CHAPTER 2 Mucosal Immunity
Mucosal immunity refers to immune responses that occur at mucosal sites. The demands on the mucosal immune system are distinct from their systemic counterparts. At mucosal sites, the outside world is typically separated from the inner world by a single layer of epithelium. The mucosal immune system exists at a number of sites, including the respiratory tract (especially the upper respiratory tract), the urogenital tract, the mammary glands, and even the eye and ear. Regardless of the site, unique lymphoid and other cell populations are required to handle a wide array of environmental stimuli. Together, these sites are called mucosa-associated lymphoid tissue, or MALT.1–5 However, the site that is most often associated with mucosal immunity is the intestinal tract.
DISTINCT IMMUNE RESPONSES IN GUT-ASSOCIATED LYMPHOID TISSUE
CONTROLLED/PHYSIOLOGIC INFLAMMATION
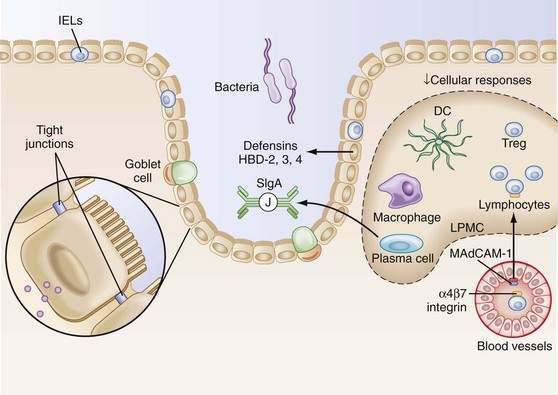
Within the lamina propria exist billions of activated plasma cells, memory T cells, memory B cells, macrophages, and dendritic cells.6,7 In fact, given the large surface area of the gastrointestinal (GI) tract and the resident cell populations that inhabit this space, the gut is the largest lymphoid organ in the body. Still, in contrast to activated lymphocytes in the peripheral immune system, significant inflammation is not present in the intestine. This phenomenon has been termed controlled/physiologic inflammation (Fig. 2-1).
The entry and activation of the cells into the lamina propria is antigen-driven. Germ-free mice have few cells in their lamina propria. However, within hours to days following colonization with normal intestinal flora (without pathogens), there is a massive influx and activation of cells.8–11 Despite the persistence of an antigen drive (luminal bacteria), the cells fail to develop into aggressive inflammation-producing lymphocytes and macrophages. Bacteria or their products play a role in this persistent state of activation12 and likely contribute to the controlled inflammatory process as well.
The failure to produce gastrointestinal pathology, despite the activation state of intestinal lymphocytes, is probably the consequence of regulatory mechanisms. The failure of lamina propria lymphocytes (LPLs) to generate “normal” antigen-receptor mediated responses is an important factor in controlled inflammation (e.g., lack of expansion, despite a state of activation). LPLs respond poorly when activated via their T cell receptor (TCR), failing to proliferate although they still can produce cytokines.13,14 This is key to the maintenance of controlled inflammation.
ORAL TOLERANCE
The most recognized phenomenon equated with mucosal immunity and associated with suppression is oral tolerance.15–20 Oral tolerance can be defined as the active, antigen-specific nonresponse to antigens administered orally.18,21,22 Disruption of oral tolerance may result in food allergies (see Chapter 9) and food intolerances such as celiac disease (see Chapter 104). Part of the explanation for oral tolerance relates to the properties of digestion per se, where large potentially antigenic macromolecules are degraded so that potentially immunogenic substances are rendered nonimmunogenic. However, approximately 2% of dietary proteins enter the draining enteric vasculature intact.23 How does the body regulate the response to these potential antigens that have bypassed complete digestion? This is achieved by oral tolerance. Factors affecting the induction of oral tolerance are the age of the host, genetic factors, the nature of antigen, the tolerogen’s form, and dose. In addition, the state of the intestinal barrier affects oral tolerance and, when barrier function is reduced, oral tolerance decreases as well.
Oral tolerance is difficult to achieve in the neonate, probably because of the rather permeable intestinal barrier that exists in the newborn, as well as the immaturity of the mucosal immune system. Within 3 months of age (in the mouse), oral tolerance can be induced and many previous antibody responses to food antigens are suppressed. The limited diet in the newborn may further serve to protect the infant from generating a vigorous response to food antigens. Furthermore, the intestinal flora has been demonstrated to affect the development of oral tolerance. Probiotics such as Lactobacillus GG given to mothers before delivery and during lactation have provided protection against the development of atopic eczema in their offspring.24 The effects of probiotics on oral tolerance are probably mediated through modulation of cytokine responses,25 a positive effect on intestinal barrier function and restitution of tight junctions,26–27 suppression of intestinal inflammation via down-regulation of Toll-like receptor (TLR) expression,28,29 and secretion of metabolites that may inhibit inflammatory cytokine production by mononuclear cells. The role of genetic factors in oral tolerance has been suggested in murine models in which certain strains develop tolerance more easily than others.30
The nature and form of the antigen also play a significant role in tolerance induction. Protein antigens are the most tolerogenic whereas carbohydrates and lipids are much less effective at inducing tolerance.31 The way the antigen is delivered is also critical. For example, a protein delivered in soluble form (e.g., ovalbumin) is tolerogenic, whereas aggregation of this protein reduces its potential to induce tolerance. This phenomenon may be associated with an alteration in the sites of antigen sampling.6 Exposure or prior sensitization to an antigen through an extraintestinal route also affects the development of tolerance responses. Finally, the dose of antigen administered is critical to the form of oral tolerance generated. In mouse models, high doses of antigen are associated with clonal deletion or anergy.32 In this setting, tolerance is not transferable; transfer of T cells from tolerized animals does not lead to the transfer of tolerance. The mechanism underlying T cell deletion is possibly Fas-mediated apoptosis.33 On the other hand, low doses of antigen activate regulatory-suppressor T cells.34,35 Increasing numbers of such T cells occur, both in CD4 and CD8 lineages. Th3 cells were the initial regulatory-suppressor cells described as mediators of oral tolerance.35–37 These cells appear to be activated in Peyer’s patches and secrete transforming growth factor-β (TGF-β). This cytokine plays a dual role in mucosal immunity; it is the most potent suppressor of T and B cell responses while also promoting the production of IgA (TGF-β is the IgA switch factor).38–41 The production of TGF-β by Th3 cells elicited by low-dose antigen administration helps explain an associated phenomenon of oral tolerance, namely, bystander suppression. Whereas oral tolerance is antigen specific, the effector arm is antigen non-specific. If an irrelevant antigen is coadministered systemically with the tolerogen, suppression of T and B cell responses to that irrelevant antigen will also occur (hence, bystander suppression). Secreted TGF-β suppresses the response to the coadministered antigen.
T regulatory 1 (Treg1, or Tr1) cells may also participate in bystander suppression and oral tolerance by producing interleukin-10 (IL-10), another potent immunosuppressive cytokine.42–44 Evidence for the activation of CD4+, CD25+ regulatory T cells during oral tolerance also exists, although their exact role in this process is still being investigated.45–49 Tolerance studies carried out in mice depleted of CD4+, CD25+ T cells coupled with neutralization of TGF-β have demonstrated that CD4+, CD25+ T cells and TGF-β together are involved in the induction of oral tolerance, partly through regulation of the expansion of antigen-specific CD4+ T cells.50 The ability to identify regulatory CD4+, CD25+ T cell subpopulations was enhanced by the recognition that these cells express the transcription factor FoxP3. Because not every cell within the CD4+, CD25+ population is a naturally occurring Treg cell, the ability to use FoxP3 as a marker of these Treg cells has been a major breakthrough in our ability to study these cells.51,52 Importantly, the absence of CD4+ T regulatory cell activity in mice results in inflammatory bowel disease (IBD), although this has not been demonstrated in humans.53–56
Preliminary data also support a role for antigen-specific CD8+ T cells in oral tolerance,57–61 as well as in the regulation of mucosal immune responses. Specifically, in vitro activation of human CD8+ peripheral blood T cells by normal intestinal epithelial cells (IECs) results in the expansion of CD8+, CD28− T cells with regulatory activity. Moreover, in the lamina propria of IBD patients, such CD8+, CD28− cells were significantly reduced, supporting a role for these epithelial-induced T regulatory (TrE) cells in the control of intestinal inflammation.62
Another factor affecting tolerance induction is the state of the intestinal barrier. In addition to the failure to generate tolerance in the neonatal period (when intestinal permeability is higher), several other states of barrier dysfunction are associated with aggressive inflammation and a lack of tolerance. During anaphylaxis, increased intestinal permeability caused by the disruption of tight junctions allows luminal antigens to pass through paracellular spaces.63–65 Treatment with interferon-γ (IFN-γ) can disrupt the mucosal barrier in mice and they fail to develop tolerance in response to ovalbumin feeding. Even more interesting is the failure of N-cadherin–dominant negative mice to suppress mucosal inflammation (loss of controlled inflammation),66 possibly because of the enormous antigenic exposure resulting from the leaky barrier in these mice.
Lastly, oral tolerance may also be associated with the cells serving as the antigen-presenting cell (APC; see later), as well as the site of antigen uptake. Orally administered reovirus type III is taken up in mice by microfold (M) cells expressing reovirus type III–specific receptors (Fig. 2-2).67 This epithelial uptake by M cells induces an active IgA response to the virus. Reovirus type I, on the other hand, infects intestinal epithelial cells (IECs) adjacent to M cells and this uptake induces tolerance to the virus. Thus, the route of entry (M cell versus IEC) of a specific antigen may dictate the type of immune response generated (IgA versus tolerance). Interestingly, poliovirus vaccine, one of the few oral vaccines effective in humans, binds to M cells, which may account for its ability to stimulate active immunity in the gut.68
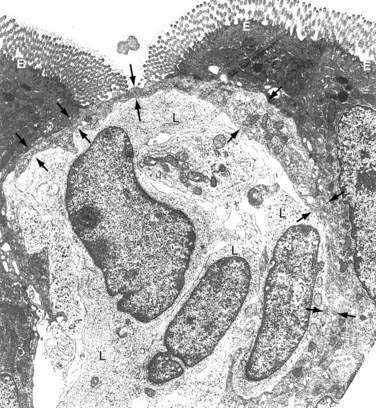
UNUSUAL IMMUNOGLOBULINS OF GUT-ASSOCIATED LYMPHOID TISSUE
IMMUNOGLOBULIN A
The unique antibody, secretory IgA (SIgA), is the hallmark of MALT-GALT immune responses (Fig. 2-3). Although IgG is the most abundant isotype in the systemic immune system, IgA is the most abundant antibody in mucosal secretions.69–71 In fact, given the numbers of IgA-positive plasma cells and the size of the MALT system, IgA turns out to be the most abundant antibody in the body. SIgA is a dimeric form of IgA produced by plasma cells in the lamina propria and transported into the lumen by a specialized pathway through the intestinal epithelium (Fig. 2-4). Two IgA monomers are bound together by a J chain (also produced by plasma cells). Subsequently, the dimer binds to secretory component (SC), also known as the polymeric immunoglobulin receptor (pIgR), a highly specialized 55-kd glycoprotein produced by IECs. SC (pIgR) is expressed on the basolateral aspect of the IEC and binds only to dimeric IgA or to IgM (also polymerized with J chain; see later). Once bound to SC, SIgA is actively transported within vesicles to the apical membrane of the IEC. The vesicle fuses with the apical membrane and the SC-IgA complex is released into the lumen. Once in the lumen, SC serves its second function, protection of the SIgA dimer from degradation by luminal proteases and gastric acid. SIgA and SIgM are the only antibodies that can bind SC and therefore withstand the harsh environment of the GI tract.
