Rheumatology and Musculoskeletal Emergencies
Edited by Anthony Brown
14.1 Rheumatological emergencies
Michael J Gingold, Adam B Bystrzycki and Flavia M Cicuttini
Introduction
Rheumatological conditions are common and encompass inflammatory or connective tissue diseases and mechanical/musculoskeletal conditions. Life-threatening emergencies are rare and relate to either the underlying condition or its treatment. The challenge is in making the distinction between the two, as treatment is frequently diametrically opposed; for example, the difference between administering further immunosuppression and giving antibiotics.
The most common rheumatological emergency seen in the emergency department (ED) is acute monoarthritis (see Chapter 14.2). This chapter discusses the important general emergencies associated with rheumatological conditions. Many of these are multisystem diseases and emergencies relate to either a primary joint problem, an extra-articular manifestation or sometimes to the drugs used in management.
Many of these conditions are autoimmune, thus immunosuppression is usually central to their management making infection a frequent complication. More targeted, so-called biological therapies, which inhibit proinflammatory cytokines as well as B- and T-cell activity have been developed. They carry their own set of potential complications, again including infection.
Rheumatoid arthritis
Rheumatoid arthritis (RA) affects 1–2% of the population across most ethnic subgroups and is two to three times more common in females than males. RA is a systemic inflammatory condition of unknown aetiology characterized by widespread synovitis resulting in joint erosions and destruction. It may also produce extra-articular manifestations including vasculitis and visceral involvement.
Management typically involves symptom relief with non-steroidal anti-inflammatory drugs (NSAIDs) and/or corticosteroids and early initiation of conventional disease modifying antirheumatic drugs (DMARDs). These include methotrexate, leflunomide, sulphasalazine and hydroxychloroquine and, if these agents fail to control disease progression, biological agents are commenced. The latter act by inhibiting tumour necrosis factor-α (TNF-α) (infliximab, etanercept, adalimumab, golimumab, certolizumab pegol), interleukin-6 (IL-6) (tocilizumab), T-cell co-stimulation (abatacept) or by depleting B cells (rituximab). See Chapter 14.3 for further details on the clinical features of RA, its diagnosis, investigation and management.
Articular manifestations of rheumatoid arthritis
Acute monoarthritis
A patient with established RA may present with an acutely painful, hot, swollen joint that may be due to the underlying condition or, alternatively it may be due to a septic arthritis. Patients with RA are two to three times more susceptible than matched controls [1]. The risk is approximately twofold higher again in RA patients on TNF-α inhibitors compared to RA patients on conventional DMARDs [2]. Thus, the possibility of septic arthritis must be considered in a patient with RA who has acute monoarthritis out of keeping with their disease activity. See Chapter 14.2 for an approach to acute monoarthritis.
Cervical spine involvement
Cervical spine involvement in RA is common with a prevalence of up to 61%. It is more likely in those with long-standing, erosive disease and disease of greater severity and activity [3]. Cervical spine involvement is associated with increased mortality [4] and may manifest as atlanto-axial subluxation (most commonly anterior movement on the axis) or subluxation of lower cervical vertebrae. Either of these can result in cervical myelopathy.
Cervical spine subluxation is often asymptomatic – up to 44% in one study [3]. The most common symptom is neck pain that may radiate towards the occiput. Other suggestive symptoms include sensory loss in hands or feet, paraesthesia or weakness in the distribution of cervical nerve roots and slowly progressive spastic quadriparesis.
Important ‘red flags’ suggesting cervical myelopathy are listed in Table 14.1.1.
Table 14.1.1
Symptoms and signs of cervical myelopathy
Symptoms
Pain
Weakness
Peripheral paraesthesia
Gait disturbance
Sphincter dysfunction
Changes in consciousness
Respiratory dysfunction
Signs
Spasticity
Weakness
Hyperreflexia of deep tendon reflexes
Extensor plantar response
Gait ataxia
Respiratory irregularity
Imaging
Plain X-rays of the cervical spine (lateral view) may demonstrate an increase in separation between the odontoid and arch of C1. Prior to taking flexion–extension films, perform plain ‘peg’ X-rays through the open mouth to exclude odontoid fracture or severe atlanto-axial subluxation. Computed tomography (CT) can provide additional useful information, although if there is concern regarding myelopathy, magnetic resonance imaging (MRI) is more sensitive.
Management
The main implication of RA of the cervical spine in the ED is when endotracheal intubation is required. Excessive manipulation (neck flexion with head extension) of the rheumatoid cervical spine in preparation for intubation can result in significant morbidity and even mortality from atlanto-axial and subaxial subluxation.
Whenever possible, a patient with RA who is scheduled to undergo surgery should have imaging of his or her cervical spine prior to endotracheal intubation. Anaesthetic consultation is recommended.
Patients with subluxation and signs of spinal cord compression represent a neurosurgical emergency and prompt referral is essential.
Extra-articular manifestations of rheumatoid arthritis
Rheumatoid vasculitis
Vasculitis in RA can occur in both small- and medium-sized vessels. Patients typically have long-standing, aggressive joint disease. This presentation, although important, is becoming less frequent.
Clinical features
Rheumatoid vasculitis presentations are varied and non-specific. Patients frequently have constitutional symptoms and fatigue. The most common manifestation is cutaneous vasculitis with deep skin ulcers on the lower limbs [5], digital ischaemia and gangrene (medium vessels) or palpable purpura (small vessels). Mononeuritis multiplex is another frequent presentation resulting from vasculitic infarction of the vasa nervorum, which typically has an acute onset.
Medium vessel rheumatoid vasculitis may cause organ infarction and necrosis. Rheumatoid vasculitis can mimic polyarteritis nodosa (PAN) with involvement of the renal arteries and, less commonly, the mesenteric circulation. Pericarditis may accompany rheumatoid vasculitis, but coronary vasculitis is rare. Ocular manifestations include episcleritis and peripheral ulcerative keratitis. Central nervous system (CNS) involvement is rare.
Investigations
Rheumatoid factor titre is typically elevated in rheumatoid vasculitis, although this is a non-specific finding. Rheumatoid vasculitis in the absence of rheumatoid factor is rare. Erythrocyte sedimentation rate (ESR) and C-reactive protein (CRP) are also elevated. Check a full blood count, urea and electrolytes and a midstream urine specimen for active urinary sediment including abnormal red cells or casts, as well as infection.
Further investigations are directed at the relevant organ system involved usually after specialist consultation:
Angiography findings are non-specific and not always diagnostic.
Diagnosis
Suspect rheumatoid vasculitis in patients with a long-standing history of seropositive RA who presents with constitutional symptoms and one of the above features, such as a typical rash, digital gangrene, red eye, neurological complaint or an active urinary sediment.
Management
Systemic rheumatoid vasculitis has a poor prognosis without immune-suppressive therapy. Urgent rheumatology consultation is required as treatment usually consists of high-dose corticosteroids as well as cyclophosphamide, often necessitating hospital admission.
Other extra-articular manifestations of RA
Pulmonary disease
Pulmonary manifestations include pleural-based disease, such as pleurisy or pleural effusions, or parenchymal disease, such as interstitial lung disease (the most common manifestation), organizing pneumonia and rheumatoid nodules. Caplan’s syndrome occurs when RA is associated with pneumoconiosis. Important differential diagnoses include infection due to immune suppression, treatment-related toxicity, such as methotrexate-induced pneumonitis, and other medical co-morbidities including chronic obstructive pulmonary disease.
Pleural disease often resolves without treatment, although an NSAID may be used symptomatically. Parenchymal disease documented on chest X-ray or high-resolution CT requires specialist treatment.
Cardiac disease
Pericarditis occurs in 30% of RA patients based on echocardiography, but less than 10% have clinical features. It generally presents when there is active joint and other extra-articular disease and management consists of NSAIDs or prednisolone.
Myocarditis is a rare manifestation of RA. It may be granulomatous and, depending on its location, can produce valvular (especially mitral) incompetence or conduction defects.
Sjögren’s syndrome
Sjögren’s syndrome may present in a primary form as a systemic disease, but can also occur secondary to RA and other connective tissue disorders. The classic symptoms are dry gritty eyes, dry mouth or both. Treatment is usually symptomatic in patients with no other features.
Felty’s syndrome
Felty’s syndrome is characterized by seropositive RA, splenomegaly and neutropaenia. There may be other cytopaenias, as well as leg ulcers, and infection is a risk.
Renal disease
Renal involvement with RA is rare and includes vasculitis and glomerulonephritis. Secondary amyloidosis can occur in patients with long-standing active disease. However, many medications used in RA are nephrotoxic, in particular NSAIDs and ciclosporin.
Neurological disease
Vasculitis may produce mononeuritis multiplex, otherwise central nervous system involvement is rare.
Ischaemic heart disease in RA and other connective tissue diseases
Patients with RA and other connective tissue diseases, such as systemic lupus erythematosus (SLE), have an increased risk of ischaemic heart disease (IHD) [6]. This occurs independently of traditional risk factors, such as smoking, dyslipidaemia, hypertension, etc. and is more common in those with extra- articular disease [7]. The higher incidence of IHD appears related to disease factors, such as widespread inflammation, but medications, such as NSAIDs (including selective COX-2 inhibitors) and corticosteroids, may play a role.
Thus there should be a heightened awareness when ruling out ischaemic chest pain in the RA patient. Cardiac investigations are no different to those undertaken for non-RA patients and a patient with a history suggestive of acute coronary syndrome (ACS) but negative serial cardiac biomarkers and electrocardiograms must proceed to provocative testing. Management of ACS in RA is no different (see Chapter 5.2).
Systemic lupus erythematosus
Systemic lupus erythematosus is a multisystem, autoimmune disease. It is the prototype disease of immune complex deposition resulting in tissue damage across a wide range of organ systems and one of the most common autoimmune conditions in women of childbearing age.
Clinical features
Common presenting features of SLE include general constitutional symptoms, such as fatigue, malaise and weight loss. There is a variety of skin manifestations in SLE which are lupus-specific (malar rash, discoid lupus, subacute cutaneous lupus erythematosus) or non-specific (panniculitis, alopecia, oral ulceration). Arthralgias or an acute non-erosive arthritis are the most common presenting symptoms of SLE.
Another common manifestation is serositis causing pleurisy, pericarditis or peritonitis. SLE also causes renal and CNS disease (see below) and, rarely, can involve the lung parenchyma (pneumonitis, pulmonary hypertension) and heart (myocarditis, endocarditis). Myositis may also occur.
Investigations
A full blood examination often reveals cytopaenias which are a common feature of SLE. Biochemistry may indicate renal impairment. ESR and CRP may be raised.
Clotting abnormalities can include a prolonged activated partial thromboplastin time due to the lupus anticoagulant (LA), one of the antiphospholipid antibodies along with anticardiolipin antibody (aCL) and others. Paradoxically, there is an associated predisposition to both venous and arterial blood clots when these are positive.
Serological abnormalities
The antinuclear antibody (ANA) is present in 95% of patients with SLE, but may also occur in other connective tissue and inflammatory diseases, as well as at low levels in healthy adults. The anti-Smith (Sm) and anti-dsDNA (double-stranded DNA) antibodies are more specific but less sensitive for SLE. Anti-Sm is obtained as part of a panel of antibody tests for extractable nuclear antigens (anti-ENA). Serological abnormalities also include decreased levels of complement components C3 and C4.
Other tests are directed towards the organ system involved, for example, midstream urine specimen looking for proteinuria or glomerular haematuria (>70% dysmorphic red blood cells or red-cell casts) and chest X-ray in the patient with serositis.
Assessing SLE disease activity
It is important to determine SLE disease activity in the ED. Useful symptoms of activity include mouth ulcers, alopecia and constitutional symptoms, as well as organ-specific symptoms, such as arthralgia or pleuritic chest pain.
Investigations used to assess disease activity include complement levels (low in active SLE), CRP and ESR (elevated), as well as anti-dsDNA titre. These are not diagnostic and many people with quiescent SLE may also have hypocomplementaemia or elevated anti-dsDNA titres.
A midstream urine for urinary sediment is an essential marker of renal involvement.
Management
Management of SLE is directed by the organ system involved and includes topical therapies for cutaneous lupus and NSAIDs for arthralgias and mild serositis. Most patients with SLE will be on an antimalarial, such as hydroxychloroquine, helpful for skin and musculoskeletal manifestations as well as organ involvement. Many patients will also be on corticosteroids. Those with major organ involvement will also be taking other immunosuppressants, such as methotrexate, cyclophosphamide or azathioprine. Mycophenolate mofetil is frequently now used as an alternative to cyclophosphamide for lupus nephritis.
Lupus nephritis
Early diagnosis of lupus nephritis is essential to prompt management and prevent progression of renal damage. Patients may be asymptomatic or present with nocturia, haematuria or proteinuria. Other presentations include hypertension, rapidly progressive glomerulonephritis and the nephrotic syndrome.
Urinalysis is the most useful investigation in detecting lupus nephritis and proteinuria is the most common abnormality detected. The fresh urine specimen should be sent for phase contrast microscopy in order to detect the presence of dysmorphic erythrocytes (>70% indicates glomerular disease) or cellular casts.
Urinalysis can expedite the investigation and further management of this potentially organ-threatening condition. Prompt referral to a rheumatologist or renal physician for consideration of renal biopsy and further management is indicated.
Neuropsychiatric SLE
There is a myriad of neuropsychiatric manifestations of neuropsychiatric SLE. Neurological presentations include:
Psychiatric presentations include:
These presentations are non-specific and have a broad differential diagnosis or can be subtle and progress. Unfortunately, there is no specific diagnostic test which helps differentiate SLE from other potential aetiologies. Thus, the diagnosis is made from a range of clinical features and tests. The role of the emergency department is first to exclude the more common non-SLE presentations, such as meningitis or intracranial haemorrhage.
Investigations
Imaging studies are needed as well as tests for SLE activity (see above). CT brain scan may detect changes of acute infarction, but is also useful in excluding other non-SLE causes, such as haemorrhage or tumour. MRI is more sensitive in detecting white matter abnormalities, although they are frequently non-specific.
Cerebrospinal fluid (CSF) analysis is essential to exclude infection, but may be normal in SLE. Changes, such as elevated protein, low glucose or even a positive ANA, are non-specific and do not always reflect active SLE. The electroencephalogram is occasionally useful in cases of unexplained altered conscious level with suspected non-convulsive status epilepticus.
Temporal (giant cell) arteritis and other vasculitides
Temporal (giant cell) arteritis
Giant cell arteritis (GCA) is the most frequent vasculitis and almost exclusively affects Caucasians. It is a large and medium vessel vasculitis of unknown aetiology, which predominantly affects the cranial branches of arteries originating from the aortic arch and, commonly though not exclusively, the temporal artery.
