Metabolic Emergencies
Edited by Mark Little
12.1 Acid–base disorders
David McCoubrie and Alan Gault
Introduction
Acid–base disorders are commonly encountered in the emergency department (ED) and their recognition is important for the diagnosis, assessment of severity and monitoring of many disease processes. Although these disorders are usually classified according to the major metabolic abnormality present (acidosis or alkalosis) and its origin (metabolic or respiratory), it is important to realize that acid–base disorders of a mixed type commonly occur and that the recognition and assessment of these are more complex.
CO2 produces acid when in solution and altering PaCO2 through changes in ventilation can produce or remove acid from the body. The terms respiratory acidosis/alkalosis refer to the pH shifts resulting from alterations in PaCO2 from changes in ventilation. Bicarbonate acts as a base in solution with bicarbonate accumulation resulting in a more alkaline state and its wasting or consumption indicating a more acidic state. The terms metabolic acidosis/alkalosis refer to pH shifts characterized by alterations in bicarbonate levels. By convention, the overall pH abnormality as defined by the blood gas assessment is termed alkalaemia (for pH>7.44) or acidaemia (pH<7.34).
Acid–base homeostasis
Acid–base status is one of the most tightly regulated systems in the body. The term compensation is used to describe the processes by which shifts in plasma pH are attenuated. These mechanisms include buffering, respiratory manipulation of CO2 and renal handling of bicarbonate. Buffering with plasma proteins, haemoglobin and the carbonic-acid–bicarbonate systems provide the most immediate mechanism. This is followed by respiratory compensation, which occurs within minutes and is achieved by alterations in alveolar ventilation. Renal compensation usually takes hours to days to take effect.
Acidaemia
Systemic acidaemia is defined as the presence of an increased concentration of H+ ions in the blood. An acidaemia can result from respiratory acidosis, metabolic acidosis or both. The physiological effects of acidaemia are a decrease in the affinity of haemoglobin for oxygen and an increase in serum K+ of approximately 0.4–0.6 mmol/L for each decrease in pH of 0.1 [1]. Although the presence of acidaemia is often associated with a poor prognosis, the presence of acidaemia per se usually has few clinically significant effects. It is the nature and severity of the underlying illness that principally determines outcome. A decrease in measured serum HCO3− of up to 5 mmol/L has also been reported as a result of underfilling of vacuum-type specimen tubes [2].
Metabolic acidosis
Metabolic acidosis is defined as an increase in the [H+] of the blood as a result of increased acid production or bicarbonate wasting from the gastrointestinal (GI) or renal tract. The cause is often multifactorial and can be further classified into ‘anion-gap’ and ‘non-anion-gap’ (or hyperchloraemic) metabolic acidosis.
Anion-gap metabolic acidosis (AGMA)
As electroneutrality must exist in all solutions, the anion gap represents the concentration of anions that are not commonly measured. The most commonly used formula for the calculation of the anion gap is:
< ?xml:namespace prefix = "mml" />
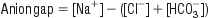
The normal value for the anion gap depends on the type of biochemical analyser used and, while the upper limit of normal has been commonly quoted as 14, the mean range with some modern analysers is only 5–12 [3]. In the normal resting state, the serum ionic proteins account for most of the anion gap, with a lesser contribution from other ‘unmeasured’ anions, such as PO4− and SO4−. In pathological conditions where there is an increase in the concentration of unmeasured anions, an AGMA results. The anions responsible for the increase in the anion gap depend on the cause of the acidosis. Lactic acid is the predominant anion in hypoxia and shock, PO4− and SO4− in renal failure, ketoacids in diabetic and alcoholic ketoacidosis, oxalic acid in ethylene glycol poisoning and formic acid in methanol poisoning.
Of the causes of an AGMA, lactic acidosis is the most commonly encountered in the ED and is defined as a serum lactate of>2.5 mmol/L (Table 12.1.1). The presence of lactic acidosis is determined by the balance between lactate production and metabolism. In the seriously ill patient, it is common for increased production and decreased metabolism to be present simultaneously.
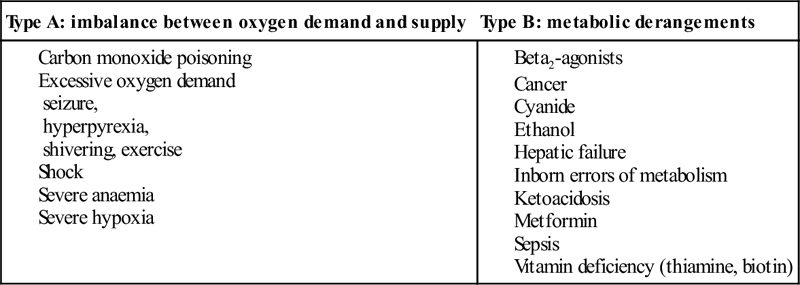
It is important to realize that, in many conditions, a variety of factors may produce the acidosis and that multiple anions may be involved in the production of an anion-gap acidosis. In a patient with an AGMA, a non-anion-gap metabolic acidosis may also exist (see below).
Non-anion-gap metabolic acidosis
Non-anion-gap metabolic acidosis results from loss of HCO3− from the body, rather than increased acid production. To maintain electroneutrality, chloride is usually retained by the renal tubules when HCO3− is lost and the hallmark of non-anion-gap acidosis is an elevation of the serum chloride. The causes of non-anion-gap metabolic acidosis are further classified according to the site of HCO3− loss. Gastrointestinal losses can occur with lower gastrointestinal tract (GIT) fluid losses that are rich in HCO3− or with cholestyramine ingestion due to binding of HCO3− in the gut. Renal losses can occur with renal tubular acidosis, carbonic anhydrase inhibitor therapy or adrenocortical insufficiency. Occasionally, direct chloride excess drives the renal bicarbonate loss (again due to electroneutrality) – which can be observed with large volume chloride rich crystalloid administration (chiefly normal saline).
Renal tubular acidosis
Renal tubular acidosis (RTA) is a group of conditions where there is an impaired ability to secrete H+ in the distal convoluted tubule or absorb HCO3− in the proximal convoluted tubule. This may result in a chronic metabolic acidosis, with hypokalaemia, nephrocalcinosis, rickets or osteomalacia. There are many subtypes of RTA and many different causes. Most commonly, it is observed in patients with chronic renal impairment but it may also be drug induced with agents such as ibuprofen, toluene and carbonic anhydrase inhibitors most often implicated.
Treatment of metabolic acidosis
The treatment of acidosis should usually be directed primarily towards correction of the underlying cause. Intravenous HCO3− is of use in the presence of acidosis and severe hyperkalaemia, severe sodium channel (e.g. tricyclic antidepressant), salicylate and methanol poisoning. The use of HCO3− in patients with diabetic ketoacidosis and lactic acidosis associated with sepsis or severe cardiorespiratory disease does not appear to improve outcome [4–6]. The potential hazards of HCO3− therapy include a high solute load, hyperosmolarity, hypokalaemia, decreased ionized serum calcium and worsening of intracellular and cerebrospinal fluid acidosis (which may precipitate hepatic encephalopathy in susceptible patients).
