CHAPTER 17
Hypoglycaemia
CHAPTER OUTLINE
GLUCOSE HOMOEOSTASIS IN THE FED AND THE POSTABSORPTIVE STATES
The neuroendocrine response to hypoglycaemia
CLASSIFICATION OF HYPOGLYCAEMIC DISORDERS
PRACTICAL APPROACH TO THE INVESTIGATION OF HYPOGLYCAEMIA
Evaluation of hypoglycaemia in persons without diabetes mellitus
Investigation of hypoglycaemia
Evaluation of hypoglycaemia in patients with diabetes mellitus
EMERGENCY TREATMENT OF HYPOGLYCAEMIA
Surreptitious administration of hypoglycaemic agents (factitious or felonious hypoglycaemia)
Islet cell tumours (insulinoma)
Non-insulinoma pancreatogenous hypoglycaemia syndrome (NIPHS)
Non-islet cell tumour hypoglycaemia (NICTH)
Hypoglycaemia associated with renal impairment
Hypoglycaemia associated with liver disease
Hypoglycaemia due to endocrine deficiencies
Hypoglycaemia due to deficient energy intake
Exercise-related hypoglycaemia
GLUCOSE HOMOEOSTASIS IN THE FED AND THE POSTABSORPTIVE STATES
In healthy subjects, the blood glucose concentration is maintained within relatively narrow limits through a tightly controlled balance between glucose production and glucose utilization. Fundamentally, glucose is derived either from dietary intake (in the fed state) or from glycogenolysis and gluconeogenesis (in the fasting or postabsorptive state). It is metabolized by oxidation or stored either in the form of glycogen or through conversion to fat (Fig. 17.1).
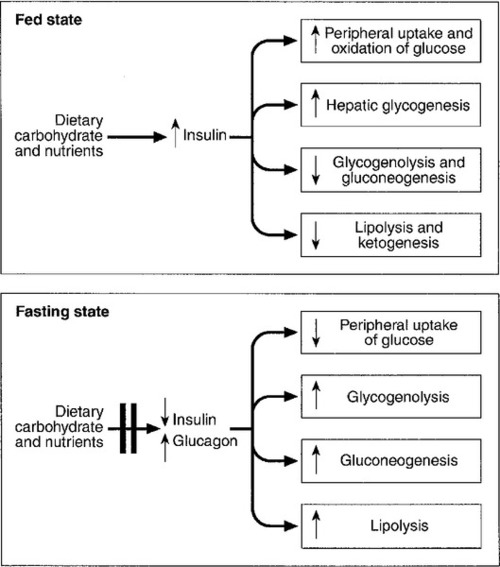
FIGURE 17.1 Glucose homoeostasis in the fed and fasting states.
Plasma glucose regulation is a complex process involving both insulin-dependent and insulin-independent mechanisms. This regulation is a multi-organ process involving the gastrointestinal tract, pancreas, liver, muscles, adipose tissue, brain and kidneys. In the gastrointestinal tract, incretin hormones (see below), secreted in response to meals, promote glucose-mediated insulin secretion and suppress glucagon production. In the liver, insulin regulates blood glucose concentrations by suppressing hepatic glucose output and increasing postprandial glucose storage in the form of glycogen. In muscle and adipose tissue, insulin binding to insulin receptors leads to increased expression of glucose transporter 4 (GLUT4) molecules in the cell membrane, facilitating glucose uptake. Insulin-independent mechanisms that contribute to glucose regulation are located in different organs, but mainly in the gastrointestinal tract and the kidneys. Sodium–glucose cotransporters (SGLT), such as SGLT1 in the gastrointestinal tract and SGLT2 in kidney, are important mediators of insulin-independent glucose transport across cell membranes.
In response to nutrient ingestion, glucagon-like peptide (GLP-1) and glucose-dependent insulinotropic peptide (GIP) are released from enteroendocrine cells. Glucagon-like peptide-1 enhances glucose-dependent insulin secretion and inhibits glucagon secretion. Glucose-dependent insulinotropic peptide also stimulates glucose-dependent insulin release, and the combined actions of GLP-1 and GIP account for up to 60% of the overall postprandial insulin secretion. The increase in glucose and insulin concentrations results in stimulation of glycogen synthase and inhibition of glycogen phosphorylase, causing a net increase in hepatic glycogen. Approximately 50% of an oral glucose load is taken up by the liver and 50% by peripheral insulin-sensitive tissues. Three to four hours following a meal, glucose and insulin concentrations decline and the liver switches from net glucose uptake to net glucose release. Hepatic glucose release continues to increase over the next several hours until it equals glucose utilization. Initially, about 75% of glucose production is attributable to glycogenolysis and 25% to gluconeogenesis, but as hepatic glycogen stores decline, the proportion from gluconeogenesis increases steadily.
During the early postabsorptive phase, glucose utilization continues at the rate of 0.10–0.14 mmol/kg per min, of which 40–50% is due to obligatory uptake by the brain and other non-insulin-dependent tissues. With prolonged fasting, the decline in plasma insulin concentration is accompanied by increases in plasma free fatty acid and ketone body concentrations. These can be used as alternative fuels, thereby decreasing the need for glucose.
HYPOGLYCAEMIA
The definition of hypoglycaemia is somewhat arbitrary since the glycaemic threshold at which symptoms occur differs between individuals, and depends on the age of the patient and the prevailing plasma glucose concentration before the hypoglycaemic episode. A venous plasma glucose concentration <3.0 mmol/L, regardless of the presence or absence of symptoms, has been recommended as the biochemical definition of hypoglycaemia by The Endocrine Society in the USA. However, many apparently healthy people, especially women, may have plasma glucose concentrations <3.0 mmol/L during a prolonged fast without experiencing any symptoms. Conversely, diabetic patients may develop hypoglycaemic symptoms at higher plasma glucose concentrations. Therefore, the diagnosis of pathological hypoglycaemia necessitates the demonstration of Whipple’s triad. This consists of: (1) either symptoms or signs, or both, consistent with hypoglycaemia in the presence of (2) a low plasma glucose concentration, with (3) the relief of symptoms or signs after the plasma glucose concentration is raised.
The neuroendocrine response to hypoglycaemia
Under normal metabolic conditions, in the unstressed state, the central nervous system (CNS) is wholly dependent upon glucose as its primary source of energy. The brain requires approximately 150 g of glucose each day, which must be continuously available. The presence of hypoglycaemia triggers a complex and well-coordinated metabolic process aimed at limiting and preventing a further fall in blood glucose concentration. The extent and duration of hypoglycaemia are primary factors in determining the magnitude of this counter-regulatory response, but other factors, such as age, sex, rate of fall of blood glucose and insulin per se have also been shown to influence the response.
In healthy subjects, the initial response to prevent a decline in blood glucose concentration is a reduction in insulin secretion, which begins while the plasma glucose concentration is falling but still within the physiological range. This is followed by an increase in glucagon and adrenaline (epinephrine) secretion as the glucose concentration declines further. Glucagon, secreted by pancreatic α-cells, rapidly raises plasma glucose concentration by stimulating hepatic glucose production via glycogenolysis and gluconeogenesis. The activation of the autonomic nervous system, mainly the sympathetic, increases the concentrations of noradrenaline (norepinephrine) at nerve terminals and adrenaline in the circulation. Adrenaline acts on α- and β-adrenergic receptors at several end organs and results in a more sustained increase in blood glucose concentration. It reduces insulin secretion from the pancreatic islets, increases glycogenolysis and gluconeogenesis in liver, increases glycolysis in muscle and increases lipolysis in adipose tissue. Hepatic glycogen stores provide glucose, particularly for the brain, whereas the mobilization of fatty acids from fat depots provides energy for tissues that can utilize fatty acids as their basic fuel, for example skeletal and cardiac muscles, renal cortex and liver, thus sparing glucose for use by the CNS.
As blood glucose concentration declines further, hypothalamic sensing of hypoglycaemia results in increased secretion of growth hormone (GH) and adrenocorticotropic hormone (ACTH) and, consequently, cortisol. The increase in GH and cortisol secretion induces metabolic changes over longer periods of time by stimulating lipolysis in adipose tissue and ketogenesis and gluconeogenesis in the liver.
The glycaemic thresholds for the activation of these responses are higher than those for the development of symptoms or the impairment of cognitive function. With falling plasma glucose concentrations, the typical sequence of responses (and their glycaemic thresholds) is as follows: decrease in endogenous insulin secretion (4.4 mmol/L); increase in glucagon and adrenaline (3.8 mmol/L); increase in GH secretion (3.7 mmol/L); increase in cortisol secretion (3.2 mmol/L); development of symptoms (3.0 mmol/L), and impairment of cognitive function (2.5 mmol/L).
