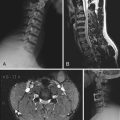
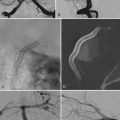
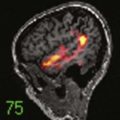
CHAPTER 123 Unusual Gliomas
Subependymal Giant Cell Astrocytoma
World Health Organization Grade I
Clinical Presentation and History
Subependymal giant cell astrocytomas generally occur only in patients with tuberous sclerosis.1 They are typically intraventricular lesions that rarely cause symptoms unless they obstruct ventricular outflow through the foramen of Monro. Tuberous sclerosis is an autosomal dominant phakomatosis associated with mental retardation, seizures, and adenoma sebaceum. Other findings include altered skin pigmentation, retinal tumors, subungual fibromas, and tumors of the spleen and pancreas. The natural history of subependymal giant cell astrocytoma is slow growth with little risk for transformation to a high-grade glioma. Consequently, treatment is not indicated unless the size and location of the tumor result in neurological findings (e.g., ventricular obstruction).1
Imaging
Subependymal giant cell astrocytomas have a characteristic appearance on magnetic resonance imaging (MRI) (Fig. 123-1). The classic finding is an intraventricular tumor in a patient with known tuberous sclerosis. These lesions are generally isointense on T1-weighted images and hyperintense on T2-weighted images. There is modest signal change in the surrounding white matter. Hypointense portions of the tumor represent calcifications, which are common. After contrast administration, the tumors commonly enhance strongly.2 Computed tomography (CT) can demonstrate calcifications as well as contrast enhancement. In infants, transcranial ultrasonography can be useful for following ventricular size and for detecting an increase in tumor size.
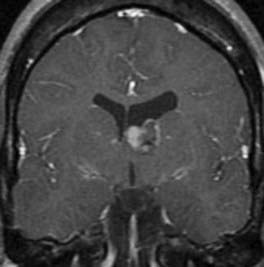
Pathology
The diagnosis of a subependymal giant cell astrocytoma is rarely difficult unless the patient has minimal evidence of tuberous sclerosis and an intraventricular tumor is found during a work-up for neurological symptoms. These tumors receive their name from their common location (periventricular) and typical pathologic appearance (giant cells mixed with astrocyte lineage cells). They are often well-demarcated tumors that show modest infiltration into the surrounding white matter but expand into the ventricular cavity. Routine histologic examination reveals not only giant cells with abundant eosinophilic cytoplasm but also spindle-shaped cells with eccentric nuclei.1,3 The presence of large eosinophilic cytoplasm can mimic gemistocytic astrocytomas; however, the location of the tumor is not consistent with that diagnosis because gemistocytic astrocytomas are found in brain parenchyma and have a high rate of malignant transformation (Fig. 123-2). Mitoses, necrosis, endothelial proliferation, and nuclear atypia may be present; however, these findings do not correlate with aggressive behavior. Anaplastic transformation is uncommon.
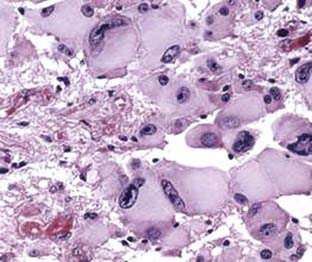
Management and Outcome
In general, subependymal giant cell astrocytomas do not require intervention unless they obstruct the foramen of Monro. The primary treatment is surgical, with total resection being the goal.4 When total resection proves to be too hazardous, subtotal resection may be adequate because these tumors often grow slowly and rarely become aggressive. There are few data regarding the role of radiation and chemotherapy in these lesions. Surgical planning is beyond the scope of this chapter, but careful planning is required to provide adequate access to the lesion; this can most often be achieved through a transcortical or transcallosal approach. In addition, tuberous sclerosis is associated with cardiac abnormalities, and a cardiac assessment is an important part of preoperative planning, particularly in children. If surgery is not possible or if hydrocephalus persists following tumor resection, shunting can relieve both ventricular obstruction and intracranial hypertension. It is appropriate to continue surveillance of these patients because recurrence is possible. An attempt to use chemotherapy in the treatment of these tumors has shown some modest early success.5
Angiocentric Glioma
World Health Organization Grade I
Clinical Presentation and History
Described in 2005,6 angiocentric glioma (AG) was recently codified by the WHO and grouped with astroblastoma and chordoid glioma.7 AG can occur at any age; however, it is most commonly seen in young adults, with a mean age of onset of 17 years. Patients typically present with a long-standing history of intractable seizures.
Imaging
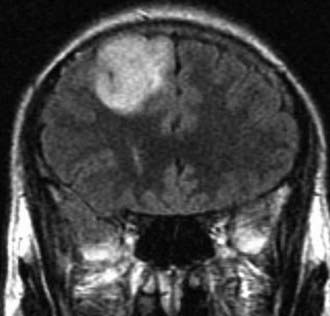
MRI of these lesions typically demonstrates a cortical lesion with expansion of a gyrus (Fig. 123-3). A stalk often extends to the ventricular surface, and this is thought to be nearly pathognomonic for AG.6 Lesions are hyperintense on T2- and fluid-attenuated inversion recovery (FLAIR)-weighted images. Contrast enhancement is either sparse or absent.
Pathology
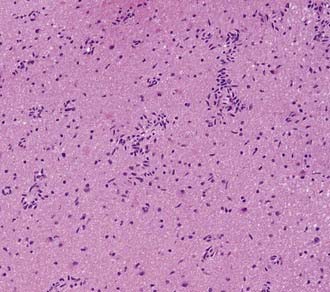
Microscopically, tumors demonstrate monomorphous, bipolar cells associated with the normal vessels of the cortex and white matter.8 Tumor cells are arranged radially akin to ependymal rosettes (Fig. 123-4). Tumor cells are uniform and spindle shaped. Mitoses are rare, with a proliferative index of less than 5%. Immunohistochemically, these tumors are positive for S-100, GFAP, and vimentin. Some cells are epithelial membrane antigen (EMA) positive as well. Cytogenetics and molecular studies are noninformative and yet to be confirmed.