Polymyalgia rheumatica (PMR) is a syndrome of inflammatory pain and stiffness in the shoulder and pelvic girdles that occurs alone or frequently in association with GCA.
Epidemiology
GCA and PMR rarely ever occur before the age of 50 years [8], with a mean age at diagnosis of approximately 72 years. The incidence of GCA is roughly 1 in 500 of people over the age of 50 years, although the incidence and prevalence of PMR are less well studied.
Clinical features
The most common symptom of GCA is new headache, usually localizing to the temporal region, although it can be more diffuse. The area is often tender and worsened by brushing the hair. Most patients complain of constitutional symptoms, such as malaise, fatigue, anorexia and weight loss. Jaw claudication (pain after a period of chewing) is the most specific symptom for GCA, although not sensitive, as it is present in only 34% [8]. On examination, the temporal arteries may be thickened, ‘ropey’ and tender with a reduced or absent pulse.
The most serious complication of GCA is anterior ischaemic optic neuropathy (AION) resulting in sudden painless loss of vision which can be bilateral, particularly if untreated. Less commonly, other branches of the aorta may be involved resulting in hemiparesis, arm claudication, aortic dissection or myocardial infarction.
Polymyalgia rheumatica
PMR usually affects the neck, shoulder and pelvic girdles resulting in stiffness and inflammatory pain, worse in the morning and after rest. The pain is often poorly localized and muscle atrophy may occur late in the disease. There may also be synovitis affecting the shoulders, knees, wrists and hands.
The relationship between onset of symptoms of GCA and PMR is highly variable. PMR symptoms may occur before, after or with GCA symptoms. At some point, 5–15% of patients with PMR will have a diagnosis of GCA and about 50% of patients with GCA have symptoms of PMR.
Differential diagnosis of GCA and PMR
GCA can mimic any of the other vasculitides. Non-arteritic anterior ischaemic optic neuropathy can also mimic GCA. The differential diagnosis of PMR includes late-onset RA, polmyositis and other myopathies, fibromyalgia, malignancy and hypothyroidism.
Investigations
The classic non-specific laboratory finding in GCA and/or PMR is a markedly elevated ESR (often>100 mm/h). CRP is also usually elevated and a full blood count often shows a mild normochromic normocytic anaemia.
A temporal artery biopsy confirms the diagnosis of GCA and is particularly useful when the diagnosis is doubtful or the presentation atypical. However, as there is a false negative rate of 10–30%, a negative biopsy does not exclude GCA.
Criteria for diagnosis
The ACR classification criteria for GCA are helpful in differentiating GCA from other forms of vasculitis [9]. They include age at onset>50 years, a new headache, temporal artery tenderness or decreased pulsation and an ESR>50. An abnormal artery biopsy showing vasculitis with mononuclear infiltrate or granulomatous inflammation with multinucleated giant cells also confirms the diagnosis.
Although various classification criteria for PMR have been published, having excluded other diagnoses (except GCA), the presence of all three of the following clinical and laboratory criteria defines the diagnosis [10]:
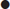
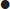
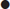
In practice, rapid response to prednisolone≤20 mg daily is also used as an additional criterion with 50–70% improvement within 72 hours.
Management
Corticosteroids are essential for GCA and should not be withheld to perform a biopsy. The initial dose for GCA is unclear, but prednisone 1 mg/kg/day is used especially for ischaemic complications. However, lower doses, such as prednisone 40–60 mg, are recommended for uncomplicated disease [10].
The dose of prednisone for PMR uncomplicated by GCA is lower at 10–20 mg/day; 15 mg is generally agreed as an appropriate standard dose [11]. Most GCA patients do not require hospital admission, provided a temporal artery biopsy can be organized within a few days. However, patients with visual loss at diagnosis require urgent treatment often with pulsed parenteral corticosteroids and inpatient admission. Patients with GCA should also be commenced on aspirin.
Approach to the other systemic vasculitides
The systemic vasculitides are a group of disorders characterized by an inflammatory infiltrate in the walls of blood vessels resulting in damage to the vessel wall. The clinical manifestations depend upon the size of vessel and location in the vascular tree and may result in systemic or organ-specific manifestations. Table 14.1.2 classifies vasculitic syndromes according to vessel size (there is much overlap).
Table 14.1.2
Classification of systemic vasculitis according to vessel size
| Vessel size | Vasculitis |
| Large | Takayasu’s arteritis |
| Temporal (giant cell) arteritis | |
| Medium | Polyarteritis nodosa |
| Kawasaki’s disease | |
| Small | Wegener’s granulomatosis (ANCA+) |
| Microscopic polyangiitis (ANCA+) | |
| Churg–Strauss syndrome (ANCA+/−) | |
| Henoch–Schönlein purpura | |
| Cryoglobulinaemic vasculitis | |
| Leucocytoclastic cutaneous vasculitis |
Clinical features
An underlying vasculitis should be considered in patients who present with one or more of the following:
 unexplained systemic illness – fatigue, fevers, night sweats, malaise
unexplained systemic illness – fatigue, fevers, night sweats, malaise
 unexplained ischaemia of an organ or limb
unexplained ischaemia of an organ or limb
 chronic inflammatory sinusitis and chronic discharge or bleeding from the nose or ears
chronic inflammatory sinusitis and chronic discharge or bleeding from the nose or ears
 microscopic haematuria, especially if dysmorphic glomerular erythrocytes
microscopic haematuria, especially if dysmorphic glomerular erythrocytes
 any of the above in the setting of atopy and peripheral blood eosinophilia.
any of the above in the setting of atopy and peripheral blood eosinophilia.
Investigations and diagnosis
Baseline investigations should include full blood count (FBC), urea and electrolytes (U&E), liver function tests (LFTs) and clotting studies as well as CRP and ESR. Blood cultures should be taken if the patient is systemically unwell.
A panel of autoimmune serological tests is carried out including ANA, ENA, rheumatoid factor, dsDNA, anticyclic citrullinated peptides (anti-CCP), complement levels (C3, C4), antineutrophil cytoplasmic antibodies (ANCA) and cryoglobulins. PAN is associated with hepatitis B and cryoglobulinaemic vasculitis with hepatitis C infection.
Collection of a midstream urine specimen to look for glomerular haematuria is mandatory when vasculitis is suspected. Imaging is indicated, such as chest X-ray, CT scan of the chest or sinuses or other areas depending on the suspected organ involved. The definitive diagnosis of vasculitis requires biopsy of affected tissue or angiography.
Differential diagnosis of systemic vasculitis
Other conditions which may mimic systemic vasculitis include:
Management of systemic vasculitis
Treatment is usually with high-dose corticosteroids and, depending on the condition, additional immunosuppression, such as cyclophosphamide. Urgent specialist referral is essential.
Ankylosing spondylitis
Ankylosing spondylitis (AS) is an inflammatory arthritis of the axial skeleton which can result in progressive spinal fusion. It affects<1% of the general population and its prevalence is linked to the prevalence of HLA-B27.
The hallmark pathological feature of AS is new bone formation and spinal fusion which, combined with the increased prevalence of low bone mineral density in these patients, makes spinal injury a particular risk. Also a range of features including reduced mobility and muscle atrophy lead to a higher falls risk.
Spinal fractures are up to four times more common in AS patients than in the general population and the risk of spinal cord injury is even higher. Fractures can occur at any point in the spine and do not have the classical appearance of wedge or endplate compression. In advanced fusion, the spine may fracture in a similar way to a long bone and not respect traditional spinal anatomical boundaries. There is also a higher rate of atlanto-axial subluxation, with similar precautions required as in those with RA. Furthermore, patients with advanced fusion may also develop cauda equina syndrome in the absence of a fracture.
Fractures in AS can be missed by plain X-ray and the onset of new spinal pain or a change in spinal pain necessitiates further imaging with either CT or MRI.
Rheumatological therapy emergencies
The medications used in rheumatology include NSAIDs, corticosteroids, DMARDs and biological DMARDs. These medications are all associated with adverse effects which occasionally result in serious morbidity.
Non-steroidal anti-inflammatory drugs
NSAIDs are commonly used for relief of arthralgia in both inflammatory and non-inflammatory conditions. They are of equal efficacy, although those with shorter half-lives appear to have less gastrointestinal toxicity [12]. NSAIDs should be used in the lowest possible dose for the shortest duration and combinations of NSAIDs (except aspirin) should be avoided [12].
The most common adverse effects of NSAIDs include peptic ulcer disease and acute renal failure, both related to inhibition of prostaglandin synthesis. Gastrointestinal (GI) toxicity is more common in the elderly, those on anticoagulants and with high doses or prolonged duration of NSAIDs. Prescribe a proton pump inhibitor when there is concern about GI toxicity. The COX-2 selective inhibitors, such as celecoxib, have a reduced incidence of peptic ulcer disease, but a similar incidence of other adverse effects including hypertension, peripheral oedema and cardiac failure. There is an increased risk of cardiovascular deaths with prolonged courses.
Corticosteroids
Corticosteroids are the mainstay of treatment for most inflammatory rheumatological conditions. At high doses, they provide rapid control of inflammatory disease and are often required for long-term management at low doses. Long-term use is associated with numerous adverse effects, such as diabetes, hypertension and osteoporosis. In addition, psychosis and mood disorders related to corticosteroid use, as well as peptic ulcer disease, may present as an emergency.
Although there is concern about infection among patients on DMARDs, prednisolone contributes considerably (possibly more) to the immune-suppressed patient’s overall infection risk.
Immunosuppressants/disease modifying antirheumatic drugs
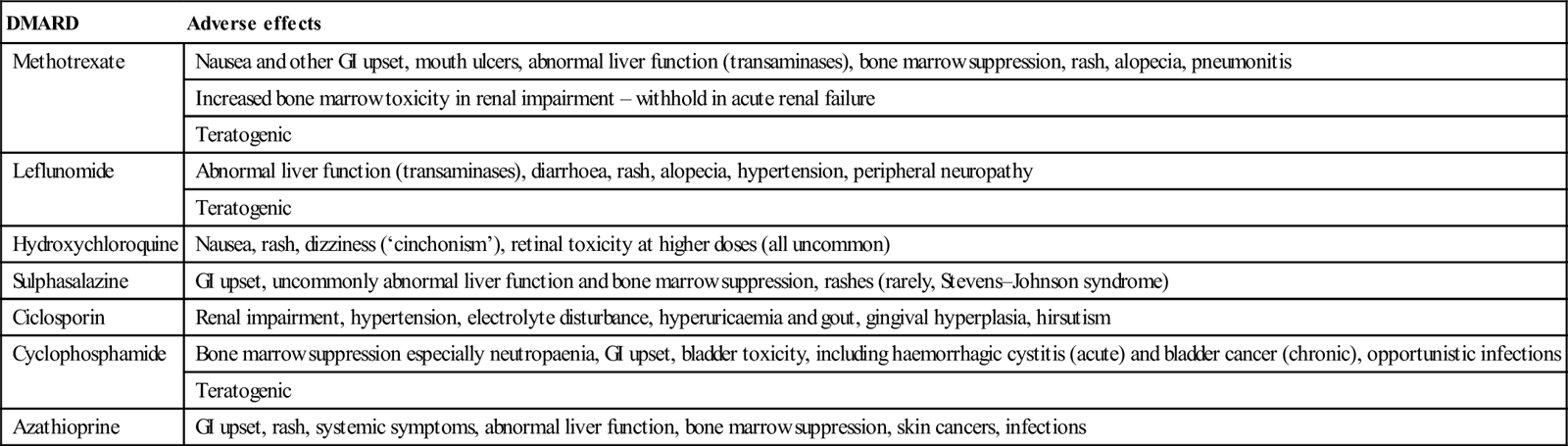
This heterogeneous group of medications is used to prevent joint destruction in the inflammatory arthritides and as steroid-sparing therapy in many connective tissue diseases. They include methotrexate, leflunomide, hydroxychloroquine, sulphasalazine, ciclosporin, azathioprine and cyclophosphamide. Each drug has its own range of adverse effects, but common adverse effects include cytopaenias, rashes including Stevens–Johnson syndrome, abnormal liver function tests, GI toxicity and heightened susceptibility to infections (Table 14.1.3).
Table 14.1.3
Adverse effects of disease modifying antirheumatic drugs (DMARDs)
| DMARD | Adverse effects |
| Methotrexate | Nausea and other GI upset, mouth ulcers, abnormal liver function (transaminases), bone marrow suppression, rash, alopecia, pneumonitis |
| Increased bone marrow toxicity in renal impairment – withhold in acute renal failure | |
| Teratogenic | |
| Leflunomide | Abnormal liver function (transaminases), diarrhoea, rash, alopecia, hypertension, peripheral neuropathy |
| Teratogenic | |
| Hydroxychloroquine | Nausea, rash, dizziness (‘cinchonism’), retinal toxicity at higher doses (all uncommon) |
| Sulphasalazine | GI upset, uncommonly abnormal liver function and bone marrow suppression, rashes (rarely, Stevens–Johnson syndrome) |
| Ciclosporin | Renal impairment, hypertension, electrolyte disturbance, hyperuricaemia and gout, gingival hyperplasia, hirsutism |
| Cyclophosphamide | Bone marrow suppression especially neutropaenia, GI upset, bladder toxicity, including haemorrhagic cystitis (acute) and bladder cancer (chronic), opportunistic infections |
| Teratogenic | |
| Azathioprine | GI upset, rash, systemic symptoms, abnormal liver function, bone marrow suppression, skin cancers, infections |
Biological disease modifying antirheumatic drugs
The so-called ‘biological’ DMARDs are a newer and expanding collection of therapies directed against molecules and cells that mediate joint destruction and help drive the inflammatory process. These therapies are being increasingly used for those who fail conventional DMARD therapy for RA and TNF inhibitors are also used for treatment-resistant psoriatic arthritis and ankylosing spondylitis, as well as other non-rheumatological conditions.
Adverse effects associated with biological DMARDs include an increased risk of infections, particularly soft-tissue and joint infections, as well as reactivation of tuberculosis. Other opportunistic infections appear more common, such as listeriosis. Patients may also develop local injection site reactions and infusion-related reactions, which can be delayed. Less common adverse effects include a form of drug-induced lupus and demyelination.
Presentations of treatment-related emergencies
Infections
As treatment of rheumatological conditions is directed at immunosuppression, infections are a common and expected adverse effect of therapy. Although studies have shown RA patients to have a de novo increased risk of infection, biological DMARDs further increase this risk. Although most of the larger studies have focused on TNF inhibitors, which have been available the longest, there is an increased risk of serious infections compared to the general RA population. This risk may be highest in the first 6 months of therapy [13].