Respiratory acidosis
Respiratory acidosis is defined as an elevation of the arterial partial pressure of carbon dioxide (PCO2) and is due to alveolar hypoventilation. This can result from central depression in respiriatory drive, neuromuscular weakness, mechanical factors, lung parenchymal disorders and ventilation/perfusion mismatch. With significant elevations in CO2, sweating, tachycardia, confusion and mydriasis occur. When the PCO2 is greater than 80 mmHg, the level of consciousness is usually depressed, known as CO2 narcosis.
Treatment
The treatment of respiratory acidosis is directed towards reversal of the causative factors while supporting and promoting ventilation. Indications for and methods of therapy are clinically determined.
Alkalaemia
Alkalaemia is defined as a decrease in [H+] in the blood. Extreme alkalaemia may cause altered mental status, tetany and seizures. These are predominantly related to a reduction in the concentration of ionized calcium, which is more commonly present in respiratory alkalosis due to anxiety, than from other causes. Alkalaemia in patients with chronic airways disease may exacerbate tissue hypoxia due to leftward shift of the oxygen-dissociation curve. Like acidaemia, there are metabolic and respiratory processes by which it occurs.
Metabolic alkalosis
Metabolic alkalosis most commonly results from loss of acid from the GIT, however, renal acid losses or accumulation of bicarbonate from exogenous sources can contribute. Diagnostically and therapeutically, metabolic alkalosis can be divided into two distinct aetiological groups – chloride-responsive and chloride-unresponsive metabolic alkalosis (Table 12.1.2).
Table 12.1.2
Low urine chloride variety (saline-responsive)
Gastric volume loss (vomiting, nasogastric suction, bulimia nervosa)
Diuretics
Licorice
Hypokalaemia
High urine chloride variety (not saline-responsive)
Primary and secondary hyperaldosteronism
Apparent mineralocorticoid excess
Liddle’s syndrome
Conn’s syndrome (aldosteronoma)
Cushing’s disease
Bartter’s syndrome
Gitelman’s syndrome
Excess bicarbonate administration – antacids, dialysis, milk-alkali syndrome
From Murray L, Daly F, Little M, Cadogan M. Toxicology Handbook, 2nd edn. Churchill Livingstone; 2011.
Chloride-responsive metabolic alkalosis arises from conditions that result in both chloride and volume loss. Reduction in extracellular volume leads to increased mineralocorticoid activity causing reabsorption of sodium and secretion of hydrogen. This, in turn, causes increased formation of bicarbonate which, ultimately, overwhelms the kidneys’ ability further to excrete it. In alkalosis, the urine is usually alkaline with higher concentrations of bicarbonate, there is minimal chloride excreted in order to maintain electroneutrality. Hence a urinary chloride<10 mmol/L is a common finding in these conditions. The commonest causes seen in the emergency department are upper GIT losses as a result of severe and prolonged vomiting and diuretic use.
Chloride-unresponsive metabolic alkalosis is typically due to disease states that either result in mineralocorticoid excess in the absence of hypovolaemia or chloride wasting, or congenital disorders with defects in the various ionic transport channels within the kidney. As extracellular volume is either normal or increased, urinary chloride is typically>10 mmol/L. These conditions are seen in the emergency department infrequently.
Treatment should be directed primarily towards correction of the underlying cause. In the presence of upper gastrointestinal fluid losses, intravenous fluids with high chloride content (such as 0.9% saline) should be used initially for rehydration and correction of hypokalaemia is also required.
Respiratory alkalosis
Respiratory alkalosis may be acute or chronic, of which the acute form is most commonly encountered in the emergency department.
Respiratory alkalosis may physiologically occur in the general population secondary to exercise, altitude-related hypoxia and stimulation of the medullary respiratory centre by progesterones during pregnancy. Disease states that give rise to respiratory alkalosis are more likely to be seen in the emergency department (Table 12.1.3). Treatment is again directed towards correction of the underlying cause.
Table 12.1.3
Causes of respiratory alkalosis
| CNS-mediated hyperventilation | Pulmonary |
| Increased intracranial pressure | Congestive cardiac failure |
| Cerebrovascular accidents | Mechanical hyperventilation |
| Psychogenic | Pneumonia |
| Pulmonary emboli | |
| Hypoxia-mediated hyperventilation | Sepsis |
| Altitude | Toxin-induced hyperventilation |
| Anaemia | Nicotine |
| V/Q mismatch | Salicylate |
| Xanthines |
Systematic acid–base interpretation
A systematic stepwise approach to acid–base interpretation is beneficial in the evaluation of disturbances as they are often multiple. What follows is an example of a conventional methodology as outlined by Whittier and Rutecki [7]:
Step 1: what is the pH (primary acid–base disturbance)?
Step 2: determine whether the primary process is respiratory, metabolic or both
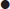
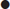
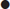
Step 3: calculate the anion gap
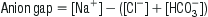
Causes of anion-gap acidosis (mnemonic ‘CAT MUDPILES’)
Causes of a low anion gap (<6)
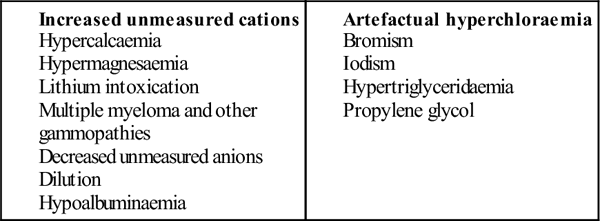
Causes of non-anion-gap metabolic acidosis

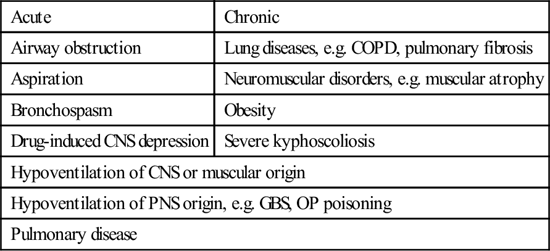
Causes of respiratory acidosis
| Acute | Chronic |
| Airway obstruction | Lung diseases, e.g. COPD, pulmonary fibrosis |
| Aspiration | Neuromuscular disorders, e.g. muscular atrophy |
| Bronchospasm | Obesity |
| Drug-induced CNS depression | Severe kyphoscoliosis |
| Hypoventilation of CNS or muscular origin | |
| Hypoventilation of PNS origin, e.g. GBS, OP poisoning | |
| Pulmonary disease | |
Step 4: check for the degree of compensation
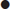
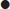
Step 5: determine if there is a 1:1 relationship between the anions in the blood (presence of a delta gap)
In an anion-gap metabolic acidosis, this step determines whether there is a non-anion-gap (hyperchloraemic) component as a contributing explanantion of the bicarbonate fall. There should be a 1:1 relationship between the rise in the anion gap over normal and the decrease in the bicarbonate. If the bicarbonate is higher than predicted then a metabolic alkosis is also present. If the bicarbonate is lower than predicted then a non-anion-gap acidosis is also present.
