CHAPTER 43 Gastroesophageal Reflux Disease
EPIDEMIOLOGY
On the basis of symptoms, GERD is common in Western countries. In a nationwide population-based study by the Gallup Organization in the United States, 44% of the respondents reported heartburn at least once a month.1 More convincing data were obtained from a mailing of 2200 validated self-report questionnaires to a predominantly white population living in Olmsted County, Minnesota.2 The prevalence of heartburn and acid regurgitation in the past year was 42% and 45%, respectively. Symptoms that occurred at least weekly were reported by 20% of respondents, with an equal gender distribution across all ages. Most subjects reported their heartburn as being moderately severe, with a duration of 5 years or more, and only 5.4% had seen a physician for their reflux symptoms within the past year. More varying prevalence rates for symptomatic GERD have been reported from Europe, ranging from 5% in Switzerland to 27% in Finland.3
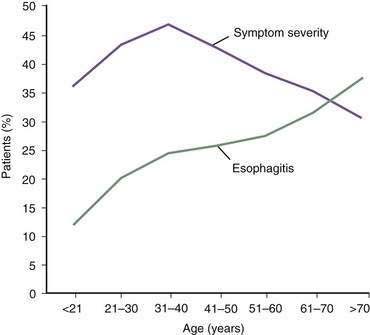
In contrast, the true prevalence of esophagitis is very difficult to define because healthy subjects rarely undergo upper endoscopy. Studies suggest that 7% of Americans have erosive esophagitis, whereas European studies identify prevalence rates ranging from 2% to 10%.4 GERD affects nearly equal proportions of men and women, but a male predominance occurs in esophagitis and Barrett’s esophagus.4 Increasing age is an important factor in the prevalence of GERD complications, probably the result of cumulative acid injury over time to the esophagus (Fig. 43-1).5,6
The prevalence of GERD only recently has been studied in multiracial populations. In a cross-sectional survey among employees at a Houston Veterans Affairs hospital, the prevalence of heartburn was similar (23% to 27%) across ethnic groups including African Americans, Hispanics, Asians, and whites. However, African Americans had significantly less esophagitis than whites (24% versus 50%) for the same severity of symptoms (weekly or more).7 A study from Boston reviewed endoscopic reports from nearly 2500 consecutive patients, finding complicated GERD in 12% of white patients, 3% of African American patients, and 2% of Asian patients.8
The prevalence of GERD is relatively low among residents of Africa and Asia. For example, a cross-sectional study in Singapore reported prevalence rates for reflux symptoms of 7.5% in Indians, 0.8% in Chinese, and 3% in Malays.9 There have been exceptions such as the remarkable increase in the frequency of reflux symptoms seen in Japan and Singapore.10 An endoscopic, population-based study from South Korea encompassing more than 25,000 individuals, found the prevalence of erosive esophagitis to be 8%, whereas nonerosive reflux disease occurred in 4% of examined individuals.11 More than 90 % of subjects with erosions had mild disease, consistent with previous endoscopic studies from Asia. A recent systematic review that summarized trend data from longitudinal population-based studies performed in Asia failed to demonstrate an increase in prevalence over the past decade.12 Possible reasons for the lower GERD prevalence include low dietary fat; low body mass index (BMI); and lower gastric acid output, possibly related to Helicobacter pylori infection.11,13
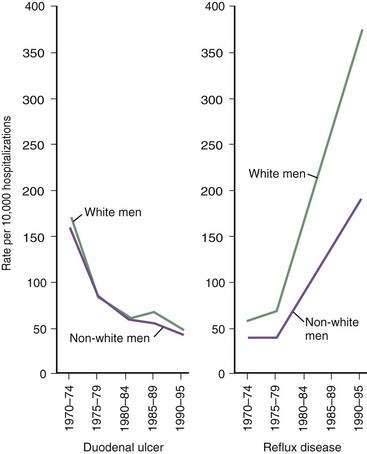
The prevalence of GERD has been increasing in Western countries over the past 30 years.14 El Serag and Sonnenberg observed opposing time trends in the prevalence of peptic ulcer disease and GERD in the United States. Rates of duodenal ulcer fell between 1970 and 1995, while the prevalence of GERD and esophageal adenocarcinoma rose significantly (Fig. 43-2).15 The authors speculated that the decreasing prevalence of H. pylori may be playing a contributory role to the increasing prevalence of GERD in these regions. Recent data suggest that many patients with H. pylori–induced gastritis have involvement of the antrum and corpus, decreasing parietal cell mass, reducing acid secretion, and elevating gastric pH.13 This may have a protective effect on the esophageal mucosa in patients susceptible to GERD.
An additional explanation for an increased prevalence of GERD in Western populations is the epidemic increase in obesity.16 In obese individuals (defined as a body mass index ≥30), epidemiologic studies suggest the prevalence of GERD is considerably higher than in the nonobese population.4,5,16 Jacobson and associates looked at the participants in the Nurses’ Health Study and found a nearly linear increase in the adjusted odds ratio for reflux symptoms for each BMI stratum.17 Interestingly, even for those participants with a normal BMI (22.5 to 24.9 kg/m2), the risk was elevated relative to a control group having a BMI in the range of 20 to 22.4 kg/m2.17 A study from the Houston VA Medical Center found a linear relationship between BMI and weekly symptoms of heartburn or regurgitation.18 Data from large population-based studies in England and Germany have been similar.19,20 A Norwegian study suggested that the odds of developing GERD were higher in obese subjects, and the risk was greater in obese women compared with male participants.21 In contrast, a nationwide case-control study from Sweden, consisting primarily of older adult men, failed to find an association between obesity and GERD.22
Central adiposity, as measured by the waist-to-hip ratio, may be more important than BMI in the pathogenesis of GERD. A large study from the Kaiser Permanente health system found a significant relationship between increased abdominal diameter and reflux symptoms independent of BMI.23 Similarly, El-Serag and colleagues found that the relationship between increasing BMI and increased acid exposure in the distal esophagus was primarily explained by the subject’s waist circumference.24
Obesity appears to be associated with complications related to long-standing GERD such as erosive esophagitis, Barrett’s esophagus, and esophageal adenocarcinoma.18,25–27 In a Swedish case-control study, researchers identified an association between esophageal adenocarcinoma and an individual’s BMI 20 years prior to the development of malignancy.28 Another case-control study demonstrated that central adiposity rather than BMI was associated with the presence of Barrett’s esophagus, particularly long-segment disease.29
Several mechanisms have been proposed to explain the association between obesity and GERD. These include an increased prevalence of esophageal motor disorders, diminished lower esophageal sphincter pressure, increased prevalence of hiatal hernia, and increased intragastric pressure (particularly with central obesity).30 In addition, visceral fat is metabolically active and produces a variety of cytokines including interleukin-6 (IL-6) and tumor necrosis factor-alpha (TNF-α), which may affect the function of the lower esophageal sphincter.
Along with environmental factors, the epidemiology of GERD may be affected by genetics. Family clustering of GERD and its complications, especially Barrett’s esophagus, have been reported.31,32 Especially exciting are the observations from two large case-control studies of twins from the United States and Sweden33,34 suggesting that genetic liability for GERD, as defined by frequent symptoms, is in the range of 30% to 45%. Although one group defined a locus on chromosome 13 associated with severe pediatric GERD,35 this has not been confirmed by other pediatric researchers36 and not yet evaluated in adults. The genetic mechanisms are unknown but may be related to a smooth muscle disorder associated with hiatal hernia, reduced lower esophageal sphincter (LES) pressure, and impaired esophageal motility.32
HEALTH CARE IMPACT
Although rarely a cause of death, GERD is associated with considerable morbidity and complications, such as esophageal ulceration (5%), peptic stricture (4% to 20%), and Barrett’s esophagus (8% to 20%).5 Not surprisingly, the burden of GERD on health care is great. In 2004, GERD was by far the most common digestive disease diagnosis during ambulatory care visits, constituting 17.5% of all GI diagnoses. There were 6 or more outpatient visits with a GERD diagnosis listed per 100 people in the United States.37 GERD was the 10th most common inpatient GI diagnosis, with an estimated total number of discharges of 95,000 per year, a two day median length of stay, and median charges of $8060.38 In summary, GERD was the second most costly GI disease in 2004, behind liver disease, with total direct and indirect costs of nearly $12.6 billion. More than 60 million prescriptions for the treatment of GERD were estimated to be filled at retail pharmacies in 2004, representing 48% of all prescriptions for GI disorders and more than 50% of their costs. The large majority of prescriptions and their costs were for proton pump inhibitors, which were the five most commonly prescribed and costliest GI medications.37 In 2004, two PPIs (lansoprazole and esomeprazole) were the second and fourth, respectively, top selling drugs of all classes in the United States.
A recent economic survey from Germany reported that 6% of individuals with established GERD missed at least one day of work per year due to this disorder. Sixty-one percent of these patients visited their physician at least once in the previous year and 2% were hospitalized specifically for GERD.39 They estimated direct and indirect costs of approximately $600 per patient per year.
Furthermore, GERD as a chronic disease significantly impairs quality of life. Compared with other chronic medical conditions, this impairment is similar to, or even greater than, that from arthritis, myocardial infarction, heart failure, or hypertension.40 GERD comorbidities are common and include irritable bowel syndrome and psychological distress in 36% and 41% of patients, respectively.41 These comorbidities potentiate the negative effect on quality of life seen with GERD, and affect the response to treatment with proton pump inhibitors.
PATHOGENESIS
ANTIREFLUX BARRIERS
The first tier of the three-tiered esophageal defense against acid damage, the antireflux barriers, is an anatomically complex region including the intrinsic lower esophageal sphincter (LES), diaphragmatic crura, the intra-abdominal location of the LES, the phrenoesophageal ligaments, and the acute angle of His (Fig. 43-3).
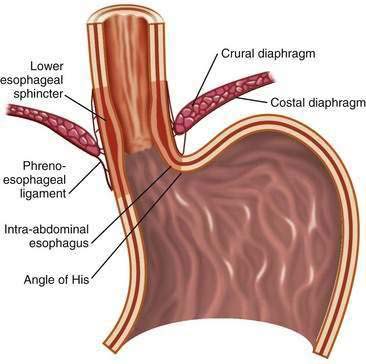
Figure 43-3. Anatomy of the gastroesophageal junction illustrating the major elements of the antireflux barrier.
The LES involves the distal 3 to 4 cm of the esophagus and at rest is tonically contracted.42 It is the major component of the antireflux barrier, being capable of preventing reflux even when completely displaced from the diaphragmatic crura by a hiatal hernia.43 The proximal portion of the LES is normally 1.5 to 2 cm above the squamocolumnar junction, whereas the distal segment, about 2 cm in length, lies within the abdominal cavity. This location maintains gastroesophageal competence during intra-abdominal pressure excursions. Resting LES pressure ranges from 10 to 30 mm Hg with a generous reserve capacity because only a pressure of 5 to 10 mm Hg is necessary to prevent GER.44 The LES maintains a high-pressure zone by the intrinsic tone of its muscle and by cholinergic excitatory neurons.45,46 There is considerable diurnal variation in basal LES pressure; it is lowest after meals and highest at night, and large increases occur with phase III of the migrating motor complex. It is also influenced by circulating peptides and hormones, foods (particularly fat), as well as a number of drugs (Table 43-1) (see also Chapter 48).
Table 43-1 Modulators of Lower Esophageal Sphincter Pressure
| INCREASE LES PRESSURE | DECREASE LES PRESSURE | |
|---|---|---|
| Hormones/peptides | Gastrin | Secretin |
| Motilin | Cholecystokinin | |
| Substance P | Somatostatin | |
| VIP | ||
| Neural agents | α-Adrenergic agonists | α-Adrenergic antagonists |
| β-Adrenergic antagonists | β-Adrenergic agonists | |
| Cholinergic agonists | Cholinergic antagonists | |
| Foods | Protein | Fat |
| Chocolate | ||
| Peppermint | ||
| Other factors | Histamine | Theophylline |
| Antacids | Prostaglandins E2 and I2 | |
| Metoclopramide | Serotonin | |
| Domperidone | Meperidine | |
| Cisapride | Morphine | |
| Prostaglandin F2α | Dopamine | |
| Baclofen | Calcium channel blockers | |
| Diazepam | ||
| Barbiturates |
LES, lower esophageal sphincter; VIP, vasoactive intestinal peptide.
The LES lies within the hiatus created by the right crus of the diaphragm and is anchored by the phrenoesophageal ligaments, which insert at the level of the squamocolumnar junction (see Fig. 43-3). Developmentally, the crural diaphragm arises from the dorsal mesentery of the esophagus and is innervated separately from the costal diaphragm. It is inhibited by esophageal distention, vomiting, and during transient LES relaxations (tLESRs), but not during swallowing. The crural diaphragm provides extrinsic squeeze to the intrinsic LES, contributing to resting pressure during inspiration and augmenting LES pressure during periods of increased abdominal pressure, such as with coughing, sneezing, or bending.47 Crural contractions impose rhythmic pressure increases of about 5 to 10 mm Hg on the LES pressure recording. During deep inspirations and some periods of increased abdominal straining, these changes may lead to pressures of 50 to 150 mm Hg.48
The oblique entrance of the esophagus into the stomach creates a sharp angle on the greater curve aspect of the gastroesophageal junction, the angle of His. This angle has been shown in cadavers to create a flap valve effect that contributes to gastroesophageal junction competency.49
MECHANISMS OF REFLUX
Transient Lower Esophageal Sphincter Relaxations
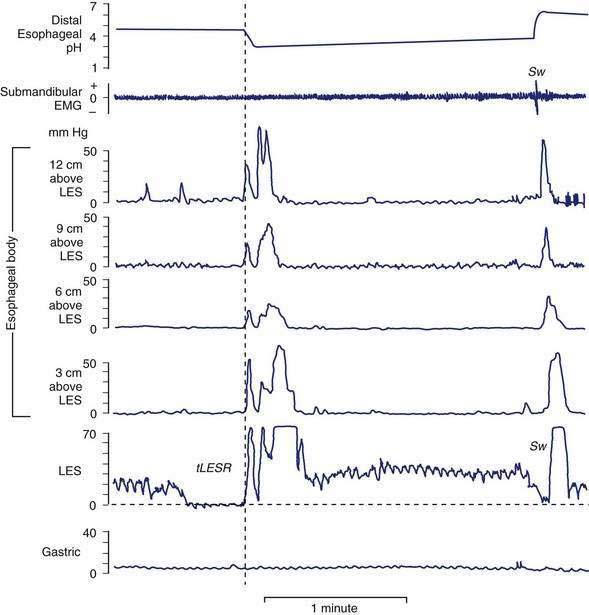
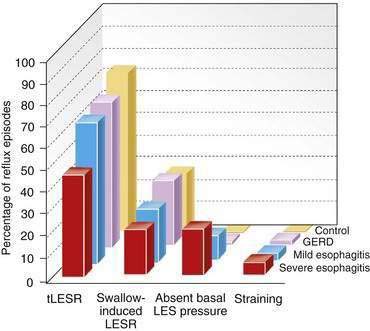
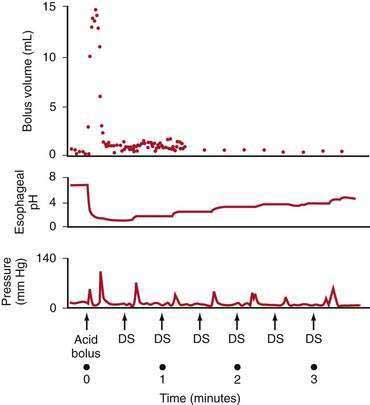
tLESRs are the most frequent mechanism for reflux in patients with healthy sphincter pressures. Figure 43-4 illustrates a transient LESR and highlights differences from swallow-induced LESRs. tLESRs occur independently of swallowing, are not accompanied by esophageal peristalsis, persist longer (>10 seconds) than swallow-induced LESRs, and are accompanied by inhibition of the crural diaphragm.50 tLESRs account for nearly all reflux episodes in healthy subjects and 50% to 80% of episodes in GERD patients, depending on the severity of associated esophagitis (Fig. 43-5).51 However, one study suggests that low basal LES pressure, rather than tLESRs, may be the primary mechanism of GER in patients with nonreducible hiatal hernias.52
tLESRs are not always associated with GER. In normal subjects 40% to 60% of tLESRs are accompanied by reflux episodes, compared with 60% to 70% in GERD patients.45,51,53 Possible factors determining whether reflux occurs include abdominal straining, presence of a hiatal hernia, degree of esophageal shortening, and duration of tLESRs. The dominant stimulus for tLESR is distention of the proximal stomach by either food or gas,54,55 which is not surprising given that a tLESR is the mechanism of belching. More varying stimuli are fat, stress, and subthreshold (for swallowing) stimulation of the pharynx.49 Various drugs may impair tLESRs including cholecystokinin A (CCK-1) receptor antagonists, anticholinergic drugs, morphine, somatostatin, nitric oxide inhibitors, 5-hydroxytryptamine (5-HT)3 antagonists, and γ-aminobutyric acid (GABAB) agonists.56
Evidence indicates that tLESRs are mediated through vagal pathways.54 Gastric distention activates mechanoreceptors (intraganglionic lamellar endings) adjacent to the gastric cardia, sending signals to the brainstem center via vagal afferent pathways.57 The structured sequence of motor events including LESR, crural diaphragm inhibition, and secondary esophageal peristalsis suggests that this process occurs in a programmed manner, probably controlled by a pattern generator within the vagal nuclei. The motor arm is the vagus nerve sharing common elements with swallow-induced LESR.56
Swallow-Induced Lower Esophageal Sphincter Relaxations
About 5% to 10% of reflux episodes occur during swallow-induced LESRs. Most episodes are associated with defective or incomplete peristalsis.53 During a normal swallow-induced LESR, reflux is uncommon because (1) the crural diaphragm does not relax, (2) the duration of LESR is relatively short (5 to 10 seconds), and (3) reflux is prevented by the oncoming peristaltic wave (see Fig. 43-4). Reflux during swallow-induced LESRs is more common with a hiatal hernia. This may be due to the lower compliance of the esophagogastric junction in hernia patients, permitting it to open at pressures equal to or lower than intragastric pressure, thereby allowing reflux of gastric juices accumulating in the hiatal hernia.58,59
Hypotensive Lower Esophageal Sphincter Pressure
GER can occur in the context of a hypotensive LES by either strain-induced or free reflux.44,51 Strain-induced reflux occurs when a relatively hypotensive LES is overcome and “blown open” by an abrupt increase in intra-abdominal pressure from coughing, straining, or bending over. This type of reflux is unlikely when the LES pressure is greater than 10 mm Hg. Free reflux is characterized by a fall in intraesophageal pH without an identifiable change in intragastric pressure, usually occurring when LES pressure is less than 5 mm Hg. Reflux due to a low or absent LES pressure is uncommon. Mostly it occurs in patients with severe esophagitis and may account for up to 25% of reflux episodes (see Fig. 43-5); it rarely occurs in patients without esophagitis.45,51,60 The mechanisms responsible for idiopathic low LES pressure (i.e., not part of a systemic disease such as scleroderma) are poorly understood. The presence of a hiatal hernia reduces the pressure measured in the LES due to losing the intrinsic support of the crural diaphragm.44 Some LES weakness may be secondary to esophagitis impairing the excitatory cholinergic pathways to the LES. Induction of experimental esophagitis in cats attenuates the release of acetylcholine and lowers LES pressures—changes that are reversible on healing of the esophagitis.60 However, healing of esophagitis in humans is rarely accompanied by an increase in LES pressure.61
HIATAL HERNIA
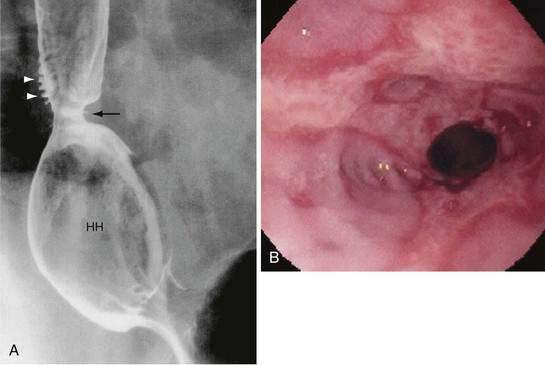
The contribution of the hiatal hernia to GERD is controversial. Opinion has shifted widely from one that virtually equated hiatal hernia with reflux disease to one that denied it a causal role. Epidemiologic and physiologic data confirm the importance of the hiatal hernia in patients with more severe esophagitis, peptic stricture, or Barrett’s esophagus.62 Hiatal hernia occurs in 54% to 94% of patients with reflux esophagitis, a rate strikingly higher than that in the healthy population.63 Two studies have also found that in individuals with reflux symptoms, the presence of hiatal hernia confers a significantly increased risk of erosive esophageal injury.64
The hiatal hernia impairs LES function through several mechanisms, as well as impairing esophageal acid clearance (Fig. 43-6).65 Reflux is worse in patients having a “nonreducible” as opposed to a “reducible” hiatal hernia. Nonreducing hernias are those in which the gastric rugal folds remain above the diaphragm between swallows.62 Statistical modeling has revealed a significant interaction between hiatal hernia and LES pressure, such that the likelihood of GER is increased as basal LES pressure decreases, an effect substantially amplified by the presence of a hernia and as the hernia size increases.43
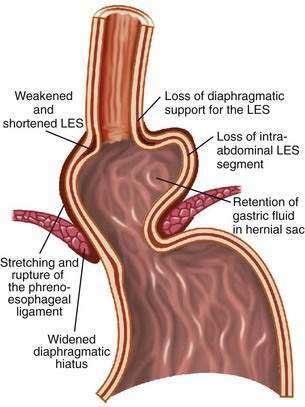
Figure 43-6. Schematic diagram showing the effect of a hiatal hernia on the antireflux barrier. LES, lower esophageal sphincter.
