CHAPTER 18
Hypothalamic, pituitary and adrenal disorders
Miles J. Levy; Trevor A. Howlett
CHAPTER OUTLINE
CLINICAL ANATOMY OF THE PITUITARY AND HYPOTHALAMUS
PHYSIOLOGY OF HYPOTHALAMO–PITUITARY–END ORGAN AXES
CLINICAL ANATOMY AND PHYSIOLOGY OF THE ADRENALS
ASSESSMENT OF NORMAL PITUITARY FUNCTION
Dynamic tests of ACTH–adrenal function
Cortisol normal ranges, borderline responses, assay precision and dynamic test reproducibility
Assessment of growth hormone reserve
Other tests of gonadotrophin secretion
Dynamic tests of posterior pituitary function
A clinical approach to assessment of the whole ACTH–adrenal axis
Monitoring of pituitary function in disease states
OTHER DIAGNOSTIC TECHNIQUES IN PITUITARY DISEASE
PITUITARY HYPERSECRETION STATES
Diagnosis and differential diagnosis of Cushing syndrome
Thyroid stimulating hormone-secreting adenomas
Gonadotrophin-secreting adenomas
HYPOTHALAMIC AND PITUITARY DEFICIENCY STATES
Diseases that may lead to generalized hypopituitarism
Other isolated anterior pituitary deficiencies
Clinical features of Addison disease
Congenital adrenal hyperplasia
Assessment of adrenal incidentaloma
INTRODUCTION
The investigation of disorders of the pituitary and hypothalamus and of adrenocortical function frequently causes great apprehension among non-endocrinologists, whether they are clinical biochemists or general physicians, because of their supposed complexity and the frequency of atypical, borderline or artefactual results. It is certainly true that the hypothalamo–pituitary axis plays the central role in the control of a large number of hormonal systems and of many other important aspects of homoeostasis. However, the clinically relevant physiology of each hormonal axis is well understood and the plasma concentrations of the majority of the hormones secreted by both the pituitary and the relevant peripheral endocrine glands can now be readily and accurately measured in most clinical biochemistry laboratories. Thus, a logical approach to the measurement of the relevant plasma hormone concentrations, applying basic physiological principles, can result in a relatively simple and efficient assessment of pituitary function in the majority of cases.
This chapter will outline a clinically relevant and efficient approach to the assessment of pituitary and adrenal function. In many cases, simple measurement of basal, unstimulated plasma concentrations of the appropriate hormone(s) will give all the information required for clinical management. Some of the traditionally used dynamic endocrine tests can now be reasonably discarded, while others remain essential, and the choice of these will be discussed critically. Recommendations for clinical management of borderline or contradictory results will also be discussed.
CLINICAL ANATOMY OF THE PITUITARY AND HYPOTHALAMUS
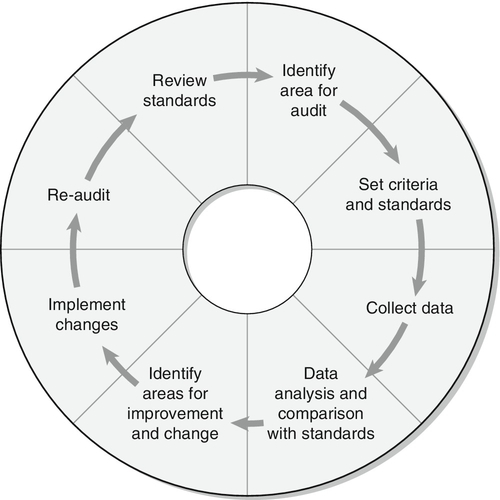
The pituitary gland (Fig. 18.1) lies within a bony compartment, the pituitary fossa or sella turcica, beneath the hypothalamus to which it is connected by the pituitary stalk. The fossa is separated from the subarachnoid space and cerebrospinal fluid by the diaphragma sellae. The adult human pituitary consists of two lobes (the third, neurointermediate lobe of other species is not present except in fetal life). The anterior lobe (adenohypophysis) is embryologically derived from cells of Rathke’s pouch. It contains a variety of cell types differentiated to secrete the majority of the peptide and glycopeptide hormones that control the function of peripheral endocrine organs: corticotrophs (adrenocorticotrophic hormone, ACTH), lactotrophs (prolactin, PRL), gonadotrophs (luteinizing hormone and follicle stimulating hormone: LH and FSH), thyrotrophs (thyroid stimulating hormone, TSH) and somatotrophs (somatotrophin or growth hormone, GH). The anterior lobe has no direct arterial blood supply but is supplied by the portal vessels that arise in the median eminence of the hypothalamus. The secretion of anterior pituitary hormones is controlled by releasing and inhibiting factors, which are predominantly peptides, released into the portal circulation from the nerve terminals of a variety of hypothalamic neurons. These neurons in turn are subject to the effects of the modulatory neurotransmitters of other neurons, within the hypothalamus and beyond.
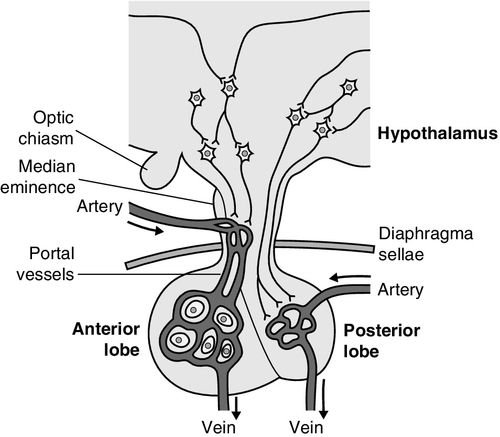
FIGURE 18.1 Functional anatomy of the human pituitary and hypothalamus.
The critical role of the portal circulation in the control of anterior pituitary function means that any disease process that interferes with this blood supply, for example non-functioning tumours of the pituitary or hypothalamus, will often result in severe pituitary dysfunction, even if the actual hormone-secreting cells of the anterior pituitary have not themselves been destroyed.
The posterior lobe (neurohypophysis), in contrast, is embryologically derived from the brain, has a direct arterial blood supply and is not controlled via a portal circulation. Hormonal secretion occurs directly from the nerve terminals of vasopressin (antidiuretic hormone, ADH) and oxytocin neurons, whose cell bodies lie within the hypothalamus, primarily in the supraoptic and paraventricular nuclei.
Both the anterior and posterior lobes contain a variety of other cell types. These are not directly responsible for hormonal secretion into the peripheral circulation, although they may have important modulatory or paracrine roles in the control of pituitary function.
Many other important neural structures lie within or adjacent to the hypothalamus, notably the optic chiasm and the neural centres controlling thirst, osmoregulation, appetite, temperature homoeostasis and circadian rhythm. Diseases of the pituitary and hypothalamus can result in dysfunction of any of these structures, in addition to the hormonal syndromes described here. In particular, visual field loss due to compression of the optic chiasm is a frequent finding with large pituitary tumours.
These anatomical relationships indicate that the pituitary gland is truly at the interface of the mind and body, with a central role in homoeostasis, explaining its source of fascination for physiologist and physician alike.
PHYSIOLOGY OF HYPOTHALAMO– PITUITARY–END ORGAN AXES
The basic physiology of the important endocrine axes is summarized diagrammatically in Figures 18.2 and 18.3. Many additional releasing, inhibiting and modulating factors have been identified, primarily in animal experiments, but none of these has yet been shown to be of relevance clinically, nor for the understanding of the principles of pituitary function testing. Hormones that can be measured in the laboratory are indicated in the figures. Peripheral plasma concentrations of hypothalamic releasing factors can also be measured (with difficulty), but for the most part do not reflect hypothalamic activity and are not of clinical relevance.
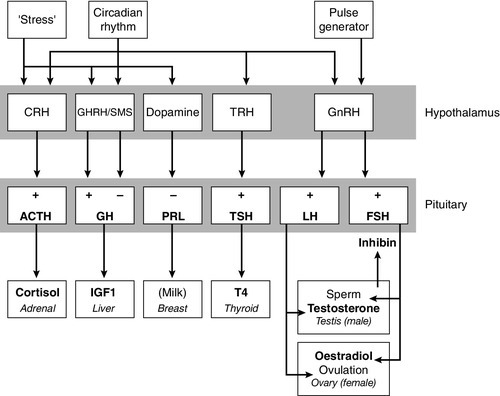
FIGURE 18.2 Physiological relationships of the anterior pituitary. Hormones (in bold) can be readily measured in most clinical biochemistry laboratories. See text for explanation of abbreviations.
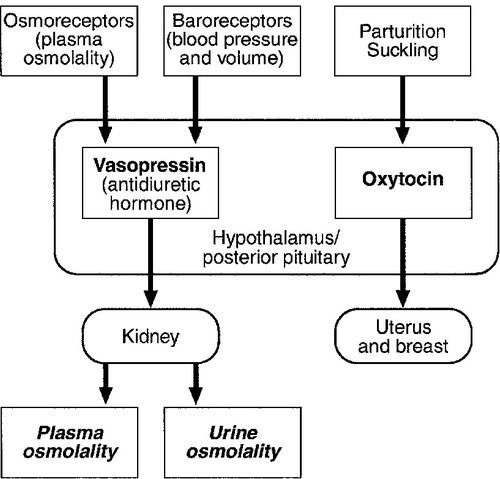
FIGURE 18.3 Physiological relationships of the posterior pituitary. Osmolalities (italicized) can be readily measured in most clinical biochemistry laboratories. Hormones (emboldened) can be measured with difficulty in a few centres.
Adrenocorticotrophic hormone (ACTH) stimulates adrenal cortisol secretion and is itself controlled by hypothalamic corticotrophin releasing factor (CRF). Corticotrophin releasing factor is now known to be a complex of factors including a 41-residue peptide (CRF-41 or corticotrophin releasing hormone, CRH) and vasopressin, which act synergistically. Adrenocorticotrophic hormone, and therefore cortisol, are secreted with a pronounced circadian rhythm: concentrations are low, typically undetectable, at midnight (if asleep), rise in the final hours of sleep to reach a peak shortly after wakening in the morning and steadily decline throughout the rest of the day with superimposed peaks of secretion with meals, exercise and stressful events. ‘Stress’ (‘fight and flight’, illness, hypoglycaemia, surgery etc.) is a major determinant of ACTH and cortisol secretion, which therefore means that basal concentrations in a well individual may not always reflect the ability to respond to illness, and conversely that high concentrations in an individual with other significant disease do not always imply oversecretion.
Secretion of growth hormone (GH) is controlled by complementary stimulatory and inhibitory factors, GH releasing hormone (GHRH, a 44-residue peptide) and somatostatin (SMS, a cyclic, 14-residue peptide), respectively. Growth hormone secretion occurs in pulses, predominantly at night during the early hours of sleep. Only infrequent, small pulses occur during the day when plasma concentrations are usually undetectable, although GH secretion also occurs in response to stress. Growth hormone’s effects on growth are mediated via synthesis of insulin-like growth factor 1 (IGF-1 – previously called somatomedin C), which is synthesized mostly in the liver but also in peripheral tissues.
Prolactin secretion is unique in being predominantly controlled by an inhibitory hypothalamic factor, dopamine. This fact explains why PRL deficiency is rare and why any disease that interferes with pituitary anatomy, and thus the flow of portal blood, may be associated with hyperprolactinaemia. Plasma PRL is raised physiologically during pregnancy and lactation, and release also occurs in response to stress. The physiological role of PRL in non-lactating women and in men is unknown.
Hypothalamic thyrotrophin releasing hormone (TRH), a tripeptide, controls TSH secretion, which in turn stimulates the synthesis and secretion of thyroid hormones thyroxine (T4) and tri-iodothyronine (T3). Secretion is regulated primarily to maintain normal circulating concentrations of thyroid hormones, although there is also a slight circadian rhythm.
Luteinizing hormone and FSH are both controlled by a single stimulatory decapeptide, gonadotrophin releasing hormone (GnRH or luteinizing hormone releasing hormone, LHRH). The major role of LH in the male is to stimulate testosterone secretion by testicular Leydig cells, and in the female to stimulate ovulation at mid-cycle. Follicle stimulating hormone controls spermatogenesis in the male and ovarian follicular development, and therefore oestradiol secretion, in the female, and in both sexes stimulates gonadal secretion of inhibin, which is responsible for some negative feedback control. The most important feature of the secretion of GnRH, and thus LH and FSH, is its pulsatile nature, imposed by the hypothalamic pulse generator. It is likely that differential secretion of LH and FSH is achieved by modulation of the frequency and amplitude of pulsatile GnRH secretion, and the peripheral effects of LH and FSH are critically dependent on appropriate pulsatile variation in concentrations. Gonadotrophin releasing hormone pulses occur, on average, every 90 min, but in the female are more rapid in the follicular phase and slower in the luteal phase of the menstrual cycle. During early puberty, the majority of pulses occur at night, during sleep.
Vasopressin (antidiuretic hormone, ADH) secretion is controlled primarily by osmotic and blood volume sensing mechanisms (osmoreceptors and baroreceptors, respectively). High plasma osmolality and hypovolaemia or hypotension are the major stimulatory factors. Vasopressin acts directly on the kidney to increase water reabsorption in the renal collecting ducts and thus reduce urine volume. Corticosteroids are necessary for the excretion of free water, so deficiency may mask the symptoms of ADH deficiency.
Oxytocin is released during labour and during suckling, but mechanisms controlling its secretion have not been studied extensively in humans and there appears to be no clinical syndrome related to oxytocin deficiency.
Secretion of all pituitary hormones is controlled, to a greater or lesser extent, by negative feedback. This occurs at all levels, including the direct inhibition by a hormone of its own secretion (‘ultra-short-loop’), but the most important negative effects are generally those of the peripheral ‘end hormone’ on hypothalamic and/or pituitary secretion. The existence of such negative feedback is of crucial importance to the interpretation of basal hormone concentrations in pituitary disease and is the basis of a number of dynamic tests of pituitary function.
