[level-membership-for-emergency-medicine-category]
Haematology Emergencies
Edited by Mark Little
13.1 Anaemia
Lindsay Murray
Introduction
Anaemia is a condition in which the absolute number of red cells in the circulation is abnormally low. The diagnosis is usually made on the basis of the full blood count (FBC). This, together with the blood film, offers qualitative as well as quantitative data on the blood components and a set of normal values is shown in Table 13.1.1.
Table 13.1.1
Full blood count: normal parameters
| Haemoglobin (Hb) | |
| Males | 135–180 g/L |
| Females | 115–165 g/L |
| Red blood cell count | |
| Males | 4500–6500×109/L |
| Females | 3900–5600×109/L |
| Haematocrit | |
| Males | 42–54% |
| Females | 37–47% |
| Other values | |
| MCH | 27–32 pg |
| MCHC | 32–36 g/dL |
| MCV | 76–98 fL |
| Reticulocytes | 0.2–2% |
| White blood cells | 4–11×109/L |
| Neutrophils | 1.8–8×109/L |
| Eosinophils | 0–0.6×109/L |
| Basophils | 0–0.2×109/L |
| Lymphocytes | 1–5×109/L |
| Monocytes | 0–0.8×109/L |
| Platelets | 150–400×109/L |

The average lifespan of a normal red blood cell in the circulation is from 100 to 120 days. Aged red cells are removed by the reticuloendothelial system but, under normal conditions, are replaced by the marrow such that a dynamic equilibrium is maintained. Anaemia develops when red cell loss exceeds red cell production. It follows that the anaemic patient is doing at least one of three things: not producing enough red cells, destroying them too quickly or bleeding.
The overriding functional importance of the red cell resides in its ability to transport oxygen, bound to the haemoglobin molecule, from the lungs to the tissues. Functionally, anaemia may be regarded as an impairment in the supply of oxygen to the tissues and the adverse effects of anaemia, from whatever cause, are a consequence of the resultant tissue hypoxia. Anaemia is not a diagnosis: rather, it is a clinical or a laboratory finding that should prompt the search for an underlying cause (Table 13.1.2).
Table 13.1.2
Haemorrhage
Traumatic
Non-traumatic
Acute
Chronic
Megaloblastic anaemia
Vitamin B12 deficiency
Folate deficiency
Aplastic anaemia
Pure red cell aplasia
Myelodysplastic syndromes
Invasive marrow diseases
Chronic renal failure
Decreased RBC survival (haemolytic anaemia)
Congenital
Spherocytosis
Elliptocytosis
Glucose-6-phosphate-dehydrogenase deficiency
Pyruvate kinase deficiency
Haemoglobinopathies: sickle cell diseases
Acquired autoimmune haemolytic anaemia, warm
Acquired autoimmune haemolytic anaemia, cold
Microangiopathic haemolytic anaemias
RBC mechanical trauma
Infections
Paroxysmal nocturnal haemoglobinuria
Anaemia secondary to haemorrhage
Aetiology
By far the most common cause of severe anaemia encountered in the emergency department (ED) is haemorrhage. Therefore, the assessment of the anaemic patient is often chiefly concerned with the search for a site of blood loss. The most common causes of haemorrhage are outlined in Table 13.1.3. However, the emergency physician must remain alert to the possibility that the patient is not bleeding but manifesting a rarer pathological condition.
Table 13.1.3
Common causes of haemorrhage in the emergency department
Trauma
Blunt trauma to mediastinum
Pulmonary contusions/haemopneumothorax
Intraperitoneal injury
Retroperitoneal injury
Pelvic disruption
Long bone injury
Open wounds: inadequate first aid
Non-trauma
Gastrointestinal haemorrhage
Oesophageal varices
Peptic ulcer
Gastritis/Mallory–Weiss
Colonic/rectal bleeding
Obstetric/gynaecological bleeding
Ruptured ectopic pregnancy
Menorrhagia
Threatened miscarriage
Antepartum haemorrhage
Postpartum haemorrhage
Other
Epistaxis
Postoperative
Secondary to bleeding diathesis
Clinical features
While it may be obvious on history and examination that a patient is bleeding, occasionally, the source of blood loss is occult and the extent of loss underestimated.
In the context of trauma, the history often gives clear pointers to both sites and extent of blood loss. Consideration of the mechanism of injury may allow anticipation of occult pelvic, intraperitoneal or retroperitoneal bleeding. Intracranial bleeding is never an explanation for hypovolaemic shock in an adult. In the context of non-trauma, it is essential to obtain an obstetric and gynaecological history in women of childbearing age. The remainder of the formal history may supply information essential in determining the aetiology of anaemia. The past medical history may point to a known haematological abnormality or to a chronic disease process. A drug and allergy history is always relevant. Many drugs cause marrow suppression, haemolytic anaemia and bleeding. The family history points to hereditary disease; the social history may alert the clinician to an unusual occupational exposure in the patient’s past or, more likely, to recreational activities liable to exacerbate an ongoing disease process. The systems review is particularly relevant to the consultation with middle-aged or elderly male patients, who must be asked about symptoms of altered bowel habit and weight loss.
The symptomatology of anaemia proceeds from vague complaints of tiredness, lethargy and impaired performance through to more sharply defined entities, such as shortness of breath on exertion, giddiness, restlessness, apprehension, confusion and collapse. Co-morbid conditions may be exacerbated (the dyspnoea of chronic obstructive airway disease) and occult pathologies unmasked (exertional angina in ischaemic heart disease).
Anaemia of insidious onset is generally better tolerated than that of rapid onset because of cardiovascular and other compensatory mechanisms. Acute loss of 40% of the blood volume may result in collapse whereas, in certain developing countries, it is not rare for patients with haemoglobin concentrations 10% of normal to be ambulant. Trauma superimposed on an already established anaemia can lead to rapid decompensation.
The cardinal sign of anaemia is pallor. This can be seen in the skin, the lips, the mucous membranes and the conjunctival reflections. Yet, not all anaemic patients are pallid and not all patients with a pale complexion are anaemic. Patients who have suffered an acute haemorrhage may show evidence of hypovolaemia: tachycardia, hypotension, cold peripheries and sluggish capillary refill. The detection of postural hypotension is an important pointer towards occult blood loss. Conversely, patients with anaemia of insidious onset are not hypovolaemic and may manifest high-output cardiac failure as a physiological response to hypoxia.
Other features of the physical examination may provide clues to the aetiology of anaemia. The glossitis, angular stomatitis, koilonychia and oesophageal web of iron- deficiency anaemia are uncommon findings. Bone tenderness, lymphadenopathy, hepatomegaly and splenomegaly may point to an underlying haematological abnormality. The rectal and gynaecological examinations can sometimes be diagnostic.
Clinical investigations
The full blood count often reveals an anaemia that has not been clinically suspected and that must be interpreted in the light of the history and examination. If the anaemia is mild it may be a chance finding with little relevance to the patient’s presenting complaint, but such a finding should never be ignored. At the very least a follow-up blood count should be arranged.
Anaemic patients have a low red cell count, a low haematocrit and a low haemoglobin, but some caveats need to be borne in mind:
 patients who are bleeding acutely may initially have a normal FBC
patients who are bleeding acutely may initially have a normal FBC
 normal or high haematocrits may reflect haemoconcentration
normal or high haematocrits may reflect haemoconcentration
 mixed pictures can be difficult to interpret, e.g. that of a polycythaemic patient who is bleeding.
mixed pictures can be difficult to interpret, e.g. that of a polycythaemic patient who is bleeding.
Red cell morphology, particularly the mean corpuscular volume (MCV), can help elucidate the cause of anaemia. The finding of a pancytopaenia suggests a problem in haematopoiesis, rather than haemolysis or blood loss. In women of childbearing age, assay of blood or urine β-HCG is important.
Treatment
The principles of management of haemorrhage are as follows:
The indications for red cell transfusion are discussed in Chapter 13.5. The faster the onset of the anaemia, the greater the need for urgent replacement. Patients who are tolerating their anaemia may require no more than an appropriate diet with or without the addition of haematinics. Elderly patients with severe bleeding often need red cells urgently. Excessive administration of colloid and/or crystalloid precipitates left ventricular failure and it can then be difficult to administer red cells.
Chronic haemorrhage
The finding of a hypochromic microcytic anaemia on blood film is usually indicative of iron deficiency and, in the absence of an overt history of bleeding, should prompt the search for occult blood loss. Iron deficiency anaemia may be due to malnutrition, but inadequate dietary intake of iron is not usually the sole cause of anaemia in developed countries: much more commonly it is the result of chronic blood loss from the gastrointestinal (GI) tract, the uterus or the renal tract. More unusual causes are haemoptysis and recurrent epistaxes.
Patients present with insidious and rather vague symptoms. They may be unaware that they are bleeding and will probably show none of the trophic skin, nail and mucosal changes of iron deficiency. The automated cell count, in addition to showing a hypochromic, microcytic picture, may also show a raised red cell distribution width, which reflects anisocytosis on the blood film.
Iron studies may confirm the diagnosis of iron deficiency without pointing to the underlying cause. Serum iron and ferritin are low and total iron-binding capacity is high.
Disposition
If the source of blood loss is obvious, for example heavy menstrual bleeding, then appropriate referral may be all that is indicated. If the source is not obvious, particularly in older patients, then sequential investigation of the GI tract and the renal tract may be indicated. Decisions to admit or discharge these patients depend on the red cell reserves, the patient’s cardiorespiratory status, home circumstances and the likelihood of compliance with follow up.
The anaemia itself can be corrected with oral iron supplements: 200 mg of ferrous sulphate three times daily is an appropriate regimen, although single daily doses are often more acceptable to the patient and have fewer GI side effects.
Anaemia secondary to decreased red cell production
Megaloblastic anaemia
The finding of a raised MCV is common in the presence or absence of anaemia. Alcohol abuse is a frequent underlying cause and other causes are listed in Table 13.1.4. MCVs greater than 115 fL are usually due to megaloblastic anaemia which, in turn, is usually due to either vitamin B12 or folate deficiency. Vitamin B12 and folate are essential to DNA synthesis in all cells. Deficiencies manifest principally in red cell production because of the sheer number of red cells that are produced. B12 deficiency is usually the result of a malabsorption syndrome, whereas folate deficiency is of dietary origin. Tetrahydrofolate is a co-factor in DNA synthesis and, in turn, the formation of tetrahydrofolate from its methylated precursor is B12-dependent. Unabated cytoplasmic production of RNA in the context of impaired DNA synthesis appears to produce the enlarged nucleus and abundant cytoplasm of the megaloblast. These cells, when released to the periphery, have poor function and poor survival.
Table 13.1.4
Some causes of a raised mass cell volume
Alcohol
Drugs
Hypothyroidism
Liver disease
Megaloblastic anaemias (B12 and folate deficiency)
Myelodysplasia
Pregnancy
Reticulocytosis
B12 deficiency is an autoimmune disorder in which autoantibodies to gastric parietal cells and the B12 transport factor (intrinsic factor) interfere with B12 absorption in the terminal ileum. Patients have achlorhydria, mucosal atrophy (a painful smooth tongue) and, sometimes, evidence of other autoimmune disorders, such as vitiligo, thyroid disease and Addison’s disease. This is so-called ‘pernicious anaemia’.
A rare, but important, manifestation of this disease is ‘subacute combined degeneration of the spinal cord’. Demyelination of the posterior and lateral columns of the spinal cord manifests as a peripheral neuropathy and an abnormal gait. The central nervous system abnormalities worsen and become irreversible in the absence of B12 supplementation. Treatment of B12 deficient patients with folate alone may accelerate the onset of this condition.
Undiagnosed untreated pernicious anaemia is not a common finding in the ED, but the laboratory finding of anaemia and megaloblastosis should prompt haematological consultation. The investigative work-up, which includes B12 and red cell folate levels, autoantibodies to parietal cells and intrinsic factor, a marrow aspirate, and Schilling’s test of B12 absorption, may well necessitate hospital admission.
The work-up for folate deficiency is similar to that for B12. Occasionally, patients require investigation for a malabsorption syndrome (tropical sprue, coeliac disease), which includes jejunal biopsy. Folate deficiency is common in pregnancy because of the large folate requirements of the growing fetus. It can be difficult to diagnose because of the maternal physiological expansion of plasma volume and also of red cell mass, but diagnosis and treatment with oral folate supplements are important because of the risk of associated neural tube defects.
Both B12 and folate deficiency are usually manifestations of chronic disease processes. Rarely, an acute megaloblastic anaemia and pancytopaenia can develop over the course of days and nitrous oxide therapy has been identified as a principal cause of this condition.
Anaemia of chronic disorders
Patients with chronic infective, malignant or connective tissue disorders can develop a mild-to-moderate normochromic normocytic anaemia. Evidence of bleeding or haemolysis is absent and there is no response to haematinic therapy. The pathophysiology of this anaemia is complex and probably involves both decreased red cell production and survival. Possible underlying mechanisms include reticuloendothelial overactivity in chronic inflammation and defects in iron metabolism mediated by a variety of acute-phase reactants and cytokines, such as interleukin-1, tumour necrosis factor and interferon γ, which impair renal erythropoietin production and function.
Anaemia of chronic disorders (ACD) is generally not so severe as to warrant emergency therapy. The importance of ACD in the ED lies in its recognition as a pointer towards an underlying chronic process. Difficulties can arise in distinguishing ACD from iron deficiency and the two conditions may coexist – in rheumatoid arthritis, for example. Iron studies generally elucidate the nature of the anaemia. In iron deficiency, iron and ferritin are low and total iron binding is high, whereas in ACD iron and total iron binding are low and ferritin is normal or high.
Other causes of decreased red cell production
Bone marrow failure is rarely encountered in emergency medicine practice. The physician must be alert to the unusual, insidious or sinister presentation and be particularly attuned to the triad of decreased tissue oxygenation, immunocompromise and a bleeding diathesis that may herald a pancytopaenia. An FBC may dictate the need for haematological consultation, hospital admission and further investigation.
Among the entities to be considered are the aplastic anaemias, characterized by a pancytopaenia secondary to failure of pluripotent myeloid stem cells. Half of cases are idiopathic, but important aetiologies are infections (e.g. non-A, non-B hepatitis), inherited diseases (e.g. Fanconi’s anaemia), irradiation, therapeutic or otherwise and, most important in the emergency setting, drugs. Drugs that have been implicated in the development of aplastic anaemia include, in addition to antimetabolites and alkylating agents, chloramphenicol, chlorpromazine and streptomycin.
Characteristic of patients with a primary marrow failure is the absence of splenomegaly and the absence of a reticulocyte response. There is a correlation between prognosis and the severity of the pancytopaenia. Platelet counts less than 20×109/L and neutrophil counts less than 500/mL equate to severe disease. Depending on the severity of the accompanying anaemia, patients may require red cell and sometimes platelet transfusion in the ED, as well as broad-spectrum antibiotic cover. It is imperative to stop all medications that might be causing the marrow failure. Other forms of marrow failure include pure red cell aplasia, where marrow red cell precursors are absent or diminished. This can be a complication of haemolytic states in which a viral insult leads to an aplastic crisis (see haemolytic anaemias).
The myelodysplastic syndromes are a group of disorders primarily affecting the elderly. In these states there is no reduction in marrow cellularity but the mature red cells, granulocytes and platelets generated from an abnormal clone of stem cells are disordered and dysfunctional. There is peripheral pancytopaenia. These disorders are classified according to observed cellular morphology (Table 13.1.5). These conditions were once termed ‘preleukaemia’ and one-third of patients progress to acute myeloid leukaemia.
Table 13.1.5
Classification of the myelodysplastic syndromes
Refractory anaemia
Refractory anaemia with ringed sideroblasts
Refractory anaemia with excess of blasts
Chronic myelomonocytic leukaemia
Two more causes of failure of erythropoiesis might be mentioned. One is due to invasion of the marrow and disruption of its architecture by extraneous tissue, the commonest cause being metastatic cancer. Finally, but not at all uncommon, is the anaemia of chronic renal failure, where deficient erythropoiesis is attributed to decreased production of erythropoietin. Most patients with chronic renal failure on dialysis treatment tolerate a moderate degree of anaemia, but occasionally require either transfusion or treatment with erythropoietin. Emergency physicians should recognize anaemia as a predictable entity in patients with chronic renal failure, usually not requiring any action.
Anaemia secondary to decreased red cell survival: the haemolytic anaemias
Patients whose main problem is haemolysis are encountered rarely in the ED. The most fulminant haemolytic emergency one could envisage is that following transfusion of ABO-incompatible blood (discussed in Chapter 13.5), a vanishingly rare event where proper procedures are followed. Haemolysis and haemolytic anaemia are occasionally encountered in decompensating patients with multisystem problems. Rarely, first presentations of unusual haematological conditions occur.
Some of the haemolytic anaemias are hereditary conditions in which the inherited disorder is an abnormality intrinsic to the red cell, its membrane, its metabolic pathways or the structure of the haemoglobin contained in the cells. Such red cells are liable to be dysfunctional and to have increased fragility and a shortened lifespan. Lysis in the circulation may lead to clinical jaundice as bilirubin is formed from the breakdown of haemoglobin. Lysis in the reticuloendothelial system generally does not cause jaundice but may produce splenomegaly. The anaemia tends to be normochromic normocytic; sometimes a mildly raised MCV is due to an appropriate reticulocyte response from a normally functioning marrow. Serum bilirubin may be raised even in the absence of jaundice. Urinary urobilinogen and faecal stercobilinogen are detectable and serum haptoglobin is depleted. The antiglobulin (Coombs’) test is important in the elucidation of some haemolytic anaemias. In this test, red cells coated in vivo (direct test) or in vitro (indirect test) with IgG antibodies are washed to remove unbound antibodies, then incubated with an antihuman globulin reagent. The resultant agglutination is a positive test.
Any chronic haemolytic process may be complicated by an ‘aplastic crisis’. This is usually a transient marrow suppression brought on by a viral infection which can result in a severe and life-threatening anaemia. Red cell transfusion in these circumstances may be life saving.