Symptoms of hypoglycaemia
The symptoms of hypoglycaemia are non-specific and vary depending on the degree of hypoglycaemia, the rapidity of the decline in blood glucose, the age of the patient, whether the hypoglycaemia is clinical or experimental, and on patients’ differing perceptions of symptoms. The symptoms differ from one person to another, but are usually consistent from episode to episode for any one person. In some patients, symptoms are mainly due to activation of the sympathetic nervous system (neurogenic or autonomic), but in others only symptoms due to brain glucose deprivation per se may be observed. All the symptoms of hypoglycaemia, however, reflect the effects of glucose deprivation on the CNS (neuroglycopenia).
Acute neuroglycopenia (neurogenic)
This is characterized by sweating, anxiety, hunger, tremor, palpitations and weakness (Box 17.1). These symptoms result mainly from activation of the sympathetic nervous system and characteristically occur when there has been a rapid decline in blood glucose concentration. It may occur in patients with diabetes owing to excessive absorption of exogenous insulin, either from overtreatment or from rapid mobilization from the injection site during exercise. In non-diabetic subjects, reactive hypersecretion of insulin may be responsible, for example after Roux-en-Y gastric bypass for obesity. Awareness of hypoglycaemia is mainly the result of the perception of neurogenic symptoms which may be lost in patients with type 1 diabetes mellitus (hypoglycaemia unawareness). The symptoms of acute neuroglycopenia may mimic those of anxiety attacks or hyperventilation.
Subacute neuroglycopenia
Symptoms range from a generalized decrease in spontaneous activity, poor concentration, episodic disorientation, confusion, incoordination, slurred speech and behavioural changes, to seizures, focal neurological signs and coma (Box 17.1). Patients with subacute neuroglycopenic symptoms have been misdiagnosed as having epilepsy, transient ischaemic attacks, psychosis, hysteria, chronic nervous exhaustion, personality disorder or inebriation.
Chronic neuroglycopenia
Chronic neuroglycopenia is very rare and may occur in patients with insulin-secreting tumours unsuspected for years. Symptoms include insidious progressive mental disturbance that may resemble personality disorders, schizophrenia, paranoid psychosis, depression or dementia (Box 17.1).
CLASSIFICATION OF HYPOGLYCAEMIC DISORDERS
Several classifications of hypoglycaemia are possible, for example aetiological, ketotic or non-ketotic, a classification based on the timing of symptoms in relation to meals (fasting or postprandial) or whether hypoglycaemia occurs in a healthy person or one who has an illness.
The usefulness of classifying hypoglycaemic disorders based on the timing of symptoms in relation to meals has been questioned because some conditions that are associated with fasting hypoglycaemia, such as insulinoma, may also produce symptoms postprandially, and post-gastric bypass patients, who typically have postprandial hypoglycaemia, may have symptoms when fasting. Also, patients with factitious hypoglycaemia may have symptoms that have no clear relationship to food intake. In view of this, a classification which is based on the clinical characteristics, i.e. on whether hypoglycaemia occurs in a seemingly well individual or an ill or medicated individual, is more useful (Box 17.2).
PRACTICAL APPROACH TO THE INVESTIGATION OF HYPOGLYCAEMIA
Evaluation of hypoglycaemia in persons without diabetes mellitus
The direction of investigation will depend essentially on the clinical presentation and whether the patient appears ill or not. Hospitalized patients are often severely ill with multisystem disease. In such cases, the underlying illnesses that can produce episodes of hypoglycaemia, such as severe renal or hepatic disease, congestive cardiac failure, sepsis and anorexia nervosa, are usually obvious clinically. A review of all medications is essential since drugs (see below) are a common cause of hypoglycaemia in hospitalized patients, particularly in the presence of renal impairment. Non-islet cell tumours causing hypoglycaemia are usually, though not invariably, large mesenchymal tumours that are clinically apparent. In these cases, confirmation of the suspected mechanism of the hypoglycaemia may be sought by measuring plasma insulin, C-peptide, proinsulin and β-hydroxybutyrate (β-OHB) concentrations during an episode of hypoglycaemia. Endocrine deficiencies such as hypopituitarism and adrenocortical insufficiency should be sought and excluded by appropriate investigations if necessary.
In a seemingly well individual, with no obvious coexisting disease, the direction and extent of evaluation will depend on the clinical presentation. A thorough and detailed history of the symptoms (frequency, type, relationship to meals and exercise) is therefore essential. If symptoms are relieved by food ingestion, it is important to enquire about the type of food and speed of recovery. All medications should be inspected to exclude a prescribing or dispensing error, and the possibility of surreptitiously induced hypoglycaemia should always be considered, especially in healthcare professionals and relatives and carers of diabetic patients. Assessment of alcohol intake and pattern of drinking is important, since alcohol can cause both fasting and reactive hypoglycaemia. A history of previous gastric surgery or Roux-en-Y gastric bypass surgery for obesity and a family history of multiple endocrine neoplasia type 1 (MEN 1) should be sought. Symptoms in individuals of Japanese or Korean ethnicity may point to autoimmune hypoglycaemia.
Investigation of hypoglycaemia
The aims of the investigation are, first, to demonstrate that hypoglycaemia is the cause of the symptoms and, second, to identify the cause of the hypoglycaemia (Fig. 17.2).
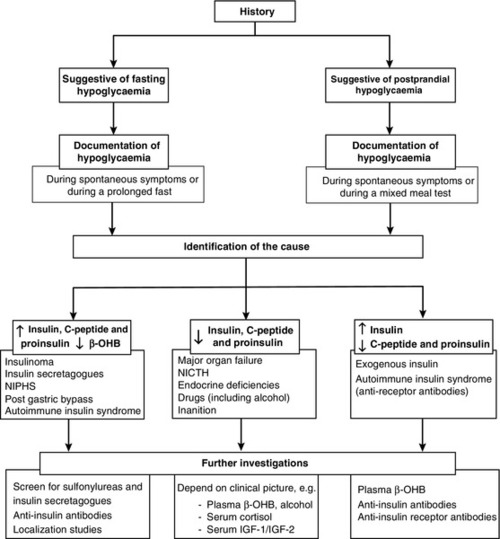
FIGURE 17.2 A practical approach to the investigation and diagnosis of hypoglycaemia (β-OHB, β-hydroxybutyrate; NIPHS, non-insulinoma pancreatogenous hypoglycaemia syndrome; NICTH, non-islet cell tumour hypoglycaemia; IGF-1 and IGF-2, insulin-like growth factors 1 and 2).
Demonstration of hypoglycaemia
Measurement of blood glucose during spontaneous symptoms
Measurement of the blood glucose concentration (and collection of a suitable blood specimen for subsequent measurement of plasma insulin, C-peptide, proinsulin and β-OHB concentrations) during spontaneous symptoms, and before glucose is given, is without doubt the best test for the diagnosis of spontaneous hypoglycaemia. The most practical way to obtain an accurate glucose concentration is to measure glucose immediately in whole blood or to separate plasma from cells within 30 min of collection, even if the specimen is collected in a tube that contains sodium fluoride. It is well recognized that the rates of decrease of glucose in the first hour after specimen collection in tubes with or without fluoride are virtually identical. The reduction in glucose after 2 h in a fluoride tube can sometimes exceed 0.5 mmol/L leading to a falsely low blood glucose concentration or ‘pseudohypoglycaemia’. In addition, although arteriovenous plasma glucose concentration differences are negligible in the fasting state, antecubital venous plasma glucose concentrations are significantly lower than arterial glucose concentrations after a glucose load owing to glucose extraction across the forearm. Therefore, arterialized blood collected from a vein at the back of the hand, which has been warmed by a heat pad, is preferred.
Often, however, patients referred for a medical opinion are asymptomatic when seen in the outpatient clinic, and measuring their blood glucose concentration at such time is usually unhelpful. In this situation, reproducing the circumstances which may lead to hypoglycaemia should be attempted. In patients with a history suggestive of fasting hypoglycaemia, a prolonged fast test should be performed, whereas in patients who only experience symptoms of hypoglycaemia within a few hours of having a meal, a mixed meal test should be performed (see provocation tests, below).
Provocation tests
Prolonged fast test
The prolonged fast (up to 72 h) is the single most useful investigation employed in the evaluation of patients with suspected spontaneous hypoglycaemia. The aim of the investigation is to demonstrate spontaneous hypoglycaemia in the presence of neuroglycopenic symptoms and the resolution of the symptoms when the plasma glucose concentration is raised to within normal limits. During a prolonged fast, normal subjects rarely develop plasma glucose concentrations <3.0 mmol/L and almost never develop neuroglycopenic symptoms.
