sacrifices previously made by other individuals.
Some have argued, in contrast, that enrolment into clinical trials ensures the absolute best care currently available, with greater involvement and scrutiny by attending healthcare teams.
If one accepts that clinical trials are morally appropriate, then the ethical challenge is to ensure a proper balance between the degree of individual sacrifice and the extent of the community benefit. However, it is a widely accepted community standard that no individual should be asked to undergo any significant degree of risk regardless of the community benefit involved, that is, the balance of risks and benefits must be firmly biased towards an individual participant.
Because of the trade-offs required and because of the spectrum of views about the degree of personal sacrifice that might be justified by a given community benefit, it is accepted that all clinical trials should be reviewed by an ethics committee that should have as a minimum:
Scientific value
It is unethical to request individuals to undergo the risks and inconvenience of a study that is unlikely to provide a scientifically worthwhile result. It is also unethical to request sacrifices from volunteers that are out of keeping with the value of the research being undertaken. In keeping with this principle, studies that suffer from substantial design errors or are susceptible to serious bias should not be approved until these deficiencies are remedied.
It is unethical to allow scientifically invalid studies to proceed. Sample-size calculations should be scrutinized because of the ethical undesirability of including too few subjects to provide an answer or many more than is needed to provide a convincing answer. Another safeguard to ensure that the research will be valuable is that the investigator should be qualified, experienced and competent, with a good knowledge of the area of study and have adequate resources to ensure its completion.
Benefits forgone
It is unethical to require any patient to forgo proven effective treatment during the course of a trial. It follows that clinical trials should only be undertaken when each of the treatments being compared is equally likely to have the more favourable outcome.
Very commonly, however, there is an expectation before a trial is commenced that one or other treatment is the more beneficial. This may be based on results of uncontrolled studies or even on biochemical or physiological expectations. The large number of times such expectations have been proven wrong can still provide strong justification for a trial.
If such an expectation of benefits is held strongly by an individual, it is probably not ethical for that individual to participate in a study. Furthermore, it is the responsibility of an ethics committee to assess the strength of the presumptive evidence facing one or other treatment and consider whether any substantial imbalance in likely outcome exists. This must be considered in relation to the importance of the question being addressed.
Informed consent
Participants in clinical trials have a fundamental right to be fully informed about the nature of a clinical trial and to be free to choose whether or not to take part. Ethical principles also dictate that prospective participants be:
 provided with a full explanation about the discomforts and inconvenience associated with the study and a description of all risks that may reasonably be considered likely to influence the decision whether or not to participate [4].
provided with a full explanation about the discomforts and inconvenience associated with the study and a description of all risks that may reasonably be considered likely to influence the decision whether or not to participate [4].
It is usual practice to provide prospective participants with a Participant Information and Consent Form that provides a simple, easy to understand account of the purposes, risks and benefits associated with participation in the study. Ethics committees are required to review these statements and confirm that they provide a reasonable account.
In practice, the procedures involved in obtaining informed consent are often problematic. Considering the dependence of sick patients on the health system, their anxiety and their desire to cooperate with their physicians, it is doubtful whether informed consent is ever freely given. When ethics committees identify situations where this scenario is likely to be a particular problem, the involvement of an independent uninvolved person to explain the study may be useful.
Controversies and future directions
24.2 Writing for publication
Anne-Maree Kelly
Introduction
Sharing of knowledge and experience through publication is an important way of improving clinical practice. In addition, researchers have an ethical obligation to publish their findings. Communication may be by way of an original research publication, brief report, case report or letter to the editor. Each of these has different requirements in terms of content, format and length and these requirements may vary between journals. It is useful to choose the intended journal for publication early. While impact factor may be a consideration in this choice, most authors are more concerned with publishing in a journal that has the appropriate target audience for the subject matter of the paper. It is important to check the Instructions for Authors for the chosen journal to ensure that your submission matches that journal’s requirements. Failure to do so reduces the chances of acceptance considerably.
Although journals may have differences in format and style, all prefer clear and concise communications. In particular, it is important for the material to be arranged logically so that clear relationships can be seen between the objective of the study or communication, the evidence and any conclusions drawn.
Important principles
Authorship, acknowledgement and competing interests
Authorship can be a contentious issue, however, there are defined requirements for qualification as an author. The International Committee of Medical Journal Editors state that authorship credit should be based on:

All three conditions must be met. Contributions that do not meet these criteria can be recognized in an Acknowledgement. To avoid misunderstandings, authorship should be decided as early as possible in the research process with the outcome clearly documented.
Most journals also require authors to disclose competing interests relevant to this paper. This is to assist readers in deciding if those interests have a potential bearing on the conduct or reporting of the research. Competing interests may be financial (e.g. external research funding, support from a company for activities potentially related to the project) or personal (authorship of guidelines, editorship of the journal of submission).
Duplicate publication
Duplicate submission and duplicate publication are unacceptable. An exception is secondary publication of material with the express approval of the editors of the relevant journals. This only occurs under special circumstances.
Sometimes studies generate large amounts of data that are difficult or unsuitable for reporting as a single paper. Authors must strike a balance between including data about a large number of secondary outcomes in a single paper (potentially causing confusion about the research question and distracting from main messages) and splitting data into a large number of small papers. In principle, separation of data into meaningful groups for separate papers is acceptable but should be acknowledged in each paper.
Readability
To get your message across, it must be accessible to the reader. This is best achieved by avoiding long sentences, using simple words and avoiding jargon. Abbreviations should be kept to a minimum and always defined in the paper at their first use.
Manuscript preparation
Original research manuscripts
Original research manuscripts aim to answer a relevant research question. Defining that question, indicating why it is important, what methods were used to answer it, key findings and their interpretation is the purpose of this type of paper. An original research manuscript is usually divided into five sections: Abstract, Introduction (or Background), Methods, Results and Discussion. In addition, some journals prefer a separate concise Conclusion, although many prefer this as the last paragraph of the Discussion. Uncommonly, journals have additional section headings, such as Theoretical Concept and Limitations. It is very important to check the journal’s preferred format for each section and ensure the manuscript complies. Most manuscripts also require a key word list of up to five words or phrases to assist with indexing.
Abstract
This is a very important part of the paper as it is the part that will appear in on-line indexing services. The usual word limit is 250 words under the headings ‘Objectives’, ‘Methods’, ‘Results’ and ‘Conclusion’. There is considerable variation between journals about how the abstract section is set out. Some prefer a single paragraph without subheadings but many of the major journals require structured abstracts with particular subheadings. These are detailed under Instructions for Authors.
The abstract should contain all key data. In particular, the specific aims, methods, outcomes of interest, main results (with numbers) and conclusions should be clear enough to be understood without the support of the text. It is important that no data or conclusions appear in the abstract that have not been presented in the main body of the paper.
Introduction
Shorter introductions are often more effective. The aim is to convince the reader why the area of study is important, what this study adds to the body of knowledge and the specific aims of the study. A lengthy review of the literature should be avoided unless it is imperative to put the study in context. A concise review of the literature is more appropriately reported in the Discussion. The last paragraph should explicitly state the aims of the study.
Methods
The Methods sections should address a number of headings. Some journals like this done explicitly, while others are happy for it to be rolled into logical paragraphs.
 Study design: what type of study is it?
Study design: what type of study is it?
 Setting: where was it conducted? What are the special features of this setting?
Setting: where was it conducted? What are the special features of this setting?

 Data collected: this should describe all the data collected.
Data collected: this should describe all the data collected.
 Outcomes measures: what was the primary outcome of interest? Were secondary outcomes also collected?
Outcomes measures: what was the primary outcome of interest? Were secondary outcomes also collected?