12.2 Electrolyte disturbances
John Pasco
Hyponatraemia
Introduction
Hyponatraemia, defined as serum sodium concentration of less than 130 mmol/L, is a common condition. The prevalence is estimated at 2.5% in hospitalized patients, of which two-thirds develop the condition while in hospital.
Pathophysiology
Hyponatraemia is almost always associated with extracellular hypotonicity, with an excess of total body water relative to sodium (hypotonic hyponatraemia). The exceptions are:
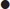
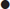
Hyponatraemia causes cellular swelling as water moves down an osmotic gradient into the intracellular fluid. Most of the symptomatology of hyponatraemia is produced in the central nervous system (CNS) by the swelling of brain cells within the rigid calvarium, causing raised intracranial pressure (hyponatraemic encephalopathy). As intracranial pressure rises, adaptive responses come into play, returning brain volume towards normal and restoring cellular function.
For this reason, chronic hyponatraemia is generally better tolerated than acute hyponatraemia. Patients can become encephalopathic when hyponatraemia develops rapidly and the adaptive responses have not had time to develop (for example, following rapid ingestion of very large amounts of water, such as in psychogenic polydipsia or post-exercise) or when the adaptive responses fail. Hyponatraemic encepaholopathy carries a high mortality (50%) if left untreated.
Aetiology and classification
Hypotonic hyponatraemia may be classified according to the volume status of the patient (hypovolaemic, euvolaemic or hypervolaemic).
Hypovolaemic hyponatraemia
These patients have deficits in both total body sodium and total body water, but the sodium deficit exceeds the water deficit. Causes include renal and extrarenal fluid losses and are listed in Table 12.2.1. Determination of the urinary sodium concentration can differentiate these two groups. Extrarenal losses are associated with low urinary sodium concentrations (<20 mmol/L) and hyperosmolar urine. The exception is with severe vomiting and metabolic alkalosis, where bicarbonaturia obligates renal sodium loss and urinary sodium is high (>20 mmol/L), despite volume depletion. However, urinary chloride, a better indicator of extracellular fluid (ECF) volume, is low.
Table 12.2.1
Causes of hypovolaemic hyponatraemia
Renal losses (urinary [Na] 20 mmol/L)
Diuretics
Mineralocorticoid deficiency – Addison’s disease
Salt-losing nephropathy
Ketonuria
Osmotic diuresis – glucose, mannitol, urea
Bicarbonaturia with metabolic alkalosis
Extrarenal losses (urinary [Na]<20 mmol/L)
Vomiting – self-induced, gastroenteritis, pyloric obstruction
Diarrhoea
Excessive sweating
Blood loss
Third-space fluid loss – burns, pancreatitis, trauma
Euvolaemic hyponatraemia
Total body water is increased with only minimal change in total body sodium. Volume expansion is mild and usually not clinically detectable. Causes are listed in Table 12.2.2.
Table 12.2.2
Causes of euvolaemic hyponatraemia
Psychogenic polydipsia
Iatrogenic water intoxication
Absorption of hypotonic irrigation fluids during TURP
Inappropriate intravenous fluid administration
Postoperative hyponatraemia (elevated ADH levels)
Non-osmotic ADH secretion
Glucocorticoid deficiency
Severe hypothyroidism
Thiazide diuretics
Drugs (ADH analogues, potentiation of ADH release, unknown mechanisms)
Psychoactive agents: phenothiazines, SSRIs, TCAs, MAOIs, ‘ecstasy’
Oxytocin
Anticancer agents: cyclophosphamine, vincristine, vinblastine
NSAIDs
Carbamazepine
Chlorpropamide
SIADH
Hypervolaemic hyponatraemia
Total body water is increased in excess of total body sodium. Causes include congestive cardiac failure, hepatic cirrhosis with ascites, nephrotic syndrome and chronic renal failure.
Clinical features
In addition to the features of the underlying medical condition and alteration in extracellular volume, clinical manifestations of hyponatraemia per se usually develop when serum sodium is less than 130 mmol/L. The severity of symptoms depends partly on the absolute serum sodium concentration and partly on its rate of fall. At sodium concentrations from 125 to 130 mmol/L, the symptoms are principally gastrointestinal whereas, at concentrations below 125 mmol/L, the symptoms are predominantly neuropsychiatric. The principal signs and symptoms of hyponatraemia are listed in Table 12.2.3.
Table 12.2.3
Clinical manifestations of hyponatraemia
Anorexia
Nausea
Vomiting
Lethargy
Muscle cramps
Muscle weakness
Headache
Confusion/agitation
Altered conscious state
Seizures
Coma
Population groups particularly prone to hyponatraemic encephalopathy have been identified (Table 12.2.4).
Table 12.2.4
Patient groups at risk of hyponatraemia
Postoperative
Menstruating females
Elderly women on thiazide diuretics
Prepubescent children
Psychiatric polydipsic patients
Hypoxaemic patients
AIDS patients
Patients taking ‘Ecstasy’ (MDMA)
Endurance athletes
Premenopausal women appear at risk of developing hyponatraemic encephalopathy because oestrogen and progesterone are thought to inhibit the brain Na-K-ATPase and increase circulating levels of antidiuretic hormone (ADH).
Psychogenic polydipsia refers to a condition in which kidney function is normal and dilute urine is produced, but free water intake overwhelms the kidney’s capabilities and the serum sodium falls. It occurs primarily in patients with schizophrenia or bipolar disorder. These patients develop hyponatraemia with a far lower fluid intake than is usually necessary (over 20 L of water/day in a 60 kg man, in the absence of elevated levels of ADH) and it may arise through a combination of factors: antipsychotics, increased thirst perception, enhanced renal response to ADH and a mild defect in osmoregulation.
Exercise-associated hyponatraemia occurs in endurance athletes and mainly relates to the consumption of excessive fluid, although non-osmotic release of vasopressin and other mechanisms may be implicated.
Hyponatraemia in AIDS is common and associated with a high mortality. It may be secondary to syndrome of inappropriate ADH (SIADH), adrenal insufficiency or volume deficiency with hypotonic fluid replacement.
The use of ‘Ecstasy’ at ‘rave’ parties has been associated with acute hyponatraemia. This may be due to a combination of drug effect and drinking large quantities of water in an attempt to prevent dehydration.
Mild chronic ‘asymptomatic’ hyponatraemia in the elderly contributes to an increased rate of falls, probably due to impairment of attention, posture and gait mechanisms.