Hereditary spherocytosis
A deficiency of the red cell wall protein, spectrin, leads to loss of deformability and increased red cell fragility. These cells are destroyed prematurely in the spleen. The condition may present at any age, with anaemia, intermittent jaundice and cholelithiasis. Patients are Coombs’ negative and show normal red cell osmotic fragility. Splenectomy radically improves general health. Hereditary elliptocytosis is a similar disease, with usually a milder course.
Glucose-6-phosphate dehydrogenase deficiency
Glucose-6-phosphate dehydrogenase (G6PD) generates reduced glutathione, which protects the red cell from oxidant stress. G6PD deficiency is an X-linked disorder present in heterozygous males and homozygous females. The disorder is commonly seen in West Africa, southern Europe, the Middle East and Southeast Asia. Oxidant stress leads to severe haemolytic anaemia. Precipitants include fava beans, antimalarial and analgesic drugs and infections. The enzyme deficiency can be demonstrated by direct assay and treatment is supportive.
Sickle cell anaemia
Whereas in the thalassaemias there is a deficiency in a given globin chain within the haemoglobin (Hb) molecule, in the haemoglobinopathies a given globin chain is present but structurally abnormal. HbS differs from normal HbA by one amino acid residue: valine replaces glutamic acid at the sixth amino acid from the N-terminus of the β-globin chain. Red cells containing HbS tend to ‘sickle’ at states of low oxygen tension. The deformed sickle-shaped red cell has increased rigidity, which causes it to lodge in the microcirculation and sequester in the reticuloendothelial system – the cause of a haemolytic anaemia.
Sickle cell disease is encountered in Afro-Caribbean people. The higher incidence in tropical areas is attributed to the survival value of the β-S gene against falciparum malaria. Heterozygous individuals have ‘sickle trait’ and are usually asymptomatic. Homozygous (HbSS) individuals manifest the disease in varying degrees. The haemolytic anaemia is usually in the range of 60–100 g/L and can be well tolerated because HbS offloads oxygen to the tissues more efficiently than HbA.
A patient with sickle cell disease may occasionally develop a rapidly worsening anaemia. This may be due to:
In any of these circumstances, transfusion may be life saving. However, these events are unusual and more commonly encountered is the vaso-occlusive crisis. A stressor – for example infection, dehydration, or cold – causes sickle cells to lodge in the microcirculation. Bone marrow infarction is one well-recognized complication of the phenomenon, but virtually any body system can be affected. Common presenting complaints include acute spinal pain, abdominal pain (the mesenteric occlusion of ‘girdle sequestration’), chest pain (pulmonary vascular occlusion), joint pain, fever (secondary to tissue necrosis), neurological involvement (transient ischaemic attacks, strokes, seizures, obtundation, coma), respiratory embarrassment and hypoxia, priapism, ‘hand–foot syndrome’ (dactylitis of infancy), haematuria (nephrotic syndrome, papillary necrosis), skin ulcers of the lower limbs, retinopathies, glaucoma and gallstones.
Most patients presenting with a vaso- occlusive crisis know they have the disease but, otherwise, the differential diagnosis is difficult. Sickle cells may be seen on the blood film and can also be induced by deoxygenating the sample. Hb electrophoresis can establish the type of Hb present. Other investigations are dictated by the presentation and may include blood cultures, urinalysis and culture, chest X-ray, arterial blood gases and electrocardiograph.
Pain relief should commence early. A morphine infusion may be required for patients with severe ongoing pain. Other supportive measures are dictated by the presentation. Intravenous fluids are particularly important for patients with renal involvement. Aim to establish a urine output in excess of 100 mL/h in adults. Antibiotic cover may be required in the case of febrile patients with lung involvement. It may be impossible to differentiate between pulmonary vaso-occlusion and pneumonia. Many patients with sickle cell disease are effectively splenectomized owing to chronic splenic sequestration with infarction and are prone to infection from encapsulated bacteria. The choice of antibiotic depends on the clinical presentation. Indications for exchange transfusion are shown in Table 13.1.6. The efficacy of exchange transfusion in painful crises remains unproven.
Table 13.1.6
Indications for exchange transfusion in sickle cell crisis
Neurological presentations: TIAs, stroke, seizures
Lung involvement (PaO2<65 mmHg with FiO2 60%)
Sequestration syndromes
Priapism
Haemoglobin S-C disease
Sickle trait or Hb S-C disease occurs in up to 10% in the black population. The clinical presentation resembles that of sickle cell disease but is usually less severe.
Haemoglobin C disease
In HbC, lysine replaces glutamic acid in the sixth position from the N terminus of the β-chain. Red cells containing HbC tend to be abnormally rigid, but the cells do not sickle. Homozygotes manifest a normocytic anaemia but there is no specific treatment and transfusion is seldom required.
Thalassaemias
There is a high incidence of β-thalassaemia trait among people of Mediterranean origin although, in fact, the region of high frequency extends in a broad band east to Southeast Asia.
Thalassaemias are disorders of haemoglobin synthesis. In the haemoglobin molecule, four haem molecules are attached to four long polypeptide globin chains. Four globin chain types (each with their own minor variations in amino acid order) are designated α, β, γ and δ. Haemoglobin A comprises two α and two β chains; 97% of adult haemoglobin is HbA. In thalassaemia, there is diminished or absent production of either the α chain (α-thalassaemia) or the β chain (β-thalassaemia). Most patients are heterozygous and have a mild asymptomatic anaemia, although the red cells are small. In fact, the finding of a marked microcytosis in conjunction with a mild anaemia suggests the diagnosis.
There are four genes on paired chromosomes 16 coding for α-globin and two genes on paired chromosomes 11 coding for β-globin. α-Thalassaemias are associated with patterns of gene deletion as follows: (−/−) is Hb-Barts hydrops syndrome, incompatible with life and (α/-) is HbH disease.
Patients who are heterozygous for β-thalassaemia have β-thalassaemia minor or thalassaemia trait. They are usually symptomless. Homozygous patients have β major.
Diagnosis of the major clinical syndromes is usually possible through consideration of the presenting features in conjunction with an FBC, blood film and Hb electrophoresis.
HbH disease patients present with moderate haemolytic anaemia and splenomegaly. The HbH molecule is detectable on electrophoresis and comprises unstable β tetramers. α Trait occurs with deletion of one or two genes. Hb, MCV and mean corpuscular haemoglobin (MCH) are low, but the patient is often asymptomatic.
β major becomes apparent in the first 6 months of life with the decline of fetal Hb. There is a severe haemolytic anaemia, ineffective erythropoiesis, hepatosplenomegaly and failure to thrive. With improved care, many of these patients survive to adulthood and may possibly present to the ED, where transfusion could be life saving. Patients with β trait may be encountered in the ED relatively frequently. They are generally asymptomatic, with a mild hypochromic microcytic anaemia. It is important not to work-up these patients continually for iron deficiency and not to subject them to inappropriate haematinic therapy.
Acquired haemolytic anaemias
Many of the acquired haemolytic anaemias are autoimmune in nature, a manifestation of a type II (cytotoxic) hypersensitivity reaction. Here, normal red cells are attacked by aberrant autoantibodies targeting antigens on the red cell membrane. These reactions may occur more readily at 37°C (warm autoimmune haemolytic anaemia, or AIHA), or at 4°C (cold AIHA). Warm AIHA is more common. Red cells are coated with IgG, complement or both. The cells are destroyed in the reticuloendothelial system. Fifty per cent of cases are idiopathic, but other recognized causes include lymphoproliferative disorders, neoplasms, connective tissue disorders, infections and drugs (notably methyldopa and penicillin). Patients have haemolytic anaemia, splenomegaly and a positive Coombs’ test. In the ED setting, it is important to stop any potentially offending drugs and search for the underlying disease. The idiopathic group may respond to steroids, other immunosuppressive or cytotoxic drugs or splenectomy.
In cold AIHA, IgM attaches to the I red cell antigen in the cooler peripheries. Primary cold antibody AIHA is known as cold haemagglutinin disease. Other causes include lymphoproliferative disorders, infections such as mycoplasma, and paroxysmal cold haemoglobinuria. Patients sometimes manifest Reynaud’s disease and other manifestations of circulatory obstruction. Symptoms worsen in winter. Red cell lysis leads to haemoglobinuria.
Microangiopathic haemolytic anaemia
In this important group of conditions, intravascular haemolysis occurs in conjunction with a disorder of microcirculation. Important causes are shown in Table 13.1.7.
Table 13.1.7
Causes of microangiopathic haemolytic anaemia
Disseminated intravascular coagulation
Haemolytic uraemic syndrome
HELLP
Malignancy
Malignant hypertension
Snake envenoming
Thrombotic thrombocytopaenic purpura
Vasculitis
Haemolytic uraemic syndrome and thrombotic thrombocytopaenic purpura
These are probably manifestations of the same pathological entity, with haemolytic uraemic syndrome occurring in children and thrombotic thrombocytopaenic purpura most commonly in the fourth decade, especially in women. The primary lesion is likely to be in the vascular endothelium. Fibrin and platelet microthrombi are laid down in arterioles and capillaries, possibly as an autoimmune reaction. The clotting system is not activated. Haemolytic anaemia, thrombocytopaenia and acute renal failure are sometimes accompanied by fever and neurological deficits.
In adults, the presentation is usually one of a neurological disturbance (headache, confusion, obtundation, seizures or focal signs). The blood film reveals anaemia, thrombocytopaenia, reticulocytosis and schistocytes. Coombs’ test is negative.
Patients require hospital admission. Adults with this condition may require aggressive therapy with prednisone, antiplatelet therapy, further immunosuppressive therapy and plasma exchange transfusions.
HELLP syndrome
HELLP stands for haemolysis, elevated liver enzymes and a low platelet count and is seen in pregnant women in the context of pre-eclampsia. Treatment is as for pre-eclampsia, early delivery of the baby being of paramount importance.
Disseminated intravascular coagulation
The introduction of procoagulants into the circulation resulting in the overwhelming of anticoagulant control systems may occur as a consequence of a substantial number of pathophysiological insults, obstetric, infective, malignant and traumatic. Disseminated intravascular coagulation has an intimate association with shock, from any cause. The widespread production of thrombin leads to deposition of microthrombi, bleeding secondary to thrombocytopaenia and a consumption coagulopathy, and red cell damage within abnormal vasculature leading to a haemolytic anaemia.
Recognition of this condition prompts intensive care admission and aggressive therapy. Principles of treatment include definitive management of the underlying cause and, from the haematological point of view, replacement therapy that may involve transfusion of red cells, platelets, fresh frozen plasma (FFP) and cryoprecipitate. There may be a role for heparin and other anticoagulant treatments if specific tissue and organ survival is threatened by thrombus.
Paroxysmal noctural haemoglobinuria
This entity is unusual in that an intrinsic red cell defect is seen in the context of an acquired haemolytic anaemia. A somatic stem cell mutation results in a clonal disorder. A family of membrane proteins (CD55, CD59 and C8 binding protein) is deficient and renders cells prone to complement-mediated lysis. Because the same proteins are deficient in white cells and platelets, in addition to being anaemic, patients are prone to infections and haemostatic abnormalities. They may go on to develop aplastic anaemia or leukaemia. Treatment is supportive. Marrow transplant can be curative.
Other causes of haemolysis
Haemolysis may be due to mechanical trauma, as in ‘March haemoglobinuria’. Artificial heart valves can potentially traumatize red cells. Historically, ball-and-cage type valves have been most prone to cause haemolysis, whereas disc valves are more thrombogenic. Improvements in design have made cardiac haemolytic anaemia very rare. Haemolysis is sometimes seen in association with a number of infectious diseases, notably malaria. Other infections that have been implicated are listed in Table 13.1.8. Certain drugs and toxins are associated with haemolytic anaemia (Table 13.1.9). The haemolytic anaemia that is commonly seen in patients with severe burns is attributed to direct damage to the red cells by heat.
Table 13.1.8
Infections associated with haemolysis
Babesiosis
Bartonella
Clostridia
Cytomegalovirus
Coxsackie virus
Epstein–Barr virus
Haemophilus
Herpes simplex
HIV
Malaria, especially Plasmodium falciparum (Blackwater fever)
Measles
Mycoplasma
Varicella
Table 13.1.9
Drugs and toxins associated with haemolysis
Antimalarials
Arsine (arsenic hydride)
Bites: bees, wasps, spiders, snakes
Copper
Dapsone
Lead (plumbism)
Local anaesthetics: lidocaine, benzocaine
Nitrates, nitrites
Sulphonamides
13.2 Neutropaenia
Mark Little
Introduction
Neutropaenia is defined as a decrease in the number of circulating neutrophils. The neutrophil count varies with age, sex and racial grouping. The severity of neutropaenia is usually graded as follows:
The risk of infection rises as the neutrophil count falls and becomes significant once the neutrophil count drops below 1.0×109/L. Recent Australian guidelines have defined febrile neutropaenia as a patient with a temperature above 38.3°C (or above 38°C on two occasions) with a neutrophil count less than 0.5×109/L or with less than 1.0×109/L and likely to fall to less than 0.5×109/L. These patients need to be examined for signs of systemic compromise (Table 13.2.1).
Table 13.2.1
Features of systemic compromise
Systolic BP≤90 mmHg or≥30 mmHg below patients usual BP or inotropic support
Room air arterial pO2≤60 mmHg ot SpO2<90% or need for mechanical ventilation
Confusion or altered mental state
Disseminated intravascular coagulation or abnormal PT/aPTT
Cardiac failure or arrhythmia, renal failure, liver failure or any major organ failure (only if new or deteriorating and not AF or CHF)
From Tam CS, OReilly M, Andersen D, et al. Use of empiric antimicrobial therapy in neutropenic fever. Int Med J 2011; 41:90-101.
Neutropaenic patients are at greater risk of overwhelming infection if the onset of the neutropaenia is acute rather than chronic and, in the case of patients receiving cancer chemotherapy, if the absolute neutrophil count is in the process of falling rather than rising.
Signs or symptoms of infection in the presence of severe neutropaenia, especially with features of systemic compromise, constitute a true emergency that mandates rapid assessment and aggressive management to prevent progression to overwhelming sepsis. In the emergency department (ED) setting, this is most commonly encountered when a patient presents with fever in the context of chemotherapy for cancer.
Pathophysiology and aetiology
Polymorphonuclear neutrophils are formed in marrow from the myelogenous cell series. Pluripotent haematopoietic stem cells are committed to a particular cell lineage through the formation of colony forming units, which further differentiate to form given white cell precursors. The mature neutrophil has a multilobed nucleus and granules in the cytoplasm. The cells are termed ‘neutrophilic’ because of the lilac colour of the granules caused by the uptake of both acidic and basic dyes.
The neutrophils leave the marrow and enter the circulation, where they have a lifespan of only 6–10 h before entering the tissues. Here they migrate by chemotaxis to sites of infection and injury and then phagocytose and destroy foreign material. In health, about half of the available mature neutrophils are in the circulation. ‘Marginal’ cells are adherent to vascular endothelium or in the tissues and are not measured by the full blood count. Some individuals have fixed increased marginal neutrophil pools and decreased circulating pools; they are said to have benign idiopathic neutropaenia.
For a previously normal individual to become neutropaenic there must be decreased production of neutrophils in the marrow, decreased survival of mature neutrophils or a redistribution of neutrophils from the circulating pool. The important causes are shown in Table 13.2.2.
Table 13.2.2
Important causes of neutropaenia
Decreased production
Aplastic anaemia
Leukaemias
Lymphomas
Metastatic cancer
Drug-induced agranulocytosis
Megaloblastic anaemias
Vitamin B12 deficiency
Folate deficiency
CD8 and large granular lymphocytosis
Myelodysplasic syndromes
Decreased survival
Idiopathic immune related
Systemic lupus erythematosus
Felty syndrome
Drugs
Redistribution
Sequestration (hypersplenism)
Increased utilization (overwhelming sepsis)
Viraemia
It is a defect in neutrophil production that is most likely to prove life threatening. Consumption of neutrophils in the periphery, as occurs early in infectious processes, is likely to be rapidly compensated for by a functioning marrow. Fortunately, most of the primary diseases of haematopoiesis are rare and, in practice, many of the acquired neutropaenias are drug induced. Processes interfering with haematopoiesis, often involving autoimmune mechanisms, may affect neutrophils both in the marrow and in the periphery. Some drugs cause neutropaenia universally but many more reactions are idiosyncratic, be they dose- related or independent of dose. Some commonly implicated drugs are listed in Table 13.2.3. Cancer chemotherapy drugs are now recognized as the commonest cause of neutropaenia.
Table 13.2.3
Drugs commonly associated with neutropaenia
Antibiotics: chloramphenicol, sulphonamides, isoniazid, rifampicin, β-lactams, carbenicillin
Antidysrhythmic agents: quinidine, procainamide
Antiepileptics: phenytoin, carbamazepine
Antihypertensives: thiazides, ethacrynic acid, captopril, methyldopa, hydralazine
Antithyroid agents
Chemotherapeutic agents: especially methotrexate, cytosine arabinoside, 5-azacytidine, azathioprine, doxorubicin, daunorubicin, hydroxyurea, alkylating agents
Connective tissue disorder agents: phenylbutazone, penicillamine, gold
H2-receptor antagonists
Phenothiazines, especially chlorpromazine
Miscellaneous: imipramine, allopurinol, clozapine, ticlopidine, tolbutamide
Clinical features
Neutropaenia is frequently anticipated based on the clinical presentation, such as fever developing in the context of cancer chemotherapy, by far the most common scenario in which severe neutropaenia is seen in the ED. Alternatively, it may be identified in the course of investigation for a likely infective illness or it might be an incidental finding during investigation for an unrelated condition.
Chronic neutropaenia may be asymptomatic unless secondary or recurrent infections develop. Acute severe neutropaenia may present with fever, sore throat and mucosal ulceration or inflammation. Symptoms or signs of an associated disease process may also be present, such as pallor from anaemia or bleeding from thrombocytopaenia, as might occur in conditions causing pancytopaenia.
