Endocrine Emergencies
Edited by Anthony Brown
11.1 Diabetes mellitus and hypoglycaemia: an overview
Anthony Brown
Diabetes mellitus
Classification system and diagnostic criteria
The classification system and diagnostic criteria for diabetes were revised in 2010 by the American Diabetes Association [1]. The classification of type I and type II diabetes mellitus was retained, with the recommended criteria for the diagnosis of diabetes as a fasting plasma glucose of 7 mmol/L or greater, or a random plasma glucose of over 11 mmol/L associated with polyuria, polydipsia and weight loss. In addition, an HbA1C (glycated haemoglobin) level of≥6.5% was added. The oral glucose tolerance test is no longer routinely recommended.
Aetiology
The exact aetiology of diabetes is unclear. Type I diabetes is characterized by pancreatic beta cell destruction with an absolute insulin deficiency usually, but not exclusively, associated with autoimmune damage from a range of antibodies including to islet cells (ICA), glutamic acid decarboxylase (GAD), insulin and tyrosine phosphatases. Genetic and environmental factors are implicated, such as some human leucocyte antigen (HLA) types (most Caucasian patients are HLA-DR3 or DR4 or both), and abnormal immune responses, such as following viral infection. Certain genes are also implicated as co-contributors, particularly sites on chromosomes 6, 7, 11, 14 and 18.
Type II diabetes results from a progressive insulin secretory deficiency on the background of insulin resistance [1]. Genetic factors are implied by strong familial aggregation of cases and environmental factors in the context of genetic susceptibility, including obesity and diet. Thus, the introduction of a high fat and high calorie ‘Western’ diet rather than traditional crop foods has seen countries such as India record among the fastest growth rate of new diabetes worldwide.
Although type I diabetes occurs most frequently among Caucasians throughout the world, diabetes in Australia is three times more common in the Aboriginal community. Other groups with a high prevalence include Pacific Islanders and Native Americans.
Diabetes secondary to other conditions
Diabetes mellitus may be secondary to conditions that damage the exocrine pancreas including chronic pancreatitis, carcinoma of the pancreas and pancreatectomy, haemochromatosis, cystic fibrosis, pregnancy (gestational) and endocrinopathies, such as Cushing’s syndrome, acromegaly, phaeochromocytoma and glucagonoma [2].
Drug-induced diabetic state
Certain drugs can impair glucose tolerance or cause overt diabetes mellitus. These include glucocorticoids, the oral contraceptive pill, thiazide diuretics at higher doses, tacrolimus, sirolimus and ciclosporin, pentamadine (which may also cause hypoglycaemia) and HIV protease inhibitors.
Emergency presentations of a high blood sugar
Diabetic ketoacidosis (DKA) and hyperosmolar hyperglycaemic state (HHS) are both life-threatening acute complications of diabetes mellitus. Although important differences do exist, the pathophysiology and treatment are similar. DKA is usually seen in type I diabetes and HHS in patients with type II, but both complications can occur in type I and type II diabetes. See Chapter 11.2 for the diagnosis and management of DKA and HHS.
General management of diabetes mellitus
Aims of long-term blood sugar control
The aim of optimal long-term blood sugar control is an HbA1c (glycated haemoglobin) level of less than 7.5% without frequent disabling hypoglycaemia for the prevention of microvascular disease, and 6.5% in those at increased risk of arterial disease [3]. This should be represented by a pre-prandial blood glucose level of 4.0–7.0 mmol/L and a post-prandial blood glucose level of less than 9.0 mmol/L.
Insulins
Insulin was first administered to humans in 1922. Animal insulins (bovine, porcine) have been used for many years but, in the 1980s, human insulins became commercially available. Today, with the widespread availability of human insulins, animal insulins are of historical interest only.
Types of insulins
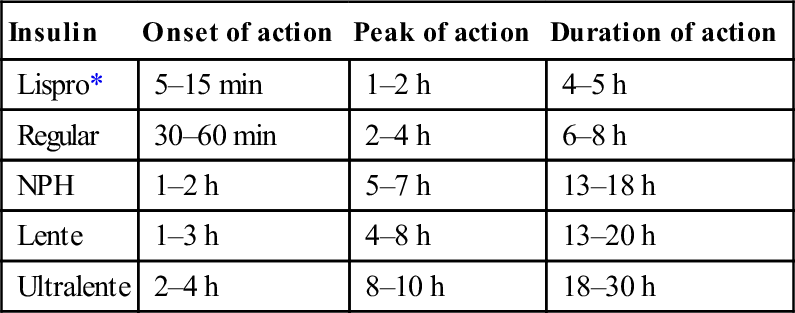
Table 11.1.1 lists the different types of insulins and the important parameters of each type. Mixtures of short- and intermediate-acting insulins are also available: 70/30 (70% NPH/30% regular) and 50/50 (50% NPH/50% regular).
Table 11.1.1
Pharmacokinetic characteristics of currently available human insulins
| Insulin | Onset of action | Peak of action | Duration of action |
| Lispro* | 5–15 min | 1–2 h | 4–5 h |
| Regular | 30–60 min | 2–4 h | 6–8 h |
| NPH | 1–2 h | 5–7 h | 13–18 h |
| Lente | 1–3 h | 4–8 h | 13–20 h |
| Ultralente | 2–4 h | 8–10 h | 18–30 h |
Antidiabetic drugs
Two major groups of oral hypoglycaemic agents used in the management of type II diabetes are the sulphonylureas and the biguanides. The sulphonylurea group of drugs acts by stimulating the pancreatic secretion of insulin, and the biguanide metformin acts by suppressing hepatic glucose production and enhancing the peripheral use of glucose. It is the first choice medication particularly in the overweight patient.
Newer types of oral agents now available for the treatment of diabetes include the alpha-glucosidase inhibitor acarbose, which acts on the gastrointestinal tract to interfere with carbohydrate digestion, but flatulence and diarrhea may be troublesome. The thiazolidinediones, such as pioglitazone and rosiglitazone, act primarily by reducing insulin resistance, thereby enhancing the effect of circulating insulin. Roziglitazone increases the risk of myocardial infarction and cardiovascular deaths and thus should be avoided in ischaemic heart disease [4]. All thiazolidinediones must be avoided in people with moderate or severe heart failure. Finally, the dipeptidyl peptidase 4 (DDP-4) inhibitors, such as sitagliptin, inhibit DDP-4 to prolong the action of the incretin hormones.