The fast must be conducted in hospital under strict medical supervision. Before initiation of the fast, a simple assessment of coordination, recent memory and calculations (e.g. counting down in sevens) should be performed as a baseline. The time for initiating the fast is determined depending on a reasonable estimate of the patient’s likely tolerance for fasting. If fasting starts after the evening meal or at midnight, the majority of patients with insulinoma will develop hypoglycaemia by the middle of the following day, when adequate staff and laboratory facilities are available. Patients should have an intravenous indwelling catheter, and an ampoule of 50% glucose solution should be readily available. During the test, the patient is allowed water and non-caloric beverages and should undertake brisk exercise under supervision. Blood, for glucose, insulin, C-peptide, proinsulin and β-OHB measurements, is collected at the beginning of the test, and every 4–6 h thereafter. Plasma glucose should be determined in the laboratory as soon as possible after collection, and plasma or serum should be stored at –20 °C for subsequent analysis of insulin, C-peptide, proinsulin and β-OHB if required. Bedside glucose meters can be used to monitor the patient’s blood glucose concentration frequently, but low values, especially in the absence of symptoms of hypoglycaemia, should always be confirmed in the laboratory. When the plasma glucose has fallen to <3.0 mmol/L, blood should be collected more frequently and the patient should be assessed for neuroglycopenia by repeating the same tests performed at the initiation of the fast. If neuroglycopenic symptoms develop and blood glucose is <3.0 mmol/L, several specimens should be obtained for plasma glucose, insulin, C-peptide, proinsulin and β-OHB measurements before any remedial action, such as glucose administration, is taken. At the end of the fast, a sample should be collected to screen for hypoglycaemic agents and to measure insulin antibodies. A glucagon test (1.0 mg intravenously) has also been recommended (see below). The criteria to stop the test before 72 h are: (1) Whipple’s triad has been observed; (2) plasma glucose concentration <3.0 mmol/L in a patient who had previously experienced Whipple’s triad, and (3) plasma β-OHB >2.7 mmol/L. The measurement of β-OHB concentrations in real-time during the fast is quite useful because a progressive rise in plasma β-OHB concentrations indicates indirectly that the circulating insulin concentration has been suppressed and that the fast can be terminated. However, blood should always be collected in this situation to confirm suppressed plasma insulin, proinsulin and C-peptide concentrations.
In patients with insulinoma, about 40% develop hypoglycaemia within 12 h, 75% within 24 h, 95% by 48 h and 99% by 72 h.
Glucagon stimulation test
This test serves as a supplemental study in the diagnosis of hypoglycaemic disorders when results from the prolonged fast are inconclusive. The rationale is that, in patients with insulinoma, there is a greater increase in plasma glucose after intravenous glucagon due to the lesser depletion of hepatic glycogen during the fast as a consequence of the higher insulin secretion. The test is performed at the termination of a prolonged fast test, either at the time of occurrence of symptomatic hypoglycaemia with concomitant plasma glucose concentration <3.0 mmol/L, or after 72 h of fast. Glucagon 1 mg is injected intravenously over 2 min and plasma glucose is measured at baseline, 10, 20 and 30 min after the glucagon injection. An increase in plasma glucose concentration of ≥1.4 mmol/L after intravenous glucagon indicates mediation of the hypoglycaemia by insulin. A disadvantage of this test is the danger of causing severe hypoglycemia after 90–180 min.
Mixed meal test
This test is used to investigate patients who experience only postprandial symptoms. The meal has not been standardized but, ideally, a meal similar in composition to the meal that usually provokes symptoms should be used. Plasma glucose concentration is monitored every 30 min for 5 h and also at any time while the patient is symptomatic. Capillary or arterialized blood (see above) should be used, since postprandial venous plasma glucose concentrations are about 10% lower than arterial concentrations owing to extraction of glucose by muscle. Samples should also be collected every 30 min for insulin, C-peptide and proinsulin measurement but should only be analysed if they have been collected at the time when plasma glucose concentration was <3.0 mmol/L and before administering carbohydrates. Patients should be observed for symptoms, and the timing of these symptoms in relation to the meal, and to the blood glucose concentrations, should be documented. If Whipple’s triad is demonstrated, a sample for the measurement of oral hypoglycaemic agents and insulin antibodies should be collected. Patients who develop neuroglycopenic symptoms during hypoglycaemia, but not at other times during the test, may be considered to have postprandial hypoglycaemia. However, in all patients with a positive mixed meal test, additional testing, including a supervised 72 h fast, is recommended. Postprandial hypoglycaemia without fasting hypoglycaemia has been documented in some patients with insulinoma and in patients with non-insulinoma pancreatogenous hypoglycaemia (see p. 343).
The diagnosis of postprandial hypoglycaemia should not be made on the basis of an oral glucose tolerance test (OGTT). At least 10% of ‘normal’ subjects may have plasma glucose nadirs of <2.6 mmol/L during an OGTT and 2.5% may have values <2.2 mmol/L. Moreover, symptoms during OGTT are often unrelated to the level of plasma glucose nadir or to the rate of decline of the glucose concentration.
Identification of the cause of hypoglycaemia
Plasma insulin, C-peptide and proinsulin
Although insulin and C-peptide are secreted in equimolar amounts by the β-Cells of the pancreas, the metabolic clearance of insulin is much more rapid than that of C-peptide. Therefore, C-peptide has a longer half-life and is present in peripheral blood in higher molar concentrations than insulin, making it less prone to marked fluctuations. Consequently, the measurement of plasma C-peptide concentrations may be more reliable as an indication of endogenous insulin production. However, as C-peptide is cleared by the kidneys, raised concentrations may occur in renal impairment.
Intact proinsulin undergoes enzymatic processing leading to production of des-31,32-proinsulin and des-64,65-proinsulin and then to insulin and C-peptide. Some proinsulin assays specifically measure intact proinsulin whereas others measure ‘total proinsulin’, i.e. intact and other molecules such as des-31,32-proinsulin. Therefore, proinsulin results depend strongly on the assay used and should be interpreted with caution. In islet cell tumours, the amount of circulating proinsulin is increased and, occasionally, tumours may secrete mainly or exclusively proinsulin. A plasma proinsulin concentration ≥5.0 pmol/L, when the plasma glucose concentration is <2.5 mmol/L during a 72 h fast test, represents the best criterion for the diagnosis of endogenous hyperinsulinaemia with 100% specificity and sensitivity. In view of this, the Endocrine Society has now recommended the measurement of plasma proinsulin as a first-line test, in addition to insulin and C-peptide.
Insulin and C-peptide can be measured in either plasma (lithium heparin) or serum, as long as the serum or plasma is separated within 15 min of collection and frozen immediately following separation. Plasma insulin concentrations can be artefactually modified by heterophilic antibodies, endogenous anti-insulin antibodies or haemolysis, as red blood cells contain an insulin-degrading enzyme, which may lead to underestimation of the actual insulin concentrations in haemolysed samples.
The measurement of plasma insulin, C-peptide and proinsulin concentrations, in the presence of hypoglycaemia, is the most useful test in identifying the cause of hypoglycaemia (see Fig. 17.2). Raised plasma insulin (≥18 pmol/L), C-peptide (≥200 pmol/L) and proinsulin concentrations (≥5.0 pmol/L) in the presence of hypoglycaemia (plasma glucose <3.0 mmol/L) indicate endogenous hyperinsulinaemia. Causes of endogenous hyperinsulinaemia include insulinoma, non-insulinoma pancreatogenous hypoglycaemia syndrome (NIPHS), autoimmune insulin syndrome, sulfonylurea- or meglitinide-induced hypoglycaemia, non-islet cell insulin-secreting tumours and reactive insulin-mediated hypoglycaemia in post-gastric bypass surgery patients. Factitious sulfonylurea-induced and meglitinide- (glinide-) induced hypoglycaemia may produce an identical clinical and biochemical picture to insulinoma. Therefore, screening for these drugs is essential if a false positive diagnosis of insulinoma is to be avoided.
Inappropriately raised plasma insulin concentrations in the presence of low or suppressed plasma C-peptide and proinsulin concentrations will identify patients with exogenous insulin administration. Therefore, it is important to use insulin assays that can detect the presence of insulin analogues. Another rare cause of raised plasma insulin but suppressed C-peptide and proinsulin concentrations is hypoglycaemia mediated by insulin receptor antibodies (IR-A). The diagnosis should be considered in patients with other autoimmune diseases and requires the demonstration of IR-A in the serum.
Other causes of hypoglycaemia are associated with suppressed plasma insulin, C-peptide and proinsulin concentrations. In many of these conditions, the diagnosis is usually obvious on clinical grounds. In others, the measurement of plasma β-OHB, GH and insulin-like growth factor (IGF-1 and IGF-2) concentrations may be required to establish the cause of hypoglycaemia (Fig. 17.2).
Plasma β-hydroxybutyrate (β-OHB)
Lipolysis is very sensitive to circulating insulin concentrations. During fasting, normal individuals will show a gradual decline in insulin concentration and a progressive increase in lipolysis and hence ketone bodies, for example β-OHB (Fig. 17.3). In patients with hypoglycaemia due to hyperinsulinaemia, lipolysis is suppressed and β-OHB concentrations are low. A plasma β-OHB concentration of ≤2.7 mmol/L during a prolonged fast, and when plasma glucose concentration is <3.0 mmol/L, indicates mediation of hypoglycaemia by insulin. Conversely, in hypoglycaemia associated with suppressed insulin secretion, such as liver disease, β-OHB concentrations are usually raised, the only exception being in severe inanition, when fat stores are depleted. Therefore, the measurement of plasma β-OHB provides an indirect measure of the prevailing insulin concentration during the hypoglycaemia. The main advantage, however, is that β-OHB is easy to measure and the result can be made available long before those of insulin or C-peptide measurements.