The history of the mode of onset and duration of the illness is important. Systems enquiry may reveal cough, headache and photophobia, a diarrhoeal illness or urinary symptoms. The past history may reveal a known haematological illness or previous evidence of immunosuppression, such as frequent and recurrent infections. A detailed drug history is vital. Most neutropaenic drug reactions occur within the first 3 months of taking a drug.
In the ED, vital signs, including pulse, blood pressure, temperature, respiratory rate and pulse oximetry, should be performed at initial assessment and monitored regularly until disposition. Attention should be paid to identifying early signs of severe sepsis and the progression to septic shock.
Physical examination may reveal necrotizing mucosal lesions, pallor, petechial rashes, lymphadenopathy, bone tenderness, abnormal tonsillar or respiratory findings, spleno- or other organomegaly. Careful examination of the skin of the back, the lower limbs and the perineum for evidence of infection is important. The presence of indwelling venous access devices should be noted and insertion sites inspected for evidence of inflammation or infection.
Clinical investigations
Investigation in the ED is first aimed at confirming and quantifying the severity of neutropaenia, identifying the cause and then at identifying the focus and severity of infection. An urgent full blood count and blood film should be ordered in any patient who is suspected of suffering febrile neutropaenia. A coagulation profile and biochemistry, including electrolytes and creatinine, serum lactate, glucose and liver function tests may be indicated once severe neutropaenia is confirmed. Anaemic patients may require a group-and-hold or cross-match.
Microbiological cultures aimed at isolating a causative organism should be taken but antibiotics should not be unreasonably delayed in the presence of fever and confirmed significant neutropaenia. Blood cultures should be taken at the time of cannulation and, if possible, prior to the instigation of antibiotic therapy. Throat swab, swabs of skin lesions and indwelling venous access device sites, urinalysis and urine culture may be indicated depending on the clinical picture. Patients with apparent central nervous system infections might require a lumbar puncture, but this should be postponed or even cancelled in the presence of an uncorrected coagulopathy, signs of raised intracranial pressure, focal neurological signs or haemodynamic instability. Antibiotics, if clinically indicated, should be commenced prior to lumbar puncture.
Treatment
Management of the patient with confirmed febrile neutropaenia in the ED involves early recognition and treatment of bacterial infection and institution of supportive care to prevent progression to overwhelming sepsis and shock. Evolving or established haemodynamic instability requires immediate, aggressive resuscitation.
Empiric broad-spectrum antibiotic therapy should be started in the ED after drawing blood for culture in any patient with fever and suspected or confirmed significant neutropaenia. This strategy has played a pivotal role in reducing mortality rates in febrile neutropaenia. Australian consensus-based clinical recommendations for the management of neutropaenic fever in adults were recently published. They reinforce the need for the administration of early antibiotics. In general, antibiotics should provide good cover for both Gram-positive and Gram-negative organisms. With increased use of indwelling venous access devices for cancer chemotherapy, there has been an increase in the incidence of sepsis due to Gram-positive organisms, such as coagulase-negative staphylococci, S. aureus and methicillin-resistant Staph. aureus (MRSA). Although occurring infrequently, bacteraemia due to Pseudomonas aeruginosa is associated with a high morbidity and mortality and therefore should also be covered.
Recent evidence has suggested that antibiotic monotherapy is as efficacious as combined therapy. Therefore, for clinically stable patients, Australian consensus guidelines recommend a beta lactam monotherapy (such as pipperacillin–tazobactam 4.5 g 6-hourly or cefepime 2 g 8-hourly or ceftazidime 2 g 8-hourly). These antibiotics should be administered within 1 hour of presentation and after at least one set of blood cultures.
For patients with systemic compromise, the Australian consensus guidlelines recommend the above beta lactam antibiotics plus gentimicin (5–7 mg/kg daily) given within 30 minutes of presentation and after at least one set of blood cultures. If the clinicians believed the shocked patient was colonized with Gram-positive organisms (e.g. MRSA or has clinical evidence of a catheter-related infection in a unit with a high incidence of MRSA), then vancomycin (1.5 g 12-hourly if normal renal function) should be added. Empiric antifungal therapy is not generally required unless there is persistent fever in high-risk patients beyond 96 hour of antibacterial therapy.
Disposition
The presence of significant neutropaenia with fever generally mandates admission to hospital. Patients with severe acute neutropaenia without an established aetiology will also generally require admission regardless of the presence or absence of fever. Both the haematological abnormality and the likely presence of infection require investigation. Sometimes the aetiology of the neutropaenia will be evident; on other occasions marrow aspiration and biopsy will be required.
There is emerging evidence that a subset of febrile neutropaenic patients can be identified who are at low risk of life-threatening complications and in whom duration of hospitalization and intensity of treatment may be safely reduced. Strategies that involve outpatient treatment of low-risk patients with oral antibiotics have also been evaluated. Such regimens are reliant upon accurate prediction of risk, as well as the availability of structured programmes and resources and are not yet in widespread use.
Prognosis
The prognosis of the neutropaenic patient is largely dependent upon the underlying aetiology of the condition. Febrile neutropaenia has in the past been associated with a significant mortality rate which varies depending on the organism causing the infection. Improvements in therapy, such as rapid treatment with empiric broad-spectrum antibiotics, have significantly reduced mortality rates from this condition. Overall mortality rates for patients with febrile neutropaenia have reduced from more than 20% to less than 4% in recent data sets.
Controversies
13.3 Thrombocytopaenia
Mark Little
Introduction
Thrombocytopaenia is defined as a reduction in the number of circulating platelets, the normal circulating platelet count being 150–400×109/L. It is the most common cause of abnormal bleeding. Like anaemia, thrombocytopaenia itself is not a diagnosis, but rather a manifestation of another underlying disease process.
In the emergency department setting, thrombocytopaenia may present as an incidental finding on a routine blood count or may be diagnosed in the context of abnormal bleeding. In most cases, the underlying aetiology can be determined by a careful history and physical examination combined with interpretation of the blood count.
Aetiology
The clinically important causes of thrombocytopaenia are outlined in Table 13.3.1. Diagnoses are classified by pathological process. It should be noted that more than one pathological process may be present. The causes can be divided into three different groups: pseudothrombocytopaenia, increased destruction of platelets or reduced production of platelets.
Table 13.3.1
Pseudothrombocytopaenia
Platelet clumping
Collection into anticoagulant (EDTA)
Platelet agglutinins
Giant platelets
Increased platelet destruction
Immune
Primary
Idiopathic thrombocytopaenic purpura (ITP)
Secondary
Autoimmune thrombocytopaenia associated with other disorders
Graves’ disease, Hashimoto’s thyroiditis, systemic lupus erythematosus
HIV-related thrombocytopaenia
Drug-induced thrombocytopaenia
Heparin, gold salts, quinine/quinidine, sulphonamides, rifampicin, H2-blockers, indomethacin, carbamazepine, valproic acid, ticlopidine, clopidogrel, monoclonal antibodies (infliximab, efalizumab, rituximab)
Post-transfusion purpura
Non-immune
Thrombotic thrombocytopaenic purpura – haemolytic uraemic syndrome
Pregnancy
Gestational benign thrombocytopaenia
Pre-eclampsia/HELLP
Disseminated intravascular coagulation
Decreased platelet production
Congenital
TAR (thrombocytopaenia with absent radius), Wiskott–Aldrich syndrome,
Fanconi anaemia
Acquired
Viral infection
Epstein–Barr virus, rubella, dengue fever
Marrow aplasia
Malignant bone marrow infiltrates
Chemotherapeutic agents
Radiation therapy
Abnormal distribution and dilution
Splenic sequestration (hypersplenism)
Splenic enlargement
Hypothermia
Massive blood transfusion
Pseudothrombocytopaenia
Pseudothrombocytopaenia results from an underestimation of the platelet count as measured by an automated particle counter. The most common mechanism is platelet clumping. Clumping is most often due to the anticoagulant EDTA, but may also result from autoantibodies, such as cold agglutinins. The presence of giant platelets and platelet satellitism may also yield falsely low automated platelet counts.
Pseudothrombocytopaenia should be suspected when the automated platelet count is low in the absence of symptoms or signs of abnormal bleeding or disorders associated with thrombocytopaenia. It is best excluded by examination of the blood film by an experienced observer. Any case of thrombocytopaenia found on an automated blood count should be confirmed by examination of the peripheral smear prior to further investigation or treatment.
Thrombocytopaenia due to increased platelet destruction
Immune-related thrombocytopaenia
Immune thrombocytopaenia
Immune thrombocytopaenia (ITP) is defined as an isolated thrombocytopaenia (low platelet count with an otherwise normal complete blood count and peripheral blood smear) in a patient with no clinically apparent associated conditions that can cause thrombocytopaenia. It is a common cause of low platelet count and abnormal bleeding in both children and adults. ITP is thought to be caused by the development of autoantibodies to platelet membrane antigens.
Treatment is aimed at modulating the immune response and reducing the rate of platelet destruction and is indicated in all patients who have counts less than 20×109/L and those with counts less than 50×109/L accompanied by significant mucous membrane bleeding. First-phase treatment includes parenteral glucocorticoids (e.g. prednisolone 1 mg/kg/day for 4–6 weeks in tapered doses) and intravenous IgG. Splenectomy is usually reserved for patients who do not respond to medical therapy and have ongoing bleeding symptoms. Platelet transfusions may cause temporary increases in platelet count and may be used in cases of life-threatening haemorrhage but are otherwise not usually indicated.
In addition to the primary idiopathic form, immune thrombocytopaenia may also accompany autoimmune disorders, such as Graves’ disease and systemic lupus erythematosus. It is the main mechanism of the thrombocytopaenia related to HIV infection.
Drug-related thrombocytopaenia
A large number of drugs have been reported to cause immune-related thrombocytopaenia. By far the most commonly implicated are quinine, quinidine and heparin. Heparin is associated with a syndrome of thrombosis due to diffuse platelet activation accompanied by a consumptive thrombocytopaenia. Some platelet inhibitors, particularly ticlopidine and, less commonly, clopidogrel, are associated with severe thrombocytopaenia and other signs and symptoms of thrombotic thrombocytopaenic purpura. Recently developed monoclonal antibodies, such as infliximab (antitumour necrosis factor-α antibody), efalizumab (anti-CD11α antibody) and rituximab (anti-CD20 antibody) are also associated with an acute, severe, but usually self-limited thrombocytopaenia.
In most cases of drug-related thrombocytopenia, recovery occurs rapidly after withdrawal of the offending agent. The exception is patients with gold sensitivity who may remain thrombocytopaenic for months due to the slow clearance of this drug.
Post-transfusion purpura
Post-transfusion purpura is clinically distinct from thrombocytopaenia due to dilution of platelets following massive transfusion. It is an acute, severe thrombocytopaenia occurring about 1 week after blood transfusion and is associated with a high titre of platelet-specific alloantibodies. It is most commonly reported in multiparous women following their first blood transfusion. The mechanism for alloantibody formation is unclear. Spontaneous recovery occurs within weeks, although fatalities from severe haemorrhage have been reported.
Non-immune platelet destruction
Thrombotic thrombocytopaenic purpura
Thrombotic thrombocytopaenic purpura (TTP) is considered to be the adult form of the haemolytic uraemic syndrome (HUS). Essentially a thrombotic microangiopathy, the classic pentad of clinical findings is: (1) fever, (2) thrombocytopaenia, (3) microangiopathic haemolytic anaemia, (4) neurological abnormalities and (5) renal involvement.
TTP can occur sporadically as an idiopathic disorder or may be associated with pregnancy, epidemics of verotoxin-producing Escherichia coli and Shigella dysenteriae, malignancy, chemotherapy, marrow transplantation and drug-dependent antibodies. Treatment with plasma exchange has dramatically influenced the outcome of TTP. Mortality has fallen from more than 90% prior to introduction of plasma exchange to less than 20% with this treatment.
Thrombocytopaenia in pregnancy
Gestational thrombocytopaenia develops during an otherwise normal pregnancy and is clinically distinct from autoimmune thrombocytopaenias such as ITP. It is thought to be due to decreased platelet survival consequent to activation of the coagulation system. Thrombocytopaenia is usually mild and there is no corresponding thrombocytopaenia in the infant. The platelet count returns to normal after delivery, although thrombocytopaenia may recur in subsequent pregnancies.
Autoimmune thrombocytopaenias, on the other hand, are often associated with more severe reductions in the platelet count. Antiplatelet antibodies are capable of crossing the placenta and may result in significant thrombocytopaenia in the fetus and newborn. This can lead to complications, such as intracranial haemorrhage, during the delivery. Treatment of the mother with autoimmune thrombocytopaenia is similar in principle to the treatment of non-pregnant cases.
In the context of pregnancy, thrombocytopaenia may also be seen as part of the HELLP (haemolysis, elevated liver enzymes, low platelets) and pre-eclampsia syndromes. The two syndromes are thought to be related. Common to both is a process of microvascular endothelial damage and intravascular platelet activation. This leads to release of thromboxane A and serotonin, which provoke vasospasm, platelet aggregation and further endothelial damage. In both syndromes, the process is terminated by delivery.
Disseminated intravascular coagulation
Thrombocytopaenia is one manifestation of the syndrome of disseminated intravascular coagulation (DIC). DIC is an acquired syndrome of diffuse intravascular coagulation up to the level of fibrin formation, accompanied by secondary fibrinolysis or inhibited fibrinolysis. It occurs in the course of severe systemic diseases or may be provoked by toxins, such as snake venoms.
Thrombocytopaenia due to impaired platelet production
Congenital disorders of impaired platelet production usually present in childhood and will not be discussed.
Of the acquired disorders of impaired platelet production, the most commonly seen in the emergency setting is the incidental finding of reduced platelet count in patients suffering viral illness. Causative viruses include Epstein–Barr virus, rubella and dengue fever. Thrombocytopaenia in these cases is reversible and requires no specific therapy other than monitoring of the platelet count to ensure normalization.
Disorders of bone marrow dysfunction, such as malignant infiltration and bone marrow suppression, cause thrombocytopaenia accompanied by reductions in numbers of other blood components. Examination of the full blood count (FBC) and blood film usually distinguishes these from other causes of isolated thrombocytopaenia. Further investigation is best referred to a haematologist.
Massive blood transfusion and thrombocytopenia
Massive blood transfusion is defined as the transfusion of a volume equivalent to the patient’s normal blood volume within a 24-h period. Thrombocytopaenia results from dilution of the patient’s remaining platelets and, where whole blood is used, decreased survival of platelets in stored blood. It is possibly the most important factor contributing to the haemostatic abnormality seen in massively transfused patients. Platelet transfusion should be reserved for cases where the platelet count falls below 50×109/L.
Hypersplenism
Hypersplenism refers to the thrombocytopaenia due to pooling in patients with splenic enlargement. It is the primary cause of thrombocytopaenia in hepatic cirrhosis, portal venous hypertension and congestive splenomegaly. In these cases, thrombocytopaenia is rarely severe and not usually of clinical importance.
Transient thrombocytopaenia has been described in patients suffering severe hypothermia and is due to splenic sequestration. Platelet counts usually return to normal within days of rewarming.
Clinical features
Thrombocytopaenia may be an incidental finding on the FBC or may be diagnosed in the context of abnormal bleeding. There are distinct differences in the patterns of abnormal bleeding associated with disorders of platelet deficiency and disorders of impaired coagulation.
Spontaneous bleeding related to thrombocytopaenia typically manifests as cutaneous petechiae and/or purpura, most commonly in dependent areas, such as the legs and buttocks. Other spontaneous manifestations include multiple small retinal haemorrhages, epistaxis, gingival and gastrointestinal bleeding. Bleeding following trauma or surgery in thrombocytopaenic patients is often immediate and may respond to local methods of haemostasis. In contradistinction to this, the bleeding associated with coagulation disorders is most commonly in the form of large haematomas or haemarthroses that occur spontaneously or develop hours to days following trauma.
In addition to the haemorrhagic manifestations of platelet insufficiency, patients with thrombocytopaenia may present with the clinical features of the underlying causative disorder. Splenic enlargement may be present in cases where thrombocytopaenia is due to hypersplenism but is not a feature of immune-related thrombocytopaenia.
The level of platelets associated with clinically significant abnormal bleeding is not precisely defined. It varies depending on the platelets’ functional integrity and with the presence or absence of other risk factors, such as coagulation disorder, trauma, and surgery. There is evidence that platelet counts above 5×109/L are sufficient to prevent bleeding when the platelets are functionally normal and there are no other risk factors. Severe haemorrhage is uncommon at platelet counts above 20×109/L and, in the setting of surgery, the risk of abnormal haemorrhage is reduced at counts above 50×109/L.
Clinical investigation
The FBC and examination of the blood film are diagnostic of thrombocytopaenia. The pattern of deficiency should be considered. Isolated thrombocytopaenia refers to a low platelet count in the presence of an otherwise normal FBC and blood film. In these cases, FBC combined with a careful clinical history and examination is often sufficient to lead to a final diagnosis. Coexistent anaemia and/or leucopaenia suggest bone marrow dysfunction as the primary aetiological process.
Other useful investigations may include coagulation studies and D-dimer (DIC, pre-eclampsia), electrolytes, urea and creatinine (TTP), liver function tests (HELLP, and liver disease) and thyroid function tests (autoimmune thyroid disorders). Platelet antibody titres are indicated in the work-up of pregnancy- related thrombocytopaenia and bone marrow aspirate may be indicated in investigation of thrombocytopaenia due to bone marrow dysfunction, but neither of these tests is useful in the emergency department setting.