Displacement of the LES from the crural diaphragm into the chest reduces basal LES pressure and shortens the length of the high-pressure zone primarily due to the loss of the intra-abdominal LES segment.62 Hiatal hernia eliminates the increase of LES pressure that occurs during straining and increases tLESRs during gastric distention with gas.65,66 Large, nonreducible hernias also impair esophageal acid clearance because of an increased tendency for reflux to occur from the hernia sac during swallow-induced LESRs.53 Finally, an alteration of esophagogastric junction compliance, especially in GERD patients with hiatal hernia, has been identified.59 For the same degree of intragastric pressure, the esophageal junction opens at a lower pressure and the cross-sectional area is greater and more symmetrical as intragastric pressure increases. These changes in compliance simulated a 10-fold increase in air and 6-fold increase in liquid reflux across the esophageal junction.
The etiology of a hiatal hernia remains unclear. Familial clustering of GERD suggests the possibilities of an inherited smooth muscle disorder. Animal studies propose that reflux itself causes esophageal shortening promoting the development of a hiatal hernia.67 Other studies find an association with obesity68 and heavy lifting,69 raising the possibilities that over time chronic intra-abdominal stressors may weaken the esophageal hiatus, causing the development of a hiatal hernia. This theory is attractive as it helps to reconcile the increased prevalence of hiatal hernias as the population grows older.63
ESOPHAGEAL ACID CLEARANCE
Volume Clearance
Esophageal peristalsis clears acid volume in the upright and supine positions but is inoperative during deep rapid-eye-movement (REM) sleep. Helm and colleagues70 showed that one or two primary peristaltic contractions completely clear a 15-mL fluid bolus from the esophagus (Fig. 43-7). Primary peristalsis is elicited by swallowing. Secondary peristalsis, initiated by esophageal distention from acid reflux, is much less effective in clearing the refluxate, thus offering only an ancillary protective role.
Peristaltic dysfunction (i.e., failed peristaltic contractions and hypotensive [<30 mm Hg] peristaltic contractions that incompletely empty the esophagus) increases in frequency with the severity of esophagitis. Kahrilas and colleagues71 found that the prevalence of peristaltic dysfunction rose from 25% in individuals with mild esophagitis to more than 50% in patients with severe esophagitis. Whether esophagitis per se leads to peristaltic dysfunction or whether an underlying smooth muscle motility disorder of the esophagus predisposes to the development of reflux disease is not clear. Animal studies have found that esophageal dysmotility associated with active esophagitis is reversible, but esophageal dysmotility associated with stricture or extensive fibrosis is irreversible.60 Clinical observations suggest that impaired motor function does not revert to normal following either effective medical or surgical therapies.61
Gravity contributes to bolus clearance when reflux occurs in the upright position. At night when supine, this mechanism is not operative unless the head of the bed is elevated. This important lifestyle change markedly improves acid clearance time and is most beneficial in patients with aperistalsis (i.e., scleroderma).72
Salivary and Esophageal Gland Secretions
Saliva is the second essential factor required for normal esophageal acid clearance. Compared with gastric acid, saliva is a weak base with a pH of 6.4 to 7.8.73 Although saliva is ineffective in neutralizing large acid volumes (5 to 10 mL), it easily neutralizes the small amount of acid remaining in the esophagus after several peristaltic contractions (see Fig. 43-7).70 The importance of saliva is supported by observation that increased salivation induced by oral lozenges or bethanechol significantly decreases acid clearance time. In contrast, suction aspiration of saliva markedly prolongs acid clearance, despite the presence of normal peristaltic contractions.73
Modulation of salivation may contribute to GERD. Decreased salivation during sleep is the reason that nocturnal reflux episodes are associated with markedly prolonged acid clearance times.74 Xerostomia (see Chapter 22) is associated with prolonged esophageal acid exposure and esophagitis.75 Cigarette smoking promotes GER. Originally attributed to nicotine’s effect on lowering LES pressure, cigarette smokers also have prolonged esophageal acid clearance times due to hyposalivation.76 Finally, the esophagosalivary reflex is impaired in patients with reflux esophagitis and individuals with strictures.77 This is a vasovagal reflex demonstrated by perfusing acid into the esophagus, which stimulates salivation. This reflex explains the symptoms of water brash (copious salivation) observed in some reflux patients.
In addition to saliva, the aqueous bicarbonate-rich secretions of the esophageal submucosal glands dilute and neutralize residual esophageal acid.78 Acid refluxing into the esophageal lumen stimulates these glands and helps neutralize the acid, even if swallowing does not occur.79
Tissue Resistance
Although clearance mechanisms minimize acid contact time with the epithelium, even healthy subjects have acid reflux during the day and sometimes at night. Nevertheless, only a few subjects experience symptomatic GER and even fewer suffer GERD. This is due to a third tier for esophageal defense, known as tissue resistance. Conceptually, tissue resistance can be subdivided into pre-epithelial, epithelial, and postepithelial factors, which act together to minimize mucosal damage from the noxious gastric refluxate.80
The pre-epithelial defense in the esophagus is poorly developed. There is neither a well-defined mucous layer nor buffering capacity by the surface cells to secrete bicarbonate ions into the unstirred water layer. This results in a lumen-to-surface pH gradient in the esophagus of only 1 : 10, in contrast with the stomach and duodenum, where the gradient can range from 1 : 1000 to 1 : 10,000.81
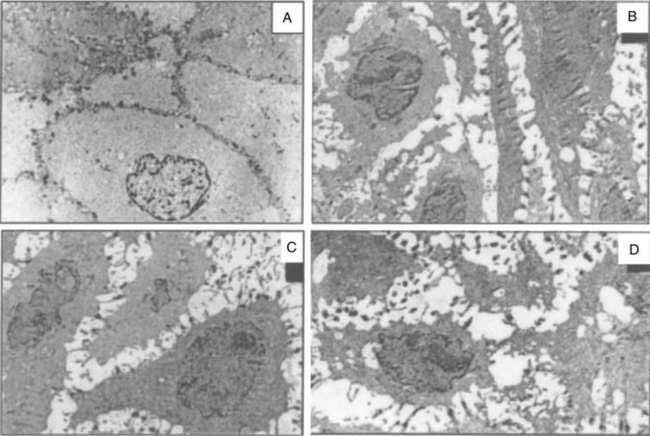
The epithelial defenses consist of structural and functional components. Structural components include the cell membranes and intercellular junctional complexes of the esophageal mucosa. As reviewed in Chapter 41, this structure is a 25- to 30-cell-thick layer of nonkeratinized squamous epithelium functionally divided into a proliferating basal cell layer (stratum basalis), a midzone layer of metabolically active squamous cells (stratum spinosum), and a 5- to 10-cell-thick layer of dead cells (stratum corneum). The esophageal mucosa is a relatively “tight” epithelium that resists ionic movement at the intercellular, as well as the cellular, level as the result of tight junctions and the matrix of lipid-rich glycoconjugates in the intercellular space.82 Luminal acid attacks the epithelial defenses by damaging the intercellular junction, allowing hydrogen ions to enter and acidify the intercellular space. As documented by transmission electron microscopy, the intercellular spaces expand and eventually the buffering capacity of this space is overwhelmed, leading to acidification of the adjacent cytosol via the basolateral membrane.80 The functional components of tissue resistance include the ability of the esophageal epithelium to buffer and extrude hydrogen ions. Intracellular buffering is accomplished by negatively charged phosphates and proteins, as well as bicarbonate ions. When the mucosal buffering capacity is exceeded and intracellular pH falls, the epithelium has the capacity to actively remove or neutralize H+. This is possibly by the action of two transmembrane proteins, one an Na+/H+ exchanger and the other an Na+-dependent Cl−/HCO3− exchanger.83,84 After reflux-induced cell acidification, these transporters restore the intracellular pH to neutrality by exchanging H+ for extracellular Na+ or by exchanging Cl− for extracellular HCO3−, respectively. Additionally, esophageal cells contain within their membrane an Na+-independent Cl−/HCO3− exchanger that extrudes HCO3− from the cytoplasm when the intracellular pH is too high.83 When the epithelial cells are no longer able to maintain intracellular pH, they lose their ability to volume regulate, edema occurs, balloon cells develop, and cell death follows. Additional contributors to the epithelial defense include salivary epidermal growth factor, transforming growth factor-α, and prostaglandin E2. These factors enhance epithelial cell turnover, enhance esophageal mucin production, and modulate bicarbonate secretion.85
Data suggest that dilated intercellular spaces are the earliest markers of esophageal epithelial cellular damage (Fig. 43-8). These alterations arise with exposure to acid and pepsin during gastroesophageal reflux, but the exact pathway of damage to the intercellular junctions remains unclear and seems to be multifactorial.86 Other noxious contents of the refluxate, such as bile acids, are harmful, and dilated intercellular spaces can be induced by acute psychological stress.87 Dilated intercellular spaces can be assessed quantitatively with electron microscopy (EM), but they also are recognizable with light microscopy (LM). In studies by Calabrese and colleagues,88,89 all controls had intercellular spaces less than 1.69 µm. Symptomatic patients had a mean intercellular space value and a mean value of the maximum dilated intracellular space at least three times greater than controls. Statistical differences were not observed between esophagitis patients and nonerosive GERD patients. The authors speculated that increased paracellular permeability could partly explain the development of heartburn in the absence of overt esophagitis. This hypothesis is supported by the presence of sensory neuron receptors within the intercellular space, only a few cell layers from the esophageal lumen.90 Importantly, aggressive acid inhibition with proton pump inhibitors leads to complete resolution of the dilated intercellular spaces in nearly all patients over three to six months. These changes correlated closely with the resolution of heartburn.89
The postepithelial defense is provided by the esophageal blood supply. Blood flow delivers oxygen, nutrients, and bicarbonate and removes H+ and CO2, thereby maintaining normal tissue acid-base balance. Blood flow to the esophageal mucosa increases in response to the stress of luminal acid.91 Cellular injury also stimulates cell proliferation, which results in thickening of the basal cell layer of the epithelium. Unlike the stomach, where superficial mucosal injury can be repaired in hours, the esophagus repairs itself more slowly, over days to weeks. Acid suppression with proton pump inhibitors has been shown to reverse the characteristic histologic changes of esophageal reflux including basal layer thickening and dilation of intercellular spaces.77,92
GASTRIC FACTORS
Gastric Acid Secretion
Acid and activated pepsin are the key ingredients of the gastric refluxate producing esophagitis. In animal studies, acid alone causes minimal injury at a pH of less than 3, primarily by protein denaturization. However, acid combined with even small amounts of pepsin disrupts the mucosal barrier, resulting in increased H+ permeability, histologic changes, and hemorrhage.93 Supporting these animal studies, clinical series find that the degree of esophageal injury, from nonerosive GERD to Barrett’s esophagus, parallels the increase in the frequency and duration of acid reflux with a pH of less than 4.94,95 Conversely, perfusing the esophagus of animals with the pepsin solution of pH 4 to 7.5 produces minimal mucosal disruption or change in mucosal permeability.93 These observations are the cornerstone of acid inhibition therapy for the treatment of GERD.
Overall, gastric acid secretion is normal in patients with GERD. For example, Hirschowitz96 compared the gastric acid secretion of 115 patients with esophagitis with more than 500 age-, gender-, and disease-matched controlled subjects without esophagitis. The average fasting basal and maximum secretions of both acid and pepsin were the same in both groups, and esophagitis severity was not related to any of these factors. On the other hand, local distribution of acid rather than total gastric secretion may be more relevant to the pathogenesis of GERD. Data suggest that the gastroesophageal junction may escape the buffering effect of meals, remaining highly acidic (median pH 1.6) compared with the body of the stomach (pH 4.7). This “proximal pocket of acid” extends from the cardia into the distal esophagus and could account for the high prevalence of disease in this location.97 A recent study confirmed these findings, demonstrating a pH less than 4 for 26.5% of the time over 24 hours when measured at the lower esophageal sphincter.98
H. pylori infection, especially with the cagA+ virulent strain, is a “biological antisecretory agent” that lowers gastric acidity, thereby possibly protecting from the development of severe esophagitis and Barrett’s esophagus.99,100 Acid output may be decreased by several mechanisms: (1) the associated severe corpus gastritis, which, over time, progresses to multifocal atrophic gastritis (see Chapter 51); (2) increased gastric alkaline (bicarbonate) secretion, which returns to normal after H. pylori eradication101; and perhaps (3) production of ammonia by the bacteria itself.102 After eradication of H. pylori, the corpus mucosa can regenerate to normal, increasing acid secretion and potentiating reflux in susceptible patients, possibly contributing to the reports of esophagitis after eradication of this organism.103,104 The consequence of returning the stomach to a healthy state is unknown but may be an underlying factor in the increasing prevalence of severe GERD, Barrett’s esophagus, and even adenocarcinoma in Western populations.13,102
Duodenogastric Reflux
Along with acid and pepsin, duodenal contents may be injurious to the esophageal mucosa. Animal studies demonstrate that conjugated bile acids produce their greatest injury in the presence of acid and pepsin, whereas trypsin and the deconjugated bile acids are damaging in a more neutral environment.105 These experiments suggest that duodenogastric reflux into the esophagus predisposes to complications of GERD.106,107 However, the accurate measurement of duodenogastric reflux is difficult. Traditionally, this phenomenon was defined indirectly by measuring the esophageal pH greater than 7 (i.e., “alkaline reflux”).107 However, on the basis of newer technology that accurately measures bilirubin, the most common pigment of bile, independent of pH, we now know that this technique is inaccurate.108 These studies show that acid and bile reflux increase in parallel across the spectrum of GERD, suggesting a synergistic role in the development of esophagitis and its complications.109,110 Additionally, aggressive acid suppression with proton pump inhibitors decreases both acid and duodenogastric reflux by decreasing the volume of gastric contents available to reflux into the esophagus.110
Delayed Gastric Emptying
The importance of delayed gastric emptying in the pathogenesis of GERD is controversial. Early studies found delay in the gastric emptying of solids in up to 50% of reflux patients.111 However, methodologic problems may have invalidated these studies. More recent investigations found only a 6% to 38% incidence of delayed gastric emptying, regardless of the severity of the esophagitis.112,113 Nevertheless, delayed gastric emptying is a major factor contributing GERD in some groups such as diabetic patients with autonomic peripheral neuropathy.
CLINICAL FEATURES
CLASSIC REFLUX SYMPTOMS
Heartburn is the classic symptom of GERD, with patients generally reporting a burning feeling, rising from the stomach or lower chest and radiating toward the neck, throat, and occasionally the back.114 It occurs postprandially, particularly after large meals or after ingesting spicy foods, citrus products, fats, chocolates, and alcohol. The supine position and bending over may exacerbate heartburn. Recent studies have suggested that sleep deprivation as well as psychological or auditory stress may lower the threshold for symptom perception.115–117 Nighttime heartburn may cause sleeping difficulties and impair next-day function.118 When heartburn dominates a patient’s complaints, it has high specificity (89%) but low sensitivity (38%) for GERD as diagnosed by 24-hour esophageal pH testing.119 GERD is usually diagnosed symptomatically by the occurrence of heartburn two or more days a week, although less frequent symptoms do not preclude the disease.120,121 Although an aid to diagnosis, the frequency and severity of heartburn do not predict the degree of esophageal damage.5
Heartburn symptoms can arise from acid reflux, weakly acidic reflux, bile reflux, and mechanical stimulation of the esophagus. The receptor that mediates the sensation of heartburn during acid perfusion has not been identified, although the capsaicin or vanilloid receptor 1 (TRPV1) is a leading candidate. TRPV1 is a cation channel that is expressed by sensory neurons, and its activation by heat, acid pH, or ethanol may trigger burning pain.122–126 Weakly acidic reflux, as detected by combined pH and impedance technology, appears to produce symptoms when there is a large proximal extent reached by the refluxate, large reflux volumes, and prolonged acid-clearance times.127 The mechanism that underlies bile-acid–induced esophageal symptoms is unknown. Bile acids are postulated to induce the release of intracellular mediators via damage to lipid membranes.128 In addition to acid-induced and bile-acid–induced esophageal damage, pepsin can cause direct damage to the esophageal mucosa, leading to dilated intercellular spaces and increased esophageal mucosal permeability.129 Esophageal distention and sustained esophageal contractions are other mechanisms proposed to explain the symptom of heartburn. Balaban and associates used high-frequency endoluminal ultrasound to demonstrate a correlation between spontaneous chest pain or chest pain induced by edrophonium chloride and sustained esophageal longitudinal muscle contractions.130 Esophageal hyperalgesia (lowered pain threshold) and psychological comorbidity have also been postulated to contribute to heartburn symptoms.131
Other common symptoms of GERD are acid regurgitation and dysphagia. The effortless regurgitation of acidic fluid, especially after meals and worsened by stooping or the supine position, is highly suggestive of GERD.119 Among patients with daily regurgitation, LES pressure is usually low; many have associated gastroparesis, and esophagitis is common, making this symptom more difficult to treat medically than classic heartburn. Dysphagia is reported by more than 30% of individuals with GERD.132 It usually occurs in the setting of long-standing heartburn with slowly progressive dysphagia for solids. Weight loss is uncommon, because patients have good appetites. The most common causes are a peptic stricture or Schatzki’s ring, but other etiologies include severe esophageal inflammation alone, peristaltic dysfunction, and esophageal cancer arising from Barrett’s esophagus (see Chapter 44).
