Chapter 66
Lymphedema
Evaluation and Decision Making
Stanley G. Rockson
Lymphedema is the clinical description of various disease states characterized by the progressive accumulation of protein-enriched interstitial fluid. These edematous states arise as a consequence of relative impairment of lymphatic drainage. Insufficient lymphatic function can result from either primary or acquired (secondary) anomalies of lymphatic outflow. Cryptogenic forms of lymphedema are often presumed to represent primary lymphatic dysfunction. Although impaired lymphatic function often is manifested as visceral involvement, particularly in the respiratory or gastrointestinal organs, upper or lower extremity edema, with or without visceral involvement, is the most common presentation of lymphatic impairment and is most predictive of the natural history of the condition.
Pathophysiology
Insufficient lymphatic outflow leads to the pathologic end result of lymphedema. In high-input failure, such as that which occurs in venous edema, increased capillary pressure leads to the accentuated production of interstitial fluid; if the production of lymph exceeds the maximal transport capacity of the lymphatic conduits, lymphedema will ensue, even if these structures are anatomically and functionally normal. By contrast, low-output failure ensues when some pathologic condition compromises lymphatic flow. Lymph stasis can accompany lymphatic hypoplasia or aplasia, functional insufficiency or anatomic absence of lymphatic valves, or, conceivably, blunted lymphatic contractility.1 Because the lymphatic circulation provides the normal conduit for the return of interstitial fluid and protein to the central circulation, abnormal lymph stasis creates an accumulation of protein and cellular metabolites in the extracellular space; with the ensuing increase in tissue colloid osmotic pressure, there is water accumulation and elevation of the interstitial hydraulic pressure (see Chapter 13).
Insufficient lymphatic transport leads to the accumulation of hyaluronan and other glycoproteins within the extracellular space. This is followed by a secondary increase in the fibroblast, keratinocyte, and adipocyte content of the affected tissues along with the accumulation of mononuclear cells, including macrophages. Ultimately, an increase in collagen deposition occurs, typically accompanied by an overgrowth of connective tissue and adipose elements in the skin and subcutaneous tissue.2 Although the contributory mechanisms are still not well understood, there is a tendency for these processes to lead to progressive subcutaneous fibrosis.
Classification and Staging
Standard clinical classifications distinguish lymphedema on the basis of cause (primary versus secondary). Primary lymphedema is further classified on the basis of genetics (familial versus sporadic) and time of onset (congenital, praecox, tarda) (Box 66-1).3–5 Although these systems are useful for categorizing lymphedema, they do not address the clinical severity of the disease and are usually not relevant to therapy. More recent classifications focus on the clinical stage of lymphedema or emphasize the underlying anatomic abnormality of the lymphatic system in an attempt to identify the best therapy.1,6,7
Primary Lymphedema
Prevalence of the heritable causes of primary lymphedema is difficult to ascertain, and estimates vary substantially. Primary lymphedema is thought to occur in approximately 1 of every 6000 to 10,000 live births. On the basis of data collected by the Rochester group study, it affects 1.15 per 100,000 persons younger than 20 years.8 Females are affected 2- to 10-fold more commonly than males, and the incidence peaks between the ages of 12 and 16 years.6,9
Of 125 patients with primary lymphedema treated at the Mayo Clinic, 97 (78%) were female and 28 (22%) were male, yielding a female-to-male ratio of 3.5 : 1.10 The ratio of unilateral to bilateral lymphedema was 3 : 1. Congenital lymphedema occurred more frequently in males than in females. In these patients, the edema was usually bilateral and involved the entire lower extremity. In contrast, the typical patient with lymphedema praecox was female and had unilateral involvement, with swelling usually extending up to the knee only.
Primary lymphedema represents a heterogeneous group of disorders; therefore, its classification schemes are numerous. Affected individuals can be classified by age at onset, morphology, or clinical setting.
Classification by Age at Onset and Inheritance
Lymphedema is termed congenital when it is apparent at birth or is recognized within the first year of life. Lymphedema praecox most commonly appears at the onset of puberty, but it may be delayed until the third decade of life. Lymphedema tarda typically begins after the age of 35 years (see Box 66-1).
Congenital.
Congenital lymphedema commonly occurs in a sporadic fashion; however, when clusters of cases occur in families, an autosomal dominant pattern of transmission is frequently observed.11 In addition to the genetic causes of isolated lymphedema, there is a strong association between intrauterine and congenital lymphatic dysfunction and heritable chromosomal abnormalities, including Turner’s syndrome, Klinefelter’s syndrome, and trisomy 21, among many others. In congenital lymphedema, the swelling can involve only a single lower extremity, but edema of multiple limbs, the genitalia, and even the face can be seen. Bilateral leg swelling and involvement of the entire lower extremity are more likely in congenital cases than in other forms of primary lymphedema.10
Lymphedema Praecox.
Lymphedema praecox is the most common form of primary lymphedema, accounting for up to 94% of cases in large series. The name Meige’s disease has historically been reserved for a specific familial form of lymphedema with a recessive pattern of inheritance and typical onset at puberty. Lymphedema praecox displays a marked gender imbalance, with an estimated 10 : 1 female-to-male prevalence.2 The edema is usually unilateral and is limited to the foot and calf in the majority of patients.10 Estrogenic hormones may play a role in the pathogenesis of this form of primary lymphedema.10
Lymphedema Tarda.
Lymphedema tarda is relatively uncommon. Appearing after the age of 35 years, it typically accounts for an estimated 10% of cases of primary lymphedema.
Classification by Morphology
It has been suggested that a morphologic classification of primary lymphedema might provide more useful prognostic information than classification by age at onset (Fig. 66-1).2 This alternative classification scheme relies on an anatomic description of the lymphatic vasculature.3,11
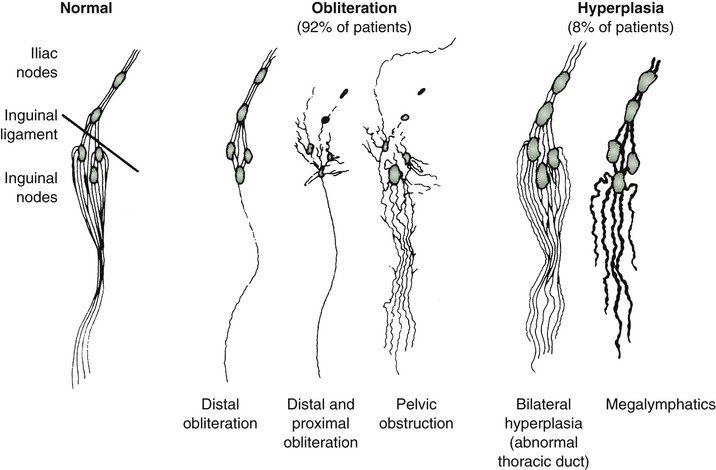
Figure 66-1 Lymphangiographic patterns of lymphatic morphology in a normal lower limb and in patients with different types of primary lymphedema. Obliteration of the lymphatic pathways may be due to aplasia, hypoplasia, or obstruction of the lymphatic channels and nodes.
Aplasia.