Management and Outcome
AGs tend to follow an indolent course with rare progression.6,7 Surgical cure is obtained by total resection, and after subtotal resections, progression is not encountered at 2.5 and 4 years follow-up without adjuvant therapy.6–8 More aggressive forms are exceedingly rare and controversial.
Astroblastoma
World Health Organization Grade I
Clinical Presentation and History
Astroblastomas are extremely rare tumors that have received little attention since their initial description by Bailey and Bucy in 1930.9 The acceptance of astroblastoma as a distinct and separate entity has been an issue of some debate because astroblastic features can be found in other neoplastic entities such as anaplastic astrocytomas, glioblastomas, and gemistocytic astrocytomas.9,10 Most astroblastomas are found in the first three decades of life; astroblastomas found later in life have been the highly malignant form. A striking female preponderance (11 : 1) has been reported in larger series.11 Astroblastomas affect the cerebral hemispheres, usually involving the cortex and the subcortical regions or the periventricular regions. No astroblastomas have been reported outside the supratentorial compartment. Patients usually present with symptoms and signs related to cortical dysfunction or from related mass effect, such as hemiparesis, seizures, or personality changes.
Great variability in the natural history of astroblastomas has been reported through case reports; these tumors can have a slow evolution with a relatively favorable outcome, or they can exhibit a rapidly progressive nature leading to fatality. In Bailey and Bucy’s9 series of 25 patients, 1 patient was alive and well 7 years after surgical resection. The relative malignancy of the remaining cases may have been related to anaplastic features. Bonnin and Rubinstein10 attempted to establish clinicopathologic correlations, reinforcing the idea that astroblastomas with malignant features indicative of anaplasia are susceptible to progression to glioblastoma. In their series, those cases that displayed benign histologic features had a benign clinical course, with no postsurgical recurrences after 12 and 20 years. The specimens that displayed malignant features appeared to correlate with a rapidly fatal clinical course, with one death 10 months and the other 16 months after surgery, with pathologic evidence of tumor progression to glioblastoma and gliosarcoma.
Imaging
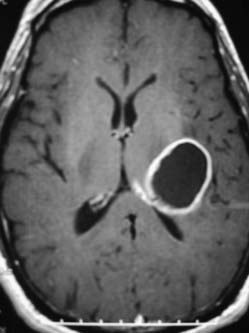
Studies of imaging characteristics of astroblastomas have been limited (Fig. 123-5). The appearance of astroblastomas on CT has ranged from a poorly defined, hypodense tumor with an irregular enhancement pattern to a well-defined tumor with intense enhancement. One case series has attempted to describe the imaging characteristics of these lesions.11 Tumors are generally hypointense to white matter on T1-weighted imaging and hyperintense on T2-weighted and FLAIR images. Frequent rim enhancement is seen around a cystic center. Calcifications can be present, and vasogenic edema is often minimal. Frequently, multiple intratumoral cysts are seen, giving the “bubbly” appearance.
Pathology
Astroblastomas are almost always well defined macroscopically, with their cut surfaces revealing a homogeneous, soft, pink-gray substance.10 Cystic areas are frequently encountered, and central areas of necrosis can be found in larger specimens. Microscopically, these tumors are richly supplied by blood vessels. The clusters of tumor cells form pseudorosettes in cross section (Fig. 123-6). The vessel walls are highly hyalinized. The nuclei of the tumor cells are found some distance away from the vessel, but long processes extend from the tumor cell to the vessels and end as an expanded footplate. The nuclei are oval and slightly irregular and contain coarse chromatin nodes. The perikaryon is angulated, club shaped, or fusiform. Cellular atypia can be present in varying degrees. High numbers of mitotic figures may be found in the high-grade variant.
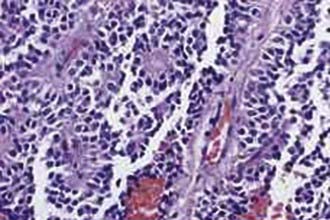
Management and Outcome
Surgical resection is the main therapeutic modality. Astroblastomas are well circumscribed and usually located in surgically accessible regions. Gross total resection can usually be achieved without difficulty. Owing to the rarity of these tumors, the therapeutic value of radiation and chemotherapy remains uncertain. Bonnin and Rubinstein10 reported one patient who had good tumor control with a biopsy and subsequent radiotherapy. Chemotherapy was also administered in five patients in the same series, without a clear change in prognosis. In Bailey and Bucy’s9 original series, most patients died within the year following surgery, owing to tumor regrowth. Outcome is difficult to predict because these tumors can remain indolent in some patients and undergo malignant degeneration to glioblastoma in others, resulting in rapid death.
Pilomyxoid Astrocytoma
World Health Organization Grade II
Clinical Presentation and History
Pilomyxoid astrocytomas (PMAs) are a recently described entity with similarities to pilocytic astrocytomas (PAs); however, the prognosis is not as favorable.12 It is possible that up until the description of PMA, these lesions were grouped together with PA and thus account for patients with PA experiencing a shorter disease-free survival. These tumors occur throughout the neuraxis, with a mean age at diagnosis of 18 months.13 Most are found in the hypothalamic-chiasmatic region and at the midline. Patients present with symptoms associated with elevated intracranial pressure or mass effect of eloquent parenchyma. In infants, increased head circumference may be the first presenting sign.
Imaging
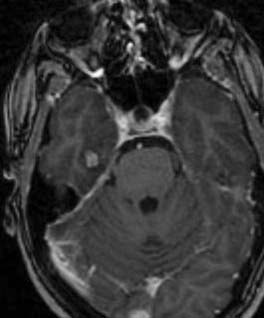
MRI findings are nonspecific and tend to be well circumscribed (noninfiltrating) and without vasogenic edema (Fig. 123-7). Lesions are hyperintense on T1- and T2-weighted images and on FLAIR sequences and demonstrate heterogeneous to homogeneous enhancement.14
Pathology
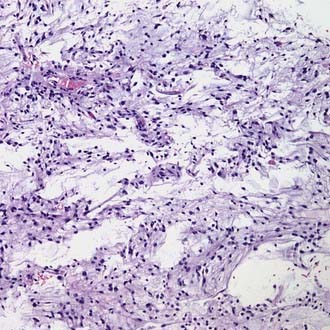
PMA histology consists of a myxoid matrix, with monomorphic piloid cells in a loose fibrillary and myxoid background without Rosenthal fibers or eosinophilic granules.12 Tumor cells are arranged radially around vessels similar to perivascular rosettes seen in ependymomas (Fig. 123-8). Mitotic figures are rare. This is in contrast to PAs, which demonstrate a biphasic architecture with Rosenthal fibers and eosinophilic granular bodies (EGBs). Molecular characterization of PMA has not been performed.