Less common symptoms associated with GERD include water brash, odynophagia, burping, hiccups, nausea, and vomiting.133 Water brash is the sudden appearance in the mouth of a slightly sour or salty fluid. It is not regurgitated fluid, but rather secretions from the salivary glands in response to acid reflux.73 Odynophagia may be seen with severe ulcerative esophagitis. However, its presence should raise the suspicion of an alternative cause of esophagitis, especially infections or injury from impacted pills (see Chapter 45).
Some patients with GERD are asymptomatic. This is particularly true in the older adults, perhaps because of decreased acidity of the reflux material in some or decreased pain perception in others.5 Many older adult patients present first with complications of GERD because of long-standing disease with minimal symptoms. For example, up to one third of patients with Barrett’s esophagus are insensitive to acid at the time of presentation.134
EXTRAESOPHAGEAL MANIFESTATIONS
GER may cause a wide spectrum of conditions including noncardiac chest pain, asthma, posterior laryngitis, chronic cough, recurrent pneumonitis, and even dental erosion.135 Some of these patients have classic reflux symptoms, but many are “silent refluxers,” contributing to problems in making the diagnosis. Furthermore, it may be difficult to establish a causal relationship even if GER can be documented by testing (e.g., pH studies) because individuals may simply have two common diseases without a cause-and-effect relationship.
Chest Pain
GER-related chest pain may mimic angina pectoris, having a squeezing or burning quality; being in a substernal location; and radiating to the back, neck, jaws, or arm. It frequently is worse after meals, can awaken the patient from sleep, and may worsen during emotional stress. Heavy exercise, even treadmill testing, may provoke GER.136 Reflux-related chest pain may last for minutes to hours, often resolves spontaneously, and may be eased with antacids. The majority of patients with GERD-induced chest pain have heartburn symptoms.137
Multiple studies since the mid-1990s identify GER, rather than spastic motility disorders, as the most common esophageal cause of noncardiac chest pain.138 The mechanism for GERD-related chest pain is poorly understood and is probably multifactorial, related to H+ ion concentration, volume, and duration of acid reflux; secondary esophageal spasm; and prolonged contractions of the longitudinal muscles.139
Asthma and Other Pulmonary Disorders
The prevalence of GERD in asthmatics is estimated between 34% and 89%, depending on the group of patients studied and how GERD is defined (e.g., symptoms or 24-hour pH monitoring).140 Symptomatic GERD is an important comorbid condition in asthma patients, being associated with greater asthma severity.141 GERD should be considered in asthmatics who present in adulthood, those without an extrinsic (allergic) component, and those not responding to bronchodilators or glucocorticoids.142 Up to 30% of patients with GERD-related asthma have no esophageal complaints. Other pulmonary diseases associated with GERD include aspiration pneumonia, interstitial pulmonary fibrosis, chronic bronchitis, and bronchiectasis. In addition, preliminary data suggest that GER may worsen the course of obstructive sleep apnea in a subset of patients.143
Proposed mechanisms of reflux-induced asthma include aspiration of gastric contents into the lungs with secondary bronchospasm and activation of a vagal reflex from the esophagus to the lungs causing bronchoconstriction. Animal144 and human145 studies report bronchoconstriction after esophageal acidification, but the response is mild and inconsistent. On the other hand, intratracheal infusion of even small amounts of acid induces profound and reproducible bronchospasm in cats.144 The reflux of acid into the trachea as compared with the esophagus alone predictably caused marked changes in peak expiratory flow rates in asthmatic patients.146 Although both mechanisms may trigger reflux-induced asthma, patients with severe asthma probably suffer from intermittent microaspiration.
Ear, Nose, and Throat Diseases
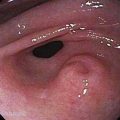
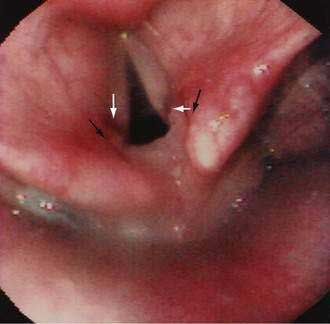
GERD may be associated with a variety of laryngeal symptoms and signs, of which “reflux laryngitis” is the most common.147,148 These patients present with hoarseness, globus sensation, frequent throat clearing, recurrent sore throat, and prolonged voice warm-up. Ear, nose, and throat signs attributed to GERD include posterior laryngitis with edema and redness (Fig. 43-9), vocal cord ulcers and granulomas, leukoplakia, and even carcinoma. These changes are usually limited to the posterior third of the vocal cords and interarytenoid areas, both in proximity to the upper esophageal sphincter. Animal studies find that the combination of acid, pepsin, and conjugated bile acids is very injurious to the larynx.149 Human studies report that proximal esophageal acid exposure, especially while sleeping, is significantly increased in patients with laryngeal symptoms and signs.150
GERD has been postulated to be a leading cause of chronic cough (after sinus problems and asthma).151 GER increases the cough sensitivity reflex (i.e., reduces the cough threshold) in patients with chronic cough.152 However, the importance of this association in humans has not been shown in treatment studies, which, in sum, have not demonstrated a superiority of proton pump inhibitors over placebo.153 Dental erosion, the loss of tooth structure by nonbacterial chemical processes, can be caused by GER in healthy subjects and patients with bulimia.154 Microaspiration of gastric contents is the most likely etiology of these complaints.
ASSOCIATED CONDITIONS
Several medical and surgical conditions discussed elsewhere in this book can predispose to GERD. The most common is pregnancy, in which 30% to 80% of women complain of heartburn, especially in the first trimester (see Chapter 38). Pregnancy increases the risk for reflux by reducing LES pressure due to the effects of estrogen and progesterone and possibly mechanical factors from the gravid uterus.155 Although symptoms may be severe, esophagitis is uncommon and this type of situational GERD is cured with childbirth. Up to 90% of patients with scleroderma have GERD due to smooth muscle fibrosis causing low LES pressure and weak or absent peristalsis (see Chapter 35). Severe disease is common, with up to 70% of patients having esophagitis. Many patients have peptic strictures, and Barrett’s esophagus and carcinoma of the esophagus have been reported.156 Acid hypersecretion and increased gastric volume are the major factors causing GERD in patients with the Zollinger-Ellison syndrome (see Chapter 32). In these patients, the esophagitis and complications are more difficult to treat than the ulcer disease.157 After Heller myotomy for achalasia, 10% to 20% of patients may develop GERD158 (see Chapter 42). In patients undergoing laparoscopic gastric banding for morbid obesity, de novo symptoms of GERD may develop postoperatively (see Chapter 7).30 Finally, prolonged nasogastric intubation may cause reflux esophagitis, in part because acid tracks orad along the tube and because the tube mechanically interferes with the LES barrier function.159
DIAGNOSIS
A large number of tests are available for evaluating patients with suspected GERD. Many times these tests are unnecessary because the classic symptoms of heartburn and acid regurgitation are sufficiently specific to identify reflux disease and begin medical treatment. However, this is not always the case, and clinicians must decide which tests to choose so as to make a diagnosis in a reliable, timely, and cost-effective manner depending on the information desired (Table 43-2).160
Table 43-2 Diagnostic Tests for Gastroesophageal Reflux Disease
EMPIRICAL TRIAL OF ACID SUPPRESSION
In empirical trials for heartburn, the initial PPI dose was high (e.g., omeprazole 40 to 80 mg/day), usually given for at least two weeks, and a positive response was defined as at least 50% improvement in heartburn. Using this approach, the PPI empirical trial has a sensitivity of 68% to 83% for determining the presence of GERD.161,162 In noncardiac chest pain, Fass and colleagues163 found that a seven-day trial of omeprazole 40 mg in the morning and 20 mg at night had a sensitivity of 78% and specificity of 86% for predicting GERD when compared with traditional tests. Likewise, Ours and colleagues164 found omeprazole 40 mg twice daily for two weeks to be a very reliable method for identifying acid-related cough.
An empirical PPI trial for diagnosing GERD offers many advantages: the test is office based, easily done, relatively inexpensive (especially with use of over-the-counter [OTC] PPIs), available to all physicians, and avoids many needless procedures. For example, Fass and colleagues163 showed a savings of greater than $570 per patient due to a 59% reduction in the number of diagnostic tests performed for noncardiac chest pain. Disadvantages are few, including a placebo response and uncertain symptomatic endpoint if symptoms do not totally resolve with extended treatment.
ENDOSCOPY
Upper endoscopy is the standard for documenting the presence and extent of esophagitis and excluding other etiologies for the patient’s symptoms. However, only 20% to 60% of patients with abnormal esophageal reflux by pH testing have esophagitis at endoscopy. Thus, the sensitivity of endoscopy for GERD is poor, but it has excellent specificity at 90% to 95%.165
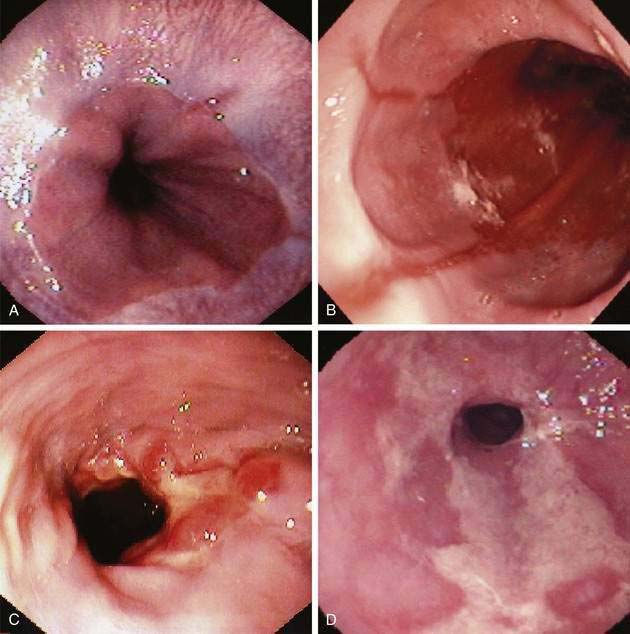
The earliest endoscopic signs of acid reflux include edema and erythema, but these findings are nonspecific and dependent on the quality of endoscopic visual images.166 More reliable signs are friability, granularity, and red streaks. Friability (easy bleeding) results from the development of enlarged capillaries near the mucosal surface in response to acid. Red streaks extend upward from the esophageal junction along the ridges of the esophageal folds.167 Erosions develop with progressive acid injury, characterized by a shallow break in the mucosa with a white or yellow exudate surrounded by erythema. Typically, erosions begin at the gastroesophageal junction, occurring along the tops of esophageal mucosal folds, where acid injury is most prone, and they may be single or multiple. Erosions can also be caused by nonsteroidal anti-inflammatory drugs, heavy smoking, and infectious esophagitis.165 Ulcers reflect more severe esophageal damage, being deeper into the mucosa or submucosa and either isolated along a fold or surrounding the esophageal junction. Multiple classification systems for esophagitis have been proposed; some are confusing and none has worldwide acceptance. In Europe the most popular scheme is the Savary-Miller classification.168 The most thoroughly evaluated esophagitis classification is the Los Angeles (LA) system, which is gaining acceptance in the United States and Europe (Table 43-3) (Fig. 43-10A to D).169
| Savary-Miller Classification | |
| Grade 0 | Not applicable |
| Grade I | Single, erosive, or exudative lesion on one longitudinal fold |
| Grade II | Multiple erosions on more than one longitudinal fold |
| Grade III | Circumferential erosions |
| Grade IV | Ulcer, stricture, or short esophagus, isolated or associated with grades I through III |
| Grade V | Barrett’s esophagus ± grades I through III |
| Los Angeles Classification | |
| Grade A | One or more mucosal breaks confined to folds, ≤5 mm |
| Grade B | One or more mucosal breaks >5 mm confined to folds but not continuous between tops of mucosal folds |
| Grade C | Mucosal breaks continuous between tops of two or more mucosal folds but not circumferential |
| Grade D | Circumferential mucosal break |
Esophageal capsule endoscopy for the evaluation of reflux symptoms has thus far been disappointing. The capsule is 11 by 26 mm and acquires video images at 14 frames per second. After swallowing images are transmitted to a portable receiver via digital radiofrequency. In one study, compared with standard upper endoscopy, the capsule has a sensitivity of 50% for erosive esophagitis, 54% for the presence of a hiatal hernia, and 79% for the presence of Barrett’s esophagus.170
As mentioned, most patients with GERD are treated initially with PPIs and without endoscopy. The important exception is the patient experiencing “alarm” symptoms: dysphagia, odynophagia, weight loss, and gastrointestinal bleeding. Here endoscopy should be performed early to diagnose complications of GERD (e.g., strictures) and to rule out other entities such as infections, ulcers, cancer, or varices. Current guidelines suggest the major role of endoscopy is to diagnose and treat GERD complications, especially peptic strictures, and to define Barrett’s esophagus.171 Using this rationale, the majority of patients with chronic GERD need only one endoscopy while on therapy.
ESOPHAGEAL BIOPSY
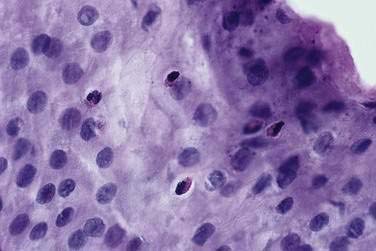
Like endoscopy, the role of esophageal biopsies in evaluating GERD has evolved over the years. Microscopic changes of reflux may occur even when the mucosa endoscopically appears normal.172 These classic changes of basal cell hyperplasia and increased height of the rete peg, both representing increased epithelial turnover of the squamous mucosa, are sensitive but not specific histologic findings for GERD.173 Acute inflammation characterized by the presence of neutrophils and often eosinophils (Fig. 43-11) is very specific for esophagitis; however, the sensitivity is low, in the range of 15% to 40%.174 Thus, there is little value for histologic examination of normal-appearing squamous mucosa to either confirm or exclude pathologic acid reflux.175 In patients with classic esophagitis, biopsies are usually not taken unless necessary to exclude neoplasm, infection, pill injury, or bullous skin disease. Therefore, the current primary indication for esophageal biopsies is to determine the presence of Barrett’s epithelium.171 When this diagnosis is suspected, biopsies are mandatory and best done when esophagitis is healed (see Chapter 44).
ESOPHAGEAL pH MONITORING
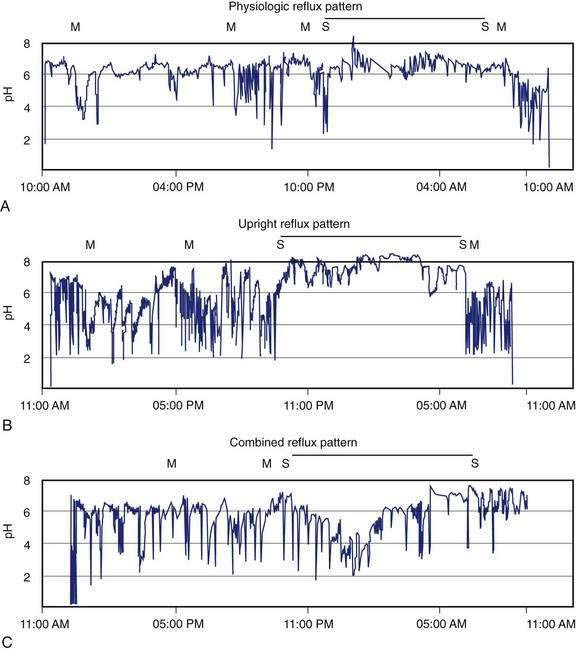
Ambulatory intraesophageal pH monitoring is the standard test for establishing pathologic reflux.176,177 For catheter-based pH testing, the probe is passed nasally, positioned 5 cm above the manometrically determined LES, and connected to a battery-powered data logger capable of collecting pH values every four to six seconds. An event marker is activated by the patient when symptoms, meals, and body position changes occur. Patients are encouraged to eat normally and engage in regular daily activities, with monitoring carried out for 18 to 24 hours. Reflux episodes are defined by a pH drop of less than 4. Conventionally measured parameters include percent of total time when pH is less than 4; percent of time, upright and supine when pH is less than 4; total number of reflux episodes; duration of longest reflux episode; and number of episodes greater than five minutes.176 The percent of total time pH is less than 4 is the most reproducible measurement for GERD, with reported upper limits of normal ranging from 4% to 5.5%.176 Ambulatory pH testing discerns positional variations in GER, meals, and sleep-related episodes and helps relate symptoms to reflux events (Fig. 43-12).
A critical limitation of esophageal pH monitoring is that there exists no absolute threshold value that reliably identifies GERD patients. Studies comparing patients with endoscopic esophagitis who underwent pH tests report sensitivities from 77% to 100% with specificities from 85% to 100%.178 However, these patients with esophagitis rarely need pH testing; rather, patients with normal endoscopy and suspected GERD might benefit most from this test. Unfortunately, data on these patients are less conclusive, with considerable overlap between controls and nonerosive refluxers.177 Other drawbacks of pH testing include possible equipment failure, pH probe missing reflux events because the probe is buried in a mucosal fold, and false-negative studies due to dietary or activity limitations from poor tolerability of the nasal probe.178
A major advance in esophageal pH testing has been the development of a catheter-free system.179 This system uses a wireless pH capsule that is affixed to the esophageal mucosa with a delivery system that drives a small needle into the epithelium. The capsule then transmits pH data to a portable receiver using radiofrequency signals. Catheter-free testing is rapidly becoming the preferred method of pH testing because monitoring can be extended beyond 24 hours and limitations on normal daily activities and meals are negligible.179
Ambulatory esophageal pH monitoring is the only test that records and correlates symptoms with reflux episodes over extended periods of time. However, because only 10% to 20% of reflux episodes are associated with symptoms, different statistical analyses have evolved, attempting to define a significant association between symptoms and reflux episodes, including the symptom index, symptom sensitivity index, and symptom association probability.146 Unfortunately, no studies have defined the accuracy of these symptom scores in predicting response to therapy. Therefore, pH testing can define an association between complaints and GER, but only treatment trials address the critical clinical issue of causality.