Syndrome of inappropriate ADH secretion
This is a diagnosis of exclusion and is characterized by inappropriately concentrated urine in the setting of hypotonicity. It accounts for approximately 50% of all cases of hyponatraemia. These patients have elevated serum ADH levels without an obvious volume or osmotic stimulus. The diagnostic criteria for SIADH secretion are shown in Table 12.2.5 and conditions associated with the syndrome are listed in Table 12.2.6.
Table 12.2.5
Hypotonic hyponatraemia
Urine osmolality>100 mmol/kg (i.e. inappropriately concentrated)
Urine sodium>20 mmol/mL while on a normal salt and water intake
Absence of extracellular volume depletion
Normal thyroid and adrenal function
Normal cardiac, hepatic and renal function
No diuretic use
Table 12.2.6
Conditions associated with SIADH
Neoplasms (ectopic ADH production)
Bronchogenic carcinoma
Pancreatic carcinoma
Lymphoma
Mesothelioma
Thymoma
Carcinoma of the bladder
Pulmonary disease
Pneumonia
Tuberculosis
Aspergillosis
Cystic fibrosis
Chronic obstructive airways disease
Positive-pressure ventilation
CNS disease
Encephalitis
Acute psychosis
Head trauma
Brain abscess
Meningitis
Hydrocephalus
Brain tumour
Delirium tremens
Guillain–Barré syndrome
Stroke
Subdural or subarachnoid bleed
HIV infection
Pneumocystis carinii pneumonia
Clinical investigations
Measurement of serum and urine sodium concentrations and osmolalities, in addition to clinical assessment of volume status, are essential for the assessment of hyponatraemia (Fig. 12.2.1).
Treatment
There is ongoing controversy over the treatment of hyponatraemia because of the risk of osmotic demyelination, which is discussed below.
Treatment should be carefully individualized and depends on the presence of symptoms, the duration of the hyponatraemia and the absolute value of sodium. Ideally, correction of the serum sodium should be of a sufficient pace and magnitude to reverse the manifestations of hypotonicity but not be so rapid and large as to pose a risk of the development of osmotic demyelination. Treatment of the underlying cause is obviously essential and may correct the hyponatraemia. For hypovolaemic hyponatraemia, adequate volume replacement is essential.
Acute symptomatic hyponatraemia (hyponatraemic encephalopathy)
Symptomatic hyponatraemia developing within 48 h is a medical emergency requiring prompt and aggressive treatment. The risks of developing osmotic demyelination are clearly outweighed by those of the encephalopathy. An immediate increase in serum sodium concentration by 8 mEq/L over 4–6 h is recommended. This can be achieved by infusing hypertonic saline (3% NaCl) at a rate of 1–2 mL/kg/h, which should raise the serum sodium by 1–2 mmol/L/h. Where neurological symptoms are severe, hypertonic saline can be infused at 4–6 mL/kg/h. Indications for ceasing rapid correction of hyponatraemia are cessation of life-threatening manifestations, moderation of other symptoms or the achievement of a serum sodium of 125–130 mEq/L. Other measures to reduce intracranial pressure, such as intubation and intermittent positive pressure ventilation (IPPV), may also be required.
Chronic symptomatic hyponatraemia
Hyponatraemia present for more than 48 h, or where the duration is unknown, presents the greatest dilemma. Care must be taken with correction of sodium as these patients are at the greatest risk of developing osmotic demyelination, yet the presence of encephalopathy mandates urgent treatment. Hypertonic saline can be infused so that a correction rate of no more than 1–1.5 mmol/L/h is maintained. Therapy with hypertonic saline should be discontinued when (1) the patient becomes asymptomatic, (2) the serum sodium has risen by 20 mmol/L or (3) the serum sodium reaches 120–125 mmol/L. Thereafter, slower correction with water restriction should follow. The serum sodium should never be acutely elevated to hypernatraemic or normonatraemic levels and should not be elevated by more than 25 mmol/L during the first 48 h of therapy.
Chronic asymptomatic hyponatraemia
In this situation, saline infusion is usually not required and patients can be managed by treating the underlying disorder, discontinuing diuretic therapy or restricting fluids. Fluid restriction is inexpensive and effective but is often limited by patient non-compliance. Other treatment options include pharmacological inhibition of ADH with demeclocycline, which is limited by its neuro- and nephrotoxic side effects, or increasing solute with the use of furosemide or urea.
Osmotic myelinolysis
This is an iatrogenic disorder which develops progressively over 3–5 days following the correction of hyponatraemia. It classically produces symmetrical lesions centred on the midline of the pons and was originally described as ‘central pontine myelinolysis’. However, about 10% of cases involve extrapontine lesions. It is reported as occurring in 25% of severely hyponatraemic patients following correction of serum sodium. Clinically, the disorder is initially manifested by dysarthria, mutism, lethargy and affective changes, which may be mistaken for psychiatric illness. Classically, pseudobulbar palsy and spastic quadriparesis are observed. Recovery is usually gradual and incomplete, although both fatalities and complete recovery are reported. Demyelination in the central pons and extrapontine sites can be demonstrated on magnetic resonance imaging (MRI) scan or at autopsy.
It appears that the risk of developing osmotic myelinolysis is associated with severity and chronicity of hyponatraemia. It rarely occurs if the serum sodium is>120 mmol/L or where hyponatraemia has been present for<48 h. Alcoholics, malnourished patients, hypokalaemic patients, burn victims and elderly patients on thiazides seem to be most at risk of developing osmotic demyelination
Both the rate and the magnitude of sodium correction appear important in the development of osmotic myelinolysis. Although there is as yet no agreed rate of correction that is regarded as completely safe, most authorities suggest that the serum sodium concentration should not rise by more than 10–14 mmol/L during any 24-h period.
Hypernatraemia
Introduction
Hypernatraemia is much less common than hyponatraemia and may be defined as a serum sodium concentration greater than 150 mmol/L.
It is important to recognize hypernatraemia because it is usually associated with severe underlying medical illness. It is a condition of hospitalized patients, elderly and dependent people. The incidence of hypernatraemia in hospitalized patients ranges from 0.3 to 1%, with from 60 to 80% of these developing hypernatraemia after admission. In-hospital mortality is high (40–55%) and may be due to a combination of hypernatraemia and the severity of the underlying disease.
Pathophysiology
Hypernatraemia is a relative deficiency of total body water compared to total body sodium, thus rendering the body fluids hypertonic. The normal compensatory response includes stimulated thirst – the most important response – and renal water conservation through ADH secretion. In the absence of ADH, water intake can match urinary losses because of increased thirst, but where the thirst mechanism is absent or defective, patients become hypernatraemic even in the presence of maximal ADH stimulation. Therefore, hypernatraemia is usually seen where water intake is inadequate, i.e. in patients too young, too old or too sick to drink, with no access to water or with a defective thirst mechanism.
Extracellular hypertonicity causes a shift of water from the intracellular space until there is osmotic equilibrium. The resultant cellular contraction may explain some of the clinical features of hypernatraemia. The brain is especially at risk from shrinkage because of its vascular attachments to the calvarium. Haemorrhage may occur if these vascular attachments tear.
