Retinitis Pigmentosa
Clinical Features:
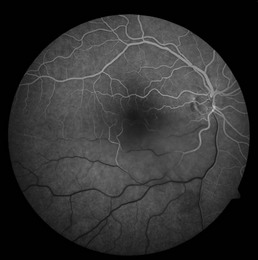
Nyctalopia is a hallmark feature of the disease. Peripheral vision is impaired early, especially in rod–cone dystrophies, and is slowly progressive. Central vision can also be lost, but typically occurs later in the disease course, although central vision can be impaired at any point by cystoid macular edema. Examination findings include characteristic bone spicule intraretinal deposits, vascular attenuation, and optic nerve pallor (Fig. 16.1.1).
OCT Features:
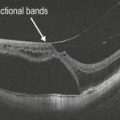
In advanced RP cases, OCT demonstrates marked attenuation of all retinal layers, particularly of the outer retina and photoreceptors (Fig. 16.1.2). Milder or earlier forms of RP can show more subtle outer retinal atrophy adjacent to a normal central macula (Figs 16.1.3 and 16.1.4). OCT can also be helpful to detect the presence of cystoid macular edema (Fig. 16.1.5), which is commonly associated with RP.

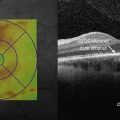
Figure 16.1.2 OCT in advanced retinitis pigmentosa shows significant attenuation of all retinal layers, most notably of the outer retina, and worse temporally. OCT thickness map (inset) shows the degree of generalized retinal thinning throughout the macula. The central macula is affected to a lesser degree than the surrounding area.

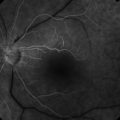
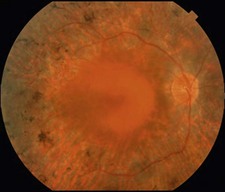
Figure 16.1.3 Color photograph of late onset retinitis pigmentosa with mild disease shows mild pigment deposition and retinal thinning temporal to the macula. There is also unrelated peripapillary atrophy around the optic nerve.

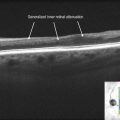
Figure 16.1.4 OCT (corresponding to Figure 16.1.3) shows outer retinal thinning just outside the fovea. OCT thickness map (inset) shows a central island of normal retinal thickness, with significant circumferential thinning outside of this area.