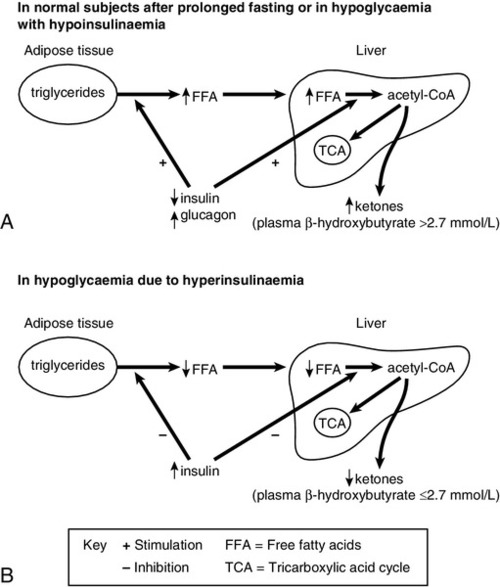
FIGURE 17.3 Changes in the concentrations of hormones and metabolites in normal subjects after prolonged fasting and in hypoglycaemia with hypoinsulinaemia (A), and in hypoglycaemia due to hyperinsulinaemia (B).
Insulin antibodies
The presence of circulating antibodies to insulin, due to previous exposure to exogenous insulin, may give falsely high plasma insulin concentrations (see p. 295). However, this is less common now with the use of human insulin, which is less antigenic than the previously used animal insulin preparations. The detection of insulin antibodies is considered to be the criterion for the diagnosis of the insulin autoimmune syndrome (see below), but antibodies may be detected in persons without hypoglycaemia and even, rarely, in patients with insulinoma. Because C-peptide does not cross-react with insulin antibodies, the measurement of C-peptide in these situations can be used as an index of β-Cell function. Autoantibodies to insulin receptors cause a particularly refractory type of fasting hypoglycaemia (see below) and the diagnosis requires the demonstration of IR-A in the serum.
Screening for oral hypoglycaemic agents
In a seemingly well individual, screening for oral hypoglycaemic agents (sulfonylureas and meglitinides) should always be considered to exclude the possibility of a prescribing or dispensing error, and of surreptitiously induced hypoglycaemia.
Evaluation of hypoglycaemia in patients with diabetes mellitus
Definition
The American Diabetes Association (ADA) has published criteria for the definition and clinical classification of hypoglycaemia in patients with diabetes mellitus (Box 17.3). In patients with diabetes, hypoglycaemia is defined as all episodes of an abnormally low plasma glucose concentration (with or without symptoms) that expose the individual to harm. The workgroup recommended that persons with diabetes become concerned about the possibility of hypoglycaemia at a self-monitored blood glucose concentration ≤3.9 mmol/L. While that value is higher than the value used to diagnose hypoglycaemia in people without diabetes, it approximates the lower limit of the physiological fasting non-diabetic range and the glycaemic threshold for activation of counter-regulatory mechanisms. This cut-off value has been debated, with some favouring a value of <3.5 mmol/L to avoid over-diagnosis of hypoglycaemia in asymptomatic patients.
Pathophysiology and risk factors
Hypoglycaemia in persons with diabetes is essentially the result of therapies that raise plasma insulin concentrations, such as insulin, sulfonylureas and non-sulfonylurea insulin secretagogues (e.g. nateglinide or repaglinide). The relative or absolute insulin excess during treatment, together with compromised physiological and behavioural defences in type 1 diabetes and longstanding type 2 diabetes, can result in hypoglycaemic episodes. In many patients with fully developed type 1 diabetes or with longstanding type 2 diabetes, the glucagon response to hypoglycaemia is lost and the adrenaline response is often reduced, leading to defective counter-regulation and ‘hypoglycaemia unawareness’.
Factors that may increase risk of hypoglycaemia include: missed or irregular meals; incorrect use of hypoglycaemic medication (dose or timing); decreased insulin clearance (e.g. renal impairment); increased glucose utilization (e.g. excessive exercise); excessive alcohol intake, and concurrent medications (Box 17.4).
Incidence
The rates of hypoglycaemia with insulin therapy vary according to the insulin regimen and the type and duration of diabetes. Overall, hypoglycaemia is less frequent in type 2 diabetes than in type 1 diabetes, but the risk of hypoglycaemia increases substantially later in the course of type 2diabetes. According to the UK Hypoglycaemia Study Group (2007), the rate of severe hypoglycaemia in insulin-treated type 1 patients of <5 years’ duration was 22%, rising to 46% in patients with duration of >15 years. In comparison, the rates in patients with type 2 diabetes treated with insulin were 7% for patients taking insulin for <2 years and 25% for patients taking insulin for >5 years. Diabetic patients who are treated with insulin are particularly prone to exercise-induced hypoglycaemia. In normal subjects, glucose uptake in skeletal muscles is increased by 20–30 times during exercise. This is compensated for by an increase in hepatic glucose release mediated by a fall in circulating insulin concentrations. In patients treated with insulin, the continuous release of insulin from subcutaneous depots inhibits glucose output from the liver. In addition, increased absorption of insulin from the injection site may occur if the site is near the muscles being exercised.
Hypoglycaemia rates with the third-generation sulfonylureas (e.g. glimepiride, glipizide and gliclazide) and the meglitinides (e.g. repaglinide and nateglinide) appear to be lower than those of previous generation sulfonylureas (e.g. glibenclamide and chlorpropamide). Elderly diabetic patients, especially those with renal or liver impairment, are particularly susceptible to hypoglycaemia and a reduction in sulfonylurea dosages is often required. The hypoglycaemia is usually prolonged, especially as the duration of action of the sulfonylureas is generally much longer that their plasma half lives, and may last between 36 h and seven days, despite continuous infusion of glucose. Additional treatment with glucagon and/or diazoxide may be required to prevent a fatal outcome.
The risk of hypoglycaemia with metformin and thiazolidinediones (e.g. pioglitazone) is negligible. These agents act by sensitizing peripheral tissues to insulin but do not directly alter insulin secretion. They rarely cause hypoglycaemia when used as monotherapy, but may contribute to hypoglycaemia when used concomitantly with insulin, sulfonylureas or other insulin secretagogues.
Glucagon-like peptide-1 analogues (such as exenatide or liraglutide) and the dipeptidyl-peptidase 4 inhibitors (such as sitagliptin, vildagliptin and saxagliptin) do not appear to be associated with increased risk of hypoglycaemia when used as monotherapy, or in combination with insulin sensitizers (such as metformin and thiazolidinediones) in short-term clinical studies, but the risk of hypoglycaemia appears to increase when GLP-1 analogues are used with sulfonylureas.
Management
In patients with documented symptomatic or asymptomatic hypoglycaemic events, excessive dosing, wrong type or ill-timed dosing of insulin or insulin secretagogues should be first considered. Conditions that may affect exogenous glucose delivery (e.g. missed meals), endogenous glucose production (e.g. alcohol), glucose utilization (e.g. exercise), insulin sensitivity (e.g. weight loss) or clearance (e.g. renal impairment) should be explored. Recurrent hypoglycaemic episodes or a history of hypoglycaemia unawareness should prompt a detailed review of the treatment regimen.
EMERGENCY TREATMENT OF HYPOGLYCAEMIA
Hypoglycaemia should be suspected in all patients presenting to the emergency department with altered mental state. In these patients, the diagnosis of hypoglycaemia should be expeditiously investigated by bedside glucose measurement; blood sampling for laboratory glucose testing should always be performed to confirm the results, though therapy should never be withheld pending laboratory confirmation. If hypoglycaemia is present, glucose should be administered immediately, preferably orally if the patient is awake and can swallow water safely with no risk of aspiration. The dose of oral glucose should be 10–20 g in an adult patient, and this dose may be repeated three times (if required) before giving a meal containing complex carbohydrates, to ensure long-lasting effects.
If the patient’s conscious level does not allow oral therapy, parenteral glucose is indicated: the dose of glucose for an average adult is 25–50 g given as an i.v. bolus of 50–100 mL of 50% dextrose. This should be followed by a maintenance dose of 10% dextrose i.v. at 3 mL/kg per h. Failure to respond to glucose is an indication to give glucagon subcutaneously, intramuscularly or intravenously (1 mg in adults). Steroids (e.g. hydrocortisone 1–2 mg/kg i.v. every 6 h) are given if the patient remains refractory to standard hypoglycaemia therapy or if adrenal insufficiency is suspected. Sulfonylureas are an important cause of refractory hypoglycaemia. They act by stimulating insulin secretion, and the administration of glucose to treat the hypoglycaemia further increases insulin release, thus tending to perpetuate the hypoglycaemia. Therefore, diazoxide or octreotide may be indicated for patients with sulfonylurea-induced hypoglycaemia.