CLINICAL ANATOMY AND PHYSIOLOGY OF THE ADRENALS
The two adrenal glands lie superior to the kidneys and are composed of the inner catecholamine-secreting medulla, which is controlled centrally by direct innervation, and the outer cortex, which is controlled by classic endocrine pathways. Glucocorticoid and androgen secretion is predominantly from the zona fasciculata and zona reticularis and controlled by pituitary ACTH secretion, while mineralocorticoid secretion is predominantly from the zona glomerulosa and controlled via the renin–angiotensin system (see Chapter 4). This means that the patterns of adrenal deficiency vary with aetiology: thus a disease that destroys the adrenal itself (e.g. autoimmune Addison disease, adrenal tuberculosis or bilateral adrenalectomy) results in deficiency of both glucocorticoid and mineralocorticoid secretion, whereas ACTH deficiency from pituitary disease results in glucocorticoid deficiency alone and, thus, less marked disturbances of salt and water balance. Even complete adrenalectomy does not seem to cause a clinical deficiency of catecholamines, since secretion by the rest of the sympathetic nervous system appears to compensate for the loss of adrenal secretion.
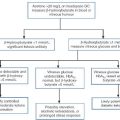
Adrenal steroid synthesis involves a complex and intersecting sequence of enzymatic steps within the cytoplasm and mitochondria of the adrenal cell. Pathways are illustrated in Figure 18.4. The initial, rate-limiting, step in steroidogenesis in the normal subject is the conversion of cholesterol to pregnenolone. Adrenocorticotrophic hormone has rapid effects on cholesterol transport into mitochondria and longer-term effects on transcription of genes encoding a number of enzymes in the synthetic pathway. These pathways are of clinical significance in congenital adrenal hyperplasia, where inherited defects in a variety of these enzymes give rise to the various subtypes and clinical syndromes (see Table 18.1), and in clinical pharmacology, since metyrapone, a drug that inhibits 11β-hydroxylase, can be used to decrease cortisol synthesis in Cushing syndrome.
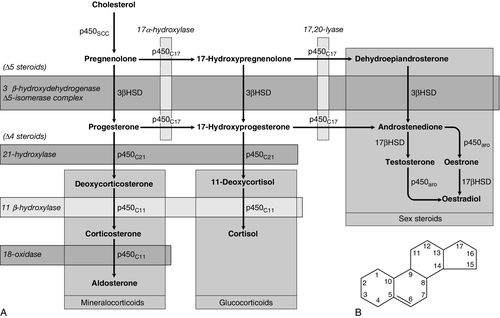
FIGURE 18.4 (A) The pathways of steroid hormone synthesis within the adrenals (and gonads). Enzymes are named by reaction catalysed (e.g. 21-hydroxylase) or by enzyme name (e.g. p450C11). p450 enzymes are in mitochondria and other enzymes are in the cytoplasm; steroid synthesis involves repeated passage of precursors between these cellular compartments. (B) Basic structure of the steroid molecule (from Kumar and Clark 2012 Clinical medicine. 8th ed. Edinburgh: Churchill Livingstone, with permission).
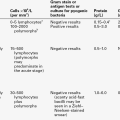
TABLE 18.1
Subtypes of congenital adrenal hyperplasia
| Enzyme defect | Frequency | Clinical features |
| 21-hydroxylase p450C21/CYP21 | 95% of cases 1 in 15 000 live births |
Ambiguous genitalia in females Virilization, some salt losing |
| 11-β-hydroxylase p450C11/CYP11B | < 5% of cases | Ambiguous genitalia in females Virilization, hypertension |
| 17-α-hydroxylase p450C17 | Rare | Ambiguous genitalia and lack of virilization in males Hypertension |
| 3β-hydroxysteroid dehydrogenase 3βHSD/HSD3B2 | Rare | Ambiguous genitalia in males Mild virilization in females Occasional Addisonian crises |
| Cholesterol desmolase p450SCC/CYP11A | Rare | Addisonian crises |
ASSESSMENT OF NORMAL PITUITARY FUNCTION
Basal hormonal investigations
A single, basal blood sample, especially if taken at 09.00 h, can give extensive and, for some purposes, complete information regarding pituitary function. The blood should ideally be taken under resting, unstressed conditions but, while the stress of major illness or surgery significantly alters results and thus might cause diagnostic errors when investigating pituitary hyperfunction, minor anxiety regarding hospital attendance or venepuncture probably has little effect.
The pituitary–thyroid axis is fully assessed by the simultaneous measurement of plasma free thyroid hormone concentrations and TSH. Thyroid stimulating hormone deficiency is characterized by a low plasma free T4 concentration without the expected elevation of plasma TSH. In hypopituitarism, TSH may be low (although rarely undetectable) or within the normal range (inappropriately for the low T4), which represents the most potent argument against the use of TSH as the sole screening test for thyroid function in a laboratory.
Basal plasma PRL gives full physiological information regarding the function of its axis. Concentrations frequently vary from day to day (particularly minor elevations), so that 2–3 basal samples are usually necessary before any treatment is instituted.
In the male, a normal basal plasma testosterone (preferably taken at 09.00 h, in view of the significant circadian rhythm) effectively demonstrates normal LH secretion, and demonstration of a normal sperm count would confirm normal FSH secretion (although this is rarely performed unless infertility is an issue). Gonadotrophin deficiency is diagnosed by the combination of a low plasma testosterone with low or inappropriately ‘normal’ (rather than elevated) concentrations of LH and FSH. The significance of borderline testosterone concentrations – near or just below the lower end of the reference range – together with normal LH and FSH, particularly in the ageing male, is an area of growing clinical interest but still with considerable controversy and is discussed more fully later in this chapter.
Assessment of the pituitary–gonadal axis in the female is complicated by the physiological changes during the menstrual cycle. A normal luteal phase plasma progesterone is the ultimate biochemical test of the adequacy of the entire axis. Spontaneous, normal, regular menstruation also excludes gonadotrophin deficiency without the need for additional biochemical tests. Gonadotrophin deficiency is suggested in the context of amenorrhoea by the presence of a consistently low plasma oestradiol in the absence of elevation of plasma LH and FSH, although a variety of ‘hypothalamic’ causes of amenorrhoea also show this combination (see below). In postmenopausal women (or, in the context of longstanding amenorrhoea, arbitrarily women aged >50 years), who are not on oestrogen replacement, the simple absence of the usual menopausal elevation of LH and FSH is sufficient to diagnose gonadotrophin deficiency.
Basal plasma cortisol is not always sufficient to diagnose ACTH deficiency or normality, because of the circadian rhythm and since concentrations that need to be achieved during physiological stress are much higher than those found under normal basal circumstances. Nevertheless, a basal cortisol >550 nmol/L in an unstressed individual certainly excludes ACTH deficiency and, in our experience, a 09.00 h cortisol <100 nmol/L is never, and >400 nmol/L is always, associated with a normal ACTH reserve (see below).
Basal concentrations of GH are usually unhelpful in the diagnosis of deficiency, since basal concentrations in normal individuals are usually undetectable. A low serum IGF-1 (using age-related normal ranges) is a good predictor of severe GH deficiency but, conversely, an IGF-1 in the normal range does not exclude severe deficiency that might respond to replacement therapy.
Plasma vasopressin and oxytocin concentrations are measured by only a few centres and are not used routinely in the assessment of basal pituitary function. Measurement of paired plasma and urine osmolality on a basal early morning sample does, however, give valuable information. A urine osmolality >600 mmol/kg excludes diabetes insipidus (DI) as long as plasma osmolality is normal (280–295 mmol/kg). Conversely, DI is suggested by an elevated plasma osmolality (>300 mmol/kg) with a urine osmolality <600 mmol/kg. Other combinations are non-diagnostic and require a water deprivation test if DI is suspected clinically. Oxytocin secretion is never investigated clinically.
The order of progression of hypopituitarism with progressively severe pituitary disease is usually predictable. Growth hormone and gonadotrophin secretion are usually most susceptible to damage, followed by ACTH and TSH, with PRL deficiency occurring only rarely and usually only after extensive pituitary damage by disease or surgery. It is thus unusual, although not impossible, for a patient with regular menstruation or normal male gonadal function to develop ACTH deficiency first in the course of their disease. Diabetes insipidus usually only occurs with inflammatory or invasive pituitary disease, after surgery or with diseases primarily involving the hypothalamus. Thus clinical background may influence the degree of investigation considered appropriate in an individual patient with borderline or equivocal results in a single axis.
Dynamic tests of ACTH–adrenal function
Insulin stress test
The insulin stress test (IST) (or insulin tolerance test, ITT) has traditionally been the ‘gold standard’ test to assess the adequacy of the ACTH–adrenal axis. After adequate hypoglycaemia (blood glucose <2.2 mmol/L), a normal rise of plasma cortisol (traditionally to >550 nmol/L) excludes ACTH and cortisol deficiency. Patients with such a response are capable of producing a normal adrenocortical response to stressful illness, without the need for replacement therapy. The increment of plasma cortisol does not add additional information. Patients with borderline peak responses (400–550 nmol/L) may only require steroid replacement during stressful illnesses, but patients with lower responses will typically be symptomatic and almost always require standard replacement therapy.
An IST is contraindicated by cardiac disease (an ECG should be normal) and by epilepsy (or other unexplained blackouts) and always requires close medical and nursing supervision to maintain safety. For this reason, alternative, simpler tests have been sought, although none has yet achieved universal acceptance. A retrospective audit of 230 ISTs in our own laboratory indicated that patients with a basal cortisol <100 nmol/L never, and >400 nmol/L always, demonstrated a normal cortisol response, so that an IST is probably unnecessary in such patients (representing 50% of ISTs performed in our study) if the only aim is to assess pituitary–adrenal reserve. In our experience, patients who have recently discontinued steroid replacement with a basal cortisol <200 nmol/L also never have a normal response.
Short tetracosactide (synacthen, tetracosactrin, ACTH) test
This test, involving administration of tetracosactide (synthetic ACTH1–24), is the cornerstone for diagnosis of primary adrenal failure, but is increasingly used in the context of pituitary disease. It has long been accepted that a normal cortisol response to tetracosactide excludes Addison disease, but to test for ACTH deficiency by administration of ACTH is clearly illogical on purely physiological grounds, since it directly tests only the adrenal response and not that of the pituitary or higher centres. However, presumably since ACTH deficiency leads to adrenal atrophy, a number of workers have shown close correlations between the cortisol responses to a simple intravenous tetracosactide test and to an IST, and have suggested that the former is a satisfactory test of the ACTH–adrenal axis as long as the test is not done within four weeks of the pituitary insult.
A majority of UK endocrinologists now accept that a normal plasma cortisol response, 30 min after tetracosactide (250 μg), adequately excludes ACTH deficiency, and several long-term clinical observational studies have confirmed the safety of such an approach. This test was originally validated with i.v. tetracosactide but responses to i.m. injection are equivalent. However, normal responses are different (typically 150 nmol/L or so higher) at 60 min, so an appropriate normal range is needed for each time point and, in practice, we recommend only measuring the 30 min value to avoid confusion.
Since the standard 250 μg dose of tetracosactide is substantially supraphysiological, many workers have advocated use of a lower dose stimulation test (typically 1 μg). However, this approach is more complicated since no suitable dose preparation exists, and it has not been demonstrated to be significantly superior in clinical practice to the traditional test.
Other tests
The glucagon test (1 mg i.m.) has been advocated to assess ACTH and GH reserve when an IST is contraindicated (usually with the same criteria for a normal response). Glucagon frequently causes nausea and vomiting, normal individuals sometimes fail to show a ‘normal’ response and sampling must continue over a 4 h period, making this test relatively unattractive as a routine alternative.
The metyrapone test (750 mg orally, four-hourly for 24 h) has been widely used as a test of ACTH reserve, particularly in North America; it measures the response of urinary cortisol precursors or, more recently, plasma 11-deoxycortisol, to blockade of adrenal cortisol synthesis by this drug. The test is time consuming, often unpleasant for the patient and does not test the response to physiological stress.
An intravenous short tetracosactide test is now recognized by most endocrinologists to be a more appropriate and simpler alternative to either of these tests, unless information on GH reserve is also required.
Cortisol normal ranges, borderline responses, assay precision and dynamic test reproducibility
Normal ranges for cortisol, particularly for dynamic tests, were predominantly developed and validated using old assay technologies (e.g. fluorimetry), which are no longer in use, and although a variety of adjustments (typically lowering of cortisol cut-off concentrations for a normal response) have been suggested, these have rarely been completely validated.
Detailed studies of normal responses to tetracosactide have shown substantial differences with gender and between commonly used cortisol assays (see Clark et al. in Further reading, below). Most workers and most assays report a lower limit of the normal response between 500 and 600 nmol/L, but some other published studies report lower peak responses than this in some apparently healthy individuals (lowest values ~400 nmol/L). Similarly, some recent studies reporting cortisol responses to IST in healthy volunteers have reported peak responses in some individuals on some occasions between 400 and 500 nmol/L.
Studies of test–re-test variability for both the tetracosactide and IST tests have mostly reported a coefficient of variation (CV) of around 10%. External quality control schemes also frequently report an interassay/intermethod CV for cortisol of 10% or higher. Therefore, if we assume conservatively that the combined variability from these two sources is represented by an overall CV of 10%, then simple statistics show that the 95% confidence limits for a test response with a ‘true’ value of 500 nmol/L actually extends from 400 to 600 nmol/L. These considerations potentially apply to both the tetracosactide test and the IST.