Other non-diabetic drugs
Angiotensin-converting enzyme inhibitors delay the onset of diabetic nephropathy even in normotensive patients with diabetes. Statins are important in the strict treatment of dyslipidaemia in diabetic patients.
Diabetic hypoglycaemia
Hypoglycaemia is more common in type I diabetes. The critical plasma level at which hypoglycaemia manifests varies between different individuals, but symptoms are likely below a plasma glucose of 3.5 mmol/L. Precipitants include exercise, a late meal, inadequate carbohydrate intake, errors of insulin dosage and ethanol ingestion.
Hypoglycaemia may also occur in the non-diabetic patient, precipitated by a variety of conditions (Table 11.1.2) [5].
Table 11.1.2
| Diabetic patients |

Clinical features
Hypoglycaemia produces neurological and mental dysfunction from tremor, sweating and anxiety to seizures and coma. Less commonly, it can present as hypothermia, depression and psychosis. In some instances, hypoglycaemia is relatively asymptomatic.
Management of hypoglycaemic coma
11.2 Diabetic ketoacidosis and hyperosmolar, hyperglycaemic state
Anthony Brown and Richard D Hardern
Essentials
3 Key management components of DKA include:
 fluids (0.9% normal saline) to replace deficits of sodium of 7–10 mmol/kg and water 100 mL/kg
fluids (0.9% normal saline) to replace deficits of sodium of 7–10 mmol/kg and water 100 mL/kg
4 Meticulous monitoring and documentation of treatment in DKA are essential.
7 Treatment of HHS is similar to DKA except:
 lower dose insulin infusion rate is used at 0.05 unit/kg/h
lower dose insulin infusion rate is used at 0.05 unit/kg/h
 this infusion rate is titrated against the serum osmolarity rather than ketoacids
this infusion rate is titrated against the serum osmolarity rather than ketoacids
 low molecular weight heparin (LMWH) thromboprophylaxis is indicated.
low molecular weight heparin (LMWH) thromboprophylaxis is indicated.
Introduction
Diabetic ketoacidosis (DKA) is an acute, potentially life-threatening complication in an insulin-dependent diabetic and in some type II diabetics. It consists of the triad of ketonaemia, hyperglycaemia and acidaemia – a high anion-gap metabolic acidosis. Although DKA is preventable, its prevalence and suboptimal management may highlight shortfalls in the quality of care for patients with diabetes. The mortality rate in developed countries has dropped to<1%.
Hyperosmolar hyperglycaemic state (HHS) is characterized by hypovolaemia, marked hyperglycaemia (>30 mmol/L) without ketonaemia or acidosis and a raised osmolality usually>320 mosmol/kg. It comes on more insidiously and has a worse prognosis with an increased mortality of around 15–20% and greater morbidity, in part related to underlying chronic medical disorders.
Epidemiology, aetiology and pathogenesis
An annual incidence of DKA of approximately 1:170 patients with type I diabetes is reported, or 2 episodes per 100 patient years of diabetes, for a prevalence of 4.6–8 episodes per 1000 patients with diabetes [1,2].
DKA may be the presenting feature of diabetes mellitus (3–25% DKA), but is usually seen in patients with autoimmune type I diabetes when there has been an insulin error with inadequate insulin or poor compliance (30% DKA), and/or an intercurrent illness (35% DKA). A variant of type II diabetes is also ketosis prone, ‘ketosis-prone (type II) diabetes’, usually in the obese with a strong family history. This was originally described in Africans and African-Americans, but is now noted worldwide.
Pathogenesis
DKA arises from an absolute or relative lack of insulin accompanied by an increase in counter-regulatory hormones, such as glucagon, cortisol and growth hormone [1,3]. Insulin absence leads to increased hepatic gluconeogenesis and glycogenolysis, with an incomplete lack of insulin related to greater hyperosmolarity (in HHS). Lack of insulin and excess counter-regulatory hormones increase lipolysis and free fatty acid production as an alternate energy source. This leads to subsequent ketone body formation produced from acetyl coenzyme A, mainly in hepatic mitochondria, including acetone, beta-hydroxybutyrate and acetoacetate and a reduced ability to prevent ketonaemia.
HHS is the other end of the hyperglycaemia spectrum from DKA, occurring with a relative rather than an absolute deficiency of insulin leading to a greater level of hyperglycaemia, therefore higher hyperosmolarity than is seen in DKA. The degree of dehydration is greater (typically 10–15% body weight), but significant ketosis does not occur. HHS is more insidious in onset than DKA and patients with HHS are typically older with pre-existing type II diabetes. However, HHS is seen in young adults and even teenagers.
Clinical features
Malaise and fatigue on a background of polyuria, polydipsia, weakness and fatigability are common, but gastrointestinal symptoms, such as nausea, vomiting and abdominal pain, may predominate [3]. A lack of history of diabetes does not rule out the diagnosis of DKA, as it may be a first presentation, often presaged by recent, unexplained rapid weight loss.
Laboured, sighing respirations with an increased rate and depth, known as Kussmaul breathing, are characteristic of DKA in association with dehydration causing decreased tissue turgor, a dry mouth and sweet foetor of pear drops (ketotic), which is not always noticed. The conscious level may be reduced, but coma is rare.
Look carefully for signs of an underlying precipitating cause including chest, urine or skin infection, such as boils, as well as meningitis or an acute abdomen, although non-surgical upper abdominal pain is common in DKA. In those with HHS, look out for the complications of acute myocardial infarction, stroke or arterial thrombosis [4,5].
The urine output should be measured regularly, which does not always require urinary catheterization. Likewise, invasive haemodynamic monitoring should not be instituted as a ‘routine’ for patients with DKA or HHS. It should be reserved for those who fail to respond or in the elderly who are at risk of fluid overload. In addition, venous blood gases are sufficient to monitor progress in DKA, rather than repeated arterial sampling.
Diagnostic criteria
DKA
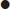
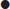
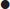
HHS
Note that there is no precise definition of HHS, but it is characterized by [5]:
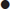
Typical deficits per body weight
DKA
HHS
Investigations
Venous blood
Measure capillary blood glucose hourly until near to the normal range, or measure venous urea and electrolytes (U&Es), glucose and pH initially hourly, then 2-hourly once the venous glucose and capillary glucose are in agreement.