 Clinical trials registry registration: many journals now require that prospective clinical studies are registered in a public trials register (e.g. www.anzctr.org.au, www.cct.cuhk.edu.hk, clinicaltrials.gov). The trial’s registration number should be included in Methods.
Clinical trials registry registration: many journals now require that prospective clinical studies are registered in a public trials register (e.g. www.anzctr.org.au, www.cct.cuhk.edu.hk, clinicaltrials.gov). The trial’s registration number should be included in Methods.
Results
It is important for the results to be presented logically and for the relationships with the objectives and methods to be obvious. A useful structure is to start by describing the study population. This should include how it was derived (a summary figure such as a Consolidated Standards of Reporting Trials (CONSORT) diagram may be very effective for this) and its features such as gender, age and so on.
This should be followed by descriptions of the results with respect to stated outcomes of interest: primary outcomes first then secondary outcomes. These should align with the stated objectives. Any subgroup or other analysis should follow this. All results should give the appropriate statistics with confidence intervals (if appropriate) and the type of test used. A significant proportion of journals are moving away from P values as a way of expressing statistical significance, instead preferring effect size with confidence intervals (or similar). Avoid any comments on what the results might mean or why they might have occurred. Interpretation of the results belongs in the Discussion section.
Tables and figures can be very effective ways of communicating results. They should not repeat what can be described adequately in the text. All tables and figures should be self-explanatory, with clear descriptive headings. Tables should be constructed so that the main comparisons of interest are horizontal and left-to-right, with number of subjects clearly shown for each column. Graphs or figures should be used to convey patterns and details that cannot be succinctly conveyed in tables or text. Figures that show the distribution of data (scatterplots, box plots, etc.) are more effective than those simply summarizing data (bar graphs, pie charts, etc.). Axes must be clearly labelled. Tables and figures should be kept to the minimum number needed to convey the information, and should be numbered in the convention of the journal.
Discussion
The Discussion should be concise and to the point and can be challenging. This structure may assist:
 Summarize the principal findings.
Summarize the principal findings.
 Comment on how it compares with other research. Where does it agree? Disagree?
Comment on how it compares with other research. Where does it agree? Disagree?

 Discuss any other results that are worthy of comment.
Discuss any other results that are worthy of comment.
 Describe any unanswered questions or directions for future research.
Describe any unanswered questions or directions for future research.


All statements throughout the Introduction, Methods and Discussion that make an assertion or refer to other evidence or methods must be referenced. Ensure that referencing is in the journal’s preferred style. Selective referencing should be avoided, that is choosing references that agree with the study findings, or worse, citing mostly the authors’ own work. Journal referees are likely to know the relevant literature and detect any selection bias.
In general, as long as the key elements are included, shorter is better than longer in manuscripts. If in doubt, shorten the Introduction and Discussion rather than Methods or Results. As Stephen Lock, former Editor of the British Medical Journal states: ‘A good paper has a definite structure, makes its point, and then shuts up’.
An alternative to the full original research manuscript is the short report. This form has a word limit of 1000–1500 words and usually has some minor formatting differences. It is, however, indexed the same as a full original research manuscript and for many studies is a good choice of format.
Case reports
Fewer journals are accepting case reports. Those accepted for publication tend to have an exceptional element or important clinical message, either in terms of an unusual diagnosis, an innovative use of tests or treatments or an unusual adverse event. It is not enough for a case to simply be ‘interesting’.
The usual structure for a case report is an abstract of about 100–150 words summarizing the case and the clinical messages, the case report itself and a discussion. The case should be described in sufficient detail for the reader to be confident of the evidence. The Discussion is the key element of a case report. It usually includes a review of the literature and uses the case to draw out important clinical messages. It needs to be logical in idea development and well referenced.
Systematic reviews and meta-analyses
A systematic review is a summary of primary studies. It aims to answer a clinical or research question by using strategies that limit bias, critically appraise and synthesize all relevant studies on a selected topic. A meta-analysis is a mathematical synthesis of the results of primary studies addressing the same question in a similar way.
A detailed description of the methods for systematic reviews and meta-analyses is beyond the scope of this chapter. That said, they share several aspects with original research including definition of a research question, development of a research protocol, literature search/review, data analysis and interpretation.
The PRISMA (Preferred Reporting Items for Systematic Reviews and Meta-Analyses; www.prisma-statement.org) checklist is an evidence-based minimum set of items aimed at assisting authors improve the quality of systematic reviews and meta-analyses. Many journals require the submission of a PRISMA checklist when review articles are submitted.
Letter to the editor
These are short communications, usually 500–600 words in length. Most often they comment on a recently published paper in the journal concerned, but they may also be used to report a case or case series, an observation or an opinion.
Manuscript submission
Most journals require online submission of manuscripts. This is good for authors as it significantly reduces turn-around time. The sites vary in the way they want material entered, so it is important to check this.
A manuscript must only be under consideration by one journal at a time. Authors will be required to attest to this and to the fact that appropriate ethics approvals were obtained.
The cover letter
Whether manuscripts are submitted electronically or in hard copy, a cover letter is usually required. This is often quite short and includes a request for consideration for publication in the journal concerned, a statement that the paper has not been published and is not under consideration by another journal, a statement regarding ethics approvals and a note of the presence or absence of author conflicts of interest.
The cover letter is also an opportunity to alert the editor to other issues that may be important. For example, if the manuscript overlaps with previously published work or another manuscript such that there might be a possibility of duplicate publication, it allows the editors to assess any overlap for themselves. Alternatively, it also provides the opportunity to identify potential reviewers that the authors believe should be avoided, usually because of actual or perceived conflicts of interest.
Feedback from journals
It is quite rare for manuscripts to be accepted ‘as is’. Usually, some revision is required and, in some cases, manuscripts are rejected. Neither of these necessarily implies that the study or material is not worth publication. It may simply be that the editors consider that their journal is not appropriate for the subject matter of the paper. Seriously consider the comments given, which are often detailed, and decide whether the issues can be addressed. If so, it is important to undertake a revision and re-submit as soon as practical. If the journal requested revisions and the concerns can be addressed, re-submit to that journal, otherwise submit to another journal after notifying the initial journal that the paper will not be re-submitted to them. Reformatting as a short report or letter to the editor are sometimes alternative ways to get your findings published.
Post-acceptance issues
There are usually several actions required post-acceptance. These include completion of assignment of copyright forms and checking the proofs of the paper. The publisher will usually manage these processes.
24.3 Principles of medical education
Debbie Paltridge
Introduction
George Bernard Shaw famously quipped, ‘He who can, does. He who cannot, teaches’. However, the emergency physician can rarely teach without doing. The tradition for doctors to teach their colleagues and students goes back to the Hippocratic Oath, where the duties of a doctor to students are outlined: ‘… to teach them this art, if they want to learn it, without fee or indenture’ [1].
Emergency physicians have been taking an increasing role in teaching and education, in part because of the need for all doctors to learn and refresh emergency skills, but also because emergency physicians are usually full time and hospital-based and have access to students, patients and teaching resources. In addition, they have a unique opportunity of seeing students progress in their chosen specialty and may have multiple inputs vertically over several years in a younger doctor’s career. This can be very satisfying and also very motivating.
The emergency environment is one of constant new learning experiences while, at the same time, being the location for patient care and critical decision making. Barriers to teaching in hospitals in general, but applicable to emergency departments (ED), have been summarized by Lake in her ‘Teaching on the Run’ series as lack of time, lack of knowledge, lack of training in teaching, criticism of teaching when given and lack of rewards, either materially or by recognition [1].
In addition, teaching in the pressure cooker environment of an ED gives further layers of difficulty, both logistically and ethically. Challenges include:
 shifts, requiring teaching at all hours of the day and night
shifts, requiring teaching at all hours of the day and night
 junior medical staff from a variety of specialties and backgrounds with varying needs
junior medical staff from a variety of specialties and backgrounds with varying needs
 numbers of junior medical staff and rostering affecting continuity for teacher and learner
numbers of junior medical staff and rostering affecting continuity for teacher and learner
 huge variation in workloads from shift to shift
huge variation in workloads from shift to shift
 administration pressures to reduce waiting times
administration pressures to reduce waiting times
 physical restraints in many ED environments caused by overcrowding [2].
physical restraints in many ED environments caused by overcrowding [2].
The ED is a teaching environment, not only for physicians at various levels, but also for nurses, allied health workers, paramedics and others. A significant component of ED teaching is procedural. It is suggested that most patients believe they should be informed if it is the first time a doctor is performing a procedure on them, but less than half of patients feel comfortable about themselves being the first patient ever for suturing (49%), intubation (29%) or lumbar puncture (15%) for a resident [3]. For non-procedural medicine the evidence is that most patients enjoy being part of the teaching process, in outpatient and ambulatory settings at least, and that no extra negative effects on patients occur from teaching [4,5]
An added component of complexity in teaching in the ED is the potential for slowing patient processing by having to stop and supervise a junior. It is often so much quicker just to do it yourself. Supervising a lumbar puncture, for example, may take both the teacher and the taught away from seeing new patients for half an hour. However, as far as it has been researched, teaching in academic EDs does not appear to slow down patient care but in fact improves quality of care [6]. Doctors who are seen by their juniors as good teachers are just as likely to see as many patients per shift as those who are not [7].
ED crowding can be seen to have positive and negative effects on emergency teaching. On the one hand, if crowding is due to patients staying for longer periods of time, it may provide increased patient contact and teaching opportunities over that time. On the other hand, the emergency doctors may have less time for teaching if the crowding is due to increased throughput and production pressure is high [8].
All emergency physicians are teachers at some stage in their career at various levels and, as in Hippocrates’ time, are mostly unpaid for it. Although most doctors become teachers, the majority of prevocational doctors in Australia have had no exposure to learning how to teach [9]. Here we present the principles of teaching and learning to assist emergency physicians, whether they are involved with medical students, residents, registrars or other health professionals.
Adult learning principles
Contemporary medical education needs to be couched in terms of contemporary education theory. Adult learning principles should underpin educational practice from the bedside, through the clinical skills laboratory to the seminar room. In addition, these principles are relevant to the education of the undergraduate, prevocational (first 2 years’ postgraduate) and vocational registrar years, as well as the continuing professional development of the mature medical practitioner.
Malcolm Knowles first introduced the notion of andragogy or adult learning in the early 1970s [10]. He described five assumptions regarding how adults learn:


Knowles and other authors have since developed principles of adult learning that can be used to guide education activities [11–15]:







However, these principles of adult learning are irrelevant to the emergency physician educator unless they are actively applied to the education of their postgraduate charges. The question remains of how these principles are put into practice. Table 24.3.1 outlines some examples of how these principles may be incorporated into education within the ED.
Table 24.3.1
Application of adult learning principles and assumptions in the ED environment
| Adult learners | Application to ED teaching |
| Have prior learning and experience | Even the most junior doctors (e.g. interns) bring experiences with them to the ED. They may have specific experience relevant to the condition that they are treating (e.g. they saw similar patients in their undergraduate course) or it may be life experience (e.g. they had relatives with that experience). Open questioning techniques (requiring a more detailed answer from the learner as opposed to a closed question requiring a yes/no answer) can be used to promote reflection on past experiences and practices. A case study with short answer questions to facilitate this reflection could be used in a small group tutorial situation. Small group discussions can also provide opportunities for learners to draw on their own experiences and to learn from each other as well as the facilitator. |
| Are self-directed learners | At the commencement of a rotation in the ED, junior staff should be asked as part of their orientation what it is they specifically want to get out of this rotation. This allows the identification of personal learning goals. This is relevant to new senior staff as well. Orientation is also very important for establishing expectations of both learner and facilitator and ground rules for how education will be carried out within the ED rotation. Learners should also be offered a choice of learning activities. This will allow learners to choose activities which will address their individual learning objectives and which will address their specific learning requirements and styles. For example, one intern may want to watch a lumbar puncture before performing one under supervision, another may want to practise a lumbar puncture on a manikin first before performing one. |
| Learn most effectively when they perceive a need for learning | The ED educator needs to help learners recognize the relevance of a learning experience. This will significantly impact on their motivation to learn. Sharing of experiences, e.g. a case example from real life, can help to establish relevancy for a learner. Additional methods may include documentation of ED presentations, participation in unit audit meetings or presentations of cases. |
| Prefer problem-centred approaches | ED presentations require sophisticated problem-solving techniques. The undifferentiated patient is the norm. Modelling of clinical reasoning from experienced practitioners can assist the novice to understand problem-solving approaches. Evidence suggests that the experienced practitioner does this subconsciously, however, verbalization is necessary to promote collaborative problem solving by the less experienced. Unit case-based discussions also encourage shared problem solving. |
| Practise self-evaluation | Adults require an opportunity for ‘reflection-on-action’1 or self-evaluation. Self-evaluation opportunities can be incorporated formally by: |
| Require feedback | Opportunities for feedback on performance should be incorporated into the ED term both formally (as part of a requirement of training, e.g. mid- and end of term feedback) and informally from supervisors or peers. Written and verbal feedback can be used. |
| Value experiential (‘hands on’) learning opportunities | There are numerous opportunities for hands on experience within the ED. Educators need to involve learners in case-based discussions and problem-solving activities. However, procedural skills may need to be practised away from patients until competence is determined. Then practice under supervision will be appropriate. |




Learner-centred education
Many traditional medical education experiences are teacher centred. The teacher is the expert and determines what, how, when and where much is learnt. The teacher is the active participant and the learner is the passive recipient [16]. However, a more effective approach to education is the learner-centred approach. Learner-centred education refers to educational events that place the learner in the pivotal position, responsible for determining learning objectives, actively engaging in learning opportunities and participating in evaluation [17]. This is more in line with adult learning principles.
So how does the ED physician become a learner-centred educationalist? The following suggestions are provided to assist:





What makes a good ED teacher?
The challenges facing the emergency physician educator, including environmental constraints, patient characteristics, administrative and production imperatives and resource availability, cannot be overstated. However, despite this, there is a consistent commitment to education by emergency physicians. What then makes a good ED teacher? Bandiera and colleagues used a qualitative research design to investigate experienced ED teachers and establish the behaviours that made them good teachers [2].
Twelve strategies were identified:



 Demonstrate a good teacher attitude – this is about being an approachable supervisor/teacher. The teacher’s affect influences the learner’s willingness to engage in the learning activity, e.g. the teacher’s positive approach to the learner, enthusiasm for teaching and openness to questioning [18].
Demonstrate a good teacher attitude – this is about being an approachable supervisor/teacher. The teacher’s affect influences the learner’s willingness to engage in the learning activity, e.g. the teacher’s positive approach to the learner, enthusiasm for teaching and openness to questioning [18].

 Provide and encourage feedback – adult learners require feedback for motivation and for learning.
Provide and encourage feedback – adult learners require feedback for motivation and for learning.
These findings are supported further in the literature with what learners want. Additional suggestions include:




The factors reported by ED teachers and ED learners reflect what is required according to adult learning theory and reinforce the applicability within the ED environment.
Types of teaching in the ED
There are a number of teaching and learning strategies available for use within the ED environment. These can include spot electronic searches on active clinical problems, formal quarantined tutorials, case discussions, demonstrations of procedures or techniques, audit meetings, self-directed learning opportunities, such as reading medical literature, online learning programmes and so on. This section deals with three strategies: ‘trolley-side’ teaching, teaching procedural skills which most ED physicians are familiar with and perform regularly, and simulation, which is developing an emergent role within teaching and learning in the ED.
‘Trolley-side’ teaching
Interactions with patients at the bedside are a crucial component for learning in medicine, the traditional apprenticeship model relying on this methodology. Bedside teaching can provide an opportunity for the experienced clinician to explain clinical reasoning and role model appropriate communication, including listening, patient questioning and respect, supervising the more junior clinicians as they practise these skills, assessing the junior clinicians’ interaction with the patient and providing feedback to them [20]. However, with the numerous environmental constraints within the ED there is a need to look at bedside teaching and determine how best to conduct this activity. In addition, the care of the patient remains paramount and ensuring that this is maintained and that the junior doctor–patient relationship is not undermined is an additional challenge.
The benefits of orientation have previously been mentioned in terms of adult learning principles and learner-centred instruction. However, they are crucial to establishing the expectations from both the learner and the teacher’s perspectives in regards to bedside teaching. Establishing up front how bedside teaching will be conducted, while remaining patient-centred, will enable the learner’s needs to be met. Briefing the patients beforehand and getting them involved in the teaching process enhances patient comfort and participation and may provide enjoyment. Expectations of patient-based teaching may include:


 Outline specific teaching approaches. There are a number of models of bedside teaching that can be used. Lake and Ryan [21] describe the use of set, dialogue and closure. This technique involves an introduction, outlining the objectives of the session (set), a discussion in which questioning techniques are used to elicit information from the learner and to discuss reasoning/rationales (dialogue) and a summation in which the main learning points are discussed and further learning required (closure). An alternative to this is the SNAPPS model described by Wolpaw et al. [22] in which the learner:
Outline specific teaching approaches. There are a number of models of bedside teaching that can be used. Lake and Ryan [21] describe the use of set, dialogue and closure. This technique involves an introduction, outlining the objectives of the session (set), a discussion in which questioning techniques are used to elicit information from the learner and to discuss reasoning/rationales (dialogue) and a summation in which the main learning points are discussed and further learning required (closure). An alternative to this is the SNAPPS model described by Wolpaw et al. [22] in which the learner:
 summarizes the case history and their findings
summarizes the case history and their findings
 narrows the differential diagnosis usually to two or three possibilities
narrows the differential diagnosis usually to two or three possibilities
 analyses the different diagnoses by comparing and contrasting them
analyses the different diagnoses by comparing and contrasting them
 probes the supervisor/teacher for opinions or any information on which they require clarification
probes the supervisor/teacher for opinions or any information on which they require clarification

This model was piloted and tested within the outpatient setting. However, it has relevance and application for a number of clinical settings. It would require the experienced clinician to explain and possibly model the process in the first instance.
Procedural skill teaching
Management of patients in the ED often involves the practitioner performing procedural skills. Some of these are to assist in formulating diagnoses in the undifferentiated patient, e.g. performance of bedside ultrasound, others are for treatment of patient conditions, e.g. application of a plaster to a fracture. Ideally, in this day and age, procedural skills should be practised in a clinical skills setting prior to implementation on a ‘real’ patient [23]. However, observation by a junior doctor of an experienced clinician performing a task is also valuable and can sometimes be overlooked in the busy ED department. Sometimes it is done before you realize you could have shown a junior doctor.
The educational theories relevant to teaching clinical skills are drawn from psychomotor theories. There are seven basic principles of the psychomotor domain [24], including:

 verbalization – where the learner needs to hear the steps of the skill verbalized
verbalization – where the learner needs to hear the steps of the skill verbalized
 practice – where the learner gets the chance to practise the skill
practice – where the learner gets the chance to practise the skill
 correction and reinforcement – where feedback is given to reinforce performance
correction and reinforcement – where feedback is given to reinforce performance
 skill mastery – where the learner can perform the skill independently in the learning environment
skill mastery – where the learner can perform the skill independently in the learning environment
Similarly, Cagne [25] describes three phases in instructional design relevant to teaching a technical skill, including a cognitive phase where the learner is developing cues from the facilitator, an associative phase where the learner is integrating the component parts and an autonomous phase where the skill has become automatic for the learner.
The issue of the relationship of the learner to the experienced clinician is further investigated within the cognitive apprenticeship model [26,27]. The emphasis in this model is on the requirement that the thinking of the expert be made visible and brought to the surface for the learner. Underpinning this model is the ability of the teacher to assess/recognize the skill level of the learner.
Another debate in the literature is around the issue of whole skill training versus part skill training. Evidence would suggest that the part skill training method be used for the more complex skills while whole skill training be used for the relatively straightforward skills. This requires the facilitator to analyse the skills to be taught and determine the level of complexity of that specific skill. Additionally, what is the whole skill? It can be argued that procedural skills do not occur in isolation. Rather, communication skills are required along with the technical expertise and should be taught together rather than in isolation to reflect the requirement in reality [28,29].
So what do these theories mean for the ED physician wanting to assist a learner in developing a procedural skill? The important requirements are:
 background knowledge is required – why, what, how?
background knowledge is required – why, what, how?

 verbalization of steps – this requires breaking the skill down into steps
verbalization of steps – this requires breaking the skill down into steps
 feedback on performance by the expert
feedback on performance by the expert
 opportunities for repeated practice under supervision, to allow for feedback and self-reflection
opportunities for repeated practice under supervision, to allow for feedback and self-reflection

A dedicated skills area within the ED is most beneficial, as this can be used in quarantined or quieter times for supervised or independent practice (once the learner is deemed relatively competent in the skill, to avoid practising incorrect technique). Highlighting the need for practice and observation to all the experienced clinicians assists in identifying these opportunities for the learner within the ED environment.
Simulation
Simulation is an emergent teaching methodology within the ED environment. In its broadest sense, it refers to any situation in which the real situation is emulated. It may involve actors playing the role of patients, who are often described as standardized patients, or manikins with computer-generated physiological responses [30,31].
The underpinning educational theory behind simulation comes from a number of theories, including adult learning. However, experiential learning theory is probably of most relevance. Kolb [32] describes experiential learning activities as opportunities for learners to acquire and apply knowledge, skills and attitudes in an immediate and relevant setting. A four-point continuous learning cycle is described:


Simulation in healthcare education is clearly an example of experiential learning. It provides the learners with a relevant and realistic patient problem to manage. Following this experience, the learners are able to observe their performance and reflect, while exploring with a facilitator hypotheses and new concepts. They can then test this experience by repeat simulations.
There are a number of ways in which ED physicians can incorporate simulation opportunities into their teaching in ED. It may be that paper-based simulations are used to explore clinical reasoning. This involves developing case scenarios and structured questions. Role-playing, using peers or expert clinicians, can be used to practise difficult communication skills, such as breaking bad news. Simple part-task trainers can be incorporated into a more complex scenario involving the practice of the skill while interacting with a patient. Kneebone and colleagues [29] describe integrating a urinary catheter manikin with an actor to ensure that the technical and communication skills are taught concurrently.
Where higher level manikins are available, whole patient scenarios can be conducted. Teams of junior medical staff can practise rarer critical situations and explore not only technical skills but non-technical skills, such as teamwork. Use of audio-visual aids to capture performance is important for providing feedback after such activities. It also allows an opportunity for reflection and peer feedback.
Not all EDs have the luxury of the highly technical simulation ‘gadgetry’, but this should not put them off using simulation as a teaching methodology. Determining the content areas appropriate for using simulation and how this fits into the overall curriculum will be important to ensure that resources are used rationally [33].
Feedback to learners
Feedback is a crucial requirement for learning and the importance of positive feedback for learning has been well established [34]. Feedback should provide the learner with information that offers ‘insight into what he or she did as well as the consequences of his or her actions’ [35]. It should allow the learner to know what went well and what could be improved or changed next time. Feedback is part of the formative assessment process that occurs throughout the learning period, rather than as a summative assessment that is to determine a grade or make a final judgment.
Effective feedback has a number of characteristics [36,37]. It should be given in a suitable environment to allow privacy and maintain confidentiality for the learner. There should be adequate time to allow the learner and the facilitator to explore the observed behaviour or skill. There should be clear goals established at the beginning of the learning so that feedback can be related to these goals. The feedback should come from direct observation of the learner’s performance where possible.
Providing learners with feedback is a specific skill in itself and requires practice to develop. A structured approach to giving feedback will assist the teacher/facilitator to provide effective and useful feedback. There are many models of feedback in the literature; however, one such model from Pendleton’s [38] work is suggested here to assist the ED physician:
 Ask the learner how he or she felt.
Ask the learner how he or she felt.
 Ask the learner what went well and why.
Ask the learner what went well and why.
 Ask the facilitator/teacher to say what went well and why.
Ask the facilitator/teacher to say what went well and why.
 Ask the learner what could have been done better and why.
Ask the learner what could have been done better and why.
 Ask the facilitator/teacher to say what could have been done better and why.
Ask the facilitator/teacher to say what could have been done better and why.
 Ask the facilitator/teacher to summarize the strengths and up to three things to concentrate on.
Ask the facilitator/teacher to summarize the strengths and up to three things to concentrate on.
Feedback, when delivered effectively, is a strong motivator to the adult learner and encourages ongoing performance review and reflection by the learner.
Conclusion
Juggling the demands of a being a busy emergency physician requires balancing clinical, administrative and teaching duties. Time is often limited and yet the rewards of being involved in teaching are obvious. Apart from personal satisfaction gained from interacting with junior colleagues, the ability to keep up to date and the opportunity to reflect on one’s own performance are enhanced. Reviewing performance as a teacher and practising education techniques to improve the effectiveness of the facilitation motivate the teacher to continue to teach and improve the satisfaction from teaching. Structuring learning experiences, considering learner needs and providing effective feedback are essential for learners in the ED environment.
Likely developments over the next 5–10 years