All of these factors lead the authors to define basal cortisol and cortisol responses to tetracosactide and other dynamic tests broadly rather than using a single precise cut-off, and to recognize a group with borderline or equivocal responses who cannot be clearly classified as ‘normal’ or ‘abnormal’:
• 400 nmol/L: a basal cortisol above this value indicates a normal hypothalamo–pituitary–adrenal (HPA) axis – unless the patient is acutely unwell or has ongoing unexplained symptoms consistent with hypoadrenalism. A cortisol response to tetracosactide or IST below this concentration usually means that routine steroid replacement is required
• 600 nmol/L: a cortisol response to tetracosactide to above this concentration at 30 min almost always means that ACTH deficiency or primary adrenal failure is excluded. A cortisol response to above this concentration in an IST certainly excludes the need for steroid replacement even during severe intercurrent illness
• 100–400 nmol/L: basal cortisol concentrations in this range need a dynamic test to confirm the abnormality of the axis; a tetracosactide or IST peak response in this range usually indicates a requirement for routine steroid replacement
• 400–600 nmol/L: peak responses to tetracosactide or IST in this range in patients with known pituitary disease may not require the patient to receive regular replacement, but do require education of the patient about the possible need for replacement and symptoms of hypoadrenalism during intercurrent illness (and often provision of an emergency supply of hydrocortisone). Responses to tetracosactide in this range can sometimes be seen in normal individuals, so if there is no other evidence of adrenal or pituitary disease, further investigation may be required (e.g. renin and ACTH concentrations, possibly followed by IST to exclude isolated ACTH deficiency if the former are normal). Interpretation of isolated responses in this range are further complicated by the fact that patients with chronic fatigue syndrome may show borderline reduced cortisol dynamic responses, although in most cases they do not respond symptomatically to steroid replacement.
Assessment of growth hormone reserve
There are several biochemical tests that are used in clinical practice to confirm growth hormone deficiency (GHD). The clinical features of GHD are non-specific, and the different GH stimulation tests have different degrees of ability to stimulate GH release. For this reason, there continues to be debate about the most appropriate test and what extent of GH response constitutes true deficiency.
Insulin stress test
The insulin stress test (IST) remains the gold standard test of GH deficiency. In adults, a peak GH response to adequate hypoglycaemia (serum glucose <2.2 mmol/L) of >7 μg/L indicates a normal GH reserve, whilst a peak GH <3 μg/L, as defined by the UK National Institute for Health and Care Excellence (NICE), indicates severe deficiency. In children, a peak GH <6.7 μg/L after two provocative tests supports a diagnosis of growth hormone deficiency, although only one dynamic test is required in children with known hypothalamo–pituitary disease. The IST is unpleasant for patients and requires close medical supervision as it is potentially hazardous and is contraindicated in ischaemic heart disease and epilepsy. As well as being a reliable GH stimulus, the IST is also a good assessment of ACTH reserve so it a useful way of assessing GH and ACTH reserve simultaneously.
Children of both sexes may show subnormal GH responses to hypoglycaemia (and other dynamic tests) in the years immediately preceding puberty (objectively when the bone age is > 10 years). At the present time, there is no consensus with respect to the practice of ‘priming’ with gonadal steroids prior to such tests.
Other pharmacological tests
The glucagon test can be useful in the assessment of GHD when an IST is contraindicated, but it is a less reliable stimulus of GH and a poorer test of ACTH reserve than the IST, and its mechanism of action is not well understood. The criteria for diagnosing GHD in the glucagon test are the same as for the IST described above.
Other provocative tests of GH reserve include the GH-releasing hormone (GHRH) + arginine and GHRH + GH-releasing hexapeptide (GHRP-6) test, both of which have now been evaluated in large numbers of patients. The GHRH + arginine test may be particularly useful for young patients with radiation-induced GHD as occasionally the IST can give false negative results. Because GHRH + arginine is a more powerful GH stimulus, the cut-off point for diagnosing GHD is <9 μg/L.
In an attempt to avoid an IST, a variety of other pharmacological tests of GH reserve have also been used, particularly in children, including clonidine (0.15 mg/m2) and arginine infusion (0.5 g/kg). Use of these tests tends to be a matter of local preference; all are associated with rather variable responses and the peak GH values used to diagnose deficiency vary from laboratory to laboratory.
Exercise testing
Growth hormone is released during exercise and some laboratories have used an exercise test to assess GH reserve. Intensive, well controlled exercise (bicycle ergometer or treadmill) may exclude GH deficiency (by a peak GH response >6.7 μg/L), but less well controlled exercise (‘run up and down the stairs’) is unreliable.
Assessment of physiological growth hormone secretion
Because GH is secreted in a pulsatile fashion with increased pulses at night, serial blood tests, which can be done overnight or over 24 h, can be used to monitor the frequency and amplitude of GH pulses. Because this approach is labour intensive, requiring hospital admission with multiple blood samples, it is mainly a research tool.
Measurement of urinary GH is not part of routine clinical practice because of its low concentration in urine, but sensitive assays may make this possible in the future, particularly for the surveillance of GH replacement therapy.
Insulin-like growth factor 1 is produced by the liver in response to GH and, although measurement of its concentration is a useful diagnostic adjunct to provocative GH stimulation tests, it should not be used alone to diagnose GHD. However, the finding of a low IGF-1 in patients with three or four other pituitary deficiencies makes the likelihood of GHD sufficiently high that a formal provocative test may not be necessary.
Re-evaluation of GH status in young adults
In patients with isolated GHD that developed in childhood, it can be difficult to know whether GH replacement should be continued into adulthood. Patients with genetic hypopituitarism or multiple hormone deficiencies are highly likely to need to continue GH replacement. However, it may be able to be discontinued in patients with isolated or less severe GHD. In this situation, patients are re-tested during the period of transition between childhood and adulthood and the current consensus guidelines suggest a cut-off of <6 μg/L during an IST to indicate a continuing need for GH replacement. In this situation, the presence of clinical symptoms can also help the decision about whether to continue GH, which is best done by evaluation of the Adult Growth Hormone Deficiency Assessment (AGHDA) score. Consensus guidelines suggest that young adults with GHD should continue GH until the age of 25 years, when peak bone mass is likely to have been achieved.
Releasing hormone tests
Thyrotrophin releasing hormone (200 μg i.v.) and LHRH (100 μg i.v.) tests have been used traditionally in combination with the IST to assess pituitary reserve in the combined pituitary function test. Growth hormone releasing hormone testing (100 μg i.v.) and CRH testing (100 μg i.v.) have also more recently been advocated as additional tests.
All these releasing hormone tests assess only the readily releasable pituitary pool of their relevant hormones and do not assess the ability of the axis to respond to appropriate physiological stimuli. When the cause of hypopituitarism is at the hypothalamic level (which is frequently the case), then ‘normal’ TRH, LHRH, GHRH or CRH responses may be seen in the presence of deficiency documented by other means. Conversely, ‘subnormal’ responses are often seen when the axis is otherwise felt to be normal. Furthermore, a ‘delayed’ TRH test response (peak TSH at 60 min rather than 20 min), initially considered to be characteristic of hypothalamic disease, is not infrequently seen in primary pituitary disease.
For these reasons, the authors believe that all these releasing hormone tests should now play no part in routine pituitary function testing, but be reserved (if used at all) for the rare cases requiring differentiation between pituitary and hypothalamic deficiency states.
Other tests of gonadotrophin secretion
Clomifene test
When the differentiation of gonadotrophin deficiency from other causes of ‘hypothalamic amenorrhoea’ is in doubt, a clomifene test (3 mg/kg, maximum 200 mg, daily for seven days) may give additional information. Patients with organic gonadotrophin deficiency show no increase in LH and FSH (measured on days 0, 4, 7 and 10), whereas some patients with hypothalamic amenorrhoea, weight-related amenorrhoea or delayed puberty show a gonadotrophin rise, usually followed by menstruation (about 28 days later). In early puberty, LH and FSH may show a characteristic fall rather than a rise.
Assessment of luteinizing hormone pulsatility
Measurement of plasma LH every 10–15 min for 6–8 h or more, when analysed by an appropriate computer algorithm, allows detailed assessment of LH pulse frequency and amplitude. This may give considerable insight into pituitary physiology but, in view of the time and number of assays involved, is essentially a research tool.
Dynamic tests of posterior pituitary function
Water deprivation test
Water deprivation is the standard physiological test of vasopressin secretion. The patient is observed, without access to water, with serial measurements of plasma and urine osmolality, urine volumes and body weight over a period of up to 8 h. If only borderline deficiency is suspected then it is wise, in order to shorten the procedure, to ask the patient to take no fluids from midnight before a morning test; otherwise the patient should be allowed free fluids prior to the test to avoid severe dehydration. Simple measurement of 24 h urine volume prior to the test may guide which approach is most appropriate; if this is <2 L/24 h then DI is unlikely in the first place. If body weight falls by more than 3% during the test, this suggests severe dehydration. The test should be stopped, and plasma osmolality measured for confirmation before allowing access to fluids.
Normal vasopressin secretion is demonstrated by a urine:plasma osmolality ratio of >2:1 (or, more simply, a urine osmolality >600 mmol/kg), in the presence of a normal plasma osmolality (280–295 mmol/kg). A high plasma osmolality (>300 mmol/kg) with a urine osmolality of <600 mmol/kg is diagnostic of DI. Intermediate responses are non-diagnostic and water deprivation must be continued or repeated, until diagnostic values are obtained. Once a diagnostic response has been obtained, then desmopressin (2 μg i.m.) is administered, free fluids are allowed and urinary osmolality is measured hourly for 2 h. In the presence of DI, full concentration of the urine after desmopressin indicates vasopressin deficiency rather than nephrogenic DI.
Patients with primary polydipsia frequently commence the test with low urine and plasma osmolalities due to water overload, and may fail to concentrate the urine adequately after the standard period of water deprivation since the plasma osmolality is frequently only increased into the low–normal range during this time. The renal response to desmopressin may also be impaired under these circumstances due to ‘washout’ of the renal concentrating gradients.
Hypertonic saline infusion
Severe DI can be diagnosed easily by a water deprivation test, but mild or borderline DI, and other more subtle defects of osmoregulation and thirst, may lead to equivocal findings despite protracted water deprivation. Under such circumstances, a hypertonic saline infusion (5% NaCl infused at 0.06 mL/kg per min for 120 min) may be used to delineate the pathology more accurately. Plasma osmolality and vasopressin are measured every 30 min but, as vasopressin measurements are not performed in most laboratories, this test is often only carried out in specialist centres. Discussion of the interpretation of this test is beyond the scope of this chapter (see Chapter 4 and Baylis, in Further reading, below).
Summary
Outline protocol for the investigation of a patient with pituitary disease
The multiple tests available and the controversies regarding their use and interpretation mean that the detail in which pituitary function is investigated remains a matter for clinical judgement. The authors propose the following protocol for the initial assessment of a patient with suspected pituitary disease.
1. Measure basal pituitary function on a single 09.00 h sample (see Appendix 18.1, p. 371).
2. If plasma cortisol is <100 nmol/L, then an IST is contraindicated and ACTH deficiency can be assumed (unless Addison disease is a possibility or the patient is receiving steroids).
3. Treat thyroid or ACTH deficiencies identified on basal concentrations (hypothyroidism reduces ACTH and GH responses to hypoglycaemia).
4. If plasma cortisol is >100 nmol/L then:
b. if cortisol is <400 nmol/L, perform a tetracosactide test – a response above 600 nmol/L confirms a normal axis (see above)
c. consider IST in the presence of equivocal responses to tetracosactide (e.g. peak 400–600 nmol/L), especially if there are unexplained symptoms, or if information on GH reserve is required.
5. Treat any ACTH deficiency identified (with hydrocortisone).
6. Consider whether investigation for GH deficiency is indicated:
b. in the presence of other pituitary deficiencies, severe GH deficiency becomes increasingly likely with an increasing number of other deficiencies. If the AGHDA score suggests possibility of the clinical syndrome of adult GH deficiency, then perform a dynamic test of GH reserve, even if IGF-1 is normal
c. at the initial assessment, or first assessment after pituitary surgery, many centres routinely perform an IST to formally document ACTH and GH reserve (except where contraindicated), even in the absence of symptoms.
7. Perform a water deprivation test if indicated by symptoms or by the basal osmolalities.
A clinical approach to assessment of the whole ACTH–adrenal axis
Assessment of the ACTH–adrenal axis is probably the most common assessment of complex endocrine function undertaken in clinical medicine, and remains the subject of ongoing controversy, variation in practice, differences in interpretation, artefact due to drugs and, frequently, the use of testing in inappropriate circumstances.
Assessment of adrenal reserve is broadly undertaken to answer the following four clinical questions.
1 Does this patient have undiagnosed hypoadrenalism?
This question is frequently asked in the clinical context of hyponatraemia, hypotension and/or hypoglycaemia. A basal 09.00 h cortisol <100 nmol/L strongly suggests hypoadrenalism, and a value of >400 nmol/L makes it very unlikely (unless the patient is seriously unwell, in which case higher concentrations might normally be expected and the patient could be relatively hypoadrenal with diminished adrenal reserve).
When basal cortisol is uninformative, a short tetracosactide test is usually performed. A normal response (defined by an appropriate normal range – see above) effectively excludes adrenal deficiency. A definitely subnormal response confirms hypoadrenalism (as long as it is certain that the patient is not receiving steroid therapy), but should not immediately attract a diagnosis of Addison disease, since the prevalence of hypopituitarism is higher than that of primary adrenal failure.
2 Is the proven hypoadrenalism primary or secondary?
In a newly diagnosed patient with hypoadrenalism, the clinical picture may contribute significantly to this differential diagnosis. Associated features may point towards primary adrenal failure: typical ‘Addisonian’ electrolytes (hyponatraemia, hyperkalaemia and slightly raised urea), typical ACTH-dependent hyperpigmentation or a personal or family history of autoimmune endocrine disease. Conversely, if the patient has visual field defects, clinical signs of hypogonadism or a pituitary mass, then ACTH deficiency is much more likely. Many patients, however, do not have clear-cut clinical clues to the diagnosis and a biochemical diagnosis is essential before embarking on life-long treatment.
Pragmatically, assessment of basal pituitary function is often the simplest and quickest approach, since it will either give evidence of other pituitary dysfunction, or, if normal, suggest primary adrenal failure (or, less likely, isolated ACTH deficiency).
Measurement of 09.00 h basal cortisol, ACTH and recumbent renin is typically slower, but diagnostic, since ACTH and renin are both elevated in primary adrenal failure.