Patients on biological therapy who develop an infection are advised temporarily to cease their treatment and to commence antibiotics. If in doubt, they should be admitted to hospital to receive parenteral antibiotics. There is also an increased risk of reactivation of tuberculosis and infections such as Listeria and Salmonella[13]. Rigorous tuberculosis screening prior to commencement of anti-TNF therapy should now be universal.
Special mention must be made of the biological agent tocilizumab directed against interleukin-6. Tocilizumab causes marked suppression of acute phase reactants, particularly CRP and, even in the presence of active infection, a patient on this may have a normal CRP. Thus if there is a clinical suspicion of infection, appropriate antibiotic therapy must be instituted regardless.
Bone marrow suppression
Anaemia, leucopaenia and thrombocytopaenia all may occur in patients taking DMARDs, such as methotrexate, cyclophosphamide, sulphasalazine and azathioprine, with neutropaenic sepsis a particular danger.
Cytopaenia in a patient taking methotrexate is uncommon but those at increased risk include the elderly and those with renal impairment, related to the drug’s mechanism of action as an inhibitor of dihydrofolate reductase. Management includes temporary cessation of treatment and administration of folinic acid, the active form of folic acid which does not require to be converted by dihydrofolate reductase.
The most common adverse effect of cyclophosphamide is myelosuppression, particularly leucopaenia. The white cell nadir occurs at 2 weeks post-infusion following intravenous therapy. Patients on oral therapy may experience a gradual decrease in white cell count, which is typically less predictable than on intravenous therapy.
Bone marrow suppression may also occur as a side effect of azathioprine treatment, especially if given in combination with allopurinol which inhibits its metabolism, thus potentiating bone marrow toxicity. Cytopaenias are also more common in patients with deficient thiopurine methyltransferase enzyme. Sulphasalazine therapy is uncommonly complicated by bone marrow suppression.
DMARD-related pneumonitis
Methotrexate and leflunomide may both result in lung toxicity. The incidence of methotrexate-induced lung toxicity is difficult to assess but uncommon, with those patients at higher risk who have prolonged duration of methotrexate treatment, pre-existing rheumatoid involvement of the lungs and pleura, increased extra-articular manifestations, diabetes mellitus, previous DMARD use and a low serum albumin [14]. Age and smoking also appear to be important. The most frequent is a hypersensitivity pneumonitis, but other forms of lung injury may occur both acute and chronic, with rapid progress to respiratory failure in more acute situations. Clinical features are non-specific and include constitutional symptoms, cough and progressive dyspnoea. Subacute presentations are more common.
Imaging reveals interstitial opacities and patchy consolidation. High-resolution CT scanning typically shows a ground-glass appearance. The main differential diagnosis is of a respiratory infection which may be due to typical pathogens or opportunistic infections, such as Pneumocystis jirovecii.
Management is supportive with empiric antibiotic therapy in case of infection. Corticosteroids are also used. Patients may become seriously ill and require intensive care, but mortality is still low (1%).
Leflunomide may also cause lung injury, typically in the first few months of therapy and usually when given in combination with methotrexate.
Allopurinol hypersensitivity syndrome
Minor hypersensitivity reactions to allopurinol occur in about 2% of patients and usually consist of a mild rash. Rarely, a severe hypersensitivity syndrome may present in an unwell patient with fever, rash including toxic epidermal necrolysis, erythema multiforme or a diffuse macropapular or exfoliative dermatitis, abnormalities of liver function, peripheral blood eosinophilia and acute renal failure due to interstitial nephritis. It is more common in those with renal impairment who do not have an appropriate dose reduction. This presentation has a mortality rate of 25%. Treatment is supportive.
14.2 Monoarthritis
Michael J Gingold, Adam B Bystrzycki and Flavia M Cicuttini
Septic arthritis
The assessment of a patient with acute monoarthritis is focused on excluding a septic arthritis. Septic arthritis can cause rapid joint destruction and mortality has been reported as high as up to 15% [1].
Pathogenesis and pathology
Non-gonococcal bacterial arthritis occurs when bacteria enter the synovial lining of a joint via the haematogenous route, local spread from nearby soft-tissue infections or following penetrating trauma or injury to a joint.
When the bacteria reach the synovium, they trigger an inflammatory response and bacteria and inflammatory cells enter the synovial fluid in the joint space, causing swelling and destruction of articular cartilage. These destructive changes may extend to subchondral bone and produce irreversible damage within days. The commonest causative organisms are staphylococci and streptococci.
Epidemiology and risk factors
The prevalence of septic arthritis ranges between 4 and 10:100,000 patients per year and appears to be rising. It is also almost seven times more common in indigenous Australians [2].
Risk factors for septic arthritis include inflammatory arthritis (especially rheumatoid arthritis), diabetes mellitus and systemic factors, such as age greater than 80 years, as well as local factors, such as recent joint surgery, joint prosthesis and overlying skin infection. These individual risk factors increase the risk of septic arthritis by two- to threefold [3]. Skin infection overlying a prosthetic joint increases the risk of infection by 15-fold [3].
Clinical features
Septic arthritis presents with joint pain and swelling in over 80% of cases, which may or may not be associated with systemic symptoms, such as sweats and rigors [3]. The hip and knee joints are the most commonly involved joints.
The patient may be febrile and the affected joint is usually swollen, warm, erythematous and tender. Classically, there is reduced ability to actively move the joint and marked pain on passive movement. Unfortunately, the symptoms and signs are not sensitive and a patient with septic arthritis may present with only certain of these features. Thus, septic arthritis cannot be excluded with confidence on the history and examination alone.
Differential diagnosis
The differential diagnosis of acute monoarthritis is shown in Table 14.2.1. Ask the patient about a history of previous rheumatological disease, such as rheumatoid arthritis, gout or other inflammatory arthritis, as well as risk factors for infection, such as immunosuppression, including diabetes and steroids. Recent trauma or history of a bleeding diathesis or anticoagulation are also relevant. Finally, ask the patient about any recent sexually transmitted infection, including gonococcal infection or non-specific urethritis, or any systemic features including uveitis and/or gastrointestinal infection, which may point towards a reactive arthritis.
Table 14.2.1
Common presentations with acute monoarthritis to an emergency department
Gout
Reactive arthritis such as post-viral, Reiter’s syndrome
Acute exacerbation of pre-existing inflammatory arthritis
Rheumatoid arthritis
Septic arthritis
Note: Orthopaedic-related joint problems, such as trauma and/or haemarthrosis, plus osteoarthritis (OA) were not included in this series [4].
Clinical investigations
Blood tests
Send blood for a full blood count, which may reveal an elevated peripheral white blood cell count, as well as C-reactive protein (CRP) and erythrocyte sedimentation rate (ESR). ESR and CRP are non-specific and not sensitive for septic arthritis, but may help in the differential diagnosis. Blood cultures are taken in the presence of fever. Finally, a serum urate may be elevated, but can be normal in acute gout.
Imaging
X-ray may be normal in septic arthritis, as it takes at least one week for destructive changes to appear on plain X-ray. Magnetic resonance imaging when available, while non-specific, is frequently helpful to determine if the pathology is in the joint or juxta-articular bone.
Joint aspiration
The single most important investigation is synovial fluid aspiration and analysis. Send the aspirate in a sterile container for Gram stain and culture, as well as for polarizing light microscopy to look for the presence of urate (strongly negative birefringent) crystals or calcium pyrophosphate crystals (weakly positive birefringent crystals). Using blood culture bottles does not appear to increase the yield of a positive culture.
Place some of the aspirate in an EDTA tube for a white cell count to be performed. The likelihood of septic arthritis increases from 2.9% with a synovial white cell count above 25,000/μL up to 28% with a synovial white cell count of greater than 100,000/μL. Synovial glucose and protein levels are unhelpful.
Criteria for diagnosis of septic arthritis
There is no ‘gold standard’ test for the diagnosis of septic arthritis. Synovial fluid Gram stain has a sensitivity of up to 50% only, while culture has a sensitivity up to 85% [3]. However, combined with an appropriate clinical presentation, the presence of microorganisms in synovial fluid on Gram stain and/or a positive synovial fluid culture with high synovial white cell count are diagnostic.
Treatment
Treatment of septic arthritis requires parenteral antibiotics and urgent referral to orthopaedics for surgical drainage with admission to hospital. Commence empirical antibiotic therapy with dicloxacillin or flucloxacillin 2 g IV 6-hourly or cephalothin 2 g IV 6-hourly (or cephazolin) if patient is allergic to penicillin to cover against Staphylococcus, until guided by microbiology results.
The patient with suspected hip sepsis or sepsis affecting a prosthetic joint must be referred to orthopaedics urgently without attempting joint aspiration.
Gout
Gout is an intra-articular inflammatory response to monosodium urate crystal deposition usually related to hyperuricaemia. It is more common in males than females, but is extremely rare in the premenopausal female.
Aetiology and pathogenesis
Uric acid is derived from purine metabolism. Hyperuricaemia is the strongest predictor for gout and relates to either overproduction or underexcretion of uric acid. Hyperuricaemia may also cause radiolucent renal calculi.
Overproduction of uric acid is due to dietary factors, such as beer, fructose-containing soft drinks, shellfish and other purine-rich foods, or endogenous factors associated with high cell turnover, such as a haematological malignancy. Reduced excretion is related to chronic kidney disease, hypovolaemia, acidosis and medications, such as diuretics, ciclosporin, pyrazinamide, ethambutol and low-dose aspirin. There is frequently a family history of gout.
Epidemiology
The peak incidence of acute gout occurs in men between the ages of 30 and 60 years and in women between 55 and 70 years. The presentation of gout in younger patients should prompt a search for a secondary cause (including lifestyle factors). Gout is more common in Maori and Polynesian populations.
Clinical features
The classic presentation is of acute onset of a hot, swollen and painful first metatarsophalangeal joint (75% of cases) known as podagra. Other commonly affected joints include joints in the foot, the ankle, knee and small joints of the hand.
Common triggers of an acute attack are binges of alcohol or purine-rich foods, dehydration, severe illness such as sepsis, trauma and surgery. Sudden cessation or the introduction (especially in an acute attack) of hypouricaemic agents, such as allopurinol or probenecid, may also precipitate gouty arthritis, as can the introduction or a dose change of a diuretic.
Untreated, the symptoms will abate over the course of several days to 2 weeks. Occasionally, the patient may appear systemically unwell during an acute attack with malaise and systemic inflammatory response features. Examination reveals a tender, warm and erythematous joint with severely restricted range of movement. The patient may also be febrile. Presentations of acute gout may also be polyarticular (see Chapter 14.3).
Recurrent untreated acute gout and hyperuricaemia results in chronic tophaceous gout, where the patient is no longer pain-free between attacks. Examination reveals tophus formation on the ears, around the elbows and in the fingers with marked joint deformity.
Investigations and diagnosis
Synovial fluid aspiration
Synovial fluid aspirate to identify monosodium urate crystals is diagnostic of acute gout. The crystals may be phagocytosed (intracellular) and the synovial fluid will have a high white cell count. Send fluid for Gram stain and culture to rule out septic arthritis, which may rarely coexist with gout. Podagra with a typical clinical scenario has a sensitivity of 96% and specificity of 95% for acute gout, so aspiration is not indicated [5].
Blood tests
Hyperuricaemia on blood testing is not diagnostic of gout as, although up to 5% of adults may have a raised serum uric acid at some point, only one-fifth (1% overall) will ever have an attack of gout. Conversely, in about one-third of patients with gout, the serum uric acid level is normal during an acute attack. Other blood tests, such as FBE, ESR and CRP are sent and may be abnormally elevated. Check the renal function with serum urea and creatinine both to identify a potential aetiology and help guide treatment, such as avoidance or reduced doses of non-steroidal anti-inflammatory drugs (NSAIDs) or colchicine.
Imaging
Plain X-ray is performed to exclude injury, but should be normal in the acute attack other than soft-tissue swelling. Punched-out periarticular erosions are seen in chronic gouty arthritis which, when associated with calcium deposition, deforming arthritis and soft-tissue swelling, are characteristic of chronic tophaceous gout.
Management
The aim is to treat acute pain and then prevent chronic relapse with hypouricaemic drugs. Colchicine is losing favour due to the frequency of its side effects and potential for serious toxicity. Educate all patients to correct lifestyle factors where appropriate.
Acute attack
Non-steroidal anti-inflammatory drugs
After excluding infection, give either an NSAID, corticosteroid and/or colchicine in the absence of contraindications. Give diclofenac 50 mg tds orally followed by 25 mg tds orally or naproxen 500 mg followed by 250 mg tds orally until symptoms subside. A selective COX-2 inhibitor, such as celecoxib 100 mg bd orally, is preferred in patients with a history of peptic ulcer disease, although there is a similar risk of renal dysfunction in the elderly or with pre-existing renal disease.
Corticosteroids
Patients with gout refractory to the above treatment or in whom other treatment is contraindicated may be given corticosteroids, such as prednisolone 25–50 mg daily for 3 days, then weaned over 1–2 weeks [6,7]. An alternative approach is intra-articular corticosteroid for monoarticular gout provided sepsis has been excluded.
Colchicine
When NSAIDs and prednisone are contraindicated, colchicine may be used. Doses of 0.5 mg 6- or 8-hourly orally have equivalent efficacy and a lower rate of gastrointestinal toxicity compared to higher doses [8]. Higher doses, such as colchicine 1.0 mg followed by 0.5 mg up to four times daily, with a maximum cumulative dose of 6–8 mg per acute attack are no longer recommended, due to increased toxicity with nausea, vomiting, diarrhoea and the risk of renal impairment. All colchicine doses should thus be less with renal impairment, as these patients are at risk of severe neuromyopathy and/or who are on statins, as this combination may increase the risk of myopathy and rhabdomyolysis.
Recurrent attacks
Urate lowering therapy
A second attack of gout usually requires urate lowering therapy, although this is not usually commenced in the emergency setting, as treatment should be delayed until the acute flare up has settled. Allopurinol, a xanthine oxidase inhibitor, prevents the production of uric acid from xanthine. It is introduced at a low dose once the acute attack has settled and gradually titrated up to a maximum of 300 mg daily [9]. Typically, the patient will remain on a low-dose NSAID (or prednisolone/low-dose colchicine) as prophylaxis against precipitating further acute attacks.
Febuxostat is a new orally administered selective xanthine oxidase inhibitor for gout that may be used to reduce urate levels, particularly in patients with poor kidney function or intolerant of allopurinol, such as due to a hypersensitivity reaction.
An alternative uricosuric agent to allopurinol is probenecid, which should be avoided in renal impairment.
Acute pseudogout
Acute pseudogout causes an acute monoarthritis and is one of the several potential presentations of calcium pyrophosphate dihydrate (CPPD) deposition disease. It is more common in females and patients over 65 years old.