24.4 Undergraduate teaching in emergency medicine
GeoffreyA Couser
Introduction
Emergency medicine now plays a central role in medical curricula in undergraduate and postgraduate medical schools in Australia and New Zealand. This is not unexpected, as the principles and practice of the specialty have much in common with the desired features of contemporary medical education: it is problem focused, interdisciplinary and integrates many aspects of community-based and hospital-based clinical practice. Clinical practice easily integrates and builds upon the biomedical sciences traditionally taught early in the medical course. Much growth in academic emergency medicine has occurred in the last two decades, with the establishment of academic departments and dedicated university positions. However, the majority of medical student teaching is performed by emergency physicians in clinical practice in public and an increasing number of private emergency departments (EDs). This chapter will address the issues surrounding medical student teaching at the departmental and the broader faculty level.
Overview of undergraduate medical education in Australia
It is important for emergency physicians interested in teaching medical students to be aware of recent trends and developments in medical education in Australasia. The CanMEDS 2000 Project and the World Health Organization, among others, have listed the key outcomes expected of a doctor [1]. These outcomes have been adopted by many schools worldwide as a basis for reform and reorganization and are listed in Table 24.4.1. Of interest, these principles have been embraced at the early postgraduate level through the Australian Curriculum Framework for Junior Doctors [2] and at a specialty college level, such as the Australasian College for Emergency Medicine’s Curriculum Revision Project [3]. Some schools have adopted problem-based learning as a tool to achieve these educational outcomes, with others using case-based and outcomes-based learning to place content in a clinical context. In 2010, the US-based Carnegie Foundation (which published the landmark Flexner Report in 1910) published a comprehensive review of current physician education and the need for reform which should be essential reading for any prospective medical educator [4].
Table 24.4.1
Essential roles and key competencies of specialist physicians – the CanMEDS criteria [1]
Medical expert
Communicator
Collaborator
Manager
Health advocate
Scholar
Professional
Copyright © 1996 The Royal College of Physicians and Surgeons of Canada. http://www.royalcollege.ca/portal/page/portal/rc/canmeds. CanMEDS 2000 Project skills for the new millennium: Report of the societal needs working group. Reproduced with permission.
Curriculum reform has occurred in parallel with significant changes in the health system, with a growth in information technology, changing patient expectations and a massive increase in medical knowledge. Models of healthcare delivery are changing, with increased pressures on public hospitals, such as access block, the unsustainable increase in health spending in relation to all other public spending, declining numbers of inpatient beds and the increasing complexity of medical conditions. The shift to the home and community management of many conditions has altered the patient mix available for student teaching. University salaries have not kept pace with the growth in public and private medical salaries, which has contributed to a decline in numbers of academic faculty and core medical school functions often shifting to sometimes reluctant specialists within the public hospital system. Despite this, there has been an increasing number of medical students and medical schools, with 3770 medical students commencing study in Australia in 2011 [5]. It is in this changing environment that emergency medicine has established itself as a key part of modern medical curricula and the specialty is poised to play an even greater role in the training of future doctors. Both the need and the opportunity exist for such expansion.
The importance of medical student teaching
Many clinicians feel that the provision of clinical care is the core business of an emergency department (ED) and hence may not be initially willing to allocate resources for teaching students. Similarly, university medical schools may not be aware of the growth of emergency medicine as a specialty and hence may not be aware of what it can offer students or, indeed, may not even be aware that it is an independent specialty with its own body of knowledge. However, once established, a strong academic presence can contribute to departmental morale, quality of care, performance and the standing of the department within the hospital and community. Table 24.4.2 lists the benefits of emergency medicine teaching to students, EDs and medical schools. These points may be used to argue for an increased presence and accompanying resources within a curriculum [6]. Resources and a formal place in the curriculum often come only after years of hard work in establishing the bona fides of emergency medicine. This may require much time and effort from a dedicated individual.
Table 24.4.2
Benefits of medical student teaching in emergency departments















Curriculum development
Undergraduate emergency medicine in the past was ad hoc, unstructured and highly selective, in that clinical exposure was based around what the students themselves thought was interesting and useful in the department at the time. With the growth of the specialty as an academic discipline, departments have been able to take a more active role in education, control student entry to the department, ensure appropriate orientation and attempt to take advantage of the rich and broad clinical experience on offer. As faculties have become aware of the learning opportunities on offer in EDs, as well as the teaching abilities of staff, emergency physicians have been able to negotiate a greater role in university affairs and integrate emergency medicine into the broader undergraduate curriculum. With this comes a responsibility for emergency physicians to understand the function of universities and the requirements which come with running an academic term. Table 24.4.3 provides suggestions for developing a university teaching presence.
Table 24.4.3
Minimum requirements for developing student placements in emergency departments