As with hyponatraemia, the rate and magnitude of the rise in sodium determine the severity of the symptoms, which is a reflection of the brain’s capacity to adapt to the deranged osmotic conditions.
Aetiology and classification
The clinical causes of hypernatraemia are listed in Table 12.2.7. Population groups at particular risk of developing hypernatraemia are listed in Table 12.2.8.
Table 12.2.7
Altered perception of thirst
Osmoreceptor damage/destruction
Exogenous: trauma
Endogenous: vasculitis, carcinoma, granuloma
Idiopathic: psychogenic, head injury
Drugs
Normal perception of thirst
Poor intake
Confusion
Coma
Depression
Dysphagia
Odynophagia
Increased water loss and decreased intake
Diuresis
Renal loss
Diabetes insipidus
Chronic renal failure
Diuretic excess
GIT loss: fistulae, diarrhoea
Exogenous increase in salt intake
Table 12.2.8
Groups at particular risk for hypernatraemia
Elderly or disabled, unable to obtain oral fluids independently
Infants
Inpatients receiving: hypertonic infusions tube feedings osmotic diuretics lactulose mechanical ventilation
Altered mental status
Uncontrolled diabetes mellitus
Underlying polyuric disorders
Hypernatraemia is classified into these categories based on extracellular volume status: hypovolaemic, hypervolaemic and euvolaemic.
Hypovolaemic hypernatraemia
This occurs where there is loss of both total body water and sodium, but with a greater loss of water. Renal causes include osmotic diuresis and diuretic excess. Urinary sodium is usually>20 mmol/L. Extrarenal losses include profuse diarrhoea, sweating, burns and fistulae. Urinary sodium is usually<20 mmol/L.
Euvolaemic hypernatraemia
This is the most common form of hypernatraemia. Patients have pure water losses, with intracellular dehydration as water shifts according to the osmotic gradient. Hypernatraemia in these patients occurs only when there is no accompanying water intake, i.e. restricted access to water or a defect in thirst sensation.
Extrarenal losses are usually seen in skin losses in burns patients and via the respiratory system in respiratory infections and at high altitude. Renal water loss is usually due to diabetes insipidus – a failure of ADH production or secretion (central diabetes insipidus) or a failure of the collecting duct of the kidney to respond to ADH (nephrogenic diabetes insipidus).
Hypervolaemic hypernatraemia
This is not very common. These patients are typically extracellular volume expanded but intracellular volume depleted. It is seen following resuscitation with sodium bicarbonate, with the use of hypertonic saline solutions, with excess salt intake, in primary hyperaldosteronism and in Cushing’s syndrome.
Clinical features
In addition to the features of the underlying medical condition and alteration in extracellular volume, the clinical features of hypernatraemia per se are primarily CNS. Early symptoms are anorexia, nausea and vomiting; lethargy, hyperreflexia, confusion, seizures and coma occur later.
Treatment
The speed at which hypernatraemia is corrected should take into account the rate of development and severity of symptoms. Too rapid correction, especially in chronic hypernatraemia, can cause cerebral oedema or isotonic water intoxication. The rate of correction of chronic hypernatraemia should not exceed 0.5–0.7 mmol/L/h.
Treatment is based on clinical assessment of the patient’s volume status.
Hypovolaemic hypernatraemia
These patients require restoration of the volume deficit with isotonic saline, colloid or blood in the first instance, to prevent peripheral vascular collapse and treatment of the underlying cause. Following this, the water deficit is corrected with 0.45% saline, 5% dextrose or oral water.
The water deficit is calculated as follows:
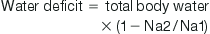
where Na2=desired sodium, Na1=actual sodium and total body water is usually 60% of the body weight. The calculated normal daily maintenance fluids should be added to the above volumes.
Euvolaemic hypernatraemia
Calculate the water deficit as above and replace the deficit and ongoing losses with 5% dextrose, 0.45% saline or oral water. To avoid cerebral oedema, particularly in chronic hypernatraemia, 50% of the water deficit should be replaced over the first 6–12 h and the rest given slowly over 1–2 days. Serum sodium estimations should be repeated at regular intervals.
Hypervolaemic hypernatraemia
Removal of sodium is required with the use of diuretics, such as furosemide, and discontinuation of causative agents. Furosemide causes excretion of more water than sodium, so a hypotonic fluid, such as 5% dextrose, may need to be infused. In severe cases or in renal failure, dialysis may be required.
Hypokalaemia
Introduction
Hypokalaemia may be defined as a serum potassium concentration of less than 3.5 mmol/L. It is usually considered to be severe when this is less than 2.4 mmol/L.
Pathophysiology
Hypokalaemia may develop as a consequence of potassium depletion or a shift of potassium into cells. In either case, there is an increase in the ratio of intracellular to extracellular potassium concentrations. This, in turn, produces hyperpolarization across excitable membranes and is responsible for the effects of hypokalaemia on striated muscle and the cardiac conducting system.
Aetiology
The causes of hypokalaemia are listed in Table 12.2.9.
Table 12.2.9
Inadequate dietary intake
Abnormal losses
Gastrointestinal
Vomiting, nasogastric aspiration
Diarrhoea, fistula loss
Villous adenoma of the colon
Laxative abuse
Renal
Mineralocorticoid excess
Conn’s syndrome
Bartter syndrome
Ectopic ACTH syndrome
Small cell carcinoma of the lung
Pancreatic carcinoma
Carcinoma of the thymus
Renal tubular acidosis
Magnesium deficiency
Drugs
Diuretics
Corticosteroids
Gentamicin, amphotericin B
Cisplatin
Compartmental shift
Alkalosis Insulin
Na-K-ATPase stimulation
Sympathomimetic agents with β2 effect
Methylxanthines
Barium poisoning
Hypothermia
Toluene intoxication
Hypokalaemic periodic paralysis
Clinical presentation
Hypokalaemia commonly produces no symptoms in otherwise healthy subjects.
Clinical features may include weakness, constipation, ileus and ventilatory failure. Myopathy may develop, with weakness of the extremities which characteristically worsens with exercise. If the hypokalaemia is severe and untreated, rhabdomyolysis may occur. Polyuria and polydipsia may result from the effect of hypokalaemia on the distal renal tubule (nephrogenic diabetes insipidus of hypokalaemia). Cardiac effects include ventricular tachycardias and atrial tachycardias, with or without block. Characteristic electrocardiogram (ECG) changes include PR prolongation, T-wave flattening and inversion and prominent U waves
Treatment
Oral replacement is safe for asymptomatic patients and 40–60 mmol of potassium every 1–4 h is usually well tolerated.
Intravenous administration of potassium is recommended when hypokalaemia is associated with cardiac arrhythmias, familial periodic paralysis or severe myopathy. Usual infusion rates are 10–20 mmol/h. Rates greater than 40 mmol/h are not recommended. Potassium is a sclerosant and should, therefore, be given via a large peripheral or central vein. Serum potassium estimations every 1–4 h and continuous cardiac monitoring are mandatory.