In aplasia, no collecting vessels can be identified.
Hypoplasia.
In hypoplasia, a diminished number of vessels are seen.
Numerical Hyperplasia.
In numerical hyperplasia (as defined by Kinmonth3), an increased number of vessels are seen.
Hyperplasia.
In addition to an increase in number, the vessels have valvular incompetence and display tortuosity and dilatation (megalymphatics, lymphangiectasia). Megalymphatics and lymphatic hyperplasia are less common than hypoplasia or aplasia. This pattern demonstrates a male predominance. These patients most often have unilateral edema involving the entire lower extremity. Cutaneous angiomas and chylous reflux can also be seen (Fig. 66-2). Megalymphatics are associated with a greater extent of involvement and a worse prognosis.
Classification by Anatomy
Aplasia and hypoplasia have a different natural history, depending on whether they involve the distal or proximal portion of the leg.
Distal Obstruction.
Approximately one third of all cases are secondary to agenesis, hypoplasia, or obstruction of the distal lymphatic vessels, with relatively normal proximal vessels (see Fig. 66-1).11 In these cases, the swelling is usually bilateral and mild, and females are affected much more frequently than males. The prognosis is good. In general, after the first year of symptoms, there is little extension in the same limb or to uninvolved extremities. Although the maximal extent of involvement is established early in the disease in about 40% of patients, the girth of the limb continues to increase. Distal hypoplasia or aplasia of the lymphatics most often correlates with the presence of bilateral peripheral edema of the lower extremities. Familial occurrence, female predominance, and indolent progression characterize this pattern of lymphatic disturbance.
Proximal Obstruction.
In more than half the cases, the defect primarily involves obstruction of the proximal lymphatics or nodes, with an initial lack of involvement of distal lymphatic vessels. Pathologic studies reveal intranodal fibrosis.12 In these cases, the swelling tends to be unilateral and severe, and there may be a slight female predominance.11 In patients with proximal involvement, the extent and degree of the abnormality are likely to progress, requiring surgical intervention. Initially uninvolved distal lymphatic vessels may become obliterated over time. A minority of patients have a pattern of bilateral hyperplasia of the lymphatic channels. In these less common forms of primary lymphedema, there is a slight male predominance. When isolated proximal obstructive hypoplasia is observed, clinical involvement of the entire limb is more likely, with relentless worsening of edema.
Classification by Clinical Setting
Alternatively, primary lymphedema can be classified by abnormal phenotype or associated clinical anomalies (see Box 66-1).13
Inheritance.
Although sporadic cases of primary lymphedema are more common,11 the tendency for congenital lymphedema to cluster in families is significant (Fig. 66-3). A familial predisposition for congenital lymphedema, which was ultimately determined to have an autosomal dominant form of inheritance with variable penetrance, was first described by Milroy in 1892.14 He reported “hereditary edema” affecting 22 individuals in 1 family over 6 generations. Although Milroy studied not only congenital lymphedema but also the praecox and tarda variants of the syndrome that bears his name, lymphedema praecox is better known as Meige’s disease.15
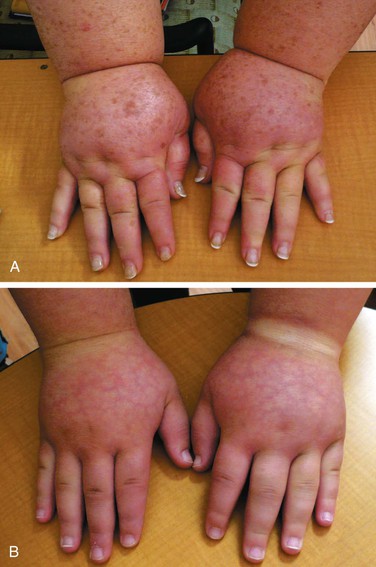
Figure 66-3 A, Adult patient with congenital lymphedema. In addition to the bilateral arm lymphedema depicted, she has edema of both legs and the face. B, Upper extremities of this patient’s 18-year-old son, who has a similar distribution of lymphedema. This is an example of Milroy’s disease.
In general, congenital lymphedema with autosomal or sex-linked recessive forms of inheritance is less common than that with dominant forms of inheritance.11,16,17 Nevertheless, the list of heritable lymphedema-associated syndromes is long and growing.18 Primary lymphedema has been described in association with various forms of chromosomal aneuploidy, such as Turner’s and Klinefelter’s syndromes; with various dysmorphogenic genetic anomalies, such as Noonan’s syndrome and neurofibromatosis; and with a variety of as yet unrelated disorders, such as yellow nail syndrome, intestinal lymphangiectasia, lymphangiomyomatosis, and arteriovenous malformation.19–24 The association of lymphedema with vascular anomalies suggests a common developmental origin of the lymphatic and blood vasculature.
Associated Disorders.
Numerous disorders are associated with heritable forms of lymphedema. Increasingly, these disorders have yielded to chromosomal mapping techniques. Lymphedema-cholestasis, or Aagenaes syndrome, has been mapped to chromosome 15q.25 In several family cohorts of Milroy’s disease, it has been determined that the disorder reflects missense inactivating mutations in the tyrosine kinase domain of vascular endothelial growth factor receptor 3 (VEGFR-3),26,27 thus underscoring the likelihood that this condition reflects an inherited defect in lymphatic vasculogenesis. Several additional lymphedema syndromes have recently lent themselves to successful genetic mapping.7 Lymphedema-distichiasis, an autosomal dominant dysmorphic syndrome in which lymphedema presents in association with a supplementary row of eyelashes arising from the meibomian glands, has been linked to truncating mutations in the forkhead-related transcription factor FOXC228; mutations in FOXC2 have subsequently been associated with a wide variety of primary lymphedema presentations.29 Similarly, a more unusual form of congenital lymphedema, hypotrichosis-lymphedema-telangiectasia, has been ascribed to both recessive and dominant inheritance of mutations in the transcription factor gene SOX18.30 It is plausible that further elucidation of the molecular pathogenesis of these diseases linked to FOXC2 and SOX18 mutations will lead to enhanced insights into the mechanisms of normal and abnormal lymphatic development.
Secondary Lymphedema
Acquired (secondary) lymphedema is the most commonly encountered form of lymphatic dysfunction (Fig. 66-4). In the United States, iatrogenic causes predominate among the acquired forms of lymphedema owing to the common occurrence of lymphatic trauma after surgery or radiotherapy for cancer.9
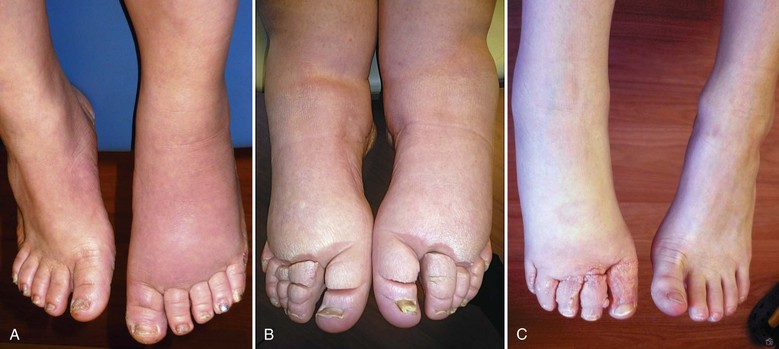
Figure 66-4 Chronic acquired lymphedema of the lower extremities. Note severe skin changes (A) and swelling of the foot (B) associated with squaring of the toes (Stemmer’s sign) and the typical peau d’orange. C, Severe lymphedema with subcutaneous lymph cysts and chronic verrucous superinfection.
Cancer
Of the various clinical settings that predispose patients to lymphedema, treatment of breast cancer is most commonly associated with acquired lymphatic insufficiency (of the upper extremity). Lymph node dissection and adjuvant radiation therapy independently and synergistically predispose to lymphatic vascular insufficiency.20 According to the most recent estimates, 20% to 30% of breast cancer survivors experience clinically significant arm lymphedema after axillary intervention. Despite the benefits of recent surgical and radiotherapeutic enhancements, the problem of lymphedema has not been eradicated.18
Similar lymphatic sequelae are encountered in the lower extremities and pelvis after interventions for gynecologic or urologic malignant neoplasms. Malignant melanoma can cause either upper or lower extremity lymphedema when radical dissection is required in the axilla or groin, respectively.
Filariasis
Filariasis, caused by infestation with parasites such as Wuchereria bancrofti, Brugia malayi, and Brugia timori, is by far the most frequent cause of secondary lymphedema in third-world countries. Of the estimated 90.2 million people in the world who are infected, more than 90% have bancroftian filariasis.31 The disease is most frequent in subtropical and tropical countries such as China, India, and Indonesia. It is transmitted by different types of mosquitoes, and transmission is closely related to poor urban sanitation.32
Perilymphatic inflammation, fibrosis, and sclerosis of the lymph nodes are caused by the indwelling adult worms. Lymph node fibrosis, reactive hyperplasia, and dilatation of the lymphatic collecting channels are caused by the worm products, by physical injury to the valves and vessel walls caused by the live worms, and by the immune response of the host.33 Eosinophilia is found in the peripheral blood smear, and microfilariae can be demonstrated in peripheral nocturnal blood, centrifuged urine sediment, or lymphatic fluid.34 Filarial lymphedema rapidly develops into grossly incapacitating elephantiasis that is extremely difficult to treat.
Other Causes
Lymphedema can also be acquired from other types of lymphatic vascular trauma, including burns and large or circumferential wounds to the extremity. Additional causes of acquired lymphedema include pregnancy, bacterial and fungal infections, infections after snake or insect bites, contact dermatitis, and rheumatoid arthritis.9 Autoimmune destruction of the lymphatics has been hypothesized but not directly demonstrated.
Clinical Staging
Because none of the classification schemes addresses the clinical stage of the disease, the Working Group of the 10th International Congress of Lymphology in 1985 suggested staging chronic lymphedema, regardless of cause. A latent, subclinical stage and three clinical grades were established,35 and each grade was subclassified as mild, moderate, or severe:
Grade III: Edema is irreversible and develops from repeated inflammatory attacks, fibrosis, and sclerosis of the skin and subcutaneous tissue. This is the stage of lymphostatic elephantiasis.
The advantage of this classification is that it permits the evaluation of treatment effectiveness and the comparison of different treatment modalities. One drawback is that appropriate staging may be difficult in some cases without a biopsy of the subcutaneous tissue.
Clinical Presentation
History
A careful history frequently reveals the cause of the swelling and suggests the diagnosis of lymphedema. A family history that is positive for leg swelling may indicate familial lymphedema. The development of painless leg swelling in a teenage girl without any identifiable cause strongly suggests primary (idiopathic) lymphedema. A history of diarrhea and weight loss is a clue to mesenteric lymphangiectasia, whereas intermittent drainage of milky fluid from skin vesicles indicates reflux of chyle. In patients with secondary lymphedema, the cause of limb swelling should be evident from the history, such as previous lymph node dissection, irradiation, tumor, trauma, or infection. In patients who have traveled in tropical countries, filariasis is suspected. Although the causes of primary and secondary lymphedema are different, the clinical presentation and characteristic physical findings are frequently similar.
Signs and Symptoms
The clinical signs and symptoms of lymphedema largely depend on the duration and severity of the disease.
Edema
Initially, the interstitial space is expanded by an excess accumulation of relatively protein rich fluid. The swelling produced by the fluid collection is typically soft, is easily displaced with pressure (pitting edema), and may substantially decrease with elevation of the limb. In the lower extremities, the edema typically extends to the distal aspects of the feet, resulting in the characteristic “square toes” (Stemmer’s sign) seen in this condition. The dorsum of the forefoot is usually involved, resulting in the typical appearance of a “buffalo hump.” During a period of years, the limb may take on a woody texture as the surrounding tissue becomes indurated and fibrotic.
Skin Changes
In the early stage of lymphedema, the skin usually has a pinkish red color and a mildly elevated temperature owing to the increased vascularity. In long-standing lymphedema, the skin becomes thick and shows areas of hyperkeratosis, lichenification, and development of peau d’orange. The term pigskin reflects the reactive changes of the dermis and epidermis in response to the chronic inflammation caused by lymphatic stasis.36 Recurrent chronic eczematous dermatitis or excoriation of the skin may occur, but frank ulcerations are rare. Unlike the situation in venous stasis, the skin maintains a higher degree of hydration and elasticity for a long time in lymphedema, and ischemic changes due to high skin tension and disruption of the circulation to the skin and subcutaneous tissue are rare.37
Additional skin changes in chronic lymph stasis, primarily in patients with hyperplasia of the lymphatics and valvular incompetence, include verrucae or small vesicles, which frequently drain clear lymph (lymphorrhea). In patients with lymphangiectasia and reflux of chyle, drainage from the vesicles is milky (chylorrhea; see Fig. 66-2).
Primary lymphedema may be associated with yellow discoloration of the nails.38–40 In the yellow nail syndrome, pleural effusion is also present. The pale yellow color of the nails is most likely caused by impaired lymphatic drainage. Severe clubbing, transverse ridging, friability, and decreased rate of nail growth are also observed.39,40
Pain
Although some aching or heaviness of the limb is a frequent complaint, significant pain is rare. If a patient with lymphedema complains of marked pain, infection or neuritic pain in the area of scar tissue or radiation treatment should be suspected. Other possible causes of leg swelling, such as venous edema or reflex sympathetic dystrophy, should also be considered (see the later discussion of differential diagnosis).
Complications
Infection
The propensity for recurrent soft tissue infection is one of the most troublesome aspects of long-standing lymphedema. Accumulated fluid and proteins provide a good substrate for bacterial growth. Lymphatic dysfunction impairs local immune responses, which plays a permissive role in the propagation of bacterial and fungal invasion. Furthermore, once it is established, soft tissue infection exacerbates the existing lymphatic dysfunction, sometimes irreversibly. With recurrent infection, there is progressive damage of lymphatic capillaries. In primary lymphedema, the reported infection rate is as high as 31%.10
The clinical presentation of soft tissue infection in lymphedema varies substantially—from the acute manifestation of rapidly progressive infection to only modest exacerbations of edema accompanied by subtle cutaneous erythema in the absence of fever. Recurrent attacks of cellulitis can damage the existing cutaneous lymphatics, exacerbate the skin disease, and further aggravate existing edema.
Malnutrition and Immunodeficiency
Lymphangiectasia with protein-losing enteropathy or chylous ascites or chylothorax may result in severe loss of proteins, long-chain triglycerides, cholesterol, and calcium.41,42 Loss of lymphocytes, immunoglobulins, polypeptides, and cytokines results in a state of immunodeficiency that decreases the patient’s ability to resist infections or malignant disease.
Malignant Tumors
In rare cases, chronic lymphedema of any cause may be complicated by the development of malignant tumors in the involved limb. Lymphangiosarcoma after long-standing secondary lymphedema, originally described by Stewart and Treves,43 is a rare malignant disease that frequently results in limb loss or even death. Lymphangiosarcoma is manifested as multicentric lesions with bluish nodules, sclerotic plaques, or bullous changes. Other malignant tumors that appear with increased frequency in lymphedematous limbs include Kaposi’s sarcoma, squamous cell carcinoma, malignant lymphoma, and melanoma.
Diagnosis
In most cases of advanced, sustained lymphedema, the characteristic clinical presentation, history, and physical findings establish the diagnosis with near certainty.44 In more subtle presentations, it may be difficult to distinguish primary lymphedema from other edematous conditions. Additional objective data may be required to confirm the presence of impaired lymphatic flow or the typical pattern of abnormal fluid distribution within tissues. The diagnosis is more difficult to establish in the early stages of disease, particularly when edema is mild or intermittent.
Physical Examination
The physical examination of a patient with lymphedema should include inspection for cutaneous and subcutaneous fibrosis and peau d’orange and attempts to elicit the pathognomonic Stemmer sign, in which the examiner is unable to tent the skin of the interdigital webs, based on the characteristic loss of cutaneous elasticity in lymphedema (see Fig. 66-4). In some cases, particularly early in the disease, the pitting edema of this condition may be indistinguishable from other local or systemic causes of edema.
Testing
Objective documentation of lymphatic dysfunction is sometimes useful. Available tests include isotopic lymphoscintigraphy, indirect and direct lymphography, lymphatic capillaroscopy, magnetic resonance imaging, axial tomography, and ultrasonography. Direct lymphography is rarely used today and should be restricted to those patients who are potential candidates for lymphatic surgery. Lymphatic capillaroscopy is available only in specialized centers.
Lymphoscintigraphy
Isotopic lymphoscintigraphy is a reliable and reproducible method to confirm the diagnosis of lymphedema. A radiolabeled macromolecular tracer is injected intradermally or subdermally within one of the interdigital spaces of the affected limb. The lymphatic transport of the radiolabeled macromolecule is tracked with a gamma camera. The biokinetic behavior of interstitially applied colloid particles depends on their surface charge and particle size. Particles with small diameters are absorbed into capillaries, whereas those in the 10-nm range, such as antimony trisulfide (Sb2S3), are absorbed into the lymphatic system. The time needed for activity to appear in the regional lymph nodes varies according to the physical characteristics of the imaging agent. For example, small particles such as technetium Tc 99m–labeled human serum albumin may appear in the pelvic nodes within 10 minutes,45 whereas relatively large agents, including rhenium and Sb2S3 colloid, should arrive within 30 minutes46 to 1 hour,47 respectively. In most centers, 99mTc-Sb2S3 or 99mTc-labeled human serum albumin is used for lymphoscintigraphy.45,47–49
Interpretation.
Lymphoscintigraphy provides a semiquantifiable assessment of lymphatic function as well as visualization of major lymphatic trunks and lymph nodes. The data can be recorded in a standardized report format, which is helpful for creating reproducible reports when many physicians review these tracings. A sample report form, shown in Figure 66-5, is an adaptation of one proposed by Kleinhans and colleagues for the estimation of a transport index.50
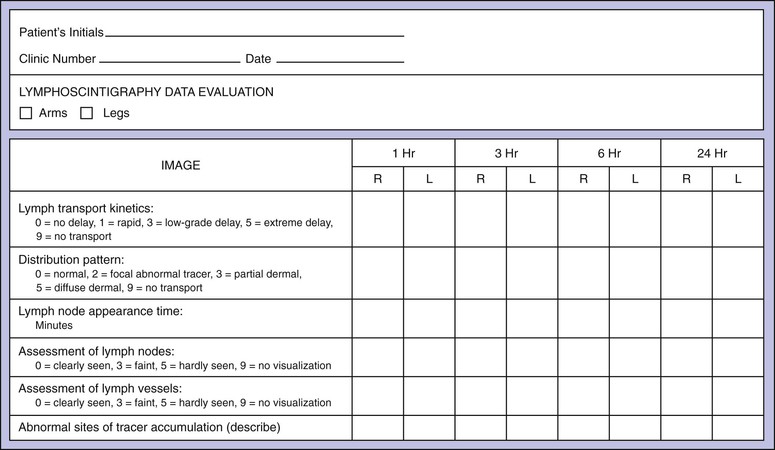
Figure 66-5 Evaluation form for calculation of the lymphatic transport index. (Modified from Kleinhans E, et al: Evaluation of transport kinetics in lymphoscintigraphy: follow-up study in patients with transplanted lymphatic vessels. Eur J Nucl Med 10:349, 1985. Courtesy Springer-Verlag.)
In normal limbs, lymphoscintigraphy shows several lymph vessels as the tracer is visualized along the anteromedial aspect of the leg. The injection site, because of the relatively large tracer dose given, does not show details, and no information about lymph distribution in the feet is obtainable. Several lymph channels may be identified in the calf. In the thigh, however, the lymph vessels run close to each other, and separate activity in each larger channel is seldom seen on lymphoscintigrams (Fig. 66-6).
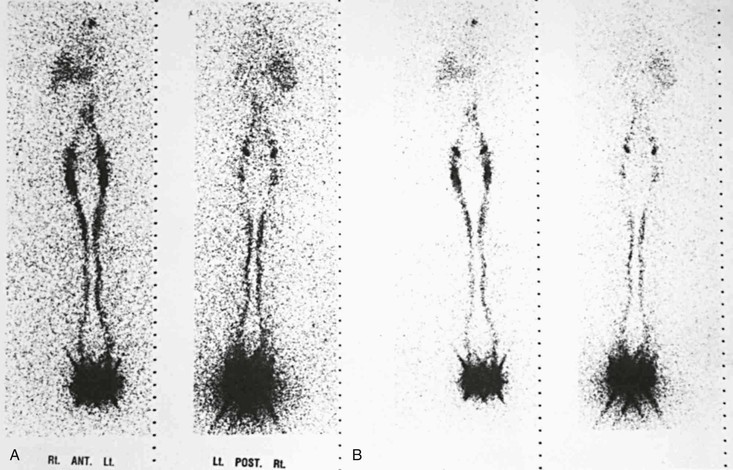
Figure 66-6 Anterior and posterior images in two intensity settings from a total-body scan with a dual-headed gamma camera. A, Normal lymphoscintigram. B, Higher intensity settings in the same patient. A large area of high activity and scatter is seen at the feet, where the injection was made. The single well-outlined band in each leg represents the main lymphatic channels. The lymph nodes in the groin and liver, the pelvic and para-aortic nodes, and an area at the site of the upper thoracic duct are visualized.
Tracer activity is clear in the inguinal lymph nodes by 60 minutes (range, 15 to 60 minutes). A faint hepatic uptake, activity in the bladder, and faint traces in the para-abdominal nodes are visible at 1 hour. Three-hour images show intense uptake in the liver; symmetrical and good uptake in the lymph nodes of the groin, pelvis, and abdomen; and occasionally a tracer focus in the left supraclavicular area at the site of the distal thoracic duct.
The qualitative interpretation of images has resulted in excellent sensitivity (92%) and specificity (100%) for the diagnosis of lymphedema.48 Quantitative lymphoscintigraphy, with measurement of lymphatic clearance, may improve the detection of early disease,45 but the results obtained in some studies have been equivocal.47,48 Neither the image pattern nor the quantitative parameters can reliably distinguish primary from secondary lymphedema.47–49
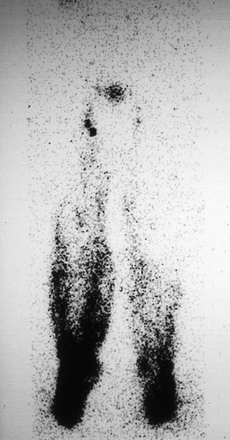
Lymphedema.
Typical abnormalities observed in lymphedema include dermal backflow (Fig. 66-7), absent or delayed transport of tracer, crossover filling with retrograde backflow, and either absent or delayed visualization of lymph nodes. In primary lymphedema, channels are obliterated or absent; in a smaller percentage of cases, they become ectatic and incompetent. The asymmetry or delayed appearance of radiocontrast material in the proximal nodal tissue can be used as a semiquantitative measure of the severity of lymphatic vascular insufficiency. The density of subcutaneous accumulation of radiotracer (dermal backflow) can also be quantitated, as can the ratio of radioactivity in ipsilateral versus contralateral nodal tissues in the setting of unilateral limb edema. Quantitation has the greatest utility in predicting the likelihood of a beneficial response to therapeutic intervention.
Lymphangiectasia.
Scintigraphic findings in lymphangiectasia consist of dilated lymph channels with only mild or no delay in lymph transport (Fig. 66-8). Colloid injected into the unaffected lower extremity may reflux into the affected lymphedematous leg because of lymphatic valvular incompetence. Similar reflux of the colloid may be seen in the dilated mesenteric lymphatics (Fig. 66-9) or in the retroperitoneum, perineum, or scrotum. Ruptured lymphatics cause extravasation of the colloid into the abdominal cavity or the chest in patients with chylous ascites or chylothorax. The images are generally not helpful in determining the exact site of the lymphatic leak.
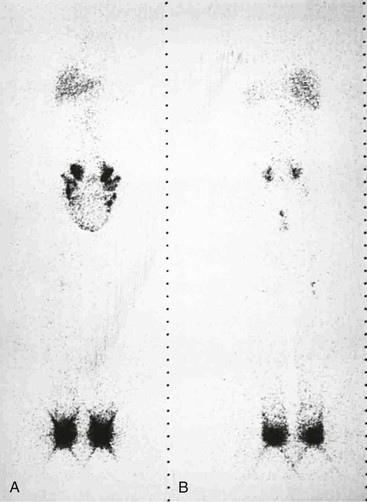
Figure 66-8 Bilateral leg scintigraphy with anterior (A) and posterior (B) views in a 24-year-old man outlines the swollen scrotum in the 6-hour image. Colloid reflux resulted from dilatation and valvular incompetence of the lymphatics.
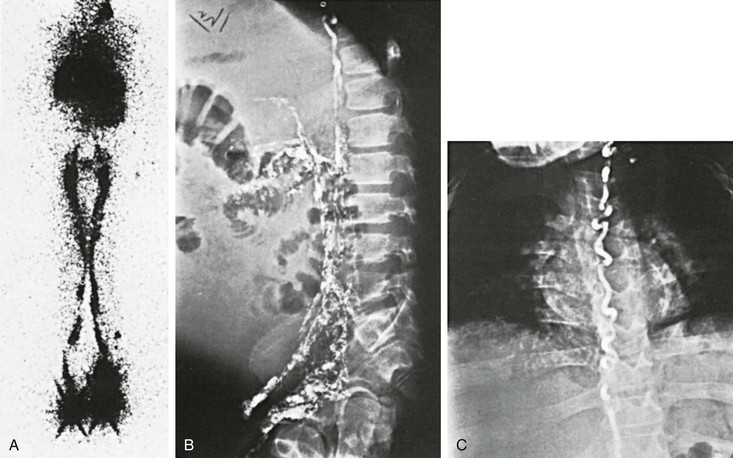
Figure 66-9 A-C, Lymphoscintigram of an 18-year-old man with lymphangiectasia, protein-losing enteropathy, and chylous ascites. Note the large leg lymphatics (A) and reflux of colloid into the mesenteric lymph vessels (B), filling almost the entire abdominal cavity. Note the large thoracic duct (C).
Computed Tomography and Magnetic Resonance Imaging
Lymphedema is typically confined to the epifascial space of the skin and subcutaneous tissue, sparing muscle. This characteristic absence of muscle involvement produces distinctive changes that can be observed with computed tomography (CT) or magnetic resonance imaging (MRI). These typical imaging features facilitate the differentiation of lymphedema from other edematous entities. In lymphedema, the images reveal a characteristic honeycomb distribution of edema within the epifascial structures along with thickening of the skin. In venous edema, both the epifascial and subfascial compartments are affected, whereas in lipedema, there is fat accumulation without fluid. MRI is also helpful in the identification of lymph nodes and enlarged lymphatic trunks and in the differentiation of various potential causes of lymphatic obstruction in secondary lymphedema. The anatomic information derived from MRI can complement the functional assessment provided by lymphoscintigraphy. The greatest value of CT and MRI in the evaluation of a patient with a swollen leg is to exclude any obstructing mass that may result in decreased transport capacity of the lymphatic system.
Direct Contrast Lymphangiography
Contrast lymphography is used primarily before reconstructive lymphatic surgery. Imaging is accomplished through the direct injection of iodine-based, lipid-soluble agents into the subcutaneous lymphatics, which are first identified by the subcutaneous injection of dye (methylene blue) and then cannulated. Contrast lymphography poses distinct technical difficulties and may, in fact, exacerbate lymphatic malfunction through the accumulation and pooling of the oil-based contrast medium. For these reasons, its use should be limited to preoperative evaluation in specialized centers.
Differential Diagnosis
The differential diagnosis (Box 66-2) frequently includes lipedema; a lipodystrophy that typically causes symmetrical enlargement of the lower extremities, particularly in obese females; and venous insufficiency, a hydrostatic cause of lower extremity edema. In lipedema, there may be a component of pitting edema; but in contradistinction to lymphedema, there is sparing of the feet despite pronounced enlargement of the calves and thighs. Venous stasis has relatively distinctive cutaneous attributes, including chronic deposits of hemosiderin in the skin. The patient’s history and the clinical setting often determine the degree to which chronic venous insufficiency plays a role in the differential diagnosis. However, even when the clinical setting and physical examination suggest the presence of venous stasis, the accompanying venous hypertension may chronically elevate the lymphatic load and thus predispose to the secondary development of lymphatic edema. In practice, therefore, such patients often have a mixed lymphatic-venous form of chronic edema.
Systemic Causes
During the evaluation of patients with chronic limb swelling, a systemic cause should be excluded first. Underlying cardiac diseases, such as congestive heart failure, chronic constrictive pericarditis, and severe tricuspid regurgitation, are the most frequent systemic causes leading to pitting or bilateral leg swelling. Hepatic or renal failure, hypoproteinemia, malnutrition, and endocrine disorders (myxedema) are other possible causes of leg swelling. Allergic reactions, hereditary angioedema, and idiopathic cyclic edema are rare systemic causes that should be considered. Chronic use of diuretics may lead to generalized swelling that most frequently affects the extremities and the face. Other drugs that may cause swelling include corticosteroids, some antihypertensive drugs, and anti-inflammatory agents.
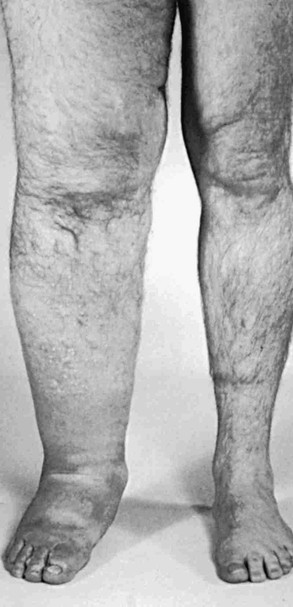
Venous Insufficiency
Among the local or regional causes of limb swelling, chronic venous insufficiency is much more common than lymphedema. In some patients with chronic iliac or iliocaval obstruction, massive swelling of the entire extremity can develop (Fig. 66-10). The usual causes of proximal venous occlusion are deep venous thrombosis or external compression of the vein by tumor or retroperitoneal fibrosis. Whereas lymphedema is usually painless, venous hypertension results in marked pain and cramps after prolonged standing or at the end of the day. Patients with proximal venous obstruction may complain of typical claudication, which is manifested as throbbing pain in the thigh or calf after walking. The pain resolves with rest, although elevation of the extremity provides the fastest relief. The presence of varicosity, pigmentation, induration, or venous ulcers makes the diagnosis of venous insufficiency easier. Chronic inflammation in the subcutaneous tissue due to venous stasis may result in destruction of the collecting lymph channels; a mixed venous-lymphatic edema develops in these patients.
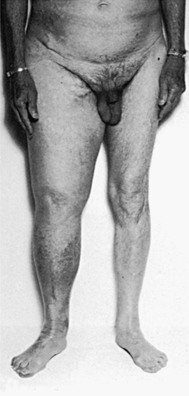
Vascular Malformation
Patients with congenital vascular malformations frequently have a larger than normal extremity that may be difficult to distinguish from lymphedema (Fig. 66-11). An increase in the length of the affected extremity, the presence of atypical lateral varicosity, and a port-wine stain with underlying developmental abnormality of the deep venous system are characteristic of Klippel-Trénaunay syndrome.51 Although hypertrophy of the soft tissues and bones is caused by an abnormality in mesenchymal development, congenital lymphedema may also be present in these patients. In patients with high-shunt, high-flow arteriovenous malformations, the extremity is larger than normal and frequently longer as well.52 A bruit and thrill are present, the superficial veins are dilated and frequently pulsatile, and the distal arterial pulses may be diminished.
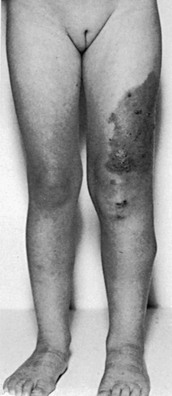
Lipedema
Lipedema is characterized by deposition of a large amount of fatty tissue in the subcutaneous layers. Most of these patients have morbid obesity; some, mostly females, have fat deposition localized to the lower half of the body. Evaluation of the lymphatic system with lymphoscintigraphy or lymphangiography shows essentially normal findings.
Other Causes
Trauma and subsequent reflex sympathetic dystrophy may result in painful swelling of the extremity. Because of disuse, a varying degree of osteoporosis can be observed, and increased sympathetic activity occurs in the limbs of these patients. The swelling is usually the result of high-output lymphatic failure, and increased lymphatic transport may be demonstrated on lymphoscintigraphy (Fig. 66-12). Baker’s cyst, soft tissue tumor, hematoma, and inflammation such as tenosynovitis or arthritis are additional local causes of limb swelling that should be considered in the differential diagnosis of lymphedema.
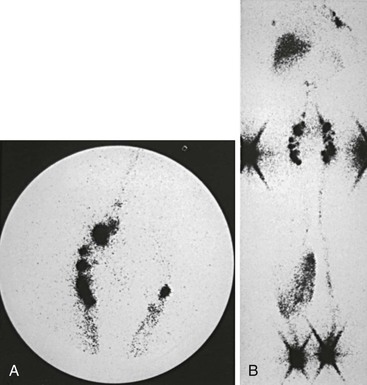
Figure 66-12 Lymphoscintigraphy in high-output lymphatic failure due to reflex sympathetic dystrophy of the right leg. A, Fast lymphatic transport in the affected right leg compared with the normal left leg is evident in the image of the inguinal nodes 20 minutes after injection. B, Total-body image at 3 hours shows a dermal pattern on the right but no evidence of proximal lymphatic obstruction.
Decision Making
Clinical examination of the patient frequently reveals the correct cause of limb swelling. Initial laboratory examinations should include routine blood tests to look for signs of renal or hepatic failure, eosinophilia, or hypoproteinemia. Urinalysis may indicate proteinuria. Once a systemic cause of edema is excluded, the local or regional cause should be confirmed.
Patients at risk for the development of secondary lymphedema (e.g., cancer survivors) pose a distinct challenge for decision making because there is an imperative to recognize the evolving condition at its earliest stages. Within the last decade, there has been introduction of a new technology, bioimpedance spectroscopy, that provides the requisite sensitivity and specificity to detect stage 0 disease.53 Increasingly, this technique, which is rapidly and efficiently performed at the bedside, will have applicability to the serial monitoring of the patient’s treatment response as well.54
Venous duplex scanning confirms or excludes venous occlusion or valvular incompetence in the leg. CT has become routine for most adult patients with leg swelling to exclude underlying malignant disease. MRI provides the most accurate information in patients with clinical signs of congenital vascular malformation, soft tissue tumor, or retroperitoneal fibrosis.
Lymphoscintigraphy is the test of choice for the confirmation of lymphedema, and a normal finding on lymphoscintigraphic examination essentially excludes the diagnosis of lymphedema. Patients with chronic venous insufficiency may have abnormal results on lymphoscintigraphic examination, with delayed transport because of mixed lymphatic and venous edema. As mentioned earlier, direct contrast lymphangiography should be performed selectively and should not be part of the routine evaluation of patients with chronic limb swelling.
For all patients with chronic lymphedema, conservative management through physical means is the mainstay of therapy. Current indications and patient selection for conservative and surgical therapies of both primary and secondary lymphedema are discussed in detail in Chapters 67 and 68.
Prospects for Molecular Therapy
Although current therapeutic strategies for lymphedema effectively reduce excess volume, minimize complications, and optimize function, the disease currently lacks a cure. For these reasons, there has been an emphasis on the possible application of effective molecular therapies. Among these, one of the most exciting prospects is therapeutic lymphangiogenesis, a molecular approach based on growing insights into the mechanisms of lymphatic vascular development.
Among the mitogenic substances that initiate and regulate the growth of vascular structures, the vascular endothelial growth factor (VEGF) family plays a central role.55,56 VEGF-C and VEGF-D direct the development and growth of the lymphatic vasculature in embryonic and postnatal life by binding to VEGFR-3, which is nearly exclusively expressed on lymphatic endothelia.57,58 In transgenic mice that overexpress VEGF-C, the lymphatic vessels demonstrate a hyperplastic, proliferative response, with secondary cutaneous changes.59 Exogenous administration of VEGF-C upregulates VEGFR-3, leading to a lymphangiogenic response.60,61
These molecular observations have helped elucidate the mechanisms that contribute to disease expression in the most common heritable form of lymphedema, the autosomal dominant condition known as Milroy’s disease. In many affected family cohorts, this disease has been linked to the flt4 locus, encoding VEGFR-3.26 Disease-associated alleles contain missense mutations that inactivate the tyrosine kinase signaling mechanism, thereby preventing downstream cellular activation. It is believed that the mutant form of the receptor is not only functionally inactive but also excessively stable, serving as a potential “sink” for activating ligands. Thus, the normal signaling mechanism is blunted, leading to hypoplastic development of lymphatic vessels.62,63
The prospects for therapeutic lymphangiogenesis in human lymphedema have been underscored by the recent description of a mouse model of inherited limb edema that features a mutation in the VEGFR-3 signaling mechanism and a pathologic process that resembles human disease.63 In this model, therapeutic overexpression of VEGF-C with use of a viral vector induces the generation of new functional lymphatics and the amelioration of lymphedema. Similarly, in a rodent model of acquired postsurgical lymphatic insufficiency (resembling postmastectomy lymphedema), the exogenous administration of human recombinant VEGF-C restores lymphatic flow (as assessed by lymphoscintigraphy),64,65 increases lymphatic vascularity, and reverses the hypercellularity that characterizes the untreated lymphedematous condition.
Intensive future investigation will be required to verify the therapeutic potential of such approaches and to establish dose-response relationships and the durability of the therapeutic response. As with other forms of angiogenic therapy, the relative virtues of growth factor therapy versus gene therapy must be established.66
Selected Key References
Cambria RA, Gloviczki P, Naessens JM, Wahner HW. Noninvasive evaluation of the lymphatic system with lymphoscintigraphy: a prospective, semiquantitative analysis in 386 extremities. J Vasc Surg. 1993;18:773.
Casley-Smith JR, Foldi M, Ryan TJ, et al. Lymphedema: summary of the 10th International Congress of Lymphology Working Group discussions and recommendations, Adelaide, Australia, August 10-17, 1985. Lymphology. 1985;18:175.
Consensus document on the classification, evaluation, and treatment of chronic lymphedema..
Cornish BH, Chapman M, Hirst C, Mirolo B, Bunce LH, Ward LC, Thomas BJ. Early diagnosis of lymphedema using multiple frequency bioimpedance. Lymphology. 2001;34:2–11.
Rockson SG. Diagnosis and management of lymphatic vascular disease. J Am Coll Cardiol. 2008;52:799–806.
Rockson SG, Miller LT, Senie R, Brennan MJ, Casley-Smith JR, Foldi E, Foldi M, Gamble GL, Kasseroller RG, Leduc A, Lerner R, Mortimer PS, Norman SA, Plotkin CL, Rinehart-Ayres ME, Walder AL. American Cancer Society Lymphedema Workshop. Workgroup III: diagnosis and management of lymphedema. Cancer. 1998;83(Suppl American):2882–2885.
The reference list can be found on the companion Expert Consult website at www.expertconsult.com.
References
1. Browse NL, et al. Lymphedema: pathophysiology and classification. J Cardiovasc Surg. 1985;26:91.
2. Schirger A, et al. Idiopathic lymphedema. Review of 131 cases. JAMA. 1962;182:124.
3. Kinmonth JB, et al. Primary lymphoedema: clinical and lymphangiographic studies of a series of 107 patients in which the lower limbs were affected. Br J Surg. 1957;45:1.
4. Kinmonth JB. The lymphoedemas: general considerations. Kinmonth JB. The lymphatics: surgery, lymphography and diseases of the chyle and lymph systems. Edward Arnold: London; 1982:83.
5. Allen EV. Lymphedema of the extremities: classification, etiology and differential diagnosis: a study of three hundred cases. Arch Intern Med. 1934;54:606.
6. Browse NL. The diagnosis and management of primary lymphedema. J Vasc Surg. 1986;3:181.
7. Browse NL. Primary lymphedema. Ernst C, et al. Current therapy in vascular surgery. BC Decker: Philadelphia; 1987:454.
8. Kurland LT, et al. The patient record in epidemiology. Sci Am. 1981;245:54.
9. Rockson SG. Lymphedema. Am J Med. 2001;110:288.
10. Smeltzer DM, et al. Primary lymphedema in children and adolescents: a follow-up study and review. Pediatrics. 1985;76:206.
11. Wolfe JHN, et al. The prognosis of primary lymphedema of the lower limbs. Arch Surg. 1981;116:1157.
12. Mendoza E, et al. A model for mechanics of primary lymphatic valves. J Biomech Eng. 2003;125:407.
13. Rockson S. Syndromic lymphedema: keys to the kingdom of lymphatic structure and function? Lymphatic Res Biol. 2003;1:181.
14. Milroy W. An undescribed variety of hereditary oedema. N Y Med J. 1892;56:505.
15. Milroy W. Chronic hereditary edema: Milroy’s disease. JAMA. 1928;91:1172.
16. Lewis JM, et al. Lymphedema praecox. J Pediatr. 1984;104:641.
17. Dahlberg PJ, et al. Autosomal or X-linked recessive syndrome of congenital lymphedema, hypoparathyroidism, nephropathy, prolapsing mitral valve, and brachytelephalangy. Am J Med Genet. 1983;16:99.
18. Radhakrishnan K, et al. The clinical spectrum of lymphatic disease. Ann N Y Acad Sci. 2008;1131:155.
19. Mucke J, et al. Early onset lymphoedema, recessive form—a new form of genetic lymphoedema syndrome. Eur J Pediatr. 1986;145:195.
20. Henriksen HM. Turners’s syndrome associated with lymphoedema, diagnosed in the newborns. Z Geburtshilfe Perinatol. 1980;184:313.
21. White SW. Lymphedema in Noonan’s syndrome. Int J Dermatol. 1984;23:656.
22. Venencie PY, et al. Yellow nail syndrome: report of five cases. J Am Acad Dermatol. 1984;10:187.
23. Wheeler ES, et al. Familial lymphedema praecox: Meige’s disease. Plast Reconstr Surg. 1981;67:362.
24. Abe R, et al. Retroperitoneal lymphangiomyomatosis with lymphedema of the legs. Lymphology. 1980;13:62.
25. Bull LN, et al. Mapping of the locus for cholestasis-lymphedema syndrome (Aagenaes syndrome) to a 6.6-cM interval on chromosome 15q. Am J Hum Genet. 2000;67:994.
26. Karkkainen MJ, et al. Missense mutations interfere with VEGFR-3 signalling in primary lymphoedema. Nat Genet. 2000;25:153.
27. Irrthum A, et al. Congenital hereditary lymphedema caused by a mutation that inactivates VEGFR3 tyrosine kinase. Am J Hum Genet. 2000;67:295.
28. Fang J, et al. Mutations in FOXC2 (MFH-1), a forkhead family transcription factor, are responsible for the hereditary lymphedema-distichiasis syndrome [in process citation]. Am J Hum Genet. 2000;67:1382.
29. Finegold DN, et al. Truncating mutations in FOXC2 cause multiple lymphedema syndromes. Hum Mol Genet. 2001;10:1185.
30. Irrthum A, et al. Mutations in the transcription factor gene SOX18 underlie recessive and dominant forms of hypotrichosis-lymphedema-telangiectasia. Am J Hum Genet. 2003;72:1470.
31. Mak JW. Epidemiology of lymphatic filariasis. Ciba Found Symp. 1987;127:5.
32. Chernin E. The disappearance of bancroftian filariasis from Charleston, South Carolina. Am J Trop Med Hyg. 1987;37:111.
33. Case T, et al. Vascular abnormalities in experimental and human lymphatic filariasis. Lymphology. 1991;24:174.
34. Dandapat MC, et al. Management of chronic manifestations of filariasis. J Indian Med Assoc. 1986;84:210.
35. Casley-Smith JR, et al. Lymphedema: summary of the 10th International Congress of Lymphology Working Group discussions and recommendations, Adelaide, Australia, August 10-17:1985. Lymphology. 1985;18:175.
36. Schirger A. Lymphedema. Cardiovasc Clin. 1983;13:293.
37. Chant ADB. Hypothesis: why venous oedema causes ulcers and lymphoedema does not. Eur J Vasc Surg. 1992;6:427.
38. Samman PD, et al. The “yellow nail” syndrome. Br J Dermatol. 1964;76:153.
39. Taylor JS, et al. The swollen limb: cutaneous clues to diagnosis and treatment. Cutis. 1978;21:553.
40. Fields CL, et al. Yellow nail syndrome: a perspective. J Ky Med Assoc. 1991;89:563.
41. Servelle M. Congenital malformation of the lymphatics of the small intestine. J Cardiovasc Surg. 1991;32:159.
42. Kinmonth JB, et al. Protein-losing enteropathy in lymphoedema: surgical investigation and treatment. J Cardiovasc Surg. 1975;16:111.
43. Stewart FW, et al. Lymphangiosarcoma in postmastectomy lymphedema: a report of six cases in elephantiasis chirurgica. Cancer. 1948;1:64.
44. Rockson SG, et al. American Cancer Society Lymphedema Workshop. Workgroup III: diagnosis and management of lymphedema. Cancer. 1998;83:2882.
45. Weissleder H, et al. Lymphedema: evaluation of qualitative and quantitative lymphoscintigraphy in 238 patients. Radiology. 1988;167:729.
46. Stewart G, et al. Isotope lymphography: a new method of investigating the role of the lymphatics in chronic limb oedema. Br J Surg. 1985;72:906.
47. Vaqueiro M, et al. Lymphoscintigraphy in lymphedema: an aid to microsurgery. J Nucl Med. 1986;27:1125.
48. Gloviczki P, et al. Noninvasive evaluation of the swollen extremity: experiences with 190 lymphoscintigraphic examinations. J Vasc Surg. 1989;9:683.
49. Cambria RA, et al. Noninvasive evaluation of the lymphatic system with lymphoscintigraphy: a prospective, semiquantitative analysis in 386 extremities. J Vasc Surg. 1993;18:773.
50. Kleinhans E, et al. Evaluation of transport kinetics in lymphoscintigraphy: follow-up study in patients with transplanted lymphatic vessels. Eur J Nucl Med. 1985;10:349.
51. Gloviczki P, et al. Klippel-Trénaunay syndrome: the risks and benefits of vascular interventions. Surgery. 1991;110:469.
52. Gloviczki P, et al. Arteriovenous fistulas and vascular malformations. Ascher E. Haimovici’s vascular surgery. ed 5. Blackwell Publishing: Malden, Mass; 2004:991.
53. Cornish BH, et al. Early diagnosis of lymphedema using multiple frequency bioimpedance. Lymphology. 2001;34:2–11.
54. Cornish BH, et al. Bioelectrical impedance for monitoring the efficacy of lymphoedema treatment programmes. Breast Cancer Res Treat. 1996;38:169–176.
55. Olofsson B, et al. Current biology of VEGF-B and VEGF-C. Curr Opin Biotechnol. 1999;10:528.
56. Veikkola T, et al. Regulation of angiogenesis via vascular endothelial growth factor receptors. Cancer Res. 2000;60:203.
57. Joukov V, et al. A novel vascular endothelial growth factor, VEGF-C, is a ligand for the Flt4 (VEGFR-3) and KDR (VEGFR-2) receptor tyrosine kinases. EMBO J. 1996;15:1751.
58. Kaipainen A, et al. Expression of the fms-like tyrosine kinase 4 gene becomes restricted to lymphatic endothelium during development. Proc Natl Acad Sci U S A. 1995;92:3566.
59. Jeltsch M, et al. Hyperplasia of lymphatic vessels in VEGF-C transgenic mice. Science. 1997;276:1423.
61. Enholm B, et al. Adenoviral expression of vascular endothelial growth factor-C induces lymphangiogenesis in the skin. Circ Res. 2001;88:623.
62. Karkkainen MJ, et al. Vascular endothelial growth factor receptors in the regulation of angiogenesis and lymphangiogenesis. Oncogene. 2000;19:5598.
63. Karkkainen MJ, et al. A model for gene therapy of human hereditary lymphedema. Proc Natl Acad Sci U S A. 2001;98:12677.
64. Szuba A, et al. Therapeutic lymphangiogenesis with human recombinant VEGF-C. FASEB J. 2002;16:U114.
65. Cheung L, et al. An experimental model for the study of lymphedema and its response to therapeutic lymphangiogenesis. BioDrugs. 2006;20:363.
66. Rockson SG. Experimental lymphedema: can cellular therapies augment the therapeutic potential for lymphangiogenesis? J Am Heart Assoc. 2012;1:e003400.