It is essential that once a department has decided that medical student teaching should be a part of its function then a curriculum must be considered. The Australasian College for Emergency Medicine, the American College of Emergency Physicians and the International Federation of Emergency Medicine have produced documents with varying degrees of detail concerning this [7–9] and a growing number of papers are being published providing guidance to curriculum developers [10,11]. Most core curriculum statements contain elements which reflect the clinical practice of emergency medicine and an example of such a list is provided in Table 24.4.4. This provides a framework around which specific topics can then be taught. The teaching programme will need to be modified and adapted accordingly depending upon the expertise of and time available to specialists within the department, as well as the local epidemiology and patient demographics.
Table 24.4.4
Suggested core curriculum topics in emergency medicine
Assessment and management of the undifferentiated patient
Key practical skills: basic life support skills and basic procedural skills
Recognition and management of the seriously ill and injured patient
The assessment and management of common clinical problems in emergency medicine
Assessment and management of the unwell child
Acute pain management
Acute mental health
Toxinology and toxicology
Health systems management
Critical thinking and clinical decision making
Safety and quality in healthcare: medical error and handover
Professional issues: teamwork, communication, time management
Care of the elderly and end-of-life issues
An often overlooked but essential component to consider is that of the ‘hidden curriculum’. This is less well understood, but relates to what students learn by being exposed to the practice of medicine. It can cover aspects such as professionalism, ethics and physician behaviour. As the vanguard of a fair, accessible and equitable health system, emergency medicine can teach important attitudes to the next generation of doctors.
Methods of teaching emergency medicine
Once teaching content is decided upon, then it is worth spending time considering which format is the best way to deliver the material. Until recently, undergraduate emergency medicine has largely been taught in the workplace and no other teaching options have existed. However, with the growth of the specialty and the increased need for teachers, the specialty has been able to attract resources and play a greater role in all years of the medical course in some universities. Therefore, depending upon available time and resources, different formats of teaching should be considered for different situations. For example, resuscitation skills are best taught using team-based simulation and practice to reflect the reality of the workplace, once basic concepts have been covered by lectures or online modules.
In all formats, teachers should consider the basic principles of adult learning and teach accordingly. When delivering material, teachers should remember that adults learn best when the topic is meaningful, linked to experience and pitched at the correct level and the students are motivated, have clear goals, are actively involved, receive regular feedback and have time for reflection [12]. Utilizing a range of methods means that material can be delivered in a meaningful way and optimal learning conditions can be achieved. It is essential that the physician taking responsibility for undergraduate teaching within a department takes a leadership role and provides ongoing training and support for both junior and senior colleagues in effective teaching methods.
Whichever teaching method is chosen, evaluation of the process by the participants is an essential part of the quality improvement cycle. Evaluation helps ensure teaching is meeting students’ learning needs, identifies areas where teaching can be improved and provides feedback and encouragement for teachers [13]. Documenting evaluations can form part of a teaching portfolio, which can be used in academic job applications as tangible evidence of a clinician’s and a department’s commitment to and proficiency in teaching. Importantly, from a student’s perspective, being asked to evaluate a teaching session and then seeing the comments acted upon provides a strong sense that their participation is valued and, as a result, may improve the learning process overall.
Some basic pedagogical theory should be considered when choosing and applying methods of teaching: many educators refer to Miller’s triangle of clinical competence [14] and the concept of a spiral curriculum [15] when designing curricula. Figure 24.4.1 is a schematic representation of how emergency medicine as a subject could ideally progress through a 5-year course utilizing these theories of curriculum design. Most departments will only be in a position to offer clinical exposure in the final years of the course, but much progress has been made across the region in penetrating all years. The following delivery methods could be used in this framework to deliver a comprehensive and effective emergency medicine curriculum.
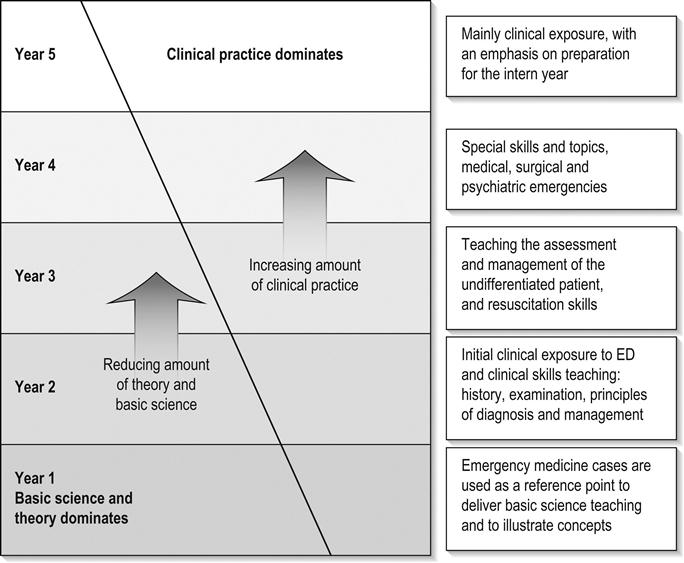
Lecture based
Lectures can be delivered at any stage of the medical course but, to maximize their effectiveness, they need to be developed in an integrated fashion and linked to other components of the course. Emergency medicine can be used effectively as a vehicle to illustrate biomedical science concepts to junior medical students [16]. Case-based learning is a popular method to use in this setting, as learning objectives and concepts can be illustrated in a ‘real-world’ setting. For example, rather than delivering a lecture on ischaemic heart disease, an emergency medicine lecture would be entitled ‘I’ve got pain in my chest’ and the lecturer would engage with students to create a genuine feel for this common emergency presentation. The content would need to be modified depending upon the seniority of students, for example, junior medical students would use the case as a reference point to illustrate anatomy and physiology, while more senior students may use the same scenario to learn about clinical decision making and evidence-based medicine.
Lectures need not be didactic and overloaded with content – a good lecture should be an efficient and entertaining means of effectively transmitting information to large groups. In general, lectures should add value. There is little point simply repeating content from a text, as students can gather the information themselves either in their own time or by using information technology during a boring lecture. In this sense, prospective lecturers should remind themselves of the qualities of an effective educator: expertise in the subject area, enthusiasm for the topic and the task, and capacity to engage the learners [17]. By utilizing these qualities, educators can turn a lecture into a valuable learning experience. Lecturers should have notes pre-prepared and available for students and be prepared for the fact that most will have mobile devices open while the lecture is taking place, will be busy annotating and linking to other resources and may not appear to be giving the lecturer their undivided attention. These are the principles of blended learning, and they will be a key part of education in the twenty-first century. Recording lectures on the university’s learning management system allows for later review and discussion.
Tutorials and small-group learning
Tutorials are small group sessions, with opportunities for interaction and reflection. This model of teaching is well suited to emergency medicine, as individual cases and experiences can be presented and discussed in a comfortable and safe environment. The learners can lead the discussion and take the topics into new and previously unconsidered areas. Nevertheless, providing a structure to the tutorial will help ensure that the time is spent wisely and learning opportunities are maximized. These are usually easily implemented in a department, as they can be integrated with other departmental activities and can run independently of any broader curriculum. This is often the only way that the specialty can deliver its core curriculum in a school without a formal emergency medicine presence. Another pragmatic way to utilize the emergency patient population and promote the department as a place of learning is to work in partnership with other disciplines who can access the department and utilize the patients and the physical space for teaching.
Web-based
This is growing in popularity as schools rely more on information technology to deliver content and this is occurring at the same time that emergency physicans worldwide are taking a leading role in developing resources and promoting the role of social media and other online resources for teaching and clinical care. At a basic level, bulletin boards, web logs (blogs) and e-mail are useful ways to maintain regular communication with students and to distribute journal articles and policies and orientation manuals can be posted online [18].
However, with faster Internet connections and a new generation of students increasingly utilizing mobile learning devices, such as smartphones and tablets, new opportunities are emerging with applications, podcasts, social media and e-books playing increasingly prominent roles in education at all levels. Specific details are provided in the next chapter.
Work-based
This is the original method of medical student education: at the bedside of the patient. Medicine has been taught this way for thousands of years and reinforces the point that medical students are essentially apprentices in a trade. Emergency medicine excels in this area because of the broad range of experiences on offer in a department. Challenges exist, as not all students will be exposed to the same conditions during a rotation. Achieving a uniform experience for students is difficult [19] and so workbooks have been developed to guide students through their rotation, alerting them to the broad range of undifferentiated conditions which regularly present. Utilizing junior staff can be helpful in the education of medical students: pairing a student with a resident or registrar allows the students to see how a doctor works and it involves junior doctors in teaching at an early stage of their careers. It helps with rostering, in that students can be allocated to medical staff with a pre-existing timetable. Medical schools offer clinical academic titles to doctors involved in teaching and all staff should be encouraged to apply for such titles.
Teaching by the bedside is an important activity and opportunities abound for clinical teaching. Clinicians being aware of a broader curriculum can be of assistance, as it can be difficult to think on the spot when confronted with a ‘teachable moment’. Some guidelines exist to maximize the value of bedside teaching and the well-developed principles of ‘set, dialogue and closure’ [20] are explained and listed with an example in Table 24.4.5.
Table 24.4.5
| Concept | Components | Example in practice |
| Set | Roles – trainers, learners, patients Objectives – what are they going to learn? Linkages – to other learning events Environment – seating, lighting, distractions |
Gather the students in a quieter part of the department where distractions will be minimal and make it clear what the session is about: ‘I’d like to talk about ways we assess headaches in the emergency department’ |
| Dialogue | Questions –– use often Understanding Eyes – two-way contact Stimulation – make it interesting Timing – finish on time |
Check what the students know about the topic and ask an open question to start, e.g. ‘What are the worrying signs of a headache?’ Encourage discussion, use first names in the discussion |
| Closure | Review – ask for questions, check understanding Eyes – contact with learner Summary Termination |
Summarize the discussion, check that the students have understood it, terminate with a comment (e.g. ‘Thunderclap headaches are a feature of subarachnoid haemorrhage and later we’ll talk about the role of a lumbar puncture’) |
Simulation
Simulation is a growing field which has been embraced by emergency physicians and the Australasian College for Emergency Medicine. A large knowledge base has been developed and, as such, it will not be discussed at length here. However, the main barrier to utilizing simulation for undergraduate teaching is cost and the available time of emergency physicians. Fortunately, universities are starting to recognize the value of this method of teaching and opportunities are being created, with simulation utilizing people factors and the principles of interprofessional learning being integrated into curricula at all levels.
Clinical skills teaching
Emergency medicine has long had a reputation among students for ‘being the place where you get to do useful things’. While proponents of the specialty as an academic discipline are keen to promote the other features of emergency medicine in an educational setting, this statement is still undoubtedly true. Many skills can be taught in the ED or a skills laboratory and emergency physicians are ideally placed to teach them. Whether it is junior medical students examining patients for the first time or learning practical skills, such as venesection and suturing, emergency physicians have a lot to offer in this regard. Like most teaching, there are useful techniques that can be employed to improve learning [21].
Assessment principles
Assessment should be considered as a tool which drives learning and should be developed in parallel with the curriculum rather than considered at the end. It is essential that the appropriate form of assessment be matched to the subject matter. Assessment tools selected should be valid, reliable and practical and have an appropriate impact on student learning [22]. For instance, when assessing competency in advanced cardiac life support, it would be more valid to use a practical-based assessment process, such as an observed objective structured clinical examination (OSCE) rather than a written examination. Figure 24.4.2 provides a graphical representation of matching assessment processes to skills and knowledge. A large number of assessment methods exist and educators should possess at least a basic understanding of their use and application. It is essential to understand that all universities have rules which govern the assessment of students and failure to adhere strictly to these rules exposes a department to academic appeals and complaints of unfairness and bias. Emergency physicians need to become intimately involved in examination processes throughout the course so that the speciality is taken seriously by students and to ensure that appropriate and realistic emergency conditions and scenarios are presented.
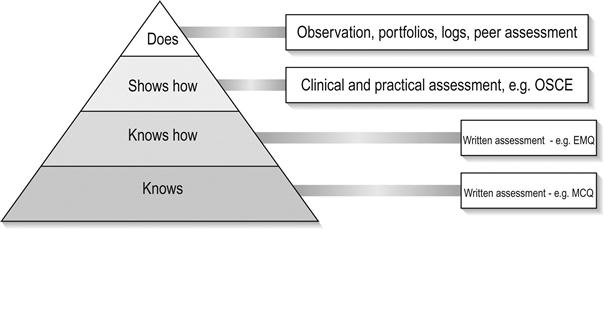
Likely developments over the next 5–10 years
Emergency medicine will continue to play a significant role in medical student education and is poised to make greater contributions in coming years. This will occur both by design and necessity: as described, the increase in student numbers is coinciding with the growth and maturation of the specialty in Australia and New Zealand. Emergency medicine will continue to expand throughout medical curricula rather than being the practice-based pre-intern term it currently occupies in the latter years of most medical courses. A recent shift in focus in the tertiary sector towards teaching ability rather than purely a research output will create opportunities for emergency medicine to gain a stronger foothold in universities. Teaching programmes will expand to deliver material, not just unique to emergency medicine, but the specialty will be opportunistic and be called upon to teach where significant gaps exist at an undergraduate level. In many universities, there is no academic presence in surgical subspecialties, such as ear, nose and throat, ophthalmology and orthopaedics. It will be left to emergency physicians to teach students the basics of these specialties, just as we currently manage many common acute conditions in these areas without specialist input.
It is now time for the specialty to move beyond just the provision of training in acute medicine as envisaged by the landmark Macy report in 1994 [23]. The specialty is developing its own body of knowledge and curriculum in important but under-represented areas of medical education, such as clinical decision making, toxicology, medical error and health systems design and management. The management of elderly patients and end-of-life decision making is becoming a significant part of most emergency physicians’ practice. Hence, emergency physicians need to take a leading role in the development and delivery of curricula in these areas. There is much to suggest that the increased sub-specialization of medicine has led to fragmentation of the health system, with the subsequent inability of society to achieve coherent and sustainable outcomes in health policy. Emergency medicine as a specialty has the opportunity to take a leading role in training doctors and other health professionals capable of understanding the key challenges facing the health system in the 21st century.
Controversies