Hyperkalaemia
Introduction
Hyperkalaemia, defined as a serum potassium concentration greater than 5.5 mmol/L, is less common than hypokalaemia. Moderate (6.1–6.9 mmol/L) and severe (>7 mmol/L) hyperkalaemia can have grave consequences, particularly if acute.
Pathophysiology
Two homeostatic mechanisms are responsible for maintaining potassium balance. The renal system maintains external potassium balance by excreting 90–95% of the average daily potassium load (100 mmol/day); the gut excretes the remainder. This is a relatively slow process: only half the administered load of potassium will have been excreted in the urine after 3–6 h. The extrarenal system involves hormonal and acid–base mechanisms that rapidly translocate potassium intracellularly. This system is critical in the management of acute hyperkalaemia.
Aetiology
The causes of hyperkalaemia are listed in Table 12.2.10.
Table 12.2.10
Pseudohyperkalaemia
Delay in separating red cells
Specimen haemolysis during or after venesection
Severe leucocytosis/thrombocytosis
Excessive intake
Exogenous: IV or oral KCl, massive blood transfusion
Endogenous: tissue damage
Burns
Trauma
Rhabdomyolysis
Tumour lysis
Decrease in renal excretion
Drugs
Spironolactone, triamterene, amiloride
Indomethacin
Captopril, enalapril
Renal failure
Addison’s disease
Hyporeninaemic hypoaldosteronism
Compartmental shift
Acidosis
Insulin deficiency
Digoxin overdose
Succinylcholine
Fluoride poisoning
Hyperkalaemic periodic paralysis
Clinical features
The clinical features of hyperkalaemia are often non-specific. Diagnosis depends on clinical suspicion, measurement of potassium concentration in the plasma and the characteristic changes on the ECG.
Generalized muscle weakness, flaccid paralysis and paraesthesia of the hands and feet are common, but there is poor correlation between the degree of muscle weakness and serum potassium concentration.
The ECG changes (Table 12.2.11) are characteristic, but are an insensitive method of evaluating hyperkalaemia.
Table 12.2.11
| Plasma potassium (mmol/L) | ECG characteristics |
| 6–7 | Tall peaked T waves (>5 mm) |
| 7–8 | QRS widening, small-amplitude P waves |
| 8–9 | Fusion of QRS complex with T wave producing sine wave |
| >9 | AV dissociation, ventricular tachycardia, ventricular fibrillation |
Serum biochemistry in almost all patients with hyperkalaemia shows some degree of renal impairment and metabolic acidosis. In dialysis patients, hyperkalaemia may develop without concomitant metabolic acidosis.
Treatment
Pseudohyperkalaemia is common and, if hyperkalaemia is an unexpected finding, the serum potassium should be remeasured.
Hyperkalaemia with ECG changes requires urgent management. The priorities are as follows:
 Antagonize potassium cardiac toxicity:
Antagonize potassium cardiac toxicity:
 IV calcium chloride 10%, 5–10 mL or
IV calcium chloride 10%, 5–10 mL or
 IV soluble insulin, 20 U with dextrose 50 g or
IV soluble insulin, 20 U with dextrose 50 g or
 salbutamol nebulized (10–20 mg) or IV (0.5 mg diluted in 100 mL over 10–15 min) or
salbutamol nebulized (10–20 mg) or IV (0.5 mg diluted in 100 mL over 10–15 min) or

The use of insulin and glucose is well supported in the literature. A response is usually seen within 20–30 min, with lowering of plasma potassium by up to 1 mmol/L and reversal of ECG changes. Transient hypoglycaemia may be observed within 15 min of insulin administration. In some patients, particularly those with end-stage renal failure, late hypoglycaemia may develop. For this reason, a 10% dextrose infusion at 50 L/h is recommended and the blood glucose should be monitored closely. The exact mechanism by which insulin translocates potassium is not known; it is thought to be stimulation of Na-K-ATPase independent of cAMP.
β2-Agonists significantly lower plasma potassium when given intravenously or via a nebulizer. Potassium levels are reduced by up to 1.00 mmol/L within 30 min following 10–20 mg of nebulized salbutamol. The effect is sustained for up to 2 h. Adverse effects of salbutamol administration include tachyarrhythmias and precipitation of angina in patients with coronary artery disease. Patients on non-selective β-blockers may not respond. Some patients with end-stage renal disease are also resistant to this therapy. The reason for this is unknown. Greater decreases in potassium have been observed when salbutamol treatment is combined with insulin and glucose. The additive effect is thought to be due to stimulation of Na-K-ATPase via different pathways. Transient hyperglycaemia may occur with combined therapy, but delayed hypoglycaemia does not occur.
Hypocalcaemia
Introduction
A reduction in serum calcium concentration manifests principally as abnormal neuromuscular function.
Pathophysiology
Calcium is involved in smooth and skeletal muscle contraction and relaxation, platelet aggregation, neurotransmission, hepatic and adipose glycogenolysis, thermogenesis and neutrophil function. In addition, most endocrine and exocrine gland function is calcium dependent.
Aetiology
Hypocalcaemia occurs when calcium is lost from the extracellular fluid at a rate greater than can be replaced by the intestine or bone. The major cause of severe hypocalcaemia is hypoparathyroidism, as a result of surgery for thyroid disease, autoimmune destruction or from developmental abnormalities of the parathyroid glands. Other causes are listed in Table 12.2.12.
Table 12.2.12
Factitious EDTA contamination
Hypoalbuminaemia
Decreased PTH activity
Hypoparathyroidism
Pseudohypoparathyroidism
Hypomagnesaemia
Decreased vitamin D activity
Acute pancreatitis
Hyperphosphataemia
Renal failure
Phosphate supplements
‘Hungry bone’ syndrome
Drugs
Mithramycin
Diuretics: furosemide, ethacrynic acid
Clinical features
Patients with acute hypocalcaemia are more likely to be symptomatic than those with chronic hypocalcaemia. Symptomatic hypocalcaemia is characterized by abnormal neuromuscular excitability and neurological sensations. Early signs are perioral numbness and paraesthesia of distal extremities. Hyperreflexia, muscle cramps and carpopedal spasm follow. Chvostek’s sign (ipsilateral contraction of the facial muscles elicited by tapping the facial nerve just anterior to the ear) and Trousseau’s sign (carpopedal spasm with inflation of a blood pressure cuff for 3–5 min) are signs of neuromuscular irritability. If muscle contractions become uncontrollable, tetany results and this can prove fatal if laryngospasm occurs. Seizures may occur when there is CNS instability. Cardiovascular manifestations include hypotension, bradycardia, impaired cardiac contractility and arrhythmias. ECG evidence of hypocalcaemia includes prolonged QT interval and possibly ST prolongation and T-wave abnormalities.