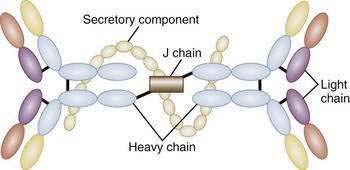
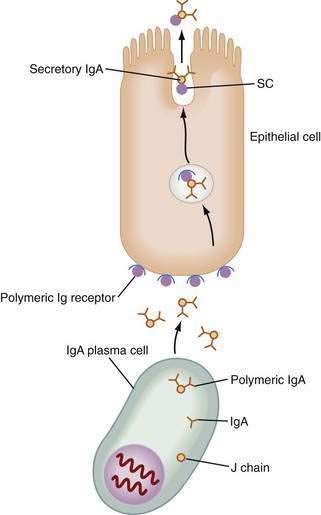
In addition to its unique form, SIgA is also unique as an immunoglobulin in that it is anti-inflammatory in nature. SIgA does not bind classic complement components but rather binds to luminal antigens, preventing their attachment to the epithelium or promoting agglutination and subsequent removal of the antigen in the mucus layer overlying the epithelium.69,72 This binding of luminal antigens by SIgA reflects immune exclusion, as opposed to the nonspecific mechanisms of exclusion exerted by the epithelium, the mucous barrier, proteolytic digestion, and other mechanisms.
OTHER IMMUNOGLOBULINS
Whereas SIgA is the major antibody isotype produced in GALT, IgG has been detected as well.73–74 The neonatal Fc receptor (FcRN), expressed by IECs, might serve as a bidirectional transporter of IgG75,76 and may be important in control of neonatal infections and IgG metabolism. In IBD, marked increases in IgG in the lamina propria and lumen have been observed.77
Even IgE production may play an important role in intestinal diseases in GALT. CD23 (low-affinity IgE Fc receptor) has been reported to be expressed by IEC. One model has suggested that CD23, or FcεRII, may play a role in facilitated antigen uptake and consequent mast cell degranulation in food allergy. In this setting, IgE transcytosis and mast cell degranulation may be associated with fluid and electrolyte loss into the lumen, an event that is intimately associated with allergic reactions in the gut and airways.78,79
PHYSIOLOGY OF GUT-ASSOCIATED LYMPHOID TISSUE: THE INTESTINAL BARRIER
The mucous coat lining the intestinal tract is composed of a mixture of glycoproteins (mucins). The protein core of mucins is enriched in serine, threonine, and proline residues, and carbohydrate moieties are attached via N-acetylgalactosamine residues. At least six different mucin species have been identified,80 each with a distinct carbohydrate and amino acid composition. Mucus protects the intestinal wall by several mechanisms. Its stickiness and competitive binding to glycoprotein receptors decrease the ability of microorganisms to penetrate the intestine.81 It also generates a stream that moves luminal contents away from epithelial cells.
Underneath the mucous layer, the physical barrier that prevents penetration of antigen across the intestinal epithelium consists of the actual epithelial cell (the transcellular route) and the tight intercellular spaces (the paracellular route) regulated by tight junction (TJ) complexes (e.g., zona occludens) and the subjunctional space.82 Of the two structures, TJs have the greater role in preventing macromolecular diffusion across the epithelium, because these junctions exclude almost all molecules present in the lumen (see Chapter 96). The barrier formed by the TJ is a dynamic structure, preserved even when epithelial cells themselves are damaged; this feature might be crucial for the prevention of intestinal inflammation (e.g., as seen in idiopathic IBD).
The epithelial cells themselves serve as a physical barrier in several ways—their microvilli are at a distance of about 25 nm from each other and are negatively charged. Thus, a negatively charged luminal molecule would be repelled from passage even if its diameter were well below 25 nm. However, intact antigens may traverse the epithelium by fluid phase endocytosis and enter the circulation.83
FUNCTIONAL ANATOMY OF GUT-ASSOCIATED LYMPHOID TISSUE
PEYER’S PATCHES AND MICROFOLD CELLS
The follicle-associated epithelium (FAE), which contains microfold (M) cells, is a specialized epithelium overlying the only organized lymphoid tissue of GALT, Peyer’s patches (PPs). The M cell, in contrast to the adjacent absorptive epithelial cell, has few microvilli, a limited mucin overlayer, a thin elongated cytoplasm, and a shape that forms a pocket surrounding subepithelial, T, B, macrophages, and dendritic cells (see Fig. 2-2). M cells are capable of taking up large particulate antigens from the lumen and transporting them intact into the subepithelial space.84–86 M cells contain few lysosomes, so little or no processing of antigen occurs.87 M cells are exposed to the lumen, thus having a larger area for contact with luminal contents than adjacent epithelial cells. The M cell expresses several unique lectin-like molecules that help promote binding to specific pathogens—the prototype being poliovirus.88 Antigens that bind to the M cell and are transported to the underlying PP generally elicit a positive (SIgA) response. Successful oral vaccines bind to the M cell and not to the adjacent epithelium. Thus, M cells appear to be critical for the positive aspects of mucosal immunity.
The M cell is a conduit to PPs. Antigens transcytosed across the M cell and into the subepithelial pocket are taken up by macrophages and DCs and carried into PPs. Once in the patch, TGF-β–secreting T cells promote B cell isotype switching to IgA.89 Importantly, there is a clear relationship between M cells and PPs. The induction of M cell differentiation has been shown to be dependent on direct contact between the epithelium and B lymphocytes in PPs.90 M cells do not develop in the absence of PPs. For example, M cells have not been identified in B cell–deficient animals in whom there are no PPs.91 Even though M cells and PPs may be involved in oral tolerance,92–94 PP-deficient mice are capable of developing tolerance after oral administration of soluble antigen.95
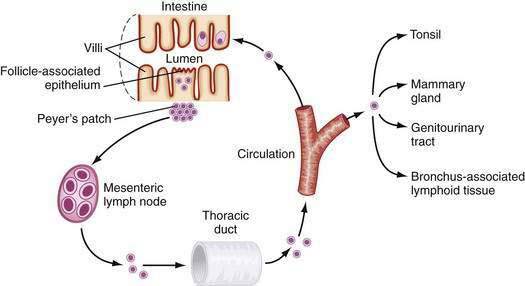
After activation in PPs, lymphocytes are induced to express specific integrins (α4β7), which provide a homing signal for mucosal sites (where the ligand is MadCAM-1).96–98 Cells then travel to MLNs and subsequently into the main intestinal lymphatic drainage system, the thoracic duct, and finally into the systemic circulation (Fig. 2-5). There, mucosally activated lymphocytes with their mucosal addressins circulate in the bloodstream to exit in high endothelial venules in various mucosal sites. Those bearing α4β7 molecules exit in the MALT-GALT lamina propria where they undergo terminal differentiation. Chemokines and their receptors (see later) as well as adhesion molecules and ligands may help direct this trafficking pattern.
INTESTINAL EPITHELIAL CELLS
The epithelium is composed of a single layer of columnar cells. These IECs are derived from the basal crypts and differentiate into absorptive villous or surface epithelium, secretory goblet cells, neuroendocrine cells, or Paneth cells (see Chapter 96). In addition to their function as a physical barrier in GALT discussed earlier, IECs contribute to innate and adaptive immunity in the gut and may play a key role in maintaining intestinal homeostasis.
Antigen Trafficking Across Intestinal Epithelial Cells
Binding to the surface of the cell depends on the structure of the antigen and the chemical composition of the microvillous membrane. For instance, bovine serum albumin binds less efficiently to the surface of the IEC than bovine milk protein and, as a consequence, is transported less efficiently.99 In addition, structural alterations in an antigen caused by proteolysis might also affect its binding, because this will change the physicochemical characteristics of the molecule.100
Several factors influence the transport of antigens from the apical to the basolateral surface of IECs. The rate of vesicular passage to the basolateral membrane depends on the rate of endocytosis, the proportion of vesicles trafficking to the lysosome, and the speed of travel of membrane-bound compartments. Lysosomally derived enzymes determine the rate of breakdown of products contained in membrane compartments. These include proteases such as cathepsins B and D (found throughout the length of the intestine, particularly in the mid and distal thirds of the small intestine), as well as those enzymes that catalyze carbohydrate breakdown, such as acid phosphatase and mannosidase. It is the degree to which the organellar contents encounter such enzymes (in the lysosome or in endocytic vesicles) that determines the rate of intracellular destruction of macromolecules.101 Although cathepsins are capable of catalyzing antigens, they may not completely digest the protein, which may require further proteolysis by peptidases in the cytoplasm.