24.5 Postgraduate emergency medicine teaching and simulation
Victoria Brazil
Introduction
Emergency departments (EDs) are fertile learning environments for postgraduate doctors. The varied clinical case mix, procedural practice and enthusiastic teaching by emergency physicians have made emergency medicine an important experience for new medical graduates [1]. Many of these same factors attract doctors to vocational training in emergency medicine.
Postgraduate training in this field, like most other specialties, traditionally followed an apprenticeship model. It was assumed that patient care experience and opportunistic bedside teaching from clinical experts provided all the knowledge, skills and attitudes requisite for emergency medicine practice. However, this approach has been changing over the last 15–20 years. Contemporary emergency medicine practice requires new knowledge and skills and encompasses different expectations from patients and healthcare systems. Emergency medicine educators need to recognize these changes in practice and employ contemporary educational approaches in developing the relevant knowledge and skills in trainee emergency physicians.
The governance of emergency medicine training, certification and credentialling varies internationally. In Australasia, training and assessment is by the Australasian College for Emergency Medicine (www.acem.org), in conjunction with hospitals and health services. Formal specialist recognition is granted by the Medical Boards of Australia (MBA) or New Zealand (MBNZ). Some ‘non-specialist’ training in emergency medicine is provided by the Australian College of Rural and Remote Medicine (ACRRM).
Other governance models of specialist training exist internationally and include oversight by universities (Malaysia), government organizations (UK Deaneries) or cross-specialty professional associations (Royal College of Physicians and Surgeons of Canada).
Curricular trends in emergency medicine
There has been a global trend in postgraduate medical education toward ‘outcomes based’ curricular models. This has resulted in a shift from curricula defining a knowledge base to be acquired, to ‘outcome’ concepts of roles and competencies for specialist physicians.
The CanMEDS model, developed in Canada and adopted by the Australasian College for Emergency Medicine (ACEM), requires training be orientated toward preparing emergency physicians (and other specialist trainees) for their roles as medical expert, communicator, collaborator, manager, health advocate, scholar and professional [2].
Integral to this curricular trend has been increased recognition of communication and professional domains of practice. Concepts of teamwork, leadership, patient safety, quality improvement and communication with patients and peers have been specified in recent curricula [3]. New domains of learning, such as medical informatics and evidence-based medicine, have become explicit curricular content.
Teaching methods in emergency medicine
Bedside teaching, or ‘teaching on the floor’, remains the foundation of most ED trainees’ educational experience. It provides the opportunity to reflect upon clinical and professional aspects of emergency medicine practice in an integrated manner. It should be facilitated by a graduated increase in patient care responsibility during training. However, the quality of this experience is dependent on the availability and skill of clinical supervisors and on time constraints in busy EDs. In this environment, clinical teaching can be ‘education by random opportunity’ and learners may not encounter important conditions or procedures and may not reflect usefully on the experiences they do have.
Didactic elements of specialist training vary in format but most programmes or institutions provide a structured element of training that consists of trainee- and supervisor-delivered presentations, procedural skill sessions, journal clubs and lectures by visitors or outside specialists. Following the trend towards competency-based curricular models, there is a general trend toward these teaching activities becoming more standardized and structured and supported by online content.
Reflective practice is an important learning skill for postgraduate trainees. Clinical audit and portfolios [4] can facilitate this and most training programmes also encourage participation in critical incident review, trauma review meetings and morbidity and mortality rounds.
There is a trend toward interprofessional and team-based learning, recognizing the team approach required for effective clinical practice. This includes multidisciplinary formal educational sessions and team-based simulation experiences [5]. These activities are focused on communication and professional domains of competence and provide trainees with a broader perspective on systems-based practice in emergency medicine.
Technology for learning in emergency medicine
Technological advances offer many potential teaching and learning applications in emergency medicine. Content-based technological adjuncts, such as textbooks and CD ROMs, are now more likely to be accessed online or in e-book formats, making published references more available and more easily updated. Electronic formats allow replication of high quality images and videos.
More significant is the change toward accessing web-based content for emergency medicine training and for continuing medical education. Websites, podcasts and blogs on emergency medicine topics number in the thousands (Table 24.5.1). As a result, the emergency medicine trainee requires effective information retrieval and quality analysis skills. Public access search engines, such as Google, have replaced traditional Medline database searches as the preferred method of information retrieval by trainees, although not always with effective results [6].
Table 24.5.1
Examples of popular emergency medicine blogs and podcasts
| Life in the Fast Lane | www.lifeinthefastlane.com | Emergency medicine | Australia |
| Academic Life in Emergency Medicine | www.academiclifeinem.blogspot.com | Emergency, research, education | USA |
| EMcrit | www.emcrit.org | Emergency, critical care, podcast | USA |
| EDExam | www.edexam.com.au | Emergency, clinical | Australia |
| St Emlyns | www.stemlynsblog.org/blog/ | Emergency, research | UK |
| ResusME | Resus.me/ | Emergency critical care, podcast | |
| The Poison Review | www.thepoisonreview.com | Toxicology | USA |
| Ultrasound Podcast | www.ultrasoundpodcast.com | Emergency ultrasound | |
| iTeachEM | www.iteachem.net | Emergency, education | USA, Australia |