A mild leucocytosis is common in DKA, which should not be interpreted as signifying infection. Likewise hyperamylasaemia is common and does not imply pancreatitis.
Urinalysis
DKA is highly likely in the presence of glycosuria and ketonuria in an unwell patient.
Electrocardiograph (ECG)
Perform an early ECG to look for T-wave changes as a first indicator of hyperkalaemia (tall and peaked) or hypokalaemia (flat or inverted), or a clinically silent myocardial infarction (Ml) as a precipitant of HHS or DKA.
Point-of-care testing
Point-of-care testing for blood ketones, such as beta-hydroxybutyrate, is helpful at triage and when monitoring the response to treatment.
Differential diagnosis of DKA
Other causes of high (greater than 16) anion-gap metabolic acidosis include:
 alcoholic or starvation ketoacidosis
alcoholic or starvation ketoacidosis
 lactic acidosis (multiple causes)
lactic acidosis (multiple causes)
 methanol, ethylene glycol (note ethanol predominantly leads to a lactic acidosis)
methanol, ethylene glycol (note ethanol predominantly leads to a lactic acidosis)
Other causes of hyperglycaemia include:
 drugs, such as corticosteroids, octreotide, thiazide diuretics, ritonavir, diazoxide and atypical antipsychotics (clozapine, olanzapine, risperidone, quetiapine – these may actually precipitate DKA soon after commencement, even in the absence of weight gain) [6]
drugs, such as corticosteroids, octreotide, thiazide diuretics, ritonavir, diazoxide and atypical antipsychotics (clozapine, olanzapine, risperidone, quetiapine – these may actually precipitate DKA soon after commencement, even in the absence of weight gain) [6]
 endocrine, such as Cushing’s, acromegaly, phaeochromocytoma, glucagonoma, VIPoma.
endocrine, such as Cushing’s, acromegaly, phaeochromocytoma, glucagonoma, VIPoma.
Management
Diabetic ketoacidosis
The treatment of DKA is rigorous and requires careful monitoring of the patient, both clinically and biochemically. Ideally, all observations and results are entered onto a purpose-designed record sheet, such as an integrated care pathway that includes data recording and guidance [7].
Severe DKA
Severe DKA necessitating intensive management includes a venous or arterial pH<7.1, bicarbonate<5 mmol/L, hypokalaemia on admission (<3.5 mmol/L), Glasgow coma scale (GCS)<12, systolic BP<90 mmHg and SaO2<92% on room air.
Fluid regimen
Intravenous fluids should be started within 30 min of the patient’s arrival in the ED. A shocked patient should receive a fluid bolus on arrival to restore perfusion, although care is needed in patients with co-morbidities, such as the elderly with heart or renal impairment, to avoid fluid overload.
Fluid rate
Give patients who are not shocked 1 L 0.9% normal saline over 1 hour, then at a rate of 500 mL/h for 4 h with added potassium, then 250 mL/h for the next 8 h, again with added potassium [1]. A suggested fluid regimen is shown in Table 11.2.1.
Table 11.2.1
| Litre | Time (hours from starting treatment) |
| First at 1000 mL/h | 0–1 |
| Second at 500 mL/h+K | 1–3 |
| Third at 500 mL/h+K | 3–5 |
| Fourth at 250 mL/h+K | 5–9 |
| Fifth at 250 mL/h+K | 9–13 |
| Reassess cardiovascular status at 12 h and adjust rate accordingly | |
| Sixth at 166 mL/h+K | 13–19 |
Most intravenous fluid regimens recommend replacing the volume deficit (often 10% of body weight) over 24 h. However, in a patient with significant co-morbidites, it is prudent (although unproven) to aim to correct half the fluid deficit in the first 24 h and the remainder in the next 24 h. Likewise, adopt a slower rate initially in a small, young adult aged 18–25 years with the total volume replaced over 24–48 h to reduce the risk of cerebral oedema (see Table 11.2.1) [1].
Cerebral oedema
The incidence of cerebral oedema is greatest in children under 12 years, possibly related to a lower pH/PaCO2 and a higher potassium and urea at presentation, and smaller increases in serum sodium. The aetiology is unclear but may result from cerebral hypoperfusion followed by reperfusion, with an increased risk associated with early (in first hour) insulin administration (odds ratio OR 12.7) and large volumes of fluid in the first 4 h (OR 6.55) [8].
Choice of fluid
There are no data from randomized controlled trials to support the choice of one crystalloid over another in the treatment of DKA. The risk of hyperchloraemic acidosis from the use of large volumes of normal saline with renal vasoconstriction and slowing of resolution of acidosis is not clinically significant. In addition, colloids are a less physiological replacement for electrolyte losses and are not recommended.
Addition of 10% dextrose
If serum [glucose] falls to<15 mmol/L, add 10% dextrose at 125 mL/h alongside the 0.9% saline infusion, with continuance of the insulin infusion until the electrolyte and volume losses have been replaced and the ketoacidosis/ketonaemia has been cleared.
Insulin
An intravenous insulin infusion should be started within 60 min of the patient’s arrival in the ED.
Insulin infusion regimen
A standard regimen is an infusion of soluble insulin, made up by adding 50 units of soluble insulin to a total of 50 mL with 0.9% saline to produce a solution containing 1 unit/mL. Remember when prescribing insulin always to write ‘units’ in full rather than as ‘u’, as the latter is too easily confused with a 0 (zero) and a 10-fold dose increase can be given in error.
Run the infusion at an initial rate of 0.1 units/kg/h (to a maximum of 6 units/h). Adjust the rate to reduce the serum [glucose] by around 3 mmol/L/h, with a rise in the serum bicarbonate of at least 3 mmol/L/h.
When the serum [glucose] is less than 15 mmol/L, halve the insulin infusion rate and then adjust it to maintain the serum [glucose] between 9 and 14 mmol/L [7].
Initial hypokalaemia
Severe hypokalaemia is associated with arrhythmias and sudden death in DKA. Do not start an insulin infusion until the serum [potassium] has been checked to make sure it is not below the lower limit of the reference range, i.e. it should be greater than 3.4 mmol/L. If it is below this, begin the IV fluid with potassium first, prior to commencing the insulin infusion.
Insulin bolus
Although there are few data on the benefits or harm of giving a bolus of insulin before starting an infusion, its short half-life makes this unnecessary. Providing the insulin infusion is prepared and started with minimal delay, there is no justification for a bolus (despite a bolus appearing in many published guidelines).