CAUSES OF HYPOGLYCAEMIA
Surreptitious administration of hypoglycaemic agents (factitious or felonious hypoglycaemia)
Surreptitious or self-induced factitious hypoglycaemia should always be considered in patients presenting with spontaneous hypoglycaemia. The diagnosis should be suspected in individuals with access to insulin, sulfonylureas or meglitinides, such as diabetic patients (and their relatives, friends and carers) and healthcare professionals. Patients often vehemently deny self-administration and rarely show signs of psychiatric disturbances. Patients with diabetes may claim to have hypoglycaemic attacks despite omitting their insulin or sulfonylurea drugs. Some have the characteristics of Munchausen syndrome, with a history of frequent hospitalizations, previous surgical procedures and extensive travel.
Once suspected, the patient should be admitted to hospital for detailed observation and blood should be collected during hypoglycaemia for the measurement of plasma insulin, C-peptide, proinsulin, insulin antibodies and oral hypoglycaemic agents. A raised plasma insulin concentration and a suppressed C-peptide concentration will confirm the diagnosis of self-administration of insulin. If plasma insulin and C-peptide concentrations are both inappropriately raised, a drug screen (ideally for all available sulfonylureas and meglitinides) should be performed, in order to avoid an incorrect diagnosis of insulinoma. Once the diagnosis of factitious hypoglycaemia is made, the patient should be referred for counselling and psychiatric therapy.
Accidental intake of sulfonylurea drugs or meglitinides is rare but should be considered in all patients presenting with spontaneous hypoglycaemia. The finding that a spouse or a relation is taking these drugs should alert the physician to the possibility of accidental intake. The possibility of a prescription or dispensing error should also be considered.
Islet cell tumours (insulinoma)
Insulinomas are rare, with an annual incidence of about four cases per million persons. They can occur at any age, with approximately 80% occurring between the ages of 20 and 60 years and with, probably, a slight preponderance in women. In approximately 80% of patients, there is a solitary benign adenoma. Benign adenomas are generally small (1–2 cm in diameter) and occur with equal frequency in the head, body and tail of the pancreas. Multiple tumours occur in about 10% of patients and approximately 10% of insulinomas are malignant, with regional or distant metastases. About 8% of insulinomas are associated with multiple endocrine neoplasia type 1 (MEN1). Diffuse islet cell hyperplasia and non-insulinoma pancreatogenous hypoglycaemia syndrome (see below) have been reported in adults. Many insulinomas contain and secrete other hormones in addition to insulin (glucagon, gastrin, somatostatin and pancreatic polypeptide), but hypersecretion of these hormones is not usually associated with clinical symptoms.
Clinical features
Hypoglycaemia is the clinical hallmark of insulinomas. Neuroglycopenic symptoms, especially confusion, weakness, slurred speech, blurred vision and irrational behaviour, typically occur before breakfast, though 50% of patients may also have symptoms late in the afternoon. More than 50% of patients have amnesia and therefore the history should always be corroborated. Nocturnal hypoglycaemia is not uncommon and can manifest as nightmares and morning headaches. Symptoms may be provoked by exercise, missing a meal or by following a restricted calorie diet. Transient hemiplegia, grand mal seizures, focal neurological signs and coma can occur, but permanent brain damage is rare and is only seen with profound hypoglycaemia that goes unrecognized and uncorrected for several hours.
Diagnosis
Insulinoma is a rare cause of spontaneous hypoglycaemia. More common causes, such as surreptitious or inadvertent administration of insulin or oral hypoglycaemic agents, should be excluded first. The diagnosis of insulinoma requires the demonstration of hypoglycaemia, unsuppressed or elevated plasma insulin concentrations during hypoglycaemia and a pancreatic tumour. Patients with suspected insulinoma should have a prolonged fast with monitoring of plasma glucose, insulin, C-peptide, proinsulin and β-OHB concentrations followed by a glucagon stimulation test (if indicated). In patients with insulinoma, plasma insulin, C-peptide and proinsulin concentrations fail to suppress despite the presence of hypoglycaemia. Diagnostic cut-off values in the presence of hypoglycaemia are plasma insulin ≥18.0 pmol/L, C-peptide ≥200 pmol/L, proinsulin ≥5.0 pmol/L, β-OHB concentrations ≤2.7 mmol/L and an increase in plasma glucose concentration of ≥1.4 mmol/L after intravenous glucagon. The plasma insulin/C-peptide molar ratio is of the order of 1:5, compared with approximately 1:10 in normal subjects, owing to decreased hepatic extraction of insulin. Plasma proinsulin concentrations are significantly higher in patients with insulinoma than in normal subjects, and the molar ratio of insulin/proinsulin is about 1:1 compared with approximately 6:1 in normal subjects.
Localization
Preoperative localization should only be attempted if a definitive biochemical diagnosis has been established. The mean diameter of insulinomas is usually <2.0 cm, and non-invasive imaging techniques may fail to localize them. Computerized tomography (CT) detects 70–80% and magnetic resonance imaging (MRI) scanning about 80% of insulinomas. A substantial percentage of insulinomas express cell surface receptors for somatostatin, and somatostatin receptor scintigraphy has been reported to have a sensitivity of 80%. However, false localization is not uncommon with non-invasive modalities (i.e. the location suggested by these procedures may not correspond to the tumour found at operation). Invasive imaging procedures, such as endoscopic pancreatic ultrasonography, selective coeliac axis angiography and trans-hepatic portal venous sampling, have been reported to have sensitivities of >90%. Selective intra-arterial calcium stimulation with hepatic venous sampling (arterial stimulation and venous sampling, ASVS), using plasma C-peptide and insulin gradients, has been shown to have a high sensitivity of 94% and is used when other imaging procedures are equivocal or negative. However, it is the procedure of choice for confirming non-insulinoma pancreatogenous hypoglycaemia syndrome and post-Roux-en-Y gastric bypass hypoglycaemia, as other imaging techniques are negative in those disorders.
Treatment
Surgical resection is the treatment of choice for insulin-secreting tumours and is curative in over 90% of patients. In about 5–10% of patients, no tumour can be found at laparotomy. In these cases, the patient needs to be sent to a centre of excellence for further imaging and re-operation. The previous practice of a blind distal two-thirds pancreatectomy is no longer recommended. In some centres, the measurement of intraoperative plasma insulin concentrations has been used to ensure complete tumour removal. This may be important in patients with MEN1, who may harbour multiple insulinomas. The recurrence rate after surgical resection is 7% but is much higher, at 21%, for those with MEN1.
If surgery is contraindicated, medical treatment using diazoxide or a somatostatin analogue, such as octreotide, can reduce insulin release. Diazoxide, a non-diuretic benzothiazide, suppresses insulin release in about 50% of patients. It can also be useful, in responsive cases, in preoperative management. It directly inhibits insulin release via its effect on the K+-ATP channel. The standard dose is 150–450 mg daily but may be increased to as much as 800 mg/day. Its main side-effect is sodium and water retention, which can be prevented by giving a thiazide diuretic. About 50% of patients with insulinoma benefit from octreotide. The usual dose is 100 μg injected three times daily, and side-effects, such as diarrhoea and steatorrhoea, are rare. However, the effect of octreotide is dependent on the presence of somatostatin receptor subtype 2 on insulinoma tumour cells. A positive OctreoScan™ is not a prerequisite before starting octreotide treatment since studies have shown that, in patients with insulinoma and a negative scan, somatostatin decreased insulin concentrations significantly and reduced the incidence of hypoglycaemic episodes. In malignant insulinoma, cytotoxic chemotherapy with streptozotocin, doxorubicin and 5-fluorouracil can be used. When systemic chemotherapy is unsuccessful in patients with unresectable metastatic disease to the liver, embolization of the hepatic artery and intra-arterial chemotherapy may be used to control symptoms, to inhibit tumour growth and to improve survival.
Non-insulinoma pancreatogenous hypoglycaemia syndrome (NIPHS)
A rare syndrome of endogenous hyperinsulinaemic hypoglycaemia has been described in adult patients who suffer from postprandial insulin-mediated hypoglycaemia. The syndrome is characterized by neuroglycopenic episodes within 4 h of meal ingestion, negative prolonged fast tests, negative perioperative localization studies for insulinoma but positive intra-arterial calcium stimulation tests. Histologically, the pancreas shows diffuse islet hypertrophy, sometimes with hyperplasia, and enlarged and hyperchromatic β-Cell nuclei (nesidioblastosis). Patients do not have mutations of the SUR1 (ABCC8) or KIR6.2 (KCNJ11) genes, which encode the subunits of the pancreatic ATP-sensitive potassium channel responsible for glucose-induced insulin secretion, and have been described as a cause of hyperinsulinaemic hypoglycaemia in children. Therefore, it has been proposed that adult patients with endogenous hyperinsulinaemic hypoglycaemia may have mutations at other genes, as yet unidentified, that are involved in the regulation of insulin secretion.