Management and Outcome
Mean progression-free survival times for PMA and PA are reported as 25 and 163 months, respectively (P < .01), and the mean overall survival times for PMA and PA are 60 and 233 months, respectively.13 Much of this reduced survival is attributed to location of the tumor and inability to achieve total resection safely.
Pleomorphic Xanthoastrocytoma
World Health Organization Grade II
Clinical Presentation and History
First described in 1979 by Kepes and colleagues,15 these rare tumors represent less than 1% of all astrocytoma cases.15 Pleomorphic xanthoastrocytoma (PXA) can occur throughout the supratentorial compartment (most commonly in the temporal lobe), with case reports of lesions in the cerebellum16 and spinal cord.17 Patients typically present in the second to third decade of life, with a median age of 14 years, although cases have been reported from infancy to the seventh decade.18 Both sexes are affected equally. Most patients (70% to 80%) present with seizures followed by headache and focal location-related deficits, or evidence of increased intracranial pressure.18 Rarely, patients present with symptomatic hemorrhage from the tumor.19
Imaging
PXAs are characterized on CT and MRI as a cystic structure with an enhancing mural nodule (Fig. 123-9). Both CT and Tl-weighted MRI typically reveal a focal, ill-defined mass with mixed intensity, isointensity, or hypointensity to gray matter.20,21 The cystic component is seen in up to 60% of cases.18 Well-defined contrast enhancement of the tumor mass or nodule is often seen on CT and is demonstrated uniformly on gadolinium-enhanced MRI. The tumor is usually of high to mixed intensity on T2-weighted imaging, and the cyst is typically hyperintense. Some authors have reported associated leptomeningeal enhancement or spread.22 The cysts may be solitary or multiloculated and are usually deep to the solid portion of the tumor. Typically, the cyst wall is nonenhancing. Mild to moderate amounts of peritumoral edema may be seen, and calcifications are rare. Peritumoral edema may be a poor prognostic sign with this tumor, although this is not universally accepted. Occasionally, erosion of the overlying calvarium may be evident.23
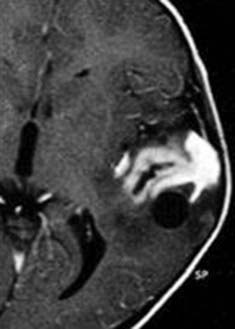
Pathology
Grossly, these tumors are usually firm, variable in color, and relatively avascular; however, recurrent tumors or tumors with more malignant features may be more vascular. They typically invade the pia-arachnoid space, and up to 13% of patients have involvement of all three layers of the meninges.23 The associated cyst fluid is invariably xanthochromic. Close to the meninges, more prominent mesenchymal features are found, whereas more glial features are found toward the interior (Fig. 123-10). These tumors rarely invade the underlying brain; normal cortex is rarely seen within tumor specimens.
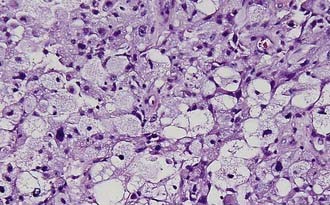
Immunohistochemical staining of these tumors reveals that the spindle cells and most of the round pleomorphic cells are positive for GFAP and S-100 protein. Staining for synaptophysin or neuron-specific enolase may also be present in some cells. These cells are not typical of ganglion cells or neurons, and the staining may occasionally colocalize with GFAP, suggesting a relationship to other tumors of developmental origin, such as gangliogliomas, dysembryoplastic neuroepithelial tumors, and infantile desmoplastic gangliogliomas. Other authors have reported these tumors in association with separate areas of ganglioglioma or cortical dysplasia.15,18,21 PXA with ganglioglioma features may indeed be a subtype of ganglioglioma; however, limits of current molecular diagnosis and the rarity of these tumors render the distinction blurred. The presence of GFAP staining in the spindle cells sets these tumors apart from fibrous histiocytomas, fibrous xanthomas, meningiomas, and other primarily sarcomatous tumor types. The age group and the presence of xanthomatous cells usually, although not always, distinguish these tumors from infantile desmoplastic astrocytomas and gangliogliomas.
Little is known about the cytogenetics or molecular biology of these lesions.24 Studies have shown associations between these tumors and neurofibromatosis type I and Sturge-Weber syndrome. Others have demonstrated loss of chromosome 9 and gain of chromosome 7 in tumor samples. Furthermore, cytogenetic changes and comparative genomic hybridization (CGH) results of PXA differ from any other glioma, suggesting a distinct genetic origin of these lesions.24
Management and Outcome
Because these tumors are quite rare, large series of patients are limited. Surgical excision appears to be associated with a favorable outcome.25 A meta-analysis of 79 published cases revealed an actuarial survival rate of 91% at 5 years, 82% at 10 years, and 77% at 15 years, with a median survival time of 18 years. Investigators found that 33% of patients who died had necrosis at presentation or recurrence, compared with only 2% of living patients. Kaplan-Meier analysis revealed this to be statistically significant. They also found statistical significance for survival based on the extent of surgical resection. For all patients, there was no difference between gross total resection and subtotal resection (91% versus 65%, P > .05); however, subgroup analysis revealed that if patients with necrosis were excluded, 95% of patients who underwent gross total resection survived, compared with 68% of patients who underwent subtotal resection (P < .05).26 The behavior of these tumors with necrosis is not as precipitous as that in patients with true glioblastomas and thus should not be grouped as such. Analysis of patients who underwent postoperative radiotherapy revealed a trend toward improved survival with treatment, but this did not reach statistical significance.