Recognition of Pathogen-Associated Molecular Patterns by Pattern Recognition Receptors
Classic APCs in the systemic immune system possess the innate capacity to recognize components of bacteria and viruses, called pathogen-associated molecular patterns (PAMPs). Receptors for these PAMPs are expressed on the cell surface (e.g., TLRs) and inside the cell (e.g., NOD2 [see later]). Despite the fact that IECs live adjacent to large numbers of luminal flora, IECs retain the ability to recognize components of these bacteria. In general, proinflammatory responses are normally down-regulated (i.e., expression of the lipopolysaccharide [LPS] receptor, TLR4, is absent) and expression of some of these pattern recognition receptors is maintained, such as TLR5, which recognizes bacterial flagellin. TLR5 is expressed basolaterally, so it is poised to identify organisms such as Salmonella species that have invaded the epithelial layer.102 Following invasion and engagement of TLR5, the IEC is induced to secrete a broad array of cytokines and chemokines that attract inflammatory cells to the local environment to control the spread of infection. In contrast to invading pathogens, some bacteria are probiotic and induce the IEC to produce anti-inflammatory cytokines (e.g., IL-10) and to increase the expression of peroxisome proliferator-activated receptor-γ (PPAR-γ).103 Furthermore, other bacterial products help promote the barrier and IEC differentiation (e.g., products of Bacteroides thetaiotaomicron).
The significance of the ability of IECs to recognize PAMPs via surface TLRs, such as TLR5, or via intracellular nuclear oligomerization domain 1, 2 (NOD1, 2), has been increasingly recognized over the past decade. The latter ability has been shown to contribute to intestinal inflammation, because about 25% of patients with Crohn’s disease have mutations in the NOD2-CARD15 gene, interfering with their ability to mount an appropriate immune response to bacterial stimuli (discussed further in Chapter 111).104–108 In addition, TLRs that are weakly expressed by normal IECs are expressed at higher levels on IECs obtained from patients with IBD.109 Expression of different TLRs by IECs, as well as their contribution to innate and adaptive T and B cell responses in intestinal inflammation and homeostasis, has been demonstrated in several murine models.110,111 TLR expression by professional APCs is also down-regulated in the lamina propria. This finding, along with others described earlier, contribute to the immunologic nonresponsiveness of GALT. The importance of TLR and NOD2-CARD15 expression and signaling in the intestine has been reviewed.112
ANTIGEN PRESENTATION IN THE GUT
Class II MHC molecules are also present on the epithelium of the normal small intestine and, to a lesser extent, colonocytes in humans113 and rodents.114 In vitro studies have demonstrated that enterocytes isolated from rat and human small intestine can present antigens to appropriately primed T cells.115–117 This raises the possibility that IECs might present peptides to GALT T cells beneath the epithelium. Thus, IECs are capable of antigen trafficking and processing as well as antigen presentation to cells in the lamina propria in the appropriate context. Importantly, increased expression of MHC class II molecules by IECs has been reported in patients with IBD.118,119 Such overexpression would be expected to increase their potential to activate T lymphocytes and this has been reported.120,121
Drugs used to treat patients with IBD, such as 5-aminosalicylic acid (5-ASA) preparations, may reduce IEC MHC class II expression on IEC.122 In addition, IECs from normal individuals or IBD patients express a variety of costimulatory molecules required for T cell activation (Fig. 2-6). These include intercellular adhesion molecule 1 (ICAM-1), which binds to leukocyte function-associated antigen 1 (LFA-1) on the T cell and to B7 (CD80) on the APC. B7, which binds CD28 and cytotoxic T-lymphocyte antigen 4 (CTLA-4),123,124 has been shown to be expressed by IECs of patients with ulcerative colitis (UC). Interestingly, unique expression of these costimulatory molecules by IECs may be involved in the distinct regulation of mucosal responses. Failure of naive T cells to engage CD28 by B7 family members may result in T cell tolerance. This may be less of an issue in GALT, in which cells express the memory phenotype.125,126 Because small intestine IECs do not express B7-1 (CD80),127 activation of naive T cells by IECs is not probable, aiding in the down-regulation of T-cell responses. On the other hand, increased expression of B7 during intestinal inflammation may serve to augment T cell stimulation.128
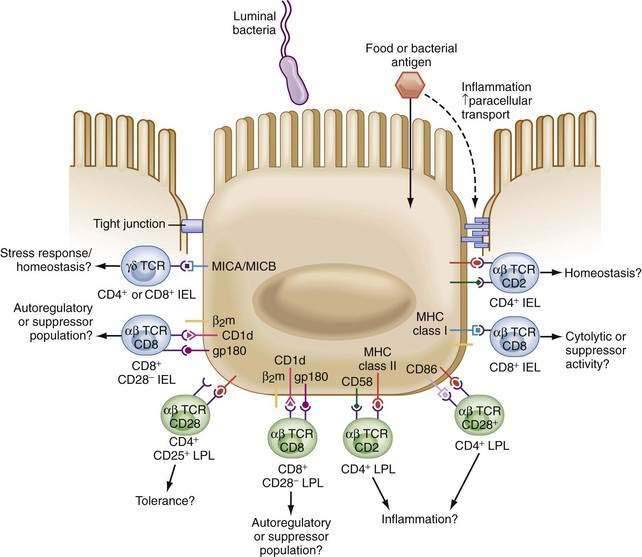
MHC class I and nonclassic class I molecules are also expressed by IECs. Thus, antigen presentation to unique T cell populations is possible and has been reported by several groups.116,129–135 Specifically, CD1d expressed on human IECs is able to present antigen, in a complex with CEACAM5, to CD8+ T cells.136–140 The role of other nonclassical class I molecules expressed by IECs (e.g., MR-1, TL, Hmt-1, MICA/B, HLA-E, HLA-G) is still unclear.141–144
In humans, IECs specifically activate CD8+ regulatory T cells.116 These regulatory cells may be involved in local tolerance and interaction with intraepithelial lymphocytes, which are CD8+ T cells (see later). The role of IECs in the regulation of mucosal immunity is best demonstrated in studies with tissues obtained from patients with IBD. IECs derived from IBD patients, in contrast to normal IECs, stimulate CD4+ T cells rather than regulatory CD8+ cells.120–121145 Furthermore, oral antigen administration does not result in tolerance in IBD patients, but instead produces active immunity.146
INTESTINAL MONONUCLEAR CELLS
INTRAEPITHELIAL LYMPHOCYTES
IELs in the small intestine are largely (>98%) T cells and are characterized by being mostly CD8+ cells,147–154 including CD8+ αα T cells and CD4+, CD8+ double-positive and CD4−, CD8− double-negative cells. Greater numbers of these cells also express the γδ TCR, in contrast to the αβ TCR expressed by T cells in the systemic immune system.155 Approximately half of murine small bowel IELs express the γδ TCR,156 whereas the murine and human large intestines contain primarily αβ CD4+ or CD8+ T cells similar to those found in the systemic immune system.
Based on their phenotype, IELs have been classified into two subsets, a and b. Subpopulation a includes TCR αβ T cells selected in the thymus with conventional MHC class I and II expression. Subpopulation b includes TCR αβ CD8+ αα, TCR γδ DP, and TCR γδ DN cells. Both subpopulations have been shown to be cytolytic, killing via granzyme or by engagement of Fas. They both also secrete Th1 cytokines such as IL-2, TNF-α, and IFN-γ. However, antigen-specific type a IELs can transfer protection against a variety of pathogenic organisms, whereas type b IELs are not able to transfer immunologic protection and do not possess immunologic memory. This is possibly caused by the activation of type b IELs by IECs in situ by nonclassical MHC molecules, rather than by the polymorphic MHC-expressed molecules on professional APCs that activate type a IELs.156 IELs express a variety of activation markers and are CD45 RO+ (memory cells). IELs also express GALT-specific integrin αEβ7.157–158 Expression of this integrin is induced by TGF-β, and its ligand on IECs is E-cadherin, which is involved in cell signaling and cytoskeletal rearrangement.158 When isolated, IELs are difficult to activate through their TCR and they barely proliferate, even in response to potent stimuli.153 On the other hand, they may be activated by alternative pathways, such as via CD2.
Type a IELs secrete cytokines such as IL-7 that are different from the ones secreted by their peripheral blood counterparts.152,159–161 Functionally it has been suggested that IELs potentially kill epithelial cells that have undergone some form of stress, such as infection, transformation, or invasion by other cells.154–156162 Alternatively, it has been proposed that IELs are active in suppressing local immunity, although the evidence that they actually function in luminal antigen recognition is weak. IELs do not travel in and out of the epithelium. Rather, the epithelial cells grow over the IELs as they move from the crypt to the surface. Thus, IELs likely serve as sentinels for epithelial integrity.
LAMINA PROPRIA MONONUCLEAR CELLS
Lamina propria mononuclear cells (LPMNCs) are a heterogeneous group of cells.163,164 The most populous cell type is the IgA-positive plasma cell, but there are more than 50% T and B cells (together comprising the LPL population), macrophages, and dendritic cells. In contrast to IELs, which express αEβ7, LPLs express the mucosal addressin α4β7. Similar to IEL, they express an activated memory phenotype and do not proliferate in response to engagement of the TCR. Alternate pathways of LPL activation are mainly via CD2 and CD28.159,165,166
Down-regulating the ability of LPLs to respond to stimulation via the TCR (i.e., antigen) may be another mechanism involved in dampening immune responses to normal luminal contents, along with the increased tendency for LPLs to undergo apoptosis if activated inappropriately. The mechanism underlying this latter apoptotic phenomenon possibly relates to engagement of the death receptor Fas and its ligand on activated LPLs, and by the imbalance between the intracellular anti- and pro-apoptotic factors, Bcl2 and Bax. Defects in this proapoptotic balance have been reported in patients with Crohn’s disease.167,168
T CELL DIFFERENTIATION
As noted, there is an organized lymphoid structure in the LP, the Peyer’s patch (see Fig. 2-5). There, B and T lymphocytes interact with antigen sampled via M cells in the FAE. Activation and maturation of T lymphocytes from naive Th0 cells to distinct biased subpopulations is strongly influenced by the microenvironment. Specifically, contact with dendritic cells (DCs), professional APCs within GALT, and their secreted mediators will skew T lymphocytes to one of several effector cells. IL-2, IFN-γ and TNF-α–secreting T helper 1(Th1) cells develop from Th0 cells when DCs secrete the IL-12/p35-40 heterodimer.169 This heterodimer induces activation and phosphorylation of the transcription factor STAT-4 (signal transducer and activator of transcription factor 4).170 STAT-4 in turn induces IFN-γ expression and production. IFN-γ then induces activation of STAT-1 and consequently of T-bet; this is the master transcription factor that induces Th1 cytokine production and IL-12 receptor β2 expression while simultaneously suppressing Th2 cytokine production. Thus, a cycle promoting Th1 and suppressing Th2 responses is created. Overactivation of T-bet is possibly an essential step for Th1-mediated mucosal diseases, such as those seen in some patients with Crohn’s disease.170 Another Th1-promoting cytokine is IL-18, mediating its effects by augmenting IL-12 receptor β2 expression on T cells and by AP-1-(c-fos/c-jun)–dependent transactivation of the IFN-γ promoter. It also activates nuclear factor κB (NF-κB) in T cells.169
In contrast, when IL-4 is secreted, STAT-6 is activated, followed by activation of the transcription factor GATA-3, which is capable of promoting the expression of several Th2 cytokines, including IL-4, IL-5, and IL-13.171 In addition to IL-4, IL-13 also plays an important role in Th2 development and IgE synthesis in an IL-4–independent fashion. These Th2 cytokines, which also include IL-6, -9, and -10, appear to play a role in the development of food allergies (see Chapter 9). IL-5 induces B cells expressing surface IgA to differentiate into IgA-producing plasma cells. IL-6 causes a marked increase in IgA secretion, with little effect on IgM or IgG synthesis.172 Thus, in the normal state in GALT, a Th2 bias might exist.