Aetiology and pathogenesis
Calcium pyrophosphate disease is characterized by deposition of CPPD crystals in cartilage causing chondrocalcinosis. When released, there may be uptake in other synovial structures and an inflammatory response producing acute synovitis, tenosynovitis or bursitis.
Advanced age is the strongest risk factor. Other associations are a family history, metabolic diseases such as haemochromatosis, Wilson’s disease, hyperparathyroidism, hypophosphataemia or hypomagnesaemia and mechanical factors, such as previous injury or osteoarthritis (OA).
Clinical features
CPPD deposition disease presents in a variety of ways. The two most common are acute pseudogout and chronic pyrophosphate arthropathy, which may mimic OA. Other presentations include tenosynovitis, bursitis or as an incidental radiographic finding of chondrocalcinosis. CPPD deposition disease may also mimic rheumatoid arthritis or ankylosing spondylitis, as well as the neuropathic joint.
Acute pseudogout typically presents in older patients and the knee is the most commonly affected joint. Other common sites include the wrist, shoulder, elbow and ankle. Occasionally, there may be an oligoarticular presentation. Presentation is with a hot, red and swollen joint. There may be systemic inflammatory response features and the patient may be febrile. Triggers include trauma, surgery or illness, but most cases are spontaneous.
Investigations and clinical diagnosis
Joint aspiration
Diagnosis of pseudogout depends on the demonstration of CPPD crystals in synovial fluid, which is frequently blood stained. Polarizing light microscopy demonstrates weakly positive birefringent rhomboid-shaped crystals.
Laboratory studies and imaging
Younger patients presenting with polyarticular chondrocalcinosis should be screened for an underlying metabolic cause, checking serum calcium, magnesium, phosphate, alkaline phosphatase, parathyroid hormone, thyroid function and iron studies.
Plain X-rays of the joint may reveal chondrocalcinosis seen in fibrocartilage, such as the knee menisci, triangular cartilage of the wrist and pubic symphysis. Other characteristic findings are of marked degenerative change in joints that are not usually affected by OA.
Management
Symptoms of acute pseudogout frequently improve once the joint has been aspirated. Intra-articular injection of corticosteroid is also appropriate for acute monoarthritis, once infection has been excluded. In addition, rest and splintage for 48–72 h is beneficial.
Give oral analgesics and NSAIDs similar to acute gout, particularly for polyarticular pseudogout as performing multiple joint injections is impractical and painful.
Take care using NSAIDs in the elderly and use the smallest doses to avoid renal impairment and precipitating heart failure.
Haemarthrosis
Haemarthrosis is bleeding into a joint which may be traumatic and related to intra-articular injury or non-traumatic related to an underlying bleeding diathesis.
Aetiology
The causes of haemarthrosis are listed in Table 14.2.2.

Clinical features
A haemarthrosis causes a painful swollen joint with a reduced range of movement. The joint is often warm. Ask about a history of trauma and, if minimal or absent, consider a bleeding disorder, such as haemophilia or anticoagulant use. Also ask about troublesome bleeding during a previous operation or following dental instrumentation and about a family history.
Investigations
Perform plain radiography to exclude a fracture. Consider a computed tomography scan if there is a high index of clinical suspicion but normal plain imaging. Send a full blood count and a coagulation screen if there is no history of significant trauma.
Haemarthrosis is diagnosed on aspiration of synovial fluid. An intra-articular fracture is indicated by observing fat globules floating on the surface of the blood.
Management
Management includes rest, immobilization, ice and compression as well as analgesia. Aspiration frequently provides pain relief if performed within 24 h of onset. NSAIDs should be avoided in patients with a bleeding diathesis.
Haemophilia or other bleeding diathesis
Haemarthrosis due to haemophilia or other disorders of clotting factor deficiency requires immediate factor replacement therapy to a level of 40–50% of normal. This should be performed as soon as possible after the presentation, in consultation with a haematology specialist.
Often the patient will be able to advise on their normal treatment (and usually knows what factor they are deficient in, their usual basal level and how much replacement is necessary in an acute bleed).
Vitamin K and administration of fresh frozen plasma may be required in patients with elevated INR related to warfarin toxicity.
Spondyloarthritis
Monoarthritis is occasionally a presentation of a spondyloarthritis, such as reactive arthritis, psoriatic arthritis or inflammatory bowel disease-associated arthritis.
Clinical features suggesting a reactive arthritis include a recent history of infective diarrhoea, uveitis or sexually transmitted infection, such as urethritis. The patient may appear ill and be febrile with a tachycardia. The patient should be asked about a history of psoriasis or inflammatory bowel disease in the past.
Check for sites of enthesitis with inflammation at a tendon insertion points, such as the Achilles tendon or plantar fascia around the heel, or dactylitis causing ‘sausage-shaped’ digits.
Guideline approach to the management of acute monoarthritis
The British Society for Rheumatology, in conjunction with other medical associations, published guidelines in 2006 regarding an approach to the hot swollen joint. No changes were recommended on review in 2012 [10]. These guidelines are summarized below:

 Other investigations should include blood cultures, CRP, ESR and full blood count.
Other investigations should include blood cultures, CRP, ESR and full blood count.
 X-ray of the affected joint should be performed as a baseline.
X-ray of the affected joint should be performed as a baseline.
 Septic joints require aspiration to dryness in addition to parenteral antibiotics.
Septic joints require aspiration to dryness in addition to parenteral antibiotics.
 Prosthetic joints and suspected hip sepsis require an urgent orthopaedic opinion.
Prosthetic joints and suspected hip sepsis require an urgent orthopaedic opinion.
14.3 Polyarthritis
Shom Bhattacharjee, Adam B Bystrzycki and Flavia M Cicuttini
Introduction
Polyarthritis is a frequent rheumatological presentation to the emergency department in adults. This chapter focuses on the initial assessment, management and most appropriate follow up of the more common conditions encountered. These include rheumatoid arthritis (RA), seronegative spondyloarthritis including psoriatic arthritis, reactive arthritis with reference to arthritides occurring in association with enteric and urogenital infections, and infectious arthritis including viral arthritis and rheumatic fever. Management principles include establishing the diagnosis, treating the acute problem and arranging appropriate follow up.
Acute polyarthritis
Polyarthritis syndromes may be difficult to diagnose accurately due to the wide range of differential diagnoses, as seen in Table 14.3.1[1]. Important principles are to rule out infection, quantify underlying inflammation and document extra-articular involvement.
Table 14.3.1
Differential diagnosis of polyarthritis syndromes
Inflammatory
Rheumatoid arthritis
Inflammatory osteoarthritis
Systemic connective tissue disease, including SLE, vasculitis, Behçet’s disease, relapsing polychondritis
Seronegative spondyloarthropathies, commonly psoriatic arthropathy
Gout
Pseudogout (calcium pyrophosphate arthropathy)
Drug induced, including lupus syndromes
Infectious arthritis – bacterial including mycobacteria, endocarditis, protozoal, viral
Reactive or post-infectious arthritis including rheumatic fever
Non-inflammatory
Neoplastic/paraneoplastic disease, including hypertrophic pulmonary osteoarthropathy
Sarcoidosis
Endocrine disease, such as haemochromatosis, acromegaly
Haematological disease, such as haemophilia, leukaemia
Clinical features and diagnosis
History
Mode of onset
Distribution
Course
Constitutional symptoms
Rheumatological systems review
Extra-articular organ involvement
Other history
History of recent sore throat, febrile illness, new sexual contact, features of a sexually transmitted disease, diarrhoea, rash or uveitis.
Past medical history of gout, rheumatic fever, inflammatory bowel disease (IBD), malignancy and juvenile polyarthritis.
Family history of gout, psoriasis, IBD, uveitis or chronic back pain suggesting ankylosing spondylitis (AS) and other seronegative arthritis.
Examination
Perform a detailed physical examination [4] and document:
Investigations
Laboratory studies
Send blood for full blood count (FBC), urea, electrolytes and liver function tests (ELFTs) and inflammatory markers including erythrocyte sedimentation rate (ESR) and C-reactive protein (CRP) [1,2].
Send serum antibody or antigen tests as indicated by the history for infectious exposure, such as hepatitis B serology, streptococcal antigen test (ASO titre) and an autoantibody panel including antinuclear antibody (ANA), rheumatoid factor (RF) and antibodies against citrullinated peptides (ACPA) usually ordered as anticyclic citrullinated peptide (anti-CCP). Antibody tests in particular should be interpreted with caution and in the context of each individual patient, due to their varying sensitivity and specificity [5].
Joint aspiration
Joint aspiration and analysis of synovial fluid are essential to diagnose septic arthritis and crystal arthropathy (see Chapter 14.2).
Imaging studies
Imaging studies, such as plain X-rays, may demonstrate diagnostic features in erosive arthropathy, but these do not occur for some time after the acute onset.
Rheumatoid arthritis
Rheumatoid arthritis (RA) is a chronic systemic inflammatory disorder of unknown aetiology characterized by symmetric synovitis, erosive polyarthritis and numerous extra-articular manifestations. It occurs in up to 2% of the general population and is two to three times more common in women [6]. The onset is often indolent and may lack the characteristic symmetrical joint involvement. Uncommonly, it presents as an acute monoarthritis.
Diagnosis
The diagnosis in adults is guided by the American College of Rheumatology/European League Against Rheumatism critera. These criteria were revised in 2010 [7] to identify features predictive of erosive disease earlier in the illness, compared with the 1987 criteria which defined the disease by its later stage features. Constitutional features, such as malaise and fatigue, are common.
‘Definite RA’ is based on the confirmed presence of:
 synovitis in at least one joint
synovitis in at least one joint
 absence of an alternative diagnosis that better explains the synovitis
absence of an alternative diagnosis that better explains the synovitis
 a total score of 6 or greater (of a possible 10) from individual scores in four domains: number and site of involved joints (score range 0–5); serologic abnormality (score range 0–3); elevated acute-phase response (score range 0–1) and symptom duration (2 levels; range 0–1) (Table 14.3.2).
a total score of 6 or greater (of a possible 10) from individual scores in four domains: number and site of involved joints (score range 0–5); serologic abnormality (score range 0–3); elevated acute-phase response (score range 0–1) and symptom duration (2 levels; range 0–1) (Table 14.3.2).
Table 14.3.2
Classification criteria for RA
A. Joint involvement
1 large joint – 0 pts
2–10 large joints – 1 pt
1–3 small joints (with or without involvement of large joints) – 2 pts
4–10 small joints (with or without involvement of large joints) – 3 pts
>10 joints (at least 1 small joint) – 5 pts
B. Serology (at least 1 test result is needed for classification)
Negative RF and negative ACPA – 0 pts
Low-positive (<3×ULN) RF or low-positive ACPA – 2 pts
High-positive (≥3×ULN) RF or high-positive ACPA – 3 pts
C. Acute-phase reactants (at least 1 test result is needed for classification)
Normal CRP and normal ESR – 0 pts
Abnormal CRP or abnormal ESR – 1 pt
D. Duration of symptoms
<6 weeks – 0 pts
≥6 weeks – 1 pt
Add score of categories A–D: a score of>6/10 classifies a patient as having definite RA
Adapted from [7]
Morning stiffness, symmetric involvement and radiographic erosions are no longer included.
Clinical features
Characteristic presentations in RA include the following [8–10].
Cervical spine
Cervical arthritis is common and may result in critical spinal problems from degeneration of the transverse ligament of the C1 vertebra that produces C1–2 instability in up to 5% of patients and can result in cervical cord compression or vertebral artery insufficiency. In addition, decreased motion and myelopathy may result from longstanding joint involvement.
Upper limb
The wrist, metacarpophalangeal and proximal interphalangeal joints are typically affected, with sparing of the distal interphalangeal joints. Swan-necking and boutonnière deformities are common, together with ulnar deviation at the metacarpophalangeal joints. Fixed flexion deformities may result in entrapment neuropathies, in particular, carpal tunnel syndrome with median nerve involvement. Tenosynovitis may lead to tendon rupture, particularly of the extensor pollicis longus, or degenerative changes in the long extensors of the middle, ring and the little fingers with rupture of these tendons.
Lower limb
The hip and knee are frequently involved. Metatarsophalangeal joint subluxation may occur. Talonavicular joint inflammation causes pronation and eversion deformity, with overlying muscle spasm. A Baker’s cyst due to posterior herniation of the joint capsule of the knee joint may occur and require differentiation from a deep vein thrombosis by Doppler ultrasound. Entrapment of the posterior tibial nerve causes burning paraesthesiae on the sole of the foot.
Extra-articular manifestations
The extra-articular manifestations of RA are protean and may involve any organ system due to local inflammation causing functional or neurological deficits, rheumatoid vasculitis or distant inflammation (see Chapter 14.1). Patients may also present with the side effects of the treatment, including sepsis related to immunosuppression. Sepsis with encapsulated organisms is of particular concern in patients with Felty’s syndrome of RA with splenomegaly and neutropaenia [11].
Investigations
Laboratory studies
Send blood for FBC and ELFTs and non-specific markers of inflammation, such as ESR, and CRP, with assays for serum rheumatoid factor and anti-CCP [12]. Anti-CCP is as sensitive but more specific than rheumatoid factor for RA and is more frequently positive early in the disease process. It is also thought to identify individuals at higher risk of erosive disease [13]. Send blood cultures as well as midstream urine for suspected sepsis.
Joint aspiration
Joint aspiration is essential to exclude coexistent or primary sepsis in any sudden hot, swollen joint.
Imaging
Initial plain imaging of affected joints at first presentation does not usually demonstrate erosive changes, but is useful in patients with longstanding disease. However, always request X-rays of the cervical spine in any patient with cervical or neurological features to look for an atlanto-dens interval of greater than 2.5 mm, which is diagnostic of instability [14]. Include a chest X-ray if there is a fever and/or any respiratory features. Request an ultrasound examination to differentiate deep vein thrombosis from a Baker’s cyst.
Emergency management
Emergency therapy aims to relieve acute pain and reduce joint inflammation. Longer-term goals include restoration and maintenance of joint function and the prevention of periarticular bony and cartilage destruction. Important principles include medication, education, rest and exercise, with the input of a multidisciplinary allied health team incorporating occupational therapy and physiotherapy.
Medication falls broadly under the categories of non-steroidal anti-inflammatory drugs (NSAIDs) and disease-modifying antirheumatic drug (DMARD) therapy. Readers are referred to Chapter 14.1 for a brief overview of these medications and common adverse effects, as well as the references at the end of this chapter [15–18].
Other long-term measures include orthopaedic and orthotic intervention. Surgery involving joint fusion, synovectomy, total joint arthroplasty and reconstruction may be required.