Treatment
Acute symptomatic hypocalcaemia
In the emergency situation where seizures, tetany, life-threatening hypotension or arrhythmias are present, IV calcium is the treatment of choice. Infusion of 15 mg/kg of elemental calcium over 4–6 h increases the total serum calcium by 0.5–0.75 mmol/L.
Administration of 10–20 mL of 10% calcium gluconate (89 mg elemental calcium per 10 mL) IV over 5–10 min is recommended. This should be followed by a continuous infusion because the effects of a single IV dose last only about 2 h. The infusion rate should be adjusted according to serial calcium measurements obtained every 2–4 h. Over-rapid infusion may cause facial flushing, headache and arrhythmias.
Calcium chloride 10% may also be used. This contains more calcium per ampoule (272 mg in 10 mL), resulting in a more rapid rise in serum calcium, but is more irritant to veins and can cause thrombophlebitis with extravasation.
Where hypcalcaemia and metabolic acidosis are present (usually in sepsis or renal failure), correction of the acidosis with bicarbonate may result in a rapid fall in ionized calcium as the number of calcium-binding sites is increased. Therefore, hypocalcaemia must be corrected before the acidosis. Bicarbonate or phosphate should not be infused with calcium because of possible precipitation of calcium salts.
Cardiac monitoring is recommended during rapid calcium administration, especially if the patient is taking digoxin, when calcium administration may precipitate digitalis toxicity.
If coexisting magnesium deficiency is suspected, or when symptoms do not improve after calcium administration, MgSO4 1–5 mmol IV over 15 min may be given.
Chronic asymptomatic hypocalcaemia
These patients are usually managed with oral calcium supplements taken between meals. Calcitriol, the active hormonal form of vitamin D, 0.5–1.5 mg daily, can also be given.
Hypercalcaemia
Introduction
The normal total serum calcium concentration is 2.15–2.55 mmol/L. Hypercalcaemia is a relatively common condition with a frequency estimated at 1:1000–1:10 000. Although there are many causes, the most frequent are malignancy and hyperparathyroidism, with the former the most likely to cause hypercalcaemia requiring urgent attention.
Pathophysiology
Total serum calcium is made up of protein-bound calcium (40%, mostly albumin and not filterable by the kidneys), ion-bound complexes (13%, bound to anions such as bicarbonate, lactate, citrate and phosphate) and the unbound, ionized fraction (47%). The ionized fraction is the biologically active component of calcium and is closely regulated by parathyroid hormone (PTH). Total serum calcium is affected by albumin and does not necessarily reflect the level of plasma ionized calcium. Normal ionized calcium levels are 1.14–1.30 mmol/L. Protein binding, in turn, is influenced by extracellular fluid pH and alterations in serum albumin. Acidaemia decreases protein binding and increases the level of ionized calcium. To correct for pH: ionized calcium rises 0.05 mmol/L for each 0.1 decrease in pH.
To correct for serum albumin:
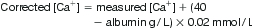
Corrected calcium is used for all treatment decisions except where direct measurement of ionized calcium using an ion-specific electrode is available.
Three pathophysiological mechanisms may produce hypercalcaemia:
Aetiology
The majority of cases of hypercalcaemia requiring urgent treatment are due to malignancy or, less commonly, primary hyperparathyroidism (parathyroid crisis). Malignant hypercalcaemia is most commonly seen with the solid tumours: lung and breast cancer, squamous cell carcinoma of the head and neck and cholangiocarcinoma and the haematological malignancies, multiple myeloma and lymphoma. Other causes of hypercalcaemia are uncommon (Table 12.2.13).
Table 12.2.13
Factitious
Haemoconcentration
Postprandial
Malignancy
Primary hyperparathyroidism
Drugs
Thiazides
Vitamin D
Lithium
Vitamin A
Hormonal
Thyrotoxicosis
Acromegaly
Hypoadrenalism
Phaeochromocytoma
Granulomas
Tuberculosis
Sarcoidosis
Renal failure
Milk alkali syndrome
Immobilization
Clinical features
Hypercalcaemia causes disturbances of the gastrointestinal, cardiovascular, renal and central nervous systems.
Gastrointestinal manifestations include anorexia, nausea, vomiting and constipation. Cardiovascular manifestations include hypertension and a shortened QT interval on the ECG. Renal manifestations include polyuria, polydipsia and nephrocalcinosis (rare). CNS symptoms include psychotic behaviour, seizures, apathy, cognitive difficulties, obtundation and coma. Renal elimination of digoxin is also impaired.
Moderately elevated total serum calcium (3.00–3.50 mmol/L) is usually associated with symptoms. Markedly elevated total serum calcium (>3.5 mmol/L) mandates urgent treatment regardless of symptoms.
Treatment
Irrespective of the cause, the management of hypercalcaemic crisis is the same. There are three primary treatment goals:


Hydration and diuresis
Hydration expands intravascular volume, dilutes calcium and increases calcium clearance. Infusion rates of 200–300 mL/h of 0.9% saline, depending on the degree of hypovolaemia and the ability of the patient to tolerate fluid, may be required and, once adequate rehydration has been achieved, the infusion rate can be adjusted to maintain a urine output of 100–150 mL/h.
This treatment, although effective, results in a relatively modest reduction in serum calcium and patients with severe hypercalcaemia usually require additional treatment with bisphosphonates.
The use of frusemide to enhance the excretion of calcium once the patient is adequately hydrated has fallen out of favour and should only be used to treate fluid overload.
Enhancement of renal excretion
Haemodialysis is the treatment of choice to decrease rapidly serum calcium in patients with heart failure or renal insufficiency
Inhibition of bone resorption
Pharmacological inhibition of osteoclastic bone resorption is the most effective treatment for hypercalcaemia, particularly hypercalcaemia of malignancy. Bisphosphonates, analogues of pyrophosphate, are the principal agents used. They inhibit osteoclast function and hydroxyapatite crystal dissolution. Unfortunately, normalization of calcium levels may take 3–6 days, which is too slow in critically ill patients.
Disodium pamidronate is currently one of the bisphosphonates of choice. The dose is 60 mg IV (in 500 mL 0.9% saline over 4 h) if serum calcium is<3.5 mmol/L, and 80 mg IV if serum calcium is>3.5 mmol/L. Calcium levels normalize in up to 80% of patients within 7 days and this effect can persist for up to a month. Common adverse reactions include a mild transient elevation in temperature, local infusion site reactions, mild gastrointestinal symptoms and mild hypophosphataemia, hypokalaemia and hypomagnesaemia.
An alternative treatment to pamidronate is zoledronic acid 4 mg/100 mL (N saline or 5% dextrose) IV over 15 minutes. It is more potent and effective than pamidronate.