Intermittent insulin
Switching from an insulin infusion to intermittent insulin is unlikely to occur until the patient is on the medical ward. If ED staff do supervise cessation of an insulin infusion, ensure the first subcutaneous insulin dose is given at least 1 h before the infusion is stopped in association with a meal, providing all the following criteria have been met before ceasing the insulin infusion:
Potassium replacement
Hyperkalaemia and then hypokalaemia are the most common life-threatening electrolyte problems seen in DKA. Therefore the serum [K] must be monitored closely and treatment planned to treat either condition rapidly, particularly the risk of hypokalaemia. A typical total body deficit of potassium in DKA is 3–5 mmol/kg [1,2].
Hyperkalaemia from intracellular shift from the acidosis and lack of insulin seen in the early phase of DKA may cause life-threatening dysrhythmias. This hyperkalaemia rapidly resolves soon after fluid and insulin commencement.
Conversely, add potassium to intravenous fluids once the serum [K] is below 5.5 mmol/L and the patient is passing urine. However, do not add potassium to the first fluid bolus infused rapidly for volume resuscitation.
Infusion rate
Replacing potassium at 10–20 mmol/h is usually sufficient, with the rate adjusted to the serum [K] measurement, with the aim of maintaining serum [K] in the range 4–5 mmol/L. Use premixed intravenous potassiuim in fluid bags with an intravenous fluid infusor to avoid dosing or infusion rate errors.
Education
Arguably, all episodes of DKA represent a failure of patient education, except for those patients in whom DKA is the first presentation of diabetes mellitus. All patients treated with insulin must understand ‘sick day rules’ to increase their normal insulin dose by 4 units or more when they have an intercurrent illness, even if they are not eating, as their insulin requirements will rise. Stopping insulin because a person is ‘not eating properly’ is all too common and an entirely avoidable precipitant of DKA.
Hyperosmolar, hyperglycaemic state
The management of HHS is similar to that of DKA, although patients are older, the water deficit is considerably greater, the sodium and potassium deficits greater and the overall mortality and moribidity higher. Focal or global neurological changes may occur, including an altered conscious level, coma, seizures and stroke (cause or effect).
Give 0.9% normal saline for initial volume resuscitation, with insulin by intravenous infusion and potassium supplementation to maintain serum [K] between 4 and 5 mmol/L, similar to DKA. In addition, the fall in serum osmolality is monitored as a marker of response to treatment.
Severe HHS
Severe HHS necessitating intensive management includes an osmolality>350 mosmol/kg, sodium>160 mmol/L, creatinine>200 μmol/L or urine output<0.5 mL/kg/h, hypokalaemia on admission (<3.5 mmol/L), GCS<12, systolic BP<90 mmHg, SaO2<92% on room air and a venous or arterial pH<7.1 (look for other causes, such as a concomitant lactic acidosis).
Management differences in HHS (to DKA)
Insulin infusion
Start fluid replacement first, before commencing an insulin infusion at 0.05 units/kg/h to a maximum of 3 units, as a patient with HHS may be more sensitive to insulin, plus the replacement of fluid alone will lead to a fall in serum glucose. Aim for a rate of decline in serum osmolarity of less than 3 mOsm/kg/h and in blood glucose of not more than 5 mmol/L/h.
Reduce the insulin infusion rate when the serum [glucose] drops to 15–18 mmol/L, to maintain serum [glucose] in the range 10–15 mmol/L until the serum osmolarity is less than 315 mOsm/kg. Complete normalization of osmolarity and electrolytes may take up to 72 h.
Fluid choice
Use 0.9% normal saline as the principal fluid to restore circulation volume [5]. An initial rise in serum sodium may occur due to a shift of water intracellularly from a lowering of the blood glucose, which is not an indication to use a more hypotonic solution (normal saline is already relatively hypotonic compared to serum in HHS).
Only change to 0.45% half-normal saline if the serum osmolality and blood sugar are not reducing. Avoid a rapid fall in serum sodium, which should not exceed 10 mmol/L per 24 h [5].
Other considerations
Miscellaneous issues
There are no data to support the use of phosphate or magnesium in the treatment of DKA or HHS, despite there often being hypophosphataemia and hypomagnesaemia.
Heparin thromboprophylaxis is not used routinely in DKA, nor are antibiotics in the absence of a focus of infection or sepsis, even though it is common for the white cell count to be mildly elevated.
Sodium bicarbonate should never be used in DKA if the pH is greater than 7.0 and even below that level its value is unproven. Significant disadvantages of giving IV bicarbonate are a rapid fall in serum [K], worsened intracellular acidosis, reduced tissue oxygen delivery, delay in clearing ketones and a possible association with cerebral oedema.
11.3 Thyroid and adrenal emergencies
Andrew Maclean
Introduction
Four conditions are covered in this chapter: thyrotoxicosis, hypothyroidism, hypoadrenal states and hyperadrenal states. Patients with the first three present relatively infrequently to emergency departments (EDs), but all four conditions are potentially fatal if they go unrecognized and untreated. The most common cause of Cushing’s syndrome is exogenous steroid administration. An inability to produce endogenous steroids in times of physiological stress and therefore the potential for adrenal insufficiency occurring with insufficient replacement therapy must be considered in such patients.
Thyrotoxicosis
Aetiology, genetics, pathogenesis and pathology
Normal secretion of thyroid hormone relies on an intact feedback loop involving the hypothalamus, pituitary gland and thyroid gland. Thyrotropin-releasing hormone (TRH) released from the hypothalamus stimulates thyroid-stimulating hormone (TSH) production in the anterior pituitary, which stimulates thyroid hormone release from thyroid follicular cells. Thyroid hormones suppress TRH and TSH production. Thyroid hormones act at a cellular level, binding with nuclear receptors to enable gene expression and protein synthesis. Thyroid hormone may also have an effect on modulating cellular metabolism.
There are a number of pathological causes of thyrotoxicosis (Table 11.3.1). Graves’ disease is an autoimmune condition related to a combination of genetic and environmental factors, including iodine intake, stress and smoking. The thyrotoxicosis of Graves’ disease is caused by autoantibodies, which stimulate the thyroid resulting in excess thyroid hormone production.