Early consultation with a rheumatologist is essential, particularly for patients with acute or first presentations. Exclusion of infection even for mild or moderate features is imperative and to control symptoms with simple analgesics with discharge for specialist follow up. Admit patients if there is evidence of multisystem involvement, severe symptoms requiring nursing or allied health management or if they are unable to tolerate oral therapy.
Prognosis
The spontaneous remission rate in RA is less than 10% [19,20]. High titres of anti-CCP or rheumatoid factor, which are present in up to 75% of patients with rheumatoid arthritis, the presence of nodules and human leucocyte antigen (HLA)-DR4 haplotype are markers of severity. A patient’s life expectancy is shortened by 10–15 years by accelerated cardiovascular disease, infection, pulmonary and renal disease and gastrointestinal bleeding.
Seronegative arthritis
The seronegative spondyloarthritis disorders are characterized by inflammation of the axial spine with sacroiliitis and spondylitis in particular, enthesitis, which is inflammation at the attachments of tendons and ligaments to bones, dactylitis, asymmetric polyarthritis often of the lower limb, eye inflammation and varied mucocutaneous features [21]. They are labelled ‘seronegative’ as the serum rheumatoid factor is negative.
Epidemiology
The term ‘seronegative spondyloarthritis’ covers conditions such as ankylosing spondylitis (AS), reactive arthritis occurring in the setting of viral or bacterial infection, psoriatic arthritis and arthritis associated with inflammatory bowel disease (IBD). It is further differentiated into axial and peripheral spondyloarthritis.
The prevalence of the seronegative spondyloarthritis disorders varies widely and may parallel the prevalence of the HLA-B27 gene.
However, the exact role of HLA-B27 in the pathogenesis of these disorders has not been clearly defined. The proportion of HLA-B27 positive individuals who develop symptomatic arthropathy varies widely from 16% in patients with AS to 70% of patients with spondylitis in the setting of IBD [22]. HLA-B27 positive individuals may be less efficient at the intracellular removal of certain inciting bacteria, although this is controversial [23].
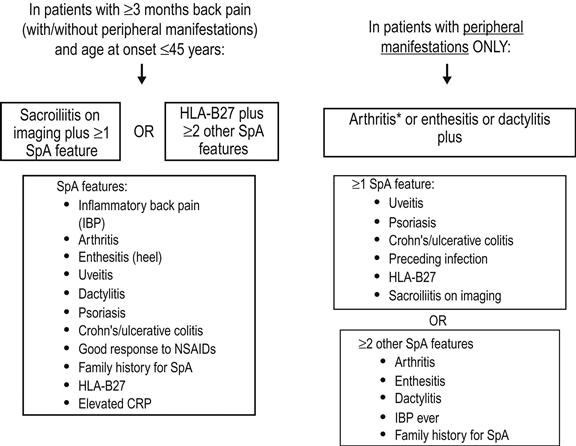
The Assessment of SpondyloArthritis international Society (ASAS) recently advanced classification criteria for axial and peripheral spondyloarthritis (Fig. 14.3.1). These criteria have better sensitivity and comparable specificity to previous criteria and are well validated [24].
Psoriatic arthritis
Psoriatic arthritis is a heterogeneous disease distinct from other inflammatory arthritides. It occurs in 10% of patients with psoriasis, but may affect up to 40% of hospitalized psoriasis patients with widespread skin involvement [25]. It occurs between the ages of 30 and 60 years, with an equal prevalence in males and females. It is thought to be inherited in a polygenic pattern significantly influenced by environmental factors, including trauma and infectious agents. Multiple studies have confirmed the important role of class I HLA, particularly B13, B16 and B27 and certain C-subclasses [26,27]. The arthropathy pattern may be pauci-articular, but more than five peripheral joints are usually involved.
Clinical features and diagnosis
The diagnosis of psoriatic arthritis is essentially clinical, requiring the demonstration of coexisting synovitis and psoriasis. A set of simple clinical diagnostic criteria, the ClASsification criteria for Psoriatic ARthritis (abbreviated to the CASPAR criteria) were proposed by a large international study group in 2006 [28].
CASPAR diagnostic criteria for psoriatic arthritis
Established inflammatory joint disease and at least three points from the following features:
 history of psoriasis (in the absence of current psoriasis) (1 point)
history of psoriasis (in the absence of current psoriasis) (1 point)
 family history of psoriasis (in the absence of current or past history) (1 point)
family history of psoriasis (in the absence of current or past history) (1 point)
 juxta-articular new bone formation (1 point)
juxta-articular new bone formation (1 point)
Five clinical subtypes are recognized, including asymmetric oligoarthritis, symmetric small joint polyarthritis, predominant distal interphalangeal joint involvement, psoriatic spondyloarthropathy and arthritis mutilans [29]. Major extra-articular organ manifestations, such as aortic insufficiency and pulmonary fibrosis, occur rarely. However, up to 30% of patients have mild inflammation at the eye, most commonly conjunctivitis.
Asymmetric oligoarthritis
This occurs in 30–50% of patients [30]. It presents as an oligoarthritis involving a single large joint, in association with a ‘sausage-shaped’ or dactylitic digit or toe. Dactylitis occurs due to a combination of arthritis and tenosynovitis. Distal interphalangeal joint involvement is typical, almost invariably associated with psoriatic nail changes of pitting, ridging and onycholysis. Enthesitis occurs most frequently with this form of the disease and commonly manifests as plantar fasciitis or epicondylitis at the elbow.
Symmetric small joint polyarthritis
This occurs in 30% of patients, in a pattern strongly resembling RA, but with more frequent distal interphalangeal joint involvement [30].
Psoriatic spondyloarthritis
This occurs in 5% of patients [30]. It is often asymptomatic, but may present with inflammatory low back pain due to sacroiliitis in up to 30% of cases.
Arthritis mutilans
‘Arthritis mutilans’ is a rare (<5% of patients), but well-characterized feature of psoriatic arthritis with severely deforming arthritis including telescoping of the fingers or toes from osteolysis of the metacarpal or metatarsal bones and phalanges [30].
Dermatological features
Dermatological features include typical erythematous, scaling plaques on the extensor surfaces of the elbows and knees, scalp and ears and nail changes. The nail changes include pitting with usually greater than 20 pits, ridging with transverse depressions and onycholysis with separation of the nail from the underlying nail bed [25]. Nodules and vasculitic features such as digital ulcers are not seen.
Psoriatic arthritis can be difficult to distinguish from the other seronegative spondyloarthritides in the absence of dermatological features or a positive family history.
Investigations
ESR and CRP are raised, but the rheumatoid factor and autoantibody screen are negative. Plain X-rays of affected joints may reveal typical radiographic features including soft-tissue swelling, bone proliferation at the base of digital phalanges coupled with resorption of the distal tufts (the ‘pencil-in-cup’ deformity) and fluffy periostitis [31]. Chest radiographs are useful as a baseline when clinical examination suggests cardiac or pulmonary involvement.
Emergency management
Emergency treatment involves the relief of pain and reduction of joint inflammation, with appropriate specialist follow up. Education, rest and exercise and referral to a multidisciplinary allied health team are the mainstay of ongoing management. Admit patients if their symptoms are severe enough to preclude oral therapy or safe discharge pending outpatient specialist follow up.
NSAIDs are useful for acute symptomatic relief and intra- or peri-articular corticosteroids may be used for short-term relief of painful arthritis or enthesitis. Long-term therapy with disease modifying agents, such as sulphasalazine or methotrexate, is instituted at specialist review [32]. Oral corticosteroids are usually avoided as their cessation often exacerbates the psoriasis. Therapy with tumour necrosis factor-α antagonists, such as infliximab, etanercept and adalimumab, has been approved for rheumatologists under strict access criteria for severe disease resistant to other DMARD therapy.
Emergency management of skin disease includes topical treatments, such as emollients and keratolytic agents [33]. Phototherapy and photo-chemotherapy may be instituted on early dermatological consultation.
Prognosis
Psoriatic arthritis generally runs a more benign course than RA but, nonetheless, patients suffer from considerable morbidity. Adverse prognostic factors include onset before 20 years of age, erosive disease and extensive skin involvement [30].
Reactive arthritis
Reactive arthritis is an aseptic peripheral arthritis following certain infections, which include bacterial infections of the urogenital tract usually by Chlamydia trachomatis, or of the gastrointestinal tract with organisms, such as Shigella, Salmonella and Campylobacter. It may also follow viral infections, such as HIV, although in the case of HIV, co-infection with sexually transmitted organisms rather than the virus itself is thought to be the cause [34]. The seroconversion illness of HIV has its own constellation of articular symptoms and is considered to be a separate entity.
Epidemiology
The prevalence of reactive arthritis is difficult to define owing to diagnostic uncertainty particularly in the setting of asymptomatic sexually transmitted infection. The male preponderance is up to 9:1 following sexually transmitted infection, but males and females are equally affected following gastrointestinal tract infection [35]. The peak incidence is around age 35 years and up to 75% of patients are HLA-B27 positive [35]. An important exception is with the reactive peripheral arthritis that occurs in 20% of patients with idiopathic IBD, a condition that may mimic gastrointestinal tract infection, but where patients are usually HLA-B27 negative.
Clinical features and diagnosis
The diagnosis of reactive arthritis is clinical. It typically manifests within a month of gastrointestinal or genitourinary infection, although the latter is frequently asymptomatic [36]. Musculoskeletal manifestations include myalgias and asymmetric polyarthritis affecting the knees, ankles and small joints of the feet in particular, although peripheral upper limb involvement is seen. Affected joints demonstrate marked inflammatory features with erythema, swelling, warmth and exquisite pain on active or passive movement. Fever and malaise are common.
Arthritis and extra-articular manifestations
Symptomatic spondylitis and sacroiliitis cause low back and buttock pain and occur frequently. Dactylitis and enthesitis are characteristic features of this disease with heel pain from plantar fasciitis or Achilles tendonitis [36].
Extra-articular features associated with reactive arthritis include keratoderma blennorrhagica, the scattered, thickened, hyperkeratotic skin lesions with pustules and crusts seen in Reiter’s syndrome and circinate balanitis. Keratoderma blennorhagica on the soles or palms may coalesce to form plaques virtually indistinguishable from those of psoriasis [37]. Circinate balanitis causes shallow meatal ulcers that are moist in uncircumcised men or hyperkeratotic and plaque-like in circumcised men [37]. An inflammatory aortitis occurs in 1% of patients and may result in aortic valvular incompetence and/or heart block
The peripheral arthritis associated with IBD is migratory and occurs in a similar distribution. Common features include large joint effusions, particularly involving the knee, and sacroiliitis or spondylitis [38]. Unlike peripheral arthritis following genitourinary infection, the spondylitis of IBD-associated arthritis does not tend to settle with treatment of the bowel inflammation. Cutaneous features associated with this form of arthritis occur mainly on the lower limbs and include erythema nodosum and pyoderma gangrenosum [38].
Investigations
Laboratory
An active inflammatory response is seen in the acute phase with a neutrophil leucocytosis and thrombocytosis and raised ESR and CRP. The presence of a mild normochromic, normocytic anaemia suggests chronic disease. Send blood for HLA-B27.
Document the preceding gastrointestinal or genitourinary organism by stool culture or cervical/urethral swabs [39]. Rheumatoid factor and ANA are negative.
Joint aspiration
Joint aspiration may be necessary to exclude intra-articular sepsis (see Chapter 14.2). The synovial fluid may be turbid, viscous and with a neutrophil leucocytosis up to 50 000/mm3, but Gram stain and bacterial culture are negative and, unlike true septic arthritis, the synovial glucose level is not significantly reduced compared to serum levels [39]. Macrophages with intracytoplasmic vacuoles containing ingested neutrophils are occasionally seen.
Imaging
X-ray changes are unusual with acute arthritis, but are seen after several months. As with psoriatic arthritis, a common finding is a ‘fluffy’ periosteal reaction, particularly at the calcaneus, and evidence of sacroiliitis or spondylitis with bridging syndesmophytes in longstanding disease.
Emergency management
Exclude infection by synovial aspiration and culture with a markedly inflamed joint and consult early with a rheumatologist, particularly in a patient with a first presentation. Admit patients with suspected septic arthritis until it is excluded or if they are unable to tolerate simple oral therapies. Request a cardiology opinion for major cardiac involvement with valvular disease or a conduction abnormality and a gastroenterology opinion when IBD is suspected, although the role of treatment and the effect on the arthropathy is unclear.
Otherwise provide symptom relief with NSAIDs as the mainstay of treatment. Corticosteroids may be given after rheumatological consultation, either systemic or topically for the skin manifestations. Disease modifying therapy with sulphasalazine is initiated at specialist follow up if NSAID therapy fails to control symptoms. Multidisciplinary physical therapy is essential on an outpatient basis.
Give antibiotics, such as doxycycline 100 mg orally bd for 7 days or azithromycin 1 g orally once for documented urethritis or cervicitis, and remember partner contact tracing and treatment. An infectious diseases opinion is useful in these cases [39].
Prognosis
Signs and symptoms usually remit within 6 months. However, up to 50% of patients suffer from recurrent arthritis and up to 30% develop chronic arthritis [40]. Post-dysenteric cases have a better prognosis than post-chlamydial cases. Poor prognostic signs include early onset under the age of 16 years, hip involvement and the presence of dactilytis.
Polyarticular crystal arthritis
Crystal-induced arthritis disorders result from the deposition of crystal in joint spaces, such as in gout or pseudogout. Both diseases cause debilitating joint inflammation resulting from the lysis of neutrophil polymorphs that have ingested monosodium urate in the case of gout, or calcium pyrophosphate crystals in pseudogout. Although usually monoarticular, polyarticular involvement can occur in up to 5% of cases. See Chapter 14.2 for a detailed discussion of these diseases.
Infectious polyarthritis
Septic bacterial arthritis is most often monoarticular, although it can present with polyarticular involvement. Infectious polyarthritis may also occur as aseptic manifestation of certain viral infections and following streptococcal infection in acute rheumatic fever (ARF).
Viral arthritis
Arthralgia affecting several joints is common in many viral infections, but few cause frank polyarthritis. In general, these are self-limiting and managed symptomatically. Viruses involved include alphaviruses, such as the Ross River virus (RRV), parvovirus B19 and hepatitis A, B and C viruses.
Alphaviruses
Alphaviruses are a mosquito-borne genus of the Togaviridae family. They are responsible for epidemics of febrile polyarthritis, including Ross River, Barmah Forest and Sindbis viruses (SINV) in Australia; West Nile virus which has recently been documented in the USA; Chikungunya virus in East Africa, South and Southeast Asia; O’nyong-nyong virus in East Africa and the Mayaro virus in South America [41].