The depot tetracosactide test has been used traditionally in this context (tetracosactide depot 1 mg i.m. with cortisol measured at intervals over 24 h). In primary adrenal failure, there is an absent or flat cortisol response, whereas in ACTH deficiency or HPA suppression by steroids, the cortisol response slowly rises to reach a near-normal peak at 24 h.
If primary adrenal failure is confirmed, measurement of adrenal antibodies may confirm autoimmune Addison disease, but if this is negative, alternative causes of adrenal destruction (tuberculosis, metastases) need to be considered and excluded. If ACTH deficiency is present, then full assessment of pituitary function and pituitary magnetic resonance imaging (MRI) are indicated.
3 Has this patient with known pituitary disease developed ACTH deficiency?
This assessment has already been outlined above.
4 Does this patient on pharmacological steroid treatment still have adrenal suppression or could they now stop treatment?
Numerically, this is probably one of the most frequent situations in which adrenal function is tested (with testing frequently being performed in inappropriate circumstances or the results being incorrectly interpreted):
• high doses of inhaled, intranasal, topical or injected (for musculoskeletal disorders) steroids will also frequently suppress cortisol responses (basal concentrations may be undetectable). This often causes difficulties in interpretation, since most synthetic steroids are not measured in cortisol assays and it is impossible to differentiate biochemically between appropriate suppression and untreated deficiency
• prednisolone cross-reacts in many cortisol assays – so measurement of cortisol after a dose of prednisolone may give artefactually higher values. As a result of this, tests are often advised ‘under dexamethasone cover’. In the authors’ view, this is both unnecessary and unhelpful since the basal cortisol concentration (which gives useful information about background spontaneous cortisol secretion) will be undetectable. It is easier simply to omit prednisolone after 18.00 h on the day before testing, perform the relevant tests at 09.00 h and administer prednisolone when the test has been completed.
The authors therefore advise the following regimen when assessing whether or not pharmacological steroids can be discontinued:
• then check the basal 08.00–09.00 h cortisol one morning 24 h after the last dose of steroid and before the steroid dose is taken that morning (tablets can be taken as usual immediately after the blood test). Interpretation:
• >400 nmol/L: the adrenal axis is probably completely normal; steroids could be stopped (or slowly reduced if the underlying disease process contraindicates complete cessation immediately); further tests of adrenal axis are unnecessary unless there are unexplained symptoms
• 200–400 nmol/L: adrenal function is not completely suppressed (and might be normal or partially suppressed); steroid doses can be slowly and cautiously reduced and the tests repeated
• 100–200 nmol/L: significant adrenal suppression is more likely but will only remit if the steroid dose is reduced. Reduce steroid dose more slowly and cautiously; repeat 09.00 h cortisol 2–3 months after each dose reduction.
• if concentrations are > 300 nmol/L, and if the patient is clinically well, then steroid replacement can safely be stopped completely for several weeks in order to carry out a dynamic test of the adrenal using tetracosactide – otherwise the test may be indicated when on half-physiological dose as reassurance (it is unlikely to be normal, but profound suppression, for example a peak cortisol <300 nmol/L, would militate against further dose reduction). It may also be worth changing from a synthetic steroid to hydrocortisone, since this is shorter acting and therefore potentially less suppressive.
Even if the results of investigation are normal, it is important to remember that patients in whom the adrenal axis has been suppressed by long-term supraphysiological steroid administration may still require replacement during severe intercurrent illness for at least a year after steroids are discontinued.
Monitoring of pituitary function in disease states
Reassessment after pituitary surgery
Pituitary surgery, usually by the trans-sphenoidal route, is the recommended therapy for many pituitary adenomas. Following surgery, residual pituitary function requires careful monitoring and reassessment.
Transient DI is very common in the immediate postoperative period. However, the presence of nasal packs (required by the surgery) frequently results in a dry mouth so that the patient may drink excessive fluid, thereby causing polyuria in the absence of DI. Patients cured of acromegaly may also show polyuria in the absence of DI owing to rapid resolution of soft tissue enlargement. It is thus essential that DI is confirmed biochemically before replacement therapy is commenced. Paired urine and plasma osmolalities should be checked daily for the first few postoperative days (and measured urgently if symptoms develop) and interpreted as outlined above. In borderline cases with persisting polyuria or nocturia, a formal water deprivation test may be required following recovery.
Most surgeons and physicians give glucocorticoid replacement therapy during the immediate perioperative period. One week postoperatively, or earlier if considered clinically appropriate and the patient is well, the evening replacement dose should be omitted and 09.00 h plasma cortisol measured the following morning prior to the next dose. If a same-day cortisol result is available, the patient can remain off replacement, which should otherwise be restarted pending the result. A 09.00 h cortisol < 100 nmol/L indicates the need for continued, possibly life-long, replacement therapy. Patients with a 09.00 h cortisol > 300 nmol/L can be safely discharged without regular replacement therapy pending further investigations, but those with cortisol <400 nmol/L require steroid cover for intercurrent illnesses until the results are known. Patients with cortisol in the 100–300 nmol/L range usually need steroid replacement until definitive assessment is possible, but this must be determined by individual clinical assessment. In a retrospective review of our own practice, in which patients were investigated one week after pituitary surgery, 13% had a cortisol < 100 nmol/L and 57% >400 nmol/L – so that only a minority of patients have equivocal responses at this stage.
Basal thyroid and gonadal function should be checked one week postoperatively. In view of the long plasma half-life of T4, thyroid function should be reassessed at one month. At one week, a frankly low plasma testosterone in the male is usually diagnostic of ongoing gonadotrophin deficiency requiring replacement therapy. Lack of elevation of gonadotrophins in the postmenopausal female is also diagnostic. In both cases, borderline results require a repeat at one month. In the premenopausal female, early results may be misleading and it is wise to wait 2–3 months to assess the return of menstrual cyclicity prior to commencing replacement therapy.
A tetracosactide test is invalid as a test for ACTH reserve until at least two weeks postoperatively: the authors’ practice is to perform this test approximately one month postoperatively in patients with a non-diagnostic basal cortisol concentration. The authors only perform a dynamic test of GH reserve in patients with symptoms suggestive of GH deficiency in whom replacement is being considered, but other centres prefer an IST at this stage to document fully postoperative pituitary function.
Monitoring after pituitary irradiation
Pituitary irradiation may be employed in the treatment of pituitary tumours (although its use is declining), and the hypothalamus and pituitary are also within the field of whole brain irradiation used for other diseases. All affected patients are at risk of slowly progressive hypopituitarism over at least the next 20 years and require regular monitoring. The order of progression is typically GH deficiency occurring early, LH and FSH deficiency next, and TSH and ACTH deficiency at a later stage. The lesion appears to be primarily at hypothalamic level and PRL deficiency therefore occurs rarely, if ever.
The development of GH deficiency in children is best assessed by careful monitoring of growth – and in adults, annual screening with the AGHDA questionnaire may be appropriate to identify patients in whom a dynamic test of GH reserve is required. For all other hormones, basal 09.00 h pituitary function (see Appendix 18.1, p. 371) should be checked annually, with a subsequent tetracosactide test when serum cortisol is <400 nmol/L. (A routine annual tetracosactide test may actually prove to be a more efficient use of both the patient’s and the laboratory’s time.)
Monitoring in other pituitary disease states
Not all pituitary diseases result in progressive pituitary failure, so the need to monitor pituitary function in such patients is a matter of clinical judgement.
Patients with large pituitary mass lesions, granulomatous disease of the pituitary or hypothalamus, or unexplained acquired partial hypopituitarism certainly require monitoring of remaining pituitary function, which should be performed annually as above, at least for the first few years of follow-up. Conversely, patients with microprolactinomas whose PRL is suppressed on therapy, or patients with clearly defined isolated pituitary deficiency syndromes such as Kallman syndrome, are at minimal risk of progressive hypopituitarism and do not require regular monitoring in the absence of symptoms. Isolated GH deficiency in childhood is sometimes followed by other pituitary deficiencies in adult life, but it is as yet unclear whether this occurs sufficiently frequently to justify regular monitoring and, conversely, some such childhood cases are shown to have normal GH reserve when retested as young adults.
OTHER DIAGNOSTIC TECHNIQUES IN PITUITARY DISEASE
Clinical assessment
Endocrinologists continue to rely on clinical assessment to determine the need for further biochemical confirmation of diseases of the pituitary and hypothalamus. Investigations for possible Cushing syndrome and acromegaly are thus rarely performed unless they are suspected clinically because of the relevant symptoms and signs. Similarly, gonadotrophin deficiency is usually suspected by the development of loss of libido in both sexes, amenorrhoea in females and impotence and loss of secondary sexual hair in males. Diabetes insipidus results in thirst and polyuria, usually manifest in the earliest stages by nocturia (since urine is normally maximally concentrated overnight). Indeed, a biochemical nihilist might reasonably argue that there is no point in assessing these axes unless these clinical symptoms or signs are present.
Thyroid stimulating hormone deficiency may lead to typical symptoms and signs of hypothyroidism (see Chapter 19), although these are usually less severe than in primary thyroid disease. Adrenocorticotrophic hormone deficiency may result in general malaise, postural hypotension, headaches and abdominal pains and weight loss (pituitary cachexia). These symptoms of deficiency are clearly non-specific and biochemical assessment is essential to confirm or deny deficiency. Growth hormone deficiency in adults results in impairment of quality of life in some patients – the pattern of symptomatology is non-specific, but can be recognized clinically or through testing with quality of life questionnaires.
Masses enlarging within the pituitary or hypothalamus frequently exert pressure on the optic chiasm and thus cause visual field loss, typically a bitemporal hemianopia. Clinical assessment of visual fields by confrontation and by formal charting is thus an essential part of the assessment of patients with pituitary disease. Large lateral extensions of pituitary tumours may more rarely result in cranial nerve deficits, the cavernous sinus syndrome or even temporal lobe epilepsy, and inferior extensions very rarely cause spontaneous cerebrospinal fluid rhinorrhoea.
Finally, panhypopituitarism is an unusual cause of unexplained hyponatraemia or hypoglycaemia and should always be considered during the investigation of these disorders.
Pituitary imaging techniques
Magnetic resonance imaging (MRI) scanning of the pituitary, with and without gadolinium enhancement, is the investigation of choice and allows detailed anatomical assessment. Computed tomography (CT) scanning of the pituitary gives less detailed information, but is used when MRI is contraindicated or impossible due to the presence of metallic implants or claustrophobia.
PITUITARY HYPERSECRETION STATES
Pituitary adenomas
Pituitary hypersecretion states are most usually associated with benign pituitary adenomas synthesizing and secreting the relevant hormone. Tumours may be subdivided into microadenomas (< 10 mm diameter) and macroadenomas (> 10 mm). Pituitary tumours may be invasive but are only very rarely malignant. Hyperplasia of the various cell types has also been described but, except when secondary to ectopic secretion of hypothalamic releasing hormones, is poorly defined. Non-functioning pituitary adenomas are more common in clinical practice.
Prolactinoma
Differential diagnosis of hyperprolactinaemia
A prolactinoma, or prolactin-secreting pituitary adenoma, is the commonest functioning pituitary adenoma. However, hyperprolactinaemia has many causes other than a prolactinoma (Fig. 18.5).
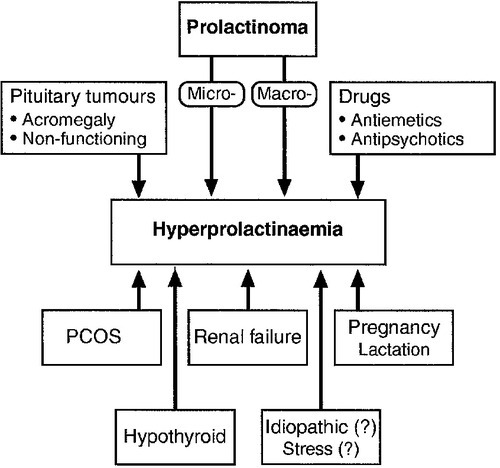
FIGURE 18.5 Causes of hyperprolactinaemia. PCOS, polycystic ovary syndrome.
Any pituitary tumour or mass that is large enough to impede pituitary portal blood flow can result in hyperprolactinaemia by preventing hypothalamic dopamine from reaching the pituitary lactotrophs. Adenomas causing acromegaly also occasionally secrete PRL directly, in addition to GH. The differentiation of a prolactinoma from a functionless pituitary tumour may therefore sometimes be difficult, although biochemical results give some assistance. A plasma PRL > 5000 mU/L almost always indicates a prolactinoma, and concentrations may reach several hundred thousands. Plasma PRL < 5000 mU/L may result from a prolactinoma, a functionless pituitary tumour (adenoma, craniopharyngioma etc.) or any of the other causes shown in Figure 18.5. It is most unlikely for a large macroprolactinoma to be associated with PRL concentrations in this range. Conversely, while a small microprolactinoma may be associated with an elevated PRL at any concentration, a small functionless microadenoma is unlikely to elevate plasma PRL into the thousands. Thus, when PRL is raised, but <5000 mU/L, the smaller the adenoma seen on CT or MRI scan, the more likely it is to be a prolactinoma and vice versa.
Any drug with dopamine antagonist effects may cause hyperprolactinaemia; this includes almost all antiemetics (e.g. metoclopramide, prochlorperazine, domperidone) and most major tranquilizers (chlorpromazine and other phenothiazines, haloperidol, risperidone etc.). A careful drug history is therefore essential in the assessment of hyperprolactinaemia. Antiemetics can often be discontinued but antipsychotic medication usually needs to be continued, although change to a newer antipsychotic with less effect on PRL may be possible (e.g. quetiapine, olanzapine and other ‘atypical antipsychotics’).
Primary hypothyroidism may be associated with hyperprolactinaemia in the low thousands (mU/L) (and with galactorrhoea and oligo- or amenorrhoea) that resolves with thyroid replacement therapy. Chronic kidney disease causes hyperprolactinaemia of a similar degree. Measurement of thyroid and renal function is thus essential in any patient with hyperprolactinaemia.