Treatment
Treatment for specific causes of thrombocytopaenia has already been discussed. Bleeding in the face of low platelet count may be responsive to local methods of haemostasis if the remaining platelets are functionally normal and there is no other disorder of coagulation present. Individual case reports provide some support for the use of recombinant Factor Vlla as an enhancer of haemostasis in the treatment of bleeding in the context of severe thrombocytopaenia, although evidence from randomized clinical trials is lacking. Platelet transfusion may be helpful in cases of severe haemorrhage and is sometimes used prophylactically to prevent bleeding in patients with very low platelet counts
Platelet transfusion is primarily indicated in patients in whom thrombocytopaenia is due to impaired platelet production and who are bleeding or have very low counts. The threshold for prophylactic transfusion in these patients is controversial. It is indicated when the platelet count is below 5×109/L, but is probably not indicated above this level unless other risk factors for bleeding are present.
Platelet transfusion is rarely indicated in immune-related thrombocytopaenias as the transfused platelets are rapidly destroyed. Transfusion of platelets may aggravate TTP. In DIC, platelet transfusion has not been proven to be effective but may be indicated in bleeding patients. There is little evidence to support the suggestion that blood component therapy aggravates DIC. In cases of massive blood transfusion, platelets are not routinely indicated unless there is ongoing bleeding and the platelet count is below 50×109/L.
Raising the platelet count to 20–50×109/L is sufficient to prevent serious bleeding. In patients undergoing surgery or other invasive procedures, counts up to 60–100×109/L may be required. A useful rule of thumb is that in a 70-kg adult, transfusion of one unit of platelets will increase the platelet count by 11×109/L.
At present, platelet preparations for transfusion are stored in liquid at 22°C. Problems include the continued risk of febrile non-haemolytic reactions, transmission of infectious agents and graft-versus-host disease. Alternatives to conventional liquid storage include frozen storage, cold liquid storage, photochemical treatment and lyophilized platelets. None of these methods is currently widely available. Several platelet substitutes have been developed but remain untested in the clinical setting. Some examples are red cells with surface-bound fibrinogen, fibrinogen-coated albumin microcapsules and liposome-based haemostatic agents.
Disposition
Disposition will depend on the presence and extent of abnormal bleeding, the degree of thrombocytopaenia and the underlying aetiology. In general, patients who present with abnormal bleeding and a low platelet count should be admitted for further evaluation and treatment. In the absence of bleeding, patients who have isolated thrombocytopaenia with counts above 20×109/L may be investigated on an outpatient basis.
Controversies
13.4 Haemophilia
Sean Arendse
Introduction
Haemophilia is a group of congenital disorders of blood coagulation that arise as a result of a deficiency of clotting factor proteins, which are essential to the normal intrinsic coagulation pathway. The classic form, haemophilia A, is attributable to deficiency of Factor VIII, while haemophilia B (also known as Christmas disease) is attributable to deficiency of Factor IX. Both these diseases have a classic X-linked pattern of inheritance and thus affect males, although female carriers may also have mild deficiency of the appropriate coagulation factor.
Haemophilia A is the commoner disease (80%), with an incidence of 1 in 8000–10 000 live male births, compared to an incidence of 1 in 25 000–30 000 for haemophilia B (20%).
Pathophysiology
The normal clotting system is activated in the presence of vascular injury to produce: (1) vascular spasm, (2) platelet plug formation, (3) coagulation: factor activation and the production of fibrin. Normal coagulation of blood is dependent on the generation of adequate thrombin via the clotting cascade. Deficiency of Factor VIII or IX reduces the amplification of the clotting cascade thus causing haemophilia. The severity of the bleeding disorder is inversely related to the level of functional factor present and is categorized into mild, moderate and severe disease:
Clinical features
Haemophilia A and B are clinically indistinguishable and symptoms vary according to the severity of the inherited disorder. Mild disease may not present until adulthood, whereas moderate to severe disease usually presents in infancy or early childhood.
Bleeding in haemophilia tends to occur spontaneously or following minor trauma and is typically delayed and persistent. This is because, although initial platelet ‘plugging’ function is normal, the subsequent coagulation ‘cascade’ response is abnormal. This delay is usually hours, and occasionally days. Once bleeding occurs it may persist for days or even weeks. Patients who are severely affected may present with bleeding episodes on a weekly basis.
The most common manifestations of haemophilia are:
 bleeding in the mouth from a cut, bitten tongue or loss of a tooth
bleeding in the mouth from a cut, bitten tongue or loss of a tooth
 bleeding into the neck (may cause airway compromise)
bleeding into the neck (may cause airway compromise)
Patients may also present with a complication of therapy. Most haemophiliac patients treated before 1985 have been exposed to pathogenic viruses, of which the most important are hepatitis C, hepatitis B and HIV. Of those who received plasma prior to the mid-1980s, 90% are hepatitis B positive, 85–100% are hepatitis C positive and 60–90% are HIV positive.
Clinical investigations
Investigations are tailored to the individual presentation. A full blood count, blood film and coagulation profiles are useful in the evaluation of first presentations, or major bleed, but unlikely to be helpful in patients with an established diagnosis. It is important to note that in an acute presentation, investigation and awaiting their results should not delay treatment with factor. The most important role for us in the emergency department is to administer factor as soon as is possible, blood results are not used to guide our use of factor in this group of patients. Generally, we always administer factor if we have any suspicion of a bleeding, regardless of how small we may think that bleed is.
Plain radiography of affected joints and computed tomography (CT) scanning of the head, chest, abdomen and pelvis may be essential to establish the presence or absence of bleeding complications.
The prothrombin time measures primarily Factors II, VII, V and X, thus patients with both haemophilia A and B have a normal prothrombin time and a normal thrombin clotting time. The partial thromboplastin time measures activation of all factors other than Factor VIII and is prolonged in haemophilia (although it can be normal if factor activity exceeds 30%). Specific factor assays are required to distinguish between haemophilias A and B.
Treatment
Treatment of haemophilia has evolved dramatically in the past 40 years with the discovery in the 1960s that coagulation Factor VIII was concentrated in cryoprecipitate. More recently, highly purified concentrates of Factor VIII and Factor IX have been developed.
Products currently available for the treatment of haemophilia include:
 ‘Recombinate’ (recombinant Factor VIII)
‘Recombinate’ (recombinant Factor VIII)
 ‘BeneFix’ (recombinant Factor IX)
‘BeneFix’ (recombinant Factor IX)
 ‘Biostate’ (plasma-derived Factor VIII, includes von Willebrand factor)
‘Biostate’ (plasma-derived Factor VIII, includes von Willebrand factor)
Treatment of acute bleeding episodes primarily involves administration of factor replacement therapy. Complications of bleeding may require specific intervention. Adjunctive therapies include pain relief, rest and immobilization. Specific treatment is influenced by:
Haemophilia patients presenting with suspected bleeds should be triaged as ATS 3 and receive prompt assessment by a senior doctor. For muscle and joint bleeds ‘RICES’ should be initiated on arrival to limit bleeding and reduce pain:
Options for adequate analgesia include:
In the context of pain relief, aspirin (or other platelet-modifying drugs) should be avoided and non-steroidal anti-inflammatory drugs (NSAIDs) used with caution. Intramuscular injections should never be administered. In major bleeds, there may be a requirement for red cell transfusion. Developing limb compartment syndromes may require surgical decompression and intracranial bleeds may require neurosurgical intervention. In all of these cases, factor replacement must commence as quickly as possible. Management of complex presentations requires a multidisciplinary approach and early consultation with the relevant state haemophilia centre, especially if the presentation is a major bleed or the patient has inhibitors.
Intravenous cannulation is best performed by a skilled practitioner to help ensure vein preservation. Invasive procedures, such as arterial puncture and lumbar puncture, must only be performed after clotting factor replacement.
Some patients with Factor VIII levels higher than 10% may be successfully treated with 1-amino-8-D-arginine vasopressin (desmopressin, DDAVP), which acts by releasing von Willebrand factor stored in the lining of the blood vessels. Von Willebrand factor is a protein that transports Factor VIII in the bloodstream and, as such, plays an important role in blood clotting. Desmopressin appears to mobilize available Factor VIII stores and may raise Factor VIII activity by a factor of three. If the patient has previously had a documented good response to desmopressin, this can be used as first-line therapy for minor bleeding, such as haemarthroses.
Desmopressin can be administered intravenously, subcutaneously or by nasal spray. The intravenous dose is 0.3 μg/kg in 100 mL saline over no less than 45 min. A response should be evident within the first hour. More rapid administration can be associated with blood pressure changes. Side effects, including facial flushing and headache, are usually well tolerated. Tachyphylaxis tends to develop after three or four doses. The antidiuretic properties of desmopressin (which can last up to 24 h after a dose) can produce fluid retention and hyponatraemia (leading to seizures) and the serum sodium levels should be measured before giving further doses. Desmopressin is useful in treating mild and very rarely moderate haemophilia A. It is not of value in severe haemophilia A or with any type of haemophilia B. In serious bleeds or major surgery, desmopressin alone will not control bleeding. In such a case, most patients should also receive Factor VIII concentrate, recombinant Factor VIII replacement and recombinant Factor IX replacement.
Most haemophiliac patients are usually well known to their state haemophilia centres, who often hold specialized treatment protocols for difficult or complex patients. Patients should have their treatment regimen cards with them. If not they should be encouraged to do so.
One unit of Factor VIII concentrate provides the amount of Factor VIII activity in 1 mL of normal plasma. Given that a 70-kg adult has a plasma volume of 3500 mL, we can expect that an infusion of 3500 units of Factor VIII will produce 100% Factor VIII activity in a haemophiliac with negligible activity prior to treatment. The half-life of Factor VIII is approximately 12 h. Accordingly, a further dose of 1750 units in 12 hours’ time will again restore 100% activity.
It is not always necessary to provide 100% Factor VIII activity in order to ensure haemostasis: levels of 30–50% may be sufficient in the context of haemarthrosis or dental extraction. Larger infusions should be reserved for life-threatening situations.
Treatment of bleeding
Minor bleed (e.g. spontaneous haemarthrosis or muscle bleed):
Moderate bleed (e.g. epistaxsis, traumatic haemarthrosis, excluding hip):
 Recombinate 30 units/kg for first dose then 20 units/kg at 12 and 24 h
Recombinate 30 units/kg for first dose then 20 units/kg at 12 and 24 h
Major bleed (e.g. intracerebral, hip, neck, throat, psoas muscle):
 Recombinate 45 units/kg stat and urgent haematology consultation
Recombinate 45 units/kg stat and urgent haematology consultation
 BeneFIX 90 units/kg stat and urgent haematology consultation.
BeneFIX 90 units/kg stat and urgent haematology consultation.
Many patients can administer Factor VIII concentrate at home ‘on demand’. Indeed, the availability of Factor VIII and the ease of administration have revolutionized the care of haemophiliac patients in the community. However, the following are indications for hospital admission:
 suspected intracranial haemorrhage
suspected intracranial haemorrhage
 suspected bleeding into the head, neck or throat
suspected bleeding into the head, neck or throat
 need for ongoing therapy, especially infusions
need for ongoing therapy, especially infusions
 suspected compartment syndrome (especially of forearm and calf)
suspected compartment syndrome (especially of forearm and calf)
 bleeding into hip or inguinal area, suspected iliopsoas haemorrhage
bleeding into hip or inguinal area, suspected iliopsoas haemorrhage
Antifibrinolytic agents, such as tranexamic acid (cyclokapron) and aminocaproic acid (amicar), have been used as adjunctive therapy in episodes of gastrointestinal and mucosal bleeding, for example, following dental extraction. Fibrin tissue adhesives containing fibrinogen, thrombin and Factor XIII have also been successfully placed in tooth sockets and similar surgical sites.
Tranexamic acid and aminocaproic acid are useful in treating both haemophilia A and B. These drugs help to hold a clot in place once it has formed. They act by stopping the activity of plasmin, which dissolves blood clots. They do not actually help to form a clot which means they cannot be used instead of desmopressin or Factor VIII or IX concentrate, but can be used to hold a clot in place on mucous membranes, including in the oral cavity, nasal cavity, intestinal and uterine walls. Tranexamic acid and aminocaproic acid are associated with minor side effects including nausea, lethargy, vertigo, diarrhoea and abdominal pain.
Oral/dental bleeds
First-line therapy should be topical tranexamic acid mouthwash (5%). Patients hold 10 mL of the solution in the mouth near the site of bleeding (without gargling) for 2 min repeated 5 times a day for a week.
Haematuria
Factor replacement and antifibrinolytic therapy is not usually recommended in these cases due to the risk of clot retention and renal tract obstruction.
Head injury
Haemophiliac patients with even apparently minor head trauma need hospital assessment and CT head scanning. Beware of subtle signs of a developing subdural haematoma. If an intracranial bleed is suspected, replacement therapy should be initiated prior to radiological investigation.
Compartment syndrome
Compartment syndromes are relatively common in patients with hereditary and acquired bleeding disorders. As compartment syndrome is a clinincal diagnosis, measuring compartment pressure is generally not needed to make the diagnosis.
Four of the classic signs of compartment syndrome, pallor, pulselessness, paraesthesia and paralysis are all (very) late signs.
Pain is the earliest sign and has the following characteristics:
 pain out of proportion to the expected
pain out of proportion to the expected
 associated with hard/tense compartment on clinical examination
associated with hard/tense compartment on clinical examination
 unremitting/increasing pain with increasing requirement of pain medications.
unremitting/increasing pain with increasing requirement of pain medications.
The treatment for this condition is urgent fasciotomy.
Patients who present to the emergency department with bleeding disorders and suspected compartment syndrome should have the usual management for these conditions plus immediate referral to the orthopaedic unit and haematology unit.
Surgical decision making and indications for fasciotomy are the same as for patients without bleeding disorders and factor replacement dosage and frequency for these patients is the same as for any major surgery.
Antibodies to Factor VIII
Some patients develop antibodies to Factor VIII, known as ‘inhibitors’. Treatment has to be modified according to the titre of inhibitor present (measured by the Bethesda inhibitor assay). Patients are classified as ‘high responders’ if their baseline inhibitor titre exceeds 10 Bethesda units (BU) or if the titre rises above 10 BU on exposure to Factor VIII. Different management strategies are employed according to the severity of the bleed. These include increasing the dose of Factor VIII or alternative therapies, such as activated prothrombin complex, porcine Factor VIII or recombinant Factor VIIa.
Most patients who develop inhibitors do so early in life and are known to have severe hereditary haemophilia, but inhibitors can also arise in previously normal individuals to produce an acquired haemophilia. The incidence of this phenomenon is from 0.2 to 1/1 000 000/year. Patients tend to be elderly and some have autoimmune disease, but there is also an association with pregnancy as well as with some drugs, notably penicillin. Patients haemorrhage into muscle and soft tissues and may present with haematemesis or with unusual postoperative bleeding. In the laboratory, the patient’s blood shows a prolonged APPT that is not corrected by ‘mixing’, that is, by the addition of normal plasma. Factor VIII levels are low. Management is directed towards control of the bleeding episode, replacement therapy and the prevention of further reactions using a variety of immunosuppressive remedies.
Disposition
von Willebrand disease
Factor VIII has an intimate association with von Willebrand factor (vWF). This is an adhesive glycoprotein, secreted by endothelium and megakaryocytes, which is required for the normal instigation of platelet plug formation and for stabilization and transport of Factor VIII within the circulation. Thus von Willebrand disease (vWD) is a result of dysfunction, reduction or a complete lack of the vWF and is often associated with low Factor VIII activity. It is the most common inherited bleeding disorder, affecting 0.1–1% of the population and affects males and females equally.
Three types of von Willebrand disease are recognized:
Treatment
If the patient has previously had a documented good response to DDAVP, this can be used as first line in type I vWD. It is occasionally also effective in type II vWD, but never effective in type III vWD. The dose is the same as used in haemophilia (0.3 μg/kg). Antifibrinolytic agents, such as tranexamic acid, are often helpful for mucosal bleeding, epistaxis and menorrhagia. ‘Biostate’ (plasma derived Factor VIII, includes von Willebrand factor) may be required in type I vWD if bleeding is severe or unresponsive to DDAVP and it can also be used to treat bleeding in patients with type II and type III vWD.
Useful contacts
Websites
Australian Haemophilia Centre Directors’ Organisation: www.ahcdo.org.au
Haemophilia Foundation Australia: www.haemophilia.org.au
Canadian Hemophilia Society: www.hemophilia.ca
Hemophilia Federation of America: www.hemophiliafed.org
Haemophilia Foundation Australia: www.haemophilia.org.au
Haemophilia Foundation of New Zealand: www.haemophilia.org.nz
Contact numbers for advice/referrals
ACT
NSW
NT
QLD
SA
TAS
VIC
WA
13.5 Blood and blood products
Sean Arendse and Biswadev Mitra
Introduction
Blood is living tissue composed of blood cells suspended in plasma; it transports nutrients and oxygen and facilitates temperature control. An average 70-kg male has a blood volume of about 5 L. The cellular elements comprise red blood cells, white blood cells and platelets and make up about 45% of the volume of whole blood. Plasma, which is 92% water, makes up the remaining 55%.
Early attempts at blood transfusion were thwarted by adverse reactions. In 1900, Karl Landsteiner demonstrated the ABO blood group system and explained many of the observed severe incompatibility reactions (Table 13.5.1). He won the Nobel prize for medicine in 1930 and went on to discover the Rhesus factor in 1940. The next major advance in transfusion medicine occurred with the development of long-term anticoagulants, such as sodium citrate, which allowed extended preservation of blood. Development of refrigeration procedures allowed storage of anticoagulated blood. The addition of a citrate–glucose solution extended the viability of collected blood to several days. The ability to preserve blood for longer than a few hours paved the way for the establishment of the first blood bank in a Leningrad hospital in 1932.
Table 13.5.1
| ABO blood group | Antigens on red cells | Antibody in serum |
| O | None | Anti-A, anti-B |
| A | A | Anti-B |
| B | B | Anti-A |
| AB | A, B | None |
Transfusion of blood and blood products is now routine and vital to the practice of emergency medicine. As with any prescribed treatment, these products are associated with potential hazards as well as advantages. The hazards are more likely to be encountered with blood products used during emergencies. The blood products available in most Australian emergency departments (EDs) are packed red blood cells, platelets, fresh frozen plasma (FFP), cryoprecipitate, activated Factor VII, prothrombin complex concentrates and other factor concentrates.