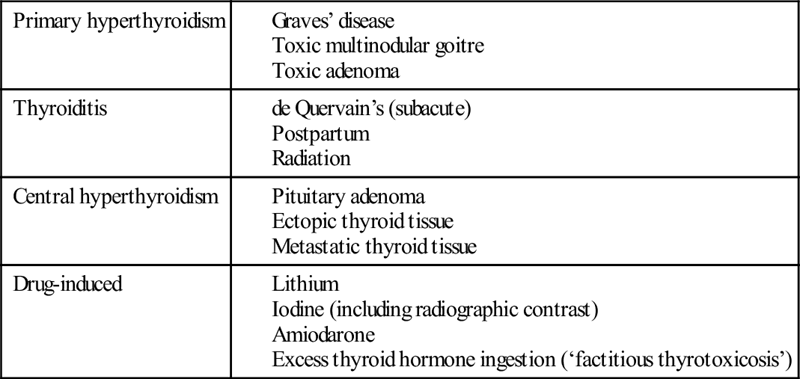
Thyroiditis may be acute (rare), subacute or chronic. Inflammation of the thyroid is associated with damage to follicles with release of thyroid hormone. Subacute thyroiditis (de Quervain’s, also known as subacute granulomatous) may follow a viral infection and is typically painful, with localized tenderness and neck pain, sometimes with odynophagia.
Multinodular goitre occurs in areas of both iodine deficiency and sufficiency, indicating that a multiplicity of genetic and environmental factors are at play. Fibrosis, hypercellularity and colloid cysts are the main pathological findings.
Epidemiology
Graves’ disease accounts for at least 80% of cases of thyrotoxicosis [1]. The prevalence increases in areas with high iodine intake. Graves’ disease has a strong female predominance, affecting up to 2% of all women [1,2]. Thyrotoxicosis due to Graves’ disease usually occurs in the second to fourth decades of life, whereas the prevalence of a toxic nodular goitre increases with age.
Clinical features
The signs and symptoms of hyperthyroidism are secondary to the effects of excess thyroid hormone in the circulation. The severity of the signs and symptoms is related to the duration of the illness, the magnitude of the hormone excess and the age of the patient. These symptoms and signs are summarized in Table 11.3.2, which illustrates the wide spectrum of possible clinical features.
Table 11.3.2
Clinical features of thyrotoxicosis
Nervousness, irritability
Heat intolerance and increased sweating
Tremor
Weight loss and alteration in appetite
Palpitations and tachycardia, particularly atrial fibrillation
Widened pulse pressure
Exertional intolerance and dyspnoea
Frequent bowel movements
Fatigue and muscle weakness
Thyroid enlargement (depending on cause)
Pretibial myxoedema (with Graves’ disease)
Menstrual disturbance and impaired fertility
Mental changes
Sleep disturbances
Changes in vision, photophobia, eye irritation, diplopia, lid lag or exophthalmos
Dependent lower extremity oedema
Sudden paralysis, with or without hypokalaemia
A comprehensive history and physical examination should be performed, with particular attention to weight, blood pressure, pulse rate and rhythm, looking specifically for cardiac failure, palpation and auscultation of the thyroid to determine thyroid size, nodularity and vascularity, neuromuscular examination and an eye examination for evidence of exophthalmos or ophthalmoplegia.
Clinical investigations and criteria for diagnosis [1–3]
The TSH level is the single best screening test for hyperthyroidism. The recent development of sensitive TSH assays has greatly facilitated the diagnosis of hyperthyroidism. Hyperthyroidism of any cause (except excess TSH production from the anterior pituitary) results in a lower than normal TSH. The reference range is 0.4–5.0 mIU/L depending on the method.
Other laboratory and isotope tests may include:
Treatment
Mild hyperthyroidism does not require any treatment in the ED and the patient may simply be referred to an appropriate outpatient clinic. Any features of thyroid storm (see below) mandate admission, as does any significant intercurrent illness. Atrial arrhythmias should be controlled by the use of β-blockers, aiming to achieve a rate of less than 100 beats per minute.
Ensure that all bloods have been collected first if thyroid-blocking drugs are to be commenced in the ED. High doses of thyroid-blocking drugs are often required to gain an initial response, after which the dose can be tapered.
Commence carbimazole 10–45 mg daily or propylthiouracil 200–600 mg daily in two or three divided doses initially, using the larger doses for more severe cases [4]. Ideally, discuss initiation of these agents with the physician who will continue managing the patient after his or her discharge from the ED. Ninety per cent of patients will be controlled within weeks using these drugs [5]. Treatment for 12–18 months will result in a long-term remission in 40–60% of patients with Graves’ disease [2].
Thyroid storm
Aetiology
Thyroid storm occurs in about 1% of patients with hyperthyroidism. It usually occurs as an acute deterioration in a patient with poorly controlled or undiagnosed hyperthyroidism, precipitated by factors such as surgery, trauma, infection, radioiodine treatment, use of iodinated contrast, exogenous thyroxine ingestion or any other significant stressor.
The diagnosis is entirely clinical, as there is no test to differentiate a thyroid storm from thyrotoxicosis. The mortality rate if untreated or if the diagnosis is missed is over 90%. Death is usually due to cardiovascular collapse.
Clinical features of a thyroid storm
The symptoms and signs of thyrotoxicosis are present and significantly exaggerated, with the abrupt onset of a combination of the following:
Differential diagnosis
The following differential diagnoses of a thyroid storm need to be considered:
Treatment
The treatment of thyroid storm is directed to blocking thyroid hormone synthesis and release, the peripheral effects of the thyroid hormones, and corticosteroids.
β-Blockers
β-Blockade is the most important factor in decreasing morbidity and mortality. Many of the peripheral manifestations of hyperthyroidism, in particular the cardiovascular effects, are reduced by the use of propranolol. Propranolol also inhibits the peripheral conversion of T4 to T3 as well as antagonizing the effects of thyroid hormones and the hypersensitivity to catecholamines.
Give intravenous increments of 0.5 mg initially up to 10 mg total with continuous cardiovascular monitoring. Subsequent doses of 40–120 mg 6-hourly orally can be given.
β-Blockers should treat the cardiac failure secondary to the tachyarrhythmia or high cardiac output, but may cause complications in patients with pre-existing heart disease or asthma.