Clinical indications for ambulatory pH monitoring are established.171 Before fundoplication, pH testing should be done in patients with normal endoscopy to ensure the presence of pathologic acid reflux. After antireflux surgery, persistent or recurrent symptoms warrant repeat pH testing. In these situations, pH monitoring is performed with the patient off antireflux medications. Esophageal pH testing is particularly helpful in evaluating patients with reflux symptoms who are resistant to treatment and who have normal or equivocal endoscopic findings. For this indication, pH testing is usually done on PPI therapy to define two populations: those with and those without continued abnormal acid exposure times. The group with persistent GER needs intensified medical therapy, whereas patients with symptoms and good acid control have another etiology for their complaints. Finally, ambulatory pH testing may help in defining patients with extraesophageal manifestations of GERD. In this situation, pH testing is often done with additional pH probes in the proximal esophagus or pharynx.147 Initially most of these studies were done off antireflux medications to confirm the coexistence of GERD; however, this does not guarantee symptom causality. Therefore, one approach is to first treat aggressively with PPIs, reserving pH testing for those patients not responding after 4 to 12 weeks of therapy.164
A relatively new method of evaluating GERD has been combined impedance and acid testing, which allows the measurement of acid and nonacid (volume) reflux. Nonacid reflux is measured by the detection of a retrograde bolus of ion-rich fluid in the esophagus. Refluxates that are a mixture of liquid and air are also readily detected. In a large group of normal subjects, roughly 40% of reflux episodes were either weakly acidic (pH 4 to 6.5) or alkaline (nadir esophageal pH during episode >6.5).180 In a multicenter study using impedance, 37% of patients experienced continued reflux symptoms despite twice-daily PPI therapy due to nonacid reflux.181 These patients would have been interpreted as negative for reflux had they been studied using conventional pH only. Another study using 24-hour ambulatory pH impedance found a temporal relationship between symptoms and nonacid reflux in 4.1% and 16.7% of subjects off and on PPI therapy, respectively.182 Regurgitation and cough were the most prevalent symptoms associated with nonacid reflux.
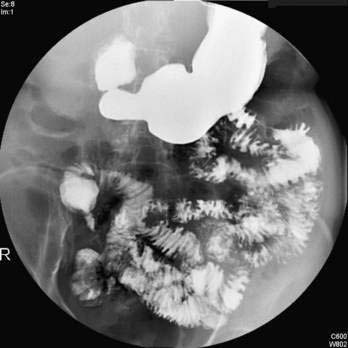
BARIUM ESOPHAGOGRAM
The barium esophagogram is an inexpensive, readily available, and noninvasive esophageal test. It is most useful in demonstrating anatomic narrowing of the esophagus and assessing the presence and reducibility of a hiatal hernia. Schatzki’s rings, webs, or minimally narrowed peptic strictures may only be seen with an esophagogram, being missed by endoscopy, which may not adequately distend the esophagus. Giving a 13-mm radiopaque pill or marshmallow along with the barium liquid can help to identify these subtle narrowings.183 The barium esophagogram allows good assessment of peristalsis and is helpful preoperatively in identifying a weak esophageal pump.184 The ability of barium esophagogram to detect esophagitis varies, with sensitivities of 79% to 100% for moderate to severe esophagitis, whereas mild esophagitis is usually missed.185,186 Barium testing also falls short when addressing the presence of Barrett’s esophagus. The spontaneous reflux of barium into the proximal esophagus is very specific for reflux, but it is not sensitive. Provocative maneuvers (e.g., leg lifting, coughing, Valsalva, or water siphon) can elicit stress reflux and improve the sensitivity of the barium esophagogram, but some argue that these maneuvers also decrease its specificity.185,186
ESOPHAGEAL MANOMETRY
Esophageal manometry allows assessment of LES pressure and relaxation, as well as peristaltic activity, including contraction amplitude, duration, and velocity. However, esophageal manometry is generally not indicated in the evaluation of the uncomplicated GERD patient because most have a normal resting LES pressure.51 (It is an integral component of pH testing to accurately define LES location; see earlier.) Esophageal manometry to document adequate esophageal peristalsis is traditionally recommended before antireflux surgery.187 If the study identifies ineffective peristalsis (low amplitude or frequent failed peristalsis),188 then a complete fundoplication may be contraindicated. However, this assumption has recently been challenged by several studies finding that reflux control was better and dysphagia no more common in patients with weak peristalsis after a complete, as opposed to a partial, fundoplication.189 An improvement of traditional manometry, combining it with impedance testing, is helping to clarify this controversy. Using this technique, a study found that less than 50% of patients with ineffective peristalsis had a significant delay in esophageal bolus transit measured by impedance.190 Therefore, potentially only these patients with a significant physiologic defect in motility will require a modified fundoplication.
CLINICAL COURSE
The clinical course of GERD depends to a great extent on whether the patient has erosive or nonerosive disease. There is controversy as to whether GERD exists as a spectrum of disease severity or as a categorical disease in three distinct groups, including Barrett’s esophagus. Patients tend not to cross over from one group to another; in follow-ups ranging from six months to longer than 22 years, less than 25% of patients with nonerosive disease evolved over time to having esophagitis, nearly all to LA grade A/B disease, or to having complications of GERD.191–194
NONEROSIVE REFLUX DISEASE
Early studies from tertiary referral centers suggested that the majority of GERD patients had esophagitis.195 However, studies carried out in community practices reveal that up to 70% of GERD patients had a normal endoscopic examination.196–198 Endoscopy-negative GERD patients are more likely to be female, younger, thin, and without hiatal hernia, and they have a higher prevalence of functional GI disorders.199 Despite their mild mucosal damage, these patients demonstrate a chronic pattern of symptoms with periods of exacerbation and remission.200 Nonerosive GERD is suspected in the patient with typical reflux symptoms and a normal endoscopy and confirmed by the patient’s response to antisecretory therapy. Esophageal pH testing identifies three distinct subsets of nonerosive GERD patients. First are the patients with excessive acid reflux who usually respond to PPI therapy. Second are the patients with normal reflux parameters but a good correlation between symptoms and acid reflux episodes. This group represents 30% to 50% of nonerosive GERD patients and has “functional heartburn.”200 These patients probably have heightened esophageal sensitivity to acid and are less likely to respond to antireflux therapy.201 The third group is characterized by normal acid exposure times and poor symptom correlation. Whether they truly represent a subset of nonerosive GERD is questionable.
EROSIVE REFLUX DISEASE
Patients with erosive esophagitis tend to be male, older, and overweight and are more likely to have hiatal hernias.199 The clinical course of these patients with erosive esophagitis is more predictable and associated with complications of GERD. Longitudinal studies have shown that up to 85% of patients with erosive GERD, on no maintenance reflux therapy, will relapse within six months of stopping PPI therapy, and the relapse rate is highest in patients with more severe grades of esophagitis (see Table 43-3).202,203 Several studies confirm that erosive esophagitis patients are prone to reflux complications, including ulcers, strictures, and Barrett’s esophagus. In a Finnish study, 20 patients with erosive GERD treated with lifestyle changes, antacids, and prokinetic drugs were followed for a median of 19 years. Fourteen patients continued to have erosions, and six new cases of Barrett’s esophagus were detected.192 In another more recent European study193 over two years, patients with LA grade C/D esophagitis developed Barrett’s esophagus at a rate of 5.8% compared with only 1.4% for LA grade A/B and 0.5% for nonerosive GERD. However, these data must be contrasted with a two-year U.S. trial in which no patient with erosive esophagitis developed Barrett’s esophagus204 and in another study in which stricture was reported in only 0.9% of 957 patients over 7.6 years of symptom driven antireflux treatment.194
COMPLICATIONS
HEMORRHAGE, ULCERS, AND PERFORATION
GERD-related non-cancer deaths are rare (0.46 per 100,000 persons). The most common fatal causes are hemorrhagic esophagitis, aspiration pneumonia, ulcer perforation, and rupture with severe esophagitis.205 Major hemorrhage and esophageal perforation are usually associated with deep esophageal ulcers or severe esophagitis.206 Esophageal perforations are very rare in the PPI era but can result in mediastinitis and death. Clinically important hemorrhage has been reported in 7% to 18% of GERD patients207 and may result in iron deficiency anemia.
PEPTIC ESOPHAGEAL STRICTURES
Strictures occur in 7% to 23% of patients with untreated reflux esophagitis, and are especially seen in older men.208 They may be linked to chronic nonsteroidal anti-inflammatory drug (NSAID) use.209 Stricture formation is complex, starting as reversible inflammation with edema, cellular infiltration, and vascular congestion, progressing to collagen deposition and ending in irreversible fibrosis. As dysphagia progresses, heartburn often decreases, reflecting the stricture acting as a barrier to further reflux. Dysphagia is usually limited to solids. Unlike malignant strictures, patients with peptic strictures have a good appetite, alter their diet, and lose little weight.
Peptic strictures are smooth-walled, tapered, circumferential narrowings in the lower esophagus, usually less than 1 cm long but occasionally extend to 8 cm (Fig. 43-13). In these unusual cases, the clinician should suspect a predisposing condition, such as Zollinger-Ellison syndrome, or another condition such as pill esophagitis or a stricture from prolonged nasogastric intubation.208 A mid- to upper esophageal stricture should raise concern for Barrett’s esophagus or malignancy. Although once controversial, today a Schatzki’s ring is considered a forme fruste of an early peptic stricture.210 All stricture patients should undergo endoscopy, at least initially, to confirm the benign nature of the lesion and, if necessary, take biopsies to exclude cancer and Barrett’s esophagus.
TREATMENT OF UNCOMPLICATED DISEASE
NONPRESCRIPTION THERAPIES
Although GERD is common, many sufferers do not seek medical care, instead choosing to change their lifestyles and self-medicate with OTC antacids, histamine-2 receptor antagonists (H2RAs), and even PPIs. These observations have led to the “iceberg” model of the GERD population. The vast majority of heartburn suffers are invisible because they self-medicate and do not seek professional help; only those at the tip of the iceberg, typically patients with severe symptoms or reflux complications, are seen by physicians.211
Lifestyle Modifications
Selective lifestyle changes, carefully explained to the patient, should be part of the initial management plan and are especially helpful in those with mild, intermittent complaints. These include elevating the head of the bed, avoiding tight-fitting clothes, losing weight if overweight, restricting alcohol and smoking, making dietary changes, refraining from lying down after meals, and avoiding bedtime snacks. Physiologic studies show that these maneuvers enhance esophageal acid clearance, decrease acid reflux–related events, or ease heartburn symptoms.212 Head-of-the-bed elevation can be done by using 6- to 8-inch blocks or a foam wedge under the mattress to elevate the upper torso. Eating several hours before retiring and avoiding bedtime snacks keeps the stomach empty at night, thereby decreasing nocturnal reflux episodes. Avoiding tight-fitting clothes and losing weight are interventions aimed at reducing the incidence of reflux by the “abdominal stress” mechanism. Targeted weight loss may be helpful, whereas discrete periods of weight gain can be associated with exacerbation of reflux symptoms.30 Cessation of smoking and alcohol reduction is valuable because both agents lower LES pressure, reduce acid clearance, and impair intrinsic squamous epithelial protective functions.76,211 Reducing meal size and avoiding fats, carminatives, and chocolate reduces reflux frequency by decreasing episodes of tLESRs, as well lowering LES pressure.211 Additionally, some patients complain of heartburn after citrus drinks, spicy foods, tomato-based products, coffee, tea, or cola drinks. Stimulation of gastric acid secretion or esophageal sensitivity to low pH (or perhaps hyperosmolar solutions) may account for these symptoms.213 However, indiscriminate food prohibition should be avoided but rather tailored to individual sensitivity to better promote compliance. Finally, patients should avoid, if possible, drugs that lower LES pressure (see Table 43-1) or promote localized esophagitis, such as certain bisphosphonates (see Chapter 45).
How good are the clinical studies assessing the efficacy of these commonly prescribed lifestyle changes? In an evidence-based review,211 studies of smoking, alcohol, chocolate, fatty foods, and citrus products had sound physiologic data that their intake can adversely effect symptoms or promote reflux on esophageal pH tests. However, there was little convincing evidence that cessation of these products predictably improved reflux symptoms. Only elevation of the head of the bed, left lateral decubitus positioning, and weight loss were associated with GERD improvement in case-controlled studies.211
Over-the-Counter Medications
These drugs are used in treating mild, infrequent heartburn symptoms triggered by lifestyle indiscretions. Antacids increase LES pressure but work primarily by buffering gastric acid, albeit for short periods. Heartburn symptoms are rapidly relieved, but patients need to take antacids frequently, usually 1 to 3 hours after meals. Gaviscon, containing alginic acid and antacids, mixes with saliva to form a highly viscous solution that floats on the gastric pool, acting as a mechanical barrier. Both antacids214 and Gaviscon215 are more effective than placebo in relieving symptoms induced by a heartburn-promoting meal. However, they do not heal esophagitis, and long-term trials suggest symptom relief in only 20% of patients.216,217 OTC H2RAs are available at doses usually one half the standard prescription dose. Although onset of relief is not as rapid as antacids, the OTC H2RAs relieve symptoms for 6 to 10 hours.218 Therefore, they are particularly useful when taken before potentially refluxogenic activities. Like antacids, OTC H2RAs are ineffective in healing esophagitis.218 OTC combinations of antacids and H2RAs are available.
The long-term safety and efficacy of PPIs led the U.S. Food and Drug Administration (FDA) to approve omeprazole at full dose (20 mg) for OTC use in 2003. Drug labeling suggested daily use for only two weeks and recommended physician follow-up for persistent symptoms. Despite initial “real world” concerns of abusing this drug, early actual-use data support that consumers accurately self-select if OTC omeprazole is appropriate for use, comply with a two-week regimen, and seek physician care for longer-term management of frequent heartburn.219
PRESCRIPTION MEDICATIONS
Prokinetic Drugs
Until recently three prokinetic drugs were available for treating GERD: bethanechol, a cholinergic agonist; metoclopramide, a dopamine antagonist; and cisapride, a serotonin (5-HT4) receptor agonist that increases acetylcholine release in the myenteric plexus. These drugs improve reflux symptoms by increasing LES pressure, acid clearance, or gastric emptying. However, none alters tLESRs, and their effectiveness decreases with disease severity.220 Current prokinetics provide modest benefit in controlling heartburn but have unreliable efficacy in healing esophagitis unless combined with acid-inhibiting drugs.220
Prokinetic drugs are limited by their side effect profiles. Bethanechol commonly causes flushing, blurred vision, headaches, abdominal cramps, and urinary frequency. Metoclopramide, which crosses the blood-brain barrier, has a 20% to 50% incidence of fatigue, lethargy, anxiety, and restlessness and rarely causes tremor, parkinsonism, dystonia, or tardive dyskinesia, especially in older patients. Side effects may be decreased by reducing the dosing regimen to twice a day, taking a larger single dose before dinner or at bedtime, or using a sustained release tablet. Domperidone, another dopamine antagonist not crossing the blood-brain barrier, has fewer side effects but is not available in the United States. Cisapride was the best prokinetic drug for treating GERD but was withdrawn from the U.S. market because of reports of serious cardiac dysrhythmias (ventricular tachycardia, ventricular fibrillation, torsades de pointes, and QT prolongation) with associated cardiac arrest and deaths related to possible drug interactions.221
Regulating the frequency of tLESRs is an attractive target for GERD treatment because of its pivotal role in most reflux episodes. Potential agents to modify this vagovagal reflex include atropine, morphine, CCKA (CCK-1) receptor antagonists, NO synthase inhibitors, and γ-aminobutyric acid (GABAB) agonists.222 Only the latter category of drug, baclofen, has been extensively studied in humans and found to be a potent inhibitory neurotransmitter in the central nervous system, antagonizing the release of neurotransmitters from vagal nerve afferents. The frequency of tLESRs, especially after meals, is decreased, reducing exposure time for acid and duodenal reflux. This correlates with improvement of acute and chronic symptoms in GERD patients.223,224 Baclofen needs to be titrated upward slowly (5 mg three or four times daily initially and increased as needed over 10 days to 40 to 60 mg per day). Side effects including drowsiness, nausea, and lowering of the threshold for seizures require discontinuation in up to 20% of patients. Potential applications could be in patients with nonerosive GERD or as adjunct therapy in patients with persistent symptoms on PPIs possibly related to nonacid duodenal reflux.224
Histamine-2 Receptor Antagonists (H2RAs) (see also Chapter 53)
These drugs (cimetidine, ranitidine, famotidine, and nizatidine) are more effective in controlling nocturnal, as compared with meal-related, acid secretion because the parietal cell is stimulated postprandially by gastrin acting via histamine and by acetylcholine (see Chapter 49).225 The four H2RAs are equally effective when used in proper doses, usually twice a day before meals. GERD trials find that heartburn can be significantly decreased by H2RAs, when compared with placebo, although symptoms are rarely abolished (Fig. 43-14). A comprehensive meta-analysis found that the overall esophagitis healing rates with H2RAs rarely exceeded 60% after up to 12 weeks of treatment, even when higher doses were used.226,227 Healing rates differ in individual trials depending primarily on the severity of esophagitis being treated: grades I and II esophagitis heal in 60% to 90% of patients, whereas grades III and IV heal in only 30% to 50% despite high-dose regimens.227
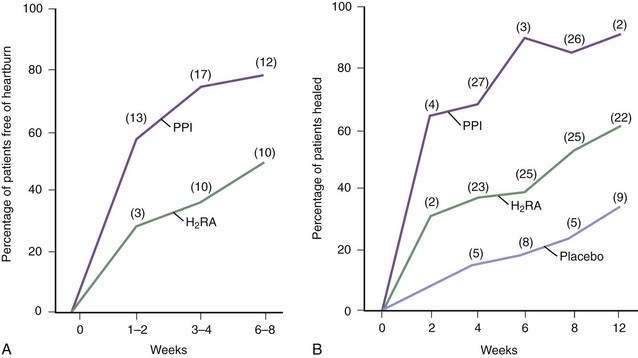
Although PPIs are more effective than H2RAs (Fig. 43-15; discussed following), nocturnal gastric acid breakthrough while on PPI therapy may cause reflux symptoms in some patients. H2RAs given at bedtime successfully eliminated this problem in one study, suggesting a new indication for H2RAs in the PPI era.228 However, this study used only a single evening dose and did not account for the tolerance that frequently develops to H2RAs over weeks to months.229 This tolerance impairs the effectiveness of chronic nocturnal dosing of H2RAs to eliminate nocturnal acid breakthrough,230 but suggests a useful role in as-needed medications in situations in which lifestyle indiscretions may promote nocturnal complaints.