Ross River virus
RRV is endemic to Australia, New Zealand and South Pacific islands and is the most common arboviral disease in Australia. RRV is transmitted by the Ochlerotatus (formerly Aedes) vigilex mosquito via a marsupial reservoir [42]. Epidemics of acute febrile polyarthritis are most common between January and May, but can occur after periods of heavy rains.
Clinical features and diagnosis
A detailed travel history is essential. There is usually low grade fever and other constitutional symptoms. A rash varying in distribution, character and duration occurs up to 2 weeks before, during or after the other features. Polyarticular symptoms are present in most patients with a symmetric arthritis or arthralgia primarily affecting the wrist, knee, ankle and small joints of the extremities. Cervical lymphadenopathy occurs frequently and paraesthesiae and tenderness of the palms and soles in a small percentage of cases [43].
The diagnosis is predominantly clinical, particularly in endemic areas in the event of a local outbreak, and confirmed by serology.
Investigations
Serology testing distinguishes RRV from other causes of febrile polyarthritis, such as Barmah Forest virus. A significant rise in IgM antibody titre to RRV indicates acute infection or the virus itself may be isolated from the serum of acutely unwell patients. Radiographs are unremarkable and unnecessary as the disease is largely self-limiting [44].
Emergency management
Patients with RRV require symptomatic treatment with simple analgesics or NSAIDs. Occasionally, a brief course of low-dose prednisolone may be used. RRV is a notifiable disease [42]. Refer to a rheumatologist if symptoms are severe or refractory to simple treatment measures.
Conventional personal preventative measures, such as protective clothing, effective mosquito repellent and avoidance of mosquito-prone areas should be recommended, as no vaccine currently exists.
Prognosis
RRV is usually self-limiting, but prolonged symptoms may occur and there may be relapses of decreasing intensity, separated by remissions for up to a year or more.
Parvovirus B19
Human parvovirus B19 infection is caused by a small, single-stranded DNA virus that has a predilection for erythroid precursor cells and is transmitted by respiratory secretions. It causes the self-limiting illness erythema infectiosum known as ‘slapped cheek disease’ or ‘fifth disease’ in children. In adults, however, parvovirus B19 manifests with severe ‘flu-like symptoms and as many as 75% develop joint symptoms. It may be responsible for up to 12% of adult patients presenting with acute polyarthritis, most notably in those who have frequent exposure to children [45].
Clinical features and diagnosis
The characteristic ‘slapped cheek’ rash is usually absent in adults. An acute polyarthritis improves over 2 weeks, with symmetric involvement of peripheral small joints, including the hands (proximal interphalangeal and metacarpophalangeal joints in particular), wrists, knees and ankle joints. Morning stiffness is prominent. These features are similar to those seen in patients with RA, with up to 50% of affected patients found to meet the (now superceded) 1987 ACR diagnostic criteria for RA [46].
Uncommon but important extra-articular features of parvovirus B19 infection include [47]:
Investigations
Send a FBC, particularly given the potential for an aplastic crisis and bone marrow suppression. Non-specific markers of inflammation are likely to be elevated. Specific serological diagnosis is made by a high IgM antibody titre specific to the virus and by isolation of the viral DNA by polymerase chain reaction (PCR). IgG antibodies to parvovirus B19 indicate past infection and are common in the adult population [48].
Transient, moderate elevations of rheumatoid factor, anti-DNA, antilymphocyte or anticardiolipin antibodies sometimes occur [48]. Radiographs of the affected joints are normal.
Emergency management
Rest and NSAIDs are the mainstay of emergency treatment, except in pregnant women when NSAIDs are contraindicated in the third trimester [49]. A short course of prednisolone may be required. Significant extra-articular manifestations may require admission and consultation with the appropriate specialist. Blood transfusion or intravenous immunoglobulin infusions may be necessary.
Prognosis
Joint symptoms are self-limited in the majority of adult patients, but up to 10% may have prolonged relapsing and remitting symptoms lasting up to 9 years [50].
Hepatitis A, B and C viruses
The hepatitis viruses A, B and C all cause viral polyarthritis. Hepatitis B virus (HBV) is responsible for 20–25%, and hepatitis A (HAV) up to 14% of causes in patients with viral polyarthritis [51]. The polyarthritis of HAV tends to occur during the infectious phase and is self-limiting. The polyarthritis of HBV occurs in early infection during a period of significant viraemia and is thought to be due to immune complexes.
Clinical features and diagnosis
HBV polyarthritis is acute and severe and manifests in a symmetric, migratory or additive fashion, most commonly involving the hand and knee joints [52]. Other large axial joints may be involved and significant early morning stiffness is often present. The arthritis may precede the development of jaundice and persist for several weeks after jaundice has developed.
Hepatitis C virus (HCV) polyarthritis is rapidly progressive and symmetrical, involving the hands, wrists, shoulders, knees and hips [53]. Carpal tunnel syndrome and tenosynovitis may occur. It is unusual for polyarthritis to be the first manifestation of the underlying disease in either HBV or HCV. Nonetheless, ask about exposure risk factors for these viruses, such as intravenous drug abuse, unprotected sexual intercourse, past blood transfusions, tattoos, as well as about previous jaundice.
Both hepatitis B and hepatitis C disease are associated with a number of important extra-articular, extra-hepatic manifestations which include:
 HBV: polyarteritis nodosa, systemic necrotizing vasculitis, membranous glomerulonephritis [54]
HBV: polyarteritis nodosa, systemic necrotizing vasculitis, membranous glomerulonephritis [54]
 HCV: mixed cryoglobulinaemia causing palpable purpura, arthritis and serum cryoglobulinaemia with cutaneous phenomena, such as Raynaud’s syndrome and digital ulcers, membranous glomerulonephritis and lymphoma [53,55].
HCV: mixed cryoglobulinaemia causing palpable purpura, arthritis and serum cryoglobulinaemia with cutaneous phenomena, such as Raynaud’s syndrome and digital ulcers, membranous glomerulonephritis and lymphoma [53,55].
Laboratory investigations and imaging
Send blood for ELFTs for raised transaminases with elevated bilirubin, hepatitis B surface antigen, antihepatitis B surface antigen IgM to indicate acute infection and/or viral DNA quantification by PCR. Check also for anti-HCV IgM and for viral DNA quantification by PCR.
Also check FBC and for ESR, CRP, cryogobulins and rheumatoid factor in the presence of a rash, ulcers or other vasculitic phenomena.
Radiographs are normal other than showing soft-tissue swelling.
Emergency management
Commence symptomatic treatment with NSAIDs and refer refractory HBV- or HCV-associated polyarthritis to a rheumatology specialist and/or combined hepatology clinic. Disease modifying agents, such as prednisolone and sulphasalazine, may be used cautiously with careful monitoring of the liver function tests and for increasing viraemia [56].
Prognosis
This varies depending on the underlying disease and on the presence of vasculitic phenomena. The polyarthritis of HBV is usually limited to the pre-icteric phase, but patients with chronic active hepatitis or chronic HBV viraemia may have recurrent arthritis.
Rheumatic fever
Acute rheumatic fever (ARF) refers to the constellation of non-infectious symptoms occurring after a pharyngeal infection with group A streptococci (GAS). Evidence suggests that it may also occur in high-risk populations following skin infections with GAS [57].
Epidemiology
ARF is characterized by inflammation of connective tissue including the joints, subcutaneous tissue, heart and blood vessels. Its prevalence has declined over time in developed countries, but it remains a major public health problem in indigenous populations in the more socially isolated parts of Australasia and in developing countries. In fact, the highest documented rates in the world occur in the Aboriginal Australian population and Torres Strait Islander populations of New Zealand and the Pacific Islands [58].
ARF is primarily a disease of children aged 5–14 years. The annual incidence may reach 350:100 000 in Aboriginal children [59]. However, the polyarthritis of ARF is most common in adolescents and young adults.
Diagnosis and clinical features
The diagnosis of ARF worldwide is made using the 1944 Jones, or more recent World Health Organization major and minor criteria. However, these criteria appear too restrictive for diagnosing ARF in Australian indigenous populations. Therefore, new criteria for use in high- and low-risk populations in Australia have been proposed (Table 14.3.3) [60].
Table 14.3.3
2005 Australian guideline for the diagnosis of acute rheumatic fever

ARF: acute rheumatic fever; GAS: group A streptococci; RHD: rheumatic heart disease.
The polyarthritis of ARF is usually the earliest symptom of the disease and is classically described as migratory, affecting several joints in quick succession for a short time, commencing with the large joints of the lower limb then the large joints of the upper limb [61]. Affected joints are painful but objective signs of inflammation, such as erythema and swelling, are not prominent.
Fever and constitutional symptoms are common. Other important extra-articular major criteria (with polyarthritis) of the disease include the following [61]:
Laboratory investigations and imaging
Measure antistreptolysin O and antideoxyribunuclease B (anti-DNase B) titres [61]. As these titres can take 6 weeks after infection to peak, interpretation in the acute phase should be cautious and serial tests should be performed. Note that antistreptococcal antibody titres are useful in low-risk populations, but are difficult to interpret in high-risk populations due to pre-existing high background titres [62].
Send a throat swab, although this is positive in less than 10% of high-risk populations. Other important tests include:
 ESR and CRP, which are almost invariably elevated
ESR and CRP, which are almost invariably elevated
 FBC, which may demonstrate a leucocytosis and, less commonly, a normochromic, normocytic anaemia
FBC, which may demonstrate a leucocytosis and, less commonly, a normochromic, normocytic anaemia
 ECG to document the P-R interval
ECG to document the P-R interval
 chest X-ray to look for cardiomegaly or symptomatic cardiac failure.
chest X-ray to look for cardiomegaly or symptomatic cardiac failure.
Synovial fluid aspirate is usually inflammatory with an elevated white cell count and sterile on microscopy and culture. Radiographs of affected joints generally demonstrate soft-tissue swelling only.
Emergency management
This depends on establishing the diagnosis and treating the manifestations. Patients are markedly symptomatic and often require admission for initial observation and management. Request rheumatology and infectious disease opinions and a neurology opinion if chorea is troublesome. The presence of heart block or, more importantly, frank cardiac failure or acute valvular regurgitation mandate cardiology admission.
The polyarthritis of rheumatic fever is exquisitely responsive to NSAID therapy, particularly aspirin, so much so that failure of NSAID therapy rapidly to relieve symptoms should prompt consideration of an alternative diagnosis [63]. Give high-dose aspirin at 80–100 mg/kg/day in 4–5 divided doses in adults, usually for 1–2 weeks [64].
Commence antibiotic therapy with phenoxymethylpenicillin 10 mg/kg up to 500 mg orally 12-hourly for 10 days to eradicate streptococcal pharyngitis, after obtaining appropriate diagnostic investigations as detailed above. Commence prophylaxis following resolution of the acute episode in high-risk indigenous communities.
Ongoing rheumatology and infectious diseases specialist follow up is recommended. Note that penicillin reduces the frequency and severity of post-streptococcal rheumatic fever, but has little effect on the course of the immune-complex mediated post-streptococcal glomerulonephritis (PSGN).
Prognosis
Recurrence of ARF commonly occurs within 2 years of the initial attack, despite prophylactic therapy. Most affected connective tissues do not sustain long-lasting damage, with the exception of the heart, which is prone to additive subclinical damage resulting in rheumatic heart disease [65].
14.4 Musculoskeletal and soft-tissue emergencies
Anthony Tzannes and Anthony Brown
Common causes of soft-tissue injuries
All injuries have a soft-tissue component. The simplest way of dividing the causes of soft-tissue injuries is into ‘acute’ (specific event that exceeds tissue tolerance) and ‘chronic’ (repetitive minor damage in excess of ability to heal). Both types may be further subdivided by the tissue affected (bone, tendon, muscle, etc.).
Acute soft-tissue trauma can also be subdivided by the mechanism:
Many of the types of trauma above are covered in other chapters.
General evaluation of a soft-tissue injury
Assessment
History
Examination
This should potentially be delayed until after an X-ray if a radiopaque foreign body (metal or glass) is suspected, after giving analgesia.
The extent of any nerve damage should be determined before using local anaesthetic, although tendon damage may be best elucidated after adequate analgesia is obtained, so the wound can be adequately explored.
Puncture injury
Management
Refer immediately to the appropriate surgical team all high-pressure gun injuries, such as from grease, paint or oil where the skin has been broken, even if no damage is apparent initially. They require extensive wound debridement and tissue plane cleaning, however innocuous they may seem.
Otherwise, clean the wound with antiseptic and evaluate the need for tetanus prophylaxis and antibiotics. A puncture wound to the sole of the foot will require exploration if it has occurred through the sole of footwear, or potentially has a foreign body. Otherwise, if there was direct skin penetration, this can be managed with a 30 min soak in an iodine/alcohol solution.
Prophylactic antibiotics are controversial as they do not adequately lower the risk of infection to offset the difficulty in managing likely resistant bacteria if infection then occurs. If prophylaxis is chosen, give amoxicillin/clavulanate 875/125 mg bd for 5 days. As these wounds are at high risk of infection, instruct the patient to return if increasing pain, redness or swelling occurs.
Acute mechanical overload injuries
These include fractures, ligament sprains, muscle strains or tears and tendon ruptures. Many are covered in Section 4 Orthopaedic Emergencies.
See Table 14.4.1 for a classification system for ligamentous sprains.
Table 14.4.1
Classification of ligament sprains/muscle strains
| Grade | Features |
| I | Small number of fibres injured, pain on loading, but no laxity or loss of strength |
| II | Significant number of fibres injured, with laxity and/or weakness and pain on loading |
| III | Complete tear with gross laxity and no strength |
Management
General principles
The initial management principles are the same for both ligament sprains and muscular strains. This includes rest, ice, compression and elevation (RICE) with analgesia, usually a combination of paracetamol 1 g orally qid, plus a non-steroidal anti-inflammatory drug (NSAID), such as ibuprofen 400 mg orally tds (in the absence of NSAID-sensitive asthma, peptic ulcer disease and renal impairment).
Ligament sprain
Ligament sprains that are grade I or II (see Table 14.4.1) are managed with a protective brace or strapping and reduction in, but not cessation of, physical activity. Consider immobilizing grade III sprains with a splint or plaster of Paris (POP) cast and/or operative repair if there is gross instability and warn the patient that they can take up to 3 months or longer to heal. This is of particular relevance to the manual labourer and high-level athlete. Proprioception retraining at physiotherapy is important in ankle injuries to prevent recurrence, especially with grade II or III injuries.
Muscle strain
Muscle strains require initial RICE to minimize bruising and haematoma formation followed by a graded return to activity. Physiotherapy may aid in return of function and prevent re-injury, although research is limited by high rates of programme non-compliance in completed studies.