In this situation, use the short-acting β-blocker esmolol, as any adverse effects will be of brief duration. Give a 250–500 μg/kg bolus followed by an infusion starting at 50–100 μg/kg/min titrated to effect. Another option is to use the combination of a β-blocker and digoxin.
Thyroid-blocking drugs
Give propylthiouracil 900–1200 mg loading dose orally or via a nasogastric tube if necessary. This is followed by 200–300 mg 4–6-hourly. Propylthiouracil acts by preventing hormone synthesis by blocking the iodination of tyrosine and also inhibits the peripheral conversion of T4 to T3.
Iodine in large doses inhibits the synthesis and release of thyroid hormones and may be given either orally as Lugol’s iodine, 30–60 drops daily in divided doses, or intravenously as sodium iodide 1 g 12-hourly. Lithium carbonate may be used in patients allergic to iodine or be added when there is difficulty with control [4].
Cholestyramine can also be considered, which acts by binding with thyroxine after biliary excretion and hence increasing elimination.
Corticosteroids
Corticosteroids are given to inhibit the peripheral conversion of T4 to T3 and as a relative deficiency may also be present. Hydrocortisone 100 mg IV 6-hourly or dexamethasone 4 mg IV 12-hourly are used.
General supportive measures
Dehydration and electrolyte disturbances need correction. Aggressive treatment of hyperthermia with cooling measures and paracetamol are necessary, but induction of shivering should be avoided. Salicylates are contraindicated as they displace T4 from binding proteins. In addition, it is essential to look for and treat any precipitating cause, which will improve the prognosis.
Prognosis
Mortality rates are high, at 10–75% despite treatment [8].
Apathetic hyperthyroidism
Patients with this condition are generally older, although it has been recorded in all age groups. The clinical picture is of a depressed mental state with cardiac complications, in particular cardiac failure. Weight loss is usually not significant and eye signs are rare. Most of the usual hyperkinetic manifestations of hyperthyroidism are absent. Treatment is as for standard hyperthyroidism.
Hypothyroidism
Aetiology, genetics, pathogenesis and pathology
Hypothyroidism results from undersecretion of thyroid hormone from the thyroid gland. Causes of primary hypothyroidism include iodine deficiency, chronic autoimmune thyroiditis (Hashimoto’s thyroiditis), congenital, surgical removal of the thyroid gland, post-radioactive iodine, thyroid gland ablation and external irradiation. A significant number of cases are idiopathic. Secondary causes of hypothyroidism include pituitary and hypothalamic disease.
Epidemiology
Iodine deficiency is the most common cause worldwide, whereas in areas of iodine sufficiency, autoimmune disease and hypothyoidism secondary to treatment of hyperthyroid disease are more common. The prevalence of hyperthyroidism in adults is around 1.4% in women and<0.1 % in men [6]. Congenital hypothyroidism is rare, occurring in about 1:4000 births.
Clinical features
The symptoms of hypothyroidism are related to the duration and severity of hypothyroidism, the rapidity with which hypothyroidism occurs and the psychological characteristics of the patient. These are summarized in Table 11.3.3.
Table 11.3.3
Clinical features of hypothyroidism
Dry skin and cold intolerance
Coarse facial features
Enlarged tongue
Coarse brittle hair or loss of hair, loss of outer third of eyebrows
Periorbital oedema
Fatigue
Constipation
Weight gain/obesity
Memory and mental impairment, decreased concentration
Depression, personality changes
Yellow skin (carotenaemia)
Swelling of ankles
Irregular or heavy menses and infertility
Hoarseness
Myalgias
Goitre
Hyperlipidaemia
Delayed relaxation phase of tendon reflexes, ataxia
Sinus bradycardia (atrioventricular block, rare)
Cardiac failure, pericardial effusion (rare)
Hypothermia (uncommon)
A complete evaluation, including a comprehensive history, physical examination and appropriate laboratory evaluation should be performed in every patient with a goitre. Patients with chronic thyroiditis have a higher incidence of other associated autoimmune disorders, such as vitiligo, rheumatoid arthritis, Addison’s disease, diabetes mellitus and pernicious anaemia.
Clinical investigations and criteria for diagnosis
Laboratory evaluation
Perform a TSH assay as the primary test to establish the diagnosis of hypothyroidism if raised. The reference range is 0.4–5.0 mIU/L depending on the method. Additional tests may include free thyroxine assay and thyroid autoantibodies. A combination of an elevated TSH and low free thyroxine is diagnostic [3,4,6]. A patient may be hypothyroid with a TSH greater than twice the reference interval, but with a free thyroxine within the normal range. Subnormal thyroxine with a normal TSH can occur in secondary hypothyroidism.
Thyroid autoantibodies are positive in 95% of patients with autoimmune thyroiditis (Hashimoto’s thyroiditis). The high titres are of value in making this specific diagnosis.
Other investigations
A thyroid scan and/or an ultrasound are useful if structural thyroid abnormalities are suspected.
Thyroid nodules are not uncommon with chronic thyroiditis and carry a small risk of thyroid cancer.
Treatment
Start thyroxine at 50–100 μg orally daily in adults under 60 years of age without evidence of ischaemic heart disease. Too rapid commencement of full thyroid hormone replacement may cause myocardial ischaemia from increased myocardial oxygen consumption without a corresponding increase in cardiac output. The initial daily replacement dose is therefore 25 μg thyroxine in the elderly and where there is suspicion of heart disease. This dose should remain unchanged for 3–4 weeks to allow a steady state to be reached. It is appropriate to start this in the ED, when a firm diagnosis has been made and appropriate follow up arranged.
The dose of thyroxine is then increased in 25–50 μg increments at not less than 4-weekly intervals, until the optimum dose is reached as determined by clinical response and TSH level. Consider admission for any patient with coexistent unstable angina to monitor cardiac function. Any features of myxoedema coma (see below) also mandate admission.
Myxoedema coma
The clinical syndrome of altered mental state, features of hypothyroidism and hypothermia is referred to as myxoedema coma, or sometimes as myxoedema crisis. There is usually a precipitating event, such as infection, stroke, trauma, myocardial infarction or administration of drugs, particularly phenothiazines, phenytoin, amiodarone, propranolol or lithium, that initiates this terminal decompensation phase of hypothyroidism.
The mortality for myxoedema coma remains up to 50% despite aggressive treatment.
Clinical features
 Altered mental state, usually coma due to cerebral oedema, hypoxia and hypercarbia.