More detailed listing available at EMCC Blogs, www.lifeinthelastlane.com[7].
The use of social media such as Twitter (San Francisco, CA, USA) and global collaboration to ‘collate and curate’ these resources [7] offers unparalleled access to educational materials and online discussions, as well as opportunities for informal peer review.
Procedural skill training has been enhanced by the use of manikins, part trainers and virtual reality systems, reducing the use of animal labs and cadavers. Video-based instruction of procedures allows demonstration of procedural performance under ideal conditions, with rehearsed teaching scripts. Many of these are now available on personal digital assistants for ‘just-in-time learning’ for trainees.
Clinical decision support software provides educational opportunities in conjunction with solutions to clinical problems. Many are in ‘app’ format (e.g. Medcalc) for mobile access and have extensive embedded resource materials that can be utilized in clinical practice or for primarily educational purposes.
Videoconferencing, webinars and tele-education have sought to answer the challenges of distance in emergency medicine education. Improvements in technology (including ‘retail’ platforms such as Skype (Luxembourg), Google Hangouts, etc.) and bandwidth promise greater opportunities in this regard. It may help to relieve the teaching workload in smaller or more remote EDs.
Simulation-based learning
Medical simulation encompasses any kind of simulated clinical encounter. Simulations employ a range of possible modalities including simple part task trainers, such as an intravenous cannulation arm, simulated patients (SPs) or actors with a role-play script, high fidelity mannequin simulators, virtual-reality-based procedural skill trainers or combinations of modalities.
Effective simulation-based learning for emergency medicine requires clear educational objectives. These may include knowledge acquisition, procedural skill proficiency, applied physiology and pharmacology or complex teamwork skills and behaviours inherent in crisis resource management. The educational objective should then determine the nature of the equipment used, scenario design, the level of fidelity required and the approach to debriefing.
Simulators, equipment and fidelity
Mannequin technology continues to improve. The most technologically complex full body simulators operate via detailed physiological modelling software and can manifest pulses, breathing, blinking and vocalization, together with an ability to alter lung mechanics and compliance and cardiovascular parameters. Less complex simulators can manifest most of these features, but with the benefit of lower cost and increased portability.
Learner perceptions of fidelity of the simulation experience are variable. Individual learning styles, the authenticity of the scenario presented, the realism of the team composition and the physical environment all appear to be more important than the complexity of a mannequin to the learners’ perception of fidelity and the learning outcomes achieved.
‘Hybrid’ simulation – combining simple part task trainers with standardized patients – has been successfully used to integrate a procedural skill with communication performance, e.g. an actor wearing a synthetic skin pad with a laceration interacts with a doctor suturing the wound.
Teamwork and communication skills training using medical simulation
Crisis resource management (CRM) training in emergency medicine using human patient simulation draws on parallels between acute patient care and the aviation and military industries, where it has been recognized that human factors are crucial to team performance [8].
A typical scenario might involve a team of medical and nursing participants managing a patient with chest pain, complicated by a life-threatening arrhythmia. Participants would be expected to identify and manage clinical issues, while engaging in the communication, teamwork and leadership activities inherent in real clinical practice. The scenario is then followed by video-assisted, expert debriefing to reflect upon individual and team performance. This experiential learning approach enables cross-domain training in which cognitive, procedural and affective domains of practice are authentically integrated. The interprofessional nature of the learning experience provides a unique opportunity for increased understanding of the role of other healthcare disciplines.
Educational benefits
Simulation enables ‘efficient’ learning through standardized exposure to clinical challenges which may be infrequent in clinical practice. It allows practice and failure in a safe environment, without risk to patients.
Advanced mannequin and audiovisual technology, together with logistic expertise, mean that these learning experiences can be provided in situ – in clinicians’ own EDs. The use of the authentic work environment allows high levels of fidelity to be achieved. The usual work team can engage in learning together and review their everyday systems and processes, without the cost and logistical barriers of travelling to a synthetic environment. Provision of simulation-based experience and group debriefing via videoconferencing and remote control of equipment is at the boundary of present technological capacity.
In addition to their role in learning, human patient simulators can be used for competency assessment, which is made reliable by standardized challenges. However, it is important that assessment activities are clearly distinguished from those whose objective is learning, as key differences in simulation performance are observed. This reproducibility of challenges also provides opportunities to study the effect of fatigue or expertise level on emergency physician or trainee performance and for testing new medical equipment or clinical systems.
Evidence for clinical practice improvement resulting from simulation-based learning is currently lacking, despite intuitive appeal. There are methodological challenges in the reliable measurement of teamwork and communication performance and attribution issues involved in any demonstration of patient outcome improvement. Features of simulations that lead to effective learning are better understood [9].
There are limitations to simulation-based training. There are significant costs in equipment and trained personnel to run programmes. Critical care situations, especially cardiac and airway emergencies can be simulated with a high degree of fidelity, but many other emergency medicine clinical challenges are not suitable. Negative training is a recognized risk, i.e. allowing participants not to wear gloves or lead aprons or simulating overly positive clinical outcomes can allow poor behaviour patterns to develop or train learners to have unrealistic expectations in the real clinical environment.
Training for providers of simulation-based learning
Educators using simulation-based modalities require proficiency in technological issues, experiential learning principles and small group process. Training for providers of simulation-based emergency medicine education is varied. Short courses exist at many simulation centres and most providers run their own ‘in-house’ quality assurance process to ensure a standardized approach to scenario design, delivery and group debriefing. In Australia, Health Workforce Australia (HWA) is currently working on a national approach to provider training through the National Health Education and Training in Simulation (NHET-Sim) programme.
Assessment and performance appraisal for emergency medicine training
Assessment in medical education is a complex issue [10]. No single assessment format can adequately measure performance across all domains of emergency medicine practice. Within the specialty there is considerable international variation in the domains of performance formally assessed, the definition of ‘professional competence’ required and in the standardization of assessment processes undertaken.
In-training assessment by clinical supervisors is a core element in most programmes, but this may be provided by one designated supervisor of training or many clinical supervisors in a group-based assessment. Literature suggests that in-training assessment is a valid tool (i.e. measures the right thing), but the potentially subjective nature of supervisor assessments and recall bias can decrease the reliability of assessment [11].
Most training programmes also have formal examination components, generally developed and administered externally by a national training body. A variety of formats exists, including multiple-choice questions, written tests, structured interviews and clinical examination vivas.
Workplace-based assessment (WBA) is becoming more prevalent. This includes formats such as mini-CEX (clinical evaluation exercise) and DOPS (direct observation of procedural skills) in which trainees are directly observed and assessed in a clinical encounter and given immediate feedback. WBAs are valid and reliable if sufficient assessments are undertaken, but resource-intensive and challenging in implementation [12].
Other contemporary assessment tools include clinical simulations, portfolios, standardized patients and multisource ‘360 degree’ feedback assessment. Patient care quality outcomes have been suggested as an assessment tool [13]. These formats support the trend toward specific assessment of communication and professionalism domains, which mirror contemporary shifts in curricular content.
Faculty development in emergency medicine
Many emergency physicians are enthusiastic clinical teachers. However, clinician educators also require specific skills and preparation for their teaching role. Training for educators in emergency medicine might include Masters level courses, short workshops or institution-based group professional development activities. These courses typically cover topics such as curriculum development, teaching and learning processes and assessment and feedback skills.
Programmes are more formally developed in the USA, where a number of teaching fellowships exist in emergency medicine and where the Society for Academic Emergency Medicine (SAEM) has published a Faculty Development Handbook[14]. These initiatives have been reinforced by the emergence of ‘clinician educator tracks’ in academic institutions to provide an academic career structure for clinical teachers.
Continuing medical education (CME)
There is a societal expectation in developed countries that physicians maintain their skills and competence to practice. Australasian emergency physicians are required by the Medical Boards of Australia and New Zealand to demonstrate participation in a continuing professional development programme accredited by the Australian Medical Council. Internationally, the nature of CME requirements vary, but most require periodic assimilation of a portfolio of attendance at conferences and workshops, together with demonstration of participation in clinical practice, teaching, quality assurance activities, research and other special interests. Some jurisdictions require update examinations.
Controversies and future directions