However, despite the presence of classic Th1 and Th2 cells, the dominant tone in the intestine is one of regulation. Regulatory T cells, such as Th3 and Tr1 cells (see earlier, “Oral Tolerance”), develop in an environment in which IL-10 is predominant. Th3 and Tr1 cells secrete the regulatory cytokines TGF-β and IL-10, respectively. In addition to these regulatory cells, other cells such as CD4+, CD25+, Foxp3+, and even CD8+ T regulatory cells have been identified. All these cell populations may be involved in the induction of oral tolerance and controlled inflammation in GALT (see earlier).
DENDRITIC CELLS
DCs play an important role in tolerance and immunity in the gut. DCs continuously migrate within lymphoid tissues and present self antigens (likely from dying apoptotic cells to maintain self-tolerance) as well as non–self-antigens.173 Within the lamina propria of the distal small intestine, DCs express the chemokine receptor CX3CR1 and form transepithelial dendrites that allow direct sampling of luminal antigen.174 It has been suggested that IECs expressing chemokine (C-C motif) ligand 25 (CCL25), the ligand for chemokine (C-C motif) receptor 9 (CCR9) and for CCR10, attract DCs to the small bowel, whereas CCL28, the ligand for CCR3 and CCR10, attracts DCs to the colon.175–177
DCs process internalized antigens more slowly than macrophages,178 which probably contributes to local tolerance.179,180 Tolerance induction by DCs is associated with their degree of maturation at the time of antigen presentation to T cells (e.g., immature DCs activate Tregs), down-regulation of costimulatory molecules CD80 and CD86, production of the suppressive cytokines IL-10, TGF-β and IFN-α, and interaction with the costimulatory molecule CD200.181–183
GUT-ASSOCIATED LYMPHOID TISSUE: RELEVANT CHEMOKINES
Chemokine (C-C motif) ligand 5 (CCL5, formerly called RANTES), secreted predominantly by macrophages, can be produced by human IECs as well.184 CCL5 may have a role in innate as well as adaptive mucosal immunity185 and, interestingly, increased CCL5 expression has been demonstrated in the mucosa of patients with IBD.186–189
Several CXC cytokines are constitutively expressed by lymphocytes, endothelial cells, and human colonic IECs.190,191 These include CXCL9, also known as monokine induced by interferon-γ (MIG), CXCL10, also known as interferon-γ inducible protein 10 (IP-10), which is a chemokine that appears to promote Th1 responses and therefore may be relevant in Crohn’s disease, and CXCL11, also known as IFN-γ–inducible T cell α chemoattractant (ITAC). Expression of CXCL9, 10, and 11, and their polarized basolateral secretion, increases after IFN-γ stimulation. CXC chemokines attract Th1 cells expressing high levels of CXCR3 (CXC receptor 3).192 They also contribute to natural killer (NK) T cell chemotaxis and increased cytolytic responses193 and activate subsets of DCs.194 By attracting CD4+ Th1 cells that produce IFN-γ, up-regulation of expression and secretion of CXC chemokines occurs, because IECs express IFN-γ receptors. This appears to contribute to a positive feedback loop that may be relevant in inflammatory states, specifically in IBD and celiac disease. Importantly, blockade of the CXCR3-CXCL10 axis has been shown to be beneficial in ameliorating murine colitis195 and is currently being investigated in human IBD.
In contrast to the inflammation-related CXCR3 receptor, a tissue-specific chemokine receptor, CCR9, is constitutively expressed on small bowel IELs and LPLs.196–198 CCL25, also known as thymus-expressed cytokine (TECK), is the ligand for CCR9 and is differentially expressed in the jejunal and ileal epithelium, where levels of expression decrease from the crypt up to the villous epithelium.199 In murine models, it was shown that CCL25-CCR9 is associated with selective localization of MLN-activated CD8 αβ+ lymphocytes coexpressing αEβ7 to the small intestine.200 CCL25 expression by IECs has been shown to be increased in inflamed small bowel in Crohn’s disease patients, with increased CCR9 expression by peripheral blood lymphocytes (PBLs) and decreased expression by LPLs,197 thus supporting its role in the specific attraction of peripheral lymphocytes to the small bowel in CD. This key chemokine/chemokine receptor pair (CCL25-CCR9) has also been used as a target for therapeutic intervention in CD, with preliminary positive results.201
Fractalkine is a unique chemokine expressed by IECs. It combines the properties of chemokines and adhesion molecules. Fractalkine attracts NK cells, monocytes, CD8+ T lymphocytes and, to a lesser extent, CD4+ T lymphocytes that express CX3CR1.202 Expression of fractalkine is increased in CD, specifically in the basolateral aspect of IECs.203
Mucosa-associated epithelial chemokine (MEC) may also have a role in intestinal immunity. This chemokine and its receptors, CCR3 and CCR10, are expressed by colonic IECs. CD4+ memory lymphocytes and eosinophils are attracted by MEC in vitro, although its function in vivo has not yet been demonstrated.204
MDC-CCL2 is a chemokine that is constitutively expressed and secreted by colonic IECs. MDC-CCL2 is unique in that it attracts CCR4+ Th2 cytokine-producing lymphocytes. Polarized basolateral secretion of MDC-CCL2 from stimulated colonic IEC lines has been reported.205 The specific recruitment of lymphocytes that preferentially secrete anti-inflammatory (Th2) cytokines supports a role for the IEC in orchestrating normal mucosal homeostasis, and adds to the accumulating evidence that these cells possess the ability to regulate mucosal immune responses.
The chemokine CCL20, also known as macrophage inflammatory protein-3α (MIP-3α), is unique in its ability to attract immature DCs specifically, as well as memory CD4+ T lymphocytes.206–208 CCL20 is also expressed and produced in the human small intestine, mainly in the FAE, and by colonic IECs, and has been suggested to be the mediator of lymphocyte adhesion to the α4β7 ligand MAdCAM1.206 CCL20 expression and secretion are increased in colonic IECs derived from IBD patients.209 Its stimulated secretion is polarized to the basolateral compartment, supporting its ability to attract immune cells into the LP. Mucosal memory T cells and IECs express CCR6, the cognate receptor for CCL20. CCR6 and CCR9 are coexpressed in T cells expressing the α4β7 integrin, characteristic of mucosal lymphocytes. This may suggest that in inflammatory states, and to some extent in the normal state, CCL20 and CCL25 expression by IECs attracts CCR6- or CCR9-positive lymphocytes that are activated in mesenteric lymph nodes, enter the peripheral blood, and then are recruited to the intestinal mucosa, where they can undergo activation-induced apoptosis, if they are aberrantly activated, or terminal differentiation.
Groux H, O’Garra A, Bigler M, et al. A CD4+ T-cell subset inhibits antigen-specific T-cell responses and prevents colitis. Nature. 1997;389:737-42. (Ref 42.)
Hugot JP, Chamaillard M, Zouali H, Lesage S, et al. Association of NOD2 leucine-rich repeat variants with susceptibility to Crohn’s disease. Nature. 2001;411:599-603. (Ref 104.)
Kerneis S, Bogdanova A, Kraehenbuhl JP, Pringault E. Conversion by Peyer’s patch lymphocytes of human enterocytes into M cells that transport bacteria. Science. 1997;277:949-52. (Ref 90.)
Kraus TA, Toy L, Chan L, et al. Failure to induce oral tolerance to a soluble protein in patients with inflammatory bowel disease. Gastroenterology. 2004;26:1771-8. (Ref 146.)
MacDonald TT. T cell immunity to oral allergens. Curr Opin Immunol. 1998;10:620-7. (Ref 20.)
Mowat AM, Viney JL. The anatomical basis of intestinal immunity. Immunol Rev. 1997;156:145-66. (Ref 2.)
Neurath MF, Finotto S, Glimcher LH. The role of Th1/Th2 polarization in mucosal immunity. Nat Med. 2002;8:567-73. (Ref 169.)
Neurath MF, Weigmann B, Finotto S, et al. The transcription factor T-bet regulates mucosal T cell activation in experimental colitis and Crohn’s disease. J Exp Med. 2002;195:1129-43. (Ref 170.)
Neutra MR. Current concepts in mucosal immunity. V. Role of M cells in transepithelial transport of antigens and pathogens to the mucosal immune system. Am J Physiol. 1998;274:G785-91. (Ref 85.)
Niess JH, Reinecker HC. Lamina propria dendritic cells in the physiology and pathology of the gastrointestinal tract. Curr Opin Gastroenterol. 2005;21:687-91. (Ref 182.)
Ogura Y, Bonen DK, Inohara N, et al. A frameshift mutation in NOD2 associated with susceptibility to Crohn’s disease. Nature. 2001;411:603-6. (Ref 105.)
Perera L, Shao L, Patel A, et al. Expression of nonclassical class I molecules by intestinal epithelial cells. Inflam Bowel Dis. 2007;13:298-307. (Ref 141.)
Sakaguchi S, Toda M, Asano M, et al. T cell-mediated maintenance of natural self-tolerance: Its breakdown as a possible cause of various autoimmune diseases. J Autoimmun. 1996;9:211-20. (Ref 45.)
Singh UP, Venkataraman C, Singh R, et al. CXCR3 axis: Role in inflammatory bowel disease and its therapeutic implication. Endocr Metab Immune Dis Drug Targets. 2007;7:111-23. (Ref 195.)