Some patients who have undergone Roux-en-Y gastric bypass surgery for obesity develop postprandial neuroglycopenic symptoms, as a result of endogenous hyperinsulinaemia. These symptoms are different from those ascribed to the dumping syndrome, which are characterized by sweating, dizziness and flushing, but not neuroglycopenia. The precise mechanisms of hypoglycaemia in these patients have not yet been determined but increased secretion of GLP-1 and decreased ghrelin concentrations have been implicated. Nesidioblastosis was reported in some patients but not in others and, therefore, whether gastric bypass-induced hypoglycaemia is the result of a structural pancreatic abnormality or a functional dysregulation of postprandial insulin secretion remains to be established. Some patients respond to dietary measures or medications (α-glucosidase inhibitor, diazoxide, calcium channel blockers, octreotide) but others require partial pancreatectomy.
Non-islet cell tumour hypoglycaemia (NICTH)
Non-islet cell tumour hypoglycaemia (NICTH) is the term applied to hypoglycaemia caused by tumours other than insulinomas. A wide variety of tumours have been associated with hypoglycaemia (Box 17.5). Most patients are elderly and present with predominantly neuroglycopenic symptoms that may antedate the recognition of the existence of the tumour by many years. In most cases, especially those due to large mesenchymal tumours, the tumour is readily detectable by physical or radiological examination, but in others the tumour may be small and difficult to detect.
The pathogenesis of hypoglycaemia has been related to the secretion of incompletely processed insulin-like growth factor 2 (pro-IGF-2) by these tumours. This form of IGF-2 binds poorly to its binding proteins and penetrates more freely into tissue spaces. Normally, both IGF-1 and IGF-2 circulate almost completely bound to specific IGF-binding proteins (IGFBPs). They can be found as binary complexes (IGF-1 or IGF-2 with IGFBP-3) or ternary complexes (IGF-1 or IGF-2 with IGFBP-3 and an acid-labile subunit, ALS). In normal human plasma, 70–80% of IGF-2 is associated with a ternary 150 kD IGFBP complex, consisting of IGF-2, IGFBP-3 and an ALS, while 20–30% is bound to a binary complex of <60 kD consisting of IGF-2 and IGFBP-3, and <2% is present in the free form. Ternary complexes appear to be confined to the circulation, whereas binary complexes appear to leave the circulation almost as rapidly as the free form. In NICTH, IGFBP-3 and ALS production is decreased and the ability of the binary complex to form the more stable ternary complex is impaired; thus tumour-derived IGF-2 appears mainly as binary IGF-IGFBP complexes, which have greatly increased turnover compared with ternary complexes. Although circulating concentrations of IGF-2 are usually normal, the ratio of pro-IGF-2 to IGF-2 is raised. Pro-IGF-2 interacts with insulin receptors and IGF-1 receptors and inhibits GH secretion, which in turn leads to a decreased plasma IGF-1 concentration. The low GH concentrations also result in a reduction in IGFBP-3, contributing to the greater bioavailability of pro-IGF-2. The hypoglycaemia of tumours secreting pro-IGF-2 is therefore characterized by suppressed insulin and C-peptide, inappropriately low GH and β-OHB concentrations, and the molar ratio of total plasma IGF-2/IGF-1 is usually >10.
Surgical removal of a benign or locally invasive tumour usually results in complete remission of hypoglycaemia. Treatment with human GH (hGH) and/or prednisolone has also been used with good effect. Growth hormone is thought to exert its beneficial effect by stimulating gluconeogenesis and hepatic glycogenolysis, and by increasing IGFBP-3 production and redistribution of plasma IGFBP-3 from binary to ternary form, potentially reducing the bioavailability of pro-IGF-2. However, this effect is induced only at supraphysiological GH doses. Prednisolone, at daily doses of 30 mg or more, alleviates hypoglycaemia by direct stimulation of gluconeogenesis and glycogenolysis, and may also suppress tumour production of pro-IGF-2. Although both treatments are effective in relieving hypoglycaemia, prednisolone may have greater long-term benefit through suppression of pro-IGF-2.
Autoimmune hypoglycaemia
Two autoimmune syndromes that can cause hypoglycaemia have been described. In one of these, antibodies bind insulin receptors and mimic the action of insulin. The hypoglycaemia may be either fasting or postprandial and is often severe. Some patients may have a preceding phase of hyperglycaemia and significant insulin resistance, whereas in other patients, the onset of hypoglycaemia may be the first manifestation of the syndrome. Most patients with this syndrome also have evidence, either in the laboratory (elevated titres of antinuclear, antithyroid, antimitochondrial or antiplatelet antibodies) or clinically, of other autoimmune diseases such as systemic lupus erythematosus, primary biliary cirrhosis or Hashimoto thyroiditis (Box 17.6). Laboratory investigations show high plasma insulin concentrations (as a result of interference with the insulin assay) but suppressed C-peptide and proinsulin concentrations, similar to the findings with surreptitious administration of insulin. Therefore, the demonstration of the presence of antibodies directed against the insulin receptor (IR-A) is essential to make the diagnosis. The hypoglycaemia seems to respond rapidly to high doses of steroids but not to immunosuppression or plasmapheresis. Prognosis is poor, but in those who survive, the antireceptor antibodies disappear and remission occurs over several months or years.
The other syndrome, the autoimmune insulin syndrome, in which antibodies are directed towards insulin, has been reported mainly in Japan and is extremely rare. Most patients are middle-aged, with no history of previous administration of insulin, and may have evidence of other autoimmune diseases such as Graves disease, rheumatoid arthritis or systemic lupus erythematosus (see Box 17.6). An association with administration of certain drugs such as carbimazole, hydralazine, procainamide, penicillamine, glutathione and captopril has also been reported. The hypoglycaemia is most often postprandial, although fasting hypoglycaemia has been reported. Presentation can often be dramatic owing to severe neuroglycopenia with confusion and even coma. Paradoxically, hyperglycaemia may occur immediately following a meal or oral glucose challenge. Several possible mechanisms for the hypoglycaemia have been suggested. Sudden dissociation of insulin from the antibodies is the most plausible one. Laboratory investigations reveal high plasma insulin concentrations but, because the insulin is produced endogenously, C-peptide concentrations are not completely suppressed. The majority of circulating insulin is bound to antibodies, and the demonstration of the presence of anti-insulin antibodies is essential to make the diagnosis. In most cases, the hypoglycaemia is usually transient and resolves spontaneously. In severe cases, dietary treatment with frequent low-carbohydrate meals and acarbose has produced some benefit in alleviating the hypoglycaemia.
Hypoglycaemia associated with renal impairment
Renal impairment is a common predisposing factor for hypoglycaemia and is probably the second most common cause of hypoglycaemia, after insulin therapy, in hospitalized patients. The most important predisposing factor for hypoglycaemia in chronic kidney diseases (CKD) is caloric restriction, whether acute, due to anorexia and vomiting, or chronic. Other predisposing factors include concomitant liver disease, congestive heart failure, sepsis and drug therapy such as insulin, oral hypoglycaemic agents and β-blockers. The pathogenesis of hypoglycaemia in renal failure is complex and several mechanisms have been proposed. In normal subjects, the kidneys play a major role in gluconeogenesis and may supply as much as 45% of glucose during prolonged starvation. In uraemic patients, who are often malnourished, renal gluconeogenesis may not be able to maintain an adequate glucose supply, even if hepatic gluconeogenesis is normal. Other mechanisms include impaired hepatic glycogenolysis and gluconeogenesis; increased insulin half-life due to decreased renal degradation; diminished availability of alanine, and impaired counter-regulatory mechanisms.
Therapy for diabetes mellitus, with insulin or oral hypoglycaemic drugs, is by far the most common cause of hypoglycaemia in patients with renal impairment. Decreased renal degradation of insulin, frequently leading to a reduction in insulin or sulfonylurea requirements, is the main reason. Iatrogenic hypoglycaemia may be a consequence of haemodialysis or peritoneal dialysis. In patients undergoing haemodialysis, the mechanism may be due, in part, to post-dialysis glucose-induced hyperinsulinaemia caused by high glucose content of the dialysate and partly due to impaired insulin degradation by the kidneys.
The symptoms of uraemic hypoglycaemia are mainly neuroglycopenic (drowsiness, headache, lethargy, confusion, convulsions and coma) and may be confused with the symptoms of the dialysis dysequilibrium syndrome. Spontaneous hypoglycaemia in renal failure, not associated with hypoglycaemic agents or dialysis, carries a grave prognosis, with over half of the patients dying within months of onset.
Since the clearance of insulin, C-peptide and proinsulin is impaired in CKD, these parameters are unreliable in excluding the diagnosis of insulinoma in such patients. In these situations, β-OHB, measured during a prolonged fast test, and the glucose response to glucagon have been reported to be the best diagnostic parameters.