The H2RAs are very safe with a side effect rate (most of which are minor and reversible) of about 4%.225 Serum concentrations of phenytoin, procainamide, theophylline, and warfarin are higher after the administration of cimetidine and, to a lesser degree, ranitidine, whereas these interactions are not reported with the other two H2RAs.
Proton Pump Inhibitors (see also Chapter 53)
PPIs inhibit meal-stimulated and nocturnal acid secretion to a significantly greater degree than H2RAs231 but rarely make patients achlorhydric. After oral ingestion, acid inhibition is delayed because PPIs need to accumulate in the parietal cell secretory canaliculus to bind irreversibly to actively secreting proton pumps.232 Therefore, the slower a PPI is cleared from plasma, the more it is available for delivery to the proton pumps. PPIs should be taken before the first meal of the day, when most proton pumps become active. Because not all pumps are active at any given time, a single PPI dose will not inhibit all pumps. A second dose, if necessary, can be taken before the evening meal.
PPIs (omeprazole, lansoprazole, rabeprazole, pantoprazole, and esomeprazole) have superior efficacy compared with H2RAs on the basis of their ability to maintain an intragastric pH greater than 4 from 10 to 14 hours daily compared with approximately 6 to 8 hours daily with the H2RAs.233,234 PPIs are superior to H2RAs in completely relieving heartburn symptoms in patients with severe GERD, usually within one to two weeks (see Fig. 43-15A).227 PPI therapy has been shown in a Cochrane meta-analysis to be superior to placebo and H2RAs in nonerosive GERD and for undiagnosed reflux symptoms in primary care, although the effect is 20% to 30% lower than in patients with esophagitis.235 Controlled studies and a large meta-analysis report complete healing of even severe ulcerative esophagitis after eight weeks in more than 80% of patients taking PPIs compared with 51% on H2RAs and 28% receiving placebo (see Fig. 43-15B).227,236–239 In another recent Cochrane review involving 4064 patients in 26 trials,240 PPIs were superior to H2RAs in healing esophagitis at four to eight weeks (risk ratio, 0.47) with a number-to-treat of 3. In patients not healing initially, prolonged therapy with the same dose or an increased PPI dose usually resulted in 100% healing.241 Until recently, therapeutic efficacy among PPIs was similar. However, large studies have found the newest PPI esomeprazole 40 mg superior to omeprazole 20 mg and to lansoprazole 30 mg in healing esophagitis.242,243 A meta-analysis of 10 randomized clinical trials244 comparing esomeprazole to all other PPIs found the therapeutic advantage is minimal with LA grade A/B esophagitis (number-to-treat 50 and 33, respectively), and greater with severe LA grade C/D esophagitis (number-to-treat 14 and 8, respectively). This superiority is related to higher systemic bioavailability and less interpatient variability with esomeprazole. Several PPIs are available in the United States for intravenous use.245
PPIs are well tolerated, with headaches and diarrhea described as the most common side effects. Increased fasting serum gastrin levels are reported with all the PPIs, but the elevations generally do not exceed the normal range for gastrin and return to normal values within one to four weeks of drug discontinuation. Omeprazole decreases the clearance of diazepam and warfarin due to competition for the cytochrome P-450 isoenzyme P2C19.246 The four newer PPIs have minimal or no important drug-drug interactions.
MAINTENANCE THERAPIES
GERD may be a chronic relapsing disease, especially in patients with low LES pressure, severe grades of esophagitis, and difficult-to-manage symptoms.217 After esophagitis is healed, recurrence within six months of stopping medication occurs in more than 80% of patients with severe esophagitis and in 15% to 30% of those with milder esophagitis.202,247
Cochrane reviews have identified the superiority of PPIs over H2RAs in maintaining the remission of esophagitis over 6 to 12 months.248 Among 10 randomized trials, the relapse rate for esophagitis was 22% on PPIs versus 58% with H2RAs, with a number-to-treat of 2.5. The FDA has approved all the PPIs, sometimes at one half the acute dose, for maintenance therapy, but only ranitidine 150 mg twice a day among the H2RAs has maintenance indications for mild esophagitis. Many clinicians now place their patients with severe disease (daily symptoms, severe esophagitis, or complications) on chronic PPI therapy indefinitely. The efficacy of this approach is supported by open, compassionate use data primarily from the Netherlands and Australia.249 In a study of 230 patients with severe esophagitis healed with 40 mg omeprazole, all subjects remained in remission for up to 11 years. More than 60% were maintained on omeprazole 20 mg a day, whereas higher doses of 60 mg or more were necessary in only 12% of patients, confirming a lack of tolerance to PPIs. Relapses were rare (one per 9.4 years of follow-up), strictures did not occur, and Barrett’s esophagus did not progress.245
Although PPIs offer the best symptom relief and esophagitis healing, many patients do well long term on lesser treatments after having their complaints initially alleviated with PPIs. Using this “step-down approach,” a recent Veterans Affairs study reported that 58% of 71 patients on chronic PPIs could be switched to H2RAs and/or prokinetics or taken off medication completely.250 Younger age and severe heartburn symptoms predicted PPI requirement. Overall, this approach saved money for the health care system. A similar study by the same investigators found that 80% of patients using multiple-dose PPIs could be stepped down to single-dose PPI, remaining symptom free for six months with considerable cost savings.251 Hence the adage “once on a PPI, always on a PPI” is not true.
The initial safety concerns with the PPIs was the potential to develop stomach or other cancers with long-term use. This was initiated by reports of omeprazole producing hypergastrinemia and gastric carcinoid tumors in rats, changes also subsequently demonstrated with chronic ranitidine therapy and subtotal resection of the gastric fundus.252 However, rats have a high density of enterochromaffin-like (ECL) cells and an exaggerated gastrin response to achlorhydria; chronic omeprazole therapy in other species with lower densities of ECL cells (mice, dog, man) has not caused carcinoid tumors. Another review found no evidence of an increased risk of colon cancer with chronic PPI use.253
Fundic gland polyps are the most common gastric polyp found at endoscopy. Their association with chronic PPI use has been a topic of debate since these drugs were first described. A recent study evaluated 599 patients of whom 322 used PPIs and 107 had fundic gland polyps.254 Long-term PPI use was associated with up to a four-fold increase in the risk of fundic gland polyps. Low-grade dysplasia was found in one fundic gland polyp. Etiologically, these polyps seem to arise because of parietal cell hyperplasia and parietal cell protrusions resulting from acid suppression.
Recent studies confirm that chronic acid suppression may be associated with an increased risk of community-acquired pneumonias and enteric infections. In a large Scandinavian population-based study,255 the adjusted relative risk for pneumonia among current PPI users, compared with those who stopped using PPIs, was 1.89. Current users of H2RAs had a 1.63-fold increased risk of pneumonia compared with those who stopped. A significant positive dose-response relationship was observed in the PPI users. Likewise, a recent systematic review found an increased risk of enteric infections with acid suppression.256 The correlation was stronger with Salmonella, Campylobacter, and other enteric infections, compared with Clostridium difficile, and greater with PPI compared with H2RA therapy.
Chronic high-dose use of PPIs may also affect the absorption of calcium and vitamin B12. A nested case-controlled study from the United Kingdom among 13,556 patients found that the risk of hip fractures increased with chronic PPI use over one year (adjusted odds ratio, 1.44), especially in those patients receiving high-dose PPIs (adjusted odds ratio, 2.65). A smaller but still significant risk was observed in chronic H2RA users.257 A large Canadian study258 reached similar conclusions, but found the risk for hip fractures became apparent after five years of treatment (adjusted odds ratio, 1.62) and after seven years for all osteoporotic fractures (adjusted odds ratio, 1.92). The mechanism for this association is unknown and cause-and-effect is not proven as yet. It has been suggested that if PPIs cause osteoporosis, they may interfere with insoluble calcium absorption or possibly inhibit the osteoclastic proton transport system, potentially reducing bone resorption. PPIs could retard the absorption of vitamin B12 by decreasing gastric acidity; reducing the release of cobalamin from dietary protein; or by promoting small bowel bacterial overgrowth, thereby increasing luminal cobalamin consumption. However, cohort and case-control studies have not shown a convincing link between PPI use and vitamin B12 deficiency.259
Finally, a study suggested that patients on long-term omeprazole who are infected with H. pylori develop atrophic gastritis, a precursor to gastric adenocarcinoma, at a more rapid rate than noninfected patients.260 Nevertheless, a subsequent FDA panel determined that the available data were insufficient for recommending screening and treatment of H. pylori infection in patients on long-term PPI therapy.261
SURGICAL THERAPY
Only surgical fundoplication can correct the physiologic factors contributing to GERD and potentially eliminate the need for long-term medications. Antireflux surgery reduces GER by increasing basal LES pressure, decreasing episodes of tLESRs, and inhibiting complete LESR.262 This is done by reducing the hiatal hernia into the abdomen, reconstructing the diaphragmatic hiatus, and reinforcing the LES.263 Before laparoscopic surgery, the three most common operations were the Nissen fundoplication, Belsey Mark IV repair, and Hill posterior gastropexy. Since the introduction of minimally invasive surgery, the two most popular procedures, performed laparoscopically through the abdomen are the Nissen 360-degree fundoplication and the Toupet partial fundoplication (see Fig. 43-15).264 The former is a superior operation with better long-term durability, but it causes more postoperative dysphagia and gas bloat symptoms.265,266 The typical hospital stay is 1 to 2 days, and many patients return to normal activity in 7 to 10 days. Patients with more severe disease and a short esophagus suggested by a large nonreducible hernia, tight stricture, or long-segment Barrett’s esophagus will require a Collis lengthening procedure to create a 3- to 5-cm neoesophagus that allows the fundoplication to be placed in the abdomen under minimal tension.267
The popularity of antireflux surgery has undergone an explosion since the advent of the laparoscopic operation. The numbers of antireflux operations performed in the United States nearly tripled from 11,000 per year in 1985 (open surgery) to a peak of nearly 32,000 in 1999 but have leveled off at around 24,000 cases per year (11 cases per 100,000 adult population).268,269 A systematic review identified six randomized controlled trials involving 449 patients that compared open and laparoscopic fundoplication.270 There was no significant difference in recurrence rates between the procedures, and laparoscopic fundoplication was associated with lower operative morbidity (number-to-treat to prevent complication, 8) and shorter hospital stay.
In the PPI era, symptom resolution on treatment helps predicate the success of antireflux surgery for classic as well as atypical symptoms.271 Antireflux surgery is a reasonable option in (1) the healthy patient with typical or atypical GERD symptoms well controlled on PPIs desiring alternative therapy because of drug expense, poor medication compliance, or fear of unknown long-term side effects; (2) patients with volume regurgitation and aspiration symptoms not controlled on PPIs; and (3) recurrent peptic strictures in younger patients.265 Patients recalcitrant to PPI therapy may well have another etiology for their complaints (e.g., pill esophagitis, gastroparesis, functional heartburn) and should be approached cautiously with surgery.
Testing must be done before antireflux surgery. Endoscopy is necessary to exclude stricture, Barrett’s esophagus, and dysplasia or carcinoma. A barium esophagogram can help define a nonreducible hiatal hernia, a shortened esophagus, and poor esophageal motility. Esophageal manometry, possibly combined with impedance if available, will identify ineffective esophageal peristalsis and previously misdiagnosed achalasia or scleroderma. Twenty-four-hour pH testing is necessary in patients with nonerosive GERD or those with esophagitis not responding to PPI therapy. Gastric analysis and gastric emptying studies may be indicated in select patients. Careful testing will result in modification of the original operation or an alternative diagnosis in approximately 25% of patients.187
Antireflux surgery relieves reflux symptoms and reduces the need for stricture dilation in more than 90% of patients,272 but Barrett’s esophagus rarely regresses and the risk of developing esophageal cancer is unchanged.273 Older studies found antireflux surgery superior to antacids, H2RA, and prokinetic therapy,204 but not PPI therapy, especially when dose titration is permitted.274 Mortality is rare (<1%) after antireflux surgery, but new postoperative complaints occur in up to 25% of patients including dysphagia, gas bloat, diarrhea, and increased flatus.272 Most symptoms improve over 1 year, but persistent complaints suggest too tight a wrap, a displaced fundoplication, or inadvertent damage to the vagus nerve. Successful antireflux surgery does not guarantee a permanent cure. Best surgical results are obtained by experienced surgeons in high-volume centers who report long-term symptom recurrence in only 10% to 15% of patients.265 However, many operations are performed in lower-volume community hospitals where the results are more variable. A study from community hospitals in a midsize Midwest city275 found that 32% of patients had symptom relapses after two years and 7% required repeat surgery. In contrast, a review of more than 3000 patients undergoing antireflux surgery at VA medical centers between 1990 and 2001 was more reassuring.269 Postoperative dysphagia was recorded in 19.4% of patients, 6.4% required esophageal dilations, and a repeat antireflux operations was needed in only 2.3% of patients. There seems to be increasing consensus from long-term follow-up studies that 25% to 62% of patients are back on some type of acid suppressive medication 5 to 15 years after their antireflux surgery.269,275–277 However, the evidence for recurrent GERD by pH testing is infrequent.
Tertiary specialized centers are seeing an increased rate of fundoplication failures. The most common reason for failure are herniation of the intact fundoplication into the chest, “slipped” fundoplication with a recurrent hiatal hernia possibly due to a short esophagus, paraesophageal hernia through an intact fundoplication, too tight a fundoplication, and malpositioned fundoplication usually on the cardia of the stomach. Total breakdown of the fundoplication is now rare.278 Revisional antireflux surgery needs to be performed by very experienced surgeons and can be done laparoscopically, although many prefer an open approach. Reoperation has increased morbidity and mortality compared with the initial operation.
ENDOSCOPIC THERAPY
A variety of endoscopic techniques for the treatment of GERD have been developed as alternatives to antisecretory therapy or antireflux surgery.279–281 These techniques include the delivery of radiofrequency energy to the gastroesophageal junction (Stretta), injection of bulking agents (Enteryx), or implantation of a bioprosthesis (Gatekeeper) into the LES, and suture plication of the proximal gastric folds (EndoCinch endoscopic plication system). Studies to date have primarily enrolled PPI-dependent patients without severe esophagitis or large hiatus hernia.
All of these techniques decrease reflux symptoms, improve quality of life, and decrease the need for antisecretory medications. However, physiologic studies are much less impressive, with LES pressure rarely increasing, pH normalizing in only 30% of patients, and even mild esophagitis infrequently healing.279–281 Controlled studies with Stretta,282 Enteryx,281 and the Plication283 system likewise show a decrease in heartburn symptoms and improved quality of life after the active therapy compared with the sham-treated group after three to six months. Only the Plication study showed a significant decrease in pH values, but only by 18%, whereas the other techniques observed no change in pH or LES parameters.283
Most studies of endoscopic therapy have only limited follow-up information on a relatively small number of patients. The durability of these techniques beyond one to two years remains unclear and seems to gradually decrease over time. The cost-effectiveness of these techniques is difficult to define. Most importantly, safety issues have haunted these procedures, especially when used in the broader community of gastroenterologists. Chest pain, bleeding, esophageal perforations, mediastinitis, and at least eight deaths to date have been attributed to these endoscopic techniques. Serious adverse events, including deaths, led to the voluntary withdrawal of Enteryx by the manufacturer in September 2005 and suspension of the Gatekeeper clinical program in late 2005. A recent AGA Institute medical position statement recommended that “current data suggest that there are no definite indications for endoscopic therapy for GERD at this time.”284
TREATMENT OF COMPLICATIONS
CHEST PAIN AND EXTRAESOPHAGEAL MANIFESTATIONS
The efficacy of acid suppression, especially by PPI treatment, in the extraesophageal presentations of GERD is variable. There are two systemic reviews,285,286 both suggesting that patients with noncardiac chest pain respond to PPIs better than placebo. These reports identified eight randomized controlled trials that assessed 321 patients, with a pooled relative risk for continued chest pain after PPI therapy compared with placebo of 0.54, with a number-to-treat of 3. Systematic reviews and randomized trials, however, do not support the efficacy of aggressive acid suppression, particularly with PPIs, in other extraesophageal disorders, such as chronic cough,287 asthma,288 or ear, nose, and throat disorders.289 Some patients definitely respond, usually over several months, but the heterogeneous etiology of these groups and the lack of pretreatment predictors of response confound any treatment approach to these patients.
Sleep disturbances may occur in up to 75% of patients with GERD, impairing quality of life. In a large multicenter study, patients with GERD-associated sleep disturbances and nighttime heartburn were randomized to two doses of esomeprazole (40 mg and 20 mg) or placebo for four weeks.290 GERD-related sleep disturbances resolved in significantly more patients on esomeprazole 40 mg (73.7%) or 20 mg (73.2%) than those who received placebo (41.2%). These changes were associated with improved sleep quality and daytime productivity.
PEPTIC ESOPHAGEAL STRICTURES
Dysphagia in patients with esophageal strictures and rings is related to stricture diameter and severity of esophagitis.291 When the esophageal lumen diameter is less than 13 mm, dysphagia is common and esophageal dilation is required. Simple short strictures can be dilated by blind peroral passage of rubber Hurst (rounded ends) or Maloney (tapered ends) mercury-filled dilators of increasing sizes (16 to 60 French, 3 French equals 1 mm). Complicated longer, tighter, or more irregular strictures will require bougienage over a guidewire using hollow-centered, Savary, plastic-covered polyvinyl dilators or balloon (Gruentzig) dilators.292 The extensive use of PPIs has markedly impacted our treatment of peptic strictures and esophageal rings. PPIs are superior to H2RAs in relieving the symptoms of heartburn and dysphagia experienced by stricture patients while reducing the frequency of repeat dilations and the cost for treating these patients.293 Several studies in community and veterans hospitals note an approximate 33% decline in the incidence of recurrent strictures. The timeline for this decrease parallels the marked increase in PPI use since 1995.294 Another study convincingly shows that in patients with symptomatic Schatzki’s rings, maintenance PPI therapy after bougienage markedly decreases future relapses of the rings.295 In a randomized study, 30 patients with symptomatic rings without esophagitis were dilated and randomized to placebo or omeprazole 20 mg per day. In the treated group, one patient relapsed after 13 months, whereas seven patients relapsed on placebo after a mean of 20 months.
American Lung Association Asthma Clinical Research CentersMastronarde JG, Anthonisen NR, et al. Efficacy of esomeprazole for treatment of poorly controlled asthma. N Engl J Med. 2009;360:1487-99. (Ref 288.)
Dent J. Microscopic esophageal mucosal injury in nonerosive reflux disease. Clin Gastroenterol Hepatol. 2007;5:4-16. (Ref 172.)
DeVault KR, Castell DO. Updated guidelines for the diagnosis and treatment of gastroesophageal reflux disease. Am J Gastroenterol. 2005;100:190. (Ref 171.)