Steinman RM, Hawiger D, Nussenzweig MC. Tolerogenic dendritic cells. Annu Rev Immunol. 2003;21:685-711. (Ref 173.)
1. Brandtzaeg P, Farstad IN, Haraldsen G, et al. Cellular and molecular mechanisms for induction of mucosal immunity. Dev Biol Stand. 1998;92:93-108.
2. Mowat AM, Viney JL. The anatomical basis of intestinal immunity. Immunol Rev. 1997;156:145-66.
3. Moldoveanu Z, Clements ML, Prince SJ, et al. Human immune responses to influenza virus vaccines administered by systemic or mucosal routes. Vaccine. 1995;13:1006-12.
4. Bloom PD, Boedeker EC. Mucosal immune responses to intestinal bacterial pathogens. Semin Gastrointest Dis. 1996;7:151-66.
5. Brandtzaeg P. The human intestinal immune system: Basic cellular and humoral mechanisms. Baillieres Clin Rheumatol. 1996;10:1-24.
6. Mayer L, Sperber K, Chan L, et al. Oral tolerance to protein antigens. Allergy. 2001;56(Suppl 67):12-15.
7. Mayer L. Mucosal immunity and gastrointestinal antigen processing. J Pediatr Gastroenterol Nutr. 2000;30(Suppl):S4-12.
8. Anderson JC. The response of gut-associated lymphoid tissue in gnotobiotic piglets to the presence of bacterial antigen in the alimentary tract. J Anat. 1977;124:555-62.
9. Ishikawa K, Satoh Y, Tanaka H, et al. Influence of conventionalization on small-intestinal mucosa of germ-free Wistar rats: quantitative light microscopic observations. Acta Anat (Basel). 1986;127:296-302.
10. Cebra JJ, Periwal SB, Lee G, et al. Development and maintenance of the gut-associated lymphoid tissue (GALT): The roles of enteric bacteria and viruses. Dev Immunol. 1998;6:13-18.
11. Rothkotter HJ, Ulbrich H, Pabst R. The postnatal development of gut lamina propria lymphocytes: number, proliferation, and T and B cell subsets in conventional and germ-free pigs. Pediatr Res. 1991;29:237-42.
12. Saparov A, Kraus LA, Cong Y, et al. Memory/effector T cells in TCR transgenic mice develop via recognition of enteric antigens by a second, endogenous TCR. Int Immunol. 1999;11:1253-64.
13. Qiao L, Schurmann G, Betzler M, et al. Activation and signaling status of human lamina propria T lymphocytes. Gastroenterology. 1991;101:1529-36.
14. De Maria R, Fais S, Silvestri M, et al. Continuous in vivo activation and transient hyporesponsiveness to TcR/CD3 triggering of human gut lamina propria lymphocytes. Eur J Immunol. 1993;23:3104-8.
15. Xiao BG, Link H. Mucosal tolerance: A two-edged sword to prevent and treat autoimmune diseases. Clin Immunol Immunopathol. 1997;85:119-28.
16. Whitacre CC, Gienapp IE, Meyer A, et al. Treatment of autoimmune disease by oral tolerance to autoantigens. Clin Immunol Immunopathol. 1996;80:S31-9.
17. Weiner HL, Mayer LF. Oral tolerance: Mechanisms and applications. Introduction. Ann N Y Acad Sci. 1996;778:xiii-xviii.
18. Titus RG, Chiller JM. Orally induced tolerance. Definition at the cellular level. Int Arch Allergy Appl Immunol. 1981;65:323-38.
19. Strober W, Kelsall B, Marth T. Oral tolerance. J Clin Immunol. 1998;18:1-30.
20. MacDonald TT. T cell immunity to oral allergens. Curr Opin Immunol. 1998;10:620-7.
21. Challacombe SJ, Tomsai TB. Systemic tolerance and secretory immunity after oral imunization. J Exp Med. 1980;152:1459-72.
22. Mattingly JA, Waksman BH. Immunologic suppression after oral administration of antigen. I. Specific suppressor cells formed rat Peyer’s patches after oral administration. J Immunol. 1978;121:1878-83.
23. Webb KEJr. Amino acid and peptide absorption from the gastrointestinal tract. Fed Proc. 1986;45:2268-71.
24. Kalliomaki M, Salminen S, Poussa T, et al. Probiotics and prevention of atopic disease: 4-year follow-up of a randomized placebo-controlled trial. Lancet. 2003;361:1869-71.
25. Pohjavuori E, Viljanen M, Korpela R, et al. Lactobacillus GG effect in increasing IFN-gamma production in infants with cow’s milk allergy. J Allergy Clin Immunol. 2004;114:131-6.
26. Otte JM, Cario E, Podolsky DK. Mechanisms of cross hyporesponsiveness to Toll-like receptor bacterial ligands in intestinal epithelial cells. Gastroenterology. 2004;126:1054-70.
27. Rosenfeldt V, Benfeldt E, Valerius NH, et al. Effect of probiotics on gastrointestinal symptoms and small intestinal permeability in children with atopic dermatitis. J Pediatr. 2004;145:612-16.
28. Bullock NR, Booth JC, Gibson GR. Comparative composition of bacteria in the human intestinal microflora during remission and active ulcerative colitis. Curr Issues Intest Microbiol. 2004;5:59-64.
29. Gosselink MP, Schouten WR, van Lieshout LM, et al. Delay of the first onset of pouchitis by oral intake of the probiotic strain Lactobacillus rhamnosus GG. Dis Colon Rectum. 2004;47:876-84.
30. Lamont AG, Mowat AM, Browning MJ, Parrott DM. Genetic control of oral tolerance to ovalbumin in mice. Immunology. 1988;63:737-9.
31. Garside P, Mowat AM. Mechanisms of oral tolerance. Crit Rev Immunol. 1997;17:119-37.
32. Whitacre CC, Gienapp IE, Orosz CG, et al. Oral tolerance in experimental autoimmune encephalomyelitis. III. Evidence for clonal anergy. J Immunol. 1991;147:2155-63.
33. Chen Y, Inobe J, Marks R, et al. Peripheral deletion of antigen-reactive T cells in oral tolerance. Nature. 1995;376:177-80.
34. Friedman A, Weiner HL. Induction of anergy or active suppression following oral tolerance is determined by antigen dosage. Proc Natl Acad Sci U S A. 1994;91:6688-92.
35. Hafler DA, Kent SC, Pietrusewicz MJ, et al. Oral administration of myelin induces antigen-specific TGF-beta 1 secreting T cells in patients with multiple sclerosis. Ann N Y Acad Sci. 1997;835:120-31.
36. Fukaura HS, Kent C, Pietrusewicz MJ, et al. Induction of circulating myelin basic protein and proteolipid protein- specific transforming growth factor-beta1-secreting Th3 T cells by oral administration of myelin in multiple sclerosis patients. J Clin Invest. 1996;98:70-7.
37. Inobe J, Slavin AJ, Komagata Y, et al. IL-4 is a differentiation factor for transforming growth factor-beta secreting Th3 cells and oral administration of IL-4 enhances oral tolerance in experimental allergic encephalomyelitis. Eur J Immunol. 1998;28:2780-90.
38. Strobel SS. Neonatal oral tolerance. Ann N Y Acad Sci. 1996;778:88-102.
39. Kunimoto DY, Ritzel M, Tsang M. The roles of IL-4, TGF-beta and LPS in IgA switching. Eur Cytokine Netw. 1992;3:407-15.
40. Kim PH, Kagnoff MF. Transforming growth factor beta 1 increases IgA isotype switching at the clonal level. J Immunol. 1990;145:3773-8.
41. Coffman RL, Lebman DA, Shrader B. Transforming growth factor beta specifically enhances IgA production by lipopolysaccharide-stimulated murine B lymphocytes. J Exp Med. 1989;170:1039-44.
42. Groux H, O’Garra A, Bigler M, et al. A CD4+ T-cell subset inhibits antigen-specific T-cell responses and prevents colitis. Nature. 1997;389:737-42.
43. Levings MK, Roncarolo MG. T-regulatory 1 cells: A novel subset of CD4 T cells with immunoregulatory properties. J Allergy Clin Immunol. 2000;106:S109-12.
44. Roncarolo MG, Bacchetta R, Bordignon C, et al. Type 1 T regulatory cells. Immunol Rev. 2001;182:68-79.
45. Sakaguchi S, Toda M, Asano M, et al. T cell-mediated maintenance of natural self-tolerance: Its breakdown as a possible cause of various autoimmune diseases. J Autoimmun. 1996;9:211-20.
46. Sakaguchi S, Sakaguchi N, Shimizu J, et al. Immunologic tolerance maintained by CD25+ CD4+ regulatory T cells: Their common role in controlling autoimmunity, tumor immunity, and transplantation tolerance. Immunol Rev. 2001;182:18-32.
47. Shevach EM, Thornton A, Suri-Payer E. T lymphocyte-mediated control of autoimmunity. Novartis Found Symp. 1998;215:200-11.
48. Nakamura K, Kitani A, Strober W. Cell contact-dependent immunosuppression by CD4(+)CD25(+) regulatory T cells is mediated by cell surface-bound transforming growth factor beta. J Exp Med. 2001;194:629-44.
49. Zhang X, Izikson L, Liu L, Weiner HL. Activation of CD25(+)CD4(+) regulatory T cells by oral antigen administration. J Immunol. 2001;167:4245-53.
50. Chung Y, Lee SH, Kim DH, et al. Complementary role of CD4+CD25+ regulatory T cells and TGF-beta in oral tolerance. J Leukoc Biol. 2005;77:906-13.
51. Bluestone JA, Abbas AK. Natural versus adoptive regulatory T cells. Nat Rev Immunol. 2003;3:253-7.
52. Hai S, Namura T, Sakaguchi S. Control of regulatory T cell development by the transcription factor Foxp3. Science. 2003;299:1057-61.
53. Veltkamp C, Ruhwald R, Giesem T, et al. CD4+CD25+ cell depletion from the normal CD4+ T cell pool prevents tolerance toward the intestinal flora and leads to chronic colitis in immunodeficient mice. Inflamm Bowel Dis. 2006;12:437-46.
54. Morrissey PJ, Charrier K, Braddy S, et al. CD4+ T cells that express high levels of CD45RB induce wasting disease when transferred into congenic severe combined immunodefficient mice. Disease development is prevented by cotransfer of purified CD4+ T cells. J Exp Med. 1993;178:237-44.