An additional retrospective series of 71 patients with a mean follow-up of 5 years revealed a similar result of overall survival rates of 81% at 5 years and 70% at 10 years, whereas recurrence-free survival rates were 71% at 5 years and 61% at 10 years.18 Multivariate analysis showed extent of surgical resection and mitotic index to be significant predictors of relapse-free survival.
It is generally accepted that total resection in all patients should be attempted if it is possible without unacceptable deficits resulting. It appears that resection of the cyst lining may not be indicated. In some cases, a more malignant tumor may be suspected preoperatively, and frozen-section diagnosis at the time of resection may be equivocal. In these cases, when a subtotal resection is performed owing to the assumption of greater malignancy, an attempt at complete resection may be justified at the time of definitive histologic diagnosis. The data regarding adjuvant therapy for these tumors are not helpful at this time and are limited to single case reports. Adjuvant therapy for classic PXA should be reserved until observation of disease progression. Patients who have subtotally resected tumors or a high mitotic index or necrosis on pathologic examination should at least undergo vigilant, routine gadolinium-enhanced MRI as surveillance for progression. Patients without these negative predictors should also undergo surveillance, but perhaps at less frequent intervals. At the time of progression, in patients with favorable anatomy, repeat resection may be attempted. We believe that progression is an indicator of a more aggressive tumor, and these patients should also be considered for adjuvant therapy. Furthermore, although rare, malignant transformation may necessitate adjuvant treatment with chemotherapy and radiation in the same vein as a malignant glioma.27
Ganglioglioma
World Health Organization Grade I
Clinical Presentation and History
Ganglioglioma is an infrequent tumor of the central nervous system, initially characterized by Perkins in 1926.28 These lesions are histopathologically benign, well-differentiated neuroepithelial tumors. They can occur anywhere in the central nervous system but are most commonly found in the temporal lobe (up to 85%), often affecting young patients with seizure disorders (they are the most common tumor found in temporal lobe epilepsy).29 Other reports have described gangliogliomas in the spinal cord,30 brainstem,31 and cerebellum.32 The WHO has classified these lesions as grade I.1 Gangliogliomas are thought to account for about 1% (reported range of 0.4% to 7%) of all central nervous system neoplasms, most frequently affecting children and young adults with a slight male predominance.33 Because of its low incidence, most of what we understand about the behavior of this lesion is derived from case reports and small series. The most common presenting signs and symptoms are seizures (temporal lobe and other supratentorial locations), followed by headache, dizziness, ataxia (posterior fossa), and progressive weakness (spinal cord). The duration of symptoms before diagnosis ranges from months to decades.
Imaging
Gangliogliomas display highly variable imaging characteristics on MRI, and thus there is no typical imaging description (Fig. 123-11).34 For example, one large series of gangliogliomas reported that half of these tumors showed both cystic and solid components; 43% of the tumors were entirely solid, and 5% were entirely cystic.29 Enhancement after administration of gadolinium has also been found to vary from no enhancement to marked, heterogeneous enhancement.29,35 Generally speaking, gangliogliomas are isointense to hypointense on T1-weighted imaging, are hyperintense and heterogeneous on T2-weighted imaging, and can contain solid, cystic, and calcified components. Lesions are rarely associated with vasogenic edema but can exhibit mass effect, as seen in gangliogliomas of the posterior fossa. Malignant gangliogliomas tend to demonstrate more contrast enhancement and have marked vasogenic edema surrounding the tumor.
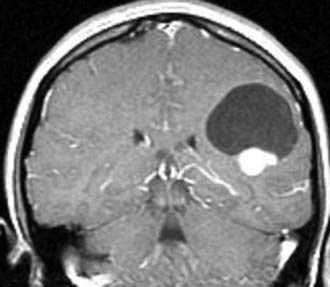
FIGURE 123-11 T1-weighted contrast-enhanced magnetic resonance image of a ganglioglioma in the left frontal lobe.
Fluorodeoxyglucose positron emission tomography (FDG-PET) of WHO grade I gangliogliomas shows tumor hypometabolism or eumetabolism. Increased perfusion is seen on thallium-201 single-photon emission computed tomography (SPECT) of high-grade (anaplastic) lesions. Some authors advocate either of these imaging modalities before surgical consideration for the purpose of preoperative prognosis.34,36
Pathology
Gangliogliomas are characterized by an intimate mixture of abnormal neuronal and glial elements.1,37 Macroscopically, gangliogliomas appear solid or cystic, or a combination of both. Cystic gangliogliomas are well delineated from surrounding tissue, often showing a mural nodule protruding into the cystic cavity. Solid tumors are also generally well circumscribed; however, poorly delineated, infiltrative tumors are occasionally encountered.38 Calcification is common. Gangliogliomas may vary considerably in the degree to which the neuronal and glial components participate in the neoplastic process (Fig. 123-12). The involved neurons are often irregularly oriented, exhibiting anomalous processes with early branching. Binucleated or multinucleated forms are found frequently and are thought to be characteristic.
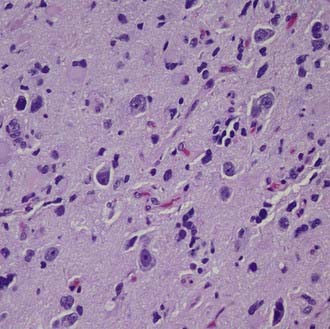
FIGURE 123-12 Gangliogliomas are characterized by the presence of neoplastic glial cells and dysplastic, occasionally binucleated neurons.
Ultrastructural analysis with electron microscopy demonstrates characteristic homogeneity.37 The neurons are large and polygonal, containing round or oval nuclei with some intervening irregularities, such as infolding of the nuclear membrane. The nuclei have prominent nucleoli, with sparsely distributed chromatin. The cytoplasm contains well-formed organelles, including variable numbers of dense core granules that were 100 to 230 nm in diameter, found in both the perikarya and the neuronal processes. Chromosomal imbalances are common with amplification of all or some of chromosome 7 seen in up to 30% of lesions.39 Expression arrays, mutational analysis, and CGH have identified numerous upregulated and downregulated proteins ranging from cell-cycle checkpoint genes to p53. No study to date has correlated molecular biology and cytogenetics to prognosis.