Assess the functional limitations imposed by these injuries, particularly in patients who live alone and/or who are elderly and infirm, as loss of independence is likely. A complete muscle tear, especially in an active individual, may benefit from operative repair following referral to an orthopaedic specialist. Consider the need for community services, respite care or admission in those who are initially unable to care for themselves.
Tendon rupture
Evaluation
Acute rupture of the supraspinatus tendon, long head of biceps and Achilles tendon are the most common serious tendon injuries that present to an emergency department (ED). Injury may be secondary to an acute event or chronic overload that is often asymptomatic until a tear occurs, and the extent of the rupture may be partial or complete.
Management
Request an ultrasound to confirm the diagnosis. Magnetic resonance imaging (MRI) is equally or more sensitive and specific depending on which tendons are being imaged, although is much less readily available.
Treatment is aimed at the earliest return to normal function, with the least likelihood of recurrence. Refer a complete tear, particularly in active people, for orthopaedic surgery repair. Manage partial tears conservatively, but they too may have a better outcome if repaired surgically, depending on local hospital practice and surgical availability.
Pretibial laceration
These are most common in elderly patients, often from trivial trauma that tears a flap of skin, particularly if taking steroids. Ask about general mobility and safety issues at home.
Management
Clean the wound, remove blood clots, trim obviously necrotic tissue and unfurl the rolled edges of the wound to determine actual skin loss. Refer the patient immediately for consideration of early skin grafting if there is significant skin loss or marked skin retraction preventing alignment of the skin edges.
Otherwise, lay the flap back over the wound and hold in place with adhesive skin-closure strips (Steristrips). Then cover the wound with a non-adhesive dressing, and apply a firm crêpe bandage and instruct the patient to keep the leg elevated whenever possible. Determine the need for tetanus immunization or booster.
Arrange follow up in 5 days for review and a dressing change, or earlier if blood or serum has seeped through the wound dressing, known as ‘strike-through’, which increases the risk of secondary infection.
Expect the wound edges to heal by granulation tissue, with new epidermal tissue laid down at the rate of approximately 1 mm per week. If healing has not started to occur by the time of review, then skin grafting may be necessary and appropriate referral made. Otherwise, arrange follow up with the GP, with community nurse input as necessary.
Degloving injury
Evaluation
Degloving injuries are caused by either a shearing or traction force on the skin, causing it to be torn from its underlying capillary blood supply. When the skin actually peels off it leaves an obvious exposed open injury, or the skin may remain intact causing a closed injury.
A closed degloving injury is much harder to diagnose. It may only cause the skin to feel less tethered than prior to injury but with poor capillary return with the failure to blanch on pressure and, most importantly, altered cutaneous sensation. This is most accurately assessed by 2-point discrimination. If 2-point discrimination is normal, a significant degloving injury has not occurred. Pain may or may not be prominent and/or may relate to an underlying bony injury.
Management
Arrange specialist assessment and admission for all degloving injuries by the appropriate surgical team, usually plastic and/or orthopaedic surgery. Keep any degloved skin, as it may be used as a skin graft.
Do not be tempted simply to replace the skin into its original position and hold it there with sutures or adhesive skin-closure strips (Steristrips), as this is inadequate. Degloving injuries are also a high-risk wound for tetanus.
Chronic overuse (overload) injuries
Chronic overuse injuries develop wherever tissue microtrauma occurs at a rate that exceeds the body’s natural ability to heal. Few require emergency treatment, but general knowledge of these conditions is valuable to advise patients on cause and management.
Classification
Bony overuse injuries follow a continuum from pain on activity only, through local tenderness to pain at rest, with loss of function. Many will have led to a stress fracture by the time of presentation to an ED.
Other overuse injuries are classified by the tissue type and extent of injury and are often best diagnosed by the timing of the pain in relation to physical activity. They are further classified by the presence or absence of inflammation. See Table 14.4.2 for a classification of chronic overuse syndromes.
Table 14.4.2
Classification of chronic overuse syndromes
| Grade | Symptom |
| I | Pain after activity |
| II | Pain early on and after activity; activity not limited |
| III | Pain throughout activity, which is limited |
| IV | Pain at rest |
Management
Most chronic overuse injuries are managed with a decrease in activity and NSAIDs. Arrange referral to a physiotherapist or specialty physician, such as sports or performing arts physician, as appropriate. Tendon-related injuries may benefit from steroid injection, which should only be performed by doctors trained in the technique (orthopaedic surgeons, rheumatologists, sports physicians or some ED doctors).
Specific chronic overuse injuries that require more extensive management are summarized in Table 14.4.3.
Table 14.4.3
Stress fractures which require active specialist management
| Injury | Associated with | Symptoms | X-ray | Other imaging | Management |
| Pars interarticularis | Gymnasts, ballet dancers, fast bowlers | Unilateral low back pain, worse on extension | Pars # often seen | CT or MRI definitive.XR+Bone scan alternative option | Avoiding hyper extension for 6/52, consider brace for 6-12/52. Core stability retraining once healed |
| Femoral neck | Athletes/military increased activity | Vague thigh/groin pain with loading | Often normal | Bone scan or MRI, CT less sensitive | if<50% of bone fractured, decrease activity, if>50% ORIF |
| Femoral shaft | Dancers | Vague thigh/knee pain with loading | # Usually visible | CT or bone scan | Lateral cortex – ORIF, medialcortex (much rarer) non-weightbearing 6/52 |
| Anterior cortex of midtibia | Distance runners, ballet dancers | Progressive anterior leg pain with activity | Anterior # line, thickened cortex | Bone scan? Non-union vs recent injury | Decrease activity, intermedulary nail if progresses |
| Talus | Repeated falls/jumping from height | Foot/ankle pain worse with weightbearing | Usually normal | Bone scan, CT or MRI | 6/52 Non-weightbearing in POP |
| Navicular | Increased running/marching | Vague midfoot pain with point tenderness over navicular | May show # | Bone scan, CT or MRI | 6-8/52 Non-weightbearing, ORIF if fails to heal |
| Base 2nd metatarsal | Ballet dancers | Forefoot pain on exercise | # Usually visible | Bone scan, CT or MRI but usually not needed | Non-weightbearing on crutches for 4-6/52 |
| Base 5th metatarsal | Ballet dancers | Midfoot pain with activity | # Usually visible | Bone scan, CT or MRI but usually not needed | Non-weightbearing with POP for 6/52 or direct ORIF as often fail to heal |
| Sesamoid bone of hallux | Increased running/marching | Forefoot pain, tender/swelling over ball of foot | Often hard to interpret | Bone scan or MRI | 6/52 Non-weightbearing with crutches then orthotics to correct biomechanics |
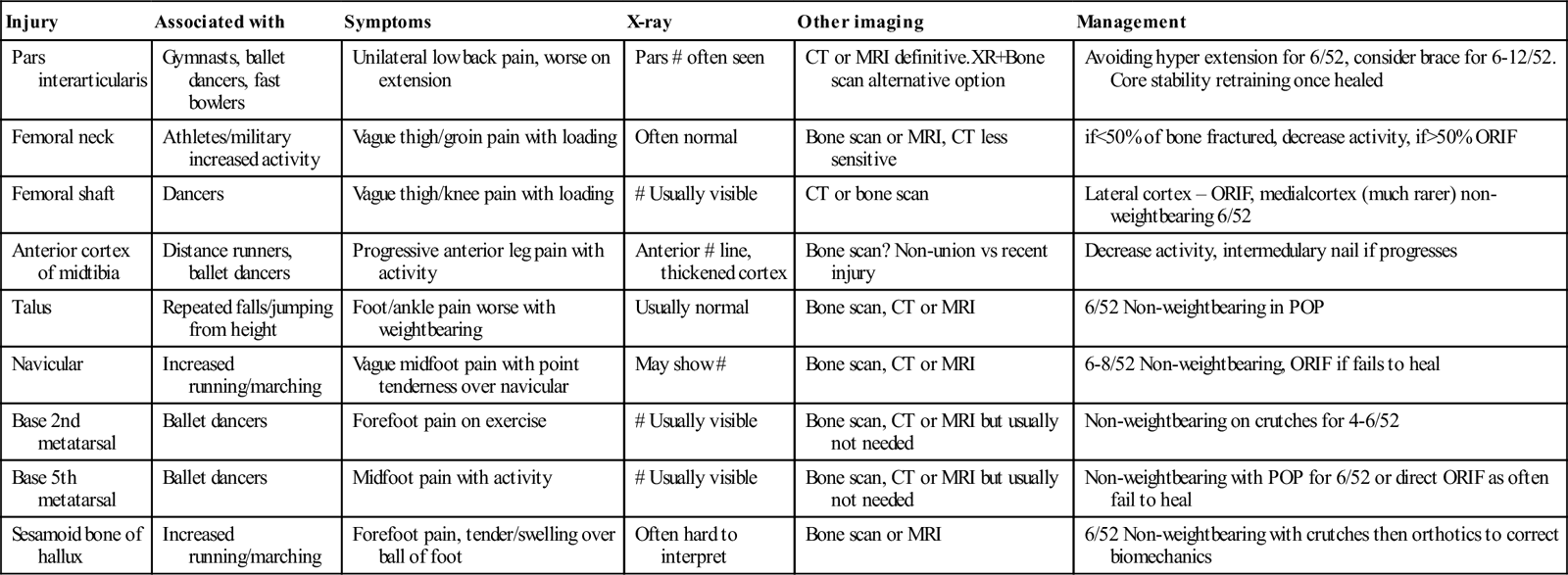
Non-arthritic joint and soft-tissue disorders
General management of non-arthritic joint and soft-tissue disorders
Joint pain, swelling and tenderness mimicking arthritis may be due to inflammation of periarticular structures. Most patients can be treated with NSAIDs, such as ibuprofen 200–400 mg orally tds or naproxen 250 mg orally tds and/or with paracetamol in combination with codeine, usually as paracetamol 500 mg plus codeine 8 mg.
Underlying or secondary true arthritis may also be present and complicate the presentation. Joint aspiration is indicated to rule out a septic arthritis and, when this is suspected, follow local guidelines as to who performs it. Refer the patient back to their GP or outpatients unless mobility is so significantly affected that they require admission.
Do not perform steroid injection in the emergency department, as complications, such as septic arthritis and joint destruction do occur. This is best left to the specialist who undertakes long-term care. Some of the more common presentations include the following.
Torticollis (‘wry neck’)
Diagnosis
Torticollis is abnormal unilateral neck muscle spasm, resulting in the head being held in a bent or twisted position. The aim of the history and examination is to exclude a serious underlying cause such as local sepsis, from a quinsy or submandibular abscess, recent trauma, cervical disc prolapse, acute drug dystonia or even a carotid artery dissection.
Management
Benign ‘wry neck’ most commonly occurs on waking after sleeping in an awkward position or follows unaccustomed activity or minor trauma. Arrange for cervical imaging if there is a history of possible bony trauma or cervical pathology. Give benztropine 1–2 mg IV when drug-induced dystonia is suspected.
Once serious causes have been excluded, use NSAIDs or paracetamol in combination with codeine 8 mg. Recommend gentle manipulation or muscle energy techniques to slowly work loose the muscles in spasm. Discharge the patient back to their GP with analgesia and ongoing exercises/stretches to maintain neck alignment.
Frozen shoulder (adhesive capsulitis)
Diagnosis
Frozen shoulder (adhesive capsulitis) has a natural history lasting 1–5 years, with an average duration of 2.5 years. It begins with an acutely painful period of 3–9 months with a progressively decreasing range of motion at the glenohumeral joint, ‘freezing phase’, over 4–12 months starting soon after the pain. Pain tends to be worse at night or when lying flat. The decreased range of motion usually resolves in the ‘thawing’ phase, but this may take from 1 to 4 years.
A frozen shoulder may occur spontaneously, but more commonly follows local trauma (which can be trivial), immobilization, a cerebrovascular accident or shingles. There is an increased risk in diabetic patients where the condition may present bilaterally, in smokers, with hyperlipidaemia and in those on treatment with protease inhibitors. It is more common in females with a peak incidence age of 55 years and in the non-dominant arm.
On examination, the most sensitive sign is loss of external rotation at the glenohumeral joint. Test for this by first immobilizing the scapula by placing a hand over the top of the shoulder to exclude scapulothoracic movement.
Management
Treatment includes high-dose intra-articular steroid injection that reduces early pain but without effect on the range of motion and physical disruption of the joint capsule, for instance by arthroscopic capsule release. There is no evidence that NSAIDs or physiotherapy alone have an effect on outcome, with physiotherapy in the painful and freezing phases specifically found to increase pain with no effect on range of movement.
Rotator cuff tear (usually rupture of supraspinatus)
Diagnosis
Sudden traction on the arm may tear the muscles that make up the ‘rotator cuff’ which include supraspinatus, infraspinatus, subscapularis and teres minor. Although the onset of pain may be insidious, a traumatic incident may complete a tear, causing sudden severe pain and reduced shoulder function.
The vast majority (95%) of tears occur in patients over 40 years with a component of chronic tendon damage. Any movement of the shoulder may be markedly painful after an acute injury, limiting assessment. Where able, evaluate active and passive ranges of motion, with limitation of abduction (supraspinatus) most commonly found. The combination of weakness of supraspinatus (tested with downwards pressure on the abducted, 30° forward flexed arm) and impingement in both internal (shoulder 90° flexion) and external (shoulder 90° abduction) rotation is highly sensitive (95%) for a rotator cuff tear.
Radiography
Shoulder X-ray may show a decrease in the space between the head of the humerus and the acromion in a large chronic tear. Ultrasound is best at characterizing the extent of a full thickness rotator cuff tear and/or a biceps tendon dislocation, but is less sensitive for a partial-thickness tear. MRI is highly sensitive and specific for delineating the degree, location and characteristics of rotator cuff pathology, when available.
Management
Refer a young patient with an acute full thickness tear to the orthopaedic specialist for consideration of early operative repair, as it becomes technically more difficult after 2–3 weeks due to tissue retraction. Otherwise, conservative management consists of analgesia, an immobilizing sling and referral to the physiotherapy department for a physical therapy rehabilitation programme.
Supraspinatus tendonitis
Diagnosis and management
Supraspinatus tendonitis is one of the causes of the ‘painful arc’ occurring between 60° and 120° of shoulder abduction. Perform a shoulder X-ray, which may reveal calcification in the supraspinatus tendon and/or ‘hooking’ of the acromion, decreasing the subacromial space and predisposing to this condition. Ultrasound is used for both diagnosis and to facilitate aspiration and local steroid injection.