55. Powrie F, Leach MW, Mauze S, et al. Phenotypically distinct subsets of CD4+ T cells induce or protect from chronic intestinal inflammation in C. B-17 scid mice. Int Immunol. 1993;5:1461-7.
56. Huilbregtse IL, van Lent AU, van Deventer SJH. Immunopathogenesis of IBD: Insufficient suppressor function in the gut? Gut. 2007;56:584-92.
57. Mowat AM, Lamont AG, Strobel S, Mackenzie S. The role of antigen processing and suppressor T cells in immune responses to dietary proteins in mice. Adv Exp Med Biol. 1987;216A:709-20.
58. Mowat AM, Lamont AG, Parrott DM. Suppressor T cells, antigen-presenting cells and the role of I-J restriction in oral tolerance to ovalbumin. Immunology. 1988;64:141-5.
59. Mowat AM. Depletion of suppressor T cells by 2′-deoxyguanosine abrogates tolerance in mice fed ovalbumin and permits the induction of intestinal delayed-type hypersensitivity. Immunology. 1986;58:179-84.
60. Mowat AM. The role of antigen recognition and suppressor cells in mice with oral tolerance to ovalbumin. Immunology. 1985;56:253-60.
61. Allez M, Mayer L. Regulatory T cells: Peace keepers in the gut. Inflamm Bowel Dis. 2004;10:666-76.
62. Brimnes J, Allez M, Dotan I, et al. Defects in CD8+ regulatory T cells in the lamina propria of patients with inflammatory bowel disease. Immunology. 2005;174:5814-22.
63. Brandt EB, Strait RT, Hershko D, et al. Mast cells are required for experimental oral allergen-induced diarrhea. J Clin Invest. 2003;112:1666-77.
64. Li XM, Schofield BH, Huang CK, et al. A murine model of IgE-mediated cow’s milk hypersensitivity. J Allergy Clin Immunol. 1999;103:206-14.
65. Berin MC, Kiliaan AJ, Yang PC, et al. The influence of mast cells on pathways of transepithelial antigen transport in rat intestine. J Immunol. 1998;161:2561-6.
66. Hermiston ML, Gordon JI. Inflammatory bowel disease and adenomas in mice expressing a dominant negative N-cadherin. Science. 1995;270:1203-7.
67. Chen YJ, Inobe R, Marks P, et al. Peripheral deletion of antigen-reactive T cells in oral tolerance. Nature. 1995;376:177-80.
68. Brandt EB, Strait RT, Hershko D, et al. Mast cells are required for experimental oral allergen-induced diarrhea. J Clin Invest. 2003;112:1666-77.
69. Mestecky J, McGhee JR, Elson CO. Intestinal IgA system. Immunol Allergy Clin North Am. 1988;8:349-8.
70. Kaetzel CS, Robinson JK, Chintalacharuvu KR, et al. The polymeric immunoglobulin receptor (secretory component) mediates transport of immune complexes across epithelial cells: A local defense function for IgA. Proc Natl Acad Sci U S A. 1991;88:8796-800.
71. Goldblum RM, Ahlstedt S, Carlson B, et al. Antibody-forming cells in human colostrum after oral immunization. Nature. 1975;257:797-8.
72. Challacombe SJ, Rahman D, O’Hagan DT. Salivary, gut, vaginal and nasal antibody responses after oral immunization with biodegradable microparticles. Vaccine. 1997;15:169-75.
73. Raux M, Finkielsztejn L, Salmon-Ceron D, et al. IgG subclass distribution in serum and various mucosal fluids of HIV type 1-infected subjects. AIDS Res Hum Retroviruses. 2000;16:583-94.
74. Schneider T, Zippel T, Schmidt W, et al. Increased immunoglobulin G production by short-term cultured duodenal biopsy samples from HIV infected patients. Gut. 1998;42:357-61.
75. Israel EJ, Taylor S, Wu Z, et al. Expression of the neonatal Fc receptor, FcRn, on human intestinal epithelial cells. Immunology. 1997;92:69-74.
76. Dickinson BL, Badizadegan K, Wu Z, et al. Bidirectional FcRn-dependent IgG transport in a polarized human intestinal epithelial cell line. J Clin Invest. 1999;104:903-11.
77. MacDermott RP, Nash GS, Bertovich MJ, et al. Alterations of IgM, IgG, and IgA synthesis and secretion by peripheral blood and intestinal mononuclear cells from patients with ulcerative colitis and Crohn’s disease. Gastroenterology. 1981;81:844-52.
78. Berin MC, Kiliaan AJ, Yang PC, et al. Rapid transepithelial antigen transport in rat jejunum: Impact of sensitization and the hypersensitivity reaction. Gastroenterology. 1997;113:856-64.
79. Berin MC, Yang PC, Ciok L, et al. Role for IL-4 in macromolecular transport across human intestinal epithelium. Am J Physiol. 1999;276:C1046-52.
80. Podolsky DK. Oligosaccharide structures of isolated human colonic mucin species. J Biol Chem. 1985;260:15510-15.
81. Leitch GJ, Dickey AE, Udezulu IA, et al. Entamoeba histolytica trophozoites in the lumen and mucus blanket of rat colons studied in vivo. Infect Immunol. 1985;47:68-73.
82. Madara JL. Loosening tight junctions. Lessons from the intestine. J Clin Invest. 1989;83:1089-94.
83. Warshaw AL, Walker WA, Isselbacher KJ, et al. Protein uptake by the intestine: Evidence for absorption of intact macromolecules. Gastroenterology. 1974;66:987-92.
84. Bhalla DK, Owen RL. Migration of B and T lymphocytes to M cells in Peyer’s patch follicle epithelium: An autoradiographic and immunocytochemical study in mice. Cell Immunol. 1983;81:105-7.
85. Neutra MR. Current concepts in mucosal immunity. V. Role of M cells in transepithelial transport of antigens and pathogens to the mucosal immune system. Am J Physiol. 1998;274:G785-91.
86. Owen RL. Sequential uptake of horseradish peroxidase by lymphoid follicle epithelium of Peyer’s patches in the normal unobstructed mouse intestine: An ultrastructural study. Gastroenterology. 1977;72:440-51.
87. Allan CH., Mendrick DL, Trier JS. Rat intestinal M cells contain acidic endosomal-lysosomal compartments and express class II major histocompatibility complex determinants. Gastroenterology. 1993;104:698-708.
88. Sicinski P, Rowinski J, Warchol JB, et al. Poliovirus type 1 enters the human host through intestinal M cells. Gastroenterology. 1990;98:56-8.
89. Frossard CP, Hauser C, Eigenmann PA. Antigen-specific secretory IgA antibodies in the gut are decreased in a mouse model of food allergy. J Allergy Clin Immunol. 2004;114:377-82.
90. Kerneis S, Bogdanova A, Kraehenbuhl JP, Pringault E. Conversion by Peyer’s patch lymphocytes of human enterocytes into M cells that transport bacteria. Science. 1997;277:949-52.
91. Gonnella PA, Waldner HP, Weiner HL. B cell-deficient (mu MT) mice have alterations in the cytokine microenvironment of the gut-associated lymphoid tissue (GALT) and a defect in the low dose mechanism of oral tolerance. J Immunol. 2001;166:4456-64.
92. Fujihashi K, Dohi T, Rennert PD, et al. Peyer’s patches are required for oral tolerance to proteins. Proc Natl Acad Sci U S A. 2001;98:3310-5.
93. Spahn TW, Fontana A, Faria M, et al. Induction of oral tolerance to cellular immune responses in the absence of Peyer’s patches. Eur J Immunol. 2001;31:1278-87.
94. Spahn TW, Weiner HL, Rennert PD, et al. Mesenteric lymph nodes are critical for the induction of high-dose oral tolerance in the absence of Peyer’s patches. Eur J Immunol. 2002;32:1109-13.
95. Kunkel D, Kirchhoff D, Nishikawa S, et al. Visualization of peptide presentation following oral application of antigen in normal and Peyer’s patches-deficient mice. Eur J Immunol. 2003;33:1292-301.
96. Butcher EC. Leukocyte endothelial cell recognition: Three or more steps to specificity and diversity. Cell. 1991;67:1-4.
97. Chin YH, Sackstein R, Cai JP. Lymphocyte-homing receptors and preferential migration pathways. Proc Soc Exp Biol Med. 1991;196:374-80.
98. Picker LJ, Butcher EC. Physiological and molecular mechanisms of lymphocyte homing. Ann Rev Immunol. 1992;10:561-91.
99. Stern M, Walker WA. Food proteins and gut mucosal barrier I. Binding and uptake of cow’s milk proteins by adult rat jejunum in vitro. Am J Physiol. 1984;246:G556-62.
100. Stern M. Food proteins and maturation of small intestinal microvillus membranes (MVM). II. Binding of gliadin hydrolysate fractions and of the gliadin peptide B3142. J Pediatr Gastroenterol Nutr. 1988;7:122-7.
101. Dinsdale D, Healy PJ. Enzymes involved in protein transmission by the intestine of the newborn lamb. Histochem J. 1982;14:811-21.
102. Maaser C, Eckmann L, Paesold G, et al. Ubiquitous production of macrophage migration inhibitory factor by human gastric and intestinal epithelium. Gastroenterology. 2002;122:667-80.
103. Voltan S, Martines D, Elli M, et al. Lactobacillus crispatus M247-derived H2O2 acts as a signal transducing molecule activating peroxisome proliferator activated receptor-gamma in the intestinal mucosa. Gastroenterology. 2008;135:1216-27.
104. Hugot JP, Chamaillard M, Zouali H, et al. Association of NOD2 leucine-rich repeat variants with susceptibility to Crohn’s disease. Nature. 2001;411:599-603.
105. Ogura Y, Bonen DK, Inohara N, et al. A frameshift mutation in NOD2 associated with susceptibility to Crohn’s disease. Nature. 2001;411:603-6.
106. Ogura Y, Lala S, Xin W, et al. Expression of NOD2 in Paneth cells: A possible link to Crohn’s ileitis. Gut. 2003;52:1591-7.
107. Inohara N, Ogura Y, Fontalba A, et al. Host recognition of bacterial muramyl dipeptide mediated through NOD2. Implications for Crohn’s disease. J Biol Chem. 2003;278:5509-12.
108. Van Heel DA, Ghosh S, Hunt KA, et al. Synergy between TLR9 and NOD2 innate immune responses is lost in genetic Crohn’s disease. Gut. 2005;54:1553-7.