Management and Outcome
After radiographic identification of a lesion is made, gross total surgical excision, when possible, is the “gold standard.” Seizure control after surgery can be as high as 80%.40 Survival rates after surgery are high. Lang found no survival differences between low-grade (WHO grade I) ganglioglioma patients (84% survival after 10 years) and high-grade (WHO grade II and III) ganglioglioma patients (83% survival after 10 years).38 However, the event-free survival analysis strongly suggested a difference in the propensity for recurrence between these two groups; the event-free survival rate for low-grade gangliogliomas was 57%, compared with only 15% for high-grade gangliogliomas.
The optimal management of a patient with ganglioglioma may best be based on the eloquence of the involved neural tissues and the histologic features of the tumor. Surgery alone has been widely advocated as the optimal treatment for patients with cerebral hemisphere tumors. The use of chemotherapy is limited to case reports, most of which describe anaplastic lesions. Typically, these lesions are treated like malignant gliomas and thus command similar chemotherapy and radiotherapy regimens. The use of adjuvant radiotherapy after subtotal resection or for unresectable low-grade lesions is controversial, with evidence to suggest therapeutic radiation as a cause of malignant transformation.41,42 The issue of radiotherapy is more relevant in cases in which gross total resection is not possible because of the location of the tumor or in which the histopathology harbors anaplastic features. Because there is no proven benefit of radiotherapy for gangliogliomas, and because of the potential harm that may be associated with malignant degeneration and damage to normal surrounding neural tissues, adjuvant therapy with radiation must be approached with caution.43
Papillary Tumor of the Pineal Region
World Health Organization Grades II and III
Clinical Presentation and History
Papillary tumor of the pineal region (PTPR) is a relatively new addition to the WHO classification.44 The name indicates the common site of origin. These tumors have been identified across a large age range in both men and women. As with most mass lesions in the pineal region, they reach medical attention because of headache, mental status change, and altered vision.
Imaging
PTPRs are generally found to be diffusely contrast-enhancing mass lesions in the pineal region and posterior third ventricle often associated with hydrocephalus (Fig. 123-13). They appear well demarcated from the thalamus and cerebellum.45 This appearance is common to many tumors in this region, and the diagnosis cannot be established on imaging alone. Peritumoral signal changes on MRI in the diencephalon and midbrain likely reflect edema rather than tumor infiltration.
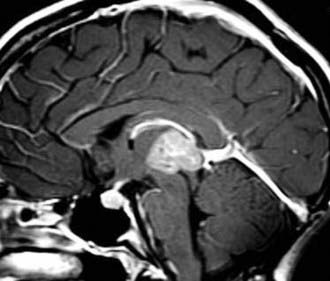
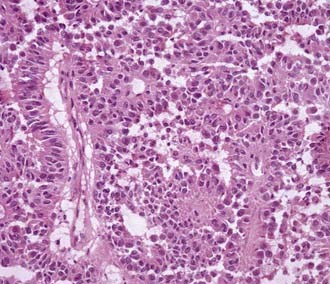
Pathology
These tumors contain eosinophilic cells with indistinct borders and large pleomorphic nuclei, but without necrosis or vascular proliferation.46 They derive their name from perivascular pseudorosettes (Fig. 123-14). Tumor cells are positive for cytokeratin and synaptophysin and stain variably for GFAP, vimentin, S-100 protein, and neuron-specific enolase. Because of the rarity of these tumors, genetic testing remains unknown.
Management and Outcome
The optimal treatment appears to be surgery.46 Most reported cases suggest that PTPRs grow slowly and do not transform into a higher grade tumor. However, local recurrence is possible, and although insufficient data are available for advising adjuvant treatment, radiotherapy appears to provide good local control for incompletely removed tumors.
Dysembryoplastic Neuroepithelial Tumor
World Health Organization Grade I
Clinical Presentation and History
Dysembryoplastic neuroepithelial tumors (DNTs) are benign, typically supratentorial, cortically based lesions initially described in 1988.47 They are found in young adults with a history of epilepsy. They share morphologic similarities with oligodendrogliomas and likely were misclassified as such before their description. Most DNTs are found in the temporal lobe, but reports have documented these lesions in a variety of locations in the central nervous system. Although the incidence is rare, they are more commonly found at centers that focus on the surgical treatment of epilepsy. Symptomatic mass effect is not found with these tumors.48
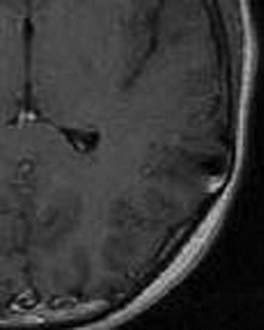
Imaging
MRI demonstrates a cortically based lesion that expands a gyrus, commonly in the temporal lobe (Fig. 123-15). Their long-standing presence may result in thinning of the inner table of the skull. Enhancement is variable, and calcification is common. One significant imaging characteristic is the absence of mass effect.49 These tumors tend to be radiographically stable over an extended period, indicating their indolent growth rate.
Pathology
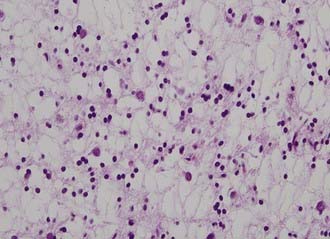
DNTs have a characteristic appearance marked by a columnar organization of axons draped by small oligodendroglioma-like cells.47,48 These cells are GFAP and S-100 positive. Synaptophysin positivity is diffusely present in the neuronal elements of the tumor. DNTs have a very low labeling index. This pattern results in a nodular organization of the tumor within the cortex (Fig. 123-16). Simple and complex forms of DNTs have been described, and these tumors are often associated with cortical dysplasia. The WHO classifies DNT as a mixed neuronal-glial tumor based on the mixed cell population, with phenotypes of both lineages. These tumors are often associated with cortical dysplasia, indicating a dysembryoplastic origin.