Hypoglycaemia associated with liver disease
Although the liver plays a central role in normal glucose homoeostasis, hypoglycaemia is rare in patients with liver disease. Glucose homoeostasis can be maintained with the mass of functioning hepatocytes reduced to <20% of normal, and hypoglycaemia does not occur until the liver is extensively damaged. Hypoglycaemia is not usually a feature of chronic hepatic failure nor is it implicated in the production of hepatic coma. Conversely, it may occasionally be associated with mild liver disease, and has been reported in a wide variety of liver disease such as fatty infiltration, portal cirrhosis, infective hepatitis and hepatocellular carcinoma.
Frank hypoglycaemia is also uncommon in acute (fulminant) hepatic failure, whether due to viral infection or hepatotoxic drugs. When it does occur, it can be severe and persistent. The diagnosis of hepatogenous hypoglycaemia may be difficult since the extent of liver disease, as assessed by standard liver function tests, does not correlate with the degree of hypoglycaemia. Therefore, other causes of hypoglycaemia should always be sought in a patient with hypoglycaemia and abnormal liver function tests. The hypoglycaemia associated with congestive heart failure, sepsis and Reye syndrome is thought to be due to hepatic mechanisms.
Hypoglycaemia due to endocrine deficiencies
Endocrine deficiencies, especially those involving the pituitary and the adrenal glands, can result in low blood glucose concentrations but these are rarely <3.0 mmol/L unless associated with other disorders. Spontaneous hypoglycaemia due to pituitary insufficiency is more commonly seen in neonates and children than in adults but may occasionally be the presenting feature, especially in the elderly. The hypoglycaemia is mainly due to GH deficiency, but the associated deficiency of ACTH accentuates the tendency to hypoglycaemia. Spontaneous hypoglycaemia is also recognized in isolated ACTH deficiency and isolated GH deficiency, particularly after prolonged fasting. The diagnosis is usually suspected on clinical examination and is confirmed by appropriate combined pituitary function tests (see Chapter 18). Plasma insulin and C-peptide concentrations are both appropriately suppressed, but β-OHB concentrations are not, indicating that ketogenesis may not be impaired in the absence of growth hormone. The hypoglycaemia in this setting should be treated with both glucose and hydrocortisone.
Hypoglycaemia is uncommon in primary adrenal insufficiency (Addison disease). It can be precipitated by missing a meal and by exercise and is mainly due to impaired gluconeogenesis. Alcohol, by further impeding gluconeogenesis, may also provoke hypoglycaemia in these patients. A plasma cortisol concentration at the lower end of the reference range, during a spontaneous hypoglycaemic episode, is not sufficient evidence of adrenocortical insufficiency as it may be the result of a lower glycaemic threshold to stimulate cortisol secretion in patients with recurrent hypoglycaemia but without adrenal insufficiency. Plasma insulin and C-peptide concentrations are appropriately low and β-OHB concentrations are high. Hypoglycaemia occurring in acute adrenal insufficiency (Addisonian crisis) is a medical emergency and requires immediate correction by intravenous infusion of glucose in addition to hydrocortisone and other measures.
Congenital adrenal hyperplasia is a rare cause of neonatal hypoglycaemia. In the majority of patients, cortisol deficiency is not severe owing to a compensatory increase in ACTH secretion.
Although untreated hypothyroidism is associated with some lowering of fasting blood glucose concentrations, symptomatic hypoglycaemia has only been reported in a few cases and its existence has been questioned. Deficiency of other glucogenic hormones, such as adrenaline and glucagon, does not seem to cause hypoglycaemia.
Drug-induced hypoglycaemia
Insulin, insulin analogues or insulin secretagogues (sulfonylureas and meglitinides) are by far the most common causes of hypoglycaemia. Precipitating factors are restricted carbohydrate intake and liver and renal impairment. Sulfonylurea drugs can cross the placental barrier and stimulate insulin secretion in the fetus. Life-threatening hypoglycaemia has been reported in newborn infants of diabetic mothers who were treated with chlorpropamide during the third trimester. Sulfonylurea-induced hypoglycaemia may be prolonged, especially in those with renal impairment, lasting for up to seven days and necessitating continuous treatment. Mortality is high, ranging from 7.5 to 8.4%, and in attempted suicide with sulfonylurea drugs, the mortality is even higher with seven deaths out of 20 reported cases.
Salicylate overdose has been associated with hypoglycaemia in children. In adults, therapeutic doses of salicylates have been shown to lower blood glucose concentration in both diabetic and non-diabetic patients. The mechanisms by which salicylates produce hypoglycaemia are unknown. Enhancing insulin secretion and inhibiting hepatic gluconeogenesis are possible mechanisms.
Non-selective β-blockers (e.g. propranolol), in therapeutic doses, may induce hypoglycaemia, particularly in the presence of other precipitating factors such as liver disease, fasting or strenuous exercise. Hypoglycaemia has also been reported in newborn infants of mothers who were treated with propranolol until hours before delivery. The mechanism is thought to be through the prevention of the normal glucagon-mediated glycogenolytic and gluconeogenic responses by the liver. In diabetic patients, β-blockers may change the pattern of adrenergic symptoms of hypoglycaemia by diminishing the occurrence of tremor and palpitations and by increasing the occurrence of sweating. In addition, because glycogenolysis and gluconeogenesis in liver are stimulated through β2-receptors, blockade of these receptors by non-selective β-blockers could prolong recovery time from hypoglycaemia. Selective β1-blockers (e.g. atenolol and bisoprolol), however, do not impair the recovery from hypoglycaemia in patients treated with insulin or oral hypoglycaemic agents.
Quinine stimulates insulin secretion and its intravenous use in the treatment of falciparum malaria has been associated with profound hypoglycaemia. Plasma insulin and C-peptide concentrations are both elevated, indicating increased endogenous secretion. Falciparum malaria, especially in children, can itself cause hypoglycaemia. This, however, is associated with suppressed plasma insulin concentrations and is probably due to high glucose uptake by the parasitized erythrocytes. Acute kidney injury, hepatic dysfunction and starvation are common in malaria and may be additional factors in precipitating the hypoglycaemia. Symptoms of hypoglycaemia may be mistaken for those of cerebral malaria, and blood glucose should always be monitored in these patients. Life-threatening, quinine-induced hypoglycaemia can be treated with octreotide, which blocks the insulinotropic effect of quinine.
Pentamidine, a drug used for the treatment of trypanosomiasis and leishmaniasis, has a direct toxic effect on β-Cells, causing release of preformed insulin and hence hypoglycaemia. This can be followed by β-Cell destruction with ultimate insulin deficiency and diabetes. Pentamidine is also used for the treatment of Pneumocystis carinii infection, and there are at least 30 case reports of severe pentamidine-induced hypoglycaemia in undernourished patients with AIDS (acquired immune deficiency syndrome).
Disopyramide, an antiarrhythmic drug, has also been associated with severe hypoglycaemia, particularly in elderly patients with renal or hepatic impairment. Patients with non-functional endocrine tumours and liver metastases may be at risk of developing hypoglycaemia when treated with long-acting somatostatin analogues, as a result of reduced glucagon and GH secretion.
Other drugs that have occasionally caused hypoglycaemia include angiotensin-converting-enzyme (ACE) inhibitors, angiotensin–receptor antagonists, haloperidol, lidocaine, p-aminobenzoic acid, phenylbutazone, propoxyphene, saquinavir, tricyclic antidepressants, tyrosine kinase inhibitors and sulphonamides.
Alcohol-induced hypoglycaemia
Alcohol can be associated with both fasting and reactive hypoglycaemia. Several mechanisms may be operational in alcohol-induced fasting hypoglycaemia but the most important is direct inhibition of gluconeogenesis. This is mainly due to accumulation of NADH and increased NADH/NAD+ ratio resulting from the oxidation of ethanol.
Alcohol-induced fasting hypoglycaemia characteristically occurs 6–36 h after ingestion of moderate to large amounts of alcohol. Most patients are aged between 20 and 40 years, but occasionally it can occur in children after a relatively small amount of alcohol. Patients present with neuroglycopenic symptoms including stupor and coma. The patient’s breath may smell of alcohol and the symptoms may be mistaken for acute alcoholic intoxication. Therefore, delayed recovery from a presumed alcoholic intoxication should alert the doctor to the possibility of hypoglycaemia.
The blood glucose concentration is usually <2.2 mmol/L and alcohol is nearly always detectable in the blood, though the concentration is not necessarily very high. Severe metabolic acidosis with high blood lactate concentration is a characteristic feature. Hyperketonaemia and ketonuria are almost invariably present but predominantly in the form of β-OHB, since the accumulation of NADH suppresses the conversion of β-OHB to acetoacetate. Ketosis may, therefore, go unrecognized if methods that only detect acetoacetate are employed (e.g. Ketostix®). Plasma insulin and C-peptide concentrations during the hypoglycaemia are usually appropriately suppressed. Prompt diagnosis and treatment, with intravenous glucose, is essential since mortality is relatively high. Glucagon is not effective because hepatic glycogen stores are depleted by the time hypoglycaemia ensues.