Mild and often intermittent hyperprolactinaemia (usually <1000 mU/L) is often seen in polycystic ovary syndrome (PCOS). In the appropriate clinical context (hirsutism, acne, obesity), assessment of plasma androgen concentrations and sex hormone-binding globulin (SHBG) may therefore be indicated (see Chapter 22). Major ‘stress’, for example hypoglycaemia, also elevates plasma PRL but it is far from clear whether psychological stress or fear of venepuncture is able to do so.
Finally, it should be remembered that by far the commonest causes of a state of hyperprolactinaemia and amenorrhoea are pregnancy and lactation: a pregnancy test may be indicated.
Role of dynamic tests of PRL secretion
In order to assist the differential diagnosis of hyperprolactinaemia, a number of dynamic tests of PRL secretion have been advocated and used, particularly in continental Europe. Such tests include the TRH test, metoclopramide test and domperidone test. None of these tests gives clear differentiation between prolactinomas and other causes of hyperprolactinaemia and they have no role in routine biochemical assessment.
Assessment of remaining pituitary function
Few patients with small microprolactinomas have deficiencies of other pituitary hormones, but, conversely, a functionless macroadenoma can cause severe hypopituitarism associated with mild hyperprolactinaemia. Clearly not every patient with a high PRL requires full pituitary function testing. The authors’ own (arbitrary) practice is to perform basal anterior pituitary function tests and a pituitary MRI whenever the plasma PRL is consistently >1000 mU/L, but not otherwise unless there are relevant symptoms or signs.
Outline of presentation and management of prolactinoma
In females, hyperprolactinaemia causes oligo- or amenorrhoea, galactorrhoea and breast tenderness, poor libido and infertility. Patients may present with any combination of these symptoms, with or without other signs of pituitary enlargement such as visual field defects. In males, poor libido and impotence are the only early symptoms and patients frequently present at a later stage with visual field loss, with or without headaches. Development of a prolactinoma in the peripubertal period can also result in delayed or arrested puberty in both sexes, and a serum PRL measurement is thus an essential part of the assessment of such patients.
The management of prolactinomas is primarily medical, using dopamine agonist therapy, which can suppress plasma PRL to normal and cause clinical remission of all symptoms. The long-acting dopamine agonist cabergoline is now most commonly used – bromocriptine and quinagolide are alternatives. Bromocriptine may be preferred when pregnancy is planned, in view of its longer safety record, although there is no evidence of fetal harm with the newer drugs. In addition to lowering PRL and relieving symptoms, dopamine agonist therapy also causes dramatic shrinkage of macroprolactinomas in a majority of cases, with resolution of pressure effects such as visual field loss. Such tumour shrinkage is the ultimate non-invasive method to distinguish a prolactinoma from other pituitary lesions causing hyperprolactinaemia. In a majority of prolactinomas the hyperprolactinaemia will recur if dopamine agonist therapy is stopped, so that treatment is usually long term, although a significant minority may be able to discontinue therapy.
Alternative therapies for a prolactinoma include trans-sphenoidal surgery and, rarely, pituitary irradiation.
Monitoring the response to dopamine agonist therapy
The aim of dopamine agonist therapy is to suppress plasma PRL concentrations into the normal range. The dose of dopamine agonist is usually slowly increased with sequential PRL estimations until this is achieved. Thereafter, plasma PRL would normally be measured at each clinic visit, although these need only be annual in patients on long-term therapy. Fibrotic reactions have been described as rare side-effects of long-term dopamine agonist therapy (pulmonary, retroperitoneal and pericardial); some form of monitoring is advised and the authors’ practice is to monitor for symptoms and measure urea and electrolytes, full blood count and C-reactive protein (CRP) (or plasma viscosity or erythrocyte sedimentation rate (ESR)) annually.
Macroprolactinaemia
In some patients, PRL circulates bound to immunoglobulin forming an immunoreactive PRL of high molecular weight (macroprolactin). This is biologically inactive but in molar terms may considerably exceed the concentration of free PRL. Commonly used immunoassays for PRL cross-react with macroprolactin to a variable extent, and with some assays macroprolactin is a common cause of hyperprolactinaemia. This can usually be detected by polyethylene glycol (PEG) precipitation, although gel filtration chromatography is more reliable. Laboratories should know the susceptibility of their own assay to this artefact, and always arrange to screen for macroprolactinaemia if persistent hyperprolactinaemia does not correlate with clinical symptoms or expected responses to treatment.
Hook effect
Extremely high PRL concentrations (say >100 000 mU/L) in patients with large pituitary macroprolactinomas may, rarely, exceed the usual linear range of the assay calibration curve when homogeneous assays are used, and give apparently only modest elevations not suggestive of a large prolactinoma. This can be detected by assays of doubling dilutions of serum (when measured PRL concentration fails to fall as expected and may begin to rise).
Acromegaly
Diagnosis of acromegaly
The diagnosis of acromegaly is usually suspected clinically before any biochemical tests are performed. However, by the time acromegaly is clinically obvious, the patient may have already suffered considerable morbidity from soft tissue changes, arthritis, hypertension, diabetes and consequent cardiovascular disease. Because of this, the aim should be to diagnose acromegaly at an early stage, when clinical signs are less clear cut and differentiation from normality can only be confirmed by biochemical investigations.
Basal GH and IGF-1 estimation
Random plasma GH measurements are never diagnostic of acromegaly, since high concentrations may occur with normal pulses of secretion and with stress. Nevertheless, a low plasma GH (<0.4 μg/L), together with a normal IGF-1 essentially excludes the diagnosis, whereas a high IGF-1 with consistent GH concentration makes the diagnosis very likely. It is therefore the authors’ practice to measure a random GH and IGF-1 concentration as a simple first test when acromegaly is suspected. An age-related normal range for IGF-1 must be used for interpretation since concentrations vary considerably throughout life.
Glucose tolerance test
The 75 g glucose tolerance test (GTT) remains the gold standard test for the diagnosis of acromegaly. After glucose, plasma GH in normal individuals is suppressed to <0.4 μg/L. Patients with acromegaly fail to show complete suppression and may show a paradoxical rise of plasma GH. Failure of GH to fall to <0.4 μg/L is highly suggestive of acromegaly, even if substantial suppression of GH has occurred, and an elevated IGF-1 will normally confirm the diagnosis.
Failure of normal glucose suppression of GH may occur in the absence of acromegaly in liver disease, poorly controlled diabetes, chronic kidney disease, malnutrition (including anorexia nervosa) and primary growth hormone insensitivity (Laron syndrome). Diabetes can be a particular diagnostic challenge, since a GTT is inappropriate in established disease. Insulin-like growth factor 1 concentrations, however, are typically normal in uncomplicated diabetes, and, if elevated, the authors usually recommend a four-point GH day curve to confirm circulating GH concentrations consistently above the ‘safe’ range.
Other diagnostic tests
Patients with acromegaly may show a paradoxical rise in plasma GH during standard TRH or LHRH tests. When acromegaly has been clearly diagnosed by other means, these tests give no additional clinically useful information, but they may help confirm mild acromegaly in patients when the results of other tests are equivocal.
In normal individuals, plasma GH shows a transient rise during a dopamine infusion (4 μg/kg per min for 3 h), whereas patients with acromegaly characteristically show a fall in plasma GH. This test can also be used to confirm an equivocal diagnosis but is not used routinely.
Ectopic secretion of GHRH, for example from a pancreatic endocrine tumour, is a rare cause of clinical acromegaly. The diagnosis is not usually suspected until somatotroph hyperplasia is found at trans-sphenoidal surgery rather than the expected adenoma. In these circumstances, peripheral plasma GHRH concentration (available only in a few specialist centres) is elevated and diagnostic.
Outline of presentation and management of acromegaly
Acromegaly is usually diagnosed clinically when the patient presents with an incidental illness to a doctor who recognizes the typical physical appearance, which is frequently longstanding and only slowly progressive.
Growth hormone-secreting tumours that develop in childhood result in gigantism, but after puberty, fusion of the major epiphyses means that growth in height is no longer possible. Patients with acromegaly do, however, develop excessive growth of soft tissues and small bones of the skull, face, hands and feet, which results in the characteristic clinical features. They have large, ‘spade-like’ hands and large, broad feet, usually with a history of increasing ring size and shoe size over a number of years. Facial appearance is characteristic with prognathism, prominent supraorbital ridges, large nose and tongue and generalized coarsening of the features. Examination of old photographs will frequently demonstrate that these changes have been progressive over 5–10 years or more. Headaches, often severe, and troublesome, inappropriate sweating are common symptoms; other findings frequently include a multinodular goitre, kyphosis, hypertension and diabetes mellitus, the last two presumably contributing to the observed increase in cardiovascular morbidity and mortality in this condition.
Patients with acromegaly often develop deficiencies of other pituitary hormones, and full investigation of pituitary function is mandatory once the diagnosis has been made.
Although acromegaly frequently goes undiagnosed for many years, treatment is almost always indicated since, if left untreated, the condition results in disfigurement, morbidity (primarily due to kyphosis and osteoarthritis) and mortality (primarily cardiovascular, but possibly also an increase in malignancies).
First-line therapy in almost all patients is trans-sphenoidal surgery, which is able to cure at least 75% of microadenomas but a much smaller proportion of large macroadenomas, some of which are locally invasive. A number of studies have shown substantial differences in success rates between surgeons and centres, and an experienced pituitary surgeon is essential. Unsuccessful pituitary surgery is traditionally followed by pituitary irradiation, which steadily lowers GH secretion, but may require 10–20 years to normalize GH concentrations in patients where the initial GH is very high (over 40 μg/L). Medical therapy with dopamine agonists such as high-dose cabergoline (e.g. 0.5 mg daily) is useful in a proportion of patients. The potent, long-lasting somatostatin analogues octreotide and lanreotide are able to normalize GH concentrations in the majority of patients (but not all), albeit at the expense of monthly injections and a high economic cost. Both classes of drugs may allow significant shrinkage of a somatotroph adenoma preoperatively. More recently, the GH antagonist pegvisomant has been shown to be capable of normalizing IGF-1 in almost every patient, but treatment requires daily injections and is extremely expensive. The success of these drugs has led some authorities to recommend long-term medical therapy instead of radiotherapy (or even surgery), but this is not currently routine practice in most centres.
Monitoring the response to therapy
There has been considerable controversy over the years regarding the appropriate biochemical criteria to diagnose remission or cure of acromegaly after therapy, but results of large retrospective studies and consensus meetings have achieved much greater uniformity. The current consensus criteria for disease control in acromegaly define active and controlled disease. The criteria for active disease are a random GH > 1 μg/L and nadir GH after OGTT ≥0.4 μg/L with an elevated IGF-1. Patients with active disease usually require closer monitoring to ensure the clinical and biochemical picture does not deteriorate, and would be considered for additional treatment if the clinical picture were to deteriorate or the GH increases to >2 μg/L. The criteria for controlled disease are a random GH <1 μg/L or nadir GH after OGTT <0.4 μg/L and an age-sex normalized IGF-1 for age and sex. Occasionally after treatment for acromegaly there is a discrepancy between GH and IGF-1 concentrations, most commonly involving a normal GH and elevated IGF-1 concentration. Although the aim of management should be to normalize both GH and IGF-1, it is currently felt that a normal GH level concentration is probably the more important determinant of cardiovascular risk reduction. The current literature suggests that a GH <2 μg/L is probably within the safe range for cardiovascular disease, although there remains debate about whether adjuvant treatment is indicated for patients who have discordantly elevated IGF-1 despite acceptable growth hormone concentrations.
Diagnosis and differential diagnosis of Cushing syndrome
Endogenous Cushing syndrome is rare, with an incidence of approximately 5 per million population per year. However, many of the clinical features of the syndrome are very common (weight gain, hypertension, hirsutism, diabetes) so that biochemical tests are frequently requested to exclude the diagnosis and are essential to confirm its presence. Clinical features certainly increase the suspicion of Cushing syndrome (easy bruising, thin skin, red/purple abdominal striae, proximal myopathy, specific distribution of fat, e.g. supraclavicular and dorsal fat pads in addition to central obesity). However, many patients with biochemically and surgically proven Cushing disease are not as grossly ‘cushingoid’ as seen in a patient on high-dose pharmacological steroids or as illustrated in older medical student textbooks; they are often overweight rather than obese but with a history of unexpected recent weight gain and change towards central distribution of fat.
Measurement of urinary free cortisol (UFC) is frequently used as a first-line screening test for Cushing syndrome. It has the advantage of simplicity, but the disadvantage that the diagnosis may be missed if urine collection is incomplete, and of false positive elevations of urinary free cortisol in obesity and stressful illness.
Many clinicians will therefore use some form of low-dose dexamethasone suppression test as their preferred first-line screening test.
Low-dose dexamethasone suppression test
Dexamethasone 0.5 mg is administered 6-hourly for 48 h commencing at 09.00 h, with plasma cortisol measured at 09.00 h at the end of the test. Normal response is a plasma cortisol <50 nmol/L (and undetectable in most normal individuals). Many regard this test as too complex – but it still potentially involves only one blood sample and one prescription, and patients can easily follow the administration protocol as an outpatient if given a simple information sheet. It is sensitive and much more specific than the overnight test and is the authors’ preferred screening test.
Overnight dexamethasone suppression test
Dexamethasone 1 mg is administered on going to bed at 23.00 h and plasma cortisol is measured at 09.00 h. Suppression of cortisol to <50 nmol/L is unequivocally normal and because it is a simple and sensitive investigation, it is preferred by many clinicians, although it has a higher false positive rate than a formal 48 h test.
Midnight salivary cortisol
This test is gaining increasing popularity for diagnosis of Cushing syndrome, particularly in the USA. In normal individuals, plasma cortisol concentration is low at midnight, when resting or asleep, whereas in Cushing syndrome, the circadian rhythm is lost or reduced. A midnight sleeping plasma cortisol < 100 nmol/L has traditionally been used to exclude Cushing syndrome, but the practicalities of inpatient bed and junior staff availability mean that this test is now rarely practical in the UK. More recent data suggest that an awake value > 207 nmol/L may have a higher specificity and is easier than sampling whilst the patient is asleep. A saliva sample can be collected by the patient at home and midnight salivary cortisol has been shown to be a useful screening test, although the precise cut-off value depends upon the local reference range and the assay is not yet widely available in clinical practice.