Management and Outcome
DNTs are characteristically indolent. Their association with chronic epilepsy and their favorable long-term control with surgery show that resection is the best treatment option. There are no reliable reports of the use of adjuvant therapy even in subtotally resected tumors. However, aggressive growth has rarely been reported.50
Chordoid Glioma of the Third Ventricle
World Health Organization Grade II
Clinical Presentation and History
Chordoid glioma of the third ventricle was recognized by the WHO in 1998 after the publication of several cases with similar clinical, radiologic, and pathologic findings.51 These tumors arise in the anterior third ventricle and commonly reach medical attention because of hydrocephalus. They appear to be contained within the ventricle and are thought to arise from the ventricular surface. As with other suprasellar tumors, they can cause visual loss and pituitary dysfunction. They appear to be more prevalent in women.
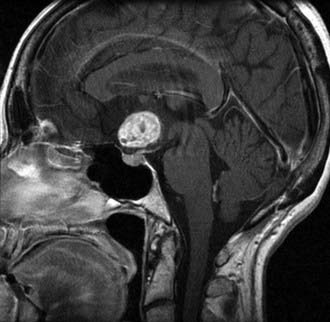
Imaging
These lesions commonly fill the third ventricle and obstruct the lateral ventricles.51 They diffusely enhance with contrast and do not appear to infiltrate into the hypothalamus (Fig. 123-17). Large tumors may be difficult to differentiate from other more common suprasellar tumors. The diagnosis should be considered in young patients with an enhancing third ventricular mass lesion. Other than colloid cysts, tumors of the third ventricle are rare. Consequently, this diagnosis should be considered in an adult patient with an enhancing third ventricular mass.
Pathology
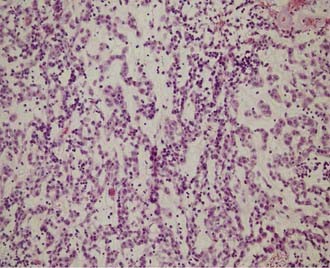
These tumors are composed of GFAP- and vimentin-positive cells in a matrix of mucinous stroma.52 Nuclei are generally uniform in size and shape; the tumors are classified as WHO grade II. They have a low labeling index suggesting indolent growth; however, aggressive behavior has been reported (Fig. 123-18). Some reports suggest that chordoid gliomas of the third ventricle arise from the lamina terminalis and perhaps from tanycytes in this region.53,54 A study of antigen expression and cytogenetics of these rare tumors shows epidermal growth factor receptor positivity but no genetic abnormalities associated with typical atrocytomas. These data indicate that chordoid glioma of the third ventricle is indeed a distinct type of glioma and not merely an astrocytoma in an unusual location.52
Management and Outcome
Surgical management of these tumors can be limited by the location.51 Access through a dilated lateral ventricle or lamina terminalis can be challenging. Consequently, subtotal resection is common in the limited number of reported cases. The usefulness of adjuvant radiotherapy remains unknown, but limited experience suggests that these tumors can progress even after radiotherapy. However, close monitoring and consideration for adjuvant treatment is required for lesions that cannot be totally removed. Ventricular shunting may be necessary when cerebrospinal fluid obstruction is not reversed with surgery or in patients who are not candidates for tumor resection.
Bonnin JM, Rubinstein LJ. Astroblastomas: a pathological study of 23 tumors, with a postoperative follow-up in 13 patients. Neurosurgery. 1989;25:6-13.
Brat DJ, Scheithauer BW, Staugaitis SM, et al. Third ventricular chordoid glioma: a distinct clinicopathologic entity. J Neuropathol Exp Neurol. 1998;57:283-290.
Daumas-Duport C. Dysembryoplastic neuroepithelial tumours. Brain Pathol. 1993;3:283-295.
Daumas-Duport C, Scheithauer BW, Chodkiewicz JP, et al. Dysembryoplastic neuroepithelial tumor: a surgically curable tumor of young patients with intractable partial seizures. Report of thirty-nine cases. Neurosurgery. 1988;23:545-556.
de Ribaupierre S, Dorfmüller G, Bulteau C, et al. Subependymal giant-cell astrocytomas in pediatric tuberous sclerosis disease: when should we operate? Neurosurgery. 2007;60:83-89.
Fèvre-Montange M, Hasselblatt M, Figarella-Branger D, et al. Prognosis and histopathologic features in papillary tumors of the pineal region: a retrospective multicenter study of 31 cases. J Neuropathol Exp Neurol. 2006;65:1004-1011.
Fouladi M, Jenkins J, Burger P, et al. Pleomorphic xanthoastrocytoma: favorable outcome after complete surgical resection. Neuro Oncol. 2001;3:184-192.
Franz DN, Leonard J, Tudor C, et al. Rapamycin causes regression of astrocytomas in tuberous sclerosis complex. Ann Neurol. 2006;59:490-498.
Giannini C, Scheithauer BW, Burger PC, et al. Pleomorphic xanthoastrocytoma: what do we really know about it? Cancer. 1999;85:2033-2045.
Kepes JJ, Rubinstein LJ, Eng LF. Pleomorphic xanthoastrocytoma: a distinctive meningocerebral glioma of young subjects with relatively favorable prognosis. A study of 12 cases. Cancer. 1979;44:1839-1852.
Kern M, Robbins P, Lee G, Watson P. Papillary tumor of the pineal region: a new pathological entity. Clin Neuropathol. 2006;25:185-192.
Lang FF, Epstein FJ, Ransohoff J, et al. Central nervous system gangliogliomas. Part 2: Clinical outcome. J Neurosurg. 1993;79:867-873.
Lellouch-Tubiana A, Boddaert N, Bourgeois M, et al. Angiocentric neuroepithelial tumor (ANET): a new epilepsy-related clinicopathological entity with distinctive MRI. Brain pathology (Zurich, Switzerland). 2005;15:281-286.
Lipper MH, Eberhard DA, Phillips CD, et al. Pleomorphic xanthoastrocytoma, a distinctive astroglial tumor: neuroradiologic and pathologic features. AJNR Am J Neuroradiol. 1993;14:1397-1404.
Louis DN, Ohgaki H, Wiestler OD, Cavenee WK, editors. World Health Organization Classification of Tumours of the Central Nervous System. Lyon: IARC, 2007.