In the Australian urban hospital setting, 50% of packed red cells are used for the treatment of anaemia, 22% pre- or perioperatively and 13% for abnormal, excessive or continued bleeding. Medical oncology uses 78% of all platelets. Forty-one per cent of all FFP is used to correct coagulopathy associated with surgery, 27% to correct coagulopathy in bleeding, 16% to reverse haemostatic disorders in patients having massive blood transfusion, 11.5% for reversal of warfarin effect and the remaining 4.5% for a number of miscellaneous conditions, including liver disease and disseminated intravascular coagulation (DIC).
In the ED setting, blood products are most often administered to patients with acute rather than chronic blood loss. In trauma centres, severely injured patients are the major consumers of blood products while, in non-trauma centres, patients with gastrointestinal haemorrhage account for the majority of transfusions. In these settings of acute blood loss, transfusion may be required rapidly and in large quantities. However, as short stay units are developed, non-time critical transfusions of blood products for other medical indications are increasingly the responsibility of ED staff.
Packed red blood cells
Packed red blood cells are produced from whole blood collections by removing most of the plasma by centrifugation and then resuspending the red cells in citrate-based anticoagulant-preservative solution to prolong storage time. Each unit of packed cells contains approximately 200 mL of red cells. Transfusion of one unit can be expected to raise the haematocrit by 3% and the haemoglobin by 10 g/L provided there is no ongoing blood loss.
Packed red cells are the blood product most commonly prescribed in the ED, the usual indication being the replacement of acute blood loss. Transfusion of packed red cells is indicated where the patient’s oxygen-carrying capacity is so impaired that control of bleeding alone, if indeed it can be readily achieved, is regarded as insufficient to prevent tissue hypoxia. In patients with primary haematological conditions, failure of erythropoiesis or a haemolysis, the indication for transfusion is usually the same as for haemorrhage: a severe reduction in oxygen-carrying capacity. In patients with assocatied complex multisystem failure, such as DIC or septic shock, red cell transfusion may be life saving by improving the oxygen debt in tissues (Table 13.5.2).
Table 13.5.2
Potential indications for red cell transfusion
Haemorrhage
Dilutional anaemia following severe burns
Iron-deficiency anaemia
Megaloblastic anaemia
Anaemia of chronic disorders
Chronic renal failure
Failure of erythropoiesis
Sickle cell disease
Septic shock
Disseminated intravascular coagulopathy
The indication of transfusion in haemorrhagic shock has been traditionally defined as persistent haemodynamic instability post-3 L of crystalloids. However, a further two patient factors must be considered. First is the concept of hypotensive resuscitation, which states that prior to definitive cessation of bleeding, relative hypotension may stabilize clots and reduce further bleeding. The clinican must therefore alter their thresholds of haemodynamic instability. Patient factors must be borne in mind, including the cardiovascular co-morbidities and the presence of head injuries. Secondly, both high volume crystalloid and blood transfusion have been associated with adverse outcomes, suggesting limitations of both, instead focusing resuscitation on the management of coagulopathy and early surgical management of haemorrhage.
Transfusion is not indicated when alternative haematinic therapy is deemed safe and appropriate. A moderately anaemic patient who is asymptomatic and not bleeding, with some reserve oxygen-carrying capacity, does not require blood transfusion. A haemoglobin of 7 g/dL is sometimes taken as the failsafe point in the decision whether to transfuse, although of course the patient’s unique circumstances need to be taken into account: treat the patient, not the number. It should be also considered that, in an acutely bleeding patient, the initial haemoglobin result, measured at a time of volume contraction, may be an inaccurate representation of circulating oxygen carrying capacity. The National Health and Medical Research Council together with the Australasian Society of Blood Transfusion have published transfusion guidelines for red blood cells and other products (Table 13.5.3).
Table 13.5.3
Guidelines for transfusion of blood components
| Indications | Considerations |
| Red blood cells | |
| Hb | |
| <70 g/L | Lower thresholds may be acceptable in patients without symptoms and/or where specific therapy is available |
| 70–100 g/L | Likely to be appropriate during surgery associated with major blood loss or if there are signs or symptoms of impaired oxygen transport |
| >80 g/L | May be appropriate to control anaemia-related symptoms in a patient on a chronic transfusion regimen or during marrow suppressive therapy |
| >100 g/L | Not likely to be appropriate unless there are specific indications |
| Platelets | |
| Bone marrow failure | At a platelet count of<10×109/L in the absence of risk factors and<20×109 in the presence of risk factors (e.g. fever, antibiotics, evidence of systemic haemostatic failure) |
| Surgery/invasive procedure | To maintain platelet count at>50×109/L. For surgical procedures with high risk of bleeding (e.g. ocular or neurosurgery), it may be appropriate to maintain at 100×109/L |
| Platelet function disorders | May be appropriate in inherited or acquired disorders, depending on clinical features and setting. In this situation, platelet count is not a reliable indicator |
| Bleeding | May be appropriate in any patient in whom thrombocytopaenia is considered a major contributory factor |
| Massive haemorrhage/transfusion | Use should be confined to patients with thrombocytopaenia and/or functional abnormalities who have significant bleeding from this cause. May be appropriate when the platelet count is<50×109/L (<100×109/L in the presence of diffuse microvascular bleeding) |
| Fresh frozen plasma | |
| Single factor deficiencies Warfarin effect | Use specific factors if available In the presence of life-threatening bleeding. Use in addition to vitamin-K-dependent concentrates |
| Acute DIC | Indicated where there is bleeding and abnormal coagulation. Not indicated for chronic DIC |
| TTP | Accepted treatment |
| Coagulation inhibitor deficiencies | May be appropriate in patients undergoing high-risk procedures |
| Following massive transfusion or cardiac bypass | Use specific factors if available May be appropriate in the presence of bleeding and abnormal coagulation |
| Liver disease | May be appropriate in the presence of bleeding and abnormal coagulation |
| Cryoprecipitate | |
| Fibrinogen deficiency | May be appropriate where there is clinical bleeding, an invasive procedure, trauma or DIC |
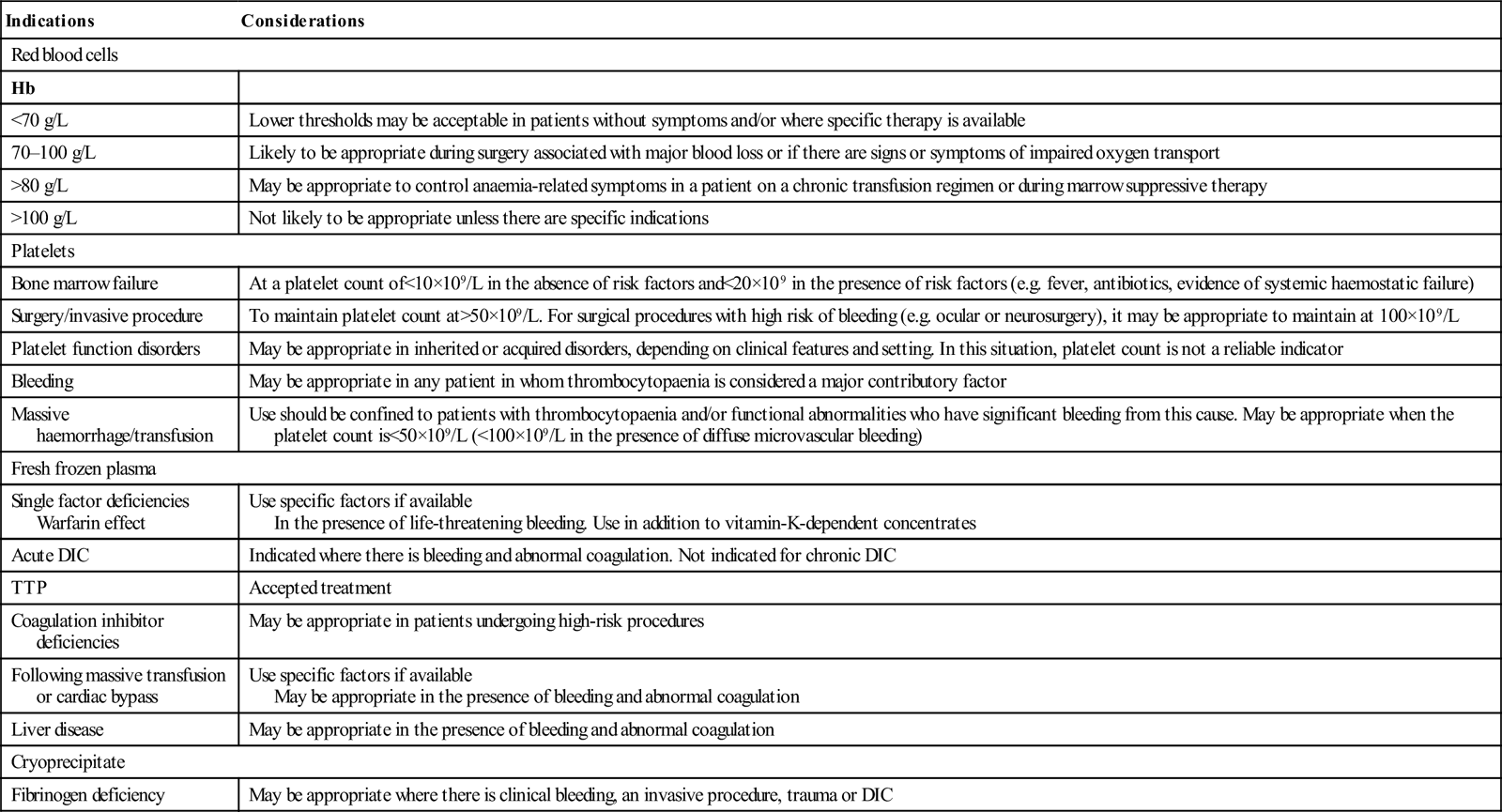
TTP: idiopathic thrombocytopaenia purpura; DIC: disseminated intravascular coagulation. Adapted from the National Health and Medical Research Council and Australasian Society Clinical Practice Guidelines on appropriate use of blood components. http://www.nhmrc.gov.au/publications/synopses/-files/cp82.pdf.
Prior to any blood product transfusion, informed consent should be sought, obtained and documented, except in emergent cases where the delays may result in substantial adverse effects. The following sections discuss the risks of red cell transfusion.
Effect of storage on red blood cells
Although it makes intuitive sense that blood loss should be replaced by blood products, there is evidence that the immediate observed benefit is from volume replacement rather than improved oxygen carriage. Red blood cells may not be fully functional until 2–6 h after transfusion because storage affects the oxygen-carrying capacity of blood. This is probably due to decreased intracellular 2,3-diphosphoglycerate (2,3-DPG), loss of red cell viability, decreased red cell deformability, relative acidosis and potassium leakage.
Storage reduces 2,3-DPG levels, leading to a leftward shift of the oxyhaemoglobin dissociation curve and increased affinity of oxygen binding. The transfused red cell does regenerate 2,3-DPG to normal levels but this can take 6–24 h post transfusion. With increasing age of stored red cells, levels of 2,3-DPG progressively fall such that by 5–6 weeks the level is 10% of normal. It is still uncertain whether this abnormality is physiologically important, even in critically ill patients. In addition, hypocalcaemia, cell lysis, release of free haemoglobin, changes in nitric oxide levels, alterations in pH and increases in lipids, complement and cytokines are other effects of red cell storage. These changes are accompanied by increased membrane fragility, which can compromise microcirculatory flow and lead to increased red cell–endothelial cell interaction and inflammatory cytokine release. Such changes could explain recent findings associating age of red blood cells with adverse outcomes and may be particularly disadvantageous to critically ill patients with a higher mortality risk.
When red cells are transfused, some of the cells are removed from the circulation within a few hours, with the rest surviving normally; as the storage time increases to 42 days, more cells are removed immediately after transfusion. This loss of viability is highly dependent upon the anticoagulant-preservative solution used.
Potassium gradually leaks out of stored red cells and this raises the plasma potassium by approximately 1 mEq/L per day. Citrate toxicity results when the citrate in the transfused blood begins to bind calcium in the patient’s body, resulting in hypocalcaemia. Clinically significant hypocalcaemia does not usually occur unless the rate of transfusion exceeds 1 unit every 5 minutes or so. Citrate metabolism is primarily hepatic – so hepatic disease or dysfunction can cause this effect to be more pronounced.
Choice of red cell product
The choice of red cell product is determined by time and safety considerations. O-negative red cells, the universal donor group, are readily available in most major hospitals. Supplies of O-negative blood are limited and the product should be used with care. The transfusion of O-negative blood is generally reserved for transfusion immediately during patient reception and the initial stages of resuscitation, switching to cross-matched blood as soon as available. It is preferable that blood be collected prior to transfusion so as to characterize the recipient’s blood group serology. Premenopausal female patients should be given group O Rhesus negative, Kell negative blood in an emergency situation in order to avoid sensitization and possibility of haemolytic disease of newborn in subsequent pregnancies. Male patients, however, can be transfused either Rhesus positive or negative blood. The incidence of adverse reaction using this type of blood is approximately 3%. By contrast, the provision of group-specific blood requires matching a blood sample to the major (ABO) and Rhesus D compatibility groups only. Group-specific blood can be available for transfusion within 35 min depending on the logistic support and staffing levels within the haematology laboratory. It has an incidence of adverse reaction similar to O-negative blood. As O-negative blood is usually in short supply, it is preferable where possible to infuse group-specific blood. A more comprehensive cross-match where there are no atypical antibodies identified in the initial screening can take 30 min or more and the incidence of adverse transfusion reaction is reduced to 0.01%.
Precautions when cross-matching and transfusing blood
Although most patients do not require transfusion in the ED, it is often appropriate to ‘group and hold’ or cross-match the patient while in the department. Many hospitals have written protocols detailing the anticipated requirements for a given surgical procedure. Documentation should be meticulous. It should be mandated that the person drawing the blood for cross-matching should also fill in and sign the laboratory request form. Most severe incompatibility reactions to blood transfusion result not from exposure to unusual antigens but from administrative errors. Any systematic change in documentation protocols, for example, the adoption of an electronic record, needs to be accompanied by obsessive risk management strategies.
The checking of the compatibility details of blood to be transfused must be meticulous. Blood products should not be left lying around workbenches. Universal precautions must be observed by staff setting up transfusions. Rapid or large transfusions should be via a blood warmer. Blood is transfused intravenously through sterile giving sets containing 170 μm filters. Alternative routes (arterial, intraperitoneal or intraosseous) are only used in exceptional circumstances. Lines for transfusion should be dedicated lines; drugs and other additives should be administered at separate sites. Normal saline is compatible with all blood components.
Pulse, blood pressure and temperature are measured at regular intervals and particular attention is paid to the patient during the first 25 min of the transfusion. The transfusion is started slowly. The rate at which it continues depends on clinical urgency. As a general rule, the faster the anaemia has developed the more rapidly it needs to be corrected. Rapid infusion techniques may be indicated in patients who appear to be exsanguinating, but over-rapid infusion may precipitate cardiac failure in the elderly. Hypothermia may be a problem if a blood warmer is not used.
Adverse reactions to transfusion
The principal adverse reactions to blood transfusion are listed in Table 13.5.4. Serious adverse reactions are relatively rare (Table 13.5.5), although some are more likely to occur when blood is administered urgently.
Table 13.5.4
Adverse effects of blood transfusion
| Immunological transfusion reactions | Transmission of infection |
| Immediate | Bacterial |
| Febrile non-haemolytic reactions | Brucella |
| Acute haemolytic transfusion reactions | Pseudomonas |
| Allergic reactions and anaphylaxis | Salmonella |
| Transfusion-related acute lung injury | Treponema pallidum |
| Delayed | Parasites |
| Delayed haemolytic transfusion reactions | Babesia |
| Alloimmunization | Plasmodium |
| Transfusion-associated graft versus host disease | Toxoplasma |
| Hypothermia | Trypanosoma |
| Dilutional coagulopathy | Viruses |
| Volume overload | Cytomegalovirus |
| Hepatitis B and delta agent | |
| Hepatitis A | |
| Hepatitis C | |
| Other hepatitis ‘non-A, non-B’ | |
| HIV-1 and HIV-2 | |
| HTLV-1 and HTLV-2 | |
| Parvovirus |
HIV: human immunodeficiency virus; HTLV: human T-cell lymphotropic virus.
Table 13.5.5
Incidence of adverse transfusion reactions (per unit packed red cells transfused)
| Adverse transfusion reaction | Incidence | Mortality |
| Bacterial sepsis | 1 in 40 000–500 000 | 1 in 4–8 million |
| Acute haemolytic reaction | 1 in 12 000–38 000 | 1 in 600 000–1.5 million |
| Delayed haemolytic reaction | 1 in 1000–12 000 | 1 in 2.5 million |
| Anaphylaxis | 1 in 20 000–50 000 | |
| Transfusion-related acute lung injury | 1 in 5000–100 000 | 1 in 5 million |
| Fluid overload | 1 in 100–700 | |
| Transfusion-associated graft versus host disease | Rare | 90% fatality |
Adapted from Australian Red Cross website: http://www.transfusion.com.au/adverse_events (Accessed April 2013).
Immunological transfusion reactions
Immunological transfusion reactions may be immediate or delayed in onset.
Immediate
Febrile non-haemolytic reactions
The most common transfusion reaction is a febrile, non-haemolytic transfusion reaction (FNHTR), which is defined as an increase in temperature of 1°C or more over baseline during a transfusion. It manifests as fever and occasionally shortness of breath 1–6 h after transfusion. FNHTRs are benign, but their presentation is very similar to acute haemolytic transfusion reaction and infection, which have a higher rate of mortality and morbidity, mandating early clinical review to exclude more serious complications.