Other tests
All tests for diagnosis and differential diagnosis are subject to false positive and false negative results. Where the result of first-line screening tests are in conflict with the clinical findings, or where doubt exists, then other tests are sometimes performed: repeated UFC measurements to establish the presence or absence of ‘cyclical Cushing syndrome’; a formal ‘circadian rhythm’ assessment of plasma cortisol as an inpatient; an insulin stress test (IST) to demonstrate suppression of the cortisol response to stress, which is typically seen in Cushing syndrome. The desmopressin stimulation test, where patients with pituitary Cushing disease typically show a response, and normal individuals do not, is no longer felt to be sufficiently useful for routine clinical practice.
At this stage, advice from a specialist tertiary centre is usually required. Scanning of the adrenals or pituitary is not advised until the diagnosis of Cushing syndrome is clearly established, since ‘incidentalomas’ of both the pituitary and adrenals are common and may lead to an inappropriate diagnosis.
Once Cushing syndrome has been diagnosed, measurement of plasma ACTH can readily distinguish primary adrenal causes from ACTH-dependent Cushing syndrome. The former can be readily confirmed on CT or MRI scan of the adrenals, but the differential diagnosis of the latter remains a challenging clinical problem (see Fig. 18.6).
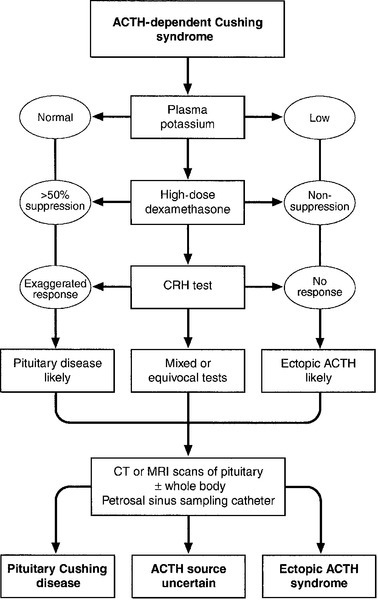
FIGURE 18.6 Strategy for the differential diagnosis of ACTH-dependent Cushing syndrome.
Clinical context of ACTH-dependent Cushing syndrome
Adrenocorticotrophic hormone-dependent Cushing syndrome may be due either to pituitary Cushing disease, usually associated with a corticotroph microadenoma, or to ectopic ACTH secretion (and very rarely ectopic CRH secretion) by a variety of tumours elsewhere.
Numerically, the commonest source of ectopic ACTH secretion is small cell carcinoma of the bronchus. In this case, the clinical presentation is usually rapid and is often with symptoms directly related to the primary tumour. Although plasma ACTH and cortisol concentrations can be extremely high, such patients are rarely clinically cushingoid but may have pigmentation (due to the ACTH), proximal myopathy and hypokalaemic alkalosis. Such patients rarely present diagnostic difficulties.
In contrast, other, often relatively benign neuroendocrine tumours, can also secrete ACTH, particularly carcinoid tumours of the lungs (the commonest), mediastinum and elsewhere; pancreatic endocrine tumours; medullary thyroid carcinomas and, rarely, phaeochromocytomas. Such tumours are small, slow growing and clinically occult and these patients may present with typical Cushing syndrome indistinguishable from that seen in pituitary-dependent disease. Such cases represent 10–15% of ACTH-dependent Cushing syndrome in most large series. Since these tumours may be only a few millimetres in diameter and thus hard to localize clinically or by conventional imaging techniques, biochemical tests are of paramount importance in their differentiation from pituitary Cushing disease.
Plasma cortisol and ACTH concentrations
In pituitary disease, plasma ACTH is often <100 ng/L, but higher values (up to at least 400 ng/L) are not infrequently seen. While, on average, plasma ACTH (usually > 100 ng/L), and thus plasma cortisol, are higher in occult ectopic ACTH secretion than in pituitary disease, there is overlap and values usually lie within the range observed in Cushing disease.
Chromatography of plasma ACTH has been employed in the past to identify large molecular weight ACTH precursors, which are frequently secreted ectopically by tumours, but also occasionally by pituitary tumours. The complexity of these techniques means that they are no longer generally available, although immunometric assays specific for ACTH precursors may sometimes provide equivalent information.
High-dose dexamethasone suppression test
The high-dose dexamethasone suppression test was initially described in 1960 for the differentiation of adrenal from pituitary causes of Cushing syndrome. While largely superseded in this role, it remains useful in the differentiation of occult ectopic ACTH secretion.
A number of dosage regimens have been described, but the authors prefer the original regimen, with which there is greatest experience. Dexamethasone is administered orally, 2 mg strictly six-hourly for 48 h, commencing at 09.00 h. Plasma cortisol is measured at 09.00 h (basal value) and after 24 h and 48 h of treatment. ‘Significant suppression’ is arbitrarily defined as a fall in plasma cortisol to 50% or less of the basal value.
Using such criteria, about 80% of patients with Cushing disease, but only 10% of patients with occult ectopic ACTH secretion, show significant suppression of plasma cortisol. Thus differentiation is far from absolute, but since pituitary disease is far more common this means that significant suppression during the test statistically favours pituitary disease by approximately 70:1, whereas patients showing no suppression at all are equally likely to have a pituitary or ectopic source.
Corticotrophin releasing hormone test
Following the isolation and synthesis of the 41-amino acid CRH, the CRH test (see Appendix 18.1, p. 372) has gained an important role in the differential diagnosis of ACTH-dependent Cushing syndrome.
Following administration of CRH (in the UK, usually human CRF-41, but in the USA, typically ovine CRF-41, in both cases 100 μg i.v.), most patients with pituitary Cushing disease show a rise in plasma ACTH and therefore cortisol (defined as an increase of 25% above basal). In about 80% of patients with pituitary disease, the rise in plasma cortisol is ‘exaggerated’, that is, above the peak value observed in normal individuals. It is of interest and of practical importance that patients who fail to suppress with high-dose dexamethasone usually do show a cortisol response to CRH. In contrast, a significant cortisol response to CRH in ectopic ACTH secretion is extremely rare and limited to one or two isolated case reports in the world literature.
Other tests
An unprovoked hypokalaemic alkalosis remains an important clue to the presence of ectopic ACTH secretion, since over 90% of such patients present with hypokalaemia, compared with only 10% of patients with pituitary Cushing disease.
In the context of ectopic ACTH, tumours frequently also secrete other peptide hormones so that one or more tumour markers may be positive in the blood of such patients. It is therefore the authors’ practice in patients with equivocal tests to screen plasma for tumour markers including calcitonin, human chorionic gonadotrophin (hCG), α-fetoprotein, carcinoembryonic antigen (CEA) and ‘gut hormones’, any of which may occasionally prove to be positive.
Petrosal sinus sampling for ACTH
It can be seen from the discussion above that no test is completely reliable in the differential diagnosis of ACTH-dependent Cushing syndrome. Because of this, techniques have been developed to ascertain directly the source of ACTH secretion via bilateral, simultaneous, inferior petrosal sinus sampling (IPSS) for ACTH after CRH stimulation.
Venous sampling catheters are passed from both femoral veins to lie in the respective inferior petrosal sinuses, which drain blood from the cavernous sinus and pituitary gland. The patient is partially heparinized prior to the final placement of the catheters to avoid the risk of venous thrombosis affecting the brain, which has been reported rarely. Blood for ACTH estimation is obtained simultaneously from both sides and from a peripheral vein, at timed intervals before and after an intravenous bolus of CRH. It is the authors’ practice to administer TRH (200 μg) at the same time to measure central secretion of TSH and PRL to confirm the equivalence of catheter placement since, in the authors’ experience, unequal sampling is frequently seen despite good radiological appearances.
After a technically successful procedure, the vast majority (all in most series) of patients with pituitary Cushing disease show a central:peripheral gradient of ACTH concentration of >2:1 basally or >3:1 after CRH, which is diagnostic of pituitary disease. Significant lateralization of ACTH secretion may also be observed and may guide the surgeon and improve the surgical cure rate. It is the authors’ practice to perform IPSS in almost every patient with ACTH-dependent Cushing disease without an obvious ectopic tumour or pituitary macroadenoma, as part of the routine differential diagnostic work-up; others reserve the test for situations where other tests are contradictory. The relative complexity of the technique means, however, that it should be restricted to those centres with experience in its use.
Venous sampling for ACTH from a wide variety of sites throughout the body, using a catheter in the central veins, occasionally may also give useful information, especially when directed towards a suspicious lesion detected on body CT, MRI or positron emission tomography (PET) scanning.
Imaging
Although imaging plays an important role in the differential diagnosis of Cushing syndrome, it is important to interpret the biochemical findings as the imaging may be misleading. An MRI scan of the pituitary is frequently normal in pituitary Cushing disease, and may be abnormal in 10% of normal individuals (and therefore in ectopic ACTH patients). In some patients, where the suspicion of ectopic ACTH secretion is high on biochemical grounds, detailed CT scanning of the entire thorax and abdomen may be necessary. An appropriate diagnostic strategy is summarized in Figure 18.6.
Outline of management
The first-line treatment of Cushing disease is now selective adenomectomy at trans-sphenoidal surgery, which results in cure in about 75% of cases, varying from series to series and with the biochemical criteria used to diagnose ‘cure’ (see below). The earlier alternatives of bilateral adrenalectomy or pituitary irradiation are now usually reserved for patients who are not cured by pituitary surgery.
Medical therapy is also possible for Cushing disease of all types, but is usually reserved for preoperative preparation or in patients where surgery has failed or is impossible. Commonly used drugs include metyrapone and ketoconazole. The aim of therapy is to maintain mean circulating cortisol concentrations between 150 and 300 nmol/L, but care must be taken to ensure that the cortisol assay used does not cross-react with precursors above the drug-induced block in cortisol synthesis (11-deoxycortisol in the case of metyrapone).
Reassessment after pituitary surgery
As with acromegaly, definition of ‘cure’ after trans-sphenoidal surgery for Cushing disease remains a controversial area. Many of the larger series reporting success of this type of surgery have failed to define their criteria for ‘cure’, and some appear to be assessed only by clinical remission or return of UFC to within the reference range.
After successful pituitary surgery, most patients have adrenal insufficiency, since the normal hypothalamo-pituitary axis is fully suppressed by the longstanding hypercortisolaemia. To be confident of ‘cure’, a 09.00 h cortisol, off replacement therapy, at the early preoperative reassessment should be subnormal, that is, < 100 nmol/L (some authors insist on undetectable concentrations). Other authors suggest that a concentration < 140 nmol/L in the context of clinical improvement may suggest remission. Occasionally, delayed remission may occur, although a basal cortisol > 140 nmol/L after six weeks post-pituitary surgery is an indication that further treatment is needed. In cases of doubt, a formal 48 h low-dose dexamethasone suppression test should be performed (0.5 mg, orally, strictly six-hourly for 48 h, commencing at 09.00 h); normal individuals suppress plasma 09.00 h cortisol to undetectable values (< 25 nmol/L in the authors’ laboratory) at the end of this test, and failure to show such complete suppression indicates residual disease, albeit mild in many cases.
Patients in remission from Cushing disease require glucocorticoid replacement therapy for several months or even years. To assess the possibility of steroid withdrawal, a 09.00 h cortisol should be measured periodically, having omitted the early morning replacement dose and the evening dose the day before. When cortisol rises to > 100 nmol/L, the replacement dose can be slowly reduced, with continued monitoring of cortisol concentrations and a tetracosactide test or insulin stress test (IST) performed once replacement is completely withdrawn. Patients successfully withdrawn from replacement therapy require periodic monitoring, with UFC and/or low-dose dexamethasone suppression testing to detect early evidence of relapse.
Thyroid stimulating hormone-secreting adenomas
Thyroid stimulating hormone-secreting pituitary adenomas (TSHomas) are rare, but occasional cases will be encountered in most centres. Patients typically present with mild clinical and biochemical thyrotoxicosis with an inappropriately ‘normal’ or slightly elevated plasma TSH.
The differential diagnosis of a patient with a raised free T4 and normal or elevated plasma TSH should include thyroid hormone resistance (THR), and assay interference. Although only patients with TSHomas should be clinically hyperthyroid, it is often difficult to distinguish TSHomas from THR or assay interference on clinical grounds alone, and other tests are often needed. Patients with THR typically show a rise in TSH after injection of TRH, whereas a flat response is usually seen with a TSHoma, aiding the differential diagnosis. The α-subunit (of TSH) can also be measured by some centres and is typically elevated with a TSHoma but not with THR. Analysis of the thyroid hormone receptor gene (or more simply measuring thyroid function in family members) may clinch the diagnosis of THR. Measurement of sex hormone binding globulin (SHBG) is helpful in the differential diagnosis because SHBG tends to be normal in the thyroid hormone resistance and elevated in the TSHoma due to the increase in SHBG produced by the liver in response to thyrotoxicosis.
Treatment of TSHoma is by somatostatin analogue therapy and/or pituitary surgery, with or without pituitary irradiation and carbimazole therapy.
Gonadotrophin-secreting adenomas
Clinically functioning gonadotrophin-secreting adenomas of the pituitary are extremely rare. In the male, they present with elevated plasma testosterone, usually with enlarged testes, in the presence of ‘normal’ or elevated LH and FSH. In the female, the condition may mimic premature ovarian failure, with amenorrhoea and elevated gonadotrophins. Of theoretical interest, however, is the experimental observation that a majority of so-called functionless pituitary adenomas are able to synthesize and secrete LH, FSH or their common α-subunit in vitro, the significance of which is unclear.