Nakajima T, Kumabe T, Shamoto H, et al. Malignant transformation of pleomorphic xanthoastrocytoma. Acta Neurochir. 2006;148:67-71.
Reifenberger G, Weber T, Weber RG, et al. Chordoid glioma of the third ventricle: immunohistochemical and molecular genetic characterization of a novel tumor entity. Brain Pathol. 1999;9:617-626.
Rumana CS, Valadka AB. Radiation therapy and malignant degeneration of benign supratentorial gangliogliomas. Neurosurgery. 1998;42:1038-1043.
Rumana CS, Valadka AB, Contant CF. Prognostic factors in supratentorial ganglioglioma. Acta Neurochir. 1999;141:63-68.
Sampetrean O, Maehara T, Arai N, Nemoto T. Rapidly growing dysembryoplastic neuroepithelial tumor: case report. Neurosurgery. 2006;59:E1337-E1338.
Tihan T, Fisher PG, Kepner JL, et al. Pediatric astrocytomas with monomorphous pilomyxoid features and a less favorable outcome. J Neuropathol Exp Neurol. 1999;58:1061-1068.
Wang M, Tihan T, Rojiani AM, et al. Monomorphous angiocentric glioma: a distinctive epileptogenic neoplasm with features of infiltrating astrocytoma and ependymoma. J Neuropathol Exp Neurol. 2005;64:875-881.
Zentner J, Wolf HK, Ostertun B, et al. Gangliogliomas: clinical, radiological, and histopathological findings in 51 patients. J Neurol Neurosurg Psychiatry. 1994;57:1497-1502.
1 Louis DN, Ohgaki H, Wiestler OD, Cavenee WK, editors. World Health Organization Classification of Tumours of the Central Nervous System. Lyon: IARC, 2007.
2 Arca G, Pacheco E, Alfonso I, et al. Characteristic brain magnetic resonance imaging (MRI) findings in neonates with tuberous sclerosis complex. J Child Neurol. 2006;21:280-285.
3 Clarke MJ, Foy AB, Wetjen N, Raffel C. Imaging characteristics and growth of subependymal giant cell astrocytomas. Neurosurg Focus. 2006;20:E5.
4 de Ribaupierre S, Dorfmüller G, Bulteau C, et al. Subependymal giant-cell astrocytomas in pediatric tuberous sclerosis disease: when should we operate? Neurosurgery. 2007;60:83-89.
5 Franz DN, Leonard J, Tudor C, et al. Rapamycin causes regression of astrocytomas in tuberous sclerosis complex. Ann Neurol. 2006;59:490-498.
6 Lellouch-Tubiana A, Boddaert N, Bourgeois M, et al. Angiocentric neuroepithelial tumor (ANET): a new epilepsy-related clinicopathological entity with distinctive MRI. Brain pathology (Zurich, Switzerland). 2005;15:281-286.
7 Brat DJ, Scheithauer BW, Fuller GN, Tihan T. Newly codified glial neoplasms of the 2007 WHO Classification of Tumours of the Central Nervous System: angiocentric glioma, pilomyxoid astrocytoma and pituicytoma. Brain Pathol (Zurich). 2007;17:319-324.
8 Wang M, Tihan T, Rojiani AM, et al. Monomorphous angiocentric glioma: a distinctive epileptogenic neoplasm with features of infiltrating astrocytoma and ependymoma. J Neuropathol Exp Neurol. 2005;64:875-881.
9 Bailey P, Bucy PC. Astroblastomas of the brain. Acta Psychiatr Neurol Scand. 1930;5:439-461.
10 Bonnin JM, Rubinstein LJ. Astroblastomas: a pathological study of 23 tumors, with a postoperative follow-up in 13 patients. Neurosurgery. 1989;25:6-13.
11 Bell JW, Osborn AG, Salzman KL, et al. Neuroradiologic characteristics of astroblastoma. Neuroradiology. 2007;49:203-209.
12 Tihan T, Fisher PG, Kepner JL, et al. Pediatric astrocytomas with monomorphous pilomyxoid features and a less favorable outcome. J Neuropathol Exp Neurol. 1999;58:1061-1068.
13 Komotar RJ, Mocco J, Carson BS, et al. Pilomyxoid astrocytoma: a review. MedGenMed. 2004;6:42.
14 Komotar RJ, Zacharia BE, Sughrue ME, et al. Magnetic resonance imaging characteristics of pilomyxoid astrocytoma. Neurol Res. 2008;30:945-951.
15 Kepes JJ, Rubinstein LJ, Eng LF. Pleomorphic xanthoastrocytoma: a distinctive meningocerebral glioma of young subjects with relatively favorable prognosis. A study of 12 cases. Cancer. 1979;44:1839-1852.
16 Lim SC, Jang SJ, Kim YS. Cerebellar pleomorphic xanthoastrocytoma in an infant. Pathol Int. 1999;49:811-815.
17 Saikali S, Le Strat A, Heckly A, et al. Multicentric pleomorphic xanthoastrocytoma in a patient with neurofibromatosis type 1. Case report and review of the literature. J Neurosurg. 2005;102:376-381.
18 Giannini C, Scheithauer BW, Burger PC, et al. Pleomorphic xanthoastrocytoma: what do we really know about it? Cancer. 1999;85:2033-2045.
19 Levy RA, Allen R, McKeever P. Pleomorphic xanthoastrocytoma presenting with massive intracranial hemorrhage. AJNR Am J Neuroradiol. 1996;17:154-156.
20 Lipper MH, Eberhard DA, Phillips CD, et al. Pleomorphic xanthoastrocytoma, a distinctive astroglial tumor: neuroradiologic and pathologic features. AJNR Am J Neuroradiol. 1993;14:1397-1404.
21 Tonn JC, Paulus W, Warmuth-Metz M, et al. Pleomorphic xanthoastrocytoma: report of six cases with special consideration of diagnostic and therapeutic pitfalls. Surg Neurol. 1997;47:162-169.
22 Passone E, Pizzolitto S, D’Agostini S, et al. Non-anaplastic pleomorphic xanthoastrocytoma with neuroradiological evidences of leptomeningeal dissemination. Childs Nerv Syst. 2006;22:614-618.