Acute haemolytic reactions
Acute haemolytic transfusion reactions (ATHRs) result from the rapid destruction of donor red cells by preformed recipient antibodies and are a medical emergency. They are usually due to ABO incompatibility and most often the result of clerical or procedural errors. ABO and Rhesus compatibility between donors and recipients is presented in Table 13.5.6. Some acquired alloantibodies, such as anti-Rh or anti-Jka, are occasionally implicated, but AHTRs more typically occur when a group O recipient is transfused with non-group O red cells. This may lead to DIC, shock and acute tubular necrosis precipitating acute renal failure. These reactions usually manifest with fever and rigors, lumbar pain, crushing chest pain, tachycardia, hypotension and haemoglobinaemia with subsequent haemoglobinuria. The symptoms usually develop within the first 30 min of transfusion.
Table 13.5.6
ABO and Rhesus compatibility between donors and recipients
| Donors | O+ | A+ | B+ | AB+ | O− | A− | B− | AB− | |
| Recipients | |||||||||
| O+ | C | C | |||||||
| A+ | C | C | C | C | |||||
| B+ | C | C | C | C | |||||
| AB+ | C | C | C | C | C | C | C | C | |
| O− | C | ||||||||
| A− | C | C | |||||||
| B− | C | C | |||||||
| AB− | C | C | C | C |
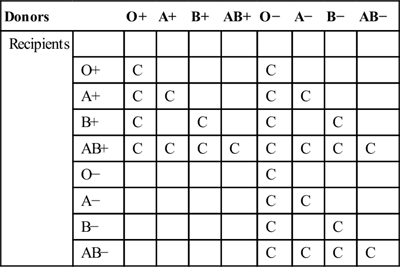
Anaphylactoid transfusion reactions
Anaphylactoid reactions usually begin within 1–45 min of the start of transfusion of blood products but less severe reactions can be delayed up to 2–3 h. Generally, a shorter time between commencement of the transfusion and onset of symptoms is associated with a more severe reaction. These reactions are manifested by rapid onset of shock, hypotension, angio-oedema and respiratory distress. They are almost always due to the presence of class-specific IgG, anti-IgA antibodies in patients who are IgA deficient. Selective IgA deficiency is not uncommon, occurring in about 1 in 300–500 people. The incidence of anaphylactic transfusion reactions can be reduced by the use of washed products (e.g. washed red cells) and by premedicating the patient with antipyretics and antihistamines.
Treatment of an anaphylactoid transfusion reaction consists of immediate cessation of transfusion and standard treatment of anaphylaxis including, oxygen fluids and adrenaline (see Chapter 28.7).
Transfusion-related acute lung injury
Transfusion-related acute lung injury (TRALI) is a syndrome characterized by acute respiratory distress following transfusion. All plasma-containing blood products have been implicated including rare reports involving IVIG and cryoprecipitate. It is a rare complication of allogeneic blood transfusion but the incidence has not been well established due to difficulty in defining the syndrome and variable reporting mechanisms worldwide. Symptoms of TRALI typically develop during, or within 6 hours of a transfusion. Patients present with the rapid onset of dyspnoea and tachypnoea. There may be associated fever, cyanosis and hypotension. Clinical exam reveals respiratory distress and pulmonary crackles may be present with no signs of congestive heart failure or volume overload. Chest X-ray (CXR) shows evidence of bilateral pulmonary oedema unassociated with heart failure (non-cardiogenic pulmonary oedema), with bilateral patchy infiltrates, which may rapidly progress to complete ‘white out’ indistinguishable from acute respiratory distress syndrome. The central venous pressure is normal, which helps distinguish the condition from transfusion-associated circulatory overload. Treatment is supportive. There is no role for diuretics or corticosteroids The blood bank needs to be notified as reporting of TRALI allows better understanding of the true incidence of this reaction, in addition to its clinical course and associated mortality
Delayed
Delayed haemolytic transfusion reaction
These reactions occur in patients who have developed antibodies from previous transfusions or pregnancy but, at the time of pretransfusion testing, the antibody in question is too weak to be detected by standard procedures. Subsequent transfusion with red cells having the corresponding antigen results in an anamnestic antibody response and haemolysis of transfused red cells. These delayed reactions are seen generally within 2–10 days after transfusion. Haemolysis is usually extravascular, gradual and less severe than with acute reactions, but rapid haemolysis can occur. A falling haematocrit, slight fever, mild increase in serum unconjugated bilirubin and spherocytosis on the blood smear may be noted.
Treatment of a delayed haemolytic transfusion reaction is usually not required unless anaemia is severe enough to require treatment. However, future transfusions containing the implicated red cell antigen need to be avoided. Alternatives to transfusion should be explored whenever possible.
Red cell alloimmunization
When antibodies are formed against foreign antigens from one’s own species, the process is termed alloimmunization and the antibodies are called alloantibodies (as opposed to forming autoantibodies to one’s own antigens or forming xenoantibodies to antigens from a foreign species). Transfused (non-leucocyte depleted) red cells and platelets contain leucocytes to which antibodies can be made. This may cause patients to become resistant to subsequent platelet transfusions. Approximately 50% of patients undergoing multiple blood transfusions become alloimmunized and are refractory to further platelet transfusions. Refractory patients require platelets matched to their specific platelet/human leucocyte antigen (HLA) type. Patients receiving leucocyte reduced blood products are at a much lower risk for refractoriness to platelet transfusion than are recipients of non-leucocyte reduced blood products. Although debate exists about its merits, for selected high-risk patients with transfusion-dependent diseases (e.g. sickle cell anaemia, thalassaemia, etc.), some transfusion services phenotype patients and provide phenotypically matched donor RBC, even to those patients without alloantibodies (i.e. to prevent the formation of antibodies). There is general agreement that this is useful for Rhesus and Kell blood group antigens in these groups of patients, but debate exists about its merits for more extensive phenotyping.
Transfusion-associated graft-versus-host disease (TA-GVHD)
Transfusion-associated graft-versus-host disease results from transfusion of viable T lymphocytes which proliferate and damage the recipient’s tissue, particularly skin, gastrointestinal tract, liver, spleen and bone marrow. This is a rare and almost always fatal complication of transfusion. Clinical manifestations typically develop 10–14 days following transfusion and consist of fever, erythematous skin rash, pancytopaenia, diarrhoea and abnormal liver function. High-risk patients for this complication include bone marrow transplant recipients, patients receiving granulocyte transfusions, transfusions from a biologically related donor (directed donation), the fetus (intrauterine transfusion), exchange transfusion, patients with Hodgkin lymphoma and patients with congenital cellular immune deficiency.
The investigation begins with the confirmation of the presence of GVHD. This is a pathological diagnosis requiring a skin or intestinal biopsy. Currently, the only method of preventing TA-GVHD is to gamma irradiate cellular components at risk of causing TA-GVHD or destined for at-risk recipients. Current techniques to leucoreduce cellular blood components are not adequate to prevent TA-GVHD
Transmission of infection
In Australia, blood is tested for ABO and Rh (D) blood groups, red cell antibodies and the following infections:
In terms of viral safety, Australia has one of the safest blood supplies in the world (Table 13.5.7).
Table 13.5.7
Risks of transfusion transmitted infection (per unit tested blood transfused)
| Infection | Residual risk |
| CMV | 1 in 127 000 |
| Hepatitis B | Approximately 1 in 660 000 |
| Syphilis | Considerably less than 1 in a million |
| Hepatitis C | <1 in 10 million |
| HIV | <1 in 10 million |
| HTLV I and II | <1 in 10 million |
| Variant CJD | Possible and cannot be excluded |
Adapted from Australian Red Cross website: http://www.transfusion.com.au/adverse_events (Accessed April 2013).
Hypothermia
Red blood cells are stored at 4°C. Rapid infusion of large volumes of stored blood can contribute to hypothermia. Blood warmers should be used during massive blood transfusion. In addition, other intravenous fluids should be warmed and other measures instituted to maintain patient body temperature (see Chapter 28.2).
Dilutional coagulopathies
Clinically significant depletion of coagulation proteins and platelets is a complication of massive transfusion, secondary to dilution and the consumptive coagulopathy of trauma. Stored red cells are deficient in platelets and clotting factors and transfusion of large amounts can complicate bleeding when not accompanied by assessment and correction of coagulation disturbances. Coagulation parameters including the prothrobin time (PT), activated partial thromboplastin time (APTT), platelet count and fibrinogen level should be monitored and corrected if deficiencies occur in the presence of abnormal bleeding. In actively bleeding patients, however, these parameters may not provide an accurate estimate of clot strength and results are often delayed by 30–60 minutes. Thromboelastography has the advantage of providing real-time assessment of clot strength and should be utilized where available.
Volume overload
This complication occurs when excessive volume of fluid is administered. Pulmonary oedema is a particular risk in the elderly, in infants and in patients with chronic severe anaemia where the red cell mass is decreased but the blood volume is normal. Abdominal compartment syndrome may result in bowel ischaemia and should be watched for.
Management of transfusion reactions
The first action to be taken in the management of any suspected transfusion reaction is to stop the transfusion immediately and assess the patient. The bag containing the transfused cells, along with all attached labels, should not be discarded so as to allow repeat typing and cross-matching of this unit by the blood bank. Management then proceeds as follows:
The laboratory which tested and issued the blood should be alerted immediately and a search for any clerical error instituted. Every hospital has a protocol for evaluating transfusion reactions, which should be rigorously followed. The haematology unit should notify the local blood bank, who have a haematologist on-call at all times and are responsible for the recall of any other implicated components from the same donor in the case of suspected infection or transfusion-related acute lung injury. If there is any suggestion (e.g. clerical mistake, hypotension, pink plasma or urine) that an AHTR is possible, oxygen should be applied to the patient and fluid resuscitation with saline to maintain a urine output of 2–3 mL/kg/h, in an attempt to prevent acute oliguric renal failure. A vasopressor, such as adrenaline, may be required. If massive intravascular haemolysis has already occurred, hyperkalaemia is likely and cardiac monitoring and acute haemodialysis may be required.
Platelets
Platelets are one of the main cellular components of blood and are central to haemostasis. Platelet products commonly available for transfusion are obtained by apheresis from a single donor or from donated blood using buff-coat or platelet-rich plasma techniques. Modifications to reduce the risk of viral transmission and prevent graft-versus-host disease include leucocyte reduction, irradiation, plasma depletion and the use of platelet additive solutions.
Platelets are transfused to prevent or treat haemorrhage in patients with thrombocytopaenia or defects in platelet function. Specific indications for platelet transfusion are shown in Table 13.5.3. Use of platelets is not generally considered appropriate in the treatment of immune-mediated platelet destruction, thrombotic thrombocytopaenic purpura, haemolytic uraemic syndrome or drug-induced or cardiac bypass thrombocytopaenia without haemorrhage.
In general, one platelet unit will raise the platelet count about 5–10×109/L in an average adult. Depending on the method of manufacture, the volume of each unit of platelets varies from 100 to 160 mL, with a storage life of about 5 days at 20–24°C.
Compatibility testing is not necessary in routine platelet transfusion, although platelet components should preferably be ABO and Rh type compatible with the recipient. ABO-incompatible platelets may be used if ABO compatible platelets are not available. The usual dose in an adult patient is 4 units, which is equivalent to 1 unit of apheresis platelets or 1 unit of pooled platelets.
Fresh frozen plasma
FFP is prepared from anticoagulated blood by separating the plasma from the blood cells through centrifugation of whole blood or apheresis. It is stored frozen until used. It contains all coagulation factors including small amounts of Factor V and approximately 200 units of Factor VIII. Fresh frozen plasma can be stored at below−25 °C for up to 12 months. Indications for use of FFP are shown in Table 13.5.3.
The appropriate dose depends on the clinical indication, patient size and results of laboratory tests. A general guide is 10–15 mL/kg per dose but, in some situations, dosages greater than this may be required (e.g. dilutional coagulopathy in the context of massive transfusion). On average 1 mL of FFP/kg patient weight will raise most coagulation factors by 1%, therefore a dose of 10–15 mL/kg would be expected to increase levels by 10–15%. Compatibility testing is not required, however, ABO compatible plasma should be used wherever possible. Group AB plasma can be used for all patients in an emergency.
Cryoprecipitate
Cryoprecipitate is prepared by thawing FFP to between 1 and 6°C and recovering the precipitable protein fraction. It contains most of the Factor VIII, fibrinogen, Factor XIII, von Willebrand factor and fibronectin from the FFP. It may be stored for up to 12 months at−25°C or below. Once thawed it must be used immediately or stored at 2–6°C for up to 24 h. Cryoprecipitate is indicated in fibrinogen deficiency with clinical bleeding or prior to an invasive procedure and in DIC. Cryoprecipitate is transfused to keep fibrinogen levels above 1.0 g/L in the acutely bleeding patient. Compatibility tests before transfusion are not necessary. It is preferable to use an ABO group compatible with the recipient’s red cells, however, ABO incompatible can be used with caution. Up to 4 units/10 kg body weight may be required to raise the fibrinogen concentration by approximately 0.5 g/L in the absence of continued haemorrhage.
The use of cryoprecipitate is not generally considered appropriate in the treatment of haemophilia, von Willebrand’s disease or deficiencies of Factor XIII or fibronectin, unless alternative therapies are unavailable.
Refusal of blood and blood product transfusion
Patients with certain religious beliefs may refuse blood and blood product transfusion, e.g. Jehovah’s witnesses. Healthy volunteers can tolerate Hb levels of 50 g/L without evidence of end-organ hypoxia. However, it is estimated that the median Hb concentration associated with mortality is about 25 g/L. The patient’s wishes must be rigorously protected and blood products avoided. Several strategies may be used to manage the anaemia. Sedation should be instituted to minimize metabolic demand. A ventilation cycle of 2 hours of 90% FiO2, followed by 2 hours of 90% SpO2 and then 20 hours of 95% SpO2 may be employed to maximize oxygen delivery while minimizing shunt from absorption atelectasis and to promote erythropoiesis. Recombinant erythropoietin (36 000 units daily), folic acid (5 mg daily), vitamin B12 (1 mg daily) and iron infusions are options to maximize haematopoiesis. In female patients, where applicable, menses should be inhibited with progesterone. Blood testing should be rationalized and performed using paediatric-sized samples.
A few case studies on the use of synthetic haemoglobin have been published. HBOC-201 is the commonest product used and is a modified lactated Ringer’s solution containing 130 g/L of polymerized Hb of bovine origin. It is compatible with all blood types, stable for 3 years when stored at 2–30°C and stable for 2 years when stored at 40°C. When fully saturated, HBOC-201 has the same oxygen-carrying capacity as whole blood with the same Hb concentration. The partial pressure of oxygen at which HBOC-201 is 50% saturated (40 mmHg) is higher than that for cellular Hb (27 mmHg), which facilitates oxygen delivery to tissues. The half-life of HBOC-201 is approximately 20 hours. Polymerization of the Hb reduces its glomerular diffusion and nephrotoxicity. The use of synthetic haemoglobin remains experimental at this stage and outcomes of further trials currently underway aim to determine the efficacy and safety profile of such products.
Controversies
[/level-membership-for-emergency-medicine-category][not-level-membership-for-emergency-medicine-category]
Haematology Emergencies
Edited by Mark Little
13.1 Anaemia
Lindsay Murray
Introduction
Anaemia is a condition in which the absolute number of red cells in the circulation is abnormally low. The diagnosis is usually made on the basis of the full blood count (FBC). This, together with the blood film, offers qualitative as well as quantitative data on the blood components and a set of normal values is shown in Table 13.1.1.
Table 13.1.1
Full blood count: normal parameters
| Haemoglobin (Hb) | |
| Males | 135–180 g/L |
| Females | 115–165 g/L |
| Red blood cell count | |
| Males | 4500–6500×109/L |
| Females | 3900–5600×109/L |
| Haematocrit | |
| Males | 42–54% |
| Females | 37–47% |
| Other values | |
| MCH | 27–32 pg |
| MCHC | 32–36 g/dL |
| MCV | 76–98 fL |
| Reticulocytes | 0.2–2% |
| White blood cells | 4–11×109/L |
| Neutrophils | 1.8–8×109/L |
| Eosinophils | 0–0.6×109/L |
| Basophils | 0–0.2×109/L |
| Lymphocytes | 1–5×109/L |
| Monocytes | 0–0.8×109/L |
| Platelets | 150–400×109/L |
The average lifespan of a normal red blood cell in the circulation is from 100 to 120 days. Aged red cells are removed by the reticuloendothelial system but, under normal conditions, are replaced by the marrow such that a dynamic equilibrium is maintained. Anaemia develops when red cell loss exceeds red cell production. It follows that the anaemic patient is doing at least one of three things: not producing enough red cells, destroying them too quickly or bleeding.
The overriding functional importance of the red cell resides in its ability to transport oxygen, bound to the haemoglobin molecule, from the lungs to the tissues. Functionally, anaemia may be regarded as an impairment in the supply of oxygen to the tissues and the adverse effects of anaemia, from whatever cause, are a consequence of the resultant tissue hypoxia. Anaemia is not a diagnosis: rather, it is a clinical or a laboratory finding that should prompt the search for an underlying cause (Table 13.1.2).
Table 13.1.2
Haemorrhage
Traumatic
Non-traumatic
Acute
Chronic
Megaloblastic anaemia
Vitamin B12 deficiency
Folate deficiency
Aplastic anaemia
Pure red cell aplasia
Myelodysplastic syndromes
Invasive marrow diseases
Chronic renal failure
Decreased RBC survival (haemolytic anaemia)
Congenital
Spherocytosis
Elliptocytosis
Glucose-6-phosphate-dehydrogenase deficiency
Pyruvate kinase deficiency
Haemoglobinopathies: sickle cell diseases
Acquired autoimmune haemolytic anaemia, warm
Acquired autoimmune haemolytic anaemia, cold
Microangiopathic haemolytic anaemias
RBC mechanical trauma
Infections
Paroxysmal nocturnal haemoglobinuria
Anaemia secondary to haemorrhage
Aetiology
By far the most common cause of severe anaemia encountered in the emergency department (ED) is haemorrhage. Therefore, the assessment of the anaemic patient is often chiefly concerned with the search for a site of blood loss. The most common causes of haemorrhage are outlined in Table 13.1.3. However, the emergency physician must remain alert to the possibility that the patient is not bleeding but manifesting a rarer pathological condition.