Altered mental state, usually coma due to cerebral oedema, hypoxia and hypercarbia.
 Seizures may precede coma in 25% of patients.
Seizures may precede coma in 25% of patients.
 Hypothermia with temperature usually less that 32.2°C. Notably, patients do not shiver.
Hypothermia with temperature usually less that 32.2°C. Notably, patients do not shiver.
 Hypoventilation resulting in hypoxia and hypercarbia.
Hypoventilation resulting in hypoxia and hypercarbia.
 Paralytic ileus, megacolon, and urinary retention.
Paralytic ileus, megacolon, and urinary retention.
 Usual clinical features of hypothyroidism (see Table 11.3.3).
Usual clinical features of hypothyroidism (see Table 11.3.3).
Treatment
Treatment should commence on clinical suspicion.
Administration of thyroid hormones
Tri-iodothyronine
Intravenous T3 may give a faster clinical response in myxoedema coma, as it is the active form of the hormone, although there is no consensus as to whether T3 or T4 replacement is preferable[7,8]. Give T3 as an initial IV bolus of 25–50 μg followed by 10–20 μg 8-hourly to a maximum of 60 μg per day. Alternatively, commence an infusion with a lower total dose of 20 μg per day, as large initial doses appear unnecessary for recovery and may in fact be harmful. Oral or nasogastric replacement of T3 is not recommended in the initial phase of management because of unreliable gastrointestinal absorption.
Thyroxine
The use of T4 is supported as the gradual delivery of T3 through the peripheral conversion of T4 is better tolerated and as the onset of action is more predictable. Give a 400–500 μg IV bolus (300 μg/m2), followed by 50 μg IV daily until oral therapy is tolerated. Combined approaches are now also described.
Corticosteroids
These are given as there is impaired response to stress and the potential for coexistent adrenal insufficiency. Give hydrocortisone 100 mg IV 6-hourly. If an ACTH stimulation test is being considered, give dexamethasone 4 mg until results are known.
General supportive measures
Requires correction of ventilatory, circulatory, temperature and metabolic abnormalities and includes the use of warm humidified oxygen. Look for and treat any precipitating cause. Finally, avoid sedative drugs and watch out for water overload.
Hypoadrenal states
Aetiology, genetics, pathogenesis and pathology
Glucocorticoids act to produce multiple effects on metabolism, including gluconeogenesis, mobilization of fatty acids and amino acids, inhibiting the effects of insulin and ketogenesis. Glucocorticoids have anti-inflammatory effects related to the inhibition of production and reduction of the effects of cytokines and reduction of cell-mediated immunity. They also maintain the normal response of the vascular system to vasoconstrictors. In addition, glucocorticoids affect the regulation of body water by increasing free water excretion. This occurs by an increase in the glomerular filtration rate as well as inhibition of migration of water into cells. Aldosterone acts primarily to cause the reabsorption of sodium and the excretion of potassium and hydrogen ions.
The adrenals normally respond within minutes by elevating corticosteroid levels in response to any physiological or pathological stress. When glucocorticoid insufficiency is present such stressors may result in hypotension, shock and ultimately death if left untreated.
Primary adrenal insufficiency (Addison’s disease)
Primary adrenal insufficiency (Addison’s disease) is due to inability of the adrenal cortex to produce adequate levels of adrenal hormones. Hyponatraemia, hyperkalaemia, acidosis and elevated serum creatinine occur mainly due to aldosterone deficiency, whereas hypoglycaemia is related to cortisol deficiency. Hypercalcaemia occurs as a result of reduction in glomerular filtration rate as well as increased proximal tubular reabsorption of calcium. There may also be some increased mobilization of calcium from bone in patients with adrenal insufficiency.
Secondary adrenal insufficiency
Secondary adrenal insufficiency is due to failure of adequate adrenocorticotrophic hormone (ACTH) from the pituitary gland (Table 11.3.4). Hyponatraemia still occurs in secondary adrenal insufficiency, but is due to cortisol deficiency [9,10].
Table 11.3.4
Causes of adrenal insufficiency
| Primary (Addison’s) | Autoimmune Surgical removal |
| Infection (TB, viral, fungal) | |
| Haemorrhage, including Waterhouse–Friedrichsen syndrome | |
| Congenital | |
| Secondary | Exogenous steroid suppression (single most common cause of adrenal insufficiency) |
| Endogenous steroid (from tumour) | |
| Pituitary failure (hypopituitarism) |
The majority of presentations of acute adrenal insufficiency occur as an exacerbation of a chronic disease process where there is a malfunctioning adrenal system. Acute precipitating factors include sepsis, major trauma, surgery and a myocardial infarct.
Causes of primary or secondary adrenal insufficiency
The cause of 80% of primary adrenal insufficiency is autoimmune. Other causes of acute adrenal gland insufficiency include primary or secondary malignancy, infection, such as tuberculosis, adrenal infarction or haemorrhage (Waterhouse–Friedrichsen syndrome) seen in meningococcaemia or severe sepsis, and drugs. Primary adrenal insufficiency also occurs in up to 20% of patients with AIDS. Up to 60% of patients with sepsis have a low baseline cortisol level, although fewer meet criteria for insufficiency on suppression testing [11,12].
The most common cause of secondary adrenal insufficiency is suppression of the adrenopituitary axis by long-term corticosteroid therapy, although other causes include pituitary failure, such as panhypopituitarism or isolated ACTH production failure.
Clinical features
Suspect adrenocortical failure in any hypotensive patient when no apparent cause is found, particularly anyone who is unresponsive to fluid therapy. Orthostatic postural hypotension is almost always present. Other common features include abdominal pain, which may be severe, with vomiting.
Less obvious findings are weakness, anorexia, diarrhoea, postural syncope, mucocutaneous pigmentation/vitiligo (only with primary adrenal disease) and a dulled mental state.
Hypercalcaemia and/or hyperkalaemia can be the first sign of adrenal insufficiency in the critically ill patient [10]. The other features of adrenal insufficiency may be masked by coexisting illness, but the possibility of adrenal insufficiency should always be considered.
Differential diagnosis
The diagnosis of adrenal insufficiency in the early stages is difficult as weakness, lethargy and gastrointestinal symptoms are common and non-specific. Consider adrenal insufficiency in any patients presenting with these symptoms once more common causes have been excluded.