23 Kros JM, Vecht CJ, Stefanko SZ. The pleomorphic xanthoastrocytoma and its differential diagnosis: a study of five cases. Hum Pathol. 1991;22:1128-1135.
24 Kaulich K, Blaschke B, Numann A, et al. Genetic alterations commonly found in diffusely infiltrating cerebral gliomas are rare or absent in pleomorphic xanthoastrocytomas. J Neuropathol Exp Neurol. 2002;61:1092-1099.
25 Fouladi M, Jenkins J, Burger P, et al. Pleomorphic xanthoastrocytoma: favorable outcome after complete surgical resection. Neuro Oncol. 2001;3:184-192.
26 Pahapill PA, Ramsay DA, Del Maestro RF. Pleomorphic xanthoastrocytoma: case report and analysis of the literature concerning the efficacy of resection and the significance of necrosis. Neurosurgery. 1996;38:822-828.
27 Nakajima T, Kumabe T, Shamoto H, et al. Malignant transformation of pleomorphic xanthoastrocytoma. Acta Neurochir. 2006;148:67-71.
28 Perkins O. Ganglioglioma. Arch Pathol Lab Med. 1926;2:11-17.
29 Zentner J, Wolf HK, Ostertun B, et al. Gangliogliomas: clinical, radiological, and histopathological findings in 51 patients. J Neurol Neurosurg Psychiatry. 1994;57:1497-1502.
30 Bevilacqua G, Sarnelli R. Ganglioglioma of the spinal cord. A case with a long survival. Acta Neuropathol. 1979;48:239-242.
31 Karamitopoulou E, Perentes E, Probst A, Wegmann W. Ganglioglioma of the brain stem: neurological dysfunction of 16-year duration. Clin Neuropathol. 1995;14:162-168.
32 Scalley JR. Ganglioglioma of the cerebellum: angiographic findings. Rocky Mountain Med J. 1976;73:80-82.
33 Zhang D, Henning TD, Zou LG, et al. Intracranial ganglioglioma: clinicopathological and MRI findings in 16 patients. Clin Radiol. 2008;63:80-91.
34 Matsumoto K, Tamiya T, Ono Y, et al. Cerebral gangliogliomas: clinical characteristics, CT and MRI. Acta Neurochir. 1999;141:135-141.
35 Johannsson JH, Rekate HL, Roessmann U. Gangliogliomas: pathological and clinical correlation. J Neurosurg. 1981;54:58-63.
36 Kincaid PK, El-Saden SM, Park SH, Goy BW. Cerebral gangliogliomas: preoperative grading using FDG-PET and 201Tl-SPECT. AJNR Am J Neuroradiol. 1998;19:801-806.
37 Hirose T, Scheithauer BW, Lopes MB, et al. Ganglioglioma: an ultrastructural and immunohistochemical study. Cancer. 1997;79:989-1003.
38 Lang FF, Epstein FJ, Ransohoff J, et al. Central nervous system gangliogliomas. Part 2: Clinical outcome. J Neurosurg. 1993;79:867-873.
39 Yin XL, Hui AB, Pang JC, et al. Genome-wide survey for chromosomal imbalances in ganglioglioma using comparative genomic hybridization. Cancer Genet Cytogenet. 2002;134:71-76.
40 Giulioni M, Galassi E, Zucchelli M, Volpi L. Seizure outcome of lesionectomy in glioneuronal tumors associated with epilepsy in children. J Neurosurg. 2005;102(Suppl):288-293.
41 Haddad SF, Moore SA, Menezes AH, VanGilder JC. Ganglioglioma: 13 years of experience. Neurosurgery. 1992;31:171-178.
42 Rumana CS, Valadka AB, Contant CF. Prognostic factors in supratentorial ganglioglioma. Acta Neurochir. 1999;141:63-68.
43 Rumana CS, Valadka AB. Radiation therapy and malignant degeneration of benign supratentorial gangliogliomas. Neurosurgery. 1998;42:1038-1043.
44 Kern M, Robbins P, Lee G, Watson P. Papillary tumor of the pineal region: a new pathological entity. Clin Neuropathol. 2006;25:185-192.
45 Chang AH, Fuller GN, Debnam JM, et al. MR imaging of papillary tumor of the pineal region. AJNR Am J Neuroradiol. 2008;29:187-189.
46 Fèvre-Montange M, Hasselblatt M, Figarella-Branger D, et al. Prognosis and histopathologic features in papillary tumors of the pineal region: a retrospective multicenter study of 31 cases. J Neuropathol Exp Neurol. 2006;65:1004-1011.
47 Daumas-Duport C, Scheithauer BW, Chodkiewicz JP, et al. Dysembryoplastic neuroepithelial tumor: a surgically curable tumor of young patients with intractable partial seizures. Report of thirty-nine cases. Neurosurgery. 1988;23:545-556.
48 Daumas-Duport C. Dysembryoplastic neuroepithelial tumours. Brain Pathol. 1993;3:283-295.
49 Parmar HA, Hawkins C, Ozelame R, et al. Fluid-attenuated inversion recovery ring sign as a marker of dysembryoplastic neuroepithelial tumors. J Comput Assist Tomogr. 2007;31:348-353.
50 Sampetrean O, Maehara T, Arai N, Nemoto T. Rapidly growing dysembryoplastic neuroepithelial tumor: case report. Neurosurgery. 2006;59:E1337-E1338.
51 Brat DJ, Scheithauer BW, Staugaitis SM, et al. Third ventricular chordoid glioma: a distinct clinicopathologic entity. J Neuropathol Exp Neurol. 1998;57:283-290.
52 Reifenberger G, Weber T, Weber RG, et al. Chordoid glioma of the third ventricle: immunohistochemical and molecular genetic characterization of a novel tumor entity. Brain Pathol. 1999;9:617-626.
53 Leeds NE, Lang FF, Ribalta T, et al. Origin of chordoid glioma of the third ventricle. Arch Pathol Lab Med. 2006;130:460-464.
54 Sato K, Kubota T, Ishida M, et al. Immunohistochemical and ultrastructural study of chordoid glioma of the third ventricle: its tanycytic differentiation. Acta Neuropathol. 2003;106:176-180.