Glucocorticoids, after rehydration, are the treatment of choice in selected patient populations where there is inappropriately high production of 1.25-dihydroxyvitamin D as the mechanism for causing hypercalcaemia. Such conditions include vitamin D toxicity, sarcoidosis, other granulomatous diseases and haematological malignancies, such as multiple myeloma and lymphoma. The usual dose is 200–300 mg hydrocortisone IV for 3–5 days. However, the maximal calcium-lowering effect does not occur for several days and glucocorticoids should only be regarded as adjunctive therapy in hypercalcaemic crises.
Treat the underlying disorder
The definitive treatment for hypercalcaemia is to treat the underlying disease: surgery for hyperparathyroidism and tumour-specific therapy for hypercalcaemia of malignancy.
Hypomagnesaemia
Introduction
The diagnosis of magnesium deficiency is difficult and often overlooked largely because the symptoms are non-specific and do not usually appear until the patient is severely deficient.
Serum magnesium concentration (normal range: 0.76–0.96 mmol/L) is not a sensitive indicator of magnesium deficiency as it may not truly reflect total body stores. However, it is commonly used in the absence of other reliable methods to estimate the ‘true’ magnesium status. A low serum magnesium concentration is usually present in symptomatic magnesium deficiency, but it is important to remember that it may be normal in the presence of significant intracellular depletion.
Pathophysiology
Magnesium plays a critical role in metabolism: as an enzyme co-factor, in the maintenance of cell membranes and in electrolyte balance. It is the fourth most common cation in the body and is predominantly an intracellular ion with the majority found in bone (>50%) and soft tissue. Only 0.3% of total body magnesium is located extracellularly, of which 33% is protein bound, 12% is complexed to anions, such as citrate, bicarbonate and phosphate, and 55% is found in the free ionized form.
Hypokalaemia is present in 40–60% of cases of magnesium deficiency, due to renal wasting of potassium. The hypokalaemia is resistant to potassium replacement alone, as a result of a combination of factors, including impaired cellular cation pump activity and increased cellular permeability to potassium.
Hypocalcaemia is usually present at serum magnesium concentrations below 0.49 mmol/L. This may be due to impaired PTH synthesis or secretion or to PTH resistance as a result of magnesium deficiency.
Aetiology
From an emergency medicine perspective, hypomagnesaemia is most frequently encountered in the context of acute and chronic diarrhoea, acute pancreatitis, diuretic use, in alcoholics and in diabetic ketoacidosis, secondary to glycosuria and osmotic diuresis. Table 12.2.14 details causes of magnesium deficiency.
Table 12.2.14
Causes of magnesium deficiency
Gastrointestinal losses
Acute and chronic diarrhoea
Acute pancreatitis
Severe malnutrition
Intestinal fistulae
Extensive bowel resection
Prolonged nasogastric suction
Renal losses
Osmotic diuresis – diabetes, urea, mannitol
Hypercalcaemia and hypercalciuria
Volume expanded states
Chronic parenteral fluid therapy
Drugs
ACE inhibitors
Alcohol
Aminoglycosides
Amphotericin B
Cisplatin
Ciclosporin
Diuretics – thiazide or loop
Other
Phosphate depletion
From Weisinger JR, Bellorin-Font E. Magnesium and phosphorus-electrolyte quintet. Lancet 1998; 352:391–6 with permission.
Hypomagnesaemia has been found in 30% of alcoholics admitted to hospital and results from a combination of the direct effect of alcohol on the renal tubule, which increases magnesium excretion, and associated malnutrition, diarrhoea and metabolic acidosis.
Clinical features
The clinical manifestations of severe magnesium deficiency include metabolic, neurological and cardiac effects (Table 12.2.15).
Table 12.2.15
Clinical manifestations of severe magnesium deficiency
| Cardiac effects | Metabolic effects | Neurological effects |
| Atrial fibrillation | Hypokalaemia | Grand mal seizures |
| Atrial flutter | Hypocalcaemia | Focal seizures |
| Supraventricular tachycardia | Hyponatraemia | Paraesthesias |
| Ventricular tachycardia | Hypophosphataemia | Dizziness |
| Torsades des pointes | Metabolic alkalosis | Vertigo |
| Coronary artery spasm | Hyperglycaemia | Ataxia |
| Hypertension | Hyperlipidaemia | Nystagmus |
| ECG changes | Tremor | |
| Atherosclerosis | Myopathy | |
| Dysphagia | ||
| Oesophageal spasm | ||
| Delirium, personality changes | ||
| Depression | ||
| Coma |
From Fawcett WJ, Haxby EJ, Male DA. Magnesium: physiology and pharmacology. Br J Anaesth 1999; 83:302–20 with permission.
The presenting symptoms are non-specific and can be attributed to associated metabolic abnormalities, such as hypocalcaemia, hypokalaemia and metabolic alkalosis. In particular, patients may present with symptoms of hypocalcaemia: neuromuscular hyperexcitability, carpopedal spasm and positive Chvostek’s and Trousseau’s signs.
Early ECG changes of magnesium deficiency include prolongation of the PR and QT intervals, with progressive QRS widening and U-wave appearance as severity progresses. Changes in cardiac automaticity and conduction, atrial and ventricular arrhythmias, including torsades des pointes, can occur. Administration of a magnesium bolus can abolish torsades des pointes, even in the presence of normal serum magnesium levels. Magnesium is a co-factor in the Na-K-ATPase system and so magnesium deficiency enhances myocardial sensitivity to digitalis and may precipitate digitalis toxicity. Digitalis-toxic arrhythmias, in turn, can be terminated with intravenous magnesium.
Treatment
Oral replacement is the preferred option in asymptomatic patients, although this route takes longer.
Symptomatic moderate-to-severe magnesium deficiency should be treated with parenteral magnesium salts. The patient should be closely monitored and therapy discontinued if deep tendon reflexes disappear or serum magnesium exceeds 2.5 mmol/L. Suggested dosing regimens are outlined in Table 12.2.16.
Table 12.2.16
Magnesium doses (in mmol magnesium)
Emergency – IV route
8–16 mmol statim
40 mmol over next 5 h
Severely ill – IM route
48 mmol on day 1
17–25 mmol on days 2–5
Asymptomatic – oral route
15 mmol/day
Hypermagnesaemia
Hypermagnesaemia (serum magnesium above 0.95 mmol/L) is rare and usually iatrogenic.
The elderly and patients with renal impairment or chronic bowel disorders are particularly at risk, especially when IV magnesium or magnesium-containing cathartics or antacids are used.
Clinical manifestations include mental obtundation progressing to coma, cardiac arrhythmias, loss of deep tendon reflexes, refractory hypotension and respiratory arrest, nausea and vomiting, muscle paralysis and flushing.
Magnesium administration should be immediately discontinued. Further management is largely supportive. Maintain urine output at greater than 60 mL/h with fluid administration to enhance renal excretion. Furosemide (40–80 mg IV) may also be given once the patient is adequately hydrated. Haemodialysis may be of benefit in severe cases, particularly if there is impaired renal function.
Controversies