Table 13.1.3
Common causes of haemorrhage in the emergency department
Trauma
Blunt trauma to mediastinum
Pulmonary contusions/haemopneumothorax
Intraperitoneal injury
Retroperitoneal injury
Pelvic disruption
Long bone injury
Open wounds: inadequate first aid
Non-trauma
Gastrointestinal haemorrhage
Oesophageal varices
Peptic ulcer
Gastritis/Mallory–Weiss
Colonic/rectal bleeding
Obstetric/gynaecological bleeding
Ruptured ectopic pregnancy
Menorrhagia
Threatened miscarriage
Antepartum haemorrhage
Postpartum haemorrhage
Other
Epistaxis
Postoperative
Secondary to bleeding diathesis
Clinical features
While it may be obvious on history and examination that a patient is bleeding, occasionally, the source of blood loss is occult and the extent of loss underestimated.
In the context of trauma, the history often gives clear pointers to both sites and extent of blood loss. Consideration of the mechanism of injury may allow anticipation of occult pelvic, intraperitoneal or retroperitoneal bleeding. Intracranial bleeding is never an explanation for hypovolaemic shock in an adult. In the context of non-trauma, it is essential to obtain an obstetric and gynaecological history in women of childbearing age. The remainder of the formal history may supply information essential in determining the aetiology of anaemia. The past medical history may point to a known haematological abnormality or to a chronic disease process. A drug and allergy history is always relevant. Many drugs cause marrow suppression, haemolytic anaemia and bleeding. The family history points to hereditary disease; the social history may alert the clinician to an unusual occupational exposure in the patient’s past or, more likely, to recreational activities liable to exacerbate an ongoing disease process. The systems review is particularly relevant to the consultation with middle-aged or elderly male patients, who must be asked about symptoms of altered bowel habit and weight loss.
The symptomatology of anaemia proceeds from vague complaints of tiredness, lethargy and impaired performance through to more sharply defined entities, such as shortness of breath on exertion, giddiness, restlessness, apprehension, confusion and collapse. Co-morbid conditions may be exacerbated (the dyspnoea of chronic obstructive airway disease) and occult pathologies unmasked (exertional angina in ischaemic heart disease).
Anaemia of insidious onset is generally better tolerated than that of rapid onset because of cardiovascular and other compensatory mechanisms. Acute loss of 40% of the blood volume may result in collapse whereas, in certain developing countries, it is not rare for patients with haemoglobin concentrations 10% of normal to be ambulant. Trauma superimposed on an already established anaemia can lead to rapid decompensation.
The cardinal sign of anaemia is pallor. This can be seen in the skin, the lips, the mucous membranes and the conjunctival reflections. Yet, not all anaemic patients are pallid and not all patients with a pale complexion are anaemic. Patients who have suffered an acute haemorrhage may show evidence of hypovolaemia: tachycardia, hypotension, cold peripheries and sluggish capillary refill. The detection of postural hypotension is an important pointer towards occult blood loss. Conversely, patients with anaemia of insidious onset are not hypovolaemic and may manifest high-output cardiac failure as a physiological response to hypoxia.
Other features of the physical examination may provide clues to the aetiology of anaemia. The glossitis, angular stomatitis, koilonychia and oesophageal web of iron- deficiency anaemia are uncommon findings. Bone tenderness, lymphadenopathy, hepatomegaly and splenomegaly may point to an underlying haematological abnormality. The rectal and gynaecological examinations can sometimes be diagnostic.
Clinical investigations
The full blood count often reveals an anaemia that has not been clinically suspected and that must be interpreted in the light of the history and examination. If the anaemia is mild it may be a chance finding with little relevance to the patient’s presenting complaint, but such a finding should never be ignored. At the very least a follow-up blood count should be arranged.
Anaemic patients have a low red cell count, a low haematocrit and a low haemoglobin, but some caveats need to be borne in mind:
patients who are bleeding acutely may initially have a normal FBC
normal or high haematocrits may reflect haemoconcentration
mixed pictures can be difficult to interpret, e.g. that of a polycythaemic patient who is bleeding.
Red cell morphology, particularly the mean corpuscular volume (MCV), can help elucidate the cause of anaemia. The finding of a pancytopaenia suggests a problem in haematopoiesis, rather than haemolysis or blood loss. In women of childbearing age, assay of blood or urine β-HCG is important.
Treatment
The principles of management of haemorrhage are as follows:
The indications for red cell transfusion are discussed in Chapter 13.5. The faster the onset of the anaemia, the greater the need for urgent replacement. Patients who are tolerating their anaemia may require no more than an appropriate diet with or without the addition of haematinics. Elderly patients with severe bleeding often need red cells urgently. Excessive administration of colloid and/or crystalloid precipitates left ventricular failure and it can then be difficult to administer red cells.
Chronic haemorrhage
The finding of a hypochromic microcytic anaemia on blood film is usually indicative of iron deficiency and, in the absence of an overt history of bleeding, should prompt the search for occult blood loss. Iron deficiency anaemia may be due to malnutrition, but inadequate dietary intake of iron is not usually the sole cause of anaemia in developed countries: much more commonly it is the result of chronic blood loss from the gastrointestinal (GI) tract, the uterus or the renal tract. More unusual causes are haemoptysis and recurrent epistaxes.
Patients present with insidious and rather vague symptoms. They may be unaware that they are bleeding and will probably show none of the trophic skin, nail and mucosal changes of iron deficiency. The automated cell count, in addition to showing a hypochromic, microcytic picture, may also show a raised red cell distribution width, which reflects anisocytosis on the blood film.
Iron studies may confirm the diagnosis of iron deficiency without pointing to the underlying cause. Serum iron and ferritin are low and total iron-binding capacity is high.
Disposition
If the source of blood loss is obvious, for example heavy menstrual bleeding, then appropriate referral may be all that is indicated. If the source is not obvious, particularly in older patients, then sequential investigation of the GI tract and the renal tract may be indicated. Decisions to admit or discharge these patients depend on the red cell reserves, the patient’s cardiorespiratory status, home circumstances and the likelihood of compliance with follow up.
The anaemia itself can be corrected with oral iron supplements: 200 mg of ferrous sulphate three times daily is an appropriate regimen, although single daily doses are often more acceptable to the patient and have fewer GI side effects.
Anaemia secondary to decreased red cell production
Megaloblastic anaemia
The finding of a raised MCV is common in the presence or absence of anaemia. Alcohol abuse is a frequent underlying cause and other causes are listed in Table 13.1.4. MCVs greater than 115 fL are usually due to megaloblastic anaemia which, in turn, is usually due to either vitamin B12 or folate deficiency. Vitamin B12 and folate are essential to DNA synthesis in all cells. Deficiencies manifest principally in red cell production because of the sheer number of red cells that are produced. B12 deficiency is usually the result of a malabsorption syndrome, whereas folate deficiency is of dietary origin. Tetrahydrofolate is a co-factor in DNA synthesis and, in turn, the formation of tetrahydrofolate from its methylated precursor is B12-dependent. Unabated cytoplasmic production of RNA in the context of impaired DNA synthesis appears to produce the enlarged nucleus and abundant cytoplasm of the megaloblast. These cells, when released to the periphery, have poor function and poor survival.
Table 13.1.4
Some causes of a raised mass cell volume
Alcohol
Drugs
Hypothyroidism
Liver disease
Megaloblastic anaemias (B12 and folate deficiency)
Myelodysplasia
Pregnancy
Reticulocytosis
B12 deficiency is an autoimmune disorder in which autoantibodies to gastric parietal cells and the B12 transport factor (intrinsic factor) interfere with B12 absorption in the terminal ileum. Patients have achlorhydria, mucosal atrophy (a painful smooth tongue) and, sometimes, evidence of other autoimmune disorders, such as vitiligo, thyroid disease and Addison’s disease. This is so-called ‘pernicious anaemia’.
A rare, but important, manifestation of this disease is ‘subacute combined degeneration of the spinal cord’. Demyelination of the posterior and lateral columns of the spinal cord manifests as a peripheral neuropathy and an abnormal gait. The central nervous system abnormalities worsen and become irreversible in the absence of B12 supplementation. Treatment of B12 deficient patients with folate alone may accelerate the onset of this condition.
Undiagnosed untreated pernicious anaemia is not a common finding in the ED, but the laboratory finding of anaemia and megaloblastosis should prompt haematological consultation. The investigative work-up, which includes B12 and red cell folate levels, autoantibodies to parietal cells and intrinsic factor, a marrow aspirate, and Schilling’s test of B12 absorption, may well necessitate hospital admission.
The work-up for folate deficiency is similar to that for B12. Occasionally, patients require investigation for a malabsorption syndrome (tropical sprue, coeliac disease), which includes jejunal biopsy. Folate deficiency is common in pregnancy because of the large folate requirements of the growing fetus. It can be difficult to diagnose because of the maternal physiological expansion of plasma volume and also of red cell mass, but diagnosis and treatment with oral folate supplements are important because of the risk of associated neural tube defects.
Both B12 and folate deficiency are usually manifestations of chronic disease processes. Rarely, an acute megaloblastic anaemia and pancytopaenia can develop over the course of days and nitrous oxide therapy has been identified as a principal cause of this condition.
Anaemia of chronic disorders
Patients with chronic infective, malignant or connective tissue disorders can develop a mild-to-moderate normochromic normocytic anaemia. Evidence of bleeding or haemolysis is absent and there is no response to haematinic therapy. The pathophysiology of this anaemia is complex and probably involves both decreased red cell production and survival. Possible underlying mechanisms include reticuloendothelial overactivity in chronic inflammation and defects in iron metabolism mediated by a variety of acute-phase reactants and cytokines, such as interleukin-1, tumour necrosis factor and interferon γ, which impair renal erythropoietin production and function.
Anaemia of chronic disorders (ACD) is generally not so severe as to warrant emergency therapy. The importance of ACD in the ED lies in its recognition as a pointer towards an underlying chronic process. Difficulties can arise in distinguishing ACD from iron deficiency and the two conditions may coexist – in rheumatoid arthritis, for example. Iron studies generally elucidate the nature of the anaemia. In iron deficiency, iron and ferritin are low and total iron binding is high, whereas in ACD iron and total iron binding are low and ferritin is normal or high.
Other causes of decreased red cell production
Bone marrow failure is rarely encountered in emergency medicine practice. The physician must be alert to the unusual, insidious or sinister presentation and be particularly attuned to the triad of decreased tissue oxygenation, immunocompromise and a bleeding diathesis that may herald a pancytopaenia. An FBC may dictate the need for haematological consultation, hospital admission and further investigation.
Among the entities to be considered are the aplastic anaemias, characterized by a pancytopaenia secondary to failure of pluripotent myeloid stem cells. Half of cases are idiopathic, but important aetiologies are infections (e.g. non-A, non-B hepatitis), inherited diseases (e.g. Fanconi’s anaemia), irradiation, therapeutic or otherwise and, most important in the emergency setting, drugs. Drugs that have been implicated in the development of aplastic anaemia include, in addition to antimetabolites and alkylating agents, chloramphenicol, chlorpromazine and streptomycin.
Characteristic of patients with a primary marrow failure is the absence of splenomegaly and the absence of a reticulocyte response. There is a correlation between prognosis and the severity of the pancytopaenia. Platelet counts less than 20×109/L and neutrophil counts less than 500/mL equate to severe disease. Depending on the severity of the accompanying anaemia, patients may require red cell and sometimes platelet transfusion in the ED, as well as broad-spectrum antibiotic cover. It is imperative to stop all medications that might be causing the marrow failure. Other forms of marrow failure include pure red cell aplasia, where marrow red cell precursors are absent or diminished. This can be a complication of haemolytic states in which a viral insult leads to an aplastic crisis (see haemolytic anaemias).
The myelodysplastic syndromes are a group of disorders primarily affecting the elderly. In these states there is no reduction in marrow cellularity but the mature red cells, granulocytes and platelets generated from an abnormal clone of stem cells are disordered and dysfunctional. There is peripheral pancytopaenia. These disorders are classified according to observed cellular morphology (Table 13.1.5). These conditions were once termed ‘preleukaemia’ and one-third of patients progress to acute myeloid leukaemia.
Table 13.1.5
Classification of the myelodysplastic syndromes
Refractory anaemia
Refractory anaemia with ringed sideroblasts
Refractory anaemia with excess of blasts
Chronic myelomonocytic leukaemia
Two more causes of failure of erythropoiesis might be mentioned. One is due to invasion of the marrow and disruption of its architecture by extraneous tissue, the commonest cause being metastatic cancer. Finally, but not at all uncommon, is the anaemia of chronic renal failure, where deficient erythropoiesis is attributed to decreased production of erythropoietin. Most patients with chronic renal failure on dialysis treatment tolerate a moderate degree of anaemia, but occasionally require either transfusion or treatment with erythropoietin. Emergency physicians should recognize anaemia as a predictable entity in patients with chronic renal failure, usually not requiring any action.
Anaemia secondary to decreased red cell survival: the haemolytic anaemias
Patients whose main problem is haemolysis are encountered rarely in the ED. The most fulminant haemolytic emergency one could envisage is that following transfusion of ABO-incompatible blood (discussed in Chapter 13.5), a vanishingly rare event where proper procedures are followed. Haemolysis and haemolytic anaemia are occasionally encountered in decompensating patients with multisystem problems. Rarely, first presentations of unusual haematological conditions occur.
Some of the haemolytic anaemias are hereditary conditions in which the inherited disorder is an abnormality intrinsic to the red cell, its membrane, its metabolic pathways or the structure of the haemoglobin contained in the cells. Such red cells are liable to be dysfunctional and to have increased fragility and a shortened lifespan. Lysis in the circulation may lead to clinical jaundice as bilirubin is formed from the breakdown of haemoglobin. Lysis in the reticuloendothelial system generally does not cause jaundice but may produce splenomegaly. The anaemia tends to be normochromic normocytic; sometimes a mildly raised MCV is due to an appropriate reticulocyte response from a normally functioning marrow. Serum bilirubin may be raised even in the absence of jaundice. Urinary urobilinogen and faecal stercobilinogen are detectable and serum haptoglobin is depleted. The antiglobulin (Coombs’) test is important in the elucidation of some haemolytic anaemias. In this test, red cells coated in vivo (direct test) or in vitro (indirect test) with IgG antibodies are washed to remove unbound antibodies, then incubated with an antihuman globulin reagent. The resultant agglutination is a positive test.
Any chronic haemolytic process may be complicated by an ‘aplastic crisis’. This is usually a transient marrow suppression brought on by a viral infection which can result in a severe and life-threatening anaemia. Red cell transfusion in these circumstances may be life saving.
Hereditary spherocytosis
A deficiency of the red cell wall protein, spectrin, leads to loss of deformability and increased red cell fragility. These cells are destroyed prematurely in the spleen. The condition may present at any age, with anaemia, intermittent jaundice and cholelithiasis. Patients are Coombs’ negative and show normal red cell osmotic fragility. Splenectomy radically improves general health. Hereditary elliptocytosis is a similar disease, with usually a milder course.
Glucose-6-phosphate dehydrogenase deficiency
Glucose-6-phosphate dehydrogenase (G6PD) generates reduced glutathione, which protects the red cell from oxidant stress. G6PD deficiency is an X-linked disorder present in heterozygous males and homozygous females. The disorder is commonly seen in West Africa, southern Europe, the Middle East and Southeast Asia. Oxidant stress leads to severe haemolytic anaemia. Precipitants include fava beans, antimalarial and analgesic drugs and infections. The enzyme deficiency can be demonstrated by direct assay and treatment is supportive.
Sickle cell anaemia
Whereas in the thalassaemias there is a deficiency in a given globin chain within the haemoglobin (Hb) molecule, in the haemoglobinopathies a given globin chain is present but structurally abnormal. HbS differs from normal HbA by one amino acid residue: valine replaces glutamic acid at the sixth amino acid from the N-terminus of the β-globin chain. Red cells containing HbS tend to ‘sickle’ at states of low oxygen tension. The deformed sickle-shaped red cell has increased rigidity, which causes it to lodge in the microcirculation and sequester in the reticuloendothelial system – the cause of a haemolytic anaemia.
Sickle cell disease is encountered in Afro-Caribbean people. The higher incidence in tropical areas is attributed to the survival value of the β-S gene against falciparum malaria. Heterozygous individuals have ‘sickle trait’ and are usually asymptomatic. Homozygous (HbSS) individuals manifest the disease in varying degrees. The haemolytic anaemia is usually in the range of 60–100 g/L and can be well tolerated because HbS offloads oxygen to the tissues more efficiently than HbA.
A patient with sickle cell disease may occasionally develop a rapidly worsening anaemia. This may be due to:
In any of these circumstances, transfusion may be life saving. However, these events are unusual and more commonly encountered is the vaso-occlusive crisis. A stressor – for example infection, dehydration, or cold – causes sickle cells to lodge in the microcirculation. Bone marrow infarction is one well-recognized complication of the phenomenon, but virtually any body system can be affected. Common presenting complaints include acute spinal pain, abdominal pain (the mesenteric occlusion of ‘girdle sequestration’), chest pain (pulmonary vascular occlusion), joint pain, fever (secondary to tissue necrosis), neurological involvement (transient ischaemic attacks, strokes, seizures, obtundation, coma), respiratory embarrassment and hypoxia, priapism, ‘hand–foot syndrome’ (dactylitis of infancy), haematuria (nephrotic syndrome, papillary necrosis), skin ulcers of the lower limbs, retinopathies, glaucoma and gallstones.
Most patients presenting with a vaso- occlusive crisis know they have the disease but, otherwise, the differential diagnosis is difficult. Sickle cells may be seen on the blood film and can also be induced by deoxygenating the sample. Hb electrophoresis can establish the type of Hb present. Other investigations are dictated by the presentation and may include blood cultures, urinalysis and culture, chest X-ray, arterial blood gases and electrocardiograph.