Clinical investigations
Laboratory findings
The classical laboratory findings are hyponatraemia (due to sodium depletion and the intracellular movement of sodium), hypochloraemia and hyperkalaemia (due to acidosis and aldosterone deficiency). Mild hypercalcaemia (in 10–20% of cases) and a non-anion-gap metabolic acidosis may be seen. Hypoglycaemia, if present, is usually mild. However, all basic laboratory investigations can be within normal limits, even in the presence of an Addisonian crisis.
Antiadrenal antibodies are positive in 70% of patients with autoimmune adrenalitis.
Criteria for diagnosis
Baseline cortisol and ACTH levels should be taken prior to treatment. The normal reference range for cortisol is 200–650 nmol/L. An ACTH level should be<50 ng/L, although interpretation needs to take into account the time of day when the sample is taken. ACTH should be high in primary adrenal disease and low in pituitary disease.
The Synacthen stimulation test is the definitive investigation and may be required if the initial test results are not diagnostic. It is usually performed as an inpatient, when Synacthen 250 μg is administered intramuscularly and cortisol levels are taken at baseline, 30 minutes and 60 minutes. A baseline or post-Synacthen cortisol level of>550 nmol/L is considered normal.
Treatment
Corticosteroid replacement
Do not delay treatment awaiting confirmatory results if acute adrenal insufficiency is suspected.
Give immediate corticosteroid replacement with either intravenous hydrocortisone or dexamethasone. Dexamethasone is recommended when the diagnosis has not been confirmed by laboratory investigations, as it does not interfere with the cortisol assay. Give 10 mg dexamethasone IV stat followed by 4 mg IV 8-hourly. Alternatively, give hydrocortisone at a dose of 250 mg stat followed by 100 mg IV 6-hourly.
Fluid replacement therapy
Give normal saline 1 L stat, then titrated to response, although the total volume deficit is rarely greater than 10% body weight. Intravenous dextrose should be given at the same time, either separately or as 5% dextrose in normal saline to avoid hypoglycaemia.
General supportive measures
These include treatment of hypoglycaemia and other electrolyte replacement abnormalities, although most will be corrected with saline rehydration alone. Mineralocorticoid replacement is usually not necessary in the acute crisis, if salt and water replacement are adequate.
Once the crisis has been successfully treated, it is important to investigate and manage the cause and to develop a maintenance regimen.
Prognosis
The patient with acute adrenal insufficiency may die if the diagnosis is not made promptly. When the diagnosis is suspected and treatment is early, the outcome is favourable depending on the nature of any precipitating illness.
Response to severe illness
The normal response to severe illness should see cortisol levels rising to at least 500 nmol/L. States of ‘relative adrenal insufficiency’ are described where glucocorticoid administration diminishes or even eliminates the requirements for vasopressor agents, even though measured cortisol levels are normal or close to normal [11]. There is no concensus on what constitutes ‘normal’ cortisol levels in severe illness.
Up to 60% of patients with severe sepsis may have some degree of adrenal insufficiency depending upon the threshold cortisol level used [12]. Moreover, it appears that it is the delta cortisol rather than the basal cortisol level that is associated with clinical outcome [13]. Repeat adrenal function testing is indicated in patients with severe illness who remain unstable or who fail to improve with aggressive supportive therapy [14].
The use of hydrocortisone has been recommended in septic shock after an abnormal 250 μg Synacthen stimulation test [15–17]. This should continue for a week if adrenal insufficiency is confirmed.
Hyperadrenal states
Aetiology, pathogenesis and epidemiology
Cushing’s disease usually refers to hyperadrenalism due to a pituitary adenoma. Cushing’s syndrome occurs as a result of hyperadrenalism from exposure to excess glucocorticoids over a prolonged period. Endogenous causes of Cushing’s syndrome are related to primary adrenal disorders, such as adrenal adenoma, carcinoma or hyperplasia, or are secondary to ACTH or CRH stimulation and ectopic ACTH production from bronchogenic carcinoma or carcinoid tumours in particular. However, by far the most common cause of Cushing’s syndrome is from the exogenous (iatrogenic) administration of steroids.
The incidence of Cushing’s syndrome ranges from 0.7 to 2.4 per million population per year, but the reported prevalence in obese patients with type II diabetes may be between 2 and 5% [18].
Clinical features
The classical clinical features of Cushing’s syndrome are increased body weight with central obesity, rounded face, hypertension, fatigue, weakness and proximal myopathy, hirsutism, striae, bruising, decreased libido, amenorrhoea, depression and/or personality changes, osteopaenia or fracture. Proximal weakness or myopathy is useful to differentiate simple obesity (strong limbs) from possible Cushing’s syndrome (relative weakness for patient’s size).
Clinical investigations and criteria for diagnosis
Laboratory tests
Full blood examination may reveal polycythaemia, neutrophilia and eosinophilia. Electrolytes may show hyperglycaemia, hypokalaemia and metabolic alkalosis.
24-h urinary cortisol level
A measured 24-h urinary cortisol level with a value more than four times the upper normal range is rare except in Cushing’s syndrome (normal range 100–300 nmol/24 h).
Overnight dexamathasone suppression test
This is an outpatient screening test for Cushing’s syndrome [3]:
 Day 1, 09:00 hours: 5 mL blood taken for baseline cortisol
Day 1, 09:00 hours: 5 mL blood taken for baseline cortisol
The baseline reference range for cortisol is 200–650 nmol/L. The day 2 cortisol level should drop to lower than 50% of the baseline level, indicating normal suppression and excluding Cushing’s syndrome.
Long dexamathasone suppression test
The long dexamathasone suppression test is performed as an inpatient, using increasing doses of dexamethasone to determine at what level suppression occurs, with testing of both cortisol and ACTH levels. Cushing’s disease only suppresses at high doses.
Other tests
A chest X-ray is important if bronchogenic carcinoma of the lung is suspected. Magnetic resonance imaging of the adrenals and/or head is used for the identification of tumours.
Treatment
Treatment will depend on the cause. When a pituitary or adrenal adenoma is identified, optimal treatment is removal of the tumour [4,18]. Glucocorticoid replacement is then required for up to 2 years following surgery to allow full recovery of the normal pituitary–adrenal axis.
Pharmacological blockade of adrenal corticosteroid production may be required in some circumstances. Ketaconazole, amino-glutethimide, metapyrone and mitotane may be used for this purpose.