109. Cario E, Podolsky DK. Differential expression in intestinal epithelial cell expression of Toll-like recptor 3 (TLR3) and TLR4 in inflammatory bowel disease. Infect Immun. 2000;68:7010-7.
110. Shang L, Fukata M, Thirunarayanan N, et al. Toll-like receptor signaling in small intestinal epithelium promotes B-cell recruitment and IgA production in lamina propria. Gastroenterology. 2008;135:529-38.
111. Lee J, Mo JH, Katakura K, et al. Maintenance of colonic homeostasis by distinctive apical TLR9 signaling in intestinal epithelial cells. Nat Cell Biol. 2006;8:1327-36.
112. Cario E. Bacterial interactions with cells of the intestinal mucosa: Toll-like receptors and NOD2. Gut. 2005;54:1182-93.
113. Wiman K, Curman B, Forsum U, et al. Occurrence of Ia antigens on tissues on non-lymphoid origin. Nature. 1978;276:711-3.
114. Kirby WN, Parr EL. The occurrence and distribution of H-2 antigens on mouse intestinal epithelial cells. J Histochem Cytochem. 1979;27:746-50.
115. Bland PW, Warren LJ. Antigen presentation by epithelial cells of the rat small intestine. II. Selective induction of suppressor T cells. Immunology. 1986;58:9-14.
116. Mayer L, Shlien R. Evidence for function of Ia molecules on gut epithelial cells in man. J Exp Med. 1987;166:1471-83.
117. Kaiserlian D, Vidal K, Revillard JP. Murine enterocytes can present soluble antigen to specific class II-restricted CD4+ T cells. Eur J Immunol. 1989;19:1513-6.
118. Selby WS, Janossy G, Mason DY, Jewell DP. Expression of HLA-DR antigens by colonic epithelium in inflammatory bowel disease. Clin Exp Immunol. 1983;53:614-8.
119. Salomon P, Pizzimenti A, Panja A, et al. The expression and regulation of class II antigens in normal and inflammatory bowel disease peripheral blood monocytes and intestinal epithelium. Autoimmunity. 1991;9:141-9.
120. Mayer L, Eisenhardt D, Salomon P, et al. Expression of class II molecules on intestinal epithelial cells in humans. Differences between normal and inflammatory bowel disease. Gastroenterology. 1991;100:3-12.
121. Dotan I, Allez M, Nakazawa A, et al. Intestinal epithelial cells from inflammatory bowel disease patients preferentially stimulate CD4+ T cells to proliferate and secrete interferon-gamma. Am J Physiol Gastrointest Liver Physiol. 2007;292:G1630-40.
122. Crotty B, Hoang P, Dalton HR, Jewell DP. Salicylates used in inflammatory bowel disease and colchicine impair interferon-gamma induced HLA-DR expression. Gut. 1992;33:59-64.
123. Linsley PS, Brady W, Grosmaire L, et al. Binding of the B cell activation antigen B7 to CD28 costimulates T cell proliferation and interleukin 2 mRNA accumulation. J Exp Med. 1991;173:721-30.
124. Azuma M, Ito D, Yagita H, et al. B70 antigen is a second ligand for CTLA-4 and CD28. Nature. 1993;366:76-9.
125. Schwartz RH. A cell culture model for T lymphocyte clonal anergy. Science. 1990;248:1349-56.
126. Harding FA, McArthur JG, Gross JA, et al. CD28-mediated signalling co-stimulates murine T cells and prevents induction of anergy in T-cell clones. Nature. 1992;356:607-9.
127. Sanderson IR, Ouellette AJ, Carter EA, et al. Differential regulation of B7 mRNA in enterocytes and lymphoid cells. Immunology. 1993;79:434-8.
128. Nakazawa A, Dotan I, Brimnes J, et al. The expression and function of costimulatory molecules B7H and B7-H1 on colonic epithelial cells. Gastroenterology. 2004;126:1347-57.
129. Bland PW. Antigen presentation by gut epithelial cells: Secretion by rat enterocytes of a factor with IL-1-like activity. Adv Exp Med Biol. 1987:219-25.
130. Bland PW, Kambarage DM. Antigen handling by the epithelium and lamina propria macrophages. Gastroenterol Clin North Am. 1991;20:577-96.
131. Hershberg RM, Framson PE, Cho DH, et al. Intestinal epithelial cells use two distinct pathways for HLA class II antigen processing. J Clin Invest. 1997;100:204-15.
132. Hershberg RM, Cho DH, Youakim A, et al. Highly polarized HLA class II antigen processing and presentation by human intestinal epithelial cells. J Clin Invest. 1998;102:792-803.
133. Hershberg RM, Mayer LF. Antigen processing and presentation by intestinal epithelial cells—polarity and complexity. Immunol Today. 2000;1:123-8.
134. Mayer L. The role of the epithelium in mucosal immunity. Res Immunol. 1997;98:498-504.
135. Kaiserlian D, Vidal K, Revillard JP. Murine enterocytes can present soluble antigen to specific class II- restricted CD4+ T cells. Eur J Immunol. 1989;19:1513-16.
136. Yio XY, Mayer L. Characterization of a 180-kDa intestinal epithelial cell membrane glycoprotein, gp180: A candidate molecule mediating T cell:epithelial cell interactions. J Biol Chem. 1997;272:12786-92.
137. Panja A, Blumberg RS, Balk SP, et al. CD1d is involved in T cell:epithelial cell interactions. J Exp Med. 1993;178:1115-20.
138. Hershberg R, Eghtesady P, Sydora B, et al. Expression of the thymus leukemia antigen in mouse intestinal epithelium. Proc Natl Acad Sci U S A. 1990;87:9727-31.
139. Bleicher PA, Balk SP, Hagen SJ, et al. Expression of murine CD1 on gastrointestinal epithelium. Science. 1990;250:679-82.
140. Blumberg RS, Terhorst C, Bleicher P, et al. Expression of a nonpolymorphic MHC class I-like molecule, CD1d, by human intestinal epithelial cells. J Immunol. 1991;147:2518-24.
141. Perera L, Shao L, Patel A, et al. Expression of nonclassical class I molecules by intestinal epithelial cells. Inflamm Bowel Dis. 2007;13:298-307.
142. Amiot M, Bernard A, Raynal B, et al. Heterogeneity of the first cluster of differentiation: Characterization and epitopic mapping of three CD1 molecules on normal human thymus cells. J Immunol. 1986;136:1752-8.
143. Shawar SM, Cook RG, Rodgers JR, et al. Specialized functions of MHC class I molecules. I. An N-formyl peptide receptor is required for construction of the class I antigen Mta. J Exp Med. 1990;171:897-912.
144. Vitetta ES, Poulik MD, Klein J, Uhr JW. Beta-2 -microglobulin is selectively associated with H-2 and TL alloantigens on murine lymphoid cells. J Exp Med. 1976;144:179-92.
145. Mayer L, Eisenhardt D. Lack of induction of suppressor T cells by intestinal epithelial cells from patients with inflammatory bowel disease. J Clin Invest. 1990;86:1255-60.
146. Kraus TA, Toy L, Chan L, et al. Failure to induce oral tolerance to a soluble protein in patients with inflammatory bowel disease. Gastroenterology. 2004;26:1771-8.
147. Viney JL, Kilshaw PJ, MacDonald TT. Cytotoxic alpha/beta+ and gamma/delta+ T cells in murine intestinal epithelium. Eur J Immunol. 1990;20:1623-6.
148. Guy-Grand D, Cerf-Bensussan N, Malissen B, et al. Two gut intraepithelial CD8+ lymphocyte populations with different T cell receptors: A role for the gut epithelium in T cell differentiation. J Exp Med. 1991;173:471-81.
149. Croitoru K, Ernst PB. Leukocytes in the intestinal epithelium: An unusual immunological compartment revisited. Reg Immunol. 1992;4:63-9.
150. Guy-Grand D, Vassalli P. Gut intraepithelial T lymphocytes. Curr Opin Immunol. 1993;5:247-52.
151. Cerf-Bensussan N, Guy-Grand D, Griscelli C. Intraepithelial lymphocytes of human gut: Isolation, characterisation and study of natural killer activity. Gut. 1985;26:81-8.
152. Ebert EC. Proliferative responses of human intraepithelial lymphocytes to various T-cell stimuli. Gastroenterology. 1989;97:1372-81.
153. Ebert EC, Roberts AI, Brolin RE, Raska K. Examination of the low proliferative capacity of human jejunal intraepithelial lymphocytes. Clin Exp Immunol. 1986;65:148-57.
154. Roberts AI, O’Connell SM, Biancone L, et al. Spontaneous cytotoxicity of intestinal intraepithelial lymphocytes: Clues to the mechanism. Clin Exp Immunol. 1993;94:527-32.
155. Guy-Grand D, Malassis-Seris M, Briottet C, Vassali P. Cytotoxic differentiation of mouse gut thymus dependent and independent intraepithelial T lymphocytes is induced locally. Correlation between functional assays, presence of perforin and granzyme transcripts, and cytoplasmic granules. J Exp Med. 1991;173:1549-52.
156. Hayday A, Theodoridis E, Ramsburg E, Shires J. Intraepithelial lymphocytes: Exploring the Third Way in immunology. Nat Immunol. 2001;2:997-1003.
157. Higgins JM, Mandlebrot DA, Shaw SK, et al. Direct and regulated interaction of integrin alphaEbeta7 with E-cadherin. J Cell Biol. 1998;140:197-210.
158. Austrup F, Rebstock S, Kilshaw PJ, Hamann A. Transforming growth factor-beta 1-induced expression of the mucosa-related integrin alpha E on lymphocytes is not associated with mucosa-specific homing. Eur J Immunol. 1995;25:1487-91.
159. Targan SR, Deem RL, Liu M, et al. Definition of a lamina propria T cell responsive state. Enhanced cytokine responsiveness of T cells stimulated through the CD2 pathway. J Immunol. 1995;154:664-75.
160. Abreu-Martin MT, Targan SR. Regulation of immune responses of the intestinal mucosa. Crit Rev Immunol. 1996;16:277-309.
161. Pirzer UC, Schurmann G, Post S, et al. Differential responsiveness to CD3-Ti vs. CD2-dependent activation of human intestinal T lymphocytes. Eur J Immunol. 1990;20:2339-42.