Pain relief should commence early. A morphine infusion may be required for patients with severe ongoing pain. Other supportive measures are dictated by the presentation. Intravenous fluids are particularly important for patients with renal involvement. Aim to establish a urine output in excess of 100 mL/h in adults. Antibiotic cover may be required in the case of febrile patients with lung involvement. It may be impossible to differentiate between pulmonary vaso-occlusion and pneumonia. Many patients with sickle cell disease are effectively splenectomized owing to chronic splenic sequestration with infarction and are prone to infection from encapsulated bacteria. The choice of antibiotic depends on the clinical presentation. Indications for exchange transfusion are shown in Table 13.1.6. The efficacy of exchange transfusion in painful crises remains unproven.
Table 13.1.6
Indications for exchange transfusion in sickle cell crisis
Neurological presentations: TIAs, stroke, seizures
Lung involvement (PaO2<65 mmHg with FiO2 60%)
Sequestration syndromes
Priapism
Haemoglobin S-C disease
Sickle trait or Hb S-C disease occurs in up to 10% in the black population. The clinical presentation resembles that of sickle cell disease but is usually less severe.
Haemoglobin C disease
In HbC, lysine replaces glutamic acid in the sixth position from the N terminus of the β-chain. Red cells containing HbC tend to be abnormally rigid, but the cells do not sickle. Homozygotes manifest a normocytic anaemia but there is no specific treatment and transfusion is seldom required.
Thalassaemias
There is a high incidence of β-thalassaemia trait among people of Mediterranean origin although, in fact, the region of high frequency extends in a broad band east to Southeast Asia.
Thalassaemias are disorders of haemoglobin synthesis. In the haemoglobin molecule, four haem molecules are attached to four long polypeptide globin chains. Four globin chain types (each with their own minor variations in amino acid order) are designated α, β, γ and δ. Haemoglobin A comprises two α and two β chains; 97% of adult haemoglobin is HbA. In thalassaemia, there is diminished or absent production of either the α chain (α-thalassaemia) or the β chain (β-thalassaemia). Most patients are heterozygous and have a mild asymptomatic anaemia, although the red cells are small. In fact, the finding of a marked microcytosis in conjunction with a mild anaemia suggests the diagnosis.
There are four genes on paired chromosomes 16 coding for α-globin and two genes on paired chromosomes 11 coding for β-globin. α-Thalassaemias are associated with patterns of gene deletion as follows: (−/−) is Hb-Barts hydrops syndrome, incompatible with life and (α/-) is HbH disease.
Patients who are heterozygous for β-thalassaemia have β-thalassaemia minor or thalassaemia trait. They are usually symptomless. Homozygous patients have β major.
Diagnosis of the major clinical syndromes is usually possible through consideration of the presenting features in conjunction with an FBC, blood film and Hb electrophoresis.
HbH disease patients present with moderate haemolytic anaemia and splenomegaly. The HbH molecule is detectable on electrophoresis and comprises unstable β tetramers. α Trait occurs with deletion of one or two genes. Hb, MCV and mean corpuscular haemoglobin (MCH) are low, but the patient is often asymptomatic.
β major becomes apparent in the first 6 months of life with the decline of fetal Hb. There is a severe haemolytic anaemia, ineffective erythropoiesis, hepatosplenomegaly and failure to thrive. With improved care, many of these patients survive to adulthood and may possibly present to the ED, where transfusion could be life saving. Patients with β trait may be encountered in the ED relatively frequently. They are generally asymptomatic, with a mild hypochromic microcytic anaemia. It is important not to work-up these patients continually for iron deficiency and not to subject them to inappropriate haematinic therapy.
Acquired haemolytic anaemias
Many of the acquired haemolytic anaemias are autoimmune in nature, a manifestation of a type II (cytotoxic) hypersensitivity reaction. Here, normal red cells are attacked by aberrant autoantibodies targeting antigens on the red cell membrane. These reactions may occur more readily at 37°C (warm autoimmune haemolytic anaemia, or AIHA), or at 4°C (cold AIHA). Warm AIHA is more common. Red cells are coated with IgG, complement or both. The cells are destroyed in the reticuloendothelial system. Fifty per cent of cases are idiopathic, but other recognized causes include lymphoproliferative disorders, neoplasms, connective tissue disorders, infections and drugs (notably methyldopa and penicillin). Patients have haemolytic anaemia, splenomegaly and a positive Coombs’ test. In the ED setting, it is important to stop any potentially offending drugs and search for the underlying disease. The idiopathic group may respond to steroids, other immunosuppressive or cytotoxic drugs or splenectomy.
In cold AIHA, IgM attaches to the I red cell antigen in the cooler peripheries. Primary cold antibody AIHA is known as cold haemagglutinin disease. Other causes include lymphoproliferative disorders, infections such as mycoplasma, and paroxysmal cold haemoglobinuria. Patients sometimes manifest Reynaud’s disease and other manifestations of circulatory obstruction. Symptoms worsen in winter. Red cell lysis leads to haemoglobinuria.
Microangiopathic haemolytic anaemia
In this important group of conditions, intravascular haemolysis occurs in conjunction with a disorder of microcirculation. Important causes are shown in Table 13.1.7.
Table 13.1.7
Causes of microangiopathic haemolytic anaemia
Disseminated intravascular coagulation
Haemolytic uraemic syndrome
HELLP
Malignancy
Malignant hypertension
Snake envenoming
Thrombotic thrombocytopaenic purpura
Vasculitis
Haemolytic uraemic syndrome and thrombotic thrombocytopaenic purpura
These are probably manifestations of the same pathological entity, with haemolytic uraemic syndrome occurring in children and thrombotic thrombocytopaenic purpura most commonly in the fourth decade, especially in women. The primary lesion is likely to be in the vascular endothelium. Fibrin and platelet microthrombi are laid down in arterioles and capillaries, possibly as an autoimmune reaction. The clotting system is not activated. Haemolytic anaemia, thrombocytopaenia and acute renal failure are sometimes accompanied by fever and neurological deficits.
In adults, the presentation is usually one of a neurological disturbance (headache, confusion, obtundation, seizures or focal signs). The blood film reveals anaemia, thrombocytopaenia, reticulocytosis and schistocytes. Coombs’ test is negative.
Patients require hospital admission. Adults with this condition may require aggressive therapy with prednisone, antiplatelet therapy, further immunosuppressive therapy and plasma exchange transfusions.
HELLP syndrome
HELLP stands for haemolysis, elevated liver enzymes and a low platelet count and is seen in pregnant women in the context of pre-eclampsia. Treatment is as for pre-eclampsia, early delivery of the baby being of paramount importance.
Disseminated intravascular coagulation
The introduction of procoagulants into the circulation resulting in the overwhelming of anticoagulant control systems may occur as a consequence of a substantial number of pathophysiological insults, obstetric, infective, malignant and traumatic. Disseminated intravascular coagulation has an intimate association with shock, from any cause. The widespread production of thrombin leads to deposition of microthrombi, bleeding secondary to thrombocytopaenia and a consumption coagulopathy, and red cell damage within abnormal vasculature leading to a haemolytic anaemia.
Recognition of this condition prompts intensive care admission and aggressive therapy. Principles of treatment include definitive management of the underlying cause and, from the haematological point of view, replacement therapy that may involve transfusion of red cells, platelets, fresh frozen plasma (FFP) and cryoprecipitate. There may be a role for heparin and other anticoagulant treatments if specific tissue and organ survival is threatened by thrombus.
Paroxysmal noctural haemoglobinuria
This entity is unusual in that an intrinsic red cell defect is seen in the context of an acquired haemolytic anaemia. A somatic stem cell mutation results in a clonal disorder. A family of membrane proteins (CD55, CD59 and C8 binding protein) is deficient and renders cells prone to complement-mediated lysis. Because the same proteins are deficient in white cells and platelets, in addition to being anaemic, patients are prone to infections and haemostatic abnormalities. They may go on to develop aplastic anaemia or leukaemia. Treatment is supportive. Marrow transplant can be curative.
Other causes of haemolysis
Haemolysis may be due to mechanical trauma, as in ‘March haemoglobinuria’. Artificial heart valves can potentially traumatize red cells. Historically, ball-and-cage type valves have been most prone to cause haemolysis, whereas disc valves are more thrombogenic. Improvements in design have made cardiac haemolytic anaemia very rare. Haemolysis is sometimes seen in association with a number of infectious diseases, notably malaria. Other infections that have been implicated are listed in Table 13.1.8. Certain drugs and toxins are associated with haemolytic anaemia (Table 13.1.9). The haemolytic anaemia that is commonly seen in patients with severe burns is attributed to direct damage to the red cells by heat.
Table 13.1.8
Infections associated with haemolysis
Babesiosis
Bartonella
Clostridia
Cytomegalovirus
Coxsackie virus
Epstein–Barr virus
Haemophilus
Herpes simplex
HIV
Malaria, especially Plasmodium falciparum (Blackwater fever)
Measles
Mycoplasma
Varicella
Table 13.1.9
Drugs and toxins associated with haemolysis
Antimalarials
Arsine (arsenic hydride)
Bites: bees, wasps, spiders, snakes
Copper
Dapsone
Lead (plumbism)
Local anaesthetics: lidocaine, benzocaine
Nitrates, nitrites
Sulphonamides
13.2 Neutropaenia
Mark Little
Introduction
Neutropaenia is defined as a decrease in the number of circulating neutrophils. The neutrophil count varies with age, sex and racial grouping. The severity of neutropaenia is usually graded as follows:
The risk of infection rises as the neutrophil count falls and becomes significant once the neutrophil count drops below 1.0×109/L. Recent Australian guidelines have defined febrile neutropaenia as a patient with a temperature above 38.3°C (or above 38°C on two occasions) with a neutrophil count less than 0.5×109/L or with less than 1.0×109/L and likely to fall to less than 0.5×109/L. These patients need to be examined for signs of systemic compromise (Table 13.2.1).
Table 13.2.1
Features of systemic compromise
Systolic BP≤90 mmHg or≥30 mmHg below patients usual BP or inotropic support
Room air arterial pO2≤60 mmHg ot SpO2<90% or need for mechanical ventilation
Confusion or altered mental state
Disseminated intravascular coagulation or abnormal PT/aPTT
Cardiac failure or arrhythmia, renal failure, liver failure or any major organ failure (only if new or deteriorating and not AF or CHF)
From Tam CS, OReilly M, Andersen D, et al. Use of empiric antimicrobial therapy in neutropenic fever. Int Med J 2011; 41:90-101.
Neutropaenic patients are at greater risk of overwhelming infection if the onset of the neutropaenia is acute rather than chronic and, in the case of patients receiving cancer chemotherapy, if the absolute neutrophil count is in the process of falling rather than rising.
Signs or symptoms of infection in the presence of severe neutropaenia, especially with features of systemic compromise, constitute a true emergency that mandates rapid assessment and aggressive management to prevent progression to overwhelming sepsis. In the emergency department (ED) setting, this is most commonly encountered when a patient presents with fever in the context of chemotherapy for cancer.
Pathophysiology and aetiology
Polymorphonuclear neutrophils are formed in marrow from the myelogenous cell series. Pluripotent haematopoietic stem cells are committed to a particular cell lineage through the formation of colony forming units, which further differentiate to form given white cell precursors. The mature neutrophil has a multilobed nucleus and granules in the cytoplasm. The cells are termed ‘neutrophilic’ because of the lilac colour of the granules caused by the uptake of both acidic and basic dyes.
The neutrophils leave the marrow and enter the circulation, where they have a lifespan of only 6–10 h before entering the tissues. Here they migrate by chemotaxis to sites of infection and injury and then phagocytose and destroy foreign material. In health, about half of the available mature neutrophils are in the circulation. ‘Marginal’ cells are adherent to vascular endothelium or in the tissues and are not measured by the full blood count. Some individuals have fixed increased marginal neutrophil pools and decreased circulating pools; they are said to have benign idiopathic neutropaenia.
For a previously normal individual to become neutropaenic there must be decreased production of neutrophils in the marrow, decreased survival of mature neutrophils or a redistribution of neutrophils from the circulating pool. The important causes are shown in Table 13.2.2.
Table 13.2.2
Important causes of neutropaenia
Decreased production
Aplastic anaemia
Leukaemias
Lymphomas
Metastatic cancer
Drug-induced agranulocytosis
Megaloblastic anaemias
Vitamin B12 deficiency
Folate deficiency
CD8 and large granular lymphocytosis
Myelodysplasic syndromes
Decreased survival
Idiopathic immune related
Systemic lupus erythematosus
Felty syndrome
Drugs
Redistribution
Sequestration (hypersplenism)
Increased utilization (overwhelming sepsis)
Viraemia
It is a defect in neutrophil production that is most likely to prove life threatening. Consumption of neutrophils in the periphery, as occurs early in infectious processes, is likely to be rapidly compensated for by a functioning marrow. Fortunately, most of the primary diseases of haematopoiesis are rare and, in practice, many of the acquired neutropaenias are drug induced. Processes interfering with haematopoiesis, often involving autoimmune mechanisms, may affect neutrophils both in the marrow and in the periphery. Some drugs cause neutropaenia universally but many more reactions are idiosyncratic, be they dose- related or independent of dose. Some commonly implicated drugs are listed in Table 13.2.3. Cancer chemotherapy drugs are now recognized as the commonest cause of neutropaenia.
Table 13.2.3
Drugs commonly associated with neutropaenia
Antibiotics: chloramphenicol, sulphonamides, isoniazid, rifampicin, β-lactams, carbenicillin
Antidysrhythmic agents: quinidine, procainamide
Antiepileptics: phenytoin, carbamazepine
Antihypertensives: thiazides, ethacrynic acid, captopril, methyldopa, hydralazine
Antithyroid agents
Chemotherapeutic agents: especially methotrexate, cytosine arabinoside, 5-azacytidine, azathioprine, doxorubicin, daunorubicin, hydroxyurea, alkylating agents
Connective tissue disorder agents: phenylbutazone, penicillamine, gold
H2-receptor antagonists
Phenothiazines, especially chlorpromazine
Miscellaneous: imipramine, allopurinol, clozapine, ticlopidine, tolbutamide
Clinical features
Neutropaenia is frequently anticipated based on the clinical presentation, such as fever developing in the context of cancer chemotherapy, by far the most common scenario in which severe neutropaenia is seen in the ED. Alternatively, it may be identified in the course of investigation for a likely infective illness or it might be an incidental finding during investigation for an unrelated condition.
Chronic neutropaenia may be asymptomatic unless secondary or recurrent infections develop. Acute severe neutropaenia may present with fever, sore throat and mucosal ulceration or inflammation. Symptoms or signs of an associated disease process may also be present, such as pallor from anaemia or bleeding from thrombocytopaenia, as might occur in conditions causing pancytopaenia.
The history of the mode of onset and duration of the illness is important. Systems enquiry may reveal cough, headache and photophobia, a diarrhoeal illness or urinary symptoms. The past history may reveal a known haematological illness or previous evidence of immunosuppression, such as frequent and recurrent infections. A detailed drug history is vital. Most neutropaenic drug reactions occur within the first 3 months of taking a drug.
In the ED, vital signs, including pulse, blood pressure, temperature, respiratory rate and pulse oximetry, should be performed at initial assessment and monitored regularly until disposition. Attention should be paid to identifying early signs of severe sepsis and the progression to septic shock.
Physical examination may reveal necrotizing mucosal lesions, pallor, petechial rashes, lymphadenopathy, bone tenderness, abnormal tonsillar or respiratory findings, spleno- or other organomegaly. Careful examination of the skin of the back, the lower limbs and the perineum for evidence of infection is important. The presence of indwelling venous access devices should be noted and insertion sites inspected for evidence of inflammation or infection.
Clinical investigations
Investigation in the ED is first aimed at confirming and quantifying the severity of neutropaenia, identifying the cause and then at identifying the focus and severity of infection. An urgent full blood count and blood film should be ordered in any patient who is suspected of suffering febrile neutropaenia. A coagulation profile and biochemistry, including electrolytes and creatinine, serum lactate, glucose and liver function tests may be indicated once severe neutropaenia is confirmed. Anaemic patients may require a group-and-hold or cross-match.
Microbiological cultures aimed at isolating a causative organism should be taken but antibiotics should not be unreasonably delayed in the presence of fever and confirmed significant neutropaenia. Blood cultures should be taken at the time of cannulation and, if possible, prior to the instigation of antibiotic therapy. Throat swab, swabs of skin lesions and indwelling venous access device sites, urinalysis and urine culture may be indicated depending on the clinical picture. Patients with apparent central nervous system infections might require a lumbar puncture, but this should be postponed or even cancelled in the presence of an uncorrected coagulopathy, signs of raised intracranial pressure, focal neurological signs or haemodynamic instability. Antibiotics, if clinically indicated, should be commenced prior to lumbar puncture.
Treatment
Management of the patient with confirmed febrile neutropaenia in the ED involves early recognition and treatment of bacterial infection and institution of supportive care to prevent progression to overwhelming sepsis and shock. Evolving or established haemodynamic instability requires immediate, aggressive resuscitation.
Empiric broad-spectrum antibiotic therapy should be started in the ED after drawing blood for culture in any patient with fever and suspected or confirmed significant neutropaenia. This strategy has played a pivotal role in reducing mortality rates in febrile neutropaenia. Australian consensus-based clinical recommendations for the management of neutropaenic fever in adults were recently published. They reinforce the need for the administration of early antibiotics. In general, antibiotics should provide good cover for both Gram-positive and Gram-negative organisms. With increased use of indwelling venous access devices for cancer chemotherapy, there has been an increase in the incidence of sepsis due to Gram-positive organisms, such as coagulase-negative staphylococci, S. aureus and methicillin-resistant Staph. aureus (MRSA). Although occurring infrequently, bacteraemia due to Pseudomonas aeruginosa
[/not-level-membership-for-emergency-medicine-category]