Falk GW, Fennerty MB, Rothstein RI. AGA Institute technical review on the use of endoscopic therapy for gastroesophageal reflux disease. Gastroenterology. 2006;131:1351-66. (Ref 284.)
Fass R. Erosive esophagitis and non-erosive reflux disease. Comparison of epidemiologic, physiologic and therapeutic characteristics. J Clin Gastroenterol. 2007;41:131-7. (Ref 200.)
Friedenberg FK, Xanthopoulos M, Foster GF, Richter JE. The association between gastroesophageal reflux disease and obesity. Am J Gastroenterol. 2008;103:2111-22. (Ref 30.)
Gralnek IM, Dulai GS, Fennerty MB, Speigel BMR. Esomeprazole versus other PPIs in erosive esophagitis: A meta-analysis of randomized clinical trials. Clin Gastroenterol Hepatol. 2006;4:1452-58. (Ref 244.)
Hirano I, Richter JE. ACG practice guidelines: Esophageal reflux testing. Am J Gastroenterol. 2007;102:668-85. (Ref 176.)
Kaltenback T, Crockett S, Gerson LB. Are lifestyle measures effective in patients with gastroesophageal reflux disease? Am J Gastroenterol. 2006;101:2128-38. (Ref 211.)
Mittal RK, Balaban DH. The esophagogastric junction. N Engl J Med. 1997;336:924-32. (Ref 47.)
Mittal RK, Holloway RH, Penagini R, et al. Transient lower esophageal sphincter relaxation. Gastroenterology. 1995;109:601-610. (Ref 54.)
Moayyedi P, Talley N. Gastroesophageal reflux disease. Lancet. 2006;367:2086-100. (Ref 240.)
Serag HB. Time trends for gastroesophageal reflux disease: A systematic review. Clin Gastroenterol Hepatol. 2007:17-26. (Ref 14.)
van Malenstein H, Farré R, Sifrim D. Esophageal dilated intercellular spaces (DIS) and nonerosive reflux disease. Am J Gastroenterol. 2008;103(4):1021-8. (Ref 86.)
Watson DI. Laparoscopic treatment of gastro-oesophageal reflux disease. Best Pract Res Clin Gastroenterol. 2004;18:19-35. (Ref 265.)
1. A Gallup Organization National Survey. Heartburn across America. Princeton, NJ: The Gallup Organization; 1988.
2. Locke GR, Talley NJ, Fett SL, et al. Prevalence and clinical spectrum of gastroesophageal reflux: A population-based study in Olmsted County, Minnesota. Gastroenterology. 1997;112:1448.
3. Stanghellini V. Three-month prevalence rates of gastrointestinal symptoms and the influence of demographic factors. Results from the Domestic/International Gastroenterology Surveillance Study (DIGEST). Scand J Gastroenterol. 1999;34:20.
4. Spechler SJ. Epidemiology and natural history of gastro-esophageal reflux disease. Digestion. 1992;51:24-9.
5. Johnson DA, Fennerty MB. Heartburn severity underestimates erosive esophagitis severity in elderly patients with gastroesophageal reflux disease. Gastroenterology. 2004;126:660-4.
6. Collen MJ, Abdulian JD, Chen YK. Gastroesophageal disease in the elderly: More severe disease that requires aggressive therapy. Am J Gastroenterol. 1995;90:1053-7.
7. El-Serag HB, Peterson NJ, Carter J, et al. Gastroesophageal reflux among different ethnic groups in the United States. Gastroenterology. 2004;126:1692-9.
8. Spechler SJ, Jain SK, Tendler DA, Parker RA. Racial differences in the frequency of symptoms and complications of gastro-esophageal reflux disease. Aliment Pharmacol Ther. 2002;16:1795-1800.
9. Ho KY, Kang JY, Seow A. Prevalence of gastrointestinal symptoms in a multiracial Asian population, with particular reference to reflux-type symptoms. Am J Gastroenterol. 1998;93:1816-22.
10. Lim LG, Ho KY. Gastroesophageal reflux disease at the turn of the millennium. World J Gastroenterol. 2003;9:2135-6.
11. Kim N, Lee SW, Cho SI, et al. The prevalence of and risk factors for erosive esophagitis prospective study in Korea. Aliment Pharmacol Ther. 2008;27:173-85.
12. Goh KL, Chang CS, Fock KM, et al. Gastroesophageal reflux disease in Asia. J Gastroenterol Hepatol. 2000;15:230-8.
13. Richter JE, Falk GW, Vaezi MF. Helicobacter pylori and gastroesophageal reflux disease: The bug may not be all bad. Am J Gastroenterol. 1998;93:1800-2.
14. Serag HB. Time trends for gastroesophageal reflux disease: A systematic review. Clin Gastroenterol Hepatol. 2007:17-26.
15. El-Serag HB, Sonnenberg A. Opposing time trends of peptic ulcer disease and reflux disease. Gut. 1998;43:327-33.
16. Mokdad A, Bowman B, Ford E, et al. The continuing epidemic of obesity and diabetes in the United States. JAMA. 2001;286:1195-1200.
17. Jacobson BC, Somers SC, Fuchs CS, et al. Body-mass index and symptoms of gastroesophageal reflux in women. N Engl J Med. 2006;354:2340-8.
18. El-Serag HB, Graham DY, Satia JA, et al. Obesity is an independent risk factor for GERD symptoms and erosive esophagitis. Am J Gastroenterol. 2005;100:1243-50.
19. Murray L, Johnston B, Lane A, et al. Relationship between body mass and gastro-oesophageal reflux symptoms: The Bristol Helicobacter project. Int J Epidemiol. 2003;32:645-50.
20. Nocon M, Labenz J, Willich SN. Lifestyle factors and symptoms of gastro-oesophageal reflux—A population-based study. Aliment Pharmacol Ther. 2006;23:169-74.
21. Nilsson M, Johnsen R, Ye W, et al. Obesity and estrogen as risk factors for gastroesophageal reflux symptoms. JAMA. 2003;290:66-72.
22. Lagergren J, Bergstrom R, Nyren O. No relation between body mass and gastro-oesophageal reflux symptoms in a Swedish population based study. Gut. 2000;47:26-9.
23. Corley DA, Kubo A, Zhao W. Abdominal obesity, ethnicity and gastro-oesophageal reflux symptoms. Gut. 2007;56:756-62.
24. El-Serag HB, Ergun GA, Pandolfino J, et al. Obesity increases oesophageal acid exposure. Gut. 2007;56:749-55.
25. Stein DJ, El-Serag HB, Kuczynski J, et al. The association of body mass index with Barrett’s oesophagus. Aliment Pharmacol Ther. 2005;22:1005-10.
26. El-Serag HB, Kvapil P, Hacken-Bitar J, et al. Abdominal obesity and the risk of Barrett’s esophagus. Am J Gastroenterol. 2005;100:2151-6.
27. Freeman HJ. Risk of gastrointestinal malignancies and mechanisms of cancer development with obesity and its treatment. Best Pract Res Clin Gastroenterol. 2004;18:1167-75.
28. Lagergren J, Bergstrom R, Nyren O. Association between body mass and adenocarcinoma of the esophagus and gastric cardia. Ann Intern Med. 1999;130:883-90.
29. Edelstein ZR, Farrow DC, Bronner MP, et al. Central adiposity and risk of Barrett’s esophagus. Gastroenterology. 2007;133:403-11.
30. Friedenberg FK, Xanthopoulos M, Foster GF, Richter JE. The association between gastroesophageal reflux disease and obesity. Am J Gastroenterol. 2008;103:2111-22.
31. Romero Y, Cameron AJ, Locke GR, et al. Familial aggregation of gastroesophageal reflux in patients with Barrett’s esophagus and esophageal adenocarcinoma. Gastroenterology. 1997;113:1149-56.
32. Orenstein R, Shalaby TM, Barmada M, Whitcomb DC. Genetics of gastroesophageal reflux disease: A review. J Pediatr Gastroenterol Nutr. 2002;34:506-10.
33. Cameron AJ, Lagergren J, Henriksson C, et al. Gastro-esophageal reflux disease in monozygotic and dizygotic twins. Gastroenterology. 2002;122:55-9.
34. Mohammed I, Cherhas LF, Riley SA, et al. Genetic influence in gastro-oesophageal reflux disease: A twin study. Gut. 2003;52:1085-9.
35. Hu FZ, Preston RA, Post JC, et al. Mapping of a gene for severe pediatric gastroesophageal reflux disease to chromosome 13q14. JAMA. 2000;284:325-34.
36. Orenstein SR, Shalaby TM, Finch R, et al. Autosomal dominant infantile gastro-esophageal reflux disease: Exclusion of a 13q14 locus in five well characterized families. Am J Gastroenterol. 2002;97:2725-32.
37. Everhart JE, Ruhl CE. Burden of digestive disease in the US. Gastroenterology. 2009;136:376-86.
38. Shaheen NJ, Hansen RA, Morgan DR, et al. The burden of gastrointestinal and liver disease 2006. Am J Gastroenterol. 2006;101:2128-8.
39. Willich SN, Nocon M, Kulig M, et al. Cost-of-disease analysis in patients with gastro-oesophageal reflux disease and Barrett’s mucosa. Aliment Pharmacol Ther. 2006;23:371-6.
40. Enck P, Dubois D, Marquis P. Quality of life in patients with upper gastrointestinal symptoms: Results from the Domestic/International Gastroenterology Surveillance Study (DIGEST). Scand J Gastroenterol. 1999;34:48-54.
41. Nojkov B, Rubinstein JH, Adlis SA, et al. The influence of co-morbid IBS and psychological distress on outcomes and quality of life following PPI therapy in patients with gastro-oesophageal reflux disease. Aliment Pharmacol Ther. 2008;27:473-82.
42. Liebermann-Meffert D, Allgower M, Schmid P, Blum AL. Muscular equivalent of the lower esophageal sphincter. Gastroenterology. 1979;76:31-8.
43. Sloan S, Rademaker AW, Kahrilas PJ. Determinants of gastroesophageal junction incompetence: Hiatal hernia, lower esophageal sphincter, or both? Ann Intern Med. 1992;117:977-82.
44. Dodds WJ, Dent J, Hogan WJ, et al. Mechanisms of gastro-esophageal reflux in normal human subjects. N Engl J Med. 1982;307:1547-52.
45. Casselbrant A, Edebo A, Wennerblom J. Actions by angiotensin II on esophageal contractility in humans.
46. Dodds WJ, Dent J, Hogan W, Arndorfer R. Effect of atropine on esophageal motor function in humans. Am J Physiol. 1981;241:G290-6.
47. Mittal RK, Balaban DH. The esophagogastric junction. N Engl J Med. 1997;336:924-32.
48. Mittal RK, Rochester DF, McCallum RW. Electrical and mechanical activity in the human lower esophageal sphincter during diaphragmatic contraction. J Clin Invest. 1988;81:1182-95.
49. Thor KB, Hill RD, Mercer DD, Kozarek RD. Reappraisal of the flap valve mechanism in the gastroesophageal junction: A study of a new valvuloplasty procedure in cadavers. Acta Chir Scand. 1987;153:25-8.
50. Holloway RH, Penagini R, Ireland AC. Criteria for objective definition of transient lower esophageal sphincter relaxation. Am J Physiol. 1995;268:G128-33.
51. Dent J, Holloway RH, Toouli J, Dodds WJ. Mechanisms of lower esophageal sphincter incompetence in patients with symptomatic gastroesophageal reflux. Gut. 1988;29:1020-8.
52. Herwaarden MV, Samsom M, Smout AJP. Excess gastro-esophageal reflux in patients with hiatus hernia is caused by mechanisms other than transient LES relaxation. Gastroenterology. 2000;119:1439-46.
53. Mittal RK, McCallum RW. Characteristics of transient lower esophageal sphincter relaxation in humans. Am J Physiol. 1987;252:G636.
54. Mittal RK, Holloway RH, Penagini R, et al. Transient lower esophageal sphincter relaxation. Gastroenterology. 1995;109:601-10.
55. Holloway RH, Kocyan P, Dent J. Provocation of transient lower esophageal sphincter relaxation by meals in patients with symptomatic gastroesophageal reflux. Dig Dis Sci. 1991;36:1034-9.
56. Holloway RH. The anti-reflux barrier and mechanisms of gastro-oesophageal reflux. Baillieres Clin Gastroenterol. 2000;14:681.
57. Zagorodynuk VP, Chen BN, Brookers SJ. Intraganglionic laminar endings are mechano-transduction sites of vagal tension receptors in the guinea pig stomach. J Physiol. 2001;534:255.
58. Mittal RK, Lange RC, McCallum RW. Identification and mechanism of delayed esophageal acid clearance in subjects with hiatus hernia. Gastroenterology. 1987;92:130.
59. Pandofino JE, Shi G, Trueworthy B, Kahrilas PJ. Esophagogastric junction opening during relaxation distinguishes non-hernia reflux patients, hernia patients and normal subjects. Gastroenterology. 2003;125:1018.
60. Salapatek AMF, Diamant NE. Assessment of neural inhibition of the lower esophageal sphincter in cats with esophagitis. Gastroenterology. 1993;104:810.
61. Singh P, Adamopoulos A, Taylor RH, Colin-James DG. Oesophageal motor function before and after healing of oesophagitis. Gut. 1992;33:1590.
62. Mattioli S, D’Ovidio F, Pilotti V, et al. Hiatus hernia and intrathoracic migration of the esophagogastric junction in gastroesophageal reflux disease. Dig Dis Sci. 2003;48:1823.
63. Koek GH, Sifrim D, Lerut T, et al. Multivariate analysis of the association of acid and duodeno-gastro-oesophageal reflux exposure with the presence of oesophagitis, the severity of oesophagitis and Barrett’s oesophagus. Gut. 2008;57:1056-64.
64. Sontag SJ, Schnell TG, Miller TQ, et al. The importance of hiatal hernia in reflux esophagitis compared with lower esophageal sphincter pressure or smoking. J Clin Gastroenterol. 1991;13:628.
65. Kahrilas PJ, Lin S, Chen J, Manka M. The effect of hiatus hernia on gastro-esophageal junction pressure. Gut. 1999;44:476.
66. Kahrilas PJ, Shi G, Manka M, Joehl RJ. Increased frequency of transient lower esophageal sphincter relaxation induced by gastric distension in reflux patients with hiatal hernia. Gastroenterology. 2000;118:688.
67. Paterson WG, Kolyn DM. Esophageal shortening induced by short-term intraluminal acid perfusion: A cause for hiatus hernia? Gastroenterology. 1994;107:1736.
68. Wilson LJ, Ma W, Hirschowitz BI. Association of obesity with hiatus hernia and esophagitis. Am J Gastroenterol. 1999;94:262.
69. Smith AB, Dickerman RD, McGuire CS, et al. Pressure overload induced sliding hiatal hernia in power athletes. J Clin Gastroenterol. 1999;28:352.
70. Helm JF, Dodds WJ, Pek LR, et al. Effect of esophageal emptying and saliva on clearance of acid from the esophagus. N Engl J Med. 1984;310:284.
71. Kahrilas PJ, Dodds WJ, Hogan WJ, et al. Esophageal peristaltic dysfunction in peptic esophagitis. Gastroenterology. 1986;91:897.
72. Johnson LF, DeMeester TR. Elevation of the head of the bed, bethanechol and antacid foam tablets on gastroesophageal reflux. Dig Dis Sci. 1976;26:673.
73. Helm JF, Dodds WJ, Hogan WJ, et al. Acid neutralizing capacity of human saliva. Gastroenterology. 1987;83:69.
74. Orr WC, Robinson MG, Johnson LF, et al. Acid clearance during sleep in the pathogenesis of reflux esophagitis. Dig Dis Sci. 1987;26:423.
75. Korstein MA, Rosman AS, Fishbein S, et al. Chronic xerostomia increases esophageal acid exposure and is associated with esophageal injury. Am J Med. 1991;90:701.
76. Kahrilas PJ, Gupta RR. The effect of cigarette smoking on salivation and esophageal acid clearance. J Lab Clin Med. 1989;114:431.
77. Helm JF, Dodds WJ, Ricdel DR, et al. Determinants of esophageal acid clearance in normal subjects. Gastroenterology. 1983;86:607.
78. Meyers RL, Orlando RC. In vivo bicarbonate secretion by human esophagus. Gastroenterology. 1992;103:1174.
79. Brown CM, Snowdon CF, Slee B, et al. Effect of topical oesophageal acidification on human salivary and oesophageal alkali secretion. Gut. 1995;36:649.
80. Orlando RC. Esophageal epithelial defenses against acid injury. Am J Gastroenterol. 1994;89:349.
81. Quigley EMM, Turnberg LA. pH of the microclimate lining human gastric and duodenal mucosa in vivo: Studies in control subjects and in duodenal ulcer patients. Gastroenterology. 1987;92:1876.
82. Orlando RC, Lacey ER, Tobey NA, Cowart K. Barriers to paracellular permeability in rabbit esophageal epithelium. Gastroenterology. 1992;102:910.
83. Tobey NA, Reddy SP, Khalbuss WE, et al. Na+ dependent and independent CL−/HCO− exchanges in cultured rabbit esophageal epithelial cells. Gastroenterology. 1993;104:185.
84. Layden TJ, Schmidt L, Agone L, et al. Rabbit esophageal cell cytoplasmic pH regulation: Role of Na+− H+ antiport and Na+− dependent HCO3− transport systems. Am J Physiol. 1992;263:G407.
85. Majewski M, Jaworski T, Sarosiek I, et al. Significant enhancement of esophageal pre-epithelial defense by tegaserod: Implications for an esophagoprotective effect. Clin Gastroenterol Hepatol. 2007;5:430-8.
86. van Malenstein H, Farré R, Sifrim D. Esophageal dilated intercellular spaces (DIS) and nonerosive reflux disease. Am J Gastroenterol. 2008;103:1021-8.
87. Farré R, De Vos R, Geboes K, et al. Critical role of stress in increased oesophageal mucosa permeability and dilated intercellular spaces. Gut. 2007;56:1191-7.
88. Calabrese C, Fabbri A, Bortolotti M, et al. Dilated intercellular spaces as a marker of oesophageal damage: Comparative results in gastro-oesophageal reflux disease with or without bile reflux. Aliment Pharmacol Ther. 2003;18:525.
89. Calabrese C, Bortolotti M, Fabbri A, Areni G. Reversibility of GERD ultrastructural alteration and relief of symptoms after omeprazole treatment. Am J Gastroenterol. 2005;100:537.
90. Rodrigo J, Hernandez DJ, Vidol MA, Pedrosa JA. Vegetative innervation of the esophagus-intraepithelial endings. Acta Anat (Basel). 1975;92:242.
91. Hollwarth ME, Smith M, Kvietys PR, et al. Esophageal blood flow in the cat: Normal distribution and effects of acid perfusion. Gastroenterology. 1986;90:622.
92. Vieth M, Kulig M, Leodolter A, et al. Histological effects of esomeprazole therapy on the squamous epithelium of the distal oesophagus. Aliment Pharmacol Ther. 2006;23:313-19.