HYPOTHALAMIC AND PITUITARY DEFICIENCY STATES
Diseases that may lead to generalized hypopituitarism
Non-functioning pituitary adenomas
Pituitary adenomas that do not result in the hypersecretion syndromes described above are described as non-functioning or functionless pituitary adenomas. As mentioned above, these can frequently be shown experimentally to be capable of synthesis of LH and FSH and occasionally of any other pituitary hormone, in subclinical amounts. Such patients usually present either with symptoms and signs of hypopituitarism or with other signs of pituitary tumour enlargement, primarily visual field loss. Treatment is by pituitary surgery, followed by irradiation where appropriate. Although such adenomas do not show dramatic shrinkage in size with dopamine agonist therapy, recent evidence suggests that these drugs may reduce the risk of recurrence after successful surgery (or during conservative observation with smaller tumours).
Other pituitary and parasellar tumours
Craniopharyngiomas, derived from remnants of Rathke’s pouch, are the only other common type of primary pituitary tumour. These usually present in childhood with poor growth, hypopituitarism and signs of pituitary enlargement, but may also present for the first time in adult life. Malignant tumours from elsewhere in the body occasionally metastasize to the pituitary; in addition to the usual signs of pituitary tumour, severe headache is a frequent symptom.
A wide variety of other tumours may also present with symptoms and signs of a pituitary or hypothalamic mass including hypothalamic germinomas, meningiomas, optic nerve and other gliomas, chordomas, hamartomas and pineal tumours.
Inflammatory diseases and disorders of unknown aetiology
Inflammatory conditions involving the hypothalamo–pituitary axis are rare and typically present with an unusual pattern of hypopituitarism in conjunction with diabetes insipidus. Sarcoidosis may cause granulomatous masses in the pituitary and hypothalamus, with or without evidence of disease elsewhere; measurement of plasma angiotensin-converting-enzyme concentration may, therefore, occasionally be helpful in the diagnosis of unexplained pituitary masses. Other granulomatous diseases that may present in the same way include tuberculosis and histiocytosis X.
Lymphocytic hypophysitis is an inflammatory disorder of unknown but possibly autoimmune aetiology; it occurs most frequently during pregnancy or the puerperium, but may occur at other times and less frequently in the male. Granulomatous hypophysitis is probably part of the same spectrum of diseases. Idiopathic pituitary fibrosis is also rarely seen and may be associated with other midline fibrosis syndromes. All these conditions may present with hypopituitarism or with signs of a pituitary mass.
Other conditions
Sheehan syndrome is hypopituitarism due to pituitary infarction secondary to massive postpartum haemorrhage and usually presents with failure of lactation and amenorrhoea in the puerperium. New cases should be rare with modern obstetric management, but some patients continue to present in later life, usually with a history of amenorrhoea since the birth of the last child, sometimes 30–40 years previously; diagnosis of such patients is usually precipitated by intercurrent illness.
Pituitary apoplexy is caused by pituitary haemorrhage or infarction, often associated with a previously undiagnosed pituitary adenoma. Patients may present with headache, visual loss, symptoms of hypopituitarism and, sometimes, meningism due to the presence of blood in the subarachnoid space; an initial working diagnosis is often meningitis or sub-arachnoid haemorrhage. Surgical decompression is frequently required. Head injury, especially when associated with skull fractures through the pituitary fossa, may also be associated with hypopituitarism, including diabetes insipidus. Congenital developmental disorders, such as septo-optic dysplasia, may also have hypopituitarism as part of a larger syndrome.
Finally, a substantial number of patients with isolated and generalized hypopituitarism have ‘idiopathic’ disease, with no lesion demonstrable on CT scanning. These presumably represent selective failure of the hypothalamic neurons that secrete the relevant releasing hormone. At least some of these cases may be autoimmune in origin, but more research is required to define their precise aetiology.
Growth hormone deficiency
Growth hormone deficiency most frequently arises in the context of other pituitary diseases discussed above. Isolated, idiopathic GH deficiency is, however, also relatively common and usually presents with short stature and poor growth in childhood. The most important feature of the management in childhood is accurate measurement of growth and growth velocity, usually followed by assessment of GH reserve if growth is falling away from the third centile on standard charts. Treatment of established GH deficiency is with biosynthetic human GH (somatotrophin).
There are, however, many other causes of short stature in the absence of GH deficiency. These include hypothyroidism, chronic kidney disease, malabsorption, Crohn disease and other inflammatory disorders (which may be otherwise asymptomatic), asthma, congenital heart disease, Turner syndrome and other congenital disorders, and familial short stature. Laboratory investigations clearly have a major role in excluding a number of these possibilities.
Primary growth hormone insensitivity (Laron syndrome) is a rare congenital cause of severe short stature caused by absence or mutation of the GH receptor. Circulating GH concentrations are high and nonsuppressible, in the presence of low or absent circulating IGF-1 concentrations. Therapy with recombinant IGF-1 is possible.
Growth hormone deficiency in adults was previously considered to be of no significance. However, many studies have now shown that treatment of such adults with somatotrophin may improve quality of life, decrease body fat and increase bone density and muscle mass and strength. In the UK, the National Institute for Health and Care Excellence (NICE) have recommended replacement therapy under strictly defined circumstances.
Gonadotrophin deficiency
Gonadotrophin deficiency may present at birth with an undervirilized male, with failure of pubertal development or with loss of previously normal gonadal function. Isolated gonadotrophin deficiency may be idiopathic, but is sometimes familial and associated with anosmia (Kallman syndrome) and sometimes cleft lip or palate.
Treatment of gonadotrophin deficiency is with the relevant gonadal steroids, testosterone or oestrogen, given by mouth, injection, implant or transdermally. If fertility is desired, complex regimens involving gonadotrophin therapy or pulsatile subcutaneous administration of GnRH are required.
Gonadotrophin deficiency is diagnosed by the combination of low plasma gonadal steroid concentrations with low or inappropriately ‘normal’ concentrations of LH and FSH. However, a large number of other conditions may also produce this biochemical pattern in the absence of organic gonadotrophin deficiency, particularly in the female (see Fig. 18.7).
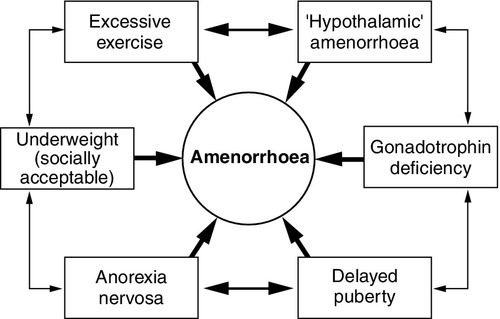
FIGURE 18.7 Inter-related conditions causing amenorrhoea and the biochemical picture of low oestradiol without elevation of gonadotrophins.
Interpretation of borderline testosterone concentrations
A common clinical and biochemical dilemma is the interpretation of an isolated testosterone that is near or below the lower end of the reference range (say 7.5–12 nmol/L), with normal gonadotrophins, in the presence of commonly occurring symptoms that might indicate hypogonadism (e.g. low libido or erectile dysfunction). The challenge is to differentiate gonadotrophin deficiency from artefact or from normal outliers, the effects of ageing, obesity and insulin resistance, or other illness.
One of the most common reasons for this pattern is blood sampling in the afternoon or evening, since testosterone concentrations show a significant circadian rhythm. The first approach is therefore to repeat the sample at 09.00 h, together with measurement of PRL (since hyperprolactinaemia may suppress LH/FSH and cause this picture). If a low testosterone is confirmed, then it is essential to assess remaining basal pituitary function (see above); measurement of SHBG and calculation of bioavailable testosterone may also assist, although total testosterone seems to be an equally effective measure in all but borderline cases (see Morris et al. in Further reading, below). Magnetic resonance imaging of the pituitary is also usually indicated to exclude a structural lesion.
However, even after such assessments, one is frequently left with a low or borderline testosterone and no other biochemical or radiological evidence of pituitary disease. In this situation, controversy remains: testosterone falls slowly with ageing and some workers (and many pharmaceutical companies) have advocated testosterone replacement for men with borderline testosterone and a wide variety of specific and non-specific symptoms of ill health, while others argue that this represents the normal pattern of ageing and does not require treatment. Long-term outcome studies are still awaited to answer this question. Low testosterone concentrations have also recently been described in patients with cardiac disease, with improvements in general health and cardiac function on replacement, but these data remain preliminary.
Delayed puberty
Prepubertal children have low or undetectable concentrations of testosterone or oestradiol, with low-normal concentrations of gonadotrophins. In the absence of a demonstrable pituitary lesion or other pituitary deficiencies it is, therefore, impossible to differentiate biochemically isolated gonadotrophin deficiency presenting with failure of sexual development from constitutional delayed puberty, the only difference being that the former does not resolve with time.
Constitutional delay of growth and puberty (see also Chapter 21) presents a combined problem of short stature and delayed puberty mimicking combined GH and gonadotrophin deficiency. Such patients, usually male, present in their mid-teens growing poorly, with height below the third centile and with minimal or no signs of pubertal development. The aetiology is unknown, but a family history is common and most patients eventually enter a delayed puberty with a growth spurt to attain normal adult height. Growth hormone reserve, assessed by standard dynamic tests, is usually normal. Treatment, if any, is usually with low doses of appropriate gonadal steroids to slowly advance puberty and initiate a normal growth spurt.
Hypothalamic amenorrhoea
As illustrated in Figure 18.7, a variety of related conditions in the female can lead to amenorrhoea associated with the typical basal biochemistry of gonadotrophin deficiency.
Weight-related amenorrhoea frequently occurs when body weight falls below the normal healthy range for height (BMI 18.5–25 kg/m2). Anorexia nervosa is an extreme example, almost always associated with amenorrhoea and regression of secondary sexual characteristics, but dieting to achieve a weight which is ‘socially acceptably skinny’ can also induce amenorrhoea. Weight gain to a normal weight will usually restore menstruation, but chances of resumption of regular cycles are < 10% with a BMI < 18 kg/m2 and only 50% at a low–normal BMI of 20 kg/m2. Low body weight is also an important cause of delayed puberty in both sexes.
A large proportion of international standard competitive athletes develop exercise-related amenorrhoea owing to their intensive training regimens. Non-competitive athletes undertaking excessive exercise may also develop amenorrhoea or delayed puberty, often exacerbated by low body weight. The incidence of amenorrhoea is directly related to the amount of exercise taken, and if exercise is discontinued for any reason, including injury, menstruation usually returns.
Psychological stress is frequently quoted anecdotally as a cause of amenorrhoea, for example in female students attending university for the first time or undertaking examinations. ‘Hypothalamic amenorrhoea’ is a descriptive label applied to such patients and to other cases of amenorrhoea with low oestradiol and gonadotrophins and no identifiable cause, in whom organic gonadotrophin deficiency is felt to be unlikely.
There is no absolute way to differentiate these causes of hypothalamic amenorrhoea from gonadotrophin deficiency, except by their reversibility. If the disorder is mild (e.g. borderline underweight), then a clomifene test (see above) may clinch the diagnosis by inducing a rise in gonadotrophins and subsequent menstruation, but lack of response to clomifene may also occur if the disorder is severe.
If the cause of the amenorrhoea cannot be reversed, then treatment is usually with oestrogen replacement therapy to prevent the long-term effects of oestrogen deficiency.
Other isolated anterior pituitary deficiencies
Isolated ACTH deficiency occurs rarely and can only be diagnosed by full pituitary function testing. The adrenals themselves may be shown to be normal either by the finding of a low plasma ACTH prior to replacement therapy or by a depot (long) tetracosactide test. A proportion of such patients show an ACTH and cortisol response to the CRH test, indicating that the deficit is one of hypothalamic CRH secretion, but this is largely of academic interest.
Isolated TSH deficiency is also a rare occurrence.
Diabetes insipidus
Since vasopressin is synthesized by cell bodies within the hypothalamus and not controlled by the portal venous system, vasopressin deficiency is rarely found in primary pituitary disease, unless treated surgically. Diabetes insipidus is therefore a most unusual presenting symptom of a pituitary adenoma and its presence should suggest that the lesion is either a craniopharyngioma or other rarer type of tumour or inflammatory disease.
Isolated central DI may occur spontaneously. Its aetiology is unknown; some cases may be due to hypothalamic autoimmunity and it may also be familial, presenting in early childhood and typically associated with mutations in the vasopressin-neurophysin-II gene. Diabetes insipidus is discussed further in Chapter 4.
ADRENAL DISEASE
Clinical features of Addison disease
Addison disease implies primary adrenal failure: it is rare, with an incidence of 3–4/million per year and overall prevalence of 40–60/million. The disease involves destruction of the whole adrenals, today most commonly caused by autoimmune adrenal failure; in Addison’s time, tuberculosis of the adrenals was also a common cause and this remains an occasional cause today, as do other infiltrations and tumours involving the adrenals.
In the absence of another acute stressful illness, the symptoms of Addison disease are often vague and non-specific, including tiredness, weight loss, anorexia, depression, abdominal pains, nausea and vomiting and other aches and pains. The rise in plasma ACTH caused by loss of feedback results in increasing pigmentation, classically in palmar creases, scars, pressure areas and buccal mucosa, but which often simply appears as an increased ‘tan’ and is ignored. Patients who are not acutely unwell may often have normal biochemistry unless plasma cortisol concentration is measured.
During acute intercurrent illness including infection, trauma or surgery but especially with vomiting and diarrhoea or other causes of fluid loss, the undiagnosed or untreated patient may become very unwell. Features of an ‘Addisonian crisis’ include dehydration, hypotension, hypoglycaemia and a typical biochemical picture of hyponatraemia, hyperkalaemia and slightly raised plasma urea and creatinine concentrations; untreated this may proceed to circulatory collapse and death. Treatment is a medical emergency and involves intravenous steroids (hydrocortisone 100 mg i.v. stat, repeated six-hourly), 0.9% saline and 5% dextrose if hypoglycaemic. Ideally, plasma cortisol concentration should be measured on the sample taken prior to steroid treatment, and this alone may be diagnostic if found to be low in the face of severe illness.