Alcohol potentiates the hypoglycaemic effect of insulin and sulfonylurea drugs. In insulin-treated patients, ingestion of alcohol may produce profound hypoglycaemia, which can be fatal. Hypoglycaemia resulting from the combined effect of alcohol and sulfonylurea drugs tends to be less profound, possibly because diabetic patients receiving these drugs tend to be obese and, therefore, are in part protected from the hypoglycaemic effects of alcohol. This is because alcohol does not inhibit the release of glucose from pre-existing glycogen stores, and in obese subjects, these stores are generally sufficient to meet the need for glucose during fasting for 12 h or more.
Alcohol has been shown to potentiate the insulin-stimulating effect of glucose and thus increases the risk for reactive hypoglycaemia. This reaction can be demonstrated in 10–20% of healthy subjects who have consumed a mixture of alcohol and sucrose, such as ‘gin and tonic’, on an empty stomach and who refrained from eating for a few hours afterward. This effect is not observed, however, when saccharin or fructose is substituted for sucrose as the sweetening agent. Starchy foods, such as nibbles and bread, increase the risk for reactive hypoglycaemia, whereas foods providing mainly fat or protein have the reverse effect.
Hypoglycaemia due to deficient energy intake
Symptomatic hypoglycaemia is well recognized in starvation and has been observed in patients with protein-calorie malnutrition and anorexia nervosa. The hypoglycaemia is usually due to reduced hepatic glycogen reserves as well as a reduced supply of gluconeogenic substrates. In addition, the reduction in ketosis due to markedly depleted fat stores deprives the central nervous system of an alternative source of energy. The major risk factors are low body weight and intercurrent infection. Compulsive excessive exercise in patients with anorexia nervosa may also be a factor. Plasma insulin and C-peptide concentrations are appropriately suppressed. Despite low circulating insulin concentrations, β-OHB concentrations are low, owing to depleted fat stores. Hypoglycaemia in anorexia nervosa has a poor prognosis.
Septicaemia
Bacterial septicaemia, especially Gram-negative, can occasionally cause hypoglycaemia. It has been postulated that cytokines produced by macrophages, in response to endotoxin stimulation, may induce hypoglycaemia by increasing insulin secretion. The endotoxins released may also have a direct hypoglycaemic effect, probably by inhibiting gluconeogenesis. Acute kidney injury, which is often associated with septicaemia, may also be an important factor in the pathogenesis of the hypoglycaemia.
Exercise-related hypoglycaemia
Exercise is associated with a marked increase in glucose uptake by muscles. During the first 5–10 min of severe exercise, glucose is supplied by the breakdown of muscle glycogen, but by 40 min, 75–90% of the glucose is supplied by the blood, mainly from increased hepatic glucose production. Initially, 75% of increased hepatic glucose output is derived from glycogenolysis and 25% from gluconeogenesis. With prolonged exercise, gluconeogenesis becomes more important and contributes up to 45% of the total hepatic glucose output. Decreased plasma insulin and increased catecholamines, glucagon, cortisol and GH concentrations all contribute to the increased hepatic glucose output. If hepatic glucose production is inadequate, blood glucose cannot be maintained during exercise and hypoglycaemia ensues.
Hypoglycaemia following excessive exercise is well recognized and may be severe enough to cause seizures. Although exercising to exhaustion can produce hypoglycaemia, the symptoms of exhaustion are not related to hypoglycaemia and glucose administration does not modify the time of exercise to exhaustion.
Postprandial (reactive) hypoglycaemia
Rarely, hypoglycaemia occurs only in response to ingestion of a meal. Symptoms are usually neurogenic or autonomic and generally occur 2–4 h after ingestion of food and last for about 10–20 min. These symptoms are different from those of the dumping syndrome (flushing, sweating, abdominal cramps and hypotension), which occur within half an hour of eating. The diagnosis of postprandial hypoglycaemia requires the demonstration of the Whipple’s triad.
Postprandial hypoglycaemia may occur in patients who have undergone major gastric surgery, such as Roux-en-Y gastric bypass, and is also a feature of the autoimmune insulin syndrome, non-insulinoma pancreatogenous hypoglycaemia, or may be alcohol induced or idiopathic (see below). Postprandial hypoglycaemia is also seen in patients with hereditary fructose intolerance after ingestion of fructose. It is worth noting that conditions that are usually associated with fasting hypoglycaemia, such as insulinoma, may occasionally produce symptoms only postprandially.
‘Idiopathic’ postprandial reactive hypoglycaemia has been documented in a small number of patients in whom no obvious other cause for hypoglycaemia was found. In these patients, a diet low in refined carbohydrates and high in soluble fibres is usually sufficient to relieve the symptoms. The addition of snacks in the middle of the morning and the afternoon may also prevent the fall in blood glucose. Patients should avoid rapidly absorbable sugars and should replace sucrose with either saccharin or fructose, a non-insulinotropic carbohydrate. If symptoms persist despite following the above dietary modifications, the use of an intestinal α-glucosidase inhibitor, such as acarbose, has been shown to be successful in alleviating symptoms in some patients with idiopathic postprandial reactive hypoglycaemia.
The postprandial syndrome
Patients who have postprandial symptoms but without documented hypoglycaemia should be referred to as having ‘the postprandial syndrome’. Symptoms are generally non-specific, such as feelings of vague ill health, lightheadedness, dizziness, anxiety, palpitations, fatigue, hunger and ‘inner trembling’, which may resemble the autonomic symptoms of hypoglycaemia. Many patients, especially those who are well informed about hypoglycaemia from the lay literature, state that symptoms are relieved by eating, thereby suggesting hypoglycaemia. However, detailed questioning often reveals that symptoms are relieved either immediately or within a few minutes of eating foods such as cheese or bread, which are unlikely to raise blood glucose concentrations in such a short period of time. Many patients complain of generalized weakness and inability to concentrate in between attacks. The symptoms are not progressive but usually persist for many years. Reliance on the use of the oral glucose tolerance test (OGTT) in the investigation of this condition in the past has led to the misdiagnosis of ‘reactive’ hypoglycaemia in many of these patients. The OGTT should not be used in this context since 10% of healthy individuals may have low blood glucose concentrations (<2.6 mmol/L) during the test. In addition, in many patients with postprandial symptoms, the symptoms do not correlate with the nadir blood glucose concentrations and some individuals may even experience symptoms after a placebo oral glucose tolerance test. Self-monitoring of blood glucose at home, to document hypoglycaemia, using glucose meters should not be encouraged, since self-diagnosis by patients themselves is seldom confirmed by accurate investigations.
Inherited metabolic disease
Several inborn errors of metabolism can produce hypoglycaemia as one of their major clinical manifestations. These disorders are almost invariably diagnosed in childhood (see Chapter 24), but in mild cases, the diagnosis may not be made until middle life. The hypoglycaemia is usually of the fasting type, but in some disorders, for example galactosaemia and hereditary fructose intolerance, it only occurs following the ingestion of certain types of food.
CONCLUSION
Hypoglycaemia literally means low blood glucose concentration. Many apparently healthy subjects may have low blood glucose concentrations during a prolonged fast, or 3–5 h after ingestion of glucose. Pathological hypoglycaemia should, therefore, be defined as a clinical syndrome in which symptoms or signs of hypoglycaemia occur in the presence of low plasma glucose concentration and that the symptoms or signs are relieved after the plasma glucose concentration is raised (Whipple’s triad). Although it is frequently suspected as a cause of symptoms, hypoglycaemia is rare, other than in diabetic patients who are being treated with insulin or oral hypoglycaemic drugs.
The diagnosis of a hypoglycaemic disorder requires a high level of suspicion, a thorough history and a careful assessment of possible underlying illnesses or drugs. A healthy-appearing patient with hypoglycaemia requires a different diagnostic approach from a patient who has a concurrent illness or is hospitalized. Generally, the measurement of plasma glucose, insulin, C-peptide and proinsulin concentrations during symptoms may be sufficient to confirm the diagnosis and establish the cause of hypoglycaemia. In the majority of patients, however, further investigations (insulin autoantibodies, screening for hypoglycaemic agents) and provocation tests (prolonged fast or a mixed meal test) may be necessary. An inability to demonstrate a low plasma glucose concentration when the patient is symptomatic virtually excludes the diagnosis of hypoglycaemia.
Further reading
Vezzosi D, Bennet A, Maiza JC, et al. Diagnosis and treatment of insulinomas in the adults. In: Akin F, ed. Basic and clinical endocrinology up-to-date. Intech Open Access Publishing; 2011. http://www.intechopen.com/books/basic-and-clinical-endocrinology-up-to-date/diagnosis-and-treatment-of-insulinomas-in-the-adults [Accessed October 2013].
An excellent and detailed up-to-date review of the diagnosis, differential diagnosis and the medical management of adult patients with insulinoma.