93. Orlando RC, Bryson JC, Powell DW. Mechanisms of H+ injury in rabbit esophageal epithelium. Am J Physiol. 1984;246:G718.
94. Stein HJ, Barlow AP, DeMeester TR, Hinder RA. Complications of gastroesophageal reflux disease. Ann Surg. 1992;216:35.
95. Gillen P, Keeling P, Byrne PJ, Hennessy TPJ. Barrett’s esophagus: pH profile. Br J Surg. 1987;74:774.
96. Hirschowitz BI. A critical analysis, with appropriate controls, of gastric acid and pepsin secretion in clinical esophagitis. Gastroenterology. 1991;101:1149.
97. Fletcher J, Wirz A, Henry E, McColl KEL. Studies of acid exposure immediately above the gastroesophageal squamocolumnar junction: Evidence of short segment reflux. Gut. 2004;53:168.
98. Mekapati J, Knight LC, Maurer AH, et al. Transsphincteric pH profile at the gastroesophageal junction. Clin Gastroenterol Hepatol. 2008;6:630-4.
99. Vicari J, Peek RM, Falk GW, et al. The seroprevalence of cagA-positive Helicobacter pylori strains in the spectrum of gastro-esophageal reflux disease. Gastroenterology. 1998;115:50.
100. Loffeld RJLF, Werdmuller BFM, Kusta JG, et al. Colonization with cagA-positive H. pylori strains is inversely associated with reflux esophagitis and Barrett’s esophagus. Digestion. 2000;62:95.
101. Feldman M, Cryer B, Lee E. Effects of Helicobacter pylori gastritis on gastric secretion in normal human beings. Am J Physiol. 1998;274:G1011.
102. Labenz J, Malfertheiner P. Hpylori in gastro-oesophageal reflux disease: Causal agent, independent or protective factor. Gut. 1997;41:277.
103. Labenz J, Blum AL, Bayerdorffer E, et al. Curing H. pylori infection in duodenal ulcer patients may provoke reflux esophagitis. Gastroenterology. 1997;112:1442.
104. Fallone CA, Barkun AN, Friedman G, et al. Is Helicobacter pylori eradication associated with gastroesophageal reflux disease? Am J Gastroenterol. 2000;95:914.
105. Lillemoe KD, Johnson LF, Harmon JW. Alkaline esophagitis: A comparison of the ability of components of the gastroduodenal contents to injure the rabbit esophagus. Gastroenterology. 1983;85:621.
106. Attwood SEA, DeMeester TR, Bremner CG, et al. Alkaline gastroesophageal reflux: Implications in the development of complications in Barrett’s columnar-lined esophagus. Surgery. 1989;106:764.
107. Pellegrini CA, DeMeester TR, Wernly JA, et al. Alkaline gastroesophageal reflux. Am J Surg. 1978;75:177.
108. Bechi P, Pucciani F, Baldini F, et al. Long-term ambulatory enterogastric reflux monitoring: Validation of a new fiberoptic technique. Dig Dis Sci. 1993;38:1297.
109. Vaezi MF, Richter JE. Role of acid and duodenogastro-esophageal reflux in gastroesophageal reflux disease. Gastroenterology. 1996;111:1192.
110. Champion G, Richter JE, Vaezi MF, et al. Duodenogastro-esophageal reflux: Relationship to pH and importance in Barrett’s esophagus. Gastroenterology. 1994;107:747.
111. McCallum RW, Berkowitz DM, Lerner E. Gastric emptying in patients with gastroesophageal reflux. Gastroenterology. 1981;80:285.
112. Shay SS, Eggli D, McDonald C, Johnson LF. Gastric emptying of solid food in patients with gastroesophageal reflux. Gastroenterology. 1987;92:459.
113. Schwizer W, Hinder RA, DeMeester TR. Does delayed gastric emptying contribute to gastroesophageal reflux? Am J Surg. 1989;157:74.
114. Carlsson R, Dent J, Bolling-Sternevold E, et al. The usefulness of a structured questionnaire in the assessment of symptomatic gastroesophageal reflux disease. Scand J Gastroenterol. 1998;33:1023.
115. Fass R, Naliboff BD, Fass SS, et al. The effect of auditory stress on perception of intraesophageal acid in patients with gastroesophageal reflux disease. Gastroenterology. 2008;134:696-705.
116. Schey R, Dickman R, Parthasarathy S, et al. Sleep deprivation is hyperalgesic in patients with gastroesophageal reflux disease. Gastroenterology. 2007;133:1787-95.
117. Wright CE, Ebrecht M, Mitchell R, et al. The effect of psychological stress on symptom severity and perception in patients with gastro-oesophageal reflux. J Psychosom Res. 2005;59:415-24.
118. Shaker R, Castell DO, Schoenfeld PS, Spechler SJ. Nighttime heartburn is an under-appreciated clinical problem that impacts sleep and daytime function: The results of a Gallup survey conducted on behalf of the American Gastroenterological Association. Am J Gastroenterol. 2003;98:1487.
119. Klauser AG, Schindlebeck NE, Muller-Lissner SA. Symptoms of gastro-oesophageal reflux disease. Lancet. 1990;335:205.
120. Dent J, Brun J, Fendrick AM, et al. An evidence-based appraisal of reflux disease management—The Genval report. Gut. 1999;44:S1.
121. Kahrilas PJ. Clinical vignette: refractory heartburn. Gastroenterology. 2003;124:1941-5.
122. Tominaga M, Caterina MJ, Malmberg AB, et al. The cloned capsaicin receptor integrates multiple pain-producing stimuli. Neuron. 1998;21:531-43.
123. Caterina MJ, Julius D. The vanilloid receptor: A molecular gateway to the pain pathway. Annu Rev Neurosci. 2001;24:487-517.
124. Hwang SW, Oh U. Hot channels in airways: Pharmacology of the vanilloid receptor. Curr Opin Pharmacol. 2002;2:235-42.
125. Trevisani M, Smart D, Gunthorpe MJ, et al. Ethanol elicits and potentiates nociceptor responses via the vanilloid receptor-1. Nat Neurosci. 2002;5:546-51.
126. Matthews PJ, Aziz Q, Facer P, et al. Increased capsaicin receptor TRPV1 nerve fibres in the inflamed human oesophagus. Eur J Gastroenterol Hepatol. 2004;16:897-902.
127. Bredenoord AJ, Weusten BL, Curvers WL, et al. Determinants of perception of heartburn and regurgitation. Gut. 2006;55:313-18.
128. Tack J. Review article: the role of bile and pepsin in the pathophysiology and treatment of gastroesophageal reflux disease. Aliment Pharmacol Ther. 2006;24:10-16.
129. Tobey NA, Hosseini SS, Caymaz-Bor C, et al. The role of pepsin in acid injury to esophageal epithelium. Am J Gastroenterol. 2001;96:3062-70.
130. Balaban D, Yamamoto Y, Liu J, et al. Sustained esophageal contraction: A marker of esophageal chest pain identified by intraluminal ultrasonography. Gastroenterology. 1999;116:29.
131. Rao SS, Gregersen H, Hayek B, et al. Unexplained chest pain: the hypersensitive, hyperreactive, and poorly compliant esophagus. Ann Intern Med. 1996;124:950-8.
132. Jacob P, Kahrilas PJ, Vanagunos A. Peristaltic dysfunction associated with non-obstructive dysphagia in reflux disease. Dig Dis Sci. 1990;35:939.
133. Brzana RJ, Koch KL. Gastroesophageal reflux disease presenting with intractable nausea. Ann Intern Med. 1997;126:704.
134. Johnson DA, Winters C, Spurling TJ, et al. Esophageal acid sensitivity in Barrett’s esophagus. J Clin Gastroenterol. 1987;9:23.
135. Extraesophageal presentations of gastroesophageal reflux disease. Am J Gastroenterol. 2000;25:S1.
136. Schofied PM, Bennett DH, Whorewell PJ, et al. Exertional gastroesophageal reflux: A mechanism for symptoms in patients with angina pectoris and normal coronary angiograms. BMJ. 1987;294:1459.
137. Hewson EG, Sinclair JW, Dalton CB, et al. Twenty-four hour esophageal pH monitoring: The most useful test for evaluating non-cardiac chest pain. Am J Med. 1991;90:576.
138. Richter JE. Approach to the patient with non-cardiac chest pain. In: Yamada T, editor. Textbook of gastroenterology. ed 2. Philadelphia: JB Lippincott; 1995:648.
139. Gustafsson U, Tibbling L. The effect of edrophonium chloride-induced chest pain on esophageal blood flow and motility. Scand J Gastroenterol. 1997;32:104-7.
140. Harding SM, Sontag SJ. Asthma and gastroesophageal reflux. Am J Gastroenterol. 2000;95:S23.
141. Liou A, Grubb JR, Schechtman KB, Hamilos DC. Causative and contributive factors to asthma severity and patterns of medication use in patients seeking specialized asthma care. Chest. 2003;124:1781.
142. Irwin RS, Curley FJ, French CL. Difficult-to-control asthma: Contributing factors and outcome of a systematic protocol. Chest. 1993;103:1662.
143. Bortolotti M, Gentilini L, Morselli C, Giovannini M. Obstructive sleep apnoea is improved by a prolonged treatment of gastrooesophageal reflux with omeprazole. Dig Liver Dis. 2006;38:78-81.
144. Tuchman DN, Boyle JT, Pack AI, et al. Comparison of airway responses following tracheal or esophageal acidification in the cat. Gastroenterology. 1984;87:872.
145. Schan CA, Harding SM, Haile JM, et al. Gastroesophageal reflux-induced bronchoconstriction: An intraesophageal acid infusion study using state-of-the-art technology. Chest. 1994;105:731.
146. Jack CIA, Calverley PMA, Donnelly RJ, et al. Simultaneous tracheal and oesophageal pH measurements in asthmatics patients with gastro-oesophageal reflux. Thorax. 1995;50:201.
147. Koufman JA. The otolaryngologic manifestations of gastro-esophageal reflux disease: A clinical investigation of 225 patients using ambulatory 24-hour pH monitoring and an experimental investigation of the role of acid and pepsin in the development of laryngeal injury. Laryngoscope. 1991;Suppl 53:1.
148. Wong RKH, Hanson DG, Waring PJ, Shaw G. ENT manifestations of gastroesophageal reflux. Am J Gastroenterol. 2000;95:S15.
149. Adhami T, Goldblum JR, Richter JE, Vaezi MF. The role of gastric and duodenal agents in laryngeal injury: An experimental canine model. Am J Gastroenterol. 2004;99:2098.
150. Jacob P, Kahrilas PH, Herzon G. Proximal esophageal pH-manometry in patients with “reflux laryngitis.”. Gastroenterology. 1991;100:305.
151. Irwin RS, Richter JE. Gastroesophageal reflux and cough. Am J Gastroenterol. 2000;95:S39.
152. Javorkova N, Varechova S, Pecova R, et al. Acidification of the oesophagus acutely increases the cough sensitivity in patients with gastro-oesophageal reflux and chronic cough. Neurogastroenterol Motil. 2008;20:119-24.
153. Chang AB, Lasserson TJ, Gaffney J, et al. Gastro-oesophageal reflux treatment for prolonged non-specific cough in children and adults. Cochrane Database Syst Rev 2006; 18:CD004823.
154. Lazarchik DA, Filler SJ. Dental erosion: Predominant oral lesion in gastroesophageal reflux disease. Am J Gastroenterol. 2000;95:S33.
155. Richter JE. Gastroesophageal reflux disease during pregnancy. Gastroenterol Clin North Am. 2003;32:235.
156. Zamost BJ, Hirschberg J, Ippoliti AF, et al. Esophagitis in scleroderma: Prevalence and risk factors. Gastroenterology. 1987;92:421.
157. Miller LS, Vinayek R, Frucht H, et al. Reflux esophagitis in patients with the Zollinger-Ellison syndrome. Gastroenterology. 1990;98:341.
158. Vaezi MF, Richter JE. Current therapies for achalasia: Comparison and efficacy. J Clin Gastroenterol. 1998;27:21.
159. Nagler R, Spiro HM. Persistent gastroesophageal reflux induced during prolonged gastric intubation. N Engl J Med. 1963;269:495.
160. Richter JE. Diagnostic tests for gastroesophageal reflux disease. Am J Med Sci. 2003;326:300.
161. Schindlebeck NE, Klauser AG, Voderholzer WA, Mueller-Lissner S. Empiric therapy for gastroesophageal reflux disease. Arch Intern Med. 1995;155:1808.
162. Fass R, Ofman JJ, Granelk I, et al. Clinical and economic assessment of the omeprazole test in patients with symptomatic suggestive of gastroesophageal reflux disease. Arch Intern Med. 1999;159:2161.
163. Fass R, Fennerty MB, Ofman JJ. The clinical and economic value of a short course of omeprazole in patients with non-cardiac chest pain. Gastroenterology. 1998;115:42.
164. Ours TM, Kavuru MS, Schilz R, Richter JE. A prospective evaluation of esophageal testing and a double blind, randomized study of omeprazole in a diagnostic and therapeutic algorithm for chronic cough. Am J Gastroenterol. 1999;94:3131.
165. Richter JE. Severe reflux esophagitis. Gastrointest Endosc Clin North Am. 1994;4:677.
166. Johnson LF, DeMeester TF, Haggitt RC. Endoscopic signs of gastroesophageal reflux objectively evaluated. Gastrointest Endosc. 1976;22:151.
167. Nayer DW, Vaezi MF. Classification of esophagitis: Who needs it? Gastrointest Endosc. 2004;60:253.
168. Ollyo JB, Lang F, Fontolliet C, Monnier P. Savary-Miller’s new endoscopic grading of reflux-oesophagitis: A simple, reproducible, logical, complete and useful classification. Gastroenterology. 1990;98:A100.
169. Lundell LR, Dent J, Bennett JR, et al. Endoscopic assessment of oesophagitis: Clinical and functional correlation and further validation of the Los Angeles classification. Gut. 1999;45:172.
170. Sharma P, Wani S, Rastogi A, et al. The diagnostic accuracy of esophageal capsule endoscopy in patients with gastroesophageal reflux disease and Barrett’s esophagus: A blinded, prospective study. Am J Gastroenterol. 2008;103:525-32.
171. DeVault KR, Castell DO. Updated guidelines for the diagnosis and treatment of gastroesophageal reflux disease. Am J Gastroenterol. 2005;100:190.
172. Dent J. Microscopic esophageal mucosal injury in nonerosive reflux disease. Clin Gastroenterol Hepatol. 2007;5:4-16.
173. Funch-Jensen P, Kock K, Christensen LA, et al. Microscopic appearance of the esophageal mucosa in a consecutive series of patients submitted to endoscopy: Correlation with gastroesophageal reflux symptoms and microscopic findings. Scand J Gastroenterol. 1986;21:65.
174. Riddell RH. The biopsy diagnosis of gastroesophageal reflux disease, “carditis,” and Barrett’s esophagus. Am J Surg Pathol. 1996;20:31.
175. Schindlbeck NE, Wiebecke B, Kaluser AG, et al. Diagnostic value of histology in non-erosive gastro-oesophageal reflux disease. Gut. 1996;39:151.
176. Hirano I, Richter JE. ACG practice guidelines: Esophageal reflux testing. Am J Gastroenterol. 2007;102:668-85.
177. Booth MI, Stratford J, Dehn TCB. Patient self-assessment of test-day symptoms in 24 hour pH metry for suspected gastroesophageal reflux disease. Scand J Gastroenterol. 2001;36:795.
178. Weusten BLAM, Snout AJPM. Symptom analysis in 24-hour esophageal pH monitoring. In: Richter JE, editor. Ambulatory esophageal pH monitoring: Practical approach and clinical application. 2nd ed. Baltimore: Williams & Wilkins; 1997:97.
179. Wenner J, Johnsson F, Johansson J, Oberg S. Wireless esophageal pH monitoring is better tolerated than the catheter-based technique: results from a randomized cross-over trial. Am J Gastroenterol. 2007;102:239-45.
180. Zerbib F, des Varannes SB, Roman S, et al. Normal values and day-to-day variability of 24-h ambulatory oesophageal impedance-pH monitoring in a Belgian-French cohort of healthy subjects. Aliment Pharmacol Ther. 2005;22:1011-21.
181. Mainie I, Tutuian R, Shay S, et al. Acid and non-acid reflux in patients with persistent symptoms despite acid suppressive therapy: A multicentre study using combined ambulatory impedance-pH monitoring. Gut. 2006;55:1398-402.
182. Zerbib F, Roman S, Ropert A, et al. Esophageal pH-impedance monitoring and symptom analysis in GERD: A study in patients off and on therapy. Am J Gastroenterol. 2006;101:1956-63.
183. Ott DJ, Kelley TF, Chen MYM, et al. Use of a marshmallow bolus for evaluating lower esophageal mucosal rings. Am J Gastroenterol. 1991;86:817.
184. Ott DJ. Gastroesophageal reflux disease. Radiol Clin North Am. 1994;32:1147.
185. Thompson JK, Koehler RE, Richter JE. Detection of gastro-esophageal reflux: Value of the barium studies compared with 24-hour pH monitoring. Am J Roentgenol. 1994;162:621.
186. Johnston BT, Troshinsky MB, Castell JA, Castell DO. Comparison of barium radiology with esophageal pH monitoring in the diagnosis of gastroesophageal reflux disease. Am J Gastroenterol. 1996;91:1181.
187. Waring JP, Hunter JG, Oddsdottir M. The preoperative evaluation of patients considered for laparoscopic antireflux surgery. Am J Gastroenterol. 1995;90:35.
188. Leite LP, Johnston BT, Barrett J, et al. Ineffective esophageal motility: The primary finding in patients with non-specific esophageal motility disorder. Dig Dis Sci. 1997;42:1853.
189. Oleynikov D, Eubanks TR, Oelschlager BK, Pellegrini CA. Total fundoplication is the operation of choice for patients with gastroesophageal reflux and defective peristalsis. Surg Endosc. 2002;16:909.
190. Tutuian R, Castell DO. Clarification of the esophageal function defect in patients with manometric ineffective esophageal motility: Studies using combined impedance-manometry. Gastroenterology. 2004;2:230.
191. Pace F, Santalucia F, Bianchi-Porro G. Natural history of gastroesophageal reflux disease without esophagitis. Gut. 1991;32:845.
192. Isolauri J, Luostarinen M, Isolauri E, et al. Natural history of gastroesophageal reflux disease: 17-22 year follow-up of 60 patients. Am J Gastroenterol. 1997;92:37.
193. Labenz J, Nocon M, Lind T, et al. Prospective follow-up data from the PROGERD study suggest that GERD is not a categorical disease. Am J Gastroenterol. 2006;101:2457-62.
194. Sontag SJ, Sonnenberg A, Schnell TG, et al. The long-term natural history of gastroesophageal reflux disease. J. Clin Gastroenterol. 2006;40:398-404.