Give an anti-inflammatory analgesic and consider referral to the orthopaedic (especially if there is a hooked acromion) or rheumatology clinic for aspiration and local steroid injection or via ultrasound by an interventional radiologist.
Subacromial bursitis
Diagnosis and management
Subacromial bursitis may follow rupture of calcific material into the subacromial bursa that again causes a ‘painful arc’ on attempted shoulder abduction or constant severe pain in the shoulder. Manage as for supraspinatus tendonitis above.
Tennis and golfer’s elbow
Diagnosis and management
Tennis elbow (incorrectly termed lateral epicondylitis) causes pain over the lateral epicondyle of the humerus from chronic angiofibroblastic tendinosis. There is disorganized tissue and neovascularization but minimal actual inflammation of the extensor origin of the forearm muscles involved in repetitive movements, such as using a screwdriver or playing tennis. Advise the patient to avoid the activity causing the pain and to rest the arm.
Give an anti-inflammatory analgesic and refer for physiotherapy. Eccentric and isometric exercises are most effective in treating and preventing recurrence. A tension strap can also be used to control symptoms, particularly when a patient presents within the first 6 weeks. Local steroid injection often reduces short-term pain and improves movement in the first 6 weeks, but has a worse longer-term outcome.
Golfer’s elbow (medial epicondylitis) is a similar condition affecting the medial epicondyle and the flexor origin. Management is the same.
Olecranon bursitis
Diagnosis and management
Painful swelling of the olecranon bursa is due to local trauma, gout or infection, usually with Staphylococcus aureus. Aspiration under sterile conditions is indicated in the presence of severe pain and/or systemic features of sepsis, with microscopy for both crystals and bacteria and culture if there is adequate fluid to allow drainage. Imaging is indicated when a foreign body is suspected.
Refer the patient for drainage of the bursa under anaesthesia if significant bacterial infection or a foreign body is confirmed, or if a septic arthritis is suspected due to markedly reduced movement at the elbow (see Chapter 14.2). Otherwise, give an antistaphylococcal antibiotic, such as di- or flucloxacillin 500 mg orally qid for 7–10 days for overlying cellulitis and/or a non-steroidal anti-inflammatory analgesic and refer back to the GP.
Prepatellar bursitis (housemaid’s knee)
Diagnosis and management
This is a prepatellar bursitis secondary to friction or, occasionally, infection. Treat by giving an anti-inflammatory analgesic, avoiding further trauma and, if necessary, by aspiration and steroid injection by an orthopaedic or rheumatology specialist, or by arrangement with the patient’s GP. When local infection is suspected, start an antistaphylococcal antibiotic, such as di- or flucloxacillin 500 mg orally qid for 7–10 days and, again, refer back to the GP.
Refer the patient to the orthopaedic specialist for intravenous antibiotics and/or local drainage if systemic infection is suspected.
De Quervain’s stenosing tenosynovitis
Diagnosis and management
This causes tenderness over the radial styloid, a palpable nodule from thickening of the fibrous sheaths of the abductor pollicis longus and extensor pollicis brevis tendons and pain on moving the thumb. Treat by resting the thumb in a splint and by using an anti-inflammatory analgesic.
Refer to a rheumatology specialist for consideration of local steroid injection, although it may require surgical release of the tendon sheaths if local steroid injection fails.
Plantar fasciitis
Diagnosis and management
Plantar fasciitis presents as a painful midfoot, especially in the sole or arch, that is worse on first weight bearing and improves after 10–15 min of walking, recurring again during load bearing for an extended period. It is one of the most common causes of recurrent foot pain and may be one manifestation of the spondyloarthropathy seen in Reiter’s syndrome, ankylosing spondylitis and psoriatic arthritis (see Chapter 14.3). On examination, there is tenderness of the plantar fascia, especially at the calcaneal attachment.
X-ray may reveal a bony spur extending along the plantar fascia, but this has no bearing on the initial management. Symptomatic relief may be obtained by a soft heel pad. Longer-term management and prevention is best achieved by a properly fitted orthotic splint.
Carpal tunnel syndrome
Diagnosis and management
This is a compressive neuropathy of the median nerve at the wrist, most commonly affecting middle-aged females. Secondary causes include rheumatoid arthritis, diabetes, post-trauma, such as a Colles’ fracture, pregnancy and, rarely, myxoedema, acromegaly and amyloidosis. Most cases though are idiopathic or related to minor repetitive trauma.
Patients complain of pain and burning paraesthesiae in the distribution of the median nerve in the hand, primarily the thumb, index, middle and lateral aspect of the ring finger. It is typically worse at night or following repetitive strain, especially with higher loads or vibrating tools.
Perform Phalen’s test by reproducing paraesthesiae in the distribution of the median nerve following 60 s of wrist hyperflexion, or look for Tinel’s sign eliciting median nerve paraesthesiae by tapping on the volar aspect of the wrist over the median nerve. Test for reduced sensation over the palmar aspect of the affected digits and weakness of thumb abduction, associated with thenar muscle wasting in chronic cases.
Treat with an anti-inflammatory analgesic and immobilize the wrist in a volar splint in the neutral position, particularly at night. Refer resistant cases to an orthopaedic specialist for consideration of carpal tunnel decompression.
Back pain
This is a common problem that usually simply requires analgesia and patient education. Assessment is targeted at determining whether concerning features, ‘red flags’, are present which mandate further investigation. Back pain may be subdivided into four major categories:
 indirect mechanical back trauma (non-specific low back pain)
indirect mechanical back trauma (non-specific low back pain)
 back pain with focal ‘hard’ neurology, or a specific serious cause suspected.
back pain with focal ‘hard’ neurology, or a specific serious cause suspected.
Direct major thoracic and lumbosacral spine trauma is covered in Chapter 3.3.
Back pain ‘red flags’
Every patient presenting to the ED with acute low back pain must be assessed for the presence of ‘red flag’ symptoms or signs suggesting a potentially serious underlying cause. These ‘red flags’ include unexplained weight loss, unexplained fever, immunosuppression, history of cancer, intravenous drug use (IVDU), duration greater than 6 weeks, focal neurological deficit/progressive or disabling symptoms, particularly pain at night, prolonged use of glucocorticoids, osteoporosis (note chronic alcoholism leads to osteopaenia) and age over 70 years.
Although the majority will still just end up having a diagnosis of musculoskeletal pain, laboratory testing with a full blood count (FBC), C-reactive protein (CRP) and erythrocycte sedimentation rate (ESR), plus imaging with CT or MRI is indicated. See Table 14.4.4 for the differential diagnosis and investigation of the possible underlying disorders from malignancy, spinal cord compression to discitis and epidural abscess.
Table 14.4.4
Differential diagnosis and investigation of serious disorders causing back pain
| Suspected diagnosis | Symptoms/findings | Investigations |
| Infection | Fever | FBC/CRP/ESR |
| osteomyelitis | IVDU | ESR most sensitive |
| discitis | Immunosuppression | MRI best imaging |
| epidural abscess | Recent infection or instrumentation | |
| Cancer | Previous cancer | X-ray |
| primary | Unexplained weight loss | +MRI if neurology |
| secondary | Age>50 (65 years some series) | |
| Failure to improve after 4 weeks | ||
| Fracture | Osteoporosis | X-ray |
| Pain still significant at rest | ||
| Steroid use | ||
| Spinal cord compression | Urinary retention | MRI |
| (includes cauda equina) | Incontinence | |
| Saddle anaesthesia | ||
| Sensory and/or motor level | ||
| Ankylosing spondylitis | Age<30 years | ESR/CRP |
| Pain worse at night | HLA-B27 | |
| Morning stiffness | Pelvis X-ray | |
| Improves with exercise |
Indirect mechanical back trauma (non-specific low back pain)
Clinical features
History
Bending, lifting, straining, coughing or sneezing may precipitate acute, severe low back pain, causing intense muscle spasm or even complete immobility. It is common for patients to have apparently minor back discomfort on one day, then wake with severe spasm the next.
Examination
This is focused on excluding any focal ‘hard’ neurology or radiculopathy. Giving adequate analgesia so that pain does not limit strength is an important part of the assessment. See Chapter 3.3 for a description of the myotomes, dermatomes and nerve roots in the leg.
Hard neurology is characterized by the loss of sensation, reflexes or true weakness. A radiculopathy is characterized by pain or subjective altered sensation following a dermatome. These should both be absent. Imaging is not usually indicated unless symptoms are continuous for greater than 6 weeks and/or are not previously investigated.
Management
The mainstay of management is to limit normal activities with some directed range of movement exercises and stretches. The ED management consists of excluding more serious causes, then education and reassurance while ensuring adequate analgesia to allow movement. Combinations, such as paracetamol 1 g qid, ibuprofen 400 mg tds±addition of oral opioids, are usually effective. The addition of diazepam 2–5 mg PO tds for patients with a particularly spasmodic and/or anxiety component is also effective.
Patients who are able to mobilize with this management may be discharged to the care of their GP for ongoing follow up and education concerning posture and lifting. Patients who fail initial management will require admission, either to an ED short-stay ward for nursing care and regular analgesia prior to physiotherapy review, or to an inpatient ward according to hospital policy, most commonly under orthopaedics or general medicine.
Back pain with radiculopathy
Clinical features
These patients have a similar presentation to those with non-specific low back pain, but also have neuropathic pain following one or more lower leg dermatomes. Examination may reveal subjectively altered sensation but with intact sharp/dull (or hot/cold), 2-point discrimination and proprioception. Straight leg raising may exacerbate radicular symptoms. Strength and reflexes are also intact, with no reported incontinence or urinary retention.
Imaging is again not indicated unless symptoms are progressing with increasing numbers of dermatomes or there are continuous symptoms for greater than 6 weeks.
Management
Management is the same as for non-specific back pain, with the addition of more specific neuropathic pain analgesia, such as tramadol (trial in ED first as may cause vomiting) 150–200 mg SR bd if effective, added to either amitriptyline 25 mg nocte or pregabalin (a GABA analogue that reduces the release of neurotransmitters by interfering with the calcium channels in nerve terminals) 75 mg bd.
Back pain with focal ‘hard’ neurology, or a specific serious cause suspected
This small group of patients with hard neurology consisting of true weakness, loss of sensation and/or reflexes requires further investigation and specialist referral (usually neurosurgery or orthopaedics). The timing of this investigation will depend on the acuity and extent of the symptoms or signs. Acute onset or ongoing progression mandate emergent investigation and referral for treatment. Subacute or chronic symptoms (especially if from a single nerve root) may be investigated and managed on a less urgent basis in discussion with the specialist team, particularly if they are unlikely to be reversible.
Other patients with ‘red flag’ symptoms or signs must also be investigated urgently (see Table 14.4.4). They may have any of the following conditions.
Spinal infection
Clinical features
Spinal infections include epidural abscess, discitis and vertebral osteomyelitis. Risk factors are recent instrumentation (prolonged epidural catheter>surgery>brief, such as obstetric epidural catheter), immunosuppression, alcoholism, diabetes, IVDU, contiguous infection or distal infection with bacteraemia. The classic progression of symptoms is from back pain to radiculopathy, to weakness, to paralysis with progression from radiculopathy to paralysis sometimes occurring over hours. Fever is absent in over one-third of cases.
Investigations
A normal WCC, ESR and CRP virtually exclude the diagnosis, with ESR being the most (110 out 117 confirmed cases in one study) and WCC the least sensitive. Blood cultures should be taken, although CT-guided or surgical specimens are more likely to culture the causative microbe. MRI is the imaging modality of choice to confirm or exclude the diagnosis.
Management
Progressive neurology requires urgent operative intervention with the decision on which antibiotic(s) prior to surgery discussed with the treating team. Patients without neurology may be treated conservatively. Empirical antibiotic therapy is targeted at skin flora including methicillin-resistant Staphylococcus aureus (MRSA) and dental flora, unless spread from a focal infection, such as from Escherichia coli or Strepotococcus pneumoniae is suspected.
Spinal cancer
Clinical features
Acute symptoms are most likely in the setting of previous cancer, particularly those that metastasize to bone (lung, breast, prostate, renal, thyroid and melanoma). Unexplained weight loss, age>50 years and symptoms failing to improve after 1 month are risk factors. Examination should thus include skin (melanoma), breasts, chest, abdomen and prostate to look for a primary tumour.
Investigations
Plain X-ray may be adequate to find a bony lesion, but more information with high sensitivity is obtained from CT scanning. An MRI is indicated if there is any focal neurology.
Management
If cancer is found or highly suspicious, admit to hospital. Spinal cord compression may respond to radiotherapy and a pathological fracture may require stabilization. Otherwise, management is with analgesia and investigation aimed at determining the primary tumour, which will dictate definitive treatment.
Fracture (vertebral compression)
Clinical features
Suspect this with significant pain at rest, long-term steroid use or known osteoporosis even with minor trauma. Examination is aimed at excluding focal neurological complications.
Investigations
Plain X-ray is the initial investigation. CT is indicated if there is greater than 50% vertebral height loss or retropulsion of fragments into spinal canal noted.
Management
Analgesia is as per non-specific back pain. It is extremely rare for these injuries to be unstable or require immobilization or surgical stabilization, although admission for analgesia and bedrest may be necessary.
Spinal cord compression or cauda equina syndrome
Clinical features
Spinal cord compression or cauda equina syndrome (lesion at or below the first lumbar vertebra) may be due to tumour, infection or central disc prolapse. Urinary retention is the most sensitive sign of cauda equina syndrome, which is seen in ≈90% of cases. A history of incontinence and perineal or perianal ‘saddle area’ anaesthesia or bilateral leg weakness may also occur. The neurology findings will correspond to a specific level in the case of spinal cord compression, but may be inconsistent and patchy in cauda equina syndrome.
Investigation
Urgent MRI is the investigation of choice, with CT of some value, particularly if bony injury is suspected or if MRI is unavailable. Imaging should not delay urgent transfer to definitive treatment under the care of a spinal surgeon.
Management
Urgent surgery. Corticosteroids are not supported unless due to a steroid-responsive tumour.
Ankylosing spondylitis
Clinical features
Ankylosing spondylitis is a chronic inflammatory enthesopathy affecting the axial skeleton. It usually occurs in males (5:1) below the age of 30 years, causing pain that improves with exercise and worsens with rest, sometimes resulting in waking in the second half of the night due to discomfort. Examination may be unremarkable or show a general decreased range of spinal motion in more advanced cases.
Investigations
The ESR and CRP are usually raised. HLA-B27 is sensitive for the disease and is present in around 95% of Caucasian and Chinese patients. Pelvic X-ray often shows sacroiliitis.
Management
Commence NSAIDs at a maximum dose and refer to rheumatology outpatients follow up. Regular physiotherapy including hydrotherapy is essential.