Patients commonly have a personal or family history of other autoimmune disease including vitiligo, primary hypothyroidism, hypoparathyroidism, premature ovarian failure, pernicious anaemia and occasionally inflammatory bowel disease. Full blood count, calcium and thyroid function should therefore always be measured both acutely and after steroid replacement (since untreated hypoadrenalism is also associated with hypercalcaemia, elevation of TSH with low free T4, and macrocytosis, all of which may resolve when appropriate steroid replacement is given).
Congenital adrenal hyperplasia
A gene mutation resulting in deficiency of an enzyme involved in steroid biosynthesis (see Fig. 18.4) will cause inability to secrete adequate circulating cortisol, increased ACTH concentrations and adrenal stimulation due to loss of feedback, and therefore increase in steroid precursors prior to the malfunctioning enzyme. These conditions are described as congenital adrenal hyperplasia (since the adrenal increases in size under ACTH stimulation) and are summarized in Table 18.1. Of the subtypes, only 21-hydroxylase deficiency is at all common.
The diagnosis is typically made in the neonatal period in the context of ambiguous genitalia in females, or evidence of acute hypoadrenalism (including a neonatal ‘salt-losing crisis’), but is sometimes delayed and, rarely, occurs at or after puberty in so-called ‘late onset’ 21-hydroxylase deficiency. Biochemical diagnosis involves demonstration of elevated concentrations of steroid precursors in plasma (17α-hydroxyprogesterone in the case of 21-hydroxylase deficiency) or in urine by gas chromatography-mass spectrometry (GC-MS). In common forms of the condition, adrenal androgen concentrations are also raised (dehydroepiandrosterone sulphate (DHEAS), androstenedione, testosterone), which causes virilization in the female, but the rarer 17α-hydroxylase deficiency prevents androgen as well as cortisol synthesis, and causes a female phenotype in genetic males as well as hypertension due to increased mineralocorticoid production.
Treatment is by steroid replacement. A variety of regimens (hydrocortisone, prednisolone and dexamethasone) is used with no hard evidence regarding the best; fludrocortisone replacement is also required in salt-losing types. In 21-hydroxylase deficiency, treatment may be monitored biochemically by measurement of 17α-hydroxyprogesterone (a low concentration indicating overtreatment) and adrenal androgens (a high concentration indicating undertreatment). In childhood, treatment success is assessed by the close clinical monitoring of growth and puberty in the setting of a specialist paediatric endocrine service.
Assessment of adrenal incidentaloma
The increasing use of CT and MRI scanning of the abdomen in clinical medicine has revealed that small lesions consistent with adenomas in the adrenal are very common. Such incidentally discovered lesions are often described as ‘adrenal incidentalomas’ and require appropriate investigation to exclude malignancy and excess hormone secretion. The risk of malignancy can be assessed on radiological grounds: evidence of invasion of surrounding tissues, calcification and heterogeneity, and the lipid content of the lesion, which, if high, increases the likelihood of the adenoma being benign. However, the risk of malignancy can also be assessed simply on the basis of size, with most authorities advising removal of lesions > 4 cm in diameter. In the absence of obvious clinical features of Cushing syndrome, however, the exclusion of a functioning lesion is primarily biochemical and includes exclusion of phaeochromocytoma (by measurement of urine catecholamines) and Conn syndrome (see p. 760), and usually exclusion of subclinical excess cortisol secretion by a formal 48 h low-dose dexamethasone suppression test. Most clinicians would advise excision of a functioning lesion, whereas non-functioning lesions < 4 cm in diameter are typically managed conservatively. There is current controversy regarding the significance of ‘subclinical Cushing syndrome’, which describes incomplete dexamethasone suppression in the absence of frank cushingoid symptoms. Some groups believe that this confers increased cardiovascular risk, while others believe it is a statistical ‘fall out’ of normal individuals.
MONITORING PITUITARY AND ADRENAL REPLACEMENT THERAPY
The clinical biochemist is often called upon to monitor the adequacy of hormone replacement therapy in pituitary disease. Clinical assessment of adequacy of replacement is imprecise and may, for example, lead to over-replacement with adrenal or thyroid hormones in response to unrelated, vague malaise.
There is no general agreement regarding the most appropriate way to monitor glucocorticoid replacement and it is difficult to validate whatever approach is taken. The authors’ practice for a patient taking hydrocortisone (which is cortisol) is to measure plasma cortisol at 09.00 h (having taken the morning dose on rising), at 12.30 h (just before any lunchtime dose) and at 17.30 h (before the evening dose) and to perform a 24 h UFC measurement beginning and ending with the morning replacement dose. The aim of therapy is then to achieve a 24 h UFC within the normal range and 09.00 h cortisol within the 09.00 h normal range (to avoid over-replacement), with neither 12.30 h or 17.30 h cortisol < 50 nmol/L (and ideally not < 100 nmol/L to avoid under-replacement). Using these criteria, most patients require thrice-daily hydrocortisone dosage, typically 10 mg on rising, 5–10 mg at lunchtime and 5 mg in the early evening, which is consistent with current estimates of normal cortisol secretion rates. Random measurements of plasma cortisol on replacement therapy are of little or no value.
In primary adrenal failure, mineralocorticoid replacement is also required, and adequacy of replacement can be assessed biochemically by measuring lying plasma renin (which is markedly elevated in untreated patients and ideally should be normal on fludrocortisone replacement). Since the renin–angiotensin system is intact in patients with pituitary disease, fludrocortisone replacement, with monitoring of plasma renin, is not necessary.
The aim of levothyroxine replacement in hypopituitarism should be to achieve a plasma free T4 in the middle- or upper-third of the normal range. Measurements of TSH are clearly unhelpful in this context, and a common mistake is to reduce the dose of thyroxine because of the incorrect conclusion that a low TSH indicates over-replacement.
Biochemical assessment of gonadal steroid replacement presents greater difficulty. In a male on testosterone ester depot preparations, plasma testosterone may reasonably be measured a few days after an injection and just prior to the next dose to ensure that adequate concentrations are being achieved. Plasma testosterone measurement is also useful in patients on transdermal or mucoadhesive testosterone preparations. In patients on oral testosterone undecanoate therapy, however, there is preferential conversion of testosterone to dihydrotestosterone, and plasma total testosterone concentrations are frequently below normal despite adequate replacement therapy. In the female, the best approach to therapy is probably to use the endometrium as a natural in vivo bioassay for oestrogen and aim simply to achieve regular withdrawal bleeds. It is possible to measure plasma oestradiol in patients receiving oral or transdermal oestradiol replacement, but other oral preparations of modified synthetic or equine oestrogens cannot logically be monitored biochemically.
Desmopressin replacement therapy can be monitored by measurement of plasma osmolality, aiming to maintain a normal osmolality and avoid dehydration or fluid overload. Patients should be instructed to allow the diuresis to resume prior to each treatment dose.
Patients with hypopituitarism and with primary adrenal failure require appropriate information and education about the nature of their replacement therapy and, in particular, the need to increase steroid hormone replacement in the face of intercurrent illness. They should be advised to carry a ‘Steroid card’ or alert bracelet and ensure that all doctors and dentists who carry out treatment are aware of their condition and the need for increased treatment during stress.
CONCLUSION
In the last decades of the 20th century, clinical neuroendocrinology was revolutionized by the introduction of reliable assays for the relevant pituitary and adrenal hormones. The relationship between endocrinologist and clinical biochemist must remain a close one, since the biochemical investigations to be performed depend closely on the clinical assessment of the patient, while the treatment which the patient will receive is equally dependent on the biochemical results obtained. In this chapter, it is hoped that a logical framework for such a harmonious relationship has been laid down.
Further reading
Baylis PH. Diabetes insipidus. J R Coll Physicians Lond. 1998;32:108–111.
National Institute for Health and Care Excellence. Growth hormone deficiency (adults) – human growth hormone (Technology Appraisal No. 64): http://www.nice.org.uk/page.aspx?o=83406; [Accessed October 2013].
APPENDIX 18.1 TEST PROTOCOLS
Assessment of basal pituitary function
A basal 09.00 h blood sample for measurement of:
• free T4
• TSH
• testosterone (male) or oestradiol (female)
• LH, FSH
• prolactin
• IGF-1 in the context of proven pituitary disease (but not simply as a screening test)
• osmolalitya.
aSimultaneous random urine for osmolality.
(aAssessment of posterior pituitary function is only required if indicated by the clinical context.)
Insulin stress test
Indications: assessment of ACTH/cortisol and GH reserve. Contraindications: ischaemic heart disease, epilepsy or unexplained blackouts, severe longstanding hypoadrenalism.
Precautions: electrocardiograph must be normal, basal 09.00 h cortisol must be > 100 nmol/L. Free T4 must be normal.
Procedure: fasting from midnight, i.v. cannula, soluble insulin, 0.15 U/kg i.v. at 09.00 h. Observe and record signs of hypoglycaemia. Hypoglycaemia only needs to be reversed with i.v. dextrose if severe and prolonged or with impending or actual loss of consciousness or fits.
Sampling: blood for glucose, cortisol, GH at 0, 30, 45, 60, 90 and 120 min.
Interpretation: blood glucose must fall below 2.2 mmol/L to achieve an adequate hypoglycaemic stress. If adequate hypoglycaemia is not achieved then cortisol or GH deficiency cannot be diagnosed. Untreated hypothyroidism can also give subnormal results.
A normal response is traditionally defined as a rise in plasma cortisol to a peak of ≥ 550 nmol/L (but see comments in text (p. 355) on normal ranges) and a rise in plasma GH to ≥ 7 μg/L (most normal individuals actually rise to a peak > 15 μg/L). ‘Severe GH deficiency’ is arbitrarily defined as a GH peak < 3 μg/L.
Patients who show such a normal cortisol response can withstand major surgery without corticosteroid replacement. Patients with responses in the range 400–550 nmol/L may not require regular replacement therapy, but do require cover for major illnesses and surgery. All other patients with subnormal responses require hydrocortisone replacement.
Children with bone age <12 years and GH peak <6.7 μg/L generally benefit from GH therapy.
Short tetracosactide (synacthen) test
Indication: screening test for adrenocortical insufficiency, including ACTH deficiency.
Procedure: synacthen (tetracosactide) 250 μg i.v or i.m. Plasma cortisol before and after 30 min.
Interpretation: plasma cortisol of ≥600 nmol/L at 30 min is highly correlated with a normal ACTH response to hypoglycaemia and a normal pituitary-adrenal axis. However, normal responses vary with gender and between commonly used cortisol assays – an appropriate normal range for the assay methodology must, therefore, be used (see p. 355).
Responses >600 nmol/L can be considered normal for almost all assays and circumstances. Peak cortisol responses between 400 and 600 nmol/L may be regarded as equivocal – they require appropriate patient education and may need confirmation by a formal IST (if not contraindicated).
Water deprivation test
Indications: diagnosis of vasopressin deficiency.
Procedure: if mild DI is suspected, the patient should have no fluids from midnight the night before (dry foods allowed). If severe DI is likely, free fluids and a light breakfast should be allowed prior to the test.
Commence test at 09.00 h or earlier if practicable. Measure plasma and urine osmolality basally, then hourly until the end of the test (after 8 h or when diagnostic osmolalities achieved). Record all urine volumes passed.
Osmolalities should be measured prospectively as the test proceeds. If this is not possible, weigh the patient every 2 h and arrange for urgent plasma osmolality if body weight falls by more than 3%.
When osmolalities diagnostic of DI have been achieved, give desmopressin 2 μg i.m., allow free fluids and measure urine osmolality hourly for 2 h.
Interpretation: diabetes insipidus is diagnosed by a rise in plasma osmolality above 300 mmol/kg, without appropriate concentration of urine osmolality (to >600 mmol/kg). Adequate concentration of the urine after desmopressin indicates central, cranial DI and lack of concentration indicates nephrogenic DI.
Normal vasopressin secretion is shown by concentration of the urine to >600 mmol/kg, in the presence of a normal plasma osmolality (280–295 mmol/kg).
Normal concentration of the urine at the expense of elevation of plasma osmolality (>295 mmol/kg) is not a strictly normal response and may indicate mild DI or other subtle defects of thirst, osmoregulation or vasopressin secretion. Further investigations may be indicated.
Failure to concentrate either plasma or urine osmolality by the end of the test is non-diagnostic, but usually represents primary polydipsia. Such patients typically start the test with very dilute urine and often dilute plasma as a result of fluid overload. If clinically indicated, more prolonged water deprivation is required to make the diagnosis.
Glucose tolerance test for the diagnosis of acromegaly
Procedure: fasting from midnight, i.v. cannula, 75 g glucose at time 0.
Sampling: blood glucose and plasma GH at 0, 30, 60, 90, 120 and 150 min.
Normal response: growth hormone suppresses to < 1 μg/L after glucose in normal subjects.
Interpretation: failure of growth hormone to suppress completely after glucose is highly suggestive of acromegaly, even if a substantial fall has occurred.
CRH test
Indications: differential diagnosis of Cushing syndrome.
Differential diagnosis of isolated ACTH deficiency.
Procedure: i.v. cannula, human CRF-41, 100 μg i.v. at 09.00 h.
Notes: in North America it is usual to administer CRH in the evening rather than early morning. Ovine CRF-41 was originally used but is not readily available in the UK. Most patients notice facial flushing after CRH, but there are no other side-effects.
Sampling: plasma cortisol and ACTH at − 15, 0, 15, 30, 45, 60, 90 and 120 min.
Normal response: plasma cortisol rises by 25% or more above the basal concentration. The peak response to hCRF-41 in normal individuals is <600 nmol/L (<800 nmol/L with ovine CRF-41).
Interpretation: patients with Cushing disease show a normal or exaggerated response to CRH. Patients with ectopic ACTH-secreting tumours and adrenal tumours show no response (see text).
Responses in ACTH deficiency remain to be defined but a response of plasma ACTH would imply hypothalamic CRH deficiency. Patients with depression show a normal cortisol but reduced ACTH response. Patients with massive obesity show a blunted cortisol response.