162. Mosley RL, Wang J, Hamad M, Klein JR. Functional heterogeneity of murine intestinal intraepithelial lymphocytes: Studies using TCR-alpha beta+ IEL lines and fresh IEL isolates reveal multiple cytotoxic subsets differentiated by CD5, CD8 alpha/alpha, and CD8 alpha/beta expression. Dev Comp Immunol. 1994;18:155-64.
163. James SP, Zeitz M, Kagnoff M, et al. Intestinal lymphocyte populations and mechanisms of cell-mediated immunity. Immunol Allergy Clin North Am. 1988;8:369-91.
164. Zeitz M, Greene WC, Peffer NJ, James SP. Lymphocytes isolated from the intestinal lamina propria of normal nonhuman primates have increased expression of genes associated with T-cell activation. Gastroenterology. 1988;94:647-55.
165. Qiao L, Braunstein J, Golling M, et al. Differential regulation of human T cell responsiveness by mucosal versus blood monocytes. Eur J Immunol. 1996;26:922-7.
166. Boirivant M, Fuss I, Fiocchi C, et al. Hypoproliferative human lamina propria T cells retain the capacity to secrete lymphokines when stimulated via CD2/CD28 pathways. Proc Assoc Am Physicians. 1996;108:55-67.
167. Boirivant M, Pica R, DeMaria R, et al. Stimulated human lamina propria T cells manifest enhanced Fas-mediated apoptosis. J Clin Invest. 1996;98:2616-22.
168. De Maria R, Boirivant M, Cifone MG, et al. Functional expression of Fas and Fas ligand on human gut lamina propria T lymphocytes. A potential role for the acidic sphingomyelinase pathway in normal immunoregulation. J Clin Invest. 1996;97:316-22.
169. Neurath MF, Finotto S, Glimcher LH. The role of Th1/Th2 polarization in mucosal immunity. Nat Med. 2002;8:567-73.
170. Neurath MF, Weigmann B, Finotto S, et al. The transcription factor T-bet regulates mucosal T cell activation in experimental colitis and Crohn’s disease. J Exp Med. 2002;195:1129-43.
171. Nakayama T, Yamashita M. Initiation and maintenance of Th2 cell identity. Curr Opin Immunol. 2008;20:265-71.
172. McGhee JR, Kyono H, Kubota M, et al. Mucosal Th1- versus Th2-type responses for antibody- or cell-mediated immunity to simian immunodeficiency virus in rhesus macaques. J Infect Dis. 1999;179(Suppl 3):S480-4.
173. Steinman RM, Hawiger D, Nussenzweig MC. Tolerogenic dendritic cells. Annu Rev Immunol. 2003;21:685-711.
174. Niess JH, Brand S, Gu X, et al. CX3CR1-mediated dendritic cell access to the intestinal lumen and bacterial clearance. Science. 2005;307:254-8.
175. Kunkel EJ, Campbell DJ, Butcher EC. Chemokines in lymphocyte trafficking and intestinal immunity. Microcirculation. 2003;10:313-23.
176. Zhao X, Sato A, Dela Cruz CS, et al. CCL9 is secreted by the follicle-associated epithelium and recruits dome region Peyer’s patch CD11b+ dendritic cells. J Immunol. 2003;171:2797-2803.
177. Caux C, Vanbervliet B, Massacrier C, et al. Regulation of dendritic cell recruitment by chemokines. Transplantation. 2002;73:S7-11.
178. Delamarre L, Pack M, Chang H, et al. Differential lysosomal proteolysis in antigen-presenting cells determines antigen fate. Science. 2005;307:1630-4.
179. Chirdo FG, Millington OR, Beacock-Sharp H, et al. Immunomodulatory dendritic cells in intestinal lamina propria. Eur J Immunol. 2005;35:1831-40.
180. Mowat AM, Donachie AM, Parker LA, et al. The role of dendritic cells in regulating mucosal immunity and tolerance. Novartis Found Symp. 2003;252:291-302.
181. Gorczynski RM, Lee L, Boudakov I. Augmented induction of CD4+CD25+ Treg using monoclonal antibodies to CD200R. Transplantation. 2005;79:488-91.
182. Niess JH, Reinecker HC. Lamina propria dendritic cells in the physiology and pathology of the gastrointestinal tract. Curr Opin Gastroenterol. 2005;21:687-91.
183. Yamagiwa SJ, Gray D, Hashimoto S, Horwitz DA. A role for TGF-beta in the generation and expansion of CD4+CD25+ regulatory T cells from human peripheral blood. J Immunol. 2001;166:7282-9.
184. Yang SK, Eckmann L, Panja A, et al. Differential and regulated expression of C-X-C, C-C, and C-chemokines by human colon epithelial cells. Gastroenterology. 1997;113:1214-23.
185. Lillard JWJr, Boyaka PN, Taub DD, McGhee JR. RANTES potentiates antigen-specific mucosal immune responses. J Immunol. 2001;66:162-9.
186. Yang SK, Choi MS, Kim OH, et al. The increased expression of an array of C-X-C and C-C chemokines in the colonic mucosa of patients with ulcerative colitis: Regulation by corticosteroids. Am J Gastroenterol. 2002;97:126-32.
187. Berrebi D, Banerjee A, Paris R, et al. In situ RANTES and interferon gamma gene expression in pediatric small bowel Crohn’s disease. J. Pediatr Gastroenterol Nutr. 1997;25:371-6.
188. Oki M, Ohtani H, Kinouchi Y, et al. Accumulation of CCR5+T cells around RANTES+ granulomas in Crohn’s disease: A pivotal site of Th1-shifted immune response? Lab Invest. 2005;85:137-45.
189. Scheerens H, Hessel E, de Waal-Malefyt R, et al. Characterization of chemokines and chemokine receptors in two murine models of inflammator bowel disease. IL-10-/- mice and Rag-2-/- mice reconstituted with CD4+Cd45Rbhigh T cells. Eur J Immunol. 2001;31:1465-74.
190. Farber JM. HuMig: A new human member of the chemokine family of cytokines. Biochem Biophys Res Commun. 1993;192:223-30.
191. Cole KE, Strick CA, Paradis TJ, et al. Interferon-inducible T cell alpha chemoattractant (I-TAC): A novel non-ELR CXC chemokine with potent activity on activated T cells through selective high affinity binding to CXCR3. J Exp Med. 1998;187:2009-21.
192. Bonecchi R, Bianchi G, Bordignon PP, et al. Differential expression of chemokine receptors and chemotactic responsiveness of type 1 T helper cells (Th1s) and Th2s 1998. J Exp Med. 1998;187:129-34.
193. Taub DD, Sayers TJ, Carter CR, Ortaldo JR. Alpha and beta chemokines induce NK cell migration and enhance NK-mediated cytolysis. J Immunol. 1995;155:3877-88.
194. Cella M, Sallusto F, Lanzavecchia A. Origin, maturation and antigen presenting function of dendritic cells. Curr Opin Immunol. 1997;9:10-16.
195. Singh UP, Venkataraman C, Singh R, et al. CXCR3 axis: role in inflammatory bowel disease and its therapeutic implication. Endocr Metab Immune Dis Drug Targets. 2007;7:111-23.
196. Zabel BA, Agace WW, Campbell JJ, et al. Human G protein-coupled receptor GPR-9-6/CC chemokine receptor 9 is selectively expressed on intestinal homing T lymphocytes, mucosal lymphocytes, and thymocytes and is required for thymus-expressed chemokine-mediated chemotaxis. J Exp Med. 1999;190:1241-56.
197. Papadakis KA, Prehn J, Moreno ST, et al. CCR9-positive lymphocytes and thymus-expressed chemokine distinguish small bowel from colonic Crohn’s disease. Gastroenterology. 2001;121:246-54.
198. Papadakis KA, Prehn J, Nelson V, et al. The role of thymus-expressed chemokine and its receptor CCR9 on lymphocytes in the regional specialization of the mucosal immune system. J Immunol. 2000;165:5069-76.
199. Kunkel EJ, Campbell JJ, Haraldsen G, et al. Lymphocyte CC chemokine receptor 9 and epithelial thymus-expressed chemokine (TECK) expression distinguish the small intestinal immune compartment: Epithelial expression of tissue-specific chemokines as an organizing principle in regional immunity. J Exp Med. 2000;192:761-8.
200. Svensson M, Marsal J, Ericsson A, et al. CCL25 mediates the localization of recently activated CD8alphabeta(+) lymphocytes to the small-intestinal mucosa. J Clin Invest. 2002;110:1113-21.
201. Keshav S, Ungashe S, Zheng W, et al. Ccx282-B, an orally active inhibitor of chemokine receptor Ccr9, shows anti-inflammatory and clinical activity in the treatment of Crohn’s disease [abstract]. Gastroenterology. 2007;132:A157.
202. Imai T, Hieshima K, Haskell C, et al. Identification and molecular characterization of fractalkine receptor CX3CR1, which mediates both leukocyte migration and adhesion. Cell. 1997;91:521-30.
203. Muehlhoefer A, Saubermann LJ, Gu X, et al. Fractalkine is an epithelial and endothelial cell-derived chemoattractant for intraepithelial lymphocytes in the small intestinal mucosa. J Immunol. 2000;164:3368-76.
204. Pan J, Kunkel EJ, Gosslar U, et al. A novel chemokine ligand for CCR10 and CCR3 expressed by epithelial cells in mucosal tissues. J Immunol. 2000;165:2943-9.
205. Kim JM, Cho SJ, Oh YK, et al. Nuclear factor-kappa B activation pathway in intestinal epithelial cells is a major regulator of chemokine gene expression and neutrophil migration induced by Bacteroides fragilis enterotoxin. Clin Exp Immunol. 2002;130:59-66.
206. Campbell JJ, Hedrick J, Zlotnik A, et al. Chemokines and the arrest of lymphocytes rolling under flow conditions. Science. 1998;279:381-4.
207. Dieu MC, Vanbervliet B, Vicari A, et al. Selective recruitment of immature and mature dendritic cells by distinct chemokines expressed in different anatomic sites. J Exp Med. 1998;188:373-86.
208. Liao F, Rabin RL, Smith CS, et al. CC-chemokine receptor 6 is expressed on diverse memory subsets of T cells and determines responsiveness to macrophage inflammatory protein 3 alpha. J Immunol. 1999;162:186-94.
209. Kwon JH, Keates S, Bassani L, et al. Colonic epithelial cells are a major site of macrophage inflammatory protein 3alpha (MIP-3alpha) production in normal colon and inflammatory bowel disease. Gut. 2002;51:818-26.