195. Winters C, Spurling TJ, Chobanian SJ, et al. Barrett’s esophagus: A prevalent, occult complication of gastroesophageal reflux disease. Gastroenterology. 1987;92:118.
196. Lind T, Havelund T, Carlsson R, et al. Heartburn without oesophagitis: Efficacy of omeprazole therapy and features determining therapeutic response. Scand J Gastroenterol. 1997;32:974.
197. Jones RH, Hungin ADS, Phillips J, et al. Gastroesophageal reflux disease in primary care in Europe: Clinical presentation and endoscopic findings. Eur J Gen Pract. 1995;1:149.
198. Robinson M, Earnest D, Rodriguez-Stanley S, et al. Heartburn requiring frequent antacid use may indicate significant illness. Arch Intern Med. 1998;156:2373.
199. Wu JC, Chenng CMY, Wong VWS, Sung JJY. Distinct clinical characteristics between patients with non-erosive reflux disease and those with reflux esophagitis. Clinical Gastroenterology and Hepatology. 2007;5:690-5.
200. Fass R. Erosive esophagitis and non-erosive reflux disease. Comparison of epidemiologic, physiologic and therapeutic characteristics. J Clin Gastroenterol. 2007;41:131-7.
201. Trimble KC, Douglas S, Pryde A, Heading RC. Clinical characteristics and natural history of symptomatic but not excess gastroesophageal reflux. Dig Dis Sci. 1995;40:1098.
202. Hetzel DJ, Dent J, Reed WD, et al. Healing and relapse of severe peptic esophagitis after treatment with omeprazole. Gastroenterology. 1988;95:903.
203. Vigneri S, Termini R, Leandro G, et al. A comparison of five maintenance therapies for reflux esophagitis. N Engl J Med. 1995;333:1106.
204. Spechler SJ. Comparison of medical and surgical therapy for complicated gastroesophageal reflux disease in veterans. N Engl J Med. 1992;326:786.
205. Rantanen TK, Silvio EI, Rasanen JV, Salo JA. Gastroesophageal reflux as a cause of death is increasing: Analysis of total cases after medical and surgical treatment. Am J Gastroenterol. 2007;102:246-53.
206. Higuchi D, Sagawa C, Shab SH, et al. Etiology, treatment and outcome of esophageal ulcers: A 10 year experience in an urban emergency hospital. J Gastrointest Surg. 2003;7:836.
207. DaCosta N, Guillaume C, Merle C, et al. Bleeding reflux esophagitis: A prospective 1-year study in a university hospital. Am J Gastroenterol. 2001;96:47.
208. Richter JE. Peptic strictures of the esophagus. Gastroenterol Clin North Am. 1999;28:875.
209. El-Serag HB, Sonnenberg A. Association of esophagitis and esophageal strictures with diseases treated with non-steroidal anti-inflammatory drugs. Am J Gastroenterol. 1997;92:52.
210. Marshall JB, Kretschman JM, Kiaz-Arias AA. Gastro-esophageal reflux as a pathogenic factor in the development of symptomatic lower esophageal rings. Arch Intern Med. 1990;150:1669.
211. Kaltenback T, Crockett S, Gerson LB. Are lifestyle measures effective in patients with gastroesophageal reflux disease? Am J Gastroenterol. 2006;101:2128-38.
212. Meining A, Classen M. The role of diet and lifestyle measures in the pathogenesis and treatment of gastroesophageal reflux disease. Am J Gastroenterol. 2000;95:2692.
213. Feldman M, Barnett C. Relationship between the acidity and osmolality of popular beverages and reported postprandial heartburn. Gastroenterology. 1995;108:125.
214. Weberg R, Berstad A. Symptomatic effect of a low-dose antacid regimen in reflux oesophagitis. Scand J Gastroenterol. 1989;24:401.
215. Buts JP, Barudi C, Otte JB. Double-blind controlled study on the efficacy of sodium alginate (Gaviscon) in reducing gastroesophageal reflux assessed by 24h continuous pH monitoring in infants and children. Eur J Pediatr. 1987;146:156.
216. Behar J, Sheahan DG, Biancani P, et al. Medical and surgical management of reflux esophagitis: A 38-month report on a prospective clinical trial. N Engl J Med. 1975;293:263.
217. Lieberman DA. Medical therapy for chronic reflux esophagitis: Long term follow-up. Arch Intern Med. 1987;147:717.
218. Gonzalez ER, Grillo JA. Over-the-counter histamine2-blocker therapy. Ann Pharmacother. 1994;28:392.
219. Fendrick AM, Shaw M, Schachtel B, et al. Self-selection and use patterns of over-the-counter omeprazole for frequent heartburn. Clin Gastroenterol Hepatol. 2004;2:17.
220. Ramirez B, Richter JE. Review article: Promotility drugs in the treatment of gastro-oesophageal reflux disease. Aliment Pharmacol Ther. 1993;7:5.
221. Wysowski DK, Corken A, Gallo-Torres H, et al. Postmarketing reports of QT prolongation and ventricular arrhythmia in association with cisapride and Food and Drug Administration regulatory action. Am J Gastroenterol. 2001;96:1698.
222. Richter JE. New investigational therapies for gastroesophageal reflux disease. Thorac Surg Clin. 2005;15:377-84.
223. Ciccaglione AF, Marzio I. Effect of acute and chronic administration of the GABAB agonist baclofen on 24 hour pH-metry and symptoms in control subjects and in patients with gastro-oesophageal reflux disease. Gut. 2003;52:464.
224. Koek GH, Sifrim D, Lerut T, et al. Effect of the GABAB agonist baclofen in patients with symptoms and duodeno-gastroesophageal reflux refractory to proton pump inhibitors. Gut. 2003;52:1397.
225. Lipsy RJ, Fennerty B, Fagan TC. Clinical review of histamine -2 receptor antagonists. Arch Intern Med. 1990;150:745.
226. Sontag SJ. Gastroesophageal reflux disease. In: Brandt LJ, editor. Clinical practice of gastroenterology. Philadelphia: Churchill Livingstone; 1999:21.
227. Chiba N, Gara CJ, Wilkinson JM, Hunt RH. Speed of healing and symptom relief in grade II to IV gastroesophageal reflux disease: A meta-analysis. Gastroenterology. 1997;112:1798.
228. Peghini PL, Katz PO, Castell DO. Ranitidine controls nocturnal gastric acid breakthrough on omeprazole: A controlled study in normal subjects. Gastroenterology. 1998;115:1335.
229. Wilder-Smith CH, Merki HS. Tolerance during dosing with H2-receptor antagonists: An overview. Scand J Gastroenterol. 1992;27:14.
230. Fackler WK, Ours TM, Vaezi MF, Richter JE. Long-term effect of H2RA therapy on nocturnal gastric acid breakthrough. Gastroenterology. 2002;122:625.
231. Klinkenberg-Knol EC, Festen HPM, Meuwissen SGM. Pharmacological management of gastro-oesophageal reflux disease. Drugs. 1995;49:697.
232. Wolfe MM, Sachs G. Acid suppression: Optimizing therapy for gastroduodenal ulcer healing, gastroesophageal reflux disease, and stress-related erosive syndrome. Gastroenterology. 2000;118:9.
233. Hunt RH. Importance of pH control in the management of GERD. Arch Intern Med. 1999;159:649.
234. Miner P, Katz PO, Chen Y, Sostek M. Gastric acid control with esomeprazole, lansoprazole, omeprazole, pantoprazole and rabeprazole: A five-way crossover study. Am J Gastroenterol. 2003;98:2616.
235. vanPinxteren B, Numans ME, Bonis PA, Lau J. Short term treatment with proton pump inhibitors, H2RAs and prokinetics for gastro-oesophageal reflux disease-like symptoms and endoscopy negative reflux disease. Cochrane Database Syst Rev 2004; 3:CD002095.
236. Sontag SJ, Hirschowitz BH, Holt S, et al. Two doses of omeprazole versus placebo in symptomatic erosive esophagitis: The U.S. multicenter study. Gastroenterology. 1992;102:109.
237. Castell DO, Richter JE, Robinson M, et al. Efficacy and safety of lansoprazole in the treatment of erosive reflux esophagitis. Am J Gastroenterol. 1996;91:1749.
238. Cloud ML, Enas N, Humphries TJ, et al. Rabeprazole in treatment of acid peptic diseases. Dig Dis Sci. 1998;43:993.
239. Richter JE, Bochenek W, Pantoprazole US GERD Study Group. Oral pantoprazole for erosive esophagitis: A placebo-controlled, randomized clinical trial. Am J Gastroenterol. 2000;95:3071.
240. Moayyedi P, Talley N. Gastroesophageal reflux disease. Lancet. 2006;367:2086-100.
241. Bianchi Porro G, Pace F, Peracchia A, et al. Short-term management of refractory reflux esophagitis with different doses of omeprazole or ranitidine. J Clin Gastroenterol. 1992;15:192.
242. Richter JE, Kahrilas PJ, Johanson J, et al. Efficacy and safety of esomeprazole compared with omeprazole in GERD patients with erosive esophagitis: A randomized controlled trial. Am J Gastroenterol. 2001;96:656.
243. Castell DO, Kahrilas PJ, Richter JE, et al. Esomeprazole (40 mg) compared with lansoprazole (30 mg) in the treatment of erosive esophagitis. Am J Gastroenterol. 2002;97:575.
244. Gralnek IM, Dulai GS, Fennerty MB, Speigel BMR. Esomeprazole versus other PPIs in erosive esophagitis: A meta-analysis of randomized clinical trials. Clin Gastroenterol Hepatol. 2006;4:1452-8.
245. Metz D, Pratha V, Martin P, et al. Oral and intravenous dosage forms of pantoprazole are equivalent in their ability to suppress gastric acid secretion in patients with GERD. Am J Gastroenterol. 2000;95:626.
246. Garnett WR. Consideration for long-term use of proton pump inhibitors. Am J Health Syst Pharm. 1998;55:2269.
247. Toussaint J, Gossuin A, Deruyttere M, et al. Healing and prevention of relapse of reflux esophagitis by cisapride. Gut. 1991;35:590.
248. Donnellan C, Sharma N, Preston C, Moayyedi P. Medical treatments for the maintenance therapy of reflux esophagitis. Cochrane Database Syst Rev 2004; 3:CD003245.
249. Klinkenberg-Knol EC, Nelis F, Dent J, et al. Long-term omeprazole treatment in resistant gastroesophageal reflux disease: Efficacy, safety, and influence on gastric mucosa. Gastroenterology. 2000;118:661.
250. Inadomi JM, Jamal R, Murata GH, et al. Step-down management of gastroesophageal reflux disease. Gastroenterology. 2001;121:1095.
251. Inadomi JM, McIntyre L, Bernard L, Fendrick AM. Step-down from multiple-to-single dose PPIs: A prospective study of patients with heartburn or acid regurgitation relieved by PPIs. Am J Gastroenterol. 2003;98:1940.
252. Freston JW. Omeprazole, hypergastrinemia and gastric carcinoid tumors. Ann Intern Med. 1994;121:232.
253. Singh M, Dhindsa G, Friedland S, Triadafilopoulos G. Long-term use of proton pump inhibitors does not affect the frequency, growth, or histologic characteristics of colon adenomas. Aliment Pharmacol Ther. 2007;26:1051-61.
254. Jalving M, Koornstra JJ, Wesseling J, et al. Increased risk of fundic gland polyps during long-term proton pump inhibitor therapy. Aliment Pharmacol Ther. 2006;24:1341-8.
255. Laheij RJF, Sturkenboom MCJM, Hassing RJ, et al. Risk of community-acquired pneumonia and use of gastric acid-suppression drugs. JAMA. 2004;292:1955-60.
256. Leonard J, Marshall JK, Moayyedi P. Systematic review of the risk of enteric infections in patients taking acid suppression. Am J Gastroenterol. 2007;102:2047-56.
257. Yang YX, Lewis JD, Epstein S, Metz DC. Longterm proton pump inhibitor therapy and risk of hip fracture. JAMA. 2006;296:2947-53.
258. Targownik LE, Lix LM, Metge CJ, et al. Use of proton pump inhibitors and risk of osteoporosis-related fractures. CMAJ. 2008;179:319-26.
259. Howden CW, Coté GA. Potential adverse effects of proton pump inhibitors. Current Gastroenterology Reports. 2008;10:208-14.
260. Kuipers E, Lundell L, Klinkenberg-Knol EC, et al. Atrophic gastritis and Helicobacter pylori infection in patients with reflux esophagitis treated with omeprazole or fundoplication. N Engl J Med. 1996;334:1018.
261. Proton pump inhibitors relabeling for cancer risk not warranted. FDA report, November 11, 1996.
262. Ireland AC, Holloway RH, Toouli J, Dent J. Mechanisms underlying the antireflux action of fundoplication. Gut. 1993;34:303.
263. Rice TW. Why antireflux surgery fails. Digest Dis. 2000;18:43.
264. Oelschlager BK, Eubanks TR, Pellegrini CA. Hiatal hernia and gastroesophageal reflux disease. In Townsend CM, Beauchamp RD, Foshee JC, et al, editors: Sabiston Textbook of Surgery: The Biological Basis of Modern Surgical Practice, 18th ed, Saunders, 2007.
265. Watson DI. Laparoscopic treatment of gastro-oesophageal reflux disease. Best Pract Res Clin Gastroenterol. 2004;18:19-35.
266. Horvath KD, Jobe BA, Herron DM, Swanstrom LL. Laparoscopic Toupet fundoplication is an inadequate procedure for patients with severe reflux disease. J Gastrointest Surg. 1999;3:583.
267. Gastal OL, Hagen JA, Peters JH. Short esophagus: Analysis of predictors and clinical implications. Arch Surg. 1999;134:633.
268. Fink JF, Wei Y, Birkmeyer JD. The rise and fall of antireflux surgery in the United States. Surg Endosc. 2006;20:1698-701.
269. Dominitz JA, Dire CA, Billingsley KG, Todd-Stenberg JA. Complications and anti-reflux medication use after antireflux surgery. Clin Gastroenterol Hepatol. 2006;4:299-305.
270. Allgood PC, Bachmann M. Medical or surgical treatment for chronic gastroesophageal reflux? A systematic review of the published evidence of effectiveness. Eur J Surg. 2000;166:713-21.
271. So JBY, Zeitel SM, Rattner DW. Outcomes of atypical symptoms attributed to gastroesophageal reflux treated by laparoscopic fundoplication. Surgery. 1998;124:28.
272. Perdikis G, Hinder RA, Lund RJ, et al. Laparoscopic Nissen fundoplication: Where do we stand? Surg Laparosc Endosc. 1997;7:17.
273. Corey KE, Schmitz SM, Shaheen NJ. Does a surgical antireflux procedure decrease the incidence of esophageal adenocarcinoma in Barrett’s esophagus? A meta-analysis. Am J Gastroenterol. 2003;98:2390-4.
274. Lundell L, Miettinen P, Myrvold HE, et al. Continued (5-year) follow-up of a randomized clinical study comparing antireflux surgery and omeprazole in gastroesophageal reflux disease. J Am Coll Surg. 2001;192:172.
275. Vakil N, Shaw M, Kirby R. Clinical effectiveness of laparoscopic fundoplication in a U.S. community. Am J Med. 2003;114:1.
276. Spechler SJ, Lee EL, Ahnen D, et al. Long-term outcome of medical and surgical therapies for gastroesophageal reflux disease: Follow-up of a randomized controlled trial. JAMA. 2001;285:2331.
277. Oelschlager BK, Quiroga E, Parras JD, et al. Long-term outcomes from laparoscopic antireflux surgery. Am J Gastroenterol. 2008;103:280-7.
278. Hatch KF, Daily MF, Christiansen BJ, Glasgow RE. Failed fundoplication. Am J Surg. 2004;188:786-91.
279. Kahrilas PJ. Radiofrequency therapy for the lower esophageal sphincter for treatment of GERD. Gastrointest Endosc. 2003;57:723.
280. Fennerty MB. Endoscopic suturing for treatment of GERD. Gastrointest Endosc. 2003;57:390.
281. Edmundowicz SA. Injection therapy of the lower esophageal sphincter for the treatment of GERD. Gastrointest Endosc. 2004;59:545.
282. Corley DA, Katz P, Wo JM, et al. Improvement of gastro-esophageal reflux symptoms after radiofrequency energy: A randomized, sham-controlled trial. Gastroenterology. 2003;125:668.
283. Rothstein R, Filipi C, Caca K, et al. Endoscopic full-thickness plication for the treatment of gastroesophageal reflux disease: a randomized, sham-controlled trial. Gastroenterology. 2006;131:704-12.
284. Falk GW, Fennerty MB, Rothstein RI. AGA Institute technical review on the use of endoscopic therapy for gastroesophageal reflux disease. Gastroenterology. 2006;131:1351-66.
285. Cremmini F, Wise J, Moayyedi P, Talley N. Diagnostic and therapeutic use of proton pump inhibitors in non-cardiac chest pain: A meta-analysis. Am J Gastroenterol. 2005;100:1226-32.
286. Wang WH, Huang JQ, Zheng GK, et al. Is proton pump inhibitor testing an effective approach to diagnose gastroesophageal reflux disease in patients with non-cardiac chest pain? A meta-analysis. Arch Intern Med. 2005;165:1222-8.
287. Chang AB, Lasserson TJ, Gaffney J, et al. Gastroesophageal reflux disease for prolonged non-specific cough in children and adults. Cochrane Database Systemic Review 2005;2:CD004823.
288. American Lung Association Asthma Clinical Research CentersMastronarde JG, Anthonisen NR, et al. Efficacy of esomeprazole for treatment of poorly controlled asthma. N Engl J Med. 2009;360:1487-99.
289. Qadeer MA, Philips CO, Lopez AR, et al. Proton pump inhibitor therapy suspected GERD-related chronic laryngitis: A meta-analysis of randomized controlled trials. Am J Gastroenterol. 2006;101:2646-54.
290. Johnson DA, Orr WC, Crawley JA, et al. Effect of esomeprazole on nighttime heartburn and sleep quality in patients with GERD: A randomized, placebo-controlled trial. Am J Gastroenterol. 2005;100:1914-22.
291. Dakkak M, Hoare RC, Maslin SC, et al. Oesophagitis is as important as oesophageal stricture diameter in determining dysphagia. Gut. 1993;34:152.
292. Riley SA, Attwood SEA. Guidelines on the use of oesophageal dilatation in clinical practice. Gut. 2004;55:1.
293. Marks RD, Richter JE, Rizzo J, et al. Omeprazole vs H2RAs in treating patients with peptic stricture and esophagitis. Gastroenterology. 1994;106:907.
294. Guda NM, Vakil N. Proton pump inhibitors and the time trends for esophageal dilation. Am J Gastroenterol. 2004;99:797.
295. Sgouros SN, Vlachogiannakos J, Karamanolis G, et al. Long-term acid suppression therapy may prevent the relapse of lower esophageal (Schatzki’s) rings: A prospective, randomized, placebo-controlled study. Am J Gastroenterol. 